

Abstract
Idiosyncratic drug reactions are unpredictable adverse reactions. Although most such adverse reactions appear to be immune mediated, their exact mechanism(s) remain elusive. The idiosyncratic drug reaction most associated with serious consequences is idiosyncratic drug-induced liver injury (IDILI). We have developed a mouse model of amodiaquine (AQ)-induced liver injury that reflects the clinical characteristics of IDILI in humans. This was accomplished by impairing immune tolerance by using PD-1–/– mice and an antibody against CTLA-4. PD-1 and CTLA-4 are known negative regulators of lymphocyte activation, which promote immune tolerance. Immune checkpoint inhibitors have become important tools for the treatment of cancer. However, as in our model, immune checkpoint inhibitors increase the risk of IDILI with drugs that have an incidence of causing liver injury. Agents such as 1-methyl-d-tryptophan (D-1-MT), an inhibitor of the immunosuppressive indoleamine 2,3-dioxygenase (IDO) enzyme, have also been proposed as anti-cancer treatments. Another possible risk factor for the induction of an immune response is the release of danger-associated molecular patterns (DAMPs). Acetaminophen (APAP) is known to cause acute liver injury, and it is likely to cause the release of DAMPs. Therefore, either of these agents could increase the risk of IDILI, although through different mechanisms. If true, then this would have clinical implications. We found that co-treatment with D-1-MT paradoxically decreased liver injury in our model, and although APAP appeared to slightly increase AQ-induced liver injury, the difference was not significant. Such results highlight the complexity of the immune response, which makes potential interactions difficult to predict.
Introduction
Idiosyncratic drug reactions (IDRs) are adverse drug reactions that do not occur in most patients treated with a drug, and in general, do not involve the therapeutic effect of the drug. However, they can be life threatening and represent a significant source of morbidity and mortality. Their unpredictable nature also results in a significant risk to drug development. Mechanistic studies are exceedingly difficult because it is virtually impossible to perform prospective clinical studies. In addition, IDRs are also idiosyncratic in animals, which has precluded most practical animal models. Although the mechanisms of IDRs are not well understood, multiple lines of evidence indicate that most IDRs are mediated by the adaptive immune system.1 This provides one explanation for their idiosyncratic nature. The most common immune IDR leading to drug candidate failure is idiosyncratic drug-induced liver injury (IDILI). The major immune response in the liver is immune tolerance presumably because it is exposed to many “foreign” and inflammatory molecules from the intestine.2 We were able to develop an animal model of IDILI with characteristics very similar to IDILI in humans by the inhibition of immune tolerance. This was accomplished by blocking specific immune checkpoints: programmed cell death protein-1 (PD-1) and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), two molecules that inhibit T cell activation.3,4 This strategy was originally developed to promote an immune response to tumors, and it represents a major development in cancer chemotherapy. In our model, we used the combination of PD-1–/– mice and anti-CTLA-4 antibodies. In this model, amodiaquine (AQ) produces delayed-onset liver injury that is immune mediated and blocked by anti-CD8 T cell antibodies. This model also unmasks the ability of other drugs to cause liver injury, although the injury is milder with other drugs.3,5,6
Much of the idiosyncratic nature of IDILI is presumably due to interindividual differences in the immune response including human leukocyte antigen (HLA) genotypes and different T cell receptor repertoires; however, it is likely that other factors play a role. It is somewhat surprising that few genetic factors other than HLA genotypes have been associated with a clear increased risk of IDILI.7 One recent finding is an increased risk of IDILI in patients with a missense variant in PTPN22, rs2476601.8 PTPN22 encodes the protein lymphoid protein tyrosine phosphatase, which is involved in immune tolerance, and the same missense variant is associated with an increased risk of various autoimmune diseases.
