

Abstract

Two series of novel 4-oxoquinazoline-based N-hydroxypropenamides (9a–m and 10a–m) were designed, synthesized, and evaluated for their inhibitory and cytotoxicity activities against histone deacetylase (HDAC). The compounds showed good to potent HDAC inhibitory activity and cytotoxicity against three human cancer cell lines (SW620, colon; PC-3, prostate; NCI-H23, lung cancer). In this series, compounds with the N-hydroxypropenamide functionality impeded at position 7 on the 4-oxoquinazoline skeleton (10a–m) were generally more potent than compounds with the N-hydroxypropenamide moiety at position 6 (9a–m). Also, the N3-benzyl-substituted derivatives (9h–m, 10h–m) exhibited stronger bioactivity than the N3-alkyl-substituted ones (9a–e, 10a–e). Two compounds 10l and 10m were the most potent ones. Their HDAC inhibitory activity (IC50 values, 0.041–0.044 μM) and cytotoxicity (IC50 values, 0.671–1.211 μM) were approximately 2- to 3-fold more potent than suberoylanilide hydroxamic acid (SAHA). Some compounds showed up to 10-fold more potent HDAC6 inhibition compared to their inhibitory activity in total HDAC extract assay. Analysis of selected compounds 10l and 10m revealed that these compounds strongly induced both early and late apoptosis and arrested SW620 cells at the G2/M phase. Docking studies were carried out on the HDAC6 isoform for series 10a–m and revealed some important features contributing to the inhibitory activity of synthesized compounds.
Introduction
Histone deacetylases (HDACs) and histone acetyltransferases (HATs) are two groups of enzymes that tightly control the epigenetic balance in cells.1 HATs catalyze the acetylation of the amino groups on the histone tails, causing chromatins to open and allowing transcription factors to access DNA, allowing gene expression.2 HDACs act in an opposite way by removal of the acetyl groups on the histone lysine amino tails, causing chromatins to close. When chromatins are closed, transcription factors cannot access DNA, leading to suppression of gene expression, especially the expression of tumor suppressor oncogenes.2,3 To date, 18 isoforms of HDACs have been identified in mammalians and their implication in the initiation and development of various cancer have been clearly demonstrated.3 HDACs, therefore, have become a validated and very attractive target for anticancer drug design and development.3 Consequently, intensive effort of research groups worldwide has led to the discovery of hundreds of HDAC inhibitors, which can be classified into several main classes, including hydroxamic acids (e.g., SAHA), short-chain fatty acids (e.g., valproic acid, phenylbutyric acid), cyclic peptide (e.g., Depsipeptide), and benzamides.4−9
In 2006, suberoylanilide hydroxamic acid (SAHA, Zoinza) was approved by the U.S. FDA as the first HDAC inhibitor to treat cutaneous T cell lymphoma. To date, several more HDAC inhibitors have also been approved by the U.S. FDA to treat different types of cancers. These include belinostat (PXD101), Romidepsin (Istodax), and Panobinostat (LBH-589, Farydak).7−10 In 2015, the Chinese FDA also approved chidamide (Epidaza) for the treatment of relapsed or refractory peripheral T cell lymphoma.11 Research on the development of novel HDAC inhibitors as anticancer agents is one of the most intense and attractive fields nowadays in anticancer drug discovery. As a result, a number of promising HDAC inhibitors, such as Givinostat (ITF2357), Mocetinostat (MGCD0103), and Entinostat (MS-27-527) (Figure 1) among others, are undergoing clinical trials at different phases.12−14
Figure 1.

Structures of some HDAC inhibitors approved or under clinical trials.
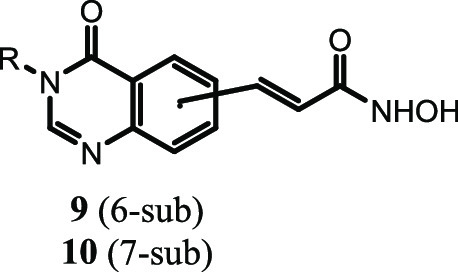
In our research program to discover novel HDAC inhibitors as antitumor agents, we have previously screened large-structure databases and synthesized several series of hydroxamic acids as analogues of SAHA, which incorporated heterocyclic systems, e.g., benzothiazole-, 5-aryl-3,4,5-thiadiazole-, or 2-oxoindoline.15−21 All of those compound series were found to exhibit very potent HDAC inhibitory activity as well as cytotoxicity and in vivo antitumor effects in nude mice inoculated with PC-3 human prostate cancer cells.19,21 Especially, a series of N-hydroxypropenamides as analogues of LBH-589 (Panbinostat) or PXD101 (Belinostat) were also found to be potential as HDAC inhibitors and antitumor agents.15,22 Inspired by these results, we expanded our design to the new series of N-hydroxypropenamides incorporating 4-oxoquinazoline system (Figure 2). The quinazoline heterocycle has been well known as a common scaffold embedded in the structures of diverse biological active compounds, especially anticancer agents (e.g., Gelfitinib, Erlotinib, Lapatinib, Sorafetinib).23 Recently, quinazoline-based hydroxamic acids designed as hybridized anticancer agents (e.g CUDC-101) have also been reported.24 We previously have also described several series of hydroxamic acids incorporating quinazoline or 4-oxoquinazoline systems with potent HDAC inhibitory and cytotoxic activities.16,25−28 The addition of the 4-oxo functional group on the quinazoline rings was aimed at creating more hydrogen-bond interaction of the compounds with the amino acid chains located at the entrance of the enzyme active binding pocket. This paper describes the results obtained from the synthesis, biological evaluation, and docking study of these novel N-hydroxypropenamides.
Figure 2.

Rational design of 4-oxoquinazoline-based N-hydroxypropenamides as HDAC inhibitors.
Results and Discussion
Chemistry
The target N-hydroxybenzamides incorporating substituted 4-oxoquinazolin-4(3H)-ones (9a–m and 10a–m) were synthesized via a four-step pathway, as illustrated in Scheme 1. The reaction between bromo-substituted anthranilic acid (1 or 2) and excess formamide at 120 °C yielded the corresponding 6- and 7-bromo-4-oxoquinazolines (3 or 4) in good yields (87–95%). The selective N-alkylation of 6/7-bromo-4-oxoquinazolines (3 or 4) was carried out using corresponding alkyl halide in the presence of potassium carbonate (K2CO3) and a catalytic amount of potassium iodide (KI). Introduction of the ethyl propenoate moiety at position 6 or 7 on the quinazoline ring was a key step. The α,β-unsaturated ester moiety was installed at C6 or C7 position from the corresponding bromo compound (5 or 6) and ethyl acrylate using Pd-catalyzed Heck cross-coupling strategy to yield precursor 7 or 8 in good yields (>80%).28b In the final step, the hydroxamic acids were obtained by reacting hydroxylamine hydrochloride with the ester intermediates 7 and 8. This was a nucleophilic acyl substitution reaction, which readily occurred under alkaline conditions (NaOH). The overall yields of compounds 9a–m and 10a–m were moderate to high (65-87%).
Scheme 1. Synthesis of 4-Oxoquinazoline-Based N-Hydroxypropenamides 9a–m and 10a–m.

The structures of the synthesized compounds were easily elucidated by analysis of spectroscopic data, including IR, MS, and 1H and 13C NMR. Theoretically, 4-oxoquinazolines (3 and 4) could react with alkyl bromides or benzyl bromides to form both N3- and O4-alkylated products, depending on the reaction conditions. However, it has been demonstrated that, using acetone as the reaction solvent, the alkylation gave only N3-alkylated products (5, 6).16,26,29−31 The NMR spectroscopic data also proved the formation of the N3-alkylated products. In the 13C NMR spectra of compounds 9a–m and 10a–m, there were peaks at around 162.5 ppm, attributable to the carbonyl carbons at position 4 of the 4-oxoquinazoline skeletons. For the O-alkylated products, the carbon at this position should appear at around 167–168 ppm. In the 1H NMR spectra of compounds 9h–m and 10h–m, always two protons appeared as a singlet around 5.15 ppm, corresponding to the benzyl’s methylene protons of the N3-alkylated compounds.16,26,29−31 In the 1H NMR spectra of compounds 9a–g and 10a–g, there were multiplet peaks around 3.90 ppm, which were also typical of the N3-alkylated compounds.32
Bioactivity
The target N-hydroxypropenamides 9a–m and 10a–m were subjected to Hela cell nuclear extract and the sulforhodamine B (SRB) assays to evaluate their HDAC inhibitory activity and cytotoxicity. In the SRB assays, there were three human cancer cell lines, including SW620 (colon cancer), PC3 (prostate cancer), and NCI-H23 (lung cancer). SAHA was used as a positive control. The results are summarized in Table 1.
Table 1. Inhibition of HDAC Activity and Cytotoxicity of the Synthesized Compounds against Several Cancer Cell Lines.

Calculated by ChemDraw 9.0 software.
Concentration (μM) of compounds that produces a 50% reduction in enzyme activity or cell growth; the numbers represent the averaged results from triplicate experiments.
Cell lines: SW620, colon cancer; PC3, prostate cancer; NCI-H23, lung cancer.
SAHA, suberoylanilide acid, a positive control.
Belinostat, a positive control.
Within series 9a–m, two compounds 9a and 9b appeared to be the least potent HDAC inhibitors. These compounds showed IC50 values of 1.354 and 2.385 μM, approximately 10- and 20-fold higher than that of SAHA. However, replacement of the ethyl group by 2-hydroxyethyl, n-propyl, n-butyl, cyclopentylmethyl, and especially cyclohexylmethyl substituents (compounds 9c–g) significantly enhanced the inhibition of HDAC, as evidenced from the IC50 values of these compounds in Hela cell nuclear extract assay (0.974–0.391 μM). From the above results, it appeared that bulkier substituents, such as cyclopentylmethyl and cyclohexylmethyl, were more favorable for HDAC inhibition. The benzyl and substituted benzyls even enhanced HDAC inhibitory activity more significantly (compounds 9h–m, IC50 values of 0.588–0.285 μM).
