

Abstract
The serum iron level in humans is tightly controlled by the action of the hormone hepcidin on the iron efflux transporter ferroportin. Hepcidin regulates iron absorption and recycling by inducing ferroportin internalization and degradation1. Aberrant ferroportin activity can lead to diseases of iron overload, like hemochromatosis, or iron limitation anemias2. Here, we determined cryogenic electron microscopy (cryo-EM) structures of ferroportin in lipid nanodiscs, both in the apo state and in complex with cobalt, an iron mimetic, and hepcidin. These structures and accompanying molecular dynamics simulations identify two metal binding sites within the N- and C-domains of ferroportin. Hepcidin binds ferroportin in an outward-open conformation and completely occludes the iron efflux pathway to inhibit transport. The carboxy-terminus of hepcidin directly contacts the divalent metal in the ferroportin C-domain. We further show that hepcidin binding to ferroportin is coupled to iron binding, with an 80-fold increase in hepcidin affinity in the presence of iron. These results suggest a model for hepcidin regulation of ferroportin, where only iron loaded ferroportin molecules are targeted for degradation. More broadly, our structural and functional insights are likely to enable more targeted manipulation of the hepcidin-ferroportin axis in disorders of iron homeostasis.
Introduction
Iron is essential for life. Despite this central role in biology, free ferrous iron (Fe2+) is toxic. In excess, iron can catalyze the production of free radicals, leading to cellular damage. Iron levels are therefore tightly controlled, both at the cellular and organism level.
In mammals, iron levels are regulated by the action of hepcidin, a peptide hormone, on ferroportin (FPN), the only known iron efflux transporter2–4 (Fig. 1a). FPN mediates absorption of dietary iron by transporting ferrous iron across the basolateral surface of intestinal enterocytes. FPN also enables iron recycling from hepatocytes and macrophages5. Synthesis of FPN is transcriptionally regulated by cellular hypoxia, iron and heme concentrations, and inflammatory signaling6. In settings of elevated serum iron levels, liver-derived hepcidin negatively regulates cell surface FPN by acutely blocking iron transport7 and inducing FPN ubiquitination, internalization, and degradation1,8–10. Therefore, hepcidin decreases serum iron levels by suppressing FPN-mediated dietary iron absorption and release of iron from cellular stores.
Fig. 1 |. Structures of human ferroportin.
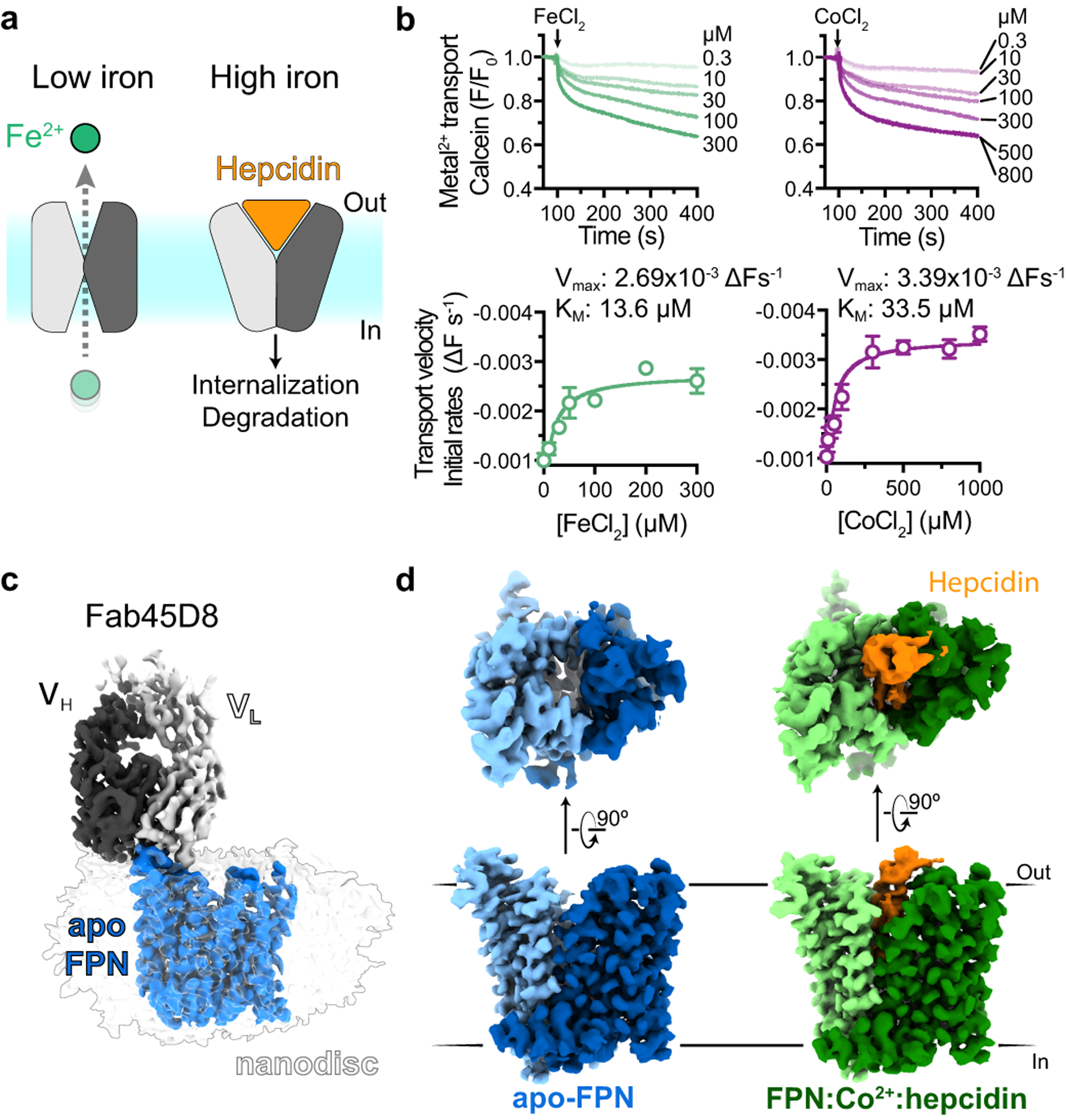
a, Ferroportin effluxes cellular iron (Fe2+) by an alternating access mechanism. Hepcidin binds to outward-open ferroportin and induces ubiquitination and degradation. b, Transport of Fe2+ and Co2+ by human ferroportin reconstituted into calcein containing liposomes. Time-course experiments were performed by addition of ions (arrow) and the steady-state kinetic analysis was performed by fitting initial rates, obtained from the linear phase of transport, to the Michaelis-Menten function. Time-course data are representative experiments, and data points are means +/− s.e.m. from n = 3 independent experiments. c, Cryo-EM map of apo-FPN-Fab45D8 complex in lipid nanodisc. d, Cryo-EM density of apo and Co2+/hepcidin bound FPN. The N and C domains are colored in different shades of blue for apo-FPN and green for Co2+/hepcidin bound FPN. Hepcidin (orange) binds to an extracellular facing cavity in FPN.
Iron disorders in humans can result from dysregulation of hepcidin or FPN. Deficits in hepcidin-mediated regulation of FPN, often due to hereditary hemochromatoses, lead to iron overload and widespread tissue damage affecting the liver, heart, pancreas, and joints11–13. By contrast, inappropriate elevation of hepcidin levels yields iron-restricted anemia14,15. Although several approaches to restore aberrant FPN function have been evaluated in clinical trials16–18, none have thus far succeeded.
The molecular mechanism of FPN regulation by hepcidin remains incompletely defined. A confluence of human genetics studies and structure-function evaluations have identified key regions of FPN important in hepcidin regulation7,19–22. To understand how FPN transports iron, and how this process is regulated by hepcidin, we used a combination of cryogenic electron-microscopy (cryo-EM), molecular dynamics simulations, and in vitro biochemical assays. These studies reveal the molecular recognition of iron and hepcidin by FPN, demonstrate acute inhibition of iron transport by hepcidin, and suggest a new regulatory mechanism enabling hepcidin to selectively target actively transporting ferroportin molecules for degradation.
Structures of apo and hepcidin-bound FPN
We screened the antigen-binding fragments (Fabs) of antibodies previously raised against FPN23 for use as a fiducial mark to guide image alignment of a small membrane protein embedded in a lipid nanodisc. This approach leverages a strategy we previously proposed for structure determination by single particle cryo-EM24. Fab45D8 was previously determined to be non-competitive with hepcidin and does not induce FPN internalization23. Indeed, Fab45D8 did not alter the binding properties of hepcidin to nanodisc-reconstituted FPN. (Extended Data Fig. 1).
We determined cryo-EM structures of FPN reconstituted in a NW9 nanodisc25 and bound to Fab45D8, both in the apo state (3.2 Å, Extended Data Table 1, Extended Data Fig. 3) and bound to hepcidin and Co2+ (2.5 Å, Extended Data Fig. 4). We reconstituted FPN metal transport in a liposome-based calcein quenching assay, which yielded a Vmax of −0.00269 ΔFs−1 and Km of 13.6 μM for FeCl2 (Fig. 1b, Extended Data Fig. 2). Unlike Fe2+, Co2+ is not readily oxidized and therefore provides a tractable surrogate divalent metal for FPN biochemical and structural studies. CoCl2 was also robustly transported by FPN with a Vmax of −0.00339 ΔFs−1 and Km of 33.5 μM, supporting its use as a substrate mimetic. Fab45D8 decreased both the Vmax and Km of CoCl2 transport (Extended Data Fig. 2), suggesting that Fab45D8 prefers a subset of conformations within the FPN ensemble, but does not inhibit transitions required for metal transport.
The cryo-EM density map of FPN enabled building of an atomic model of FPN regions important for iron transport and hepcidin binding2,7, and a portion of the intracellular loop 3 (ICL3) important in hepcidin-induced FPN internalization8–10(Extended Data Fig. 5). The FPN extracellular loop 5 (ECL5) remains unresolved, likely due to conformational flexibility. We separately obtained an X-ray crystal structure of Fab45D8 at 2.1 Å to enable modeling (Extended Data Table 2).
Both cryo-EM structures reveal a monomeric FPN bound to a single Fab45D8 molecule, which binds FPN extracellular loop 2 (ECL2) (Fig. 1b, Extended Data Fig. 6). Similar to other MFS transporters, FPN contains twelve transmembrane (TM) helices arranged in two domains (Fig. 2a), with a large central cavity open to the extracellular side in both structures (Fig. 2b). Ferroportin shares structural similarity with a previously characterized bacterial homolog, the bbFPN transporter26 (Extended Data Fig. 6). Unlike most other MFS transporters, the FPN TM7 is interrupted by a short non-helical stretch of six residues. This unique feature, previously posited to be important in iron binding27, is shared between FPN and bbFPN. Similar to bbFPN, FPN metal transport is stimulated by transmembrane pH gradients, but addition of valinomycin had no significant effect (Extended Data Fig. 7).
Fig. 2 |. Structure of apo-FPN.
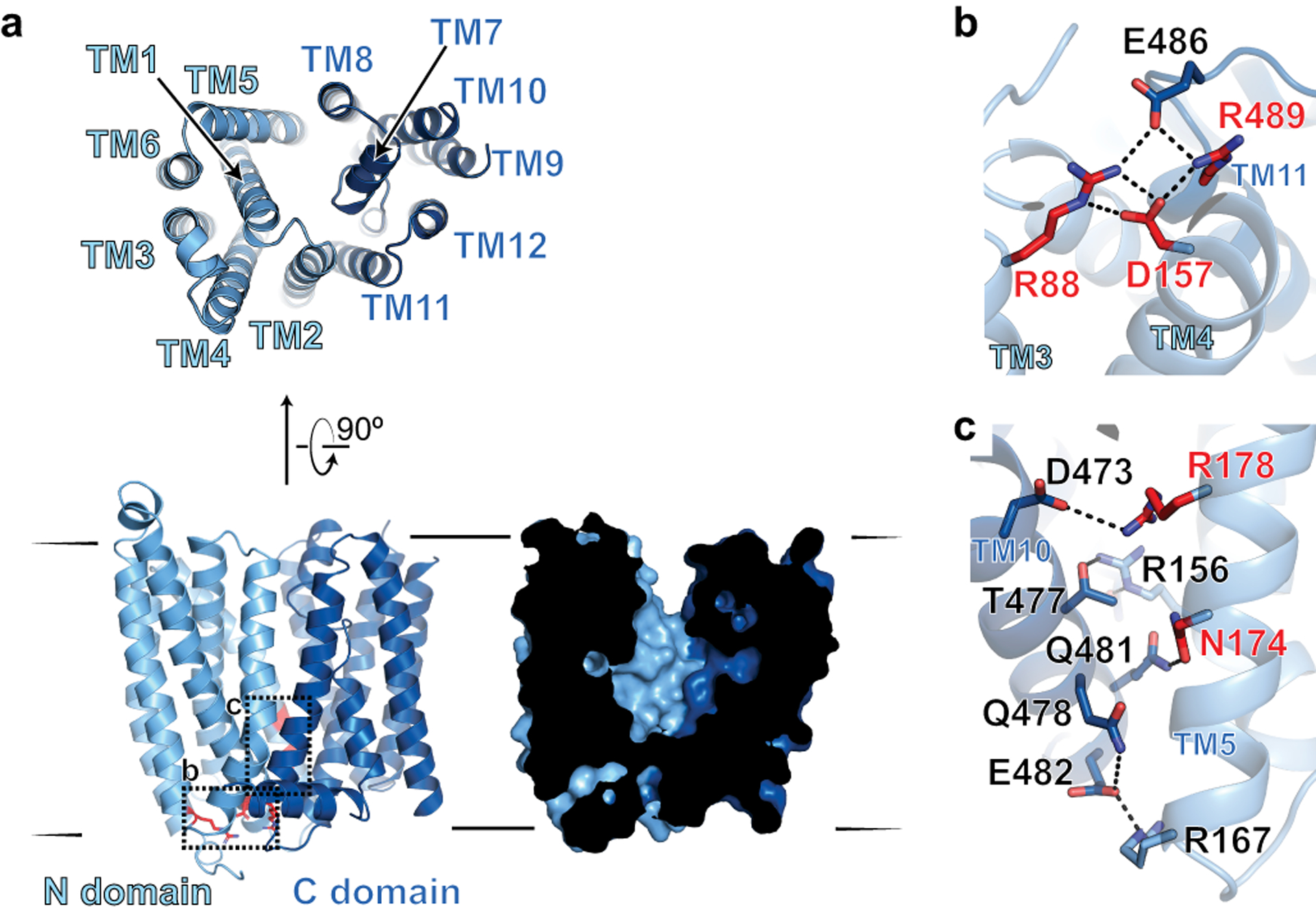
a, Ribbon diagram of FPN reveals 12 transmembrane helices. The N- and C-domains are colored in different shades of blue. Cutaway surface view (right) shows outward open conformation. b, Intracellular gating residues are shown as sticks. c, TM10 and TM5 form an extensive network of interactions, further stabilizing the outward open conformation. Residues highlighted in red in (b) and (c) are known loss-of-function mutations that lead to ferroportin disease in humans.
