

Abstract
The ability to track extracellular vesicles (EVs) in vivo without influencing their biodistribution is a key requirement for their successful development as drug delivery vehicles and therapeutic agents. Here, we evaluated the effect of five different optical and nuclear tracers on the in vivo biodistribution of EVs. Expi293F EVs were labeled using either a noncovalent fluorescent dye DiR, or covalent modification with 111indium-DTPA, or bioengineered with fluorescent (mCherry) or bioluminescent (Firefly and NanoLuc luciferase) proteins fused to the EV marker, CD63. To focus specifically on the effect of the tracer, we compared EVs derived from the same cell source and administered systemically by the same route and at equal dose into tumor-bearing BALB/c mice. 111Indium and DiR were the most sensitive tracers for in vivo imaging of EVs, providing the most accurate quantification of vesicle biodistribution by ex vivo imaging of organs and analysis of tissue lysates. Specifically, NanoLuc fused to CD63 altered EV distribution, resulting in high accumulation in the lungs, demonstrating that genetic modification of EVs for tracking purposes may compromise their physiological biodistribution. Blood kinetic analysis revealed that EVs are rapidly cleared from the circulation with a half-life below 10 min. Our study demonstrates that radioactivity is the most accurate EV tracking approach for a complete quantitative biodistribution study including pharmacokinetic profiling. In conclusion, we provide a comprehensive comparison of fluorescent, bioluminescent, and radioactivity approaches, including dual labeling of EVs, to enable accurate spatiotemporal resolution of EV trafficking in mice, an essential step in developing EV therapeutics.
Keywords: exosomes, extracellular vesicles, vesicle tracers, biodistribution, delivery, optical imaging, nuclear imaging
Introduction
Extracellular vesicles (EVs) are cell-derived nanoparticles secreted into the extracellular environment by nearly all cell types.1 Cells release EVs of different sizes and subcellular origins, including exosomes, which are relatively small (∼30–200 nm) and derived from endosomes.1,2 EVs are important mediators of physiological and pathological processes as they can carry and transfer a wide range of biomolecules such as lipids, proteins, and nucleic acids to regulate the functions and properties of recipient cells.2,3 These features, and their low toxicity and immunogenicity in vivo,4,5 make EVs promising candidates for delivery of multiple types of therapeutic agents. Nucleic acids including siRNA,6,7 microRNA,8 mRNA,9 CRISPR/Cas9,10 and small chemotherapeutic drugs such as paclitaxel11 and doxorubicin12 have been successfully loaded into EVs and delivered to target cells. Additionally, EVs can be engineered for targeting specific cell types by either the display of targeting ligands at their surface, such as a fusion with platelet-derived growth factor receptor,13 lactadherin,14 and lysosomal associated membrane protein 2b15 or by decorating the purified vesicles with targeting molecules including nanobodies,16 anchor peptides,17 and aptamers.18 While promising, these approaches depend on accurate prediction, monitoring, and control of EV biodistribution in vivo, which have not yet been achieved.
Monitoring the traffic of exogenous EVs in vivo remains extremely challenging due to their complex composition, small size, and short half-life. Indeed, there are only a few examples of optical, nuclear, and magnetic resonance imaging being explored in vivo, as recently reviewed.19 For in vivo tracking of EVs, several different approaches have been tested, including directly labeling EVs with lipophilic fluorescent dyes, such as PKH26/67,20 DiD,21 and DiR,22,23 or radioisotopes, such as 99mTc-HMPAO24 and [111indium] ([111In]),25 or using more complex cell-engineering approaches involving encoding luminescent proteins in EVs such as Renilla(26) or Gaussia luciferases.20,27,28 However, no study has yet compared the effect of different approaches directly. Notably, several studies exploring the biodistribution of exogenous EVs tagged using a single approach conclude that their retention is particularly robust in organs with high blood perfusion and well-developed phagocytic systems, such as liver, spleen, kidney, and lung.19,29 Therefore, these organ sites represent an attractive reference point to help compare the effect of different EV tagging and bioimaging technologies on quantitative detection in vivo.
Given that all EV tracers are biologically inert, the choice of tracer can be largely guided by instrumental and technical parameters. However, in this study, we question this assumption and show that the choice of the EV tag can dramatically influence the selection of the right imaging application to accurately reflect EV localization in vivo and ultimately may also contribute to changes in the physiological biodistribution of EVs. Thus, we evaluated different methodologies to analyze the in vivo/ex vivo biodistribution, blood kinetics, and renal clearance of systemically administered engineered and naïve EVs in mice. For that, we performed a side-by-side comparison of three different bioimaging modalities using the fluorescent tracers DiR dye and mCherry fluorescent protein, the bioluminescent tracers Firefly (Fluc) and NanoLuc (Nluc) luciferases, as well as nuclear imaging using the [111In] radioisotope. Our findings offer insights into the impact of the labeling technique on the intrinsic properties of the EVs, which are highly relevant to further understand their potential targeting ability, toxicity, and therapeutic dose.
Results and Discussion
Generation of Reporter EVs by Membrane Labeling and Cell-Engineering Methods
Isolation and Characterization of Naïve and Engineered Expi293F EVs
For the in vivo comparative biodistribution study, we used Expi293F cells to generate small EVs with exosome-like features including size, morphology, and protein markers. Diverse approaches were selected to label the EVs: membrane or surface labeling of naïve vesicles using the lipophilic dye XenoLight DiR or the radionuclide [111In], and genetic modification of the donor cells to introduce fusions of the EV marker protein CD63 with fluorescent mCherry or bioluminescent Fluc and Nluc proteins for expression in the vesicle lumen (Figure 1A). Differential centrifugation coupled with high-resolution density separation resulted in nine fractions (F1–F9) containing different EV subsets (Figure 1A and Supplementary Figure S1A). Analysis of the presence of canonical EV proteins and nonvesicular protein markers in all fractions across gradients confirmed the separation of low-density vesicles from higher-density materials and protein aggregates (Figure 1B). The low-density fractions (F1–F3) from naïve EV gradients were characterized by the absence of the nuclear protein Lamin B1 and the presence of the EV-specific markers Alix, Flotillin-1, CD63, CD81, and CD9 (Figure 1B). The overexpression of CD63 in the engineered EVs as a fusion protein with mCherry, Fluc, and Nluc led to an increased signal for CD63 and CD9 and a significant reduction of the levels of CD81 in F1–F3 compared to naïve EVs. Low-density fractions measured by NanoSight tracking analysis (NTA) showed a similar narrow particle size distribution centered around 126–154 nm for all EVs (Supplementary Figure S1B), demonstrating that the introduction of reporter proteins in the lumen of the vesicles by genetic engineering does not affect the particle size. Notably, we observed a significant increase, up to 10-fold, in yield of purified EVs upon the overexpression of CD63 fusion proteins compared to that from unmodified cells, when a similar number of cells and identical volume of cell-conditioned media was used for EV production (Figure 1C). The presence of Alix and CD9 proteins and the increased particle count upon CD63 overexpression in F7 and F8 could indicate nonvesicular high-density protein material or EV aggregation, as shown by transmission electron microscopy (TEM) analysis of high-density fractions (Supplementary Figure S1C). Other published studies also showed that CD9 and Alix were not exclusively present in fractions of densities classically described for EVs of endosomal origin but also appeared in nonexosomal density fractions, indicating a heterogeneous vesicle subset whose biogenesis is not fully understood.30,31 It could also indicate that the genetic modification of cells changes the biology of the EVs, and that changes in the protein composition of vesicles after CD63 overexpression, including CD81 reduction and CD9 increase, are possibly the result of a shared mechanism of EV biogenesis that operates both at the plasma membrane and endosome membrane level. Negative staining TEM of low-density fractions from all gradients revealed highly pure and clean small EV preparations with abundant material of preserved shape, small size (<100 nm), and round cup-shaped morphology typically observed by TEM (Figure 1D). No morphological differences were observed between naïve and engineered EVs; however, mCherry EVs had a diameter significantly smaller than that of the rest of the particles when EM images were analyzed (Supplementary Figure S1D). Overall, these results indicate that small EVs are enriched in low-density fractions (F1–F3) across gradients, and therefore, those fractions were pooled and used for the comparative in vivo biodistribution studies.
Figure 1.

Generation and characterization of membrane-labeled and engineered EVs. (A) Schematic illustrations representing the EV production process. Expi293F cells were either used unmodified to produce naïve EVs or were transiently transfected with plasmids coding for mCherry, Firefly luciferase (Fluc), and NanoLuc luciferase (Nluc) fused to the C-terminal of human CD63. Cell supernatant from naïve or engineered cells was collected 48 h post-transfection and subjected to differential centrifugation for the isolation of small EVs (also called exosomes). Small EVs (100,000g pellets) were subsequently bottom-loaded in high-resolution iodixanol density gradients (Optiprep) with decreasing densities (50–10%, bottom to top). Nine fractions of 1 or 2 mL each were collected from top to bottom and analyzed. Naïve EVs were membrane-labeled with XenoLight DiR (lipophilic dye) or 111indium [111In]-DTPA (chemical labeling) or genetically modified to carry mCherry, Fluc, or Nluc proteins. PDB ID codes (2H5Q, 1LCI, and 5IBO) were used for illustrations of the protein structures. (B) Western blot analysis of the density fractions (F1–F9) (12 μL/each). Membranes were blotted with the following antibodies: Lamin B1, Alix, Flotillin, CD63, CD81, CD9. Low-density fractions (F1–F3) are represented with a box. (C) Representative nanoparticle tracking analysis graphs of EV concentration as the total number of particles per milliliter in each fraction (F1–F9). Bars represent the mean ± standard error of mean. (D) Representative negative staining transmission electron microscopy and zoomed-in images of low-density EVs (F1–F3). Five microliters was loaded to the grids. Scale bars are 200 nm in the wide-field images and 100 nm in the magnifications.
Labeling Efficiency and In Vitro Detection of EVs
We first examined the labeling efficiency and stability of the EVs for the various tagging approaches. Labeling efficiency for fluorescent and bioluminescent tracers was defined as the average number of tracer molecules per EV particle. Due to the decaying nature of radioisotopes, radiolabeling efficiency of EVs was defined as the fraction of radioisotope (in terms of radioactivity in MBq) bound to the bulk EVs and relative to the initial activity of the radioisotope used for labeling. The labeling stability assessed in this work refers to the fraction of the tracer that remained undegraded and associated with the EVs. Fluorescent and luminescent plate readouts of EVs were performed to determine their optical detection threshold. The estimated labeling efficiency of EVs with DiR dye or mCherry protein based on the number of fluorescent molecules per vesicle showed that EVs had on average ∼6.745 molecules of DiR dye and ∼314 molecules of mCherry protein per particle (Supplementary Figure S2A). Analysis of the optical fluorescence of mCherry- and DiR-labeled EVs at 10 and 50% of the in vivo dose confirmed good sensitivity and labeling efficiencies as well as bright in vitro fluorescent signals for both EV types (Supplementary Figure S2B). Western blot analysis of EVs detected mCherry protein at the highest levels in F1 and F2, where small EVs were found to be enriched (Supplementary Figure S2C). The stability of the fluorescently labeled particles was also evaluated by incubation with phosphate-buffered saline (PBS) or fetal bovine serum (FBS) at 37 °C for 24 h. The results show that while mCherry tracer was stable within the vesicles both in PBS and serum, the stability of DiR-labeled EVs was lower in PBS compared to that in serum (Supplementary Figure S2D). Interestingly, the introduction of serum influenced the total fluorescence intensity of the solution with increased background compared to that of fresh EV samples. Both mCherry and DiR EVs were internalized by HepG2 cells with a rapid increase in the percentage of DiR and mCherry positive cells up to 12 h after stimulation (Supplementary Figure S2E–G). These results suggest that EVs were internalized and accumulated intracellularly with time and that neither DiR incorporation into the EV membrane nor EV membrane modification with CD63–mCherry fusion protein disrupted vesicle–cell interactions. When analyzing the bioluminescent properties of the EVs in a cell-free assay to determine the in vivo detection threshold, we observed that Nluc EVs generated a very bright and 106-fold higher signal intensity per particle compared to that of Fluc EVs after addition of their corresponding substrates (Supplementary Figure S3A). Weak Fluc signal intensities were detected in vitro even with the highest EV concentration analyzed (1011 particles), corresponding to 100% of the dose tested in vivo. In contrast, the Nluc system offered a highly sensitive readout for in vitro detection of EVs, detecting down to 105–106 total Nluc EVs, which corresponds to ∼105-fold lower dose than that assayed in vivo. Western blotting confirmed the high-intensity signals of Nluc EVs compared to that of Fluc EVs are due to the higher levels of CD63 fusion protein (Supplementary Figure S3B). The labeling efficiency of Nluc EVs from low-density fractions was estimated to be ∼304 molecules per particle based on the semiquantitative analysis of CD63 and Nluc immunoblots (Supplementary Figure S3C). The enzymatic activity of Nluc EVs after incubation in serum for 24 h was around 60% of the initial activity of an equal number of fresh Nluc EVs. These results suggest that Nluc particles are stable in serum up to 24 h (Supplementary Figure S3D). The bioluminescent signal of HepG2 cells exposed to Fluc EVs was similar to that of control cells incubated with nonlabeled EVs (Supplementary Figure S3E). This is attributed to the low labeling efficiency of Fluc EVs, as shown earlier rather than the EVs not being internalized by the cells, thus impairing a reliable detection of Fluc EVs in vitro. Based on the low sensitivity of detection of Fluc EVs in vitro, these vesicles were deemed unsuitable for in vivo biodistribution studies. On the contrary, the uptake of Nluc EVs by HepG2 cells was detectable with a high luminescent signal (Supplementary Figure S3E). Interestingly, the signal intensity decreased with time after EV stimulation, suggesting EV processing and enzymatic degradation by the recipient cells.