Regarding the results from SRB assays, it was found that all compounds showed good cytotoxicity in three human cancer cell lines tested. There was relatively fine correlation between HDAC inhibitory activity and cytotoxicity within the series. Two compounds 9a and 9b with the highest IC50 values in HDAC inhibition assays were also the least cytotoxic ones (IC50 values, 3.204–7.044 μM). Compounds 9e–m were more potent with cytotoxic IC50 values in low, even submicromolar range. The cytotoxic potency of these compounds was comparable to that of SAHA (IC50 values, 1.448–1.658 μM). Compound 9i was the most potent in the series in terms of cytotoxicity (IC50 values, 0.639–1.122 μM).
Compounds in series 10a–m generally exhibited better HDAC inhibitory activity in comparison to series 9a–m. The IC50 values of these compounds in Hela cell nuclear extract assays were in the range of 0.041–0.247 μM, much lower than that of series 9a–m. Nine out of 13 compounds in series, including 10a, 10c, 10d, 10f, and 10i–m, were more potent than SAHA in terms of HDAC inhibitory activity, as manifested by their lower IC50 values (0.041–0.098 μM) compared to SAHA’s IC50 value (0.121 μM). The cytotoxicity of compounds 10a–m was also generally stronger than compounds in series 9a–m. 3-Benzyl-substituted derivatives (10h–m) displayed significantly higher cytotoxicity in all three human cancer cell lines tested. Two compounds 10l and 10m were the most potent ones. Their HDAC inhibitory activity (IC50 values, 0.041–0.044 μM) and cytotoxicity (IC50 values, 0.671–1.211 μM) were approximately 2- to 3-fold more potent than SAHA (IC50 values, 0.121 μM/HDAC and 1.448–1.658 μM/cancer cell lines).
A fluorogenic HDAC Assay Kit using an HDAC Hela cell nuclear extract, a rich source of HDAC activity, contains predominantly class-I isoforms (HDAC1, 2, 3, and 8).33 Recently, great interest is focused on HDAC6 inhibition since in contrast to most other HDACs, which are transiently or permanently localized in the nucleus, HDAC6 is localized exclusively in the cytoplasm. HDAC6 has two main substrates, including α-tubulin (a protein involved in cytoskeletal structural integrity and cellular motility) and Hsp90 (heat shock protein, a protein that helps client proteins fold properly and maintain their function).34 Therefore, it is expected that inhibition of HDAC6 would regulate α-tubulin and Hsp pathways without affecting DNA modification, thus causing minimal side effects. We therefore decided to evaluate compounds 10a–m for their inhibitory effects against HDAC6 isoform to primarily understand the role of HDAC6. The results are summarized in Table 2. It is very interesting to note that six compounds, including 10e–j, showed slightly potent inhibitory activity against HDAC6 (IC50 values of 0.012–0.040 μM), compared to HDAC inhibition using a Hela cell nuclear extract (IC50 values of 0.072–0.247 μM). However, this small difference indicates that the compounds are nonselective inhibitors. Noteworthy, compound 10g inhibited HDAC6 with an IC50 value of 0.012 μM, 10-fold lower than its IC50 value in Hela nuclear extract assay (0.124 μM). It should be noted that Hela cell nuclear extract contains predominantly class-I isoforms (HDAC1, 2, 3, and 8); therefore, the synthesized compounds exhibited mainly as pan-HDAC inhibitors. Among them, compound 10g proved to be potential as a good template for further development of HDAC6 selective inhibitors.
Table 2. Inhibition of HDAC6 Activity by Compounds 10a–m.
| cpd. code | HDAC (Hela extract) inhibition (IC50,a μM) | HDAC6 inhibition (IC50,a μM) | cpd. code | HDAC (Hela extract) inhibition (IC50,a μM) | HDAC6 inhibition (IC50,a μM) |
|---|---|---|---|---|---|
| 10a | 0.068 ± 0.001 | 0.080 ± 0.000 | 10h | 0.247 ± 0.021 | 0.040 ± 0.001 |
| 10b | 0.113 ± 0.000 | 0.322 ± 0.013 | 10i | 0.098 ± 0.002 | 0.025 ± 0.001 |
| 10c | 0.091 ± 0.005 | 0.096 ± 0.006 | 10j | 0.096 ± 0.001 | 0.023 ± 0.000 |
| 10d | 0.067 ± 0.002 | 0.048 ± 0.002 | 10k | 0.048 ± 0.000 | 0.042 ± 0.001 |
| 10e | 0.124 ± 0.012 | 0.029 ± 0.000 | 10l | 0.041 ± 0.003 | 0.033 ± 0.000 |
| 10f | 0.072 ± 0.001 | 0.022 ± 0.000 | 10m | 0.044 ± 0.000 | 0.047 ± 0.001 |
| 10g | 0.124 ± 0.004 | 0.012 ± 0.000 | SAHAb | 0.121 ± 0.031 | 0.131 ± 0.001 |
Concentration (μM) of compounds that produces a 50% reduction in enzyme activity.
SAHA, suberoylanilide acid, a positive control.
The results from the above SRB assays demonstrate that two compounds 10l and 10m exhibited the most potent HDAC inhibition and cytotoxicity against three human cancer cell lines. Therefore, we selected these two compounds to analyze their effects on the cell cycle and apoptosis. In these experiments, SW620 human colon cancer cells were used. Flow cytometry was employed to analyze the cell cycle. Initially, SW620 cells were preincubated for 24 h. Then, the cells were treated with each compound (10 μM) or SAHA (1 μM) for 24 h. After that, the DNA contents were analyzed. It was found that two compounds 10l and 10m killed significant population of SW620 cells at the G0/G1 phase (30.60 and 32.20%, respectively). Within viable cell population, compounds 10l and 10m substantially arrested SW620 cells at the G2/M phase (26.70 and 30.11%, respectively, vs. 6.29% of the VH). In contrast, SAHA caused cells (61.95%) more accumulated at the G0/G1 phase (Figure 3). This observation might suggest some differences in the cytotoxic mechanism of compounds 10l and 10m in comparison to that of SAHA.
Figure 3.

Cell cycle analysis of representative compounds 10l and 10m. SW620 cells (human colon cancer) (2 × 105 cells) were treated with compounds (10 μM) or SAHA (1 μM) for 24 h. The harvested cells were stained with propidium iodide (PI) in the presence of RNase and then were analyzed for DNA content. UN: untreated, VH: vehicle (dimethyl sulfoxide, DMSO 0.05%). Data were represented as histograms (left) and bar graphs (right).
To investigate the effects of the compounds on the cell apoptosis, we carried out an Annexin V-FITC/propidium iodide (PI) dual staining assay. This assay is based on phosphatidylserine (PS), one component of the cell membrane, which plays an important role in cell cycle signaling, especially pathway related to cellular apoptosis. During early apoptosis, PS, which is located on the cytosolic (inner) side of the cell membrane, translocates to the extracellular (outer) side of the cell membrane. Annexin V exhibits a high affinity for PS. Therefore, annexin V fluorescently labeled with fluorescein isothiocyanate (Annexin V-FITC) has been used to identify early apoptotic cells. Meanwhile, propidium iodide (PI), a fluorescent intercalating agent, cannot cross the membrane of live cells. Therefore, the membranes of dead cells or cells in the later stages of apoptosis are permeable to PI. SW620 cells were treated with compounds at 10 μM for 24 h, and the cells were stained with Annexin V-FITC and PI. The results demonstrate that compounds 10l and 10m substantially induced both early and late apoptosis in SW620 cells (Figure 4).
Figure 4.

Apoptosis (Annexin V/PI) analysis of representative compounds 10l and 10m. SW620 cells (human colon cancer) (2 × 105 cells) were treated with compounds (10 μM) or SAHA (1 μM) for 24 h. The harvested cells were stained with propidium iodide (PI) in the presence of RNase and then were analyzed for DNA content. UN: untreated, VH: vehicle (DMSO 0.05%). Area 1 = PI positive population, Area 2: Annexin V-positive population. Data were represented as histograms (left) and bar graphs (right).
Regarding cell morphology, it was found that compounds 10l and 10m caused similar morphological changes in the shape of SW620 cells as SAHA did (Figure 5).
Figure 5.

Morphology changes of cells treated with representative compounds 10l and 10m. SW620 (human colon cancer) cells (2 × 105 cells/well in a six-well plate, preincubated for 2 h) were treated with compounds (10 μM) or SAHA (1 μM) for 24 h. Then, the cells were photographed using an imaging device: Biostation with 20× lens. Scale bar: 50 μm.
Docking Studies
As can be seen in the experimental evaluations, compounds from series 10 appeared to be potential inhibitors of class-I and HDAC6 isoform equally. Some of them, e.g., 10h, 10l, and 10m exhibited higher cytotoxicity, and HDAC inhibitory activities in comparison to SAHA, a well-known nonselective pan-HDAC inhibitor. Human HDAC6 (Uniprot Q9UBN7) is the largest member of the metal-dependent HDAC family, which comprises two catalytic domains, namely, CD1 and CD2,35 in addition to a ubiquitin binding domain. Even though both domains are required for deacetylase activity, only CD2 is a tubulin deacetylase and a tau deacetylase, and the development of HDAC6 selective inhibitors has focused exclusively on this domain. To gain more insight into the structure–activity relationships, we decided to perform comparative docking experiments for compounds of series 10 against zebrafish HDAC6 CD2 (PDB ID: 5EEN).36
To validate the docking protocol, redocking simulations were performed with the co-crystal Belinostat. The snapshot from the superimposition of the redocked and co-crystallized Belinostat shown in Figure 6B revealed the suitability of the docking procedures applied. The redocked orientation was highly overlapped with the native ligand with the root-mean-square deviation of 0.8110Å and similar interactions such as H-bonds with residues His573, His574, Tyr745, and hydrophobic interactions with Pro464, Phe583, and Phe643. In the same manner, SAHA was docked into the active site of HDAC6 and showed key interactions with zinc ion and important catalytic residues His573, His574, and Tyr745 at the bottom of the pocket.22,25,36 The lower binding energy of SAHA-HDAC6 in comparison to Belinostat could be attributed to the lack of multiple stacking contacts with backbone Phe583 and Pro464. The target binding energies of SAHA and Belinostat were calculated via the London and affinity scoring function implemented into MOE software (see Table 3).37
Figure 6.