Several interacting residues define an intracellular gate that keeps the N- and C- domains of FPN in an outward open conformation. Similar to an interaction in bbFPN26, R489 in TM11 of the C-domain forms an ionic interaction with D157 in TM4 of the N-domain (Fig. 2b), which is further supported by an extended ionic and hydrogen-bonding network including residues E486 (TM11) and R88 (TM3). An additional cluster of ionic and hydrogen bonding interactions between TM5 in the N-domain and TM10 in the C-domain of FPN further stabilizes the outward open conformation (Fig. 2c). Mutation of several residues within the intracellular gate leads to FPN loss of function in ferroportin disease, highlighting the importance of the gate in coordinating the conformational steps necessary for iron efflux19,28.
Iron binds to the N and C domains of FPN
Cryo-EM maps from a Co2+-hepcidin-FPN complex revealed two new densities in the central cavity of FPN, corresponding to single metal binding sites in the N and in the C domain respectively (Fig. 3a–c). Within the C domain, Co2+ directly interacts with C326 in TM7b and H507 in TM11 while making a water-mediated contact with D325 and the backbone carbonyl of T320 (Fig. 3b). Intriguingly, the tetrahedral coordination geometry for Co2+ is fulfilled by the carboxy terminus of hepcidin. Within the N domain, we observe density for Co2+ coordinated directly by TM1 residues D39 and H43 (Fig. 3c). Mutation of D39 to alanine or glutamate abolishes iron transport22,27, which was previously ascribed to disruption of a putative calcium binding site within the N domain of FPN27. Our structure, by contrast, suggests that D39 directly binds the substrate. Transport assays with reconstituted FPN, however, revealed clear modulatory effects of calcium (Extended Data Fig. 7). We observed a modest decrease in both Km and Vmax for Co2+ with intra-liposomal CaCl2, whereas the rate of transport was increased by ~40% for Fe2+.
Fig. 3 |. Iron binds to the N and C domains of FPN.
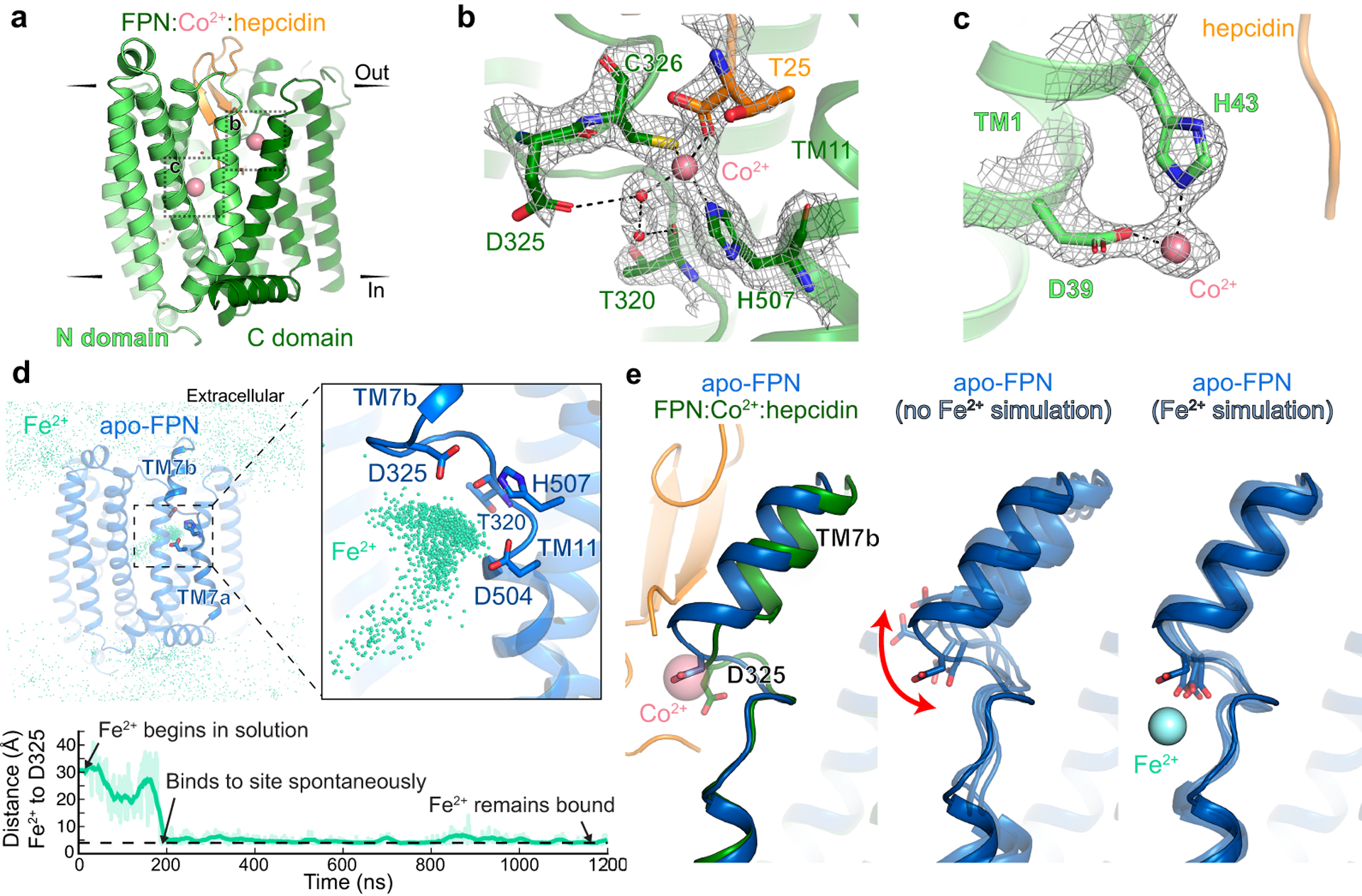
a, Ribbon diagram of FPN-Co2+-hepcidin complex. Closeup view of cryo-EM density for Co2+ ion (pink) in the FPN C domain (b) and in the FPN N domain (c). d, Top: In molecular dynamics simulations with Fe2+ initially positioned randomly in bulk water surrounding FPN, the Fe2+ ions spontaneously bind to a region near H507, D325, and D504. The aggregated position of Fe2+ ions from six simulations, each 2 μs in length, is shown superimposed with apo FPN. Bottom: In one representative simulation, an Fe2+ ion binds within 200 ns and remains localized at this site for >1000 ns. Distance shown is from the ion to the nearest oxygen atom of D325. Thick trace is a 15-ns sliding mean. e, Comparison of TM7b conformation in apo-FPN and FPN bound to Co2+ and hepcidin. In simulations without Fe2+, TM7b is dynamic, with significant fluctuation of D325. D325 coordinates Fe2+ in simulations and leads to decreased TM7b motion.
The identification of two metal binding sites within FPN raises the question of whether both sites are required for iron efflux. Disease causing mutations and experimental mutagenesis studies suggest that the N domain metal site is critical for iron efflux, while the C domain site may not be required for transport. Several hereditary hemochromatosis mutations in FPN directly coordinate Co2+ in the C domain, including C326S/F/Y13,29–31 and H507R32. These mutations are predicted to disrupt the precise tetrahedral coordination geometry required for metal binding within the C domain. However, these mutants are fully capable of iron transport, and lead to iron overload even in the presence of hepcidin. While mutation of the C domain residue D325 leads to decreased iron efflux22, recent modeling studies of an inward open conformation of FPN suggests that D325 may interact with the N domain in the outward open state; loss of iron efflux in D325 mutants may therefore be caused by disruption of the extracellular gate33. By contrast, mutation of D39 to alanine in the N domain metal binding site of FPN completely abolishes iron efflux from HEK293 cells22, suggesting that the N domain may be a primary site for effluxed iron. Because the C domain metal-binding site is important for hepcidin binding, it may primarily serve an important iron-dependent regulatory function in hepcidin control of FPN activity.
Hepcidin binding may influence interactions between FPN and the metal. To understand divalent metal binding to FPN in the absence of hepcidin, we used all-atom molecular dynamics simulations. We first performed six simulations of apo-FPN in a hydrated lipid bilayer with Fe2+ ions initially positioned randomly in bulk solvent. In all six independent simulations, Fe2+ ions bound spontaneously to the C domain, localizing to the unwound region of TM7 near residues D325, D504, and H507 within hundreds of nanoseconds (Fig. 3d and Extended Data Fig. 8). The Fe2+ ion also occasionally moves closer to TM1 to interact with D39 in the N domain. In parallel simulations of apo-FPN without Fe2+ ions, we observed mobility of TM7b with significant fluctuations of D325 (Fig. 3e), consistent with comparatively weaker cryo-EM density for TM7b as compared to other transmembrane helices in apo-FPN (Extended Data Fig. 8). By contrast, both D325 and TM7b are less mobile in simulations with iron bound at the C domain site (Fig. 3e). Divalent metal binding to the C domain may therefore stabilize an otherwise dynamic TM7b in a conformation that favors hepcidin binding.
Hepcidin occludes outward open FPN
Hepcidin binds FPN in a central cavity between the N and C domains, acting as a molecular cork to occlude the iron efflux pathway (Fig. 4a). Consistent with acute inhibition of iron transport by hepcidin7, we observed a decrease in Co2+ transport by reconstituted FPN when hepcidin is included within the liposome. By contrast, hepcidin added to the external solution had minimal effect on metal transport (Fig. 4b), suggesting that the calcein assay primarily measures transport in the forward direction (cellular efflux). Indeed, constitutively inhibited transport of a liposome-reconstituted hepcidin-FPN complex could be rescued by addition of membrane permeable β-mercaptoethanol, which likely denatures internally bound hepcidin by reducing its disulfide bonds (Fig. 4c).
Fig. 4 |. Hepcidin binding to FPN requires iron.
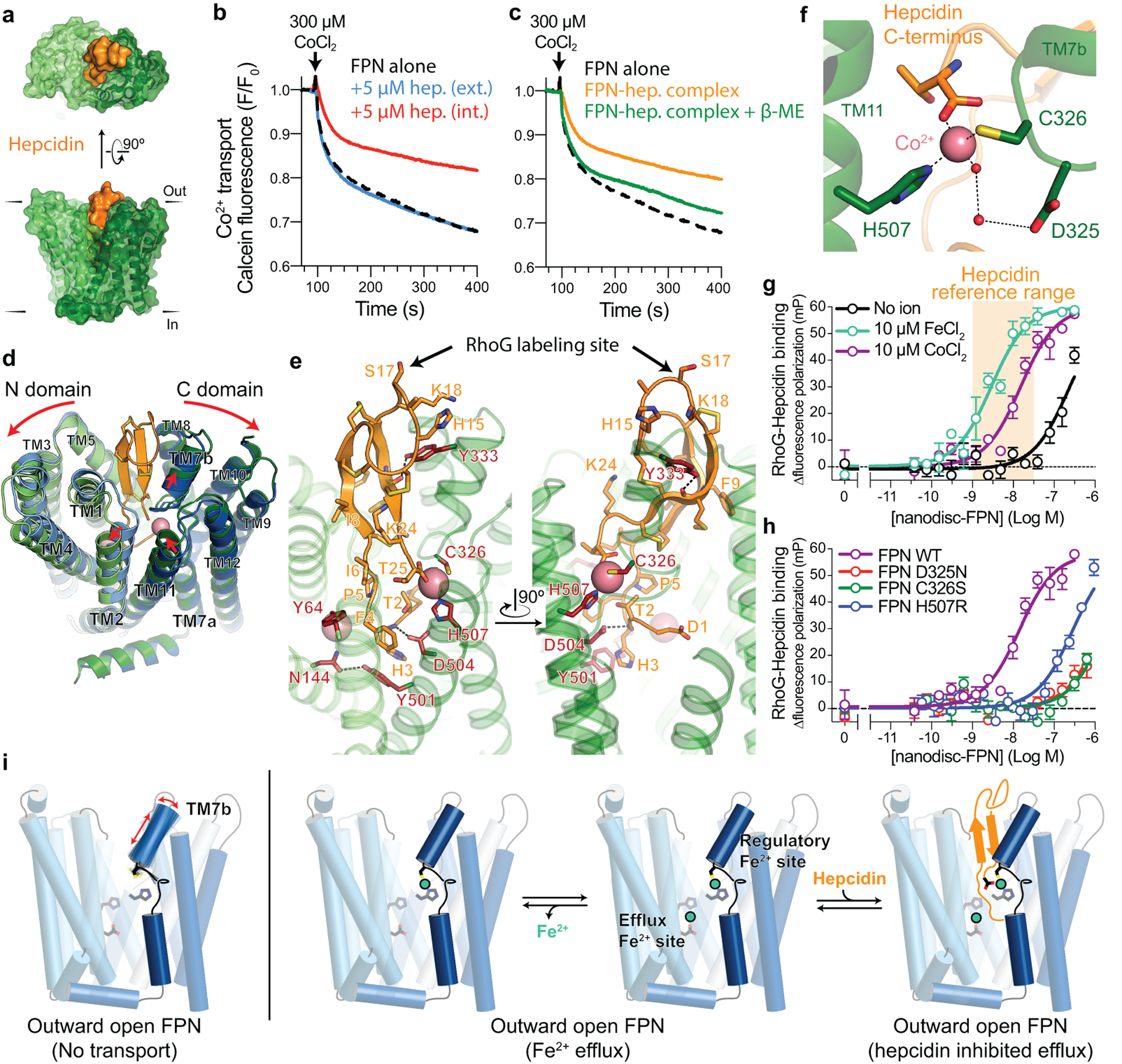
a, Surface representation of the FPN-Co2+-hepcidin complex. b, Hepcidin (hep.) added internally (int.) to calcein-loaded liposomes inhibits FPN transport. External (ext.) hepcidin has no effect on transport. c, Reconstitution of a ferroportin-hepcidin (FPN-hep.) complex shows inhibited transport. Addition of 1 mM β-mercaptoethanol (β-ME) rescues transport activity. All transport traces are averaged values from n = 3 independent experiments. d, Ribbon diagrams of apo-FPN (blue) aligned to FPN-Co2+-hepcidin (green, orange, and pink spheres. Red arrows highlight structural differences. e, Closeup views of the hepcidin binding site. Residues in red are known hepcidin resistance mutations. Rhodamine green (RhoG) - hepcidin labeling site on position 17 is highlighted. f, Closeup of C domain metal binding site. g, Fluorescence polarization increase in rhodamine green-labeled hepcidin (RhoG-hepcidin) as nanodisc-reconstituted FPN is titrated with a KD of 210 nM. Addition of 10 μM FeCl2 or CoCl2 increases the affinity of hepcidin to 2.5 nM and 7.7 nM (pKD = −8.11 ± 0.16), respectively. Hepcidin concentration range in healthy human adults is shown in orange. Data points are means ± s.e.m. from n = 3 independent experiments. h, C domain metal binding site mutants decrease RhoG-hepcidin binding affinity at FPN, even in the presence of 50 μM CoCl2. Data points are means ± s.e.m. from n = 3 independent experiments. i, Model for iron-coupled hepcidin regulation of FPN function. In settings of iron efflux, TM7b is conformationally stabilized by iron coordination in the C domain regulatory site. High affinity hepcidin binding to outward open FPN depends on the direct coordination of iron in the C domain.