Next, we examined the radiolabeling efficiency and radiochemical stability of the [In111]-DTPA EVs. Membrane radiolabeling was performed by covalent attachment of the bifunctional chelator DTPA-anhydride to the EV surface in an amine-dependent reaction followed by the chelation of 111In3+ by the DTPA on the surface of the EVs. This method resulted in an EV radiolabeling efficiency of 73.5 ± 4.7% (Supplementary Figure S3F), significantly higher than that we previously reported for melanoma EVs,25 and a high EV radiochemical stability of 82.1 ± 6.3% in 50% serum at 24 h (Supplementary Figure S3G). Interestingly, DiR, Nluc, and [111In]-DTPA EVs showed lower stability in PBS compared to that in 50% serum, and these results are consistent with those reported for other types of synthetic nanoparticles.32,33 It is postulated that the formation of protein corona on the surface of EVs and synthetic nanoparticles in serum helps maintain their integrity, thus conferring protection from dissociation of the tracer for labeled EVs or enzyme degradation in the case of Nluc EVs in vivo. Taken together, the characterization of the tagged EVs demonstrates practical in vitro detection thresholds for DiR, mCherry, [111In], and Nluc tracers, which were carried forward for the subsequent in vivo studies.
In Vivo Biodistribution of EVs by Fluorescence Imaging
We first explored the effect of EVs carrying mCherry red fluorescent protein as a tracer for biodistribution studies in mice. mCherry is a robustly fluorescent protein widely used as a vesicle tracer for in vitro studies34−36 and has also been explored for live tracking of endogenous EVs and interorgan communication studies in zebrafish.37 Murine colorectal CT26 tumor-bearing syngeneic mice were used as a model of a highly vascularized solid tumor38 to identify the most suitable and sensitive imaging technique to track low levels of EV accumulation into tumors. Tumor-bearing BALB/c mice were intravenously (i.v.) injected with 1011 mCherry EVs, and their distribution was real-time visualized with a noninvasive small animal in vivo imaging system 24 h postadministration. Fluorescent signals were detected throughout the animal with higher levels originating from the abdominal and thoracic regions in both ventral and dorsal positions for EV-treated as well as PBS control animals (Figure 2A). Ex vivo imaging of the excised major organs confirmed the fluorescence signal in the stomach and intestines (Figure 2B). High levels of background autofluorescence of mammalian tissues within the wavelength range required for mCherry imaging limited the detection sensitivity of the EVs. The change to a low-chlorophyll diet before imaging to improve the sensitivity resulted in a small reduction of background fluorescence (Supplementary Figure S4A). However, the mCherry signals were not sufficiently bright for real-time in vivo and ex vivo detection of EVs. The results were validated by image analysis of excised organs (Figure 2C) and quantitative fluorescent analysis of tissue lysates (Figure 2D and Supplementary Figure S4B), whereby stomach and intestine recorded the highest signals with similar radiance in both control and treated mice. Notably, negligible signals were detected in the liver and spleen where EVs have been shown to accumulate due to the high blood perfusion and phagocytic systems.19,29 Thus, the value of the mCherry red fluorescent tracer for robust EV tracking in vivo is limited by a poor signal-to-noise ratio and the effect on the physiological biodistribution of EVs.
Figure 2.

In vivo tracking of mCherry- and XenoLight DiR-labeled Expi293F EVs in mice. CT26 tumor-bearing BALB/c mice were intravenously injected with 1011 mCherry EVs or DiR-labeled EVs or PBS via the tail vein. In vivo and ex vivo imaging analyses and tissue quantifications were performed at 24 h postadministration. (A) Representative in vivo ventral and dorsal images of PBS-treated or mCherry EV-treated mice. (B) Following in vivo imaging of PBS-treated or mCherry EV-treated mice, whole major organs (brain, heart, lungs, liver, spleen, kidneys, pancreas, stomach, intestine, and tumors) were excised and imaged ex vivo. Representative ex vivo images are shown. Organs are annotated on the left side of the panel. Tumors: right (R), left (L). (C) Semiquantitative analysis of the ex vivo imaging data of organs from PBS-treated (white) and EV-treated (black) animals. Data were analyzed using the Living Image 4.7.2 software. Individual regions of interest (ROIs) were drawn for each organ to obtain their respective fluorescence signals. Fluorescent signal is represented as total radiant efficiency [p/s]/[μW/cm2] per grams of tissue (gT); n = 3 for all groups. (D) Quantitative organ biodistribution profile from tissue lysates of mice treated with PBS or mCherry EVs. Organs were homogenized using a lysis buffer and cleared of tissue debris before mCherry fluorescence detection using the IVIS Lumina III system. Relative fluorescence signals (RFU) are expressed per gT. All values are represented as mean ± standard error of mean; n = 3 for all groups. (E) Representative real-time in vivo ventral and dorsal images of mice treated with DiR-labeled EVs or PBS. (F) Major organs were excised and imaged ex vivo. Representative ex vivo images of whole organs are shown. Organs are annotated on the left side of the panel. Tumors: right (R), left (L). (G) Semiquantitative analysis of the organ biodistribution profile from ex vivo imaging of DiR EV-treated mice. Individual ROIs were drawn for each organ to obtain their respective DiR fluorescence signals. Background signals from the PBS-treated mice were subtracted from the data. Fluorescent signal is represented as total radiant efficiency [p/s]/[μW/cm2] per gT. Data were analyzed using the Living Image 4.7.2 software. Values are expressed as mean ± standard error of mean; n = 3 for all groups. (H) Quantitative organ biodistribution profile from tissue lysates of mice treated with DiR-labeled EVs. Organs were homogenized and analyzed as described above. RFU signals are expressed per gT after background tissue subtraction of PBS-treated animals. Values are expressed as mean ± standard error of mean; n = 3 for all groups.
Fluorescent lipophilic near-infrared dye DiR could be regarded as an acceptable alternative for mCherry tagging of exogenous EVs. To explore the performance of the fluorescent dye DiR as an exogenous EV tracer in vivo, DiR-labeled EVs were administered i.v. to tumor-bearing BALB/c mice at 1011 vesicles (identical to the mCherry EV dose) and imaged at 24 h postinjection. Noninvasive real-time live imaging of the EV-treated animals detected DiR signals exclusively in the upper abdominal section, corresponding to the location of the liver and the spleen, with undetectable levels in other regions including the tumors (Figure 2E). No background fluorescence was detected in the PBS control mice, confirming that the signals in the treated animals were originating from DiR-labeled EVs, as also reported by others.17 In contrast to mCherry, DiR allowed noninvasive detection of the vesicles in vivo with better sensitivity, signal-to-noise ratio, and no background fluorescence at expected tissue locations. To ascertain the accuracy of this detection, the animals were sacrificed, and major organs were excised and imaged ex vivo. Strong DiR signals were observed in the liver and spleen, correlating with the in vivo imaging data but with higher fluorescent intensity. A weak signal was observed in the lungs, and no further signals were detected in any other tissue including the tumors (Figure 2F). Semiquantitative analysis from ex vivo imaging of organs (normalized to organ weights) (Figure 2G) and quantitative analysis of tissue homogenates (Figure 2H) supported the qualitative in vivo observations, whereby liver and spleen recorded the highest DiR signal, with low signals in the lungs and negligible signals in other organs (Figure 2F). We observed a ∼2-fold reduction in the fluorescence of the tissue lysates compared to that of full organs, which could be attributed to the decrease in DiR signals following the freeze–thaw process to produce the lysates. The relative fluorescence units per organ are shown in Supplementary Figure S5. In summary, our results indicate that DiR, unlike mCherry protein or other non-near-infrared probes previously used for EV tracking, is an effective EV tracer (in the fluorescent tracers category) for both in vivo and ex vivo tracking of EVs. We show that DiR provides improved sensitivity and low levels of autofluorescence in the infrared range. Our findings also show that following systemic administration of DiR-labeled EVs, fluorescent signals tend to accumulate mostly in the liver, followed by the spleen, and to a lesser extent the lungs and kidney with consistent results across in vivo and ex vivo imaging and tissue quantifications techniques.
In Vivo Biodistribution of [111In]-DTPA EVs by Nuclear Imaging
To overcome limitations of sensitivity and tissue penetration depth associated with in vivo detection of fluorescent tracers, we evaluated the biodistribution of radiolabeled EVs by nuclear imaging using single-photon emission computed tomography (SPECT) coupled with computed tomography (CT) for anatomical information on EV localization. We selected SPECT/CT as it enables acquisition of both anatomic and functional information that is very accurately fused in a single examination. Radiolabeled EVs ([111In]-DTPA EVs) at a dose of 1011 vesicles were i.v. administered to BALB/c tumor-bearing mice. The animals were imaged real-time at 0.5–1, 4, and 24 h post-EV administration. Whole-body in vivo live imaging revealed a rapid accumulation of [111In]-DTPA EVs in the peri-abdominal area including the liver, spleen, and kidneys (Figure 3A). The accumulation in those organs was retained at later time points of 4 and 24 h. Given the high serum stability of [111In]-DTPA EVs (Supplementary Figure S3G), the weaker signals observed in the abdominal region at 24 h were due to the decay of 111In rather than its clearance. The direct and specific real-time monitoring of radiolabeled EVs demonstrates the superb sensitivity and depth penetration of SPECT/CT imaging for in vivo tracking. To determine the accuracy of the live imaging results in a quantitative manner, major organs were excised and subjected to ex vivo gamma counting. The liver recorded the highest signals, which reached ∼45% of the injected dose (ID) per gram of tissue (gT) at 1 and 4 h, with a significant increase to ∼78% ID/gT at 24 h (Figure 3B). Spleen recorded the second-highest accumulation with ∼43% ID/gT at 1 h, which decreased to ∼33% and finally ∼25% ID/gT at 4 and 24 h, respectively. Notably, unlike with the DiR EVs, kidneys recorded the third-highest signal with ∼7% ID/gT at 1 and 4 h, which then increased to ∼12% ID/gT at 24 h. Similar EV biodistribution profiles have been previously reported by our group and others with EVs entrapping radioisotopes such as 99mTc-HMPAO in their lumen24 or by displaying [111In] on their surface.25 Of note, in contrast to optical imaging methods, the high specificity and sensitivity of the radioactive signal permitted an accurate ex vivo quantification of EV accumulation in the different organs without the need for further tissue homogenization. In addition, the high sensitivity and unlimited tissue depth penetration of nuclear imaging also enabled the detection of tumor accumulation of [111In]-DTPA EVs, which was initially ∼1% ID/gT at 1 h, increasing to ∼2% ID/gT at 24 h. Given the high microvessel density and vessel pore cutoff of subcutaneous CT26 tumors,38 it is likely that the gradual increase in tumor accumulation of [111In]-DTPA EVs over time was due to the enhanced permeability and retention effect.39 The % ID of [111In]-DTPA EV accumulation per organ is shown in Supplementary Figure S6A. We also evaluated whether the overall biodistribution of radiolabeled EVs in tumor-bearing BALB/c mice significantly differed from that of wild-type BALB/c animals (without tumors). Analysis at 24 h post-EV administration revealed a similar EV biodistribution in wild-type and tumor-bearing mice (Supplementary Figure S6B). The highest signals were detected in the liver with ∼54% ID/gT, followed by the spleen with ∼48% ID/gT, and to a lesser extent the kidneys with ∼17% ID/gT. These results indicate that the presence of xenograft tumors does not substantially change the overall organ distribution of exogenously administered EVs.