(A) Binding orientation of reference compounds (Belinostat and SAHA); superposition of (B) redocked (stick representation with yellow carbons) and co-crystal Belinostat (green carbons); and (C) between co-crystal Belinostat and docked SAHA (blue carbon), in the active site of HDAC6. Zinc ions are shown as gray spheres.
Table 3. Docking Scores of All Compounds with HDAC6 Enzyme.
| distance
to Zn2+a |
distance
to Zn2+ |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| cpd. code | E_score1b | E_score2b | –OH | =O | cpd. code | E_score1 | E_score2 | –OH | =O |
| 10a | –17.348 | –10.056 | 1.97 | 2.33 | 10h | –17.456 | –11.116 | 1.97 | 2.34 |
| 10b | –11.083 | –9.090 | 1.97 | 2.33 | 10i | –16.092 | –11.130 | 1.97 | 2.34 |
| 10c | –14.148 | –10.153 | 1.97 | 2.34 | 10j | –16.649 | –10.895 | 1.97 | 2.33 |
| 10d | –14.908 | –9.914 | 1.97 | 2.33 | 10k | –16.321 | –10.740 | 1.97 | 2.13 |
| 10e | –16.303 | –11.231 | 1.97 | 2.33 | 10l | –18.326 | –11.157 | 1.97 | 2.35 |
| 10f | –15.676 | –10.930 | 1.97 | 2.32 | 10m | –17.566 | –10.428 | 1.97 | 2.33 |
| 10g | –17.793 | –10.944 | 1.96 | 2.32 | SAHA | –14.436 | –9.447 | 1.96 | 2.31 |
| Belinostat | –16.147 | –10.424 | 1.97 | 2.31 | |||||
The docking score (kcal/mol) calculated from the London (with refinement) and affinity scoring function from MOE software.
Distances (Å) from oxygen atoms (−O and =OH) of hydroxamate group to zinc ion.
In the subsequent steps, the same protocol mentioned above was applied for investigating the binding modes of active compounds 10a–m against HDAC6 enzyme. In general, all of the compounds appropriately bound to the active site of HDAC6 with the key interactions such as bidentate hydroxamate-Zn2+ coordination mode, and H-bonding networks between hydroxamates toward His573, His574, Asp612, and Tyr745 (Figure 7A). By incorporating 4-oxoquinazoline moieties into the linker region, most of the compounds displayed two pi–pi stacking contacts with Phe583 and Phe643, which are similar to Belinostat.36 In addition, the carboxyl group of 4-oxoquinazoline moiety appeared to be important for substrate recognition as it formed one more H-bond with Ser531 (e.g., 10l and 10m). The role of Ser531 in target binding has recently been demonstrated in several reports.36,38 Interestingly, 10b and 10c could not participate in the interactions with Ser531. We observed that their 4-oxoquinazoline moieties were attracted by pi–pi interaction with Phe583 and pi-alkyl contact with Leu712. These stacking interactions could contribute to the different binding mode of 10b from SAHA and the other derivatives in the active site of HDAC6.38 On the other hand, substitutions at phenyl capping groups showed little impact on the binding mode of 10h–m. The structural superimposition in Figure 7A of 10h–m confirmed a high similarity of binding modes of 3-benzyl-substituted derivatives at the HDAC6 CD2 active site.
Figure 7.

(A) Alignment of docking poses of compounds 10a–m toward SAHA; docking conformation and interactions of compounds 10l (B) and 10m (C) in HDAC6 active site.
Finally, we continued analyzing the correlation between experimental values and theoretical binding energies estimated through docking simulations. In this study, the binding energies were estimated for the complex using London (E_score1, kcal/mol) and affinity (E_score2, kcal/mol) scoring functions. A more detailed description of these functions can be found elsewhere.16,37 As can be seen in Figure 8, for 10a–m and SAHA, E_score1 and E_score2, having acceptable correlation coefficients (R2 = 0.76–0.77), showed similar rank-order relationships with experimental IC50 values, suggesting their ability to quantitatively predict experimental in vitro HDAC6 inhibitory activities.
Figure 8.

Correlation graph between experimental IC50 values and docking scores of 10a–m and SAHA.
Conclusions
In conclusion, we have synthesized two series of novel 4-oxoquinazoline-based N-hydroxypropenamides (9a–m and 10a–m). Their HDAC inhibitory effects and cytotoxicity against three human cancer cell lines, including SW620 (human colon cancer), PC-3 (prostate cancer), and NCI-H23 (lung cancer), have been evaluated. The compounds were found to exhibit good HDAC inhibitory activity as well as cytotoxicity. Compounds with the N-hydroxypropenamide functionality impeded at position 7 on the 4-oxoquinazoline skeleton (10a–m) were generally more potent than compounds with the N-hydroxypropenamide moiety at position 6 (9a–m). Also, the N3-benzyl-substituted derivatives (9h–m and 10h–m) exhibited stronger bioactivity than the N3-alkyl-substituted ones (9a–e, 10a–e). Among the compounds in two series, 10l and 10m were two most potent compounds, in terms of both HDAC inhibition and cytotoxicity. Their HDAC inhibitory activity (IC50 values, 0.041–0.044 μM) and cytotoxicity (IC50 values, 0.671–1.211 μM) were approximately 2- to 3-fold more potent than SAHA. Compounds 10l and 10m also strongly induced both early and late apoptosis and arrested SW620 cells in the G2/M phase. Docking studies were carried out with the series 10a–m and showed some important features for HDAC6-substrate recognition as well as to quantitatively predict experimental in vitro HDAC6 inhibitory activities. Based on the results obtained, two inhibitors 10l and 10m were identified as potential hit compounds for further evaluation and development as HDAC targeting anticancer agents.
Experimental Section
Chemistry
The synthesis procedure of 4-oxoquinazoline-based N-hydroxypropenamide series (9a–m and 10a–m) was carried out as illustrated in Scheme 1. The details are described below.
General Procedures for the Synthesis of Compounds 9a–m
The mixtures of 5-bromo-2-aminobenzoic acid (1) (3 mmol) and formamide (5 mmol) were stirred at 120 °C for 6 h. Upon completion, the resulting mixture was cooled and poured into ice-cold water (50 mL). A solution of NaHCO3 5% was gradually added to adjust pH to 7, which led to the formation of light brown solids. The solids were filtered and washed with water (3 times) to give the corresponding quinazolinone derivative 3, which was used for the next step without further purification.
To a respective solution of 4-oxoquinazoline derivatives 3 (2 mmol) in dimethylformamide (DMF) (5 mL) was added K2CO3 (342.5 mg, 2.5 mmol). The resulting mixture was stirred at 60 °C for 1 h; then, a catalytic amount of KI (49.8 mg, 0.3 mmol) was added. After stirring for further 15 min, alkyl bromide (2 mmol) was added. The reaction mixture was again stirred at 60 °C for 5 h until the reaction completed. After completion of the reaction, the resulting mixtures were cooled and poured into ice-cold water (20 mL). The precipitates were obtained, washed with water, and dried to give a respective compound (intermediates 5a–m, yields: 80–94%).
In the next step, the respective solution of compounds 5a–m (1.5 mmol) was dissolved in 5 mL of DMF; then, dried triethylamine (0.5 mL) and ethyl acrylate (0.5 mL) were added. After stirring for further 15 min, a solution of triphenylphosphine (105 mg, 0.4 mmol in 0.5 mL of DMF) and palladium diacetate (0.2 mmol, 45 mg) was added and the reaction mixtures were again stirred at 110 °C until the reaction completed (6 h, checked by thin-layer chromatography, TLC). After that, the reaction mixtures were poured into ice-cold water (10 mL). The obtained brown precipitates were washed with water and dried at 40 °C under vacuum for 24 h. The crude products were then purified by column chromatography (silica gel; DCM/methanol = 100/5) to give the corresponding intermediate esters 7a–m in yields of 64–77%.
Each of the intermediate esters 7a–m was dissolved in methanol (10 mL). Then, hydroxylamine. HCl (685 mg, 10 mmol) was added, followed by dropwise addition of a solution of NaOH (400 mg in 1 mL of water). The mixture was stirred at −5 °C until the reaction completed (1–2 h, checked by TLC). At the end of this reaction, the resulting reaction mixture was poured into ice-cold water, neutralized to pH ∼ 7, and acidified by dropwise addition of a solution of HCl 5% to induce the maximum precipitation. The precipitates were filtered, dried, and recrystallized in methanol to give the target compounds 9a–m.
(E)-3-(3-Ethyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9a)
White solid; Yield: 62%. mp: 168-169 °C. Rf = 0.45 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3277 (OH); 3046, 2968 (CH, aren); 2922, 2851 (CH, CH2); 1655 (C=O); 1599, 1551, 1539 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.71 (s, 1H, NHOH); 9.03 (s, 1H, NHOH); 8.35 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.91 (dd, J = 8.50 Hz, J′ = 1.50 Hz, 1H, H-7′); 7.60 (d, J = 8.50 Hz, 1H, H-8′); 7.50 (d, J = 16.00 Hz, 1H, H-3); 6.51 (d, J = 15.50 Hz, 1H, H-2); 3.96-3.91 (m, 2H, H-1″a, H-1″b); 1.21 (t, J = 7.25 Hz, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.90, 160.31, 149.05, 148.88, 143.45, 133.94, 133.27, 128.36, 125.28, 122.35, 120.94, 49.07, 14.94. HRMS (ESI) m/z calculated for C13H14N3O3, [M + H]+ 260.1035. Found, 260.1028.