Although the conformations of apo- and Co2+-hepcidin-bound FPN are highly similar (RMSD 0.73 Å), the Co2+-hepcidin-bound structure shows a ~3 Å rigid body separation of the N and C domains on the extracellular side of FPN (Fig. 4d). Within the N domain, hepcidin binding leads to displacement of TM2 from the central cavity, driven in part by a specific contact between I6 of hepcidin and T61 and Y64. Within the C domain, the largest conformational changes occur around the Co2+ binding site, leading to changes in the conformation of TM7b and the extracellular side of TM11.
Hepcidin makes extensive polar and hydrophobic contacts with FPN with a total buried surface area of ~1300 Å2. Our structure of hepcidin bound to FPN provides insight into disease-causing mutations associated with FPN gain of function in hereditary hemochromatoses. Several FPN mutations decrease hepcidin binding to FPN, including N144H/D/T34–36, C326S/F/Y13,29–31, Y333H37, Y501C7,38, D504N7,39 and H507R32. Of these, C326 and H507 directly coordinate the cobalt ion and mutations therefore indirectly affect hepcidin affinity or alter the atomic basis of binding specificity (Fig. 4e). Other interactions between FPN and hepcidin are either hydrophobic or depend on hepcidin amide backbone atoms, which is consistent with the relatively high tolerance of amino acid substitutions within hepcidin40. For example, Y333 fits into a hydrophobic cavity in hepcidin and hydrogen bonds with the backbone carbonyl of hepcidin residue M21 (Fig. 4e). D504 in FPN coordinates the backbone amide of hepcidin H3 while Y501 π-stacks with the imidazole side chain of H3. A further hydrogen bond between N144 and Y501 further stabilizes this interaction network. In contrast to these mutations, the Y64N/H mutants retain hepcidin binding7, but are completely resistant to hepcidin-induced FPN ubiquitination41,42. The outward displacement of TM2 near Y64 induced by hepcidin may therefore be important for FPN ubiquitination (Fig. 4d).
Hepcidin is coupled to iron binding
The direct interaction between hepcidin and Co2+ in our structure is unexpected (Fig. 4f), and suggests that divalent metals may be important for hepcidin binding to FPN. We therefore directly tested the effect of Fe2+ and Co2+ on hepcidin affinity at FPN. A fluorescently tagged version of hepcidin labeled at position 17 (Rhodamine green-hepcidin23, RhoG-Hep, Fig. 4e) bound to nanodisc-reconstituted ferroportin with an apparent KD of 210 nM (pKD = −6.67 ± 0.02) (Fig. 4g). In the presence of 10 μM FeCl2, we observed a significantly increased affinity of 2.5 nM (pKD = −8.61 ± 0.21), an almost 80-fold change in the potency of hepcidin at FPN. Addition of CoCl2 also increased hepcidin affinity. By contrast, CaCl2 did not support high affinity hepcidin binding (Extended Data Fig. 7), potentially due to an inability to form a direct interaction bridging FPN and hepcidin. Consistent with a cooperative effect, the effect of CoCl2 on hepcidin binding was saturable (Extended Data Fig. 7).
The reference range for hepcidin concentration in healthy adults is ~1–30 nM43. Our in vitro binding experiment with purified FPN indicates minimal hepcidin binding to FPN in the absence of a divalent metal, suggesting that metal binding to FPN may regulate hepcidin activity in vivo. Our structure of FPN bound to Co2+ and hepcidin revealed conformational changes in TM7b associated with hepcidin binding (Fig. 3e) and a direct contact between hepcidin and Co2+(Fig. 3b); both could be important for the observed effect of Fe2+ and Co2+ on hepcidin binding. We therefore tested whether disruption of the C domain iron binding site influences hepcidin binding. Even in the presence of 50 μM CoCl2, the D325N, C326S, and H507R mutants bound hepcidin weakly, titrating in a micromolar range similar to wild-type FPN in the absence of divalent metals (Fig. 4h). These results highlight the critical role of the C domain metal site in potent hepcidin binding to FPN, which is likely important in homeostatic control of iron levels in a physiological setting.
Discussion
Ferroportin is a central regulator of iron homeostasis in humans. Our structural and biochemical studies here reveal that hepcidin binding to FPN is greatly potentiated by iron itself, potentially due to the stabilizing effect iron has on the hepcidin-binding site of FPN (Fig. 4i). With iron, the binding affinity of hepcidin falls in a range concordant with the concentration of hepcidin observed in healthy human adults. In normal iron homeostasis, this may enable hepcidin to selectively bind and regulate FPN molecules actively transporting iron and loaded with Fe2+, while sparing FPN molecules located on cells with low transport activity. Hepcidin binding to FPN would both trap the transporter in an outward open state and limit iron egress; both actions acutely decrease iron efflux, as has been recently reported7. Elevated hepcidin levels likely inappropriately overcome this regulatory strategy and degrade FPN even in the absence of active iron efflux. The potentiation of hepcidin activity by iron may therefore have immediate consequences for the development of hepcidin mimetics currently in clinical trials16. Furthermore, hepcidin antagonism by direct targeting of FPN may require molecules with high potency to overcome the nanomolar effect of the hormone in the presence of iron. The structural and functional insights into FPN function presented here therefore provide critical foundations for the discovery of therapeutics for human disorders of iron homeostasis.
METHODS
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Expression and purification of human ferroportin
The wild-type human FPN gene was cloned into a pVL1392 vector containing an expression cassette comprised of a C terminal human rhinovirus 3C (HRV-3C) protease recognition sequence followed by a human protein C epitope tag (EDQVDPRLIDGK) and an 8x polyhistidine tag. Baculovirus was generated using Spodoptera frugiperda Sf9 insect cells (unauthenticated and untested for mycoplasma contamination, Expression Systems 94–001F) and the construct was expressed in Spodoptera frugiperda Sf9 insect cells. Cells were collected 48 h after transduction and stored at −80°C until further use. Frozen cell pellets were thawed and washed with a hypotonic buffer (20 mM HEPES pH 7.50, 1 mM EDTA, supplemented with 20 μg/mL leupeptin, and 160 μg/mL benzamidine) before solubilizing with 50 mM HEPES pH 7.5, 300 mM NaCl, 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.1% (w/v) cholesteryl hemisuccinate (CHS, Steraloids), 1 mM EDTA, and supplemented with 20 μg/mL leupeptin, and 160 μg/mL benzamidine for 1 h at 4°C. Following centrifugation, the resulting supernatant was loaded on homemade anti-protein C antibody Sepharose beads and washed extensively in 50 mM HEPES pH 7.50, 300 mM NaCl, 2 mM CaCl2, 0.1% (w/v) DDM, 0.01% (w/v) CHS. FPN was eluted with 50 mM HEPES pH 7.50, 300 mM NaCl, 0.1% (w/v) DDM, 0.01% (w/v) CHS, 0.2 mg/mL Protein C peptide (Genscript), and 5 mM EDTA. The protein was concentrated with a Vivaspin 100-kDa MWCO concentrator and the monomeric FPN fraction was collected after size-exclusion chromatography (SEC) over a Superdex S200 Increase 10/300 GL column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.50, 100 mM NaCl, 0.1% (w/v) DDM, and 0.01% (w/v) CHS.
FPN mutants used in binding studies were generated using the Q5 site-directed mutagenesis method (NEB) and cloned, along the WT FPN construct, into a pEZT vector containing an expression cassette comprised of a C terminal human rhinovirus 3C (HRV-3C) protease recognition sequence followed by an 8x polyhistidine tag and a 1D4 epitope tag (TETQSVAPA). The resulting vectors were transfected into Expi293F Human Embryonic Kidney cells (Life Technologies) with the Expifectamine transfection kit (Life Technologies) using 1 μg DNA per mL culture as per the manufacturer’s instructions. Cultures were harvested 48 h post transfection and stored at −80°C until further use. FPN mutants were purified as described for WT, except the cleared solubilized supernatant was loaded on Ni-NTA Sepharose beads and washed extensively in 50 mM HEPES pH 7.50, 300 mM NaCl, 0.1% (w/v) DDM, 0.01% (w/v) CHS, 30 mM Imidazole. FPN was eluted with 50 mM HEPES pH 7.50, 300 mM NaCl, 0.1% (w/v) DDM, 0.01% (w/v) CHS, 250 ml Imidazole. The protein was concentrated and purified by SEC as described above.
Expression and purification of MSP
Constructs encoding MSP-NW9 or MSP-NW1125 in the pET28b vector (Addgene #133442) were transformed into BL21(DE3) Rosetta Escherichia coli, and grown in terrific broth medium supplemented with 2 mM MgCl2 and 0.1% (w/v) glucose at 37°C. At OD600 of ~0.6, expression was induced by addition of 400 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and lowering the temperature to 20°C. Cells were harvested after 16 hours and resuspended into 5 mL lysis buffer (200 mM Tris pH 8.0, 500 mM NaCl, 1% (v/v) Triton X-100 (Sigma), 0.02 mg/mL leupeptin, 0.16 mg/mL benzamidine, and benzonase) per gram pellet. After stirring for 30 min at 4°C, cells were lysed by pulsed sonication on ice. The lysate was cleared by centrifugation at 15,000 × g for 25 min at 4°C and loaded on Ni-NTA Sepharose. Ni-NTA beads were washed with 50 mM Tris pH 8.0, 500 mM NaCl, 1% (v/v) Triton, then 50 mM Tris pH 8.0, 500 mM NaCl, 50 mM sodium cholate, then 50 mM Tris pH 8.0, 500 mM NaCl, and finally with 50 mM Tris pH 8.0, 500 mM NaCl, 30 mM Imidazole. MSP was eluted with 50 mM Tris-HCl pH 8.0, 500 mM NaCl, 400 mM Imidazole and dialyzed into 50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.1 mM TCEP at 4°C. The following day, MSP was concentrated on a Vivaspin 10-kDa MWCO concentrator, aliquots were flash frozen in liquid nitrogen and stored at −80°C for reconstitution.
Isolation, expression and purification of Fab45D8
The heavy and light chain sequences of mAb45D823 were separately cloned into pcDNA3.4 and the resulting vectors were transfected into Expi293F Human Embryonic Kidney cells (unauthenticated and untested for mycoplasma contamination, Life Technologies) using a 1:2 mass ratio of light and heavy chain DNA with the Expifectamine transfection kit (Life Technologies) as per the manufacturer’s instructions. Supernatant containing mAb45 was harvested 136 h after transfection and loaded on homemade Protein G Sepharose beads and extensively washed with a buffer comprising 20 mM HEPES pH 7.50, and 100 mM NaCl. mAb45D8 was eluted with 100 mM glycine (pH 3.0) and fractions were immediately neutralized with 200 mM HEPES pH 7.50. To generate the Fab fragment, 10 mg of purified mAb45D8.1 was diluted into 9.5 ml freshly prepared cleavage buffer (20 mM sodium phosphate pH 7.00, 10 mM EDTA, and 10 mM cysteine) and treated with 0.5 ml agarose immobilized papain (Thermo Scientific) at 37°C. After 16 h the cleaved Fab45D8 fragment was purified by reverse Protein A affinity chromatography, followed by SEC into buffer comprising 20 mM HEPES pH 7.50 and 100 mM NaCl. Fab45D8 was concentrated on a Vivaspin 10-kDa MWCO concentrator, and aliquots were flash frozen in liquid nitrogen and stored at −80°C for later use.
Reconstitution of FPN into lipidic nanodisc
Purified FPN (0.2–0.5 mg) was mixed with purified MSP and a lipid mixture containing a 2:3 weight ratio of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC, Avanti) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG, Avanti). For reconstitution into NW9 nanodiscs, an FPN:MSP:Lipid molar ratio of 1:20:1100 was used. For reconstitution into NW11 nanodiscs, an FPN:MSP:Lipid molar ratio of 1:20:800 was used. The reconstitution sample was nutated for 1 h at 4°C before addition of 0.2 g/mL SM2-BioBeads (BioRad), and the reconstitution sample was further nutated overnight at 4°C before removal of the biobeads. FPN containing nanodiscs for cryo-EM studies were purified by loading the reconstitution sample on anti-protein C antibody Sepharose beads and washing extensively with 20 mM HEPES pH 7.50, 100 mM NaCl, and 1 mM CaCl2 to remove empty nanodiscs. FPN containing nanodiscs were eluted with 20 mM HEPES pH 7.50, 100 mM NaCl, 0.25 mM EDTA, and 0.2 mg/mL Protein C peptide (Genscript) and concentrated on a Vivaspin 50-kDa concentrator. FPN containing nanodiscs for binding studies were purified by SEC over a Superdex S200 Increase 10/300 GL column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.50, 100 mM NaCl.
Crystallization and structure determination of Fab45D8
Purified Fab45D8 was diluted to 13.0 mg/mL in 20 mM HEPES pH 7.5, 100 mM NaCl. Fab45D8 crystals were obtained in 0.3 M trimethylamine-N-oxide (TMAO), 0.1 M Tris pH 8.5, and 30% (w/v) PEG 2000 MME at 20 °C. Individual crystals were flash frozen in liquid nitrogen after a 30 s soak in 0.3 M trimethylamine-N-oxide (TMAO), 0.1 M Tris pH 8.5, and 30% (w/v) PEG 2000, and 20% v/v ethylene glycol. A full diffraction dataset was collected at the Advanced Photon Source GM/CA-CAT beamline 23ID-B, and processed using xia2dials44 implementation of XDS45. The structure of the Fab was solved by molecular replacement using Phaser46, with a search model of a closely related germline mouse monoclonal antibody (PDB ID: 6BZV47) lacking complementarity determining regions (CDRs). The model was iteratively improved by refinement in Coot48 and Phenix49 Data collection and refinement statistics are summarized in Extended Data Table 2. The final model contained 96.77%, 2.23% and 0% in the favored, allowed and outlier regions of the Ramachandran plot, respectively as assessed by MolProbity50.