Figure 3.

In vivo tracking of radiolabeled 111indium-DTPA Expi293F EVs. Membrane-radiolabeled 111indium [111In]-DTPA Expi293F EVs were intravenously administered into subcutaneous CT26 tumor-bearing BALB/c mice at a dose of 1011 vesicles per animal. Mice were imaged by single-photon emission computed tomography (SPECT) coupled with computed tomography (CT) for anatomical information. (A) Whole-body SPECT/CT live imaging of [111In]-DTPA EVs. Representative whole-body ventral view images of all time points are shown. Imaging was performed at 30 min, 4 h, and 24 h post-EV injection. Representative images of sagittal, coronal, and transverse views of EV-treated animals at 24 h are shown. Scale bar represents low (black) to high (yellow) signals. (B) Ex vivo quantification of organ biodistribution of [111In]-DTPA EVs by gamma counting. Animals were culled at 1 h, 4 and 24 h post-EV injection, perfused with saline, and whole organs were excised for quantitative analysis. Inset shows the tumor accumulation values over time. Values were normalized to the grams of tissue and expressed as mean ± standard error of mean, where n = 3 for each group. Two-way ANOVA with Tukey’ multiple comparison test;****p value <0.0001, *p value <0.05.
In Vivo Tracking and Biodistribution of EVs by Bioluminescence Imaging
Practicalities of isotopic tracers often complicate their use in preclinical studies, a limitation that could be overcome by tagging EVs with luciferases. Moreover, unlike fluorescent imaging in the red part of the spectrum, bioluminescent imaging offers very low background levels with enzymatic amplification of signal and enhanced sensitivity for detection.40 To assess the properties of this bioimaging technology, we evaluated Nluc as a bioluminescent tracer for in vivo monitoring of EVs. Nluc was selected since it has smaller size, enhanced stability, and provides >150-fold increase in luminescence over more traditional systems.41,42 Nluc EVs were systemically administered at 1011 vesicles into tumor-bearing mice, and in vivo and ex vivo imaging was performed at 1, 4, and 24 h post-EV administration. Noninvasive real-time live imaging exclusively recorded weak signals in the area corresponding to the spleen at 1 and 4 h (Figure 4A). Thus, Nluc imaging of deep organs in living mice, including the liver, spleen, and lungs, is challenging as the short wavelength of the blue-shifted Nluc does not readily penetrate mammalian tissues.42 Moreover, the low signals detected could be attributed to the restricted diffusion of the Nluc substrate furimazine across intact cell membranes in the body and the excretion of the substrate from living mice. However, ex vivo imaging of excised major organs showed high bioluminescence and a somewhat different biodistribution profile of Nluc EVs compared to that of DiR EVs and [111In]-DTPA EVs (Figure 4B). The highest bioluminescence was detected in the lungs at all time points, with a signal increase from 1 to 4 h and then decrease at 24 h (Figure 4B). The next strongest bioluminescence was detected in the spleen, followed by reduction of the signal at 24 h. Much weaker bioluminescence was also detected in the liver at 1 h postinjection, with no detectable levels in other organs including the tumors. The accuracy of the results was confirmed with semiquantitative analyses of the ex vivo images of whole organs (Figure 4C) and with quantitative analyses of tissue lysates (Figure 4D). An increase in the detection sensitivity of Nluc in the tissue homogenates was observed, which is possibly due to the more efficient penetration of furimazine into the tissues. The lysate analysis on the percentage of ID per individual organ showed that EVs tend to accumulate in the liver, followed by lungs, and spleen (Supplementary Figure S7). When analyzing the values of ID per milligram of tissue, lungs recorded the highest Nluc EV signals increasing from ∼20 to ∼77% ID/gT from 1 to 4 h and then decreasing to ∼34% ID/gT at 24 h (Figure 4D). The spleen was the second highest with ∼27% ID/gT at 1 h, which steadily decreased to ∼16 and ∼8% ID/gT at 4 and 24 h, while only minimal Nluc EV accumulation was observed in the liver. Tumors showed minimal bioluminescent signals with only ∼0.6% ID/gT, which remained constant between 4 and 24 h. Accumulation of EVs in the lung could result from the presence of aggregates in the sample that are then trapped in the microvascular blood vessels in the lung. However, we did not observe any signs of aggregation or changes in the size distribution of our EV samples before i.v. administration (Supplementary Figure S1B). A similar pattern of prominent lung accumulation of EVs has been previously reported using Gaussia luciferase as a fusion construct with lactadherin20,43 or platelet-derived growth factor receptor.27 This is likely to be a consequence of the alteration of the repertoire of EV surface proteins, including tetraspanins, following the exogenous high-level expression of the fusion proteins (Figure 1B). The variation of the EV surface protein composition and possibly the glycosylation pattern has been shown to modulate the in vivo biodistribution of EVs and uptake to various cells44,45 and could also be the result of high EV signal in the lungs. In our study, Nluc luciferase-tagged EVs yielded very strong signal intensities ex vivo. However, due to the tissue attenuation of the bioluminescent signal, in vivo noninvasive real-time monitoring of the EVs in deep tissues using Nluc remained challenging. It is also interesting to note that the tumor accumulation of Nluc EVs did not show a steady increase over time as observed with [111In]-DTPA EVs, suggesting the lower detection sensitivity of luminescence as compared to radioactivity.
Figure 4.

In vivo tracking of NanoLuc Expi293F EVs. Engineered CD63-NanoLuc (Nluc) Expi293F EVs were intravenously administered into subcutaneous CT26 tumor-bearing BALB/c mice at a dose of 1011 vesicles per mice via the tail vein. (A) Representative real-time in vivo live imaging of Nluc EVs. The substrate furimazine was injected intravenously at 1, 4, and 24 h post-EV administration, and the mice were imaged within 2 min of substrate administration. (B) Following in vivo imaging, animals were sacrificed, perfused with saline, and major organs (brain, heart, lungs, liver, spleen, kidneys, pancreas, stomach, intestine, and tumors) were excised, immersed in furimazine for 30 s, blotted on tissue paper, and imaged within 2 min. A representative panel of ex vivo imaging of organs is shown. Organs are annotated on the left side of the panel. (C) Semiquantitative analysis of Nluc EVs from ex vivo images of whole organs analyzed using the Living Image 4.7.2 software. Values are normalized to organ weight as total flux per gram of tissue (gT). (D) Quantitative analysis of Nluc EV signals from tissue lysates. Organs were homogenized and cleared of tissue debris before bioluminescence quantification as above. Values are normalized to organ weight and expressed as the percentage of injected dose (ID) per gT. Inset shows the tumor accumulation values of Nluc EVs. For all graphs, values are expressed as mean ± standard error of mean, where n = 3 for each group. Two-way ANOVA with Tukey’s multiple comparison test; ***p value <0.001, **p value <0.001.
To confirm whether the different biodistribution pattern of CD63-Nluc EVs was the result of changes in their protein composition and tropism due to the engineering process and to exclude the possibility that the labeling technique accounts for the observed characteristics, CD63-Nluc engineered EVs were labeled with DiR fluorescent dye and their in vivo biodistribution was dually tracked using both fluorescent and bioluminescent approaches (Supplementary Figure S8). The DiR-labeled Nluc EVs at a dose of 1011 particles were i.v. injected into CT26 tumor-bearing BALB/c mice, and their biodistribution was analyzed at 1, 4, and 24 h post-EV administration. Ex vivo quantification analysis of DiR-labeled Nluc EVs revealed that their in vivo biodistribution corresponded to that observed by single tracking of Nluc EVs or DiR EVs using either bioluminescence or fluorescence approaches, respectively (Supplementary Figure S8, Figure 2G, and Figure 4C). Surprisingly, fluorescence signals did not completely colocalize with bioluminescence signals despite imaging been carried out in the same animals. Bioluminescence tracking of dual-labeled particles depicted EV accumulation in the lungs with a signal increased from 1 to 4 h and then decreased at 24 h, followed by the spleen and minimal signal detected in the liver (Supplementary Figure S8A). On the other hand, DiR fluorescence tracking of the dual-labeled EVs showed the highest fluorescence signals in the liver and spleen (Supplementary Figure S8B), following a similar biodistribution pattern as that observed for nonengineered EVs labeled postisolation (Figure 2E–H and Figure 3). The different biodistribution profiles observed for the dual DiR-labeled Nluc EVs could be explained by the intrinsic heterogeneity of the vesicle landscape. The overexpression of CD63–Nluc fusion protein could result in one subset of vesicles significantly distinct from the rest with in vivo tropism toward the lung in contrast to the liver accumulation observed when the bulk EVs are analyzed. Overall, our results show that upon genetic modification of the parental cells for endogenously tagging EVs with bioluminescent tracers such as Nluc, changes in the characteristics of the EVs might occur resulting in different protein composition and altered in vivo biodistribution.
Blood Kinetics and Urine Clearance of EVs
To investigate the circulation kinetics of exogenously administered EVs in vivo, blood from [111In]-DTPA EV- and Nluc EV-treated animals was collected over time, and the radioactivity or luminescence was measured. Blood kinetic analysis from DiR EV-treated animals was not possible as the recorded signals were below the limit of detection. Nluc EVs and [111In]-DTPA EV showed relatively short circulating half-lives with less than 10% of ID remaining in the blood at 10 min postadministration (Figure 5A). For [111In]-DTPA EVs, ∼21% ID was detected in the blood at 2 min postinjection, decreasing to ∼10% ID at 10 min. The signal slowly decreased with time to ∼4 and ∼1.5% ID at 4 and 24 h, respectively. For Nluc EVs, a higher signal of ∼52% ID was detected at 2 min postinjection, with only ∼6% ID detected in blood within 15 min. The detected dose decreased to ∼1.4% at 1 h and ∼0.4% ID at 4 h and was undetectable at 24 h. Interestingly, both EV types showed distinct circulation kinetics with an initial short half-life phase likely reflecting the rapid distribution of EVs in the tissues and a long terminal phase possibly indicating the slow release of EVs from the tissues. Our results show that Nluc EVs were more rapidly cleared from the blood than [111In]-DTPA EVs. The rapid clearance of the EVs was consistent with that of other reports using EVs from various cellular sources and different luminescent and nuclear modalities.20,25,27,43 We confirmed that [111In]-DTPA EV and Nluc EVs were stable in serum up to 24 h (Supplementary Figure S3D,G), ruling out the possibility that the degradation or lysis of EVs caused a rapid decline of the signal in serum but rather reflect rapid tissue distribution or uptake by the mononuclear phagocytic system. Tissue-resident and monocyte-derived macrophages have been shown to play substantial roles in the clearance of EVs. Specifically, studies have shown decreased clearance of EVs from the blood in macrophage-depleted mice20 and increased blood circulation of EVs overexpressing CD55, CD59, and CD47 “do not eat me” signals.46,47 Rapid clearance of EVs has also been associated with the display of phosphatidylserine.48 It is possible that the exogenous overexpression of CD63 on Nluc EVs alters their surface protein composition or phosphatidylserine display, resulting in a slightly slower initial tissue distribution phase of Nluc EVs compared to [111In]-DTPA EVs. Notably, we observed durable retention of EVs in the tissues with detectable levels of the radiotracer up to 24 h. Urine and feces of mice administered with [111In]-DTPA EVs were also collected at 24 h to study the excretion profile (Figure 5B). Minimal renal excretion was observed for [111In]-DTPA EVs with only ∼0.8 and ∼1.4% ID detected in the urine and feces, respectively, at 24 h. These results show that renal excretion does not contribute to the rapid clearance of radiolabeled EVs from the circulation.