(E)-3-(3-(2-Chloroethyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9b)
White solid; Yield: 61%. mp: 169-170 °C. Rf = 0.46 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3213 (OH); 2986, 2901 (CH, aren); 1678 (C=O); 1603, 1555 ( (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.69 (s, 1H, NHOH); 9.02 (s, 1H, NHOH); 8.21 (s, 1H, H-5′); 8.13 (s, 1H, H-2′); 7.91 (d, J = 9.00 Hz, 1H, H-7′); 7.58 (d, J = 8.50 Hz, 1H, H-8′); 7.48 (d, J = 15.50 Hz, 1H, H-3); 6.49 (d, J = 16.00 Hz, 1H, H-2); 4.04 (t, J = 10.00 Hz, 2H, H-2″a, H-2″b); 3.68 (t, J = 10.50 Hz, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.86, 160.79, 149.02, 148.95, 137.32, 134.07, 133.30, 128.41, 125.36, 122,20, 121.08, 49.05, 45.60. HRMS (ESI) m/z calculated for C13H11ClN3O3, [M + H]− 292.0489, 294.0459. Found, 292.0486, 294.0457.
(E)-N-Hydroxy-3-(3-(2-hydroxyethyl)-4-oxo-3,4-dihydroquinazolin-6-yl)acrylamide (9c)
White solid; Yield: 57%. mp: 174-175 °C. Rf = 0.35 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3358 (NH); 3240 (OH); 2918 (CH, aren); 2853 (CH, CH2); 1651, 1626 (C=O); 1607, 1553 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.86 (s, 1H, NHOH); 9.04 (s, 1H, NHOH); 8.22 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.91 (d, J = 8.00 Hz, 1H, H-7′); 7.61 (d, J = 8.50 Hz, 1H, H-8′); 7.49 (d, J = 16.00 Hz, 1H, H-3); 6.58 (d, J = 15.50 Hz, 1H, H-2); 4.95 (t, J = 5.00 Hz, 1H, 2′-OH); 3.97 (t, J = 5.00 Hz, 2H, H-1″a, H-1″b); 3.60-3.57 (m, 2H, H-2″a, H-2″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.89, 160.58, 149.62, 149.10, 137.32, 133.89, 133.19, 128.31, 125.37, 122.41, 121.09, 49.13, 34.46. HRMS (ESI) m/z calculated for C13H14N3O4, [M + H]+ 276.0984. Found, 276.0967.
(E)-N-Hydroxy-3-(4-oxo-3-propyl-3,4-dihydroquinazolin-6-yl)acrylamide (9d)
White solid; Yield: 65%. mp: 177-178 °C. Rf = 0.44 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%.IR (KBr, cm–1): 3564 (NH); 3235 (OH); 3065, 2978 (CH, aren); 2880 (CH, CH2); 1657, 1626 (C=O); 1609, 1553 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.71 (s, 1H, NHOH); 9.02 (s, 1H, NHOH); 8.33 (s, 1H, H-2′); 8.20 (d, J = 1.00 Hz, 1H, H-5′); 7.91 (dd, J = 8.50 Hz, J′ = 1.50 Hz, 1H, H-7′); 7.61 (d, J = 8.50 Hz, 1H, H-8′); 7.50 (d, J = 15.50 Hz, 1H, H-3); 6.51 (d, J = 16.00 Hz, 1H, H-2); 3.86 (t, J = 7.25 Hz, 2H, H-1″a, H-1″b); 1.67–1.60 (m, 2H, H-2″a, H-2″b); 0.81 (t, J = 7.50 Hz, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.90, 160.45, 149.11, 148.99, 137.44, 133.98, 133.30, 128.37, 125.34, 122.32, 120.96, 48.02, 22.37, 11.30. HRMS (ESI) m/z calculated for C14H16N3O3, [M + H]+ 274.1192. Found, 274.1183.
(E)-3-(3-Butyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9e)
White solid; Yield: 69%. mp: 181-182 °C. Rf = 0.45 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3215 (OH); 3096, 2955 (CH, aren); 2870 (CH, CH2); 1678, 1659 (C=O); 1603, 1549 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.71 (s, 1H, NHOH); 9.03 (s, 1H, NHOH); 8.33 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.91 (dd, J = 8.50 Hz, J′ = 1.50 Hz, 1H, H-7′); 7.61 (d, J = 8.50 Hz, 1H, H-8′); 7.50 (d, J = 16.00 Hz, 1H, H-3); 6.51 (d, J = 15.50 Hz, 1H, H-2); 3.90 (t, J = 7.25 Hz, 2H, H-1″a, H-1″b); 1.62-1.57 (m, 2H, H-2″a, H-2″b); 1.27-1.19 (m, 2H, H-3″a, H-3″b); 0.83 (t, J = 7.25 Hz, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.90, 160.47, 149.07, 148.98, 137.46, 133.98, 133.27, 128.37, 125.36, 122.31, 120.95, 46.24, 31.16, 19.75, 14.01. HRMS (ESI) m/z calculated for C15H18N3O3, [M + H]+ 288.1388. Found, 288.1339.
(E)-3-(3-(Cyclopentylmethyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9f)
White solid; Yield: 66%. mp: 191-192 °C. Rf = 0.43 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3213 (OH); 3067, 2932 (CH, aren); 2868 (CH, CH2); 1680, 1657 (C=O); 1601, 1547 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.70 (s, 1H, NHOH); 9.03 (s, 1H, NHOH); 8.35 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.91 (d, J = 8.00 Hz, 1H, H-7′); 7.61 (d, J = 8.50 Hz, 1H, H-8′); 7.50 (d, J = 15.50 Hz, 1H, H-3); 6.51 (d, J = 16.00 Hz, 1H, H-2); 3.85 (t, J = 7.50 Hz, 2H, H-1″a, H-1″b); 2.30-2.24 (m, 1H, H-2″); 1.56-1.18 (m, 8H, H-3″a, H-3″b, H-4″a, H-4″b, H-5″a, H-5″b, H-6″a, H-6″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.87, 160.60, 149.12, 148.91, 137.41, 134.02, 133.30, 128.38, 125.39, 122.30, 120.97, 50.68, 49.07, 29.97, 24.88. HRMS (ESI) m/z calculated for C17H20N3O3, [M + H]+ 314.1505. Found, 314.1494.
(E)-3-(3-(Cyclohexylmethyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9g)
White solid; Yield: 69%. mp: 201-202 °C. Rf = 0.45 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3217 (OH); 2986, 2924 (CH, aren); 2857 (CH, CH2); 1680, 1657 (C=O); 1605, 1547 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.71 (s, 1H, NHOH); 9.03 (s, 1H, NHOH); 8.28 (s, 1H, H-2′); 8.19 (s, 1H, H-5′); 7.91 (d, J = 8.00 Hz, 1H, H-7′); 7.61 (d, J = 8.00 Hz, 1H, H-8′); 7.50 (d, J = 15.50 Hz, 1H, H-3); 6.51 (d, J = 16.00 Hz, 1H, H-2); 3.75 (t, J = 6.50 Hz, 2H, H-1″a, H-1″b); 1.72-1.70 (m, 1H, H-2″); 1.58-0.90 (m, 10H, H-3″a, H-3″b, H-4″a, H-4″b, H-5″a, H-5″b, H-6″a, H-6″b, H-7″a, H-7″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.90, 160.61, 149.29, 148.91, 137.45, 134.01, 133.28, 128.37, 125.44, 122.29, 120.94, 52.06, 49.07, 30.31, 26.31, 25.61. HRMS (ESI) m/z calculated for C18H22N3O3, [M + H]+ 328.1661. Found, 328.1649.
(E)-3-(3-Benzyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9h)
White solid; Yield: 67%. mp: 189-190 °C. Rf = 0.43 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3235 (OH); 2986, 2901 (CH, aren); 1661 (C=O); 1597, 1553 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.71 (s, 1H, NHOH); 9.03 (s, 1H, NHOH); 8.51 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.93 (d, J = 7.50 Hz, 1H, H-7′); 7.63 (d, J = 8.50 Hz, 1H, H-8′); 7.49 (d, J = 15.50 Hz, 1H, H-3); 7.29-7.20 (m, 5H, H-3″, H-4″, H-5″, H-6″, H-7″); 6.50 (d, J = 16.00 Hz, 1H, H-2); 5.13 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.52, 160.43, 149.01, 148.93, 137.27, 137.19, 134.22, 133.46, 129.14, 128.48, 128.20, 128.14, 125.47, 122.43, 121.10, 49.43. HRMS (ESI) m/z calculated for C18H14N3O3, [M + H]− 320.1035. Found, 320.1030.
(E)-3-(3-(2-Fluorobenzyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9i)
White solid; Yield: 67%. mp: 206-207 °C. Rf = 0.44 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3321, 3200 (OH); 2974 (CH, aren); 2899 (CH, CH2); 1667 (C=O); 1601 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 8.46 (s, 1H, H-2′); 8.17 (s, 1H, H-5′); 7.94 (d, J = 8.00 Hz, 1H, H-7′); 7.64 (d, J = 8.50 Hz, 1H, H-8′); 7.49 (d, J = 15.50 Hz, 1H, H-3); 7.30-7.07 (m, 4H, H-4″, H-5″, H-6″, H-7″); 6.51 (d, J = 16.00 Hz, 1H, H-2); 5.17 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.78, 160.36, 149.07, 148.88, 137.27, 134.24, 133.47, 130.49, 130.46, 130.42, 130.36, 128.48, 125.47, 125.07, 125.04, 123.80, 123.69, 122.36, 121.12, 116.00, 115.84, 44.39. HRMS (ESI) m/z calculated for C18H15FN3O3, [M + H]+ 340.1097. Found, 340.1090.