Calcein transport assay for divalent cations
FPN was reconstituted into liposomes for divalent cation transport assays. Empty liposomes were prepared as a 3:1 mass ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE, Avanti) to POPG dissolved in chloroform, followed by gentle evaporation of the chloroform under a stream of nitrogen gas, and overnight desiccation. The lipids were dissolved in 20 mM HEPES pH 7.40, 100 mM KCl to a final concentration of 12.5 mg/mL, sonicated until optically clear, subjected to multiple freeze-thaw cycles, and extruded through a 400 nm polycarbonate filter (Avestin) to generate unilamellar vesicles. Subsequently 0.13% (w/v) Triton-X100 (Sigma) was added to destabilize liposomes, corresponding to the onset of solubilization as determined by turbidity measurements at 540 nm. Purified FPN was added at a 1:50 protein to lipid mass ratio and incubated for 15 min at 4°C. The FPN-Hepcidin complex was reconstituted by incubating 1μM purified FPN with 3 μM Hepcidin (Bachem) and 3μM CoCl2 for 30 min on ice before addition of liposomes. The FPN-Fab complex was reconstituted by incubating 1μM purified FPN with 2 μM purified Fab45D8 for 30 min on ice before addition of liposomes. Control liposomes devoid of FPN were prepared in parallel using the same concentration of DDM. To remove excess detergent, 0.05 g/mL of SM2-BioBeads were added to the sample and nutated for 1 hr at 4°C, then 0.05 g/mL SM2-BioBeads were added followed by incubation overnight at 4°C, and finally addition of 0.08 g/mL SM2-BioBeads followed by incubation for 2 hr at 4°C. Liposomes were harvested by ultracentrifugation at 264,500 × g for 30 min and resuspended at a concentration of 4.0 mg/mL lipids in internal buffer comprised of 100 mM KCl and 20 mM HEPES pH 7.40 or pH 7.50, before flash freezing in liquid nitrogen and storage at −80°C. On the day of the transport assay, liposomes were thawed and diluted to 2 mg/mL with internal buffer containing 250 μM calcein (Sigma), and where indicated 5 μM hepcidin (Bachem) or 1.25 mM CaCl2 was added. The samples were then subjected to three freeze-thaw cycles, and extruded through a 400 nm polycarbonate filter. The calcein containing liposomes were concentrated and washed four times by repeated ultracentrifugation and resuspension in external buffer containing 100 mM NaCl and (i) 20 mM HEPES pH 6.80, pH 7.40, pH 7.50, or pH 8.20, or (ii) 20 mM Tris/MES pH 6.00, pH 7.50 or pH 9.00. Reconstitution yields determined by the Micro BCA protein assay kit (Pierce) were 205–222 pmol/mg lipid for FPN samples, and 275 pmol/mg lipid for the FPN-Fab45D8 complex assuming 1:1 stoichiometric incorporation. Presence of FPN and Fab were further verified by SDS-PAGE. Immediately prior to the assay, liposomes were diluted to 0.125 – 0.25 mg/mL lipid in external buffer. Time-course fluorescence traces were recorded as 1–2 s integrations using a FluoroMax-4 (Horiba) or a FluoroLog (Spex) with λex of 490 nm and λem of 520 nm. Steady state fluorescence was recorded for at least 2 min, before addition of small aliquots of freshly prepared stocks of either FeCl2, CoCl2, MnCl2, NiCl2, CuCl2, or ZnCl2. To stabilize the ferrous (Fe2+) state, we prepared iron as a 1:10 ratio of sodium ascorbate:FeCl2 immediately prior to the experiment. A negative inside membrane potential was generated by incubating liposomes with 100 nM of the K+-selective ionophore valinomycin (Sigma) for 1 min prior to the experiment. To determine the full extent of the calcein quenching response, 10 μM of the divalent cation ionophore calcimycin (Sigma) was added at the end of the experiment. Transport data was normalized to the mean baseline fluorescence intensity prior to addition of ion, and initial rates of transport were obtained from linear regression and fit to the Michaelis-Menten function in GraphPad Prism 8 (GraphPad Software). Statistical significance was determined using student’s t-test. For clarity, mixing artifacts ± 5 sec from the time of addition have been removed from representative fluorescence traces.
Hepcidin binding assays
Fluorescence polarization measurements were performed using rhodamine-green labeled hepcidin labeled at position 17 (RhoG-hepcidin)23. For FPN saturation binding experiments, samples were prepared in a black 384-well plate (Greiner) containing 0–1 μM of nanodisc reconstituted NW11-FPN and 5 nM RhoG-hepcidin in sample buffer comprising 20 mM HEPES pH 7.50, 100 mM NaCl, and supplemented with FeCl2, CoCl2 or MnCl2 as indicated. For ion stimulation experiments, 100 nM NW11-FPN and 5 nM RhoG-Hepcidin was mixed with 0–600 μM of CoCl2. For Fab binding experiments, 100 nM NW11-FPN and 5 nM RhoG-Hepcidin was mixed with 0 – 3 μM of Fab45D8 in sample buffer containing 10 μM CoCl2. Binding reactions were equilibrated for 60 min at RT, and fluorescence polarization was recorded on a Biotek Synergy H4 (Agilent) in polarization mode using fixed bandpass filters with λex of 484 nm and λem of 520 nm. Binding data was processed in GraphPad Prism 8 (GraphPad Software) using 3- or 4-parameter curve fitting.
Analytical fluorescence size exclusion chromatography (FSEC) was performed by mixing 25 μg of NW11-FPN with 2 × fold molar excess of RhoG-Hepcidin in sample buffer comprised of 20 mM HEPES (pH 7.50), 100 mM NaCl and 10 μM CoCl2. Samples were incubated for 20 min on ice and 1.5 × molar excess of Fab45D8, or sample buffer, was added followed by incubation for 30 min on ice. For homologous competition, 1 μM NW11-FPN was mixed with 30 μM unlabelled hepcidin (Bachem) in sample buffer comprised of 20 mM HEPES (pH 7.50), 100 mM NaCl and 10 μM CoCl, and incubated for 30 min on ice. Then 2 μM RhoG-Hepcidin was added and the sample incubated for 30 min on ice. Samples were injected on a Superdex 200 Increase 10/300 GL column (GE Lifesciences) pre-equilibrated in 20 mM HEPES pH 7.50, 100 mM NaCl, and 10 μM CoCl2. RhoG-Hepcidin fluorescence was recorded using an FP-1520 Intelligent Fluorescence Detector (Jasco) with λex of 493 nm and λem of 524 nm.
Cryo-EM Sample Preparation and Data Collection
Nanodisc-reconstituted apo-FPN was mixed with 1.15 molar excess of Fab45D8 and incubated on ice for 30 min. The complex was purified by size-exclusion chromatography over a Superdex S200 Increase 10/300 GL column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.50, 100 mM NaCl. For Co2+/hepcidin samples, 600 μM CoCl2 and 30 μM hepcidin (Bachem) were added to nanodisc-reconstituted FPN and incubated for 20 minutes on ice prior to addition of Fab45D8. The resulting complex was purified over size-exclusion chromatography as for the apo sample but with the addition of 100 μM CoCl2 in the chromatography buffer. Collected fractions were supplemented with fresh hepcidin to 30 μM. For both preparations, fractions containing the nanodisc-FPN-Fab45D8 complex were concentrated to ~3 mg/ml on a Vivaspin 50-kDa MWCO concentrator and freshly used for electron microscopy.
For high-resolution cryo-EM, the apo-FPN-Fab45D8 complex was diluted to 0.0375 mg/mL in 20 mM HEPES pH 7.5, 100 mM NaCl directly prior to vitrification, and 2 μL sample was applied to glow-discharged gold holey carbon 1.2/1.3 300-mesh grids (Quantifoil) coated in-house with graphene oxide51–53. Grids were blotted for 2–4 seconds at 0 force and 10 seconds wait time before being plunge vitrified in liquid ethane using a MarkIV Vitrobot (ThermoFisher). The blotting chamber was maintained at 22°C and 100% humidity during freezing.
Co2+/hepcidin samples were diluted to 1.5 mg/mL in gel filtration buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 100 μM CoCl2) before vitrification on UltrAufoil 1.2/1.3 300 mesh grids. Grids were blotted for 3 seconds at 0 force and 5 seconds wait time before plunge vitrification in liquid ethane using a MarkIV Vitrobot (ThermoFisher). The blotting chamber was maintained at 22°C and 100% humidity during freezing.
FPN-Fab45D8 and Co2+/hepcidin-FPN-Fab45D8 movies were collected using a Titan Krios (ThermoFisher) outfitted with a K3 camera and Bioquantum energy filter (Gatan). The K3 detector was operated in superresolution mode and the energy filter slit width was set to 20 eV. Movies were collected at a nominal magnification of 105,000x, physical pixel size 0.834Å/pix, with a 70 μm C2 aperture and 100 μm objective aperture at a dose rate of 8 e−/pixel/second. A total dose of 66 e−/Å2 was collected as a 120-frame movie, resulting in a 6-second movie with 0.55 e−/frame. Data were collected using semi-automated imaging scripts in SerialEM54. For FPN-Fab45D8, 5009 movies were collected using a 3×3 image shift pattern at 0° tilt and 406 movies were collected on-axis with a 30° stage tilt in two separate data collection sessions. For Co2+/hepcidin-FPN-Fab45D8, 4395 movies were collected at 0° tilt in a single data collection session.
Cryo-EM Image Processing
For FPN-Fab45D8, data were motion corrected and 2x binned on-the-fly using MotionCor255 in the SCIPION pipeline56. Motion corrected micrographs were imported into cryoSPARC57 and RELION58 and contrast transfer function parameters were calculated using CTFFIND459. CTF information for tilted images were estimated using patch CTF estimation in cryoSPARC. 138,314 particles were selected from 497 micrographs using the Blob picker in cryoSPARC. 2D class averages were generated after extracting the putative particles with a 300-pixel box and binning to 64 pixels. Six of these averages were used as templates for further particle picking. Template picking yielded 4,737,795 particles. These were split into 6 groups to increase speed of processing, extracted in a 300-pixel box, and binned to 64 pixels. 2D classification was run in cryoSPARC with default settings except: number of 2D classes 200, initial classification uncertainty factor 4, number of online-EM iterations 40, batch size per class 300. Objectively “good” (showing clear Fab and receptor density) class averages were selected and exported to RELION format using csparc.py60. Class averages that were not classified as “good”, but were not clearly ice contamination or graphene oxide edges, were run through a second round of 2D classification with default settings except: number of 2D classes 200, number of online-EM iterations 40, batch size per class 300. All “saved” class averages from the second rounds of 2D classification in cryoSPARC were selected and exported to RELION format using csparc.py. Particles were extracted from CTF-corrected images in RELION at a box size of 300 pixels and binned to 128 pixels. 1,326,130 particles, in three groups (detailed in Extended Data Fig. 3) were classified in 3D with image alignment in RELION using: an initial model generated in cryoSPARC from 80,000 particles collected on a Talos Arctica filtered to 40 Å, C1 symmetry, a regularization parameter of 4, for 30–35 iterations with no mask. Particles from classes with resolved transmembrane (TM) helices were selected, extracted in a 300-pixel box, and imported back into cryoSPARC. Non-uniform refinement was run with default settings and no resolution limit, resulting in angle and shift assignments for 850,000 particles. These particles were subsequently exported to RELION format using csparc.py and run through 3D classification without image alignment. Four of the 12 classes were selected, imported into cryoSPARC, run through non-uniform alignment with an automatically generated mask, and refined to a reported global resolution of 3.2 Å. The resulting map showed signs of mild preferred orientation (Extended Data Fig. 3). The particles were exported into RELION format using csparc.py, converted into an image stack, and imported into cisTEM61 as a refinement package. The particles were reconstructed and half-maps were generated using the “generate 3D” command. These half-maps, as well as the half-maps from cryoSPARC were run through our lab’s directional Fourier shell correlation (dFSC) program62 clearly showing a more evenly weighted range of views in the map generated by cisTEM. Maps were sharpened in RELION. Average resolutions are reported using the FSC = 0.143 cut-off63 and were estimated in cryoSPARC and cisTEM. Local resolution estimates were performed through cryoSPARC, using a similar algorithm to blocres64, with an automatically generated kernel size of 24 voxels.
For Co2+/hepcidin-FPN-Fab45D8, data were motion corrected and 2x binned on-the-fly using MotionCor255 in the SCIPION pipeline56. Motion corrected micrographs were imported into cryoSPARC57 and contrast transfer function parameters were calculated using patch CTF estimation through cryoSPARC59. 3,753,516 particles were selected from 4395 micrographs using the template picker in cryoSPARC with 2D averages from the apo-FPN dataset as templates. These particles were extracted in a 360-pixel box, and binned to 64 pixels. 2D classification was run in cryoSPARC with default settings except: number of 2D classes 200, initial classification uncertainty factor 4, number of online-EM iterations 40, batch size per class 300. Objectively “good” (showing clear Fab and receptor density) class averages were selected for 3D classification. Class averages that were not classified as “good”, but were clearly not ice contamination, were run through a second round of 2D classification with default settings except: number of 2D classes 200, number of online-EM iterations 40, batch size per class 300. All “saved” class averages from the second rounds of 2D classification in cryoSPARC were sent to 3D classification. These particles were subjected to two rounds of heterogeneous refinement in cryoSPARC that serves as a “trash collector”. Four initial models were used, three generated from an early round of ab initio model generation and our final apo-FPN-Fab45D8 structure. All initial models were filtered to 30Å before refinement. Particles were unbinned and a final heterogeneous refinement was performed with three good initial models of apo-FPN-Fab45D8. Non-uniform refinement was run with default settings and no resolution limit, on the most populated class, showing clear density for hepcidin, resulting in angle and shift assignments for 310,647 particles. The particles were exported into RELION format using csparc.py, converted into an image stack, and imported into cisTEM61 as a refinement package. The particles were reconstructed and half-maps were generated using the “generate 3D” command. These half-maps, as well as the half-maps from cryoSPARC were run through our lab’s directional Fourier shell correlation (dFSC) program62 clearly showing a more distributed range of views in the map generated by cisTEM. Maps were sharpened in cisTEM. Average resolutions are reported using the FSC = 0.143 cut-off63 and were estimated in cryoSPARC and cisTEM. Local resolution estimates were performed through cryoSPARC with an automatically generated kernel size of 18 voxels. Another job was run with a kernel size of 20 voxels and results were similar.
Model building and refinement
For apo-FPN, a homology model of human FPN in the outward open state was built using Modeller65, with a previously determined X-ray crystal structure of outward open bbFPN (PDB ID: 5AYN)26 as a template. After truncating putatively flexible regions (N-terminus, ICL3, ECL5, and C-terminus), the resulting model was fit into the 3.2 Å cryo-EM map of FPN:Fab45D8 using Chimera66. The initial template was manually rebuilt in Coot48 and iteratively refined with real space refinement implemented in Phenix49 Model geometry was assessed using MolProbity50. Modeling of FPN-Co2+-hepcidin complex used the apo-FPN structure as a starting point, with iterative improvements in Coot and Phenix. We used a previously determined X-ray crystal structure of hepcidin bound to a neutralizing Fab as a starting model21 for the peptide. Further validation was performed with EMRinger67 to compare the map and final model. Map-to-model FSCs were calculated within Phenix. Figures were prepared in Chimera66 and PyMol.
Molecular dynamics simulations
The structure of the outward-open apo conformation of human FPN was used as the starting coordinates for all simulations. Three different conditions were simulated (Supplementary Table 1): (1) the iron-absent condition, where no iron was added; (2) the iron-bound condition, where an Fe2+ ion was placed in the proposed iron binding site 5.6 Å from D325 α-carbon, 6.9 Å from D504 α-carbon, and 7.8 Å from H507 α-carbon; (3) the iron-in-bulk-solvent condition, where 15 Fe2+ ions were placed randomly in the water box outside the protein using Dabble68.