Figure 5.
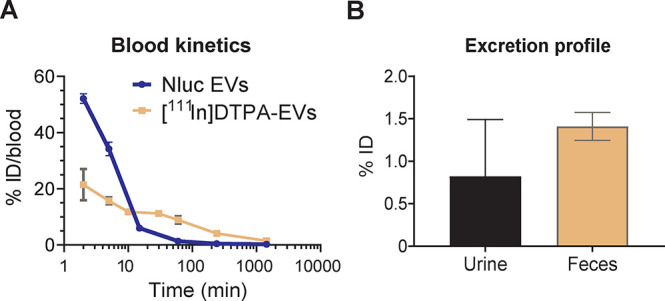
Blood clearance and excretion profile of NanoLuc and [111In]-DTPA Expi293F EVs. (A) Evaluation of the blood kinetics of EVs as a percentage of injected dose (ID) in blood over time. Blood (50 μL) from NanoLuc (Nluc) EV-treated animals was collected via tail bleeding at 2 min, 5 min, 15 min, 1 h, 4 h, and 24 h and left to clot to obtain the serum for bioluminescence quantification on a FLUOstar Omega plate reader. Blood (5 μL) from [111In]-DTPA EV-treated mice was taken via tail bleeding at 2 min, 5 min, 10 min, 30 min, 1 h, 4 h, and 24 h. Samples were analyzed for [111In]-specific activity using an automated gamma counter. (B) Excretion profile of [111In]-DTPA EVs in urine and feces collected from the animals 24 h postinjection. For all graphs, values are expressed as mean ± standard error of mean, where n = 3 for each group.
Impact of Tracking Approach on the In Vivo Organ Biodistribution of EVs
In this study, we explored the effect of different EV tracers on the in vivo biodistribution of EVs. Since all the EVs analyzed were derived from the same cell source and were administered systemically by the same i.v. route and at equal dose into subcutaneous CT26 tumor-bearing BALB/c mice, this removed any impact of the cell of origin of the EVs, the mice strain, and the administration procedure on their in vivo organ biodistribution. Analysis of tissue distribution of DiR and radiolabeled EVs by fluorescence and nuclear imaging, respectively, revealed that systemically delivered EVs mostly accumulate in the liver, spleen, and kidneys, with limited tumor accumulation. Lungs appeared to be the next highest site for accumulation specifically in the DiR EV-treated mice, consistent with a previous study using unmodified tumor-derived exosomes.22 The most likely explanation is that the residual DiR signal observed in the lungs is a consequence of dye exchange between the EVs and the high surface area in lung tissues over time. It is also possible that DiR EVs could have aggregated following systemic administration and got trapped in the lung capillaries upon first passage. Notably, the highest accumulation in the lung was observed using the genetically modified Nluc EVs, in contrast to the liver accumulation pattern observed with the unmodified DiR-labeled and radiolabeled [111In]-DTPA EVs. The small size of the EVs and the absence of aggregation in the preparation after density flotation (Figure 1 and Supplementary Figure S1) suggest that lung accumulation is unlikely due to EV aggregation. However, within the bloodstream, EVs are rapidly taken up by platelets,49 leukocytes,50 and other resident cells, especially endothelium and patrolling macrophages.37,51 Therefore, we cannot exclude the possibility that aggregation of the EVs takes place when encountering blood components, possibly triggered by slightly altered surface makeup. Additionally, the organ-specific homing potential of EVs linked to the display of specific tetraspanins has been demonstrated in several studies as previously mentioned.44,52,53 We hypothesize that the overexpression of CD63 as a fusion protein with the Nluc bioluminescent tag could modify the tetraspanin composition of EVs, which could result in the redistribution of other membrane proteins such as integrins, thereby altering the homing properties of the EVs. Interestingly, we found that the levels of other tetraspanins such as CD9 and CD81 were strongly altered in Nluc EVs compared to naïve EVs upon the overexpression of CD63 (Figure 1B). Previous reports have shown that CD63 and CD81 are present in similar intracellular locations and vesicle subsets,54,55 increasing the likelihood that a raise in the levels of CD63 in a specific EV subtype is accompanied by a reduction in other protein associated cargo, such as CD81. Moreover, it is well-known that CD63 drives vesiculation56 and in line with our results, others also reported changes in the levels of CD81 and Alix in EVs isolated from HEK293T cells overexpressing CD63 fusion proteins.35 Future studies are required to further understand the relationship between the modification of the protein composition of EVs and their homing properties in vivo. To avoid changes in EV protein composition as a result of CD63 overexpression, one could tag endogenous CD63 with Nluc by CRISPR/Cas-9-mediated knock-in. Alternatively, improved luciferase versions such as the recently discovered mutant of Nluc “teLuc” could provide a brighter signal than Nluc, with better red-shifted luminescence for deep-tissue imaging, and with a substrate that allows a higher membrane permeability and lower toxicity.57 Overall, our results show the differential effect of optical and nuclear tracers on EV biodistribution in vivo.
Comparison across EV Labeling Approaches and In Vivo Bioimaging Modalities
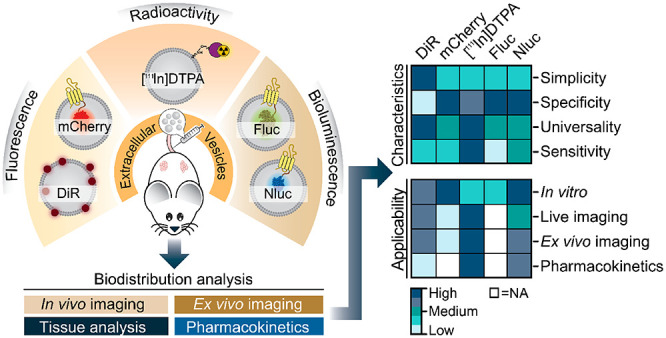
In this study, we provide key data for the selection of imaging tools for in vivo and ex vivo detection, monitoring and quantification of EV localization. The advantages and disadvantages of the different EV labeling strategies and bioimaging modalities for in vivo studies explored in this work are summarized in Table 1. EV labeling approaches were categorized based on the tags used, the labeling site, and the EV modification, with two main groups: (a) membrane/surface EV labeling by the integration of the lipophilic dye DiR or covalent binding of the111In-chelator DTPA to surface amines of EVs and (b) genetic modification-dependent labeling through the introduction of fusion proteins such as mCherry, Fluc, and Nluc in the EVs. Labeling EVs with the near-infrared dyes such as DiR has proven highly useful due to simplicity of use, high labeling efficiency, no chemical modification of the EVs, and applicability to EVs from all sources. It also offers major advantages for in vivo and ex vivo imaging such as medium signal-to-noise ratio and deeper tissue penetration with less autofluorescence over fluorescent dyes that emit at visible wavelengths.58 Optical fluorescent imagers are also widely available in different laboratories. Despite these advantages, there are several drawbacks associated with the use of lipophilic dyes including their ability to transfer between membranes,23 and their long-half-life,27 which might result in the conservation of fluorescent signal after EV degradation, thereby limiting long-term studies. Such factors can influence the reliability and accuracy of the in vivo organ biodistribution results. Nevertheless, DiR tracking and imaging of the EVs is a good option for both in vitro and short-term in vivo studies where pharmacokinetic and clearance analysis are not required. Importantly, we have shown that DiR can be successfully used for simultaneous dual labeling of particles that carry other reporter molecules such as bioluminescent proteins. The introduction of fluorescent reporter protein systems like mCherry in EVs by genetic modification allows for monitoring the fluorescent cargo intrinsically engineered in the vesicles. The labeling efficiency by genetic engineering is excellent, and no chemical modifications are required. However, due to high background fluorescence and low tissue penetration of mCherry signals, mCherry labeling is not suitable for in vivo monitoring of EVs in mice. There are other NIR fluorescent miRFP and iRFP protein families with improved excitation and emission wavelengths compared to mCherry. However, most of these proteins are dimers, which limit their use as protein tags, and are on average less bright and photostable than mCherry.59 Thus, we believe that all these aspects will not contribute to significantly improve the in vivo signal-to-ratio detection of EVs carrying far red-shifted protein variants. Instead, we selected a more versatile NIR dye that can be combined with other tracers and with a spectral region characterized by low autofluorescence and minimal phototoxicity for living cells. As an alternative, the use of genetically engineered luminescent proteins for tracking EVs in vivo has promising potential. Bioluminescence imaging with proteins such asGaussia luciferase,27,28Renilla luciferase,26 and Nluc luciferase60 has been used for real-time in vivo visualization of EVs and organ biodistribution studies. We also generated EVs carrying Fluc and Nluc as a fusion protein with CD63 by genetic engineering. In addition to the good labeling efficiency of these methods (in terms of tracer molecules per particle) and the lack of chemical modification, the amount of EVs was also increased by the overexpression of CD63. Nluc EVs emitted a significantly brighter signal than Fluc EVs with a sustained glow-type luminescence, providing a broader time window for imaging. The main disadvantage of genetic engineering for the introduction of cargo is the lack of applicability to EVs from sources such as biofluids or some primary cells. Additionally, labeling and hence, tracking can be specific to only a subpopulation of EVs, in our case CD63+, and the overexpression of certain proteins may alter the overall protein composition of EVs impacting their biodistribution. Nluc labeling of EVs was most appropriate for ex vivo monitoring of EV biodistribution. The expression of Nluc protein in EVs offered high sensitivity, high contrast ratio, and low to absent background luminescence in mammalian tissues.58 Additionally, Nluc tracking enabled partial pharmacokinetic analysis (i.e. blood circulation profile). However, real-time monitoring of EVs using Nluc was not optimal due to the attenuation of the signal by the tissues in living mice. Bioluminescent signals can be restricted by the low-spatial and temporal resolution when EVs are located in deep internal organs,58 considerably impairing EV in vivo imaging. Therefore, ex vivo analysis of whole organs or tissue lysates should be preferred, as they will provide more accurate results on tissue distribution of vesicles. Moreover, the need to inject the substrate for the generation of an optical signal restricts the long-term imaging of the animals due to the multiple injections, and also limits the throughput of bioluminescence imaging, which is normally an advantage of this technique.29 Other modalities such as nuclear imaging can overcome most of the limitations of the optical methods including high labeling efficiency of EVs from different sources with simple and stable labeling. Nuclear imaging also offers superb sensitivity and very high tissue penetration, enabling comprehensive biodistribution and pharmacokinetic analysis of EVs in vivo. SPECT/CT provides very accurate biodistribution and anatomical localization of EVs. However, due to the requirement for hazardous radioisotopes, specialized infrastructure and equipment, and the high cost of the technique, only a limited number of studies have been carried out using radioactive isotopes for both qualitative and quantitative assessment of the biodistribution of EVs in vivo.19,29
Table 1. Comparison of EV Labeling Approaches and In Vivo Bioimaging Strategies to Measure EV Biodistributiona.