(E)-3-(3-(3-Fluorobenzyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9j)
White solid; Yield: 65%. mp: 208-209 °C. Rf = 0.46 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3524 (NH); 3181 (OH); 3061, 2990 (CH, aren); 2899 (CH, CH2); 1667, 1632 (C=O); 1605, 1555 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.73 (s, 1H, NHOH); 9.04 (s, 1H, NHOH); 8.52 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.93 (d, J = 8.00 Hz, 1H, H-7′); 7.63 (d, J = 8.00 Hz, 1H, H-8′); 7.50 (d, J = 16.00 Hz, 1H, H-3); 7.35-7.03 (m, 4H, H-3″, H-5″, H-6″, H-7″); 6.51 (d, J = 15.50 Hz, 1H, H-2); 5.13 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.80, 160.37, 148.94, 142.82, 139.94, 139.88, 137.36, 134.23, 133.64, 133.49, 133.39, 131.92, 131.19, 131.13, 129.92, 129.40, 128.47, 128.43, 127.94, 127.23, 125.50, 124.19, 124.17, 122.41, 121.22, 121.10, 119.56, 115.19, 115.13, 115.02, 114.96, 49.05. HRMS (ESI) m/z calculated for C18H15FN3O3, [M + H]+ 340.1097. Found, 340.1087.
(E)-3-(3-(4-Fluorobenzyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9k)
White solid; Yield: 63%. mp: 209-210 °C. Rf = 0.45 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3547 (NH); 3227 (OH); 3061, 2988 (CH, aren); 2918 (CH, CH2); 1667, 1632 (C=O); 1603, 1555 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.73 (s, 1H, NHOH); 9.04 (s, 1H, NHOH); 8.53 (s, 1H, H-2′); 8.20 (s, 1H, H-5′); 7.93 (d, J = 8.00 Hz, 1H, H-7′); 7.62 (d, J = 8.50 Hz, 1H, H-8′); 7.50 (d, J = 16.00 Hz, 1H, H-3); 7.38-7.36 (m, 2H, H-3″, H-7″); 7.11-7.08 (m, 2H, H-4″, H-6″); 6.51 (d, J = 15.50 Hz, 1H, H-2); 5.11 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.16, 160.43, 148.90, 134.21, 133.45, 133.41, 133.38, 132.00, 131.92, 130.55, 130.49, 129.28, 129.19, 128.47, 125.49, 122.42, 121.09, 115.99, 115.82, 48.82. HRMS (ESI) m/z calculated for C18H15FN3O3, [M + H]+ 340.1097. Found, 340.1089.
(E)-3-(3-(4-Chlorobenzyl)-4-oxo-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide (9l)
White solid; Yield: 65%. mp: 210-211 °C. Rf = 0.49 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3213 (OH); 3065, 2986 (CH, aren); 2922 (CH, CH2); 1682, 1659 (C=O); 1601, 1553 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 8.52 (s, 1H, H-2′); 8.18 (s, 1H, H-5′); 7.93 (d, J = 7.00 Hz, 1H, H-7′); 7.63 (d, J = 8.00 Hz, 1H, H-8′); 7.48 (d, J = 15.50 Hz, 1H, H-3); 7.33-7.26 (m, 4H, H-3″, H-4″, H-6″, H-7″); 6.51 (d, J = 15.00 Hz, 1H, H-2); 5.11 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.72, 160.43, 149.01, 148.88, 136.99, 136.18, 134.32, 133.45, 132.85, 130.15, 129.08, 128.48, 125.39, 122.41, 121.27, 48.89. ESI-MS m/z: 356.0791 [M + H]+. HRMS (ESI) m/z calculated for C18H15ClN3O3, [M + H]+ 356.0802, 358.0772. Found, 356.0893, 358.0761.
(E)-N-Hydroxy-3-(3-(4-methylbenzyl)-4-oxo-3,4-dihydroquinazolin-6-yl)acrylamide (9m)
White solid; Yield: 66%. mp: 184-185 °C. Rf = 0.47 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3202 (OH); 3057, 2988 (CH, aren); 2911 (CH, CH2); 1694, 1649 (C=O); 1599, 1553 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.78 (s, 1H, NHOH); 9.15 (s, 1H, NHOH); 8.57 (s, 1H, H-2′); 8.28 (s, 1H, H-5′); 8.01 (d, J = 8.00 Hz, 1H, H-7′); 7.70 (d, J = 8.50 Hz, 1H, H-8′); 7.57 (d, J = 16.00 Hz, 1H, H-3); 7.26 (d, J = 8.00 Hz, 2H, H-3″, H-7″); 7.15 (d, J = 8.00 Hz, 2H, H-4″, H-6″); 6.55 (d, J = 16.00 Hz, 1H, H-2); 5.16 (s, 2H, H-1″a, H-1″b); 2.26 (s, 3H, 5″-CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.80, 160.39, 148.96, 148.91, 137.49, 137.31, 134.21, 133.41, 129.67, 128.46, 128.23, 128.14 125.47, 122.42, 121.08, 49.16, 21.14. HRMS (ESI) m/z calculated for C19H18N3O3, [M + H]+ 336.1348. Found, 336.1340.
General Procedures for the Synthesis of Compounds 10
Compounds 10a–g were synthesized via a four-step pathway, as illustrated in Scheme 1. The procedures were similar to that described for compound 9 with 4-bromo-2-aminobenzoic acid used instead of 5-bromo-2-aminobenzoic acid.
(E)-3-(3-Ethyl-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10a)
White solid; Yield: 63%. mp: 174-175 °C. Rf = 0.47 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3339 (NH); 3192 (OH); 3076, 2990 (CH, aren); 2849 (CH, CH2); 1678, 1649 (C=O); 1611, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.87 (s, 1H, NHOH); 9.17 (s, 1H, NHOH); 8.44 (s, 1H, H-2′); 8.17 (d, J = 8.50 Hz, 1H, H-5′); 7.83 (s, 1H, H-8′); 7.73 (d, J = 8.00 Hz, 1H, H-6′); 7.61 (d, J = 16.00 Hz, 1H, H-3); 6.67 (d, J = 16.00 Hz, 1H, H-2); 4.04–4.00 (m, 2H, H-1″a, H-1″b); 1.30 (t, J = 7.25 Hz, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.61, 160.12, 148.99, 148.95, 140.90, 137.44, 127.14, 126.91, 125.51, 122.88, 122.24, 41.74, 14.95. HRMS (ESI) m/z calculated for C13H14N3O3, [M + H]+ 260.1035. Found, 260.1027.
(E)-3-(3-(2-Chloroethyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10b)
White solid; Yield: 65%. mp: 171-172 °C. Rf = 0.47 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3460 (NH); 3175 (OH); 3028, 2953 (CH, aren); 2922, 2851 (CH, CH2); 1659 (C=O); 1611, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 9.17 (s, 1H, NHOH); 8.29 (s, 1H, H-2′); 8.17 (d, J = 8.00 Hz, 1H, H-5′); 7.83 (s, 1H, H-8′); 7.73 (d, J = 8.00 Hz, 1H, H-6′); 7.66 (d, J = 15.50 Hz, 1H, H-3); 6.68 (d, J = 15.50 Hz, 1H, H-2); 4.04 (d, J = 10.50 Hz, 2H, H-2″a, H-2″b); 3.68 (d, J = 10.50 Hz, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.59, 160.13, 149.01, 148.87, 141.02, 137.39, 127.18, 127.01, 125.66, 123.03, 122.28, 49.24, 45.60. HRMS (ESI) m/z calculated for C13H13ClN3O3, [M + H]+ 294.0645, 296.0616. Found, 294.0630, 296.0601.
(E)-N-Hydroxy-3-(3-(2-hydroxyethyl)-4-oxo-3,4-dihydroquinazolin-7-yl)acrylamide (10c)
White solid; Yield: 55%. mp: 175-176 °C. Rf = 0.36 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3418 (NH); 3123 (OH); 2928 (CH, aren); 2882 (CH, CH2); 1663 (C=O); 1611, 1555 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 9.82 (s, 1H, NHOH); 8.31 (s, 1H, H-2′); 8.16 (d, J = 8.00 Hz, 1H, H-5′); 7.82 (s, 1H, H-8′); 7.72 (d, J = 8.50 Hz, 1H, H-6′); 7.58 (d, J = 15.50 Hz, 1H, H-3); 6.78 (d, J = 16.00 Hz, 1H, H-2); 5.06-5.04 (m, 1H, 2″-OH); 5.06-4.05 (m, 2H, H-1″a, H-1″b); 3.67-3.65 (m, 2H, H-2″a, H-2″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.47, 160.40, 149.72, 148.99, 141.00, 137.06, 127.14, 126.76, 125.40, 123.13, 122.24, 49.05, 34.53. HRMS (ESI) m/z calculated for C13H14N3O4, [M + H]+ 276.0984. Found, 276.0968.
(E)-N-Hydroxy-3-(4-oxo-3-propyl-3,4-dihydroquinazolin-7-yl)acrylamide (10d)
White solid; Yield: 65%. mp: 178-179 °C. Rf = 0.45 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3343 (NH); 3204 (OH); 3071, 2968 (CH, aren); 2920 (CH, CH2); 1680, 1649 (C=O); 1612, 1555 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.88 (s, 1H, NHOH); 9.17 (s, 1H, NHOH); 8.42 (s, 1H, H-2′); 8.16 (d, J = 8.50 Hz, 1H, H-5′); 7.83 (s, 1H, H-8′); 7.73 (d, J = 8.50 Hz, 1H, H-6′); 7.61 (d, J = 15.50 Hz, 1H, H-3); 6.67 (d, J = 16.00 Hz, 1H, H-2); 3.94 (t, J = 7.25 Hz, 2H, H-1″a, H-1″b); 1.74-1.70 (m, 2H, H-2″a, H-2″b); 0.90 (t, J = 7.25 Hz, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.61, 160.28, 149.21, 148.89, 140.92, 137.44, 127.20, 126.93, 125.53, 122.89, 122.21, 47.95, 22.38, 11.30. HRMS (ESI) m/z calculated for C14H16N3O3, [M + H]+ 274.1192. Found, 274.1185.