Simulation coordinates were prepared by removing non-FPN molecules from the initial structure. Prime (Schrödinger) was used to model missing side chains, and neutral acetyl and methylamide groups were added to cap protein termini. The unresolved loops between TM6-TM7 and TM9-TM10 (residues 239–290 and 394–451 respectively, inclusively) were not modeled. The termini surrounding these loops were capped. PropKa was used to determine the dominant protonation state of all titratable residues at pH 769,70. The structure was internally hydrated using Dowser71. Dabble was used to additionally fill the extracellular cavity68. The structure was aligned using the Orientation of Proteins in Membranes (OPM) server72.
Using Dabble, the protein was inserted into a pre-equilibrated 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) membrane bilayer. For all simulations except condition three (iron in bulk solvent), sodium and chloride ions were added at 150 mM to neutralize the system. For condition three, chloride ions were added to neutralize the system resulting in a concentration of 108 mM. A periodic box was used with dimensions 90 × 90 Å in the x-y plane and a water buffer of 10 Å above and below the protein to the periodic boundary. We used the CHARMM36m parameters for lipids, proteins, sodium and chloride ions, and the TIP3P model for waters73–75. The Fe2+ Lennard-Jones parameters were obtained from Li et al.’s compromise model76.
All simulations were run on a single Graphical Processing Unit (GPU) using the Amber18 Compute Unified Device Architecture (CUDA) version of particle-mesh Ewald molecular dynamics (PMEMD)77,78. For each condition, 6 independent simulations were run. For each independent simulation, the system was minimized with 500 steps of steepest descent followed by 500 steps of conjugate gradient descent three times. 10 and 5 kcal mol−1 –Å2 harmonic restraints were used on the protein, lipid, and Fe2+ ions for the first and second minimization, respectively. 1 kcal mol−1 Å−2 harmonic restraints were used on the protein and Fe2+ ions for the final minimization. The system was then heated from 0 K to 100 K over 12.5 ps in the NVP ensemble with a Langevin thermostat and harmonically restraining the protein heavy atoms and Fe2+ ions with a restraint of 10 kcal mol−1 Å−2. The system was further heated with the same restraints from 100 K to 310 K in the NPT ensemble over 125 ps. The system was equilibrated with harmonic restraints on protein heavy atoms and Fe2+ ions for 30 ns. The restraint strength started at 5 kcal mol−1 Å−2 and was reduced by 1 kcal mol−1 Å−2 every 2 ns for the first 10 ns and then by 0.1 kcal mol−1 Å−2 every 2 ns for the final 20 ns. Production simulations were performed at 310 K and 1 bar using the NPT ensemble, a Langevin thermostat and a Monte Carlo barostat. Every 200 ps snapshots were saved. All simulations were run for at least 2.2 μs. These simulations used a 4-fs time step with hydrogen mass repartitioning79. Bond lengths to hydrogen atoms were constrained using SHAKE79,80. Non-bonded interactions were cut off at 9 Å.
Simulation Analysis Methods
Molecular dynamics simulation snapshots were reimaged every 1 ns and centered using CPPTRAJ package in AmberTools1881. Simulations were visualized using Visual Molecular Dynamics and figures prepared in PyMOL82. Time traces from simulation were smoothed using a moving average with a window size of 15 ns unless otherwise indicated and visualized with the PyPlot package from Matplotlib. For all analysis in the manuscript that required structural alignment, we aligned to the initial Ferroportin structure using the backbone atoms of residues 26–116, 127–228, 308–483, and 492–543.
To investigate the localization of Fe2+ ions, the iron-in-bulk-solvent simulations (condition 3) were analyzed. To visualize the density of Fe2+ ions under a given simulation condition, the position of Fe2+ ions was recorded every 10 ns for each of the 6 simulations, each 2 μs in length. Each Fe2+ ion position was then drawn as a point superimposed on the starting structure (Fig. 3d). To quantify the binding events, the distance between iron and the closest side chain oxygen atom on D325 was measured. This distance was graphed over 1.2 μs, including the equilibration time (Fig. 3d, Extended Data Fig. 8b).
To investigate the dynamics and conformation of the TM7b region, the iron-absent (condition 1) and iron-bound (condition 2) simulations were compared. The TM1-TM7b distance was measured using distance between the Cα of V51 and the Cα of Y333. For each simulation, we calculated the average of the distance over 2.2 μs, excluding equilibration. The average over the simulations for each condition was plotted with error bars representing the standard error of the mean (s.e.m.) (Extended Data Fig. 8g). The dynamics of D325 were also investigated. The dynamics were visualized by overlaying representative frames showing the movement of D325, the binding site, and TM7b. For iron-absent simulations, frames from a single replicate at 200, 350, 500, and 550 ns were overlaid (Extended Data Fig. 8d). For iron-bound simulations, frames from a single simulation at 200, 500, 725, 1000 ns were overlaid (Extended Data Fig. 8e). The conformational range of D325 was quantified by measuring the distance between Cγ of D325 and Cβ of S47. This was visualized for one simulation for each condition over a time of 1 μs inclusive of equilibration (Extended Data Fig. 8g). For each independent simulation, the mean of the Cγ D325 - Cβ S47 distance was calculated over 2.2 μs. For each condition, the average over the simulations was plotted with error bars representing the s.e.m. (Extended Data Fig. 8g). The flexibility of D325 was quantified by calculating the root-mean-square fluctuation (RMSF) of the side-chain atoms of D325 using an in-house script (Extended Data Fig. 8g). Statistical significance was determined using the Mann-Whitney U test (two-tailed).
Data Availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information. Crystallographic coordinates and structure factors for the Fab45D8 complex have been deposited in the Protein Data Bank under accession code 6W4V. Coordinates for Fab45D8-FPN complex have been deposited in the Protein Data Bank under accession code 6W4S and the maps have been deposited in the Electron Microscopy Data Bank under accession code 21539. Coordinates for the FPN-Co2+-hepcidin-Fab45D8 complex have been deposited in the Protein Data Bank under accession code 6WBV and the maps have been deposited in the Electron Microscopy Data Bank under accession code 21599.
Extended Data
Extended Data Fig. 1 |. Biochemistry of purified human ferroportin and Fab45D8 complex.
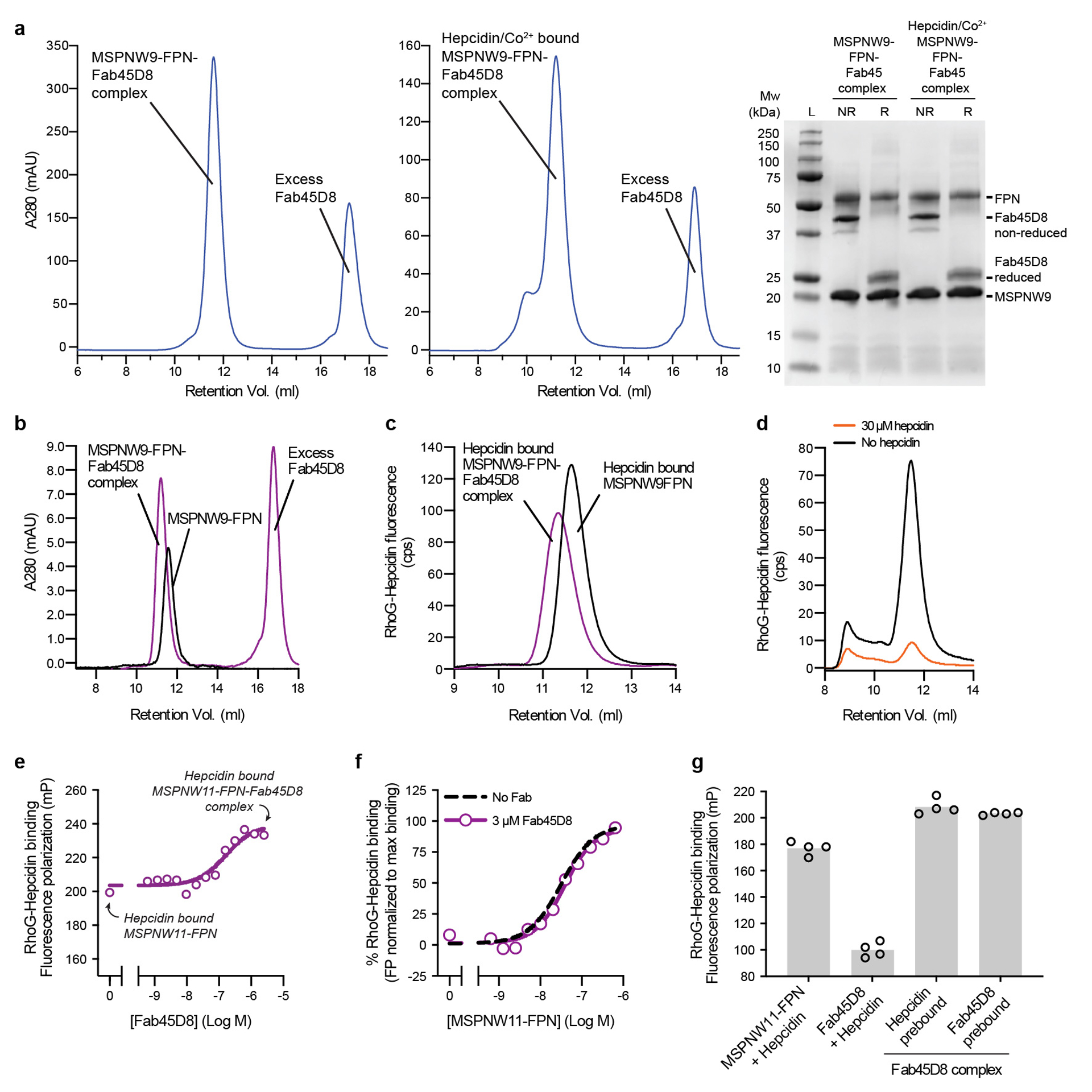
a, Size exclusion chromatography of purified apo and hepcidin/Co2+ bound FPN-Fab45D8 complex. SDS-PAGE gel of purified complex under non-reducing (NR) and reducing (R) conditions. SEC and SDS-PAGE analyses are representative and were performed four and two times for apo and hepcidin/Co2+ bound FPN-Fab45D8 samples respectively. b, Fab45D8 binds nanodisc-reconstituted FPN (nanodisc-FPN), as assessed by size-exclusion chromatography. c, Fluorescence size exclusion chromatography (FSEC) of rhodamine green hepcidin (RhoG-hepcidin). RhoG-hepcidin co-elutes with nanodisc-FPN and with a nanodisc-FPN:Fab45D8 complex in the presence of CoCl2. d, FSEC shows RhoG-hepcidin binding to nanodisc-FPN is competed by excess unlabelled hepcidin used for structural studies. e, Fluorescence polarization of RhoG-hepcidin increases further with Fab45D8, consistent with formation of a larger complex. Importantly, Fab45D8 does not decrease RhoG-hepcidin binding. Data points are means from n = 3 technical replicates. f, RhoG-hepcidin binding to nanodisc-FPN in the presence of 10 μM CoCl2 measured by fluorescence polarization in the absence (KD = 31 nM) and presence of 3 μM Fab45D8 (KD = 39 nM). Data points are means from n = 3 technical replicates. g, RhoG-hepcidin binds to FPN (FPN+hepcidin) but not to Fab45D8 alone (Fab45D8+hepcidin). Order of hepcidin and Fab45D8 addition does not influence the increase in fluorescence polarization for a Fab45D8-FPN-hepcidin complex. Data points are means from n = 4 technical replicates.
Extended Data Fig. 2 |. Calcein quenching assay for FPN metal transport.
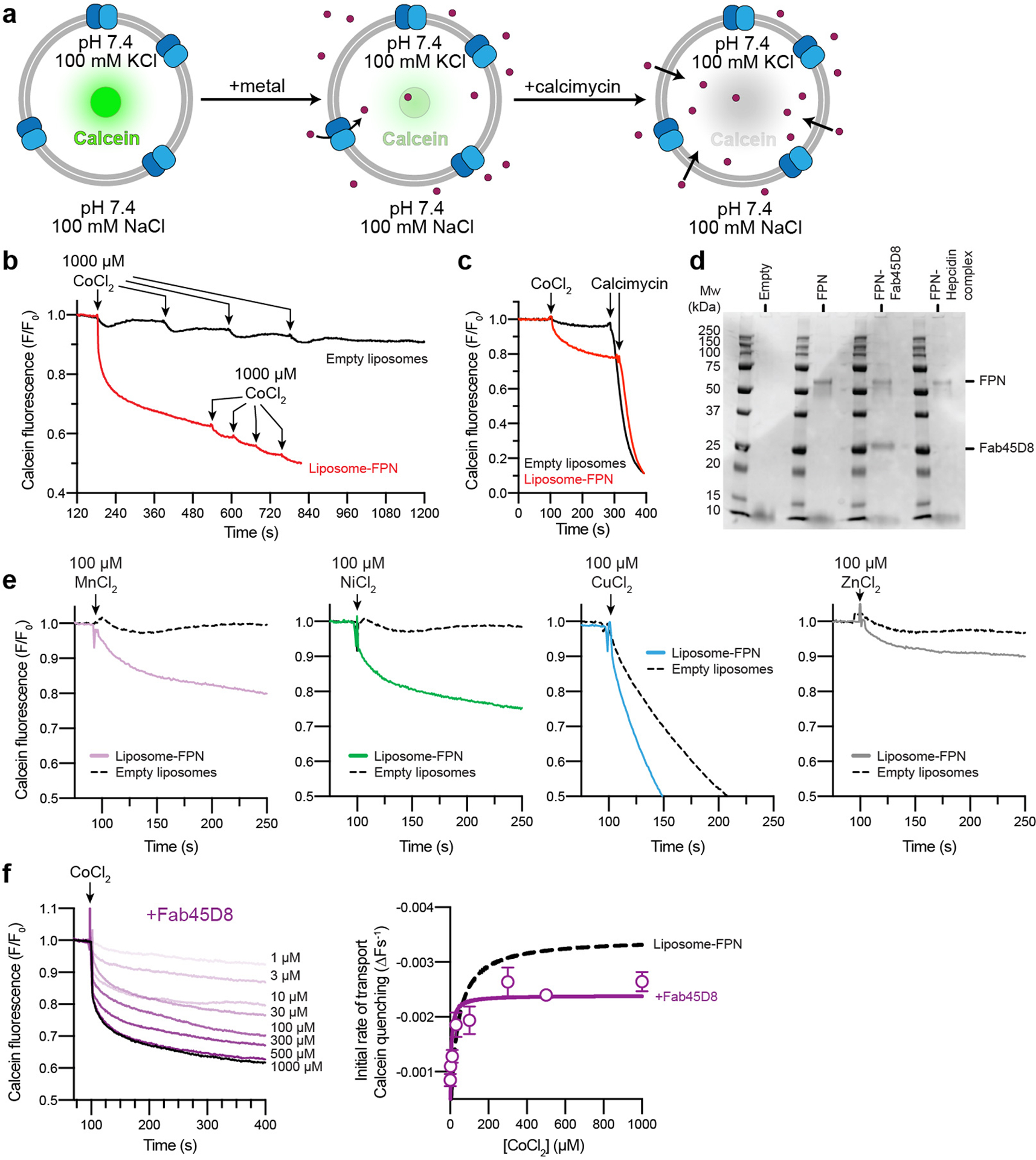
a, Schematic of calcein-based assay to measure transport of divalent cations. Purified FPN is reconstituted into proteoliposomes containing calcein. Transport of divalent cations leads to quenching of calcein fluorescence, which can also occur through the divalent cation selective ionophore calcimycin. b and c, Specificity of the calcein transport assay. FPN reconstituted liposomes facilitate uptake of 1000 μM Co2+ added (arrows, red trace) to the external bath solution, producing a calcein quenching response that follows a single exponential. Further additions of Co2+ demonstrates the transport process has reached saturation. Calcein fluorescence from empty liposomes (black trace) returns to baseline after multiple additions of 1000 μM Co2+. c, Additions of Co2+ followed by calcimycin results in rapid and almost complete quenching. Fluorescence traces are representative experiments. d, Liposome reconstitution samples analyzed by reducing SDS-PAGE, the analysis is representative and was performed three times for FPN samples and twice for FPN samples containing FPN-Hepcidin or FPN-Fab45D8. e, Calcein transport assay queried against a panel of putative transition metal ion substrates at 100 μM. Notably, the larger ions Ni2+ and Zn2+ are also reported FPN substrates83. The smaller ion Mn2+ was previously found not to be effluxed by FPN in oocytes83, but appears to be transported in liposomes. Cu2+ interferes with empty and FPN-containing liposomes, leading to non-specific quenching of calcein. Fluorescence traces are representative experiments. f, Fab45D8 decreases cobalt transport Vmax (2.40 × 10−3 ± 0.075 × 10−3 ΔF s−1) and Km (4.68 ± 0.54 μM). Data points are means ± s.e.m. from n = 3 independent experiments.