Abbreviations: bioluminescence imaging (BLI), computed tomography (CT), extracellular vesicles (EVs), Firefly luciferase (Fluc), fluorescent imaging (FLI), near-infrared (NIR), NanoLuc luciferase (Nluc), nuclear imaging (NI), single-photon emission computed tomography (SPECT), XenoLight DiR dye (DiR), 111indium (111In).
Conclusions
To conclude, we provide an important direct and comprehensive comparison of fluorescent, bioluminescent, and radioactive labeling and imaging approaches to understand how they can influence reliable and accurate monitoring and quantification of EV biodistribution in vivo. We demonstrate that the labeling method significantly impacts the sensitivity and the fidelity of the detection and tracking of EVs, and therefore their advantages and drawbacks should be deliberated thoroughly before conducting in vivo biodistribution studies.
Methods
Cell Culture and Transfection of EV Producing Expi293F Cells
Human embryonic kidney Expi293F cells (Thermo Fisher Scientific, A14527) were cultured in synthetic serum-free Expi293 expression media (Thermo Fisher Scientific) at 37 °C, 8% CO2, at 125 rpm in 2 L roller bottles (Corning, Sigma-Aldrich). Human hepatoma cell line HepG2 (ATCC, HB-8065) was cultured in Eagle’s minimum essential medium supplemented with 10% FBS, 2 mM l-glutamine, and 1× nonessential amino acids (Thermo Fisher Scientific). Expi293F cells were transiently transfected with DNA plasmids coding for mCherry, Fluc, and Nluc sequences fused to the C-terminal of the human CD63. The genes were synthesized and subcloned into modified pEBNAZ plasmids, and the sequences were codon-optimized for human expression by GeneScript. CD63 protein sequence P08962. mCherry protein sequence X5DSL3. Fluc protein sequence Q27758. Nluc protein sequence: MVFTLEDFVGDWRQTAGYNLDQVLEQGGVSSLFQNLGVSVTPIQRIVLSGENGLKIDIHVIIPYEGLSGDQMGQIEKIFKVVYPVDDHHFKVILHYGTLVIDGVTPNMIDYGRPYEGIAVFDGKKITVTGTLWNGNKIIDERLINPDGSLLFRVTINGVTGWRLCERILA. Expi293F cells at a density of 3.8–4.2 × 106 cells/mL were transiently transfected with 1 mg/mL 40 kDa PEI max (Polysciences)–DNA complexes. Fresh Expi293 expression medium was added to the cultures 24 h after transfection, and cell viability was measured with trypan blue using a Cedex HiRes analyzer (Roche). After 48 h, cells and cell-conditioned media were collected, and if cell viability surpassed 85%, the cell supernatant was then used for EV isolation. Cells were pelleted at 300g for 10 min, and cell-conditioned medium was further centrifuged at 2500g for 30 min to remove cell debris before EV isolation.
EV Isolation and Density Gradient Purification
Cell supernatant was transferred to 94 mL quick-seal polyallomer tubes (Beckman Coulter) and centrifuged at 20,000gavg for 25 min at 4 °C using a 45Ti rotor and an Optima XE-100 ultracentrifuge (Beckman Coulter) to pellet intermediate-size EVs. The supernatant was carefully removed, transferred to new tubes, and ultracentrifuged at 100,000g for 120 min at 4 °C (Type 45 Ti, k-factor 210.4) to pellet small EVs (also known as exosomes). The exosome-like pellet was resuspended in a total of 1 mL of PBS and floated in a high-resolution iodixanol density gradient (Optiprep, Sigma-Aldrich) at 120,000gavg for 16 h at 4 °C (SW 32.1 Ti, k-factor 249.1, Beckman Coulter) as previously described by our group.31 Nine fractions were collected from top to bottom (corresponding to iodixanol concentrations of 10–50%). After full characterization of each fraction, F1–F3 (corresponding to 10, 20, and 22% iodixanol) were pooled, transferred to new 94 mL PBS tubes, and ultracentrifuged at 120,000gavg for 2.5 h (Type 45 Ti, k-factor 175.3). Those EV pellets were resuspended in PBS and stored at −80 °C.
EV Protein and Particle Characterization
Samples from individual density fractions (100 μL) were collected and aliquoted: 85 μL for Western blotting, 5 μL for NTA, and 10 μL for TEM. For Western blotting, fractions were prepared and run as previously described.31 Following protein separation, transfer, and blocking, membranes were incubated overnight at 4 °C with primary antibodies, diluted 1:1,000 in TBS Odyssey blocking buffer (LI-COR Biosciences Inc., Lincoln, NE): anti-lamin B1 (D9 V6H, Cell Signaling Technology, Leiden, The Netherlands, cat# 13435s), anti-Alix (3A9, Abcam, Cambridge, UK, cat# ab117600), anti-Flotillin-1 (clone 18, BD Biosciences, San Jose CA, cat# 610820), anti-CD63 (TS63, Abcam, cat# ab59479), anti-CD81 (M38, Abcam, cat# ab79559), anti-CD9 (Abcam, cat# ab97999), anti-mCherry (Abcam, cat#167453), anti-Fluc (Novus Biologicals, cat# NB100-1677), or anti-Nluc (Promega, cat# 909747). After the membranes were washed three times with 0.1% TBS-Tween, membranes were incubated for 1 h at room temperature (RT) with the following secondary antibodies diluted 1:20,000 in 0.1% TBS-Tween/IRDye 680RD goat anti-mouse IgG (H+L) cat# 925-68070, IRDye 680RD goat anti-rabbit IgG cat# 925-68071, IRDye 800CW goat anti-mouse IgG cat# 925-32210, IRDye 800CW goat anti-rabbit IgG cat# 926-32211, or IRDye 800CW donkey anti-goat IgG cat# 925-32214 (all from LI-COR). Next, the membranes were washed, visualized with the Odyssey CLx imaging system (LI-COR), and processed in the Image Studio v.4.0 software (LI-COR). Particle concentration measurements of all fractions using NTA and TEM analysis were performed as previously described.4 Analysis of TEM micrographs to determine the size of EVs was carried out using ImageJ (Fiji), where vesicles in the images from F1–F3 were segmented and their diameter retrieved. Forty to 50 individual images per sample were analyzed.
XenoLight DiR EV Labeling
Purified EVs were labeled with XenoLight DiR fluorescent dye (PerkinElmer) before density flotation. The 1 mL PBS-EV or Nluc-CD63 EV samples were incubated with the dye at a concentration of 10 μM for 20 min at 37 °C protected from light. Next, the EV samples were bottom-loaded in the high-resolution density gradient and ultracentrifuged as described above to both isolate sEVs and remove the unbound dye. A sample of the PBS supernatant collected after the EV labeling was used as control for the in vitro experiments.
Direct In Vitro Optical Measurements on Engineered and Fluorescently Labeled EVs
Fluorescent and luminescent plate readout of EVs was performed to determine their optical detection threshold and labeling efficiency. Labeling efficiency of EVs with DiR dye or mCherry protein was determined by estimating the average number of fluorescent molecules per vesicle. Purified fluorescent recombinant protein mCherry with N-terminal HIS tag (OriGene) at a concentration ranging from 200 to 800 nM or XenoLight DiR dye at a concentration ranging from 5 to 0.2 μM was used as a reference for the standard curves. Molar concentration of DiR or mCherry was determined in bulk EV samples by comparison with the standard curves and converted into number of molecules using Avogadro’s number, followed by normalization by EV sample concentration as determined by NTA. The fluorescence of EV samples was measured using the Safire II plate reader (v 4.62n) for mCherry (Ex: 587 nm/Em: 610 nm) or DiR (Ex: 748 nm/Em: 780 nm). Fluorescence readout of mCherry- and DiR-labeled EVs, corresponding to 1010 and 5 × 1010 particles, respectively (10 and 50% ID), was prepared in a total volume of 100 μL of PBS and added to individual wells in a 96-well black plate (Sigma-Aldrich). The same volume of PBS was used as a control. The plate was then imaged using the IVIS Lumina III system with mCherry filter (Ex: 560 nm/Em: 620 nm) or DiR filter (Ex: 740 nm/Em: 790 nm), and the images obtained were analyzed using the Living Image 4.7.2 software. Bioluminescence readout of Fluc EVs (3 × 109 to 1 × 1011 particles) and Nluc EVs (9 × 103 to 2 × 107 particles) was performed using ONE-Glo and Nano-Glo luciferase assays (Promega, UK) per the manufacturer’s instructions. The luminescent signal was measured using a PHERAstar FSX (BMG Labtech). The calculations on the average number of Nluc molecules per EV to determine labeling efficiency were based on semiquantitative comparative Western blots of CD63-mCherry and CD63-Nluc.
Serum Stability Experiments
mCherry EVs (3 × 1010 particles), DiR-labeled EVs (3 × 1010 particles), and Nluc EVs (3 × 106 particles) were incubated in 50% FBS or PBS (1:1, v/v) for 24 h at 37 °C. The EV samples postincubation were then subjected to fluorescent readout using the Safire II plate reader (v 4.62n) with mCherry (Ex: 587 nm/Em: 610 nm) or DiR (Ex: 748 nm/Em: 780 nm). The Nluc EV samples were then subjected to the Nano-Glo luciferase assay (Promega). The resulting fluorescence or luminescence signal was converted to percentage activity relative to that of an equal number of fresh EVs subjected directly to the fluorescent detection or luciferase assay without any incubation (taken as 100% activity).
Membrane Radiolabeling of EVs and Radiochemical Stability Analysis
Membrane radiolabeling of EVs and radiochemical assessment were performed as previously described.25 Briefly, for membrane radiolabeling, DTPA-anhydride prepared at 1 μg/μL in dry chloroform was incubated at 1:400 (lysine/anhydride) molar ratio reaction with EVs (assuming one EV is one BSA molecule, i.e., containing 59 lysine residues). The sample was dried under a nitrogen stream before addition of EVs and incubated at 37 °C for 30 min. Excess DTPA-anhydride was purified by gel filtration using a self-packed Sepharose CL-2B column (GE Healthcare, UK). The required amount of 111In stock to achieve 5–10 or 0.5–1 MBq per mouse for whole-body imaging and gamma counting, respectively, was added to 0.2 M ammonium acetate buffer (pH 5.5) to achieve a final volume of 500 μL. The mix was then added to an equal volume of DTPA-EVs to achieve a final concentration of 0.1 M ammonium acetate. The mixture was incubated for 5 min at RT, and radiolabeled EVs ([111In]-DTPA EVs) were purified from excess 111InCl3 using gel filtration as described above. Radiolabeling efficiency was calculated as follows:
For radiochemical stability analysis, [111In]-DTPA EVs were incubated in 50% FBS or PBS (1:1, v/v) for 24 h at 37 °C and spotted on thin-layer chromatography paper strips. The strips were run on 0.1 M ammonium acetate containing 0.25 mM EDTA (pH 5.5) as the mobile phase. The strips were then placed on a multipurpose storage phosphor screen (Cyclone, Packard, Japan) and kept in autoradiography cassettes (Kodak Biomax Cassette) for 1–10 min, depending on the activity spotted on the strip. Quantitative autoradiography counting was carried out using a phosphor detector (Packard, Australia). The percentage of 111In still attached to EVs (immobile spot at the application point) was considered as the radiochemically stable [111In]-DTPA EVs.