(E)-3-(3-Butyl-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10e)
White solid; Yield: 71%. mp: 189-190 °C. Rf = 0.48 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3258 (OH); 3084, 2959 (CH, aren); 2930. 2870 (CH, CH2); 1668, 1638 (C=O); 1612, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.88 (s, 1H, NHOH); 9.17 (s, 1H, NHOH); 8.42 (s, 1H, H-2′); 8.16 (d, J = 8.50 Hz, 1H, H-5′); 7.83 (s, 1H, H-8′); 7.72 (d, J = 8.00 Hz, 1H, H-6′); 7.61 (d, J = 15.50 Hz, 1H, H-3); 6.67 (d, J = 16.00 Hz, 1H, H-2); 3.98 (t, J = 7.00 Hz, 2H, H-1″a, H-1″b); 1.71-1.65 (m, 2H, H-2″a, H-2″b); 1.34-1.30 (m, 2H, H-3″a, H-3″b); 0.93 (t, J = 7.00 Hz, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.62, 160.25, 149.16, 148.87, 140.91, 137.44, 127.18, 126.92, 125.53, 122.88, 122.19, 46.17, 31.18, 19.76, 14.01. HRMS (ESI) m/z calculated for C15H18N3O3, [M + H]+ 288.1388. Found, 288.1341.
(E)-3-(3-(Cyclopentylmethyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10f)
White solid; Yield: 72%. mp: 196-197 °C. Rf = 0.44 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3240 (OH); 3065, 2953 (CH, aren); 2864 (CH, CH2); 1684, 1659 (C=O); 1611, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 8.44 (s, 1H, H-2′); 8.16 (d, J = 8.50 Hz, 1H, H-5′); 7.83 (s, 1H, H-8′); 7.73 (d, J = 8.00 Hz, 1H, H-6′); 7.60 (d, J = 16.00 Hz, 1H, H-3); 6.68 (d, J = 15.50 Hz, 1H, H-2); 3.93 (d, J = 7.50 Hz, 2H, H-1″a, H-1″b); 2.39-2.33 (m, 1H, H-2″); 1.65-1.27 (m, 8H, H-3″a, H-3″b, H-4″a, H-4″b, H-5″a, H-5″b, H-6″a, H-6″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.51, 160.41, 149.21, 148.80, 140.99, 137.27, 127.24, 126.89, 125.55, 122.95, 122.15, 50.61, 49.07, 29.97, 24.88. HRMS (ESI) m/z calculated for C17H20N3O3, [M + H]+ 314.1505. Found, 314.1496.
(E)-3-(3-(Cyclohexylmethyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10g)
White solid; Yield: 75%. mp: 203-204 °C. Rf = 0.46 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3235 (OH); 3065, 2986 (CH, aren); 2920, 2851 (CH, CH2); 1684, 1665 (C=O); 1614, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.87 (s, 1H, NHOH); 9.17 (s, 1H, NHOH); 8.37 (s, 1H, H-2′); 8.16 (d, J = 8.00 Hz, 1H, H-5′); 7.83 (s, 1H, H-8′); 7.73 (d, J = 8.00 Hz, 1H, H-6′); 7.60 (d, J = 16.00 Hz, 1H, H-3); 6.68 (d, J = 16.00 Hz, 1H, H-2); 3.83 (d, J = 7.00 Hz, 2H, H-1″a, H-1″b); 1.81-1.78 (m, 1H, H-2″); 1.69-1.16 (m, 10H, H-3″a, H-3″b, H-4″a, H-4″b, H-5″a, H-5″b, H-6″a, H-6″b, H-7″a, H-7″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.49, 160.42, 149.40, 148.82, 140.97, 137.25, 127.25, 126.92, 125.54, 122.92, 122.16, 52.01, 37.08, 30.33, 26.32, 25.61. HRMS (ESI) m/z calculated for C18H22N3O3, [M + H]+ 328.1661. Found, 328.1650.
(E)-3-(3-Benzyl-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10h)
White solid; Yield: 65%. mp: 194-195 °C. Rf = 0.43 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3433 (NH); 3144 (OH); 3030, 2953 (CH, aren); 2845, 2783 (CH, CH2); 1655 (C=O); 1601, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.88 (s, 1H, NHOH); 9.17 (s, 1H, NHOH); 8.61 (s, 1H, H-2′); 8.16 (d, J = 8.50 Hz, 1H, H-5′); 7.86 (s, 1H, H-8′); 7.74 (d, J = 8.50 Hz, 1H, H-6′); 7.61 (d, J = 16.00 Hz, 1H, H-3); 7.39-7.29 (m, 5H, H-3″, H-4″, H-5″, H-6″, H-7″); 6.68 (d, J = 15.50 Hz, 1H, H-2); 5.21 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.59, 160.23, 149.14, 148.87, 141.16, 137.37, 137.26, 129.14, 128.18, 128.15, 127.30, 127.01, 125.76, 123.03, 122.28, 49.38. HRMS (ESI) m/z calculated for C18H16N3O3, [M + H]+ 322.1192. Found, 322.1183.
(E)-3-(3-(2-Fluorobenzyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10i)
White solid; Yield: 67%. mp: 207-208 °C. Rf = 0.45 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3287, 3177 (OH); 3061, 2994 (CH, aren); 2920, 2851 (CH, CH2); 1668, 1649 (C=O); 1607, 1555 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 8.55 (s, 1H, H-2′); 8.15 (d, J = 8.50 Hz, 1H, H-5′); 7.87 (s, 1H, H-8′); 7.74 (d, J = 8.00 Hz, 1H, H-6′); 7.61 (d, J = 15.50 Hz, 1H, H-3); 7.39-7.16 (m, 4H, H-4″, H-5″, H-6″, H-7″); 6.69 (d, J = 15.50 Hz, 1H, H-2); 5.25 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.48, 160.16, 149.21, 148.83, 141.25, 137.26, 130.51, 130.48, 130.40, 130.33, 127.22, 126.99, 125.79, 125.60, 125.04, 123.88, 123.76, 123.08, 122.19, 116.00, 115.84, 44.36. HRMS (ESI) m/z calculated for C18H15FN3O3, [M + H]+ 340.1097. Found, 340.1086.
(E)-3-(3-(3-Fluorobenzyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10j)
White solid; Yield: 66%. mp: 213-214 °C. Rf = 0.46 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3221 (OH); 3065, 3017 (CH, aren); 2920, 2851 (CH, CH2); 1680, 1647 (C=O); 1614, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 8.55 (s, 1H, H-2′); 8.16 (d, J = 8.50 Hz, 1H, H-5′); 7.85 (s, 1H, H-8′); 7.73 (d, J = 8.00 Hz, 1H, H-6′); 7.59 (d, J = 16.00 Hz, 1H, H-3); 7.42-7.12 (m, 4H, H-3″, H-5″, H-6″, H-7″); 6.68 (d, J = 16.00 Hz, 1H, H-2); 5.20 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.88, 160.68, 149.47, 149.29, 141.70, 140.44, 140.39, 137.52, 131.61, 131.55, 127.70, 127.39, 126.19, 124.61, 124.59, 123.59, 122.63, 115.62, 115.53, 115.44, 115.36, 49.41. HRMS (ESI) m/z calculated for C18H15FN3O3, [M + H]+ 340.1097. Found, 340.1087.
(E)-3-(3-(4-Fluorobenzyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10k)
White solid; Yield: 65%. mp: 211-212 °C. Rf = 0.46 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%.IR (KBr, cm–1): 3275 (OH); 3055 (CH, aren); 2920, 2849 (CH, CH2); 1653, 1612 (C=O); 1599, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 11.30 (s, 1H, NHOH); 9.59 (s, 1H, NHOH); 9.04 (s, 1H, H-2′); 8.58 (d, J = 8.50 Hz, 1H, H-5′); 8.27 (s, 1H, H-8′); 8.16 (d, J = 8.50 Hz, 1H, H-6′); 8.02 (d, J = 15.50 Hz, 1H, H-3); 7.89–7.59 (m, 4H, H-3″, H-4″, H-6″, H-7″); 7.10 (d, J = 15.50 Hz, 1H, H-2); 5.60 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.98, 160.64, 149.45, 149.27, 141.60, 137.77, 133.89, 133.87, 130.98, 130.91, 127.68, 127.41, 126.17, 123.47, 122.68, 116.40, 116.23, 49.18. HRMS (ESI) m/z calculated for C18H15FN3O3, [M + H]+ 340.1097. Found, 340.1087.
(E)-3-(3-(4-Chlorobenzyl)-4-oxo-3,4-dihydroquinazolin-7-yl)-N-hydroxyacrylamide (10l)
White solid; Yield: 70%. mp: 224-225 °C. Rf = 0.51 (DCM/MeOH/AcOH = 90:5:1). Purity > 95%. IR (KBr, cm–1): 3275 (OH); 3059, 2920 (CH, aren); 2851 (CH, CH2); 1653 (C=O); 1599, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.88 (s, 1H, NHOH); 9.21 (s, 1H, NHOH); 8.61 (s, 1H, H-2′); 8.15 (d, J = 8.00 Hz, 1H, H-5′); 7.86 (s, 1H, H-8′); 7.74 (d, J = 8.00 Hz, 1H, H-6′); 7.60 (d, J = 15.50 Hz, 1H, H-3); 7.42-7.36 (m, 4H, H-3″, H-4″, H-6″, H-7″); 6.68 (d, J = 15.50 Hz, 1H, H-2); 5.19 (s, 2H, H-1″a, H-1″b). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162.79, 160.23, 149.05, 148.86, 141.23, 137.25, 136.23, 132.84, 129.18, 128.15, 127.27, 127.00, 125.78, 123.08, 122.24, 49.38. HRMS (ESI) m/z calculated for C18H15ClN3O3, [M + H]+ 356.0802, 358.0772. Found, 356.0792, 358.0757.