Extended Data Fig. 3 |. Cryo-EM data processing for FPN-Fab45D8.
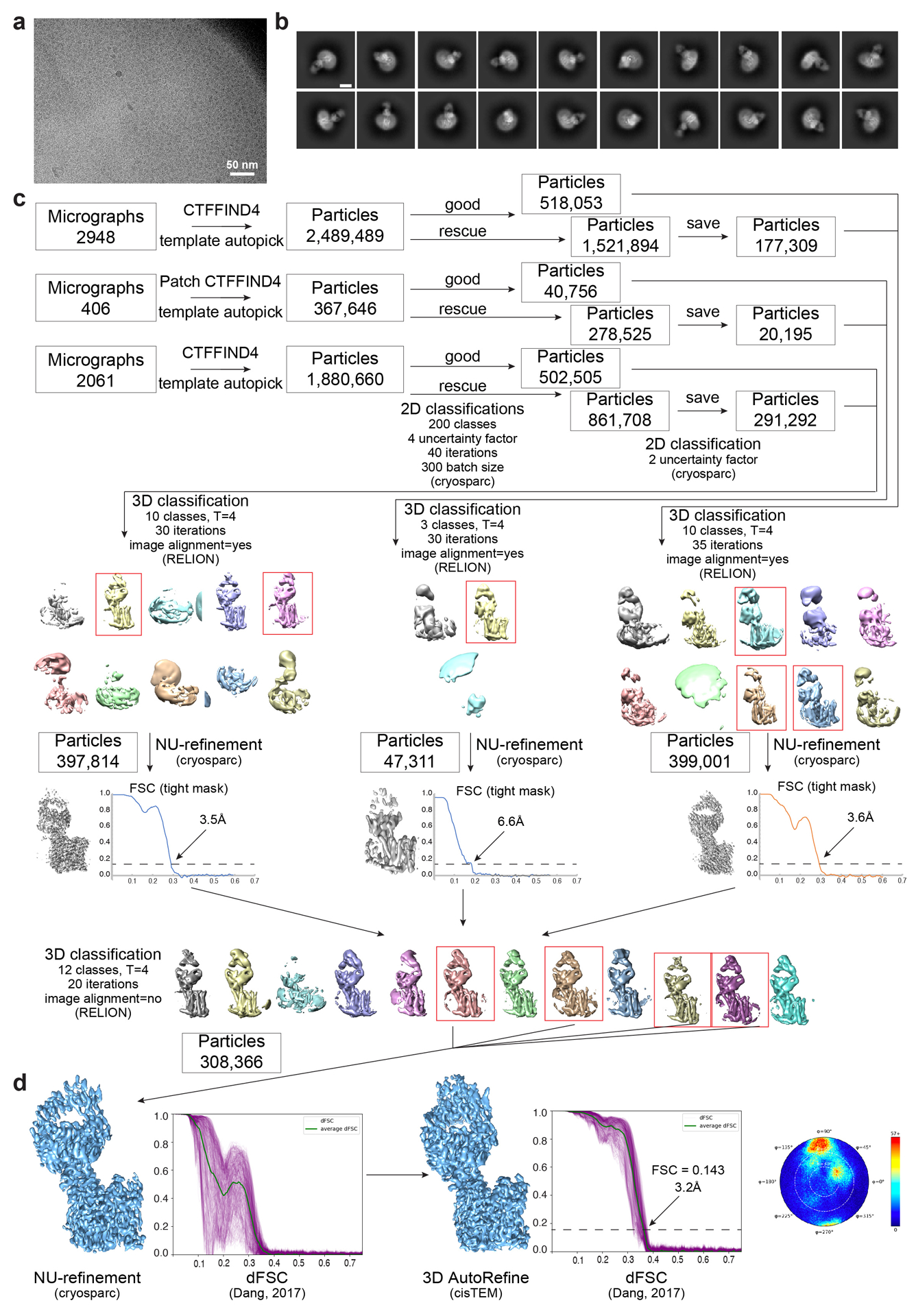
a, Representative motion-corrected micrograph collected on the Titan Krios showing monodisperse FPN-Fab45D8 nanodisc particles. Four different FPN-Fab45D8 samples were imaged on GO-coated Quantifoil grids times with similar results. b, Examples of “good” 2D class averages that were used in 3D classification. Similar quality class averages were produced when processing six unique subsets of the final particles used. Scale bar is 50 Å. c, Flowchart showing image processing pipeline for FPN-Fab45D8. Initial processing, through 2D classification, was performed in cryoSPARC. Particles were then transferred, using csparc2star.py, to RELION for 3D classification, then to cryoSPARC for a nonuniform refinement, and finally to cisTEM for refinement. The number of particles moving into each step are noted. d, Final refinements from cryosparc and cisTEM beside their directional FSC curves calculated using dfsc.0.0.1.py. Angular distribution plot from cisTEM is shown.
Extended Data Fig. 4 |. Cryo-EM data processing for Co2+-hepcidin-FPN-Fab45D8.
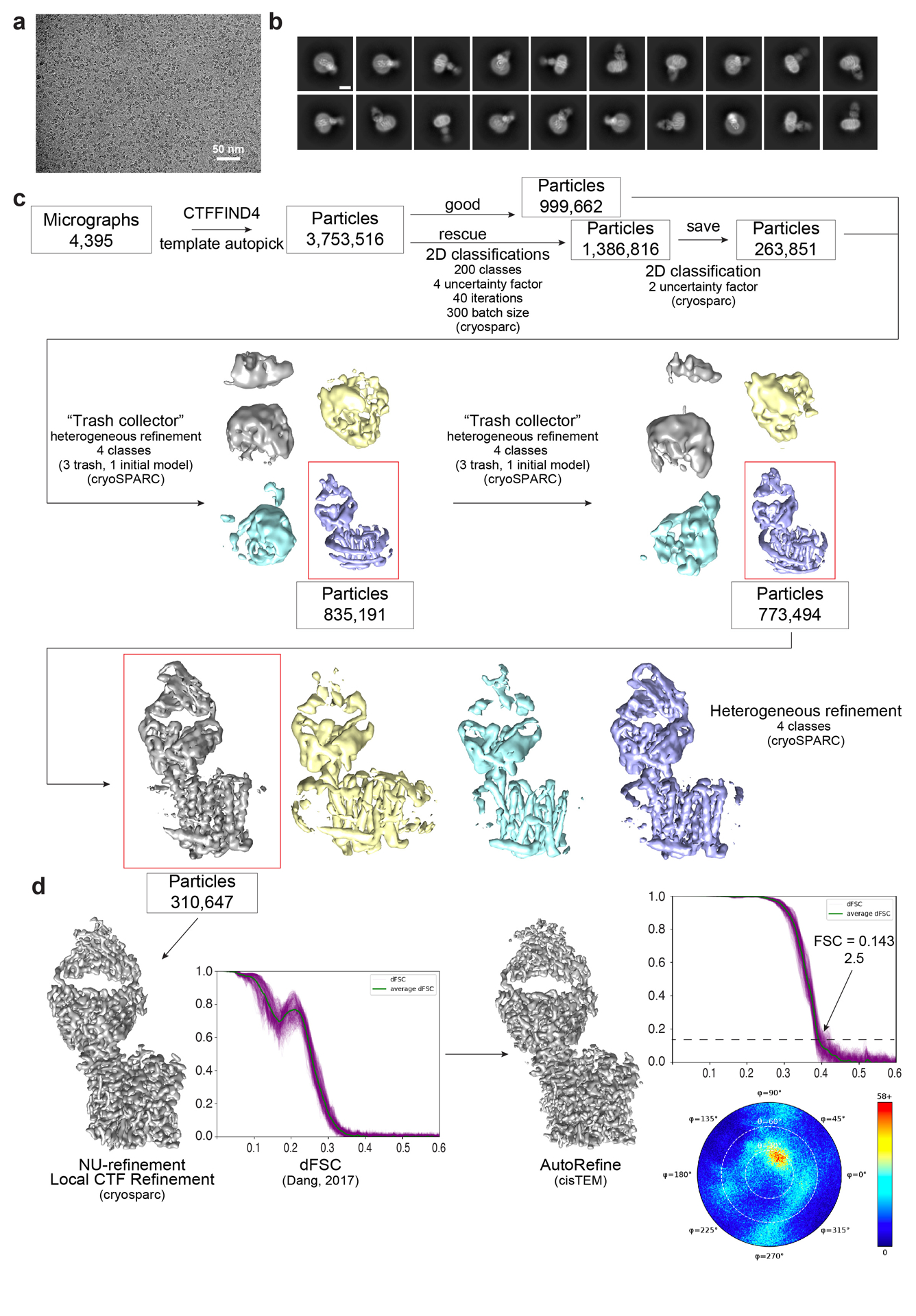
a, Representative motion-corrected micrograph collected on the Titan Krios showing monodisperse Co2+-hepcidin-FPN-Fab45D8 nanodisc particles. Two indepedent Co2+-hepcidin-FPN-Fab45D8 samples were frozen on UltrAufoil grids with similar micrograph quality. b, Examples of “good” 2D class averages that were used in 3D classification. c, Flowchart showing image processing pipeline for Co2+-hepcidinF-PN-Fab45D8. Initial processing was performed in cryoSPARC. Local CTF refinement, with a conservative high-resolution limit of 4 Å, was performed before nonuniform refinement using default parameters. A final subset of particles was transferred to cisTEM for refinement. The number of particles moving into each step are noted. d, Final refinements from cryosparc and cisTEM beside their directional FSC curves calculated using dfsc.0.0.1.py. Angular distribution plot from cisTEM is also shown.
Extended Data Fig. 5 |. Cryo-EM map density for FPN.
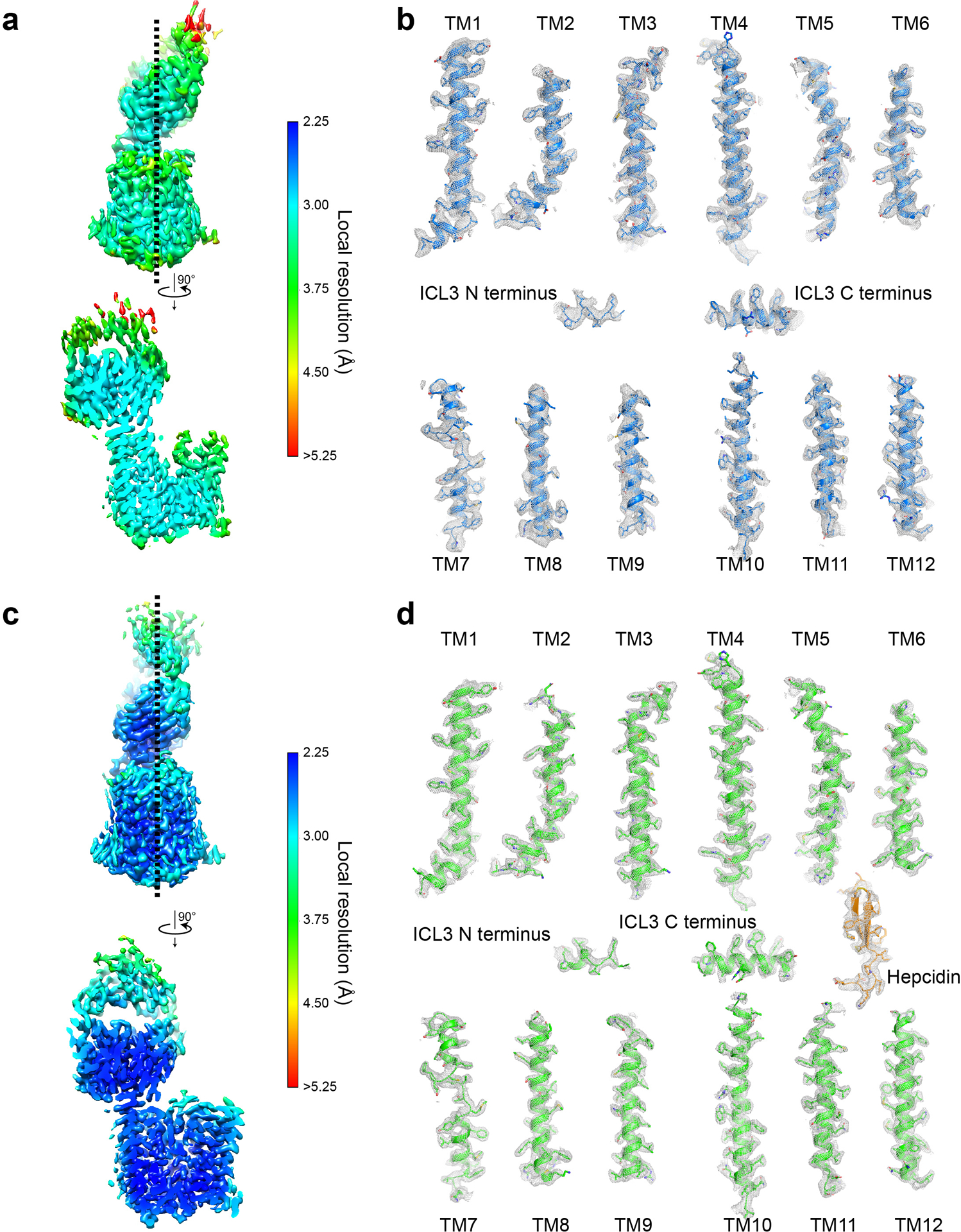
Local resolution estimation in cryoSPARC for the apo-FPN-Fab45D8 complex (a) and for the Co2+-hepcidin-FPN-Fab45D8 complex (c). Two views of each complex are shown. Shown in grey mesh is cryo-EM map density for individual FPN transmembrane helices for the apo-FPN-Fab45D8 complex (b) and for the Co2+-hepcidin-FPN-Fab45D8 complex (d). Mesh depicts density within a 2.5 Å radius of any modeled atom.