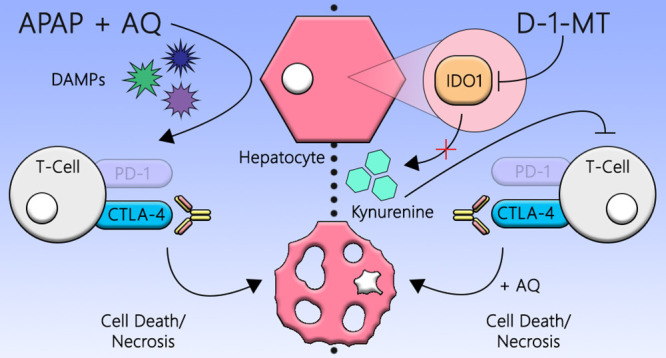
It is important to determine what risk factors make some individuals more susceptible to IDRs such as IDILI because it could improve drug safety. In the current study, we used our impaired immune tolerance model to test whether co-administration of other agents could increase the risk or severity of IDILI. One agent, 1-methyl-d-tryptophan (D-1-MT), inhibits indoleamine 2,3-dioxygenase (IDO), which is involved in immune tolerance.9 IDO is a cytoplasmic, heme-containing dioxygenase. It mediates the first and rate-limiting step in the oxidative catabolism of the essential amino acid, tryptophan, to catabolites of the kynurenine pathway.10−13 IDO has two isoforms, IDO1 and IDO2, but the former is the better-characterized isozyme. IDO is known to be involved in immunomodulation via its ability to dampen T cell responses and initiate pathways related to immune tolerance. Tryptophan deficiency and downstream kynurenine-derived analogues in the local microenvironment are hypothesized to generate immunosuppression and tolerance toward foreign antigens by blocking T cell responses and proliferation (Figure 1).14−16 Many IDO inhibitors have been proposed for the treatment of different cancers, and as such, the inhibition of IDO is a possible target for circumventing immune tolerance. Therefore, D-1-MT may increase the severity of IDILI in our impaired immune tolerance model.
Figure 1.

IDO- and cell-mediated immunosuppression. Tryptophan is an essential amino acid that is catalyzed by the IDO enzyme into kynurenine. The kynurenine pathway is immunosuppressive in nature because the catabolites have inhibitory effects on lymphocytes, and the depletion of tryptophan leads to T cell cycle arrest and reduced proliferation. PD-1 and CTLA-4 are immune checkpoint molecules expressed on T cells, which also negatively regulate T cell immunity upon recognition of their cognate ligands on antigen-presenting cells. D-1-MT is an inhibitor of the IDO enzyme.
Although the detailed steps involved in the initiation of an immune response that leads to IDILI are unknown, a prominent hypothesis is the danger hypothesis. Simply stated, if something is “foreign” but does not cause any cellular damage, then it will be ignored by the immune system. Drugs, or their reactive metabolites, have the potential to cause damage leading to the release of danger-associated molecular pattern (DAMP) molecules. DAMPs activate antigen-presenting cells, and the ability of a drug or its reactive metabolites to produce DAMPs may be one factor that determines whether it will cause IDILI. It is also possible that some other co-existing factor could cause cellular damage and increase IDILI risk. Based on the danger hypothesis, perhaps IDILI could be exacerbated by co-administered drugs that cause direct liver injury and the release of DAMPs. Acetaminophen (APAP) is one of the most widely used over-the-counter analgesics. Although it usually requires an overdose of APAP to cause liver failure, even therapeutic doses can lead to some degree of acute liver injury.17 APAP administration leads to an intrinsic form of drug-induced liver injury characterized by an immediate liver injury, which is not idiosyncratic.18 With APAP, acute liver injury occurs via the formation of its reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI). This leads to cell damage and, by extension, the release of DAMPs that may increase the risk of delayed-onset IDILI caused by drugs such as AQ. The liver injury caused by APAP can be distinguished from AQ-induced liver injury because the injury caused by AQ is delayed (Figure 2). There are also other drugs that might have similar effects as APAP and increase the risk of IDILI caused by co-administered drugs. However, the use of APAP in a wide range of clinical settings makes it a good candidate for this study. In short, the concomitant use of a drug that can cause direct liver injury may result in the release of DAMPs, which may potentiate the adaptive immune response and increase the risk of IDILI caused by co-administered drugs.
Figure 2.

APAP and liver injury. APAP is metabolized into NAPQI, which covalently binds to proteins and causes hepatocellular damage. Binding of APAP to protein has been found to correlate with APAP-induced liver injury. Damaged cells produce DAMPs, which activate antigen-presenting cells and may increase the risk of IDILI caused by co-administered drugs. Using PD-1–/– mice and anti-CTLA-4, the inhibitory signal can be blocked, leading to activation of T cells. Damaged hepatocytes can release additional DAMPs that lead to a cascade of events that may potentiate the drug-mediated liver injury.