(E)-N-Hydroxy-3-(3-(4-methylbenzyl)-4-oxo-3,4-dihydroquinazolin-7-yl)acrylamide (10m)
White solid; Yield: 68%. mp: 187-188 °C. Rf = 0.48 (DCM/MeOH/AcOH = 90:5:1). Purity > 99%. IR (KBr, cm–1): 3196 (OH); 2990 (CH, aren); 2860 (CH, CH2); 1674, 1659 (C=O); 1601, 1557 (C=C). 1H NMR (500 MHz, DMSO-d6, ppm): δ 10.89 (s, 1H, NHOH); 9.19 (s, 1H, NHOH); 8.59 (s, 1H, H-2′); 8.15 (d, J = 8.50 Hz, 1H, H-5′); 7.85 (s, 1H, H-8′); 7.74 (d, J = 8.00 Hz, 1H, H-6′); 7.60 (d, J = 15.50 Hz, 1H, H-3); 7.27 (d, J = 8.00 Hz, 2H, H-3″, H-7″); 7.16 (d, J = 8.00 Hz, 2H, H-4″, H-6″); 6.68 (t, J = 16.00 Hz, 1H, H-2); 5.15 (s, 2H, H-1″a, H-1″b); 2.27 (s, 3H, −CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 162,78, 160.19, 149.08, 148.84, 141.14, 137.47, 134.27, 129.66, 128.24, 128.13, 127.27, 126.97, 125.72, 123.03, 122.27, 49.11, 21.14. HRMS (ESI) m/z calculated for C19H18N3O3, [M + H]+ 336.1348. Found, 336.1336.
Cytotoxicity Assay
The cytotoxicity of the synthesized compounds was evaluated against three human cancer cell lines, including SW620 (colon cancer), PC3 (prostate cancer), and NCI-H23 (lung cancer). The cell lines were purchased from a Cancer Cell Bank at the Korea Research Institute of Bioscience and Biotechnology (KRIBB). The media, sera, and other reagents that were used for cell culture in this assay were obtained from GIBCO Co. Ltd. (Grand Island, New York). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) until confluence. The cells were then trypsinized and suspended at 3 × 104 cells/mL of cell culture medium. On day 0, each well of the 96-well plates was seeded with 180 μL of cell suspension. The plates were then incubated in a 5% CO2 incubator at 37 °C for 24 h. The compounds were initially dissolved in dimethyl sulfoxide (DMSO) and diluted to appropriate concentrations by culture medium. Then, 20 μL of each compound’s samples, which were prepared as described above, were added to each well of the 96-well plates, which had been seeded with cell suspension and incubated for 24 h at various concentrations. The plates were further incubated for 48 h. Cytotoxicity of the compounds was measured by the colorimetric method, as described previously39 with slight modifications.40,41 The IC50 values were calculated using a Probits method and were averages of three independent determinations (SD ≤ 10%).42
HDAC Enzymes Assay
See the SI.
Cell Cycle Analysis
SW620 human colon cancer cells (2 × 105/mL per well) were plated in six-well culture plates and allowed to grow for 24 h. The cells were treated with compounds (10 μM) or SAHA (1 μM) for 24 h and then harvested. The harvested cells were washed twice with ice-cold PBS, fixed in 75% ice-cold ethanol, and stained with propidium iodide (PI) in the presence of RNase at room temperature for 30 min. The stained cells were analyzed for DNA content using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and the data were processed using Cell Quest Pro software (BD Biosciences).
Apoptosis Assay
The Annexin V-FITC/PI dual staining assay was used to determine the percentage of apoptotic cells. SW620 human colon cancer cells (2 × 105/mL per well) were plated in six-well culture plates and allowed to grow for 24 h. The cells were treated with compounds (10 μM) or SAHA (1 μM) for 24 h and then harvested. The harvested cells were washed twice with ice-cold PBS and incubated in the dark at room temperature in 100 mL of 1 × binding buffer containing 1 μL of Annexin V-FITC and 12.5 mL of PI. After 15 min of incubation, the cells were analyzed for percentage undergoing apoptosis using a FACSCalibur flow cytometer (BD Biosciences). The data were processed using Cell Quest Pro software (BD Biosciences).
Molecular Docking Studies
The available crystal structure of HDAC6 complexed with Belinostat was taken from Protein Data Bank (PDB ID: 5EEN).36 Ligand structures were constructed using MOE 2015.10 package, and then the energy was minimized within an rms gradient of 0.1 kcal·mol–1·Å–1 applying MMFF94s force field.37 Based on the findings of Wu et al.,43 hydroxamic acids should be deprotonated upon its binding to the zinc ion. The receptor was prepared using the QuickPrep module in MOE 2015.10, similar to those reported previously.25,44 During the preparation, solvent and noncomplexed ions were deleted, Protonate3D was used for setting protonation states allowing ASN/GLN/HIS “Flips” in this function, polar hydrogen atoms were added, and all atoms were assigned AMBER FF99 force field. For the docking assays, the flexible ligand-rigid protein settings were applied using the MOE Triangle Matcher placement method, keeping the best 30 poses for conformational analysis. Only conformers showing appropriate coordination geometry with zinc ion will be considered. The finally selected poses were rescored using London (E_score1) and then by affinity dG (E_score2) scoring functions to estimate the free energy of binding of the ligands from the given poses. In the refinement stages, the energy minimization of the system was carried out using the molecular mechanics force-field method. All of the other parameters were kept as default. Binding sites were defined by all residues within a 6 Å distance from the corresponding ligands in the crystal structure. Docking poses in the individual HDAC binding pockets were analyzed using DS Visualizer (https://www.3dsbiovia.com/). See the SI for more details.
Acknowledgments
The authors acknowledge the principal financial supports from the National Foundation for Science and Technology of Vietnam (NAFOSTED, Grant number 104.01-2019.09). This work was also partly supported by a grant funded by the Korean Government (NRF, Grant number 2017R1A5A2015541). D.T.A. gratefully acknowledges the financial support from Vingroup Joint Stock Company for the scholarship (VINIF.2020.TS.16) by the Domestic Master/PhD Scholarship Programme of Vingroup Innovation Foundation (VINIF), Vingroup Big Data Institute (VINBIGDATA).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05870.
Synthesis and characterization data of all of the new compounds, and HDAC and HDAC6 enzymes assay (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hamm C. A.; Costa F. F. Epigenomes as therapeutic targets. Pharmacol. Ther. 2015, 151, 72–86. 10.1016/j.pharmthera.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Ruijter A. J. M.; Gennip A. H.; Caron H. N.; Kemp S.; Kuilenburg A. B. P. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt O.; Deubzer H. E.; Milde T.; Oehme I. HDAC family: What are the cancer relevant targets?. Cancer Lett. 2009, 277, 8–21. 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Ververis K.; Hiong A.; Karagiannis T. C.; Licciardi P. V. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics 2013, 7, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente S.; Mai A. Small-molecule inhibitors of histone deacetylase for the treatment of cancer and non-cancer diseases: a patent review (2011 - 2013). Expert Opin. Ther. Pat. 2014, 24, 401–415. 10.1517/13543776.2014.877446. [DOI] [PubMed] [Google Scholar]
- Li J.; Guangqiang L.; Wenqing X. Histone deacetylase inhibitors: an attractive strategy for cancer therapy. Curr. Med. Chem. 2013, 20, 1858–1886. 10.2174/0929867311320140005. [DOI] [PubMed] [Google Scholar]
- Zwergel C.; Valente S.; Jacob C.; Mai A. Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin. Drug Discovery 2015, 10, 599–613. 10.1517/17460441.2015.1038236. [DOI] [PubMed] [Google Scholar]
- West A. C.; Johnstone R. W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014, 124, 30–39. 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discovery 2006, 5, 769–784. 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Iyer S. P.; Foss F. F. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. Oncologist 2015, 20, 1084–1091. 10.1634/theoncologist.2015-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M. HDAC inhibitors still need a home run, despite recent approval. Nat. Rev. Drug Discovery 2015, 14, 225–226. 10.1038/nrd4583. [DOI] [PubMed] [Google Scholar]
- Qiu T.; Zhou L.; Zhu W.; Wang T.; Wang J.; Shu Y.; Liu P. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol 2013, 9, 255–269. 10.2217/fon.12.173. [DOI] [PubMed] [Google Scholar]
- Bracker T. U.; Sommer A.; Fichtner I.; Faus H.; Haendler B.; Hess-Stumpp H. Efficacy of MS-275, a selective inhibitor of class I histone deacetylases, in human colon cancer models. Int. J. Oncol. 2009, 35, 909–920. [DOI] [PubMed] [Google Scholar]
- Glaser K. B. HDAC inhibitors: clinical update and mechanism-based potential. Biochem. Pharmacol. 2007, 74, 659–671. 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Dung T. M. D.; Phan T. P. D.; Dao T. K. O.; Pham T. H.; Le T. T. H.; Vu D. L.; Hyunggu H.; Byung W. H.; Jisung K.; Sang-Bae H.; Nguyen-Hai N. Novel 3-substituted-2-oxoindoline-based N-hydroxypropenamides as histone deacetylase inhibitors and antitumor agents. Med. Chem. 2015, 11, 725–735. 10.2174/1573406411666150702130633. [DOI] [PubMed] [Google Scholar]