Extended Data Fig. 6 |. Structure of Fab45D8 and comparison of FPN with bbFPN.
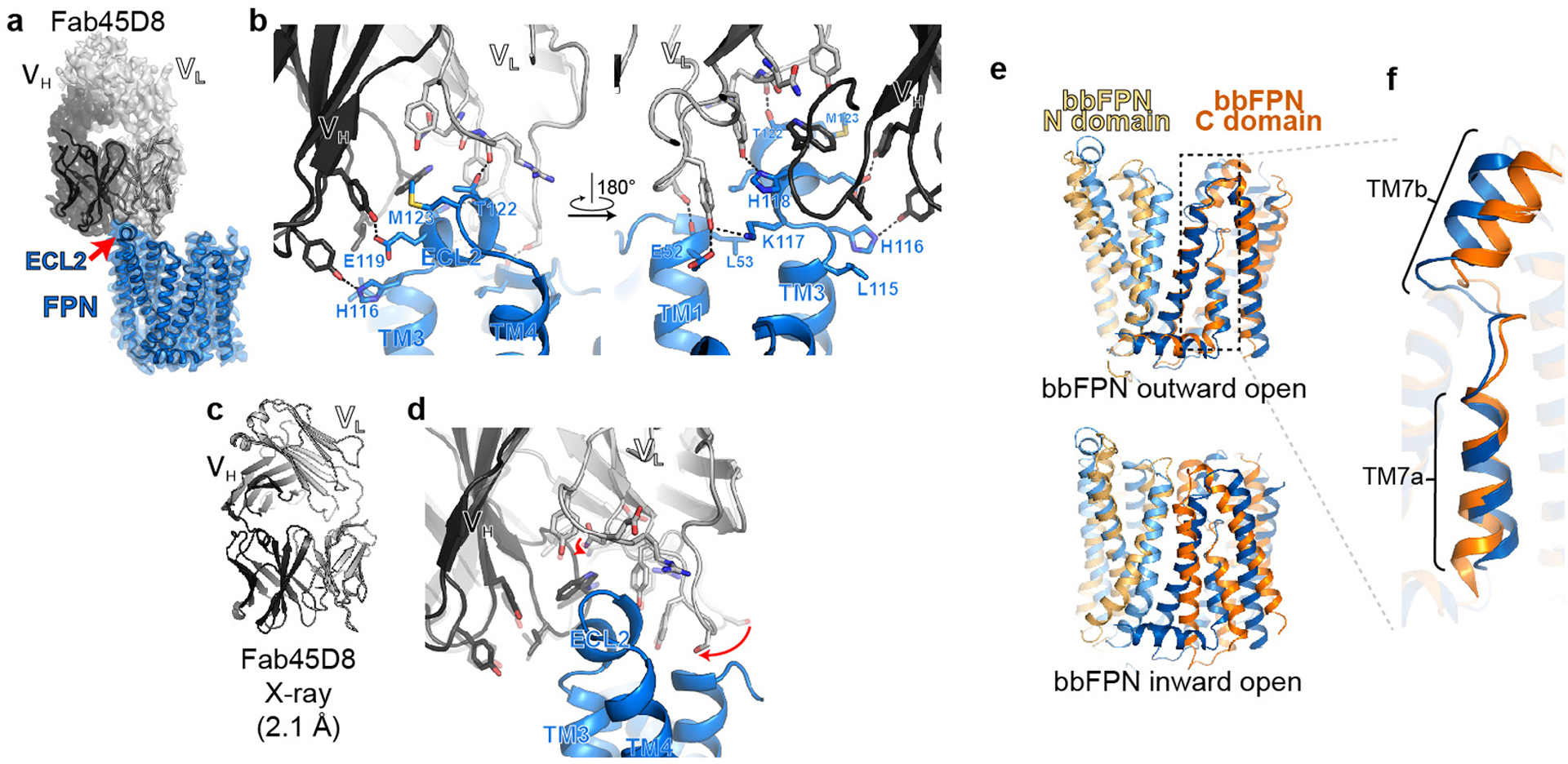
a, Cryo-EM density of FPN and Fab45D8. Although density is observed for the constant regions of Fab45D8, it is not of sufficient quality to unambiguously model. b, Fab45D8 makes extensive contacts with FPN extracellular loop 2 (ECL2) with both the heavy (VH) and light (VL) chains. c, Crystal structure of Fab45D8 at 2.1 Å. d, Comparison of Fab45D8 alone (transparent cartoon and sticks) and bound to FPN. The binding site residues of Fab45D8 change minimally upon binding FPN. e, Human FPN aligned to the outward-open (PDB: 5AYN) and inward-open (PDB: 5AYO) conformations of bbFPN. f, Unique architecture of TM7 shared between human FPN and bbFPN.
Extended Data Fig. 7 |. Effect of pH and ions on metal transport and binding.
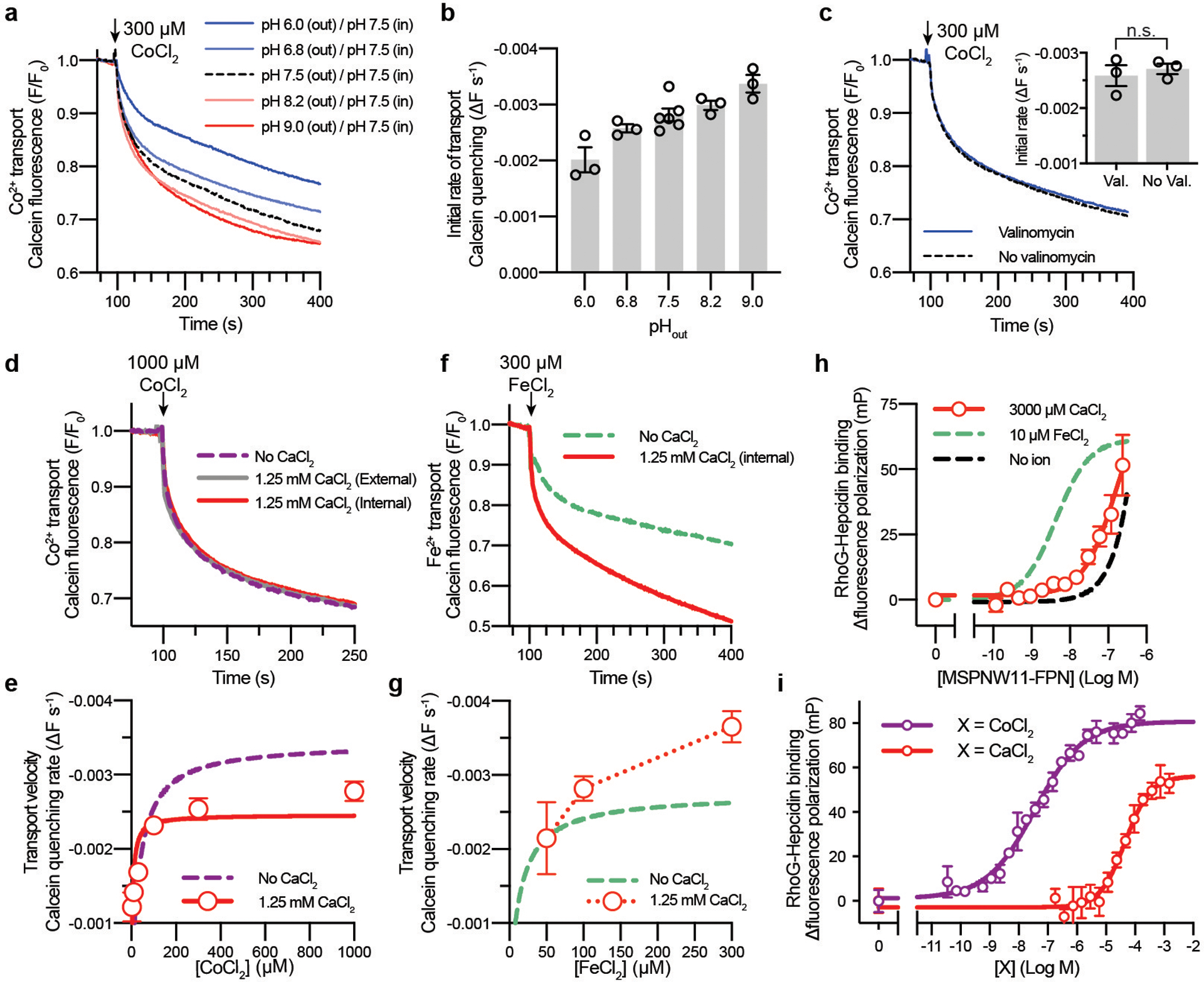
a and b, Co2+ transport by FPN is potentiated by an opposing H+-gradient. a, Time-course calcein fluorescence quenching response after addition of 300 μM CoCl2 (arrow) for FPN-liposomes containing 100 mM KCl and HEPES, pH 7.50 that were diluted into buffer comprised of 100 mM NaCl and (i) HEPES pH 6.8, (ii) HEPES pH 7.5, (iii) HEPES pH 8.2, (iv) Tris/MES pH 6.0, or (v) Tris/MES pH 9.0. b, initial rates of transport obtained from the linear phase of time-course fluorescence quenching experiments at the indicated pH values. Data points are means ± s.e.m. from n = 3 experiments. c, Calcein fluorescence responses for FPN-liposomes containing 100 mM KCl and HEPES, pH 7.50 diluted into 100 mM NaCl and HEPES, pH 7.50. A membrane potential (ΔΨ) was generated by addition of the K+ selective ionophore valinomycin prior to the experiment, which creates an outward K+ gradient. Not significant (p = 0.5986, Student’s unpaired t-test, two-tailed) difference was observed for the rate of transport by valinomycin addition. Bars are means ± s.e.m with corresponding data points from n = 3 independent experiments. d-g Calcium modulates the FPN transport mechanism. d, Time-course calcein fluorescence quenching response after addition of 1000 μM CoCl2 (arrow) for FPN-liposomes preloaded with 1.25 mM CaCl2 internally (red) or 1.25 mM CaCl2 added to the external solution (grey). e, Initial rates of Co2+ transport in the presence of internal 1.25 mM CaCl2 (red circles). Data points are means ± s.e.m from n = 3 independent experiments. Quantified transport rates in Supplementary Table 2. f, Calcein fluorescence response for FeCl2. g, Initial rates of Fe2+ transport. Data points are means ± s.e.m from n = 3 independent experiments. h-i, Calcium does not support high affinity hepcidin binding to FPN. h, Binding of 5 nM RhoG-hepcidin to nanodisc-FPN in the presence of 3 mM CaCl2 was determined by fluorescence polarization (EC50 >100 nM). Data points are means ± s.e.m from n = 3 independent experiments. i, Ca2+ stimulates the partial binding of 5 nM RhoG-hepcidin to 100 nM nanodisc-FPN with an EC50 of 47.1 ± 4.1 μM, confirming that the Ca2+ binding site in FPN titrates in the physiological range (~1.25 mM) of free ionized Ca2+. In contrast, Co2+ stimulates binding of 5 nM RhoG-hepcidin to 100 nM nanodisc-FPN with an EC50 of 35.8 ± 3.6 nM. However, depletion of RhoG-hepcidin leads to a nHill = 0.52, and the true affinity of cobalt may be higher than this value. Importantly, these results indicate that binding of physiologically relevant concentrations of hepcidin would be exceedingly ineffective to apo and Ca2+-bound FPN. Data points are means ± s.e.m from n = 3 independent experiments.
Extended Data Fig. 8 |. Molecular dynamics simulations of iron binding to apo FPN.
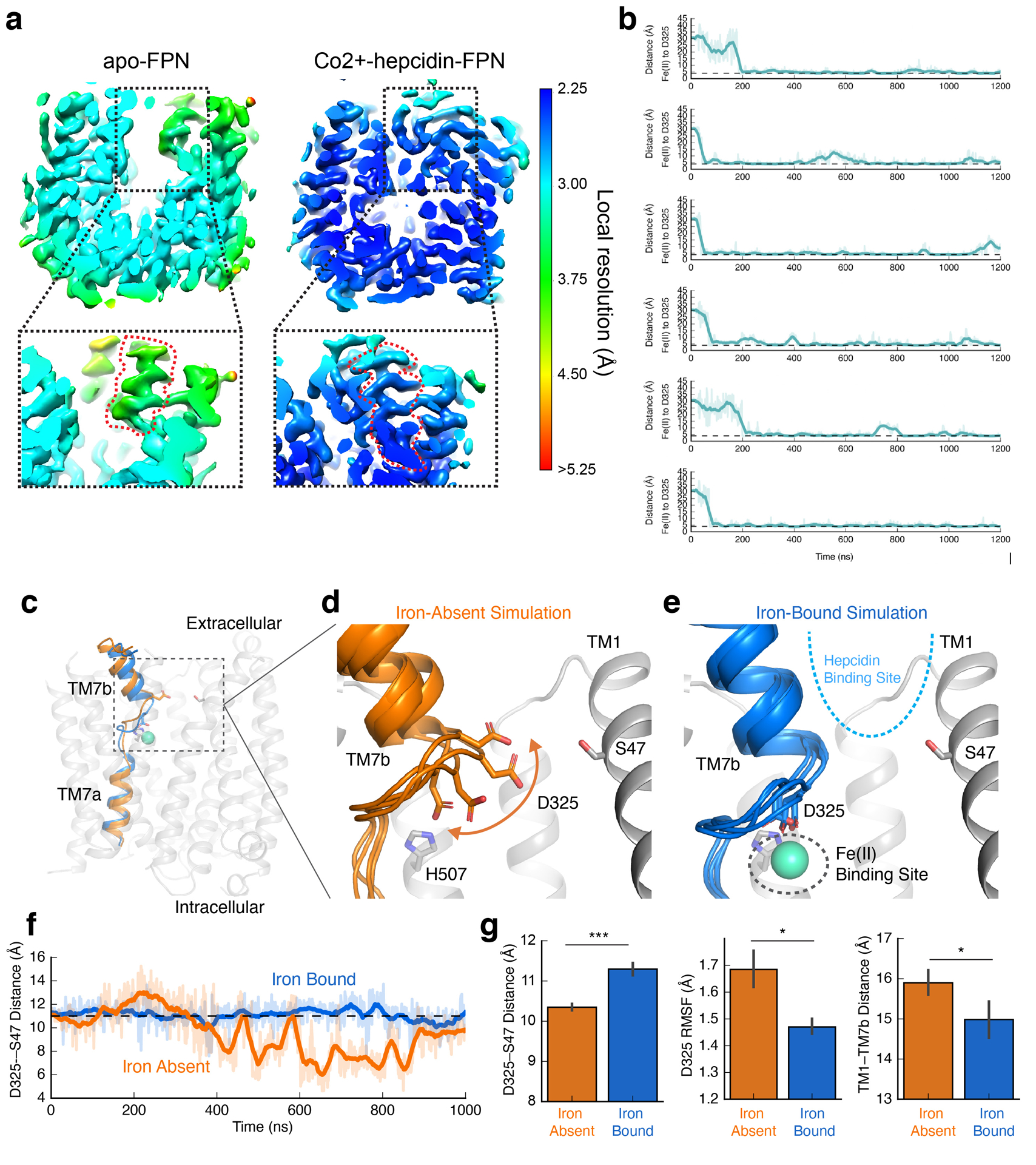
a, Local resolution of FPN cryo-EM density. Insets show local resolution around TM7b. b, Each graph corresponds to an independent simulation where Fe2+ ions start in solution and bind spontaneously to the proposed iron binding site. Distance shown is from the ion to the nearest oxygen atom of the D325 side chain. Thick traces represent a 15-ns sliding mean and thin traces represent unsmoothed values. Timetraces include 30 ns of equilibration. The average time to bind (the time from start of production simulation to when the measured distance is less) was 71 ns. c, Representative conformations of TM7 from simulations with iron bound (blue) or absent (orange). d, In the absence of bound iron, D325 is mobile and can move into the cavity between TM7b and TM1. The top of TM7b can also tilt away from TM1. Four representative frames from simulation are overlaid. e, In simulations with Fe2+ bound, interaction with the ion restricts the mobility of D325 and, in turn, TM7b. f, Iron-bound and iron-absent simulations show differences in the dynamics and position of D325, as measured by the distance between D325 Cγ and S47 Cβ. g, Comparison of conformation and dynamics with and without iron bound. With iron bound, D325 moves away from TM1 into the iron binding site (left), the root-mean-square fluctuation (RMSF) of D325 decreases (middle), and the extracellular end of TM7b moves closer to TM1 (right). For these comparisons, 6 simulations for each condition were used, each 2.0 μs in length. Error bars are s.e.m. and p-values were calculated using Mann-Whitney U test (two-tailed); the p=0.001 (left plot), 0.046 (middle plot), 0.046 (right plot).