Results
D-1-MT Decreased AQ-Induced Liver Injury in Female PD-1–/– Mice
As we had previously observed, AQ treatment led to a delayed onset increase in serum alanine transaminase (ALT) (Figure 3). Also, as we had previously observed, the increase in ALT caused by AQ was greater in the PD-1–/– mice co-treated with anti-CTLA-4 than in wild-type (WT) mice with maximal increases at the 21–35 day time points. However, addition of D-1-MT appeared to attenuate rather than accentuate this increase. Consistent with the changes in ALT and previous observations, significant hepatic necrosis was only observed in the PD-1–/– mice with the addition of anti-CTLA-4 (Figure 4), and this injury was dampened by co-administration of D-1-MT. The inflammatory infiltrates associated with the liver injury appear to be concentrated around the transitional and pericentral (zones 2 and 3) regions of the liver where CYP enzymes are heavily expressed.19
Figure 3.

D-1-MT decreases the liver injury caused by AQ in the impaired immune tolerance model but not the milder injury in wild-type mice. D-1-MT represents treatment with D-1-MT (4 mg/mL in drinking water), AQ represents treatment with AQ (0.2% w/w in the diet), and WT is short for wild-type animals. All PD-1–/– animals received weekly intraperitoneal injections of anti-CTLA-4 (300 μg/dose) along with the starting injections on days −3 and −1 prior to drug treatment with AQ and/or D-1-MT. ALT activity levels from day 21 to day 35 were significantly higher in the PD-1–/– mice treated with AQ and anti-CTLA-4 in comparison to those in which D-1-MT was added. The data represent the mean ± SEM, and statistical significance was tested using a two-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05 or **p < 0.01 between AQ + anti-CTLA-4 (PD-1–/–) and D-1-MT + AQ + anti-CTLA-4 (PD-1–/–) animals (n = 3 mice/group).
Figure 4.

Amodiaquine only caused significant histological evidence of liver injury in the impaired immune tolerance model. Treatment with D-1-MT and AQ led to a slight increase in inflammatory foci. H&E-stained histology samples of the liver (10× magnification) in wild-type animals show normal liver architecture. In both groups treated with AQ in the PD-1–/– animals, there is evidence of infiltrating lymphocytes surrounding the central vein/portal triad. The bulk of the inflammatory foci appear to be in the transitional and pericentral (zones 2 and 3) regions of the liver section.
APAP-Induced Acute Liver Injury Appeared to Increase the Subsequent AQ-Mediated Injury in PD-1–/– Mice, but the Effect was Not Significant
A preliminary study was conducted to confirm that an APAP dose of 300 mg/kg caused significant liver injury in wild-type animals (Figure 5). The first dose was administered intraperitoneally on day 0, and the second dose was given at 48 h; mice were bled for serum prior to the second injection. ALT activity levels were elevated at 24 h and decreased by day 2. The levels of ALT did not rise after the second dose of APAP; on the contrary, levels slowly returned to baseline levels.
Figure 5.

As expected, acetaminophen (300 mg/kg) caused an acute increase in serum ALT levels in wild-type female C57BL/6 mice treated with acetaminophen (300 mg/kg) at 0 and 48 h. Increases were seen on day 1 after the initial dose of acetaminophen, and this elevation in ALT levels decreased on days 2 and 3. A second dose of acetaminophen on day 2 did not produce an increase in ALT levels. The data represent the mean ± SEM, and statistical significance was tested using a one-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05 (n = 3 mice/group).
A seven-week study was subsequently conducted with the use of AQ in the diet to observe the extent of liver injury with the co-administration of APAP. APAP treatment did not accentuate the small increase in ALT caused by AQ in wild-type mice (Figure 6). Although no significant differences were detected across weeks or at any single time point, a trend toward an APAP-exacerbated increase in ALT caused by AQ in PD-1–/– mice co-treated with anti-CTLA-4 was observed. A second dose of APAP did not appear to produce any additional effect. As with the D-1-MT study, inflammatory infiltrate and hepatic necrosis were only observed in the PD-1–/– mice treated with anti-CTLA-4 and AQ in the transitional and pericentral regions of the liver (Figure 7).
Figure 6.