Cellular Uptake of EVs
HepG2 cells were seeded at a density of 20,000 cells/well in a 96-well plate in EV-depleted media. After 24 h, cells were incubated with 5 × 109 to 1 × 1010 labeled or nonlabeled EVs for 4, 8, and 12 h. PBS-DiR subjected to the same procedure was incubated with cells at the same volume as for DiR-EVs as a control. For analysis of DiR- and mCherry-EV uptake, HepG2 cells were collected, washed with wash buffer (2% FBS, 50 mM EDTA in PBS), and analyzed on an IntelliCyt iQue Screener Plus instrument equipped with ForeCyt software (v 6.2.6752) for data acquisition and analysis. Nonstimulated cells were used as a background control for gating of positive events. For analysis of Fluc- and Nluc-EV uptake, HepG2 cells were incubated with the substrates from ONE-Glo or Nano-Glo luciferase assays (Promega, UK) for 5 min under shaking, and bioluminescence analysis was performed as described above. Nonstimulated cells were used as bioluminescence background control. To visualize the cellular uptake of fluorescent EVs, HepG2 cells at a density of 10,000 cell/well were incubated with mCherry EVs up to 24 h. Next, cells were washed three times with PBS and incubated for 45 min with Cell Tracker Green CMFDA dye at 1 μM (Invitrogen). Following PBS washes, cells were incubated for 15 min with Hoechst 33342 (ThermoFisher Scientific) and live cells were next imaged on a Cell Voyager 7000 confocal microscope (CV7000, Yokogawa Inc.). Confocal fluorescent images were captured using a 60× water objective (Olympus UPLSAPO 1.2 NA) and an Andor Neo sCMOS camera. Hoechst was imaged using a 405 nm excitation laser (405 ± 5 nm, 100 mW, Coherent) with a 445/45 nm band-pass emission filter. mCherry EVs were visualized using a 561 nm excitation laser (561 ± 2 nm, 200 mW, Coherent) with a 600/37 nm band-pass emission filter, and Cell Tracker Green was imaged using a 488 nm excitation laser (488 ± 2 nm, 200 mW, Coherent) with a 525/50 nm band-pass emission filter.
Animal Model and Subcutaneous Tumor Inoculation
All animal experiments were performed in compliance with the UK Home Office Animals (Scientific Procedures) Act 1986. Female BALB/c mice aged 6–8 weeks (purchased from Charles River, UK) were used for all the experiments. The CT26 murine colon carcinoma cells were cultured in RPMI 1640 medium supplemented with 1% GlutaMax, 1% penicillin–streptomycin, and 10% FBS (Thermo Fisher Scientific) at 37 °C with 5% CO2. The harvested CT26 cells were suspended in PBS (pH 7.4) and injected subcutaneously (1 × 106 cells in 100 μL) into the left and right rear flanks of the mice. Animals were closely monitored postinoculation and were used for studies when the tumors were 200–300 mm3 in size.
Optical In Vivo Imaging of DiR- and mCherry-Labeled EVs
For real-time in vivo imaging, mice were injected i.v. in the tail vein with 1 × 1011 DiR-labeled EVs or mCherry EVs in 200 μL of PBS per animal. PBS was injected as a control (n = 3 per group). Mice were imaged after 24 h under anesthesia in dorsal and ventral positions using the IVIS Lumina III system (PerkinElmer, UK). For DiR EVs, images were obtained using sequential acquisition spectra unmixing mode with DiR filter (Ex: 740 nm/Em: 790 nm) for ventral and dorsal positions. For mCherry EVs, the mCherry filter (Ex: 560 nm/Em: 620 nm) was used. Binning factor of (HS)8, f number of 2, and field of view of E-24 cm were used for both DiR EVs and mCherry EVs. All mice were fed an alfalfa diet except for a subgroup of animals treated with mCherry EVs that were fed with a low-chlorophyll (alfalfa-free) diet for a week before imaging to minimize the autofluorescence signal originating from the gastrointestinal tract. Fluorescence signals were stored in efficiency units. For ex vivo imaging, mice were sacrificed at the 24 h time point (n = 3 for each group). Organs including brain, heart, lungs, liver, kidneys, spleen, stomach, pancreas, intestine, and tumors were collected and weighed. The collected organs were placed on black plastic spacers and imaged using the IVIS Lumina III system (PerkinElmer, UK). For DiR EVs, images were obtained using sequential acquisition spectra unmixing mode with DiR filter (Ex: 740 nm/Em: 790 nm), exposure time of 0.75 s, binning factor of (M)4, f number of 2, and field of view of E-24 cm. For mCherry EVs, images were obtained using the mCherry filter (Ex: 560 nm/Em: 620 nm), exposure time of 2 s, binning factor of (HS)8, f number of 2, and field of view of E-24 cm. Fluorescence signals were stored in efficiency units. The images obtained were analyzed using the Living Image 4.7.2 software (PerkinElmer, UK) where the regions of interest (ROIs) were drawn for each organ to obtain their individual fluorescence signals. Organs were frozen at −80 °C for tissue lysate analysis as described below. For analysis of tissue lysates, thawed organs were homogenized in 1 mL of lysis buffer (25 mM Tris-phosphate (pH 7.8), 2 mM DTT, 2 mM 1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 10% glycerol, and 1% Triton X-100) or 2 mL for the liver and intestine, in 5 s pulses on ice until no large tissue pieces were observed. The homogenates were subjected to one freeze–thaw cycle and then centrifuged at 14,000g for 30 min at 4 °C. The supernatant was collected as clarified tissue lysate and kept on ice if used immediately or stored at −80 °C. Tissue lysates were transferred into black 96-well plates (100 μL/well). For DiR EVs, the plate was imaged as described for DiR EVs ex vivo imaging above but with 3 s exposure time. For mCherry EVs, the plate was imaged using FLUOstar Omega plate reader (BMG Labtech, UK) with a gain of 2000.
Whole-Body In Vivo Imaging of Radiolabeled EVs Using SPECT/CT
Mice were injected i.v. with 1 × 1011 [111In]DTPA-EVs (5–10 MBq; in 200 μL) and imaged under anesthesia in the prone position on a heating pad at 37 °C using a nanoSPECT/CT four-head scanner (Bioscan, USA). SPECT images were obtained at 0–30 min, 4 h, and 24 h postinjection using 1.4 mm pinhole collimators (24 projections, 60 s per projection; 30 min scan), and CT images were obtained at the end of each SPECT acquisition using an X-ray source setting of 45 kVp. All data were reconstructed with proprietary Bioscan software, while SPECT and CT acquisitions were fused using PMOD software (Mediso).
Quantitative Organ Biodistribution of Radiolabeled EVs Using Gamma Counting
Mice injected i.v. with 1 × 1011 [111In]-DTPA EVs per animal were sacrificed and perfused with saline after 1, 4, and 24 h (n = 3). Organs such as brain, heart, lungs, liver, kidneys, spleen, stomach, intestine, skin, tumors, a sample of muscle (hamstring and quadriceps), and bone (femur) and the remaining carcass were collected, weighed, and placed in scintillation vials. Additionally, 5 μL blood samples were taken from the tail vein at various time points (2, 5, 10, 30, 60, 240, and 1440 min). Urine and feces were collected by housing the tumor-bearing mice in metabolic cages for 24 h to analyze the excretion profile. Each sample was analyzed for [111In]-specific activity using an automated gamma counter (LKB Wallac 1282 Compugamma, PerkinElmer, UK) together with a sample of the injected dose (ID) with dead time limit below 60%. The γ-rays emitted by the radioisotope were quantified and corrected for physical radioisotope decay by the gamma counter. Radioactivity readings (counts per minute) were expressed as percentage of ID per organ or percentage of ID per gram of tissue. Data were expressed as the mean ± standard error of mean of sample triplicates.
Bioluminescence In Vivo Imaging of Nluc EVs
For real-time in vivo imaging, mice were i.v. injected with 1 × 1011 Nluc EVs per animal (100 μL) or PBS control. Mice were injected i.v. with 100 μL of Nluc substrate furimazine (1:20 dilution in PBS, corresponding to 10 μg) (Nano-Glo luciferase assay system kit from Promega, UK), at 1, 4, and 24 h postdose (n = 3 for each time point). Animals were imaged under anesthesia in the ventral position within 2 min. Images were obtained using the bioluminescence mode with an exposure time of 60 s, binning factor of (HS)8, f number of 1.2, and field of view of E-24 cm. For ex vivo imaging, mice were sacrificed at 1, 4, or 24 h and organs including brain, heart, lungs, liver, kidneys, spleen, stomach, pancreas, intestine, and tumors were collected and weighed. The organs were then immersed in 10 mL of imazine solution (1:20 dilution in PBS) for 30 s, blotted dry on tissue paper, arranged on black plastic spacers, and imaged within 2 min using the IVIS Lumina III system. Images were obtained using the bioluminescence mode with exposure time of 2–3 s, binning factor of (HS)8, f number of 1.2, and field of view of E-24 cm. The images were analyzed using the Living Image 4.7.2 software (PerkinElmer, UK),where the ROIs were drawn for each organ to obtain their individual bioluminescence signals. Additionally, 50 μL blood samples were taken from the tail vein at various time points (2, 5, 15, 60, 240, and 1440 min). The collected blood was allowed to clot at RT for at least 2 h. Clotted blood was centrifuged at 2000g for 15 min at 4 °C, and the serum was collected. Organs and sera were frozen at −80 °C for subsequent analysis of tissue lysates and blood kinetics. Tissues were lysed as described above. Lysates were analyzed using the Nano-Glo luciferase (Promega) according to the supplier’s manual. Briefly, the assay reagent (50 μL) was then added to equal volume of diluted tissue lysate or serum samples (1:80 for liver; 1:20 for other organs and serum using lysis buffer) and mixed by pipetting. The mixture was left to stand at RT for at least 3 min before the luminescence was detected using FLUOstar Omega plate reader (BMG Labtech, UK) with gain of 3000 and 1 s exposure time. For Nluc EV standard curve preparation, 50 μL total volume of EVs of the different concentrations in PBS was used instead of tissue lysate samples.
Statistical Analysis
Statistical analyses used are detailed in the figure legends. Data were presented as mean ± standard deviation or ± standard error of mean, where “n” denotes the number of repeats. Statistical analysis (one-way and two-way ANOVA) was used to establish statistical significance using GraphPad Prism 8 software (v 8.2.1). A p value <0.05 was considered statistically significant.
All relevant EV-related methods and data for this work has been deposited in the EV-TRACK knowledgebase https://evtrack.org/ (EV-TRACK ID: EV200159).
Acknowledgments
E.L.I. and A.M.S. are fellows of the AstraZeneca postdoc programme. F.N.F. is funded by the Malaysian government agency Majlis Amanah Rakyat (MARA, 330408285430). The authors would like to thank Mario Soriano, Samantha Peel, and Jonathan Rose for their support with the in vitro and in vivo imaging of EVs. We would also like to thank Lukas Badertscher for his help with the illustrations, and Neil Henderson for his contribution to the initial tissue and blood analysis. The authors would also like to acknowledge Life Science Editors for editorial assistance. This project has received funding from the Brain Tumour Charity (GN-000398), Institutional Link-British Council (IL4337313), and Wellcome Trust (WT103913).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c09873.
In vitro characterization of genetically engineered and membrane-labeled EVs; labeling efficiency, stability, and in vitro detection of fluorescent EVs; labeling efficiency, stability, and in vitro detection of bioluminescent and radiolabeled EVs; in vivo evaluation of mCherry Expi293F EVs in mice fed an alfalfa-free diet (low-chlorophyll); organ biodistribution profile of DiR-labeled EVs in subcutaneous CT26 tumor-bearing BABL/c mice; ex vivo organ biodistribution of radiolabeled 111indium-DTPA Expi293F EVs in mice; in vivo evaluation of NanoLuc Expi293F EV biodistribution by measuring bioluminescence in tissue lysates; in vivo dual imaging biodistribution analysis of DiR-labeled NanoLuc EVs (PDF)
Author Contributions
# E.L.-I. and F.N.F. contributed equally.
The authors declare the following competing financial interest(s): E.L.-I., A.M.S., A.F.S., and N.D. are currently employed and hold equity in AstraZeneca R&D.