- Hieu D. T.; Anh D. T.; Hai P.-T.; Huong L.-T.-T.; Park E. J.; Choi J. E.; Kang J. S.; Dung P. T. P.; Han S.-B.; Nam N.-H. Quinazoline-Based Hydroxamic Acids: Design, Synthesis, and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodiversity 2018, 15, e1800027 10.1002/cbdv.201800027. [DOI] [PubMed] [Google Scholar]
- Huong T. T. L.; Cuong L. V.; Huong P. T.; Thao T. P.; Huong L.-T.-T.; Dung P. T. P.; Oanh D. T. K.; Huong N. T. M.; Quan H.-V.; Vu T. K.; Kim J.; Lee J.-H.; Han S. B.; Hai P. T.; Nam N. H. Exploration of some indole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Chem. Pap. 2017, 71, 1759–1769. 10.1007/s11696-017-0172-1. [DOI] [Google Scholar]
- Nam N. H.; Huong T. L.; Dung D. T. M.; Dung P. T. P.; Oanh D. T. K.; Park S. H.; Kim K.; Han B. W.; Yun J.; Kang J. S.; Kim Y.; Han S. B. Synthesis, bioevaluation and docking study of 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. J. Enzyme Inhib. Med. Chem. 2014, 29, 611–618. 10.3109/14756366.2013.832238. [DOI] [PubMed] [Google Scholar]
- Nam N. H.; Huong T. L.; Dung D. T. M.; Dung P. T. P.; Oanh D. T. K.; Quyen D.; Thao L. T.; Park S. H.; Kim K. R.; Han B. W.; Yun J.; Kang J. S.; Kim Y.; Han S. B. Novel isatin-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Eur. J. Med. Chem. 2013, 70, 477–486. 10.1016/j.ejmech.2013.10.045. [DOI] [PubMed] [Google Scholar]
- Pham-The H.; Casañola-Martin G.; Diéguez-Santana K.; Nguyen-Hai N.; Ngoc N. T.; Vu-Duc L.; Le-Thi-Thu H. Quantitative structure-activity relationship analysis and virtual screening studies for identifying HDAC2 inhibitors from known HDAC bioactive chemical libraries. SAR QSAR Environ. Res. 2017, 28, 199–220. 10.1080/1062936X.2017.1294198. [DOI] [PubMed] [Google Scholar]
- Tung T. T.; Oanh D. T. K.; Dung P. T. P.; Hue V. T. M.; Park S. H.; Han B. W.; Kim Y.; Hong J.; Han S. B.; Nam N. H. New benzothiazole/thiazole-containing hydroxamic acids as potent histone deacetylase inhibitors and antitumor agents. Med. Chem. 2013, 9, 1051–1057. 10.2174/15734064113099990027. [DOI] [PubMed] [Google Scholar]
- Dung T. M. D.; Nguyen V. H.; Do M. C.; Dao C. H.; Pham-The H.; Le-Thi-Thu H.; Jisung K.; Jeong E. C.; Jong S. K.; Sang-Bae H.; Nguyen-Hai N. Novel Hydroxamic Acids Incorporating 1-((1H-1,2,3-Triazol-4-yl)methyl)-3-substituted-2-oxoindolines: Synthesis, Biological Evaluation and SAR Analysis. Med. Chem. 2018, 14, 831–850. 10.2174/1573406414666180528111749. [DOI] [PubMed] [Google Scholar]
- Solyanik G. I. Quinazoline compounds for antitumor treatment. Exp. Oncol. 2019, 41, 3–6. 10.32471/exp-oncology.2312-8852.vol-41-no-1.12414. [DOI] [PubMed] [Google Scholar]
- Seo S.-Y. Multi-targeted hybrids based on HDAC inhibitors for anti-cancer drug discovery. Arch. Pharm. Res. 2012, 35, 197–200. 10.1007/s12272-012-0221-9. [DOI] [PubMed] [Google Scholar]
- Anh D. T.; Hai P. T.; Huong L. T. T.; Park E. J.; Jun H. W.; Kang J. S.; Kwon J.-H.; Dung D. T. M.; Anh V. T.; Hue V. T. M.; Han S. B.; Nam N. H. Exploration of certain 1,3-oxazole- and 1,3-thiazole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Bioorg. Chem. 2020, 101, 103988 10.1016/j.bioorg.2020.103988. [DOI] [PubMed] [Google Scholar]
- Hieu D. T.; Anh D. T.; Hai P. T.; Thuan N. T.; Huong L. T. T.; Park E. J.; Ji A. Y.; Kang J. S.; Dung P. T. P.; Han S. B.; Nam N. H. Quinazolin-4(3H)-one-Based Hydroxamic Acids: Design, Synthesis and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodiversity 2019, 16, e1800502 10.1002/cbdv.201800502. [DOI] [PubMed] [Google Scholar]
- Hieu D. T.; Anh D. T.; Tuan N. M.; Hai P. T.; Huong L. T. T.; Kim J.; Kang J. S.; Vu T. K.; Dung P. T. P.; Han S. B.; Nam N. H.; Hoa N. D. Design, synthesis and evaluation of novel N-hydroxybenzamides/N-hydroxypropenamides incorporating quinazolin-4(3H)-ones as histone deacetylase inhibitors and antitumor agents. Bioorg. Chem. 2018, 76, 258–267. 10.1016/j.bioorg.2017.12.007. [DOI] [PubMed] [Google Scholar]
- a Vu T. K.; Thanh N. T.; Minh N. V.; Linh N. H.; Thao N. T. P.; Nguyen T. T. B.; Hien D. T.; Chinh L. V.; Duc T. H.; Anh L. D.; Hai P. T. Novel Conjugated Quinazolinone-Based Hydroxamic Acids: Design, Synthesis and Biological Evaluation. Med. Chem. 2020, 16, 1–18. 10.2174/1573406416666200420081540. [DOI] [PubMed] [Google Scholar]; b Nadri S.; Rafiee E.; Jamali S.; Joshaghani M. 1,1′-Methylene-3,3′-bis[(N-(tert-butyl)imidazol-2-ylidene] and Its Effect in Palladium-Catalyzed C–C Coupling. Synlett 2015, 26, 619–624. 10.1055/s-0034-1379954. [DOI] [Google Scholar]
- Huan L. C.; Anh D. T.; Truong B. X.; Duc P. H.; Hai P. T.; Duc-Anh L.; Huong L. T. T.; Park E. J.; Lee H. J.; Kang J. S.; Tran P. T.; Hai D. T. T.; Oanh D. T. K.; Han S. B.; Nam N. H. New Acetohydrazides Incorporating 2-Oxoindoline and 4-Oxoquinazoline: Synthesis and Evaluation of Cytotoxicity and Caspase Activation Activity. Chem. Biodiversity 2020, 17, e1900670 10.1002/cbdv.201900670. [DOI] [PubMed] [Google Scholar]
- Huan L. C.; Phuong C. V.; Truc L. C.; Thanh V. N.; Pham-The H.; Huong L. T. T.; Thuan N. T.; Park E. J.; Ji A. Y.; Kang J. S.; Han S. B.; Tran P. T.; Nam N. H. (E)-N′-Arylidene-2-(4-oxoquinazolin-4(3H)-yl) acetohydrazides: Synthesis and evaluation of antitumor cytotoxicity and caspase activation activity. J. Enzyme Inhib. Med. Chem. 2019, 34, 465–478. 10.1080/14756366.2018.1555536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan L. C.; Tran P. T.; Phuong C. V.; Duc P. H.; Anh D. T.; Hai P. T.; Huong L. T. T.; Thuan N. T.; Lee H. J.; Park E. J.; Kang J. S.; Linh N. P.; Hieu T. T.; Oanh D. T. K.; Han S. B.; Nam N. H. Novel 3,4-dihydro-4-oxoquinazoline-based acetohydrazides: Design, synthesis and evaluation of antitumor cytotoxicity and caspase activation activity. Bioorg. Chem. 2019, 92, 103202 10.1016/j.bioorg.2019.103202. [DOI] [PubMed] [Google Scholar]
- Špulák M.; Novák Z.; Palát K.; Kuneš J.; Pourová J.; Pour M. The unambiguous synthesis and NMR assignment of 4-alkoxy and 3-alkylquinazolines. Tetrahedron 2013, 69, 1705–1711. 10.1016/j.tet.2012.12.031. [DOI] [Google Scholar]
- Pelzel H. R.; Schlamp C. L.; Nickells R. W. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010, 11, 62. 10.1186/1471-2202-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Shin D.; Kwon S. H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [DOI] [PubMed] [Google Scholar]
- Osko J. D.; Christianson D. W. Structural Basis of Catalysis and Inhibition of HDAC6 CD1, the Enigmatic Catalytic Domain of Histone Deacetylase 6. Biochemistry 2019, 58, 4912–4924. 10.1021/acs.biochem.9b00934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai Y.; Christianson D. W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment, 2009.10; Chemical Computing Group Inc.: 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015.
- Osko J. D.; Porter N. J.; Reddy P. A. N.; Xiao Y. C.; Rokka J.; Jung M.; Hooker J. M.; Salvino J. M.; Christianson D. W. Exploring Structural Determinants of Inhibitor Affinity and Selectivity in Complexes with Histone Deacetylase 6. J. Med. Chem. 2020, 63, 295–308. 10.1021/acs.jmedchem.9b01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Min B. S.; Huong H. T. T.; Kim J. H.; Jun H. J.; Na M. K.; Nam N. H.; Lee H. K.; Bae K. H.; Kang S. S. Furo-1,2-naphthoquinones from Crataegus pinnatifida with ICAM-1 expression inhibition activity. Planta Med. 2004, 70, 1166–1169. 10.1055/s-2004-835846. [DOI] [PubMed] [Google Scholar]
- Nam N. H.; Pitts R. L.; Sun G.; Sardari S.; Tiemo A.; Xie M.; Yan B.; Parang K. Design of tetrapeptide ligands as inhibitors of the Src SH2 domain. Bioorg. Med. Chem. 2004, 12, 779–787. 10.1016/j.bmc.2003.10.060. [DOI] [PubMed] [Google Scholar]
- Wu L.; Smythe A. M.; Stinson S. F.; Mullendore L. A.; Monks A.; Scudiero D. A.; Paull K. D.; Koutsoukos A. D.; Rubinstein L. V.; Boyd M. R.; Shoemaker R. H. Multidrug-resistant phenotype of disease-oriented panels of human tumor cell lines used for anticancer drug screening. Cancer Res. 1992, 52, 3029. [PubMed] [Google Scholar]
- Wu R.; Lu Z.; Cao Z.; Zhang Y. Zinc chelation with hydroxamate in histone deacetylases modulated by water access to the linker binding channel. J. Am. Chem. Soc. 2011, 133, 6110–6113. 10.1021/ja111104p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dung D. T. M.; Hai P. T.; Anh D. T.; Huong L. T. T.; Yen N. T. K.; Han B. W.; Park E. J.; Choi Y. J.; Kang J. S.; Hue V. T. M.; Han S. B.; Nam N. H. Novel hydroxamic acids incorporating 1-((1H-1,2,3-Triazol-4-yl)methyl)-3-hydroxyimino-indolin-2-ones: synthesis, biological evaluation, and SAR analysis. J. Chem. Sci. 2018, 130, 63. 10.1007/s12039-018-1472-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.