Treatment with intraperitoneal injections of APAP (300 mg/kg/dose) appeared to further increase serum ALT levels in PD-1–/– female C57BL/6 mice treated with amodiaquine (0.2% w/w) in the diet, but the difference was not statistically significant. “APAP only” is acetaminophen (given as two doses at 0 and 48 h) and “APAP + AQ” is acetaminophen (given as two doses at 0 and 48 h) with AQ in the diet; WT is short for wild-type mice. In the APAP-treated PD-1–/– animals, APAP was administered intraperitoneally as a single dose (at 0 h) or two doses (at 0 and 48 h) for comparison. In the PD-1–/– groups, all animals received AQ in the diet and weekly intraperitoneal injections of anti-CTLA-4 (300 μg/dose) along with the starting injections on days −3 and −1 prior to drug treatment with APAP and AQ. The data represent the mean ± SEM. No significant differences were detected in any of the groups using a two-way ANOVA with Tukey’s multiple comparisons test; n = 3 mice/group.
Figure 7.

H&E-stained histology samples of the liver (10× magnification) in wild-type and PD-1–/– animals. In both groups treated with AQ in the PD-1–/– animals, there is evidence of infiltrating lymphocytes surrounding the central vein/portal triad. Areas typical of acetaminophen-induced necrosis with variable amounts of inflammatory infiltrates and early repair in zone 3 regions were observed. No differences were seen between the single and two doses of acetaminophen; therefore, the last pane displays the results from two acetaminophen doses. The bulk of the inflammatory foci appears to be in the transitional and pericentral (zones 2 and 3) regions of the liver section.
Discussion
IDRs such as IDILI are unpredictable adverse reactions. The major genetic risk factors are HLA genotypes; however, even if a patient carries such a risk factor, it is unlikely that they will develop IDILI when treated with the associated drug. Therefore, such genetic risk factors are of limited value in preventing IDILI. There must be other risk factors, and a better understanding of such factors could be used to improve drug safety. It is known that immune checkpoint inhibitors not only unmask the ability to cause immune mediated liver injury in our model but also increase the risk of IDILI in patients being treated for cancer.20
IDO has been suggested as a target for cancer chemotherapy.21 In the present study, the addition of D-1-MT, which would be expected to further impair immune tolerance, paradoxically appeared to attenuate the liver injury in the PD-1–/– anti-CTLA-4 model with decreases in ALT levels. However, D-1-MT co-treatment did not prevent the PD-1–/–-treated animals from developing increases in ALT and inflammatory infiltrates in the liver, which appear to be concentrated around the zonal areas of the liver involved in drug metabolism. This suggests that in both studies utilizing AQ, hepatic biotransformation to the reactive imidoquinone metabolite was an important mechanism underlying its contribution to idiosyncratic hepatotoxicity involving an immune response.22 These results are consistent with more recent outcomes in the clinical development of IDO inhibitors for the treatment of cancer. A phase III trial assessing the combination of epacadostat, a small molecule inhibitor of IDO, and anti-PD-1 in patients with melanoma was halted after the combinational therapy failed to achieve its primary endpoint.23 On the other hand, a separate study reported that α-galactosylceramide-induced liver injury was exacerbated in an IDO–/– mouse.24 Furthermore, co-administration of anti-CTLA-4 and epacadostat in PD-1–/– mice was found to synergistically induce liver injury and immune cell infiltration without the use of a drug associated with IDILI.25 As the present study is exploratory in targeting different pathways in immune tolerance, it is plausible that the use of different IDO inhibitors could produce different results. Overall, the immune response has many redundant feedback mechanisms that can lead to paradoxical effects.
The APAP experiment was designed to test whether co-administration of a cytotoxic drug could increase the severity of IDILI. Most IDILI appears to involve the formation of a reactive metabolite in the liver that covalently binds to proteins. This could cause the release of DAMPs and provoke an immune response, leading to liver injury.26 APAP forms a reactive imidoquinone metabolite and causes direct liver injury; however, it strangely does not cause IDILI. In a randomized controlled trial, the treatment of healthy adults with 4 g of APAP led to elevations in serum ALT levels, which persisted in the absence of measurable APAP levels and suggests continual hepatocyte damage with inflammatory immune responses.17 PD-1–/– mice treated with an initial administration of APAP were found to have an increased trend in ALT levels at later time points, which may be indicative of an increased immune response to AQ. However, the difference was not statistically significant. It was also not clinically significant in that the injury was still not sufficient to result in liver failure. In addition, APAP did not increase the mild injury that occurs in wild-type animals, which appears to be mediated by NK cells. These results suggest that other factors that cause liver damage could increase the risk of IDILI. Hyman Zimmerman famously said that pre-existing liver disease did not increase the risk of IDILI. Inflammatory conditions such as inflammatory bowel disease also do not appear to increase the risk of IDILI. However, it is likely that the truth is very complex, and some types of liver injury may increase the risk of IDILI with some drugs. Timing of the liver injury or inflammatory condition relative to drug administration is also likely to be important. This study was designed to test the effects of injury during the period of initiation of an immune response. It is possible that administration of APAP at a later time point would have had different effects; however, the resolution phase of injury is dominated by a tolerogenic response. In addition, it would have been difficult to differentiate APAP acute toxicity from AQ-induced immune toxicity if the APAP were administered during later time points that coincide with AQ-induced liver injury. Ultimately, the major risk factor for most IDRs is probably a combination of HLA and T cell receptors with high affinities for one of the drug-modified proteins formed by the drug.