Supplementary Material
References
- Théry C.; Zitvogel L.; Amigorena S. Exosomes: Composition, Biogenesis and Function. Nat. Rev. Immunol. 2002, 2 (8), 569–579. 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- Yanez-Mo M.; Siljander P. R.-M.; Andreu Z.; Bedina Zavec A.; Borras F. E.; Buzas E. I.; Buzas K.; Casal E.; Cappello F.; Carvalho J.; Colas E.; Cordeiro-da Silva A.; Fais S.; Falcon-Perez J. M.; Ghobrial I. M.; Giebel B.; Gimona M.; Graner M.; Gursel I.; Gursel M.; Heegaard N. H. H.; Hendrix A.; Kierulf P.; Kokubun K.; Kosanovic M.; Kralj-Iglic V.; Kramer-Albers E.-M.; Laitinen S.; Lasser C.; Lener T.; Ligeti E.; Line A.; Lipps G.; Llorente A.; Lotvall J.; Mancek-Keber M.; Marcilla A.; Mittelbrunn M.; Nazarenko I.; Nolte-‘t Hoen E. N.M.; Nyman T. A.; O'Driscoll L.; Olivan M.; Oliveira C.; Pallinger E.; del Portillo H. A.; Reventos J.; Rigau M.; Rohde E.; Sammar M.; Sanchez-Madrid F.; Santarem N.; Schallmoser K.; Stampe Ostenfeld M.; Stoorvogel W.; Stukelj R.; Van der Grein S. G.; Helena Vasconcelos M.; Wauben M. H. M.; De Wever O. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4 (1), 27066. 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G.; Stoorvogel W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013, 200 (4), 373–383. 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A. F.; Lazaro-Ibanez E.; Forsgard M. A.-M.; Shatnyeva O.; Osteikoetxea X.; Karlsson F.; Heath N.; Ingelsten M.; Rose J.; Harris J.; Mairesse M.; Bates S. M.; Clausen M.; Etal D.; Leonard E.; Fellows M. D.; Dekker N.; Edmunds N. Extracellular Vesicles Induce Minimal Hepatotoxicity and Immunogenicity. Nanoscale 2019, 11 (14), 6990–7001. 10.1039/C8NR08720B. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Badawi M.; Pomeroy S.; Sutaria D. S.; Xie Z.; Baek A.; Jiang J.; Elgamal O. A.; Mo X.; Perle K. L.; Chalmers J.; Schmittgen T. D.; Phelps M. A. Comprehensive Toxicity and Immunogenicity Studies Reveal Minimal Effects in Mice Following Sustained Dosing of Extracellular Vesicles Derived from HEK293T Cells. J. Extracell. Vesicles 2017, 6 (1), 1324730. 10.1080/20013078.2017.1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlgren J.; Karlson T. D. L.; Brisslert M.; Vaziri Sani F.; Telemo E.; Sunnerhagen P.; Valadi H. Plasma Exosomes Can Deliver Exogenous Short Interfering RNA to Monocytes and Lymphocytes. Nucleic Acids Res. 2012, 40 (17), e130–e130. 10.1093/nar/gks463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujrati V.; Kim S.; Kim S.-H.; Min J. J.; Choy H. E.; Kim S. C.; Jon S. Bioengineered Bacterial Outer Membrane Vesicles as Cell-Specific Drug-Delivery Vehicles for Cancer Therapy. ACS Nano 2014, 8 (2), 1525–1537. 10.1021/nn405724x. [DOI] [PubMed] [Google Scholar]
- Villarroya-Beltri C.; Gutiérrez-Vázquez C.; Sánchez-Cabo F.; Pérez-Hernández D.; Vázquez J.; Martin-Cofreces N.; Martinez-Herrera D. J.; Pascual-Montano A.; Mittelbrunn M.; Sánchez-Madrid F. Sumoylated hnRNPA2B1 Controls the Sorting of miRNAs into Exosomes through Binding to Specific Motifs. Nat. Commun. 2013, 4, 2980. 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolukbasi M. F.; Mizrak A.; Ozdener G. B.; Madlener S.; Ströbel T.; Erkan E. P.; Fan J.-B.; Breakefield X. O.; Saydam O. miR-1289 and “Zipcode”-Like Sequence Enrich mRNAs in Microvesicles. Mol. Ther.--Nucleic Acids 2012, 1, e10. 10.1038/mtna.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Wu J.; Gu W.; Huang Y.; Tong Z.; Huang L.; Tan J. Exosome–Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. (Weinh) 2018, 5 (4), 1700611. 10.1002/advs.201700611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saari H.; Lázaro-Ibáñez E.; Viitala T.; Vuorimaa-Laukkanen E.; Siljander P.; Yliperttula M. Microvesicle- and Exosome-Mediated Drug Delivery Enhances the Cytotoxicity of Paclitaxel in Autologous Prostate Cancer Cells. J. Controlled Release 2015, 220, 727–737. 10.1016/j.jconrel.2015.09.031. [DOI] [PubMed] [Google Scholar]
- Jang S. C.; Kim O. Y.; Yoon C. M.; Choi D.-S.; Roh T.-Y.; Park J.; Nilsson J.; Lötvall J.; Kim Y.-K.; Gho Y. S. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. ACS Nano 2013, 7 (9), 7698–7710. 10.1021/nn402232g. [DOI] [PubMed] [Google Scholar]
- Ohno S.-i.; Takanashi M.; Sudo K.; Ueda S.; Ishikawa A.; Matsuyama N.; Fujita K.; Mizutani T.; Ohgi T.; Ochiya T.; Gotoh N.; Kuroda M. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor microRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185. 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rountree R. B.; Mandl S. J.; Nachtwey J. M.; Dalpozzo K.; Do L.; Lombardo J. R.; Schoonmaker P. L.; Brinkmann K.; Dirmeier U.; Laus R.; Delcayre A. Exosome Targeting of Tumor Antigens Expressed by Cancer Vaccines Can Improve Antigen Immunogenicity and Therapeutic Efficacy. Cancer Res. 2011, 71 (15), 5235–5244. 10.1158/0008-5472.CAN-10-4076. [DOI] [PubMed] [Google Scholar]
- Alvarez-Erviti L.; Seow Y.; Yin H.; Betts C.; Lakhal S.; Wood M. J. Delivery of siRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29 (4), 341–345. 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- Kooijmans S. A.; Aleza C. G.; Roffler S. R.; Van Solinge W. W.; Vader P.; Schiffelers R. M. Display of GPI-Anchored Anti-EGFR Nanobodies on Extracellular Vesicles Promotes Tumour Cell Targeting. J. Extracell. Vesicles 2016, 5 (1), 31053. 10.3402/jev.v5.31053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Ran N.; Dong X.; Zuo B.; Yang R.; Zhou Q.; Moulton H. M.; Seow Y.; Yin H. Anchor Peptide Captures, Targets, and Loads Exosomes of Diverse Origins for Diagnostics and Therapy. Sci. Transl. Med. 2018, 10 (444), eaat0195. 10.1126/scitranslmed.aat0195. [DOI] [PubMed] [Google Scholar]
- Pi F.; Binzel D. W.; Lee T. J.; Li Z.; Sun M.; Rychahou P.; Li H.; Haque F.; Wang S.; Croce C. M.; Guo B.; Evers B. M.; Guo P. Nanoparticle Orientation to Control RNA Loading and Ligand Display on Extracellular Vesicles for Cancer Regression. Nat. Nanotechnol. 2018, 13 (1), 82–89. 10.1038/s41565-017-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y. W.; Lee J. H.; Kim S.-Y.; Pack C.-G.; Ha D. H.; Park S. R.; Youn J.; Cho B. S. Advances in Analysis of Biodistribution of Exosomes by Molecular Imaging. Int. J. Mol. Sci. 2020, 21 (2), 665. 10.3390/ijms21020665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T.; Takahashi Y.; Nishikawa M.; Kato K.; Morishita M.; Yamashita T.; Matsumoto A.; Charoenviriyakul C.; Takakura Y. Macrophage-Dependent Clearance of Systemically Administered B16BL6-Derived Exosomes from the Blood Circulation in Mice. J. Extracell. Vesicles 2015, 4 (1), 26238. 10.3402/jev.v4.26238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S. W.; Sceneay J.; Lima L. G.; Wong C. S.F.; Becker M.; Krumeich S.; Lobb R. J.; Castillo V.; Wong K. N.; Ellis S.; Parker B. S.; Moller A. The Biodistribution and Immune Suppressive Effects of Breast Cancer–Derived Exosomes. Cancer Res. 2016, 76 (23), 6816–6827. 10.1158/0008-5472.CAN-16-0868. [DOI] [PubMed] [Google Scholar]
- Smyth T.; Kullberg M.; Malik N.; Smith-Jones P.; Graner M. W.; Anchordoquy T. J. Biodistribution and Delivery Efficiency of Unmodified Tumor-Derived Exosomes. J. Controlled Release 2015, 199, 145–155. 10.1016/j.jconrel.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklander O. P. B.; Nordin J. Z.; O'Loughlin A.; Gustafsson Y.; Corso G.; Mager I.; Vader P.; Lee Y.; Sork H.; Seow Y.; Heldring N.; Alvarez-Erviti L.; Smith C. E.; Le Blanc K.; Macchiarini P.; Jungebluth P.; Wood M. J. A.; Andaloussi S. E. Extracellular Vesicle in Vivo Biodistribution is Determined by Cell Source, Route of Administration and Targeting. J. Extracell. Vesicles 2015, 4 (1), 26316. 10.3402/jev.v4.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D. W.; Choi H.; Jang S. C.; Yoo M. Y.; Park J. Y.; Choi N. E.; Oh H. J.; Ha S.; Lee Y.-S.; Jeong J. M.; Gho Y. S.; Lee D. S. Noninvasive Imaging of Radiolabeled Exosome-Mimetic Nanovesicle Using 99m Tc-HMPAO. Sci. Rep. 2015, 5, 15636. 10.1038/srep15636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruqu F. N.; Wang J. T.-W.; Xu L.; McNickle L.; Chong E. M.-Y.; Walters A.; Gurney M.; Clayton A.; Smyth L. A.; Hider R.; Sosabowski J.; Al-Jamal K. T. Membrane Radiolabelling of Exosomes for Comparative Biodistribution Analysis in Immunocompetent and Immunodeficient Mice-A Novel and Universal Approach. Theranostics 2019, 9 (6), 1666. 10.7150/thno.27891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadaran P.; Li X. J.; Lee H. W.; Oh J. M.; Kalimuthu S.; Rajendran R. L.; Son S. H.; Baek S. H.; Singh T. D.; Zhu L.; Jeong S. Y.; Lee S.-W.; Lee J.; Ahn B.-C. A New Bioluminescent Reporter System to Study the Biodistribution of Systematically Injected Tumor-Derived Bioluminescent Extracellular Vesicles in Mice. Oncotarget 2017, 8 (66), 109894. 10.18632/oncotarget.22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. P.; Mardini O.; Ericsson M.; Prabhakar S.; Maguire C. A.; Chen J. W.; Tannous B. A.; Breakefield X. O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8 (1), 483–494. 10.1021/nn404945r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y.; Nishikawa M.; Shinotsuka H.; Matsui Y.; Ohara S.; Imai T.; Takakura Y. Visualization and in Vivo Tracking of the Exosomes of Murine Melanoma B16-BL6 Cells in Mice after Intravenous Injection. J. Biotechnol. 2013, 165 (2), 77–84. 10.1016/j.jbiotec.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Gangadaran P.; Hong C. M.; Ahn B.-C. An Update on in Vivo Imaging of Extracellular Vesicles as Drug Delivery Vehicles. Front. Pharmacol. 2018, 9, 169. 10.3389/fphar.2018.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrie A.; Krumeich S.; Reyal F.; Recchi C.; Moita L. F.; Seabra M. C.; Ostrowski M.; Théry C. Rab27a Supports Exosome-Dependent and -Independent Mechanisms That Modify the Tumor Microenvironment and Can Promote Tumor Progression. Cancer Res. 2012, 72 (19), 4920–4930. 10.1158/0008-5472.CAN-12-0925. [DOI] [PubMed] [Google Scholar]