Extended Data Table 1:
Cryo-EM statistics
| FPN-Fab45D8 | FPN-Co2+-hepcidin-Fab45D8 | |
|---|---|---|
| (EMDB-21539) | (EMDB-21599) | |
| (PDB 6W4S) | (PDB 6WBV) | |
| Data collection and processing | ||
| Microscope/Detector | Titan Krios/Gatan K3 with Gatan Bioquantum Energy Filter | Titan Krios/Gatan K3 with Gatan Bioquantum Energy Filter |
| Imaging software and collection | SerialEM, 3×3 image shift | Serial EM, 3×3 image shift |
| Magnification | 105,000 | 105,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e−/Å2) | 66 | 66 |
| Dose rate (e−/pix/sec) | 8 | 8 |
| Frame exposure (e−/Å2) | 0.55 | 0.55 |
| Defocus range (μm) | −0.8 to −2.0 | −0.8 to −2.0 |
| Pixel size (Å) | 0.834 (physical) | 0.834 (physical) |
| Micrographs | 5,415 | 4,395 |
| Reconstruction | ||
| Autopicked particles (template-based in cryosparc) | 4,737,795 | 3,753,516 |
| Particles in 3D classification | 1,326,130 (RELION) | 1,263,513 (cryosparc) |
| Particles in final refinement | 308,386 (cisTEM) | 310,647 (cisTEM) |
| Symmetry imposed | Cl | Cl |
| Map sharpening B factor (Å2) | −130 | −90 |
| Map resolution, global FSC (Å) | ||
| FSC 0.5, unmasked/masked | 7.1/3.4 | 4.0/2.7 |
| FSC.143, unmasked/masked | 4.0/3.2 | 3.3/2.5 |
| Refinement | ||
| Initial model used (PDB code) | 5AYN | 5AYN, 3H0T |
| Model resolution (Å) | ||
| FSC 0.5, unmasked/masked | 3.6/3.4 | 3.0/2.8 |
| Model composition | ||
| Non-hydrogen atoms | 4968 | 6869 |
| Protein residues | 650 | 892 |
| B factors (Å2) | ||
| Protein | 67.31 | 27.30 |
| Ligand | N/A | 55.15 |
| Water | N/A | 31.32 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.006 |
| Bond angles (°) | 0.563 | 0.872 |
| Validation | ||
| MolProbity score | 2.31 | 2.10 |
| Clashscore | 7.90 | 7.10 |
| Poor rotamers (%) | 4.45 | 4.26 |
| EMRinger score | 2.37 | 4.32 |
| CaBLAM score | 2.06 | 0.69 |
| Ramachandran plot | ||
| Favored (%) | 94.38 | 96.47 |
| Allowed (%) | 5.62 | 3.53 |
| Disallowed (%) | 0 | 0 |
Extended Data Table 2:
Fab45D8 crystal structure statistics
| Fab45D8 (PDB 6W4V) | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 72.19, 36.43, 84.95 |
| a, β, γ (°) | 90.0, 112.26, 90.0 |
| Resolution (Å) | 39.31 – 2.09 (2.14 – 2.09)a |
| Rsym or Rmerge | 0.087 (1.385) |
| I / σI | 7.9 (1.2) |
| Completeness (%) | 99.9 (99.9) |
| Redundancy | 3.3 (3.5) |
| CC (1/2) (%) | 99.6 (30.0) |
| Refinement | |
| Resolution (Å) | 39.31 – 2.09 |
| No. reflections | 24736 |
| Rwork / Rfree (%) | 21.6 / 24.7 |
| No. atoms | |
| Protein | 3356 |
| Ligand/ion | 20 |
| Water | 125 |
| B-factors | |
| Protein | 57.87 |
| Ligand/ion | 86.17 |
| Water | 51.43 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 0.92 |
Values for parentheses are for highest-resolution shell.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants DP5OD023048 (A.M.), R01GM098672 (Y.C.), and P50AI150476 (Y.C.). A.S.P. was supported by a National Science Foundation Graduate Research Fellowship and NIH training grant T32GM120007. R.C.K. was supported by NIH training grant T32GM008293. Cryo-EM equipment at UCSF is partially supported by NIH grants S10OD020054 and S10OD021741. C.B.B. was supported by the Alfred Benzon Foundation. R.O.D. acknowledges support from a Stanford University Bio-X seed grant. Y.C. is an Investigator of Howard Hughes Medical Institute. A.M. acknowledges support from the Pew Charitable Trusts, the Esther and A. & Joseph Klingenstein Fund and the Searle Scholars Program.
Footnotes
Competing Interests
Tara Arvedson is employed by Amgen and reports Amgen stock. The other authors declare no competing interests.
REFERENCES
- 1.Nemeth E et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Drakesmith H, Nemeth E & Ganz T Ironing out Ferroportin. Cell Metab. 22, 777–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donovan A et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191–200 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Donovan A et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Knutson MD, Oukka M, Koss LM, Aydemir F & Wessling-Resnick M Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. U. S. A 102, 1324–1328 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward DM & Kaplan J Ferroportin-mediated iron transport: expression and regulation. Biochim. Biophys. Acta 1823, 1426–1433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aschemeyer S et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood, The Journal of the American Society of Hematology 131, 899–910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Domenico I et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol. Biol. Cell 18, 2569–2578 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiao B et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 15, 918–924 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross SL et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metab. 15, 905–917 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Roetto A et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet 33, 21–22 (2003). [DOI] [PubMed] [Google Scholar]
- 12.De Domenico I et al. The molecular basis of ferroportin-linked hemochromatosis. Proc. Natl. Acad. Sci. U. S. A 102, 8955–8960 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drakesmith H et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 106, 1092–1097 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Roy CN et al. Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood 109, 4038–4044 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganz T & Nemeth E The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am. Soc. Hematol. Educ. Program 2011, 538–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manolova V et al. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of β-thalassemia. J. Clin. Invest (2019) doi: 10.1172/JCI129382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witcher DR et al. LY2928057, an antibody targeting ferroportin, is a potent inhibitor of hepcidin activity and increases iron mobilization in normal cynomolgus monkeys. (2013).
- 18.Crielaard BJ, Lammers T & Rivella S Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov 16, 400–423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vlasveld LT et al. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemeth E et al. The N-terminus of hepcidin is essential for its interaction with ferroportin: structure-function study. Blood 107, 328–333 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan JB et al. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J. Biol. Chem 284, 24155–24167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonaccorsi di Patti MC et al. A structural model of human ferroportin and of its iron binding site. FEBS J. 281, 2851–2860 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Ross SL et al. Identification of Antibody and Small Molecule Antagonists of Ferroportin-Hepcidin Interaction. Front. Pharmacol 8, 838 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu S et al. Fabs enable single particle cryoEM studies of small proteins. Structure 20, 582–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nasr ML et al. Covalently circularized nanodiscs for studying membrane proteins and viral entry. Nat. Methods 14, 49–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taniguchi R et al. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun 6, 8545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshpande CN et al. Calcium is an essential cofactor for metal efflux by the ferroportin transporter family. Nat. Commun 9, 3075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guellec J et al. Molecular model of the ferroportin intracellular gate and implications for the human iron transport cycle and hemochromatosis type 4A. FASEB J. 33, 14625–14635 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Sham RL et al. Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol. Dis 34, 157–161 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Schimanski LM et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 105, 4096–4102 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Viprakasit V et al. Molecular Diagnosis of the First Ferroportin Mutation (C326Y) in the Far East Causing a Dominant Form of Inherited Iron Overload. (2004).
- 32.Mayr R et al. Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology 140, 2056–63, 2063.e1 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Tortosa V, di Patti MCB, Brandi V, Musci G & Polticelli F An improved structural model of the human iron exporter ferroportin. Insight into the role of pathogenic mutations in hereditary hemochromatosis type 4. Bio-Algorithms and Med-Systems 13, 215–222 (2017). [Google Scholar]
- 34.Wallace DF, Clark RM, Harley HAJ & Nathan Subramaniam V Autosomal dominant iron overload due to a novel mutation of ferroportin1 associated with parenchymal iron loading and cirrhosis. Journal of Hepatology vol. 40 710–713 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Njajou OT et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet 28, 213–214 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Arden KE et al. A novel mutation in ferroportin1 is associated with haemochromatosis in a Solomon Islands patient. Gut 52, 1215–1217 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W et al. A novel SLC40A1 p.Y333H mutation with gain of function of ferroportin: A recurrent cause of haemochromatosis in China. Liver Int. 39, 1120–1127 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Létocart E et al. A novel missense mutation in SLC40A1 results in resistance to hepcidin and confirms the existence of two ferroportin-associated iron overload diseases. Br. J. Haematol 147, 379–385 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Callebaut I et al. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum. Mol. Genet 23, 4479–4490 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Preza GC et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Invest 121, 4880–4888 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rivard SR et al. Autosomal dominant reticuloendothelial iron overload (HFE type 4) due to a new missense mutation in the FERROPORTIN 1 gene (SLC11A3) in a large French-Canadian family. Haematologica 88, 824–826 (2003). [PubMed] [Google Scholar]
- 42.Raszeja-Wyszomirska J, Caleffi A, Milkiewicz P & Pietrangelo A Ferroportin-related haemochromatosis associated with novel Y64H mutation of the SCL40A1 gene. Prz Gastroenterol 9, 307–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galesloot TE et al. Serum hepcidin: reference ranges and biochemical correlates in the general population. Blood 117, e218–25 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Winter G, Lobley CMC & Prince SM Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr 69, 1260–1273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aleman F et al. Immunogenetic and structural analysis of a class of HCV broadly neutralizing antibodies and their precursors. Proc. Natl. Acad. Sci. U. S. A 115, 7569–7574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cote LJ, Kim F & Huang J Langmuir- Blodgett assembly of graphite oxide single layers. J. Am. Chem. Soc 131, 1043–1049 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Palovcak E et al. A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J. Struct. Biol 204, 80–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F et al. Amino and PEG-amino graphene oxide grids enrich and protect samples for high-resolution single particle cryo-electron microscopy. J. Struct. Biol 209, 107437 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mastronarde DN SerialEM: A Program for Automated Tilt Series Acquisition on Tecnai Microscopes Using Prediction of Specimen Position. Microsc. Microanal 9, 1182–1183 (2003). [Google Scholar]
- 55.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de la Rosa-Trevín JM et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol 195, 93–99 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Scheres SHW RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asarnow D, Palovcak E & Cheng Y asarnow/pyem: UCSF pyem v0.5. (2019). doi: 10.5281/zenodo.3576630. [DOI]
- 61.Grant T, Rohou A & Grigorieff N cisTEM, user-friendly software for single-particle image processing. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dang S et al. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 552, 426–429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Cardone G, Heymann JB & Steven AC One number does not fit all: mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol 184, 226–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eswar N et al. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci Chapter 2, Unit 2.9 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Goddard TD, Huang CC & Ferrin TE Visualizing density maps with UCSF Chimera. J. Struct. Biol 157, 281–287 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Barad BA et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Betz R Dabble. (2017). doi: 10.5281/zenodo.836914. [DOI]
- 69.Olsson MHM, Søndergaard CR, Rostkowski M & Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput 7, 525–537 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Søndergaard CR, Olsson MHM, Rostkowski M & Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput 7, 2284–2295 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Morozenko A & Stuchebrukhov AA Dowser, a new method of hydrating protein structures. Proteins: Structure, Function, and Bioinformatics vol. 84 1347–1357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lomize MA, Lomize AL, Pogozheva ID & Mosberg HI OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beglov D & Roux B Finite representation of an infinite bulk system: Solvent boundary potential for computer simulations. J. Chem. Phys 100, 9050–9063 (1994). [Google Scholar]
- 76.Li P, Roberts BP, Chakravorty DK & Merz KM Jr. Rational design of particle mesh Ewald compatible Lennard-Jones parameters for+ 2 metal cations in explicit solvent. J. Chem. Theory Comput 9, 2733–2748 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Case DA et al. AMBER 2018; 2018. University of California, San Francisco. [Google Scholar]
- 78.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S & Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 9, 3878–3888 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Hopkins CW, Le Grand S, Walker RC & Roitberg AE Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput 11, 1864–1874 (2015). [DOI] [PubMed] [Google Scholar]
- 80.Ryckaert J-P, Ciccotti G & Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 23, 327–341 (1977). [Google Scholar]
- 81.Roe DR & Cheatham TE 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph 14, 33–8, 27–8 (1996). [DOI] [PubMed] [Google Scholar]
- 83.Mitchell CJ, Shawki A, Ganz T, Nemeth E & Mackenzie B Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. Am. J. Physiol. Cell Physiol 306, C450–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Supplementary Information. Crystallographic coordinates and structure factors for the Fab45D8 complex have been deposited in the Protein Data Bank under accession code 6W4V. Coordinates for Fab45D8-FPN complex have been deposited in the Protein Data Bank under accession code 6W4S and the maps have been deposited in the Electron Microscopy Data Bank under accession code 21539. Coordinates for the FPN-Co2+-hepcidin-Fab45D8 complex have been deposited in the Protein Data Bank under accession code 6WBV and the maps have been deposited in the Electron Microscopy Data Bank under accession code 21599.