Conclusions
IDILI remains a major issue in the development of new drugs and as a source of patient morbidity/mortality. Its mechanism is not fully elucidated; however, we have developed a PD-1–/– mouse model of IDILI with the use of AQ that replicates the clinical features of mild IDILI in patients. To further our understanding of the complex interplay between the immune system and IDILI, this study investigated the use of two different compounds to modify the immune response. APAP, a widely used drug that is known to cause hepatotoxicity, was used to test if the release of DAMPs from acute liver injury would increase activation of antigen-presenting cells and synergistically increase the immune response that leads to AQ-induced liver injury. The use of D-1-MT to inhibit the production of kynurenine was used to test whether further inhibition of immune tolerance would increase AQ-induced liver injury. Results show that D-1-MT paradoxically decreased AQ-induced liver injury, whereas the co-administration of APAP and AQ led to slight increases in ALT that were statistically non-significant. In summary, the immune response is complex, and various tolerogenic mechanisms are likely in place to prevent the body from worsening its response to liver injury.
Materials and Methods
Animals
Female wild-type and PD-1–/– C57BL/6 mice, between 8 and 10 weeks of age (20–25 g), were housed in groups of three to four per experimental group. Wild-type C57BL/6 mice were purchased from Charles River Labs (Montreal, QC, Canada). PD-1–/– mice (generated by the developer, Dr. Tasuku Honjo, from Kyoto University; donated by Dr. Pamela Ohashi from the University Health Network) were bred and housed in the Division of Comparative Medicine (University of Toronto; Toronto, ON, Canada) under a 12 h lights ON/OFF cycle. Food and water were provided ad libitum. Animals were euthanized via CO2 asphyxiation at the endpoint. All animal protocols were approved by the University of Toronto Animal Care Committee and conducted in the Division of Comparative Medicine animal facility accredited by the Canadian Council on Animal Care. All procedures were in accordance with the Guide for the Humane Use and Care of Laboratory Animals.
Experimental Design
D-1-MT was obtained from Toronto Research Chemicals (Toronto, ON, Canada). It was dissolved in distilled water at a concentration of 4 mg/mL supplemented with 0.2% sucrose to increase palatability, and the pH was adjusted to 8. To avoid degradation by light, the D-1-MT solution was shielded using aluminum foil and presented ad libitum to the mice starting on day 0 of the experiments. The D-1-MT was replaced twice weekly with a freshly made solution. Groups that did not receive D-1-MT were provided with distilled water. Amodiaquine (AQ; IPCA Laboratories; Mumbai, India) was thoroughly mixed with the rodent meal (Harlan Laboratories; Indianapolis, IN, USA) at a concentration of 0.2% (w/w) using a food processor. The drug–food mix was provided in small jars ad libitum to the mice. Control mice received a regular rodent meal in the same containers. APAP was administered intraperitoneally at a dose of 300 mg/kg dissolved in warm saline solution at 0 and 48 h. PD-1–/– mice received weekly intraperitoneal injections of the anti-CTLA-4 antibody (clone 9D9 from BioXCell; West Lebanon, NH, USA) at a dose of 300 μg in phosphate buffered saline (Sigma; St. Louis, Missouri, USA) on days −3 and −1 before the start of drug treatment (i.e., day 0) and then weekly. This regimen was based on the half-life of the 9D9 antibody, which is approximately 1.5 weeks.3,27 ALT levels were determined using the Infinity ALT Liquid Stable Reagent (Thermo Scientific; Waltham, Massachusetts, USA).