- Lázaro-Ibáñez E.; Lässer C.; Shelke G. V.; Crescitelli R.; Jang S. C.; Cvjetkovic A.; García-Rodríguez A.; Lötvall J. DNA Analysis of Low- and High-Density Fractions Defines Heterogeneous Subpopulations of Small Extracellular Vesicles Based on Their DNA Cargo and Topology. J. Extracell. Vesicles 2019, 8 (1), 1656993. 10.1080/20013078.2019.1656993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J.; Wang J. T. W.; Rubio N.; Protti A.; Heidari H.; Elgogary R.; Southern P.; Al-Jamal W. T.; Sosabowski J.; Shah A. M.; Bals S.; Pankhurst Q. A.; Al-Jamal K. Triple-Modal Imaging of Magnetically-Targeted Nanocapsules in Solid Tumours in Vivo. Theranostics 2016, 6 (3), 342–356. 10.7150/thno.11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippstein R.; Wang J. T.-W.; El-Gogary R. I.; Bai J.; Mustafa F.; Rubio N.; Bansal S.; Al-Jamal W.; Al-Jamal K. Passively Targeted Curcumin-Loaded PEGylated PLGA Nanocapsules for Colon Cancer Therapy in Vivo. Small 2015, 11 (36), 4704–4722. 10.1002/smll.201403799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusermann W.; Hean J.; Trojer D.; Steib E.; von Bueren S.; Graff-Meyer A.; Genoud C.; Martin K.; Pizzato N.; Voshol J.; Morrissey D. V.; Andaloussi S. E.; Wood M. J.; Meisner-Kober N. C. Exosomes Surf on Filopodia to Enter Cells at Endocytic Hot Spots, Traffic within Endosomes, and Are Targeted to the ER. J. Cell Biol. 2016, 213 (2), 173–84. 10.1083/jcb.201506084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso G.; Heusermann W.; Trojer D.; Gorgens A.; Steib E.; Voshol J.; Graff A.; Genoud C.; Lee Y.; Hean J.; Nordin J. Z.; Wiklander O. P.B.; El Andaloussi S.; Meisner-Kober N. Systematic Characterization of Extracellular Vesicle Sorting Domains and Quantification at the Single Molecule–Single Vesicle Level by Fluorescence Correlation Spectroscopy and Single Particle Imaging. J. Extracell. Vesicles 2019, 8 (1), 1663043. 10.1080/20013078.2019.1663043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim N.; Ryu S.-W.; Choi K.; Lee K. R.; Lee S.; Choi H.; Kim J.; Shaker M. R.; Sun W.; Park J.-H.; Kim D.; Heo W. D.; Choi C. Exosome Engineering for Efficient Intracellular Delivery of Soluble Proteins Using Optically Reversible Protein–Protein Interaction Module. Nat. Commun. 2016, 7 (1), 12277. 10.1038/ncomms12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij F. J.; Revenu C.; Arras G.; Dingli F.; Loew D.; Pegtel D. M.; Follain G.; Allio G.; Goetz J. G.; Zimmermann P.; Herbomel P.; Del Bene F.; Raposo G.; van Niel G. Live Tracking of Inter-Organ Communication by Endogenous Exosomes in Vivo. Dev. Cell 2019, 48 (4), 573–589. 10.1016/j.devcel.2019.01.004. [DOI] [PubMed] [Google Scholar]
- Mei K.-C.; Bai J.; Lorrio S.; Wang J. T.-W.; Al-Jamal K. T. Investigating the Effect of Tumor Vascularization on Magnetic Targeting in Vivo using Retrospective Design of Experiment. Biomaterials 2016, 106, 276–285. 10.1016/j.biomaterials.2016.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukourakis M.; Koukouraki S.; Giatromanolaki A.; Archimandritis S.; Skarlatos J.; Beroukas K.; Bizakis J.; Retalis G.; Karkavitsas N.; Helidonis E. Liposomal Doxorubicin and Conventionally Fractionated Radiotherapy in the Treatment of Locally Advanced Non-Small-Cell Lung Cancer and Head and Neck Cancer. J. Clin. Oncol. 1999, 17 (11), 3512–3521. 10.1200/JCO.1999.17.11.3512. [DOI] [PubMed] [Google Scholar]
- Troy T.; Jekic-McMullen D.; Sambucetti L.; Rice B. Quantitative Comparison of the Sensitivity of Detection of Fluorescent and Bioluminescent Reporters in Animal Models. Mol. Imaging 2004, 10.1162/15353500200403196. [DOI] [PubMed] [Google Scholar]
- England C. G.; Ehlerding E. B.; Cai W. NanoLuc: A Small Luciferase Is Brightening up the Field of Bioluminescence. Bioconjugate Chem. 2016, 27 (5), 1175–1187. 10.1021/acs.bioconjchem.6b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol. 2012, 7 (11), 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoenviriyakul C.; Takahashi Y.; Morishita M.; Matsumoto A.; Nishikawa M.; Takakura Y. Cell Type-Specific and Common Characteristics of Exosomes Derived from Mouse Cell Lines: Yield, Physicochemical Properties, and Pharmacokinetics. Eur. J. Pharm. Sci. 2017, 96, 316–322. 10.1016/j.ejps.2016.10.009. [DOI] [PubMed] [Google Scholar]
- Rana S.; Yue S.; Stadel D.; Zöller M. Toward Tailored Exosomes: The Exosomal Tetraspanin Web Contributes to Target Cell Selection. Int. J. Biochem. Cell Biol. 2012, 44 (9), 1574–1584. 10.1016/j.biocel.2012.06.018. [DOI] [PubMed] [Google Scholar]
- Royo F.; Cossio U.; Ruiz de Angulo A.; Llop J.; Falcon-Perez J. M. Modification of the Glycosylation of Extracellular Vesicles Alters Their Biodistribution in Mice. Nanoscale 2019, 11 (4), 1531–1537. 10.1039/C8NR03900C. [DOI] [PubMed] [Google Scholar]
- Kamerkar S.; LeBleu V. S.; Sugimoto H.; Yang S.; Ruivo C. F.; Melo S. A.; Lee J. J.; Kalluri R. Exosomes Facilitate Therapeutic Targeting of Oncogenic KRAS in Pancreatic Cancer. Nature 2017, 546 (7659), 498–503. 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A.; Harris C. L.; Court J.; Mason M. D.; Morgan B. P. Antigen-Presenting Cell Exosomes Are Protected from Complement-Mediated Lysis by Expression of CD55 and CD59. Eur. J. Immunol. 2003, 33 (2), 522–531. 10.1002/immu.200310028. [DOI] [PubMed] [Google Scholar]
- Dasgupta S. K.; Abdel-Monem H.; Niravath P.; Le A.; Bellera R. V.; Langlois K.; Nagata S.; Rumbaut R. E.; Thiagarajan P. Lactadherin and Clearance of Platelet-Derived Microvesicles. Blood 2009, 113 (6), 1332–1339. 10.1182/blood-2008-07-167148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson R. J. A.; Balaj L.; Hulleman E.; van Rijn S.; Pegtel D. M.; Walraven M.; Widmark A.; Gerritsen W. R.; Verheul H. M.; Vandertop W. P.; Noske D. P.; Skog J.; Wurdinger T. Blood Platelets Contain Tumor-Derived RNA Biomarkers. Blood 2011, 118 (13), 3680–3683. 10.1182/blood-2011-03-344408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennakrishnaiah S.; Meehan B.; D'Asti E.; Montermini L.; Lee T-H.; Karatzas N.; Buchanan M.; Tawil N.; Choi D.; Divangahi M.; Basik M.; Rak J. Leukocytes as a Reservoir of Circulating Oncogenic DNA and Regulatory Targets of Tumor-Derived Extracellular Vesicles. J. Thromb. Haemostasis 2018, 16 (9), 1800–1813. 10.1111/jth.14222. [DOI] [PubMed] [Google Scholar]
- Hyenne V.; Ghoroghi S.; Collot M.; Bons J.; Follain G.; Harlepp S.; Mary B.; Bauer J.; Mercier L.; Busnelli I.; Lefebvre O.; Fekonja N.; Garcia-Leon M. J.; Machado P.; Delalande F.; López A. A.; Silva S. G.; Verweij F. J.; van Niel G.; Djouad F.; Peinado H.; Carapito C.; Klymchenko A. S.; Goetz J. G. Studying the Fate of Tumor Extracellular Vesicles at High Spatiotemporal Resolution Using the Zebrafish Embryo. Dev. Cell 2019, 48 (4), 554–572.e7. 10.1016/j.devcel.2019.01.014. [DOI] [PubMed] [Google Scholar]
- Hoshino A.; Costa-Silva B.; Shen T.-L.; Rodrigues G.; Hashimoto A.; Tesic Mark M.; Molina H.; Kohsaka S.; Di Giannatale A.; Ceder S.; Singh S.; Williams C.; Soplop N.; Uryu K.; Pharmer L.; King T.; Bojmar L.; Davies A. E.; Ararso Y.; Zhang T.; Zhang H.; Hernandez J.; Weiss J. M.; Dumont-Cole V. D.; Kramer K.; Wexler L. H.; Narendran A.; Schwartz G. K.; Healey J. H.; Sandstrom P.; Jørgen Labori K.; Kure E. H.; Grandgenett P. M.; Hollingsworth M. A.; de Sousa M.; Kaur S.; Jain M.; Mallya K.; Batra S. K.; Jarnagin W. R.; Brady M. S.; Fodstad O.; Muller V.; Pantel K.; Minn A. J.; Bissell M. J.; Garcia B. A.; Kang Y.; Rajasekhar V. K.; Ghajar C. M.; Matei I.; Peinado H.; Bromberg J.; Lyden D. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527 (7578), 329–335. 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunderson S. C.; Dunn A. C.; Crocker P. R.; McLellan A. D. CD169 Mediates the Capture of Exosomes in Spleen and Lymph Node. Blood 2014, 123 (2), 208–216. 10.1182/blood-2013-03-489732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escola J.-M.; Kleijmeer M. J.; Stoorvogel W.; Griffith J. M.; Yoshie O.; Geuze H. J. Selective Enrichment of Tetraspan Proteins on the Internal Vesicles of Multivesicular Endosomes and on Exosomes Secreted by Human B-Lymphocytes. J. Biol. Chem. 1998, 273 (32), 20121–20127. 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- Verweij F. J.; Bebelman M. P.; Jimenez C. R.; Garcia-Vallejo J. J.; Janssen H.; Neefjes J.; Knol J. C.; de Goeij-de Haas R.; Piersma S. R.; Baglio S. R.; Verhage M.; Middeldorp J. M.; Zomer A.; van Rheenen J.; Coppolino M. G.; Hurbain I.; Raposo G.; Smit M. J.; Toonen R. F.G.; van Niel G.; Pegtel D. M. Quantifying Exosome Secretion from Single Cells Reveals a Modulatory Role for GPCR Signaling. J. Cell Biol. 2018, 217 (3), 1129–1142. 10.1083/jcb.201703206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montermini L.; Meehan B.; Garnier D.; Lee W. J.; Lee T. H.; Guha A.; Al-Nedawi K.; Rak J. Inhibition of Oncogenic Epidermal Growth Factor Receptor Kinase Triggers Release of Exosome-Like Extracellular Vesicles and Impacts Their Phosphoprotein and DNA Content. J. Biol. Chem. 2015, 290 (40), 24534–46. 10.1074/jbc.M115.679217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh H.-W.; Karmach O.; Ji A.; Carter D.; Martins-Green M. M.; Ai H.-W. Red-Shifted Luciferase–Luciferin Pairs for Enhanced Bioluminescence Imaging. Nat. Methods 2017, 14 (10), 971. 10.1038/nmeth.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy P. J.; Radda G. K. Molecular Imaging Perspectives. J. R. Soc., Interface 2005, 2 (3), 133–144. 10.1098/rsif.2005.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert T. J. FPbase: A Community-Editable Fluorescent Protein Database. Nat. Methods 2019, 16 (4), 277–278. 10.1038/s41592-019-0352-8. [DOI] [PubMed] [Google Scholar]
- Hikita T.; Miyata M.; Watanabe R.; Oneyama C. Sensitive and Rapid Quantification of Exosomes by Fusing Luciferase to Exosome Marker Proteins. Sci. Rep. 2018, 8 (1), 14035. 10.1038/s41598-018-32535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.