Histology
The distal end of the left lateral lobe of the liver was collected at the endpoint. Identical portions of the liver and spleen samples isolated at necropsy were placed in 10% neutral buffered formalin (Sigma-Aldrich; Oakville, ON, Canada). Embedding, sectioning, staining with H&E, and scanning of the stained slides were conducted by the HistoCore (7-323) at the Princess Margaret Hospital/University Health Network and the University of Toronto (Toronto, ON, Canada).
Statistical Analysis
All data were presented as the means ± standard error of the mean (SEM). One-way or two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test was used to assess for statistical significance (*p < 0.05) using GraphPad Prism 6 (San Diego, CA, USA).
Acknowledgments
This research was funded by the Canadian Institutes of Health Research, Pfizer Canada, the Ontario Graduate Scholarship, and the Glaxo-Wellcome Drug Sunnybrook Drug Safety Clinic Graduate Student Fellowship. We thank Dr. Anthony Hayes for reviewing our histology sections.
Glossary
Abbreviations
- ALT
alanine transaminase
- APAP
acetaminophen
- ANOVA
analysis of variance
- AQ
amodiaquine
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- D-1-MT
1-methyl-d-tryptophan
- DAMP
danger-associated molecular pattern
- HLA
human leukocyte antigen
- IDILI
idiosyncratic drug-induced liver injury
- IDO
indoleamine 2,3-dioxygenase
- IDR
idiosyncratic drug reaction
- NAPQI
N-acetyl-p-benzoquinone imine (iminoquinone metabolite of acetaminophen)
- PD-1
programmed cell death 1
- SEM
standard error of the mean
- WT
wild-type
Author Contributions
J.U., T.C., and L.Y.K. designed the study; T.C. conducted the experiments; and T.C. analyzed the experimental data. J.U. and T.C. drafted the manuscript.
The authors declare no competing financial interest.
References
- Usui T.; Naisbitt D. J. Human leukocyte antigen and idiosyncratic adverse drug reactions. Drug Metab. Pharmacokinet. 2017, 32, 21–30. 10.1016/j.dmpk.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Ramadan A.; Land W. G.; Paczesny S. Danger Signals Triggering Immune Response and Inflammation. Front. Immunol. 2017, 8, 979. 10.3389/fimmu.2017.00979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metushi I. G.; Hayes M. A.; Uetrecht J. Treatment of PD-1–/– mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015, 61, 1332–1342. 10.1002/hep.27549. [DOI] [PubMed] [Google Scholar]
- Buchbinder E. I.; Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. 10.1097/COC.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak A.; Uetrecht J. The Role of CD8 T Cells in Amodiaquine-Induced Liver Injury in PD1–/– Mice Cotreated with Anti-CTLA-4. Chem. Res. Toxicol. 2015, 28, 1567–1573. 10.1021/acs.chemrestox.5b00137. [DOI] [PubMed] [Google Scholar]
- Mak A.; Uetrecht J. The Combination of Anti-CTLA-4 and PD1–/– Mice Unmasks the Potential of Isoniazid and Nevirapine To Cause Liver Injury. Chem. Res. Toxicol. 2015, 28, 2287–2291. 10.1021/acs.chemrestox.5b00305. [DOI] [PubMed] [Google Scholar]
- Daly A. K. Are Polymorphisms in Genes Relevant to Drug Disposition Predictors of Susceptibility to Drug-Induced Liver Injury?. Pharm. Res. 2017, 34, 1564–1569. 10.1007/s11095-016-2091-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli E. T.; Nicoletti P.; Abramson K.; Andrade R. J.; Bjornsson E. S.; Chalasani N.; Fontana R. J.; Hallberg P.; Li Y. J.; Lucena M. I.; Long N.; Molokhia M.; Nelson M. R.; Odin J. A.; Pirmohamed M.; Rafnar T.; Serrano J.; Stefansson K.; Stolz A.; Daly A. K.; Aithal G. P.; Watkins P. B.; A Missense Variant in PTPN22 is a Risk Factor for Drug-induced Liver Injury. Gastroenterology 2019, 156, 1707–1716.e2. 10.1053/j.gastro.2019.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassal N. K.; Hughes B. P.; Costabile M. Tryptophan Metabolism-Indoleamine 2, 3-Dioxygenase-Friend and Foe. J. Metabonomics Metab. 2012, 01, 1–3. 10.4172/2325-9736.1000e107. [DOI] [Google Scholar]
- Xu S.-q.; Wang C.-y.; Zhu X.-j.; Dong X.-y.; Shi Y.; Peng J.; Qin P.; Sun J.-z.; Guo C.; Ni H.; Hou M. Decreased indoleamine 2,3-dioxygenase expression in dendritic cells and role of indoleamine 2,3-dioxygenase-expressing dendritic cells in immune thrombocytopenia. Ann. Hematol. 2012, 91, 1623–1631. 10.1007/s00277-012-1451-0. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Zhang G.-X.; Gran B.; Fallarino F.; Yu S.; Li H.; Cullimore M. L.; Rostami A.; Xu H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J. Immunol. 2010, 185, 5953–5961. 10.4049/jimmunol.1001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338, 12–19. 10.1016/j.bbrc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Ball H. J.; Yuasa H. J.; Austin C. J. D.; Weiser S.; Hunt N. H. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int. J. Biochem. Cell Biol. 2009, 41, 467–471. 10.1016/j.biocel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Laviano A.; Meguid M. M.; Cascino A.; Molfino A.; Fanelli F. R. Tryptophan in wasting diseases: at the crossing between immune function and behaviour. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 392–397. 10.1097/MCO.0b013e32832b73af. [DOI] [PubMed] [Google Scholar]
- Terness P.; Bauer T. M.; Röse L.; Dufter C.; Watzlik A.; Simon H.; Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins P. B.; Kaplowitz N.; Slattery J. T.; Colonese C. R.; Colucci S. V.; Stewart P. W.; Harris S. C. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. Jama 2006, 296, 87–93. 10.1001/jama.296.1.87. [DOI] [PubMed] [Google Scholar]
- Yokoi T.; Oda S. Models of Idiosyncratic Drug-Induced Liver Injury. Annu. Rev. Pharmacol. Toxicol. 2020, 61, 247–268. 10.1146/annurev-pharmtox-030220-015007. [DOI] [PubMed] [Google Scholar]
- Oinonen T.; Lindros K. O. Zonation of hepatic cytochrome P-450 expression and regulation. Biochem. J. 1998, 329, 17–35. 10.1042/bj3290017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzman D. L.; Pelosof L.; Rosenberg A.; Avigan M. I. Hepatotoxicity of immune checkpoint inhibitors: An evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int. 2018, 38, 976–987. 10.1111/liv.13746. [DOI] [PubMed] [Google Scholar]
- Quan J.; Tan P. H.; MacDonald A.; Friend P. J. Manipulation of indoleamine 2,3-dioxygenase (IDO) for clinical transplantation: promises and challenges. Expert Opin. Biol. Ther. 2008, 8, 1705–1719. 10.1517/14712598.8.11.1705. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Vermeulen N. P. E.; Commandeur J. N. M. Characterization of human cytochrome P450 mediated bioactivation of amodiaquine and its major metabolite N-desethylamodiaquine. Br. J. Clin. Pharmacol. 2017, 83, 572–583. 10.1111/bcp.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long G. V.; Dummer R.; Hamid O.; Gajewski T. F.; Caglevic C.; Dalle S.; Arance A.; Carlino M. S.; Grob J. J.; Kim T. M.; Demidov L.; Robert C.; Larkin J.; Anderson J. R.; Maleski J.; Jones M.; Diede S. J.; Mitchell T. C. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- Ito H.; Hoshi M.; Ohtaki H.; Taguchi A.; Ando K.; Ishikawa T.; Osawa Y.; Hara A.; Moriwaki H.; Saito K.; Seishima M. Ability of IDO to attenuate liver injury in alpha-galactosylceramide-induced hepatitis model. J. Immunol. 2010, 185, 4554–4560. 10.4049/jimmunol.0904173. [DOI] [PubMed] [Google Scholar]
- Affolter T.; Llewellyn H. P.; Bartlett D. W.; Zong Q.; Xia S.; Torti V.; Ji C. Inhibition of immune checkpoints PD-1, CTLA-4, and IDO1 coordinately induces immune-mediated liver injury in mice. PLoS One 2019, 14, e0217276 10.1371/journal.pone.0217276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Murphy B. V.; Holt M. P.; Ju C. The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett. 2010, 192, 387–394. 10.1016/j.toxlet.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funt S. A.; Page D. B.; Wolchok J. D.; Postow M. A. CTLA-4 antibodies: new directions, new combinations. Oncology 2014, 28, 6–14. [PubMed] [Google Scholar]