

Abstract
DNA replication forks are constantly challenged by DNA lesions induced by endogenous and exogenous sources. DNA damage tolerance mechanisms ensure that DNA replication continues with minimal effects on replication fork elongation either by using specialized DNA polymerases, which have the ability to replicate through the damaged template, or by skipping the damaged DNA, leaving it to be repaired after replication. These mechanisms are evolutionarily conserved in bacteria, yeast, and higher eukaryotes, and are paramount to ensure timely and faithful duplication of the genome. The Primase and DNA-directed Polymerase (PRIMPOL) is a recently discovered enzyme that possesses both primase and polymerase activities. PRIMPOL is emerging as a key player in DNA damage tolerance, particularly in vertebrate and human cells. Here, we review our current understanding of the function of PRIMPOL in DNA damage tolerance by focusing on the structural aspects that define its dual enzymatic activity, as well as on the mechanisms that control its chromatin recruitment and expression levels. We also focus on the latest findings on the mitochondrial and nuclear functions of PRIMPOL and on the impact of loss of these functions on genome stability and cell survival. Defining the function of PRIMPOL in DNA damage tolerance is becoming increasingly important in the context of human disease. In particular, we discuss recent evidence pointing at the PRIMPOL pathway as a novel molecular target to improve cancer cell response to DNA-damaging chemotherapy and as a predictive parameter to stratify patients in personalized cancer therapy.
Keywords: DNA replication, DNA replication stress, DNA repair, DNA polymerases, DNA damage tolerance, PRIMPOL, repriming, genome stability
Graphical Abstract
The faithful replication of our genome is constantly challenged by a host of DNA replication obstacles from both endogenous and exogenous sources. Endogenous replication obstacles include secondary structures in the DNA, protein-DNA complexes, transcription-replication conflicts, and metabolism-induced oxidative DNA damage (Mirkin and Mirkin 2007; Chatterjee and Walker 2017; Gadaleta and Noguchi 2017; Markkanen 2017), whereas exogenous DNA lesions can be induced by UV radiation and chemotherapeutic agents (Cheung-Ong et al. 2013; Chatterjee and Walker 2017). All living organisms have evolved different mechanisms of DNA Damage Tolerance (DDT) to overcome these replication obstacles and promote efficient genome duplication. These DDT mechanisms broadly include Translesion DNA Synthesis (TLS), Template Switching (TS), and repriming (Sale 2012; Branzei and Szakal 2016).
TLS relies on specialized TLS polymerases to directly bypass DNA lesions (Waters et al. 2009; Goodman and Woodgate 2013). TLS polymerases are generally more error-prone than the high-fidelity replicative polymerases, as they can accommodate bulky lesions and distorted DNA duplex structures and lack proofreading activity (Goodman and Woodgate 2013; Sale 2013). TS is generally viewed as a higher-fidelity mechanism relative to TLS, as this mechanism utilizes a homologous template to bypass DNA lesions (Chang and Cimprich 2009). Replication fork reversal is one form of TS (Branzei and Szakal 2016) whereby the re-annealing of newly synthesized daughter strands enables the remodeling of replication forks into four-way junctions in order to cope with DNA obstacles (Leon-Ortiz et al. 2014; Zeman and Cimprich 2014; Neelsen and Lopes 2015; Berti and Vindigni 2016; Quinet et al. 2017; Berti et al. 2020).
In addition to TLS and template switching, a third mechanism of DDT is repriming, which allows DNA replication to skip the DNA lesion, leaving a single stranded DNA (ssDNA) gap between the lesion and the point where DNA synthesis resumes. Early studies showed that treatment of bacteria (Rupp and Howard-Flanders 1968) or mammalian cells (Lehmann and Kirk-Bell 1972; Meneghini 1976) with UV light promotes ssDNA gap accumulation. Follow-up studies in budding yeast confirmed that UV treatment promotes the formation of ssDNA gaps in both the lagging and leading strands of DNA replication forks (Lopes et al. 2006). While the discontinuous nature of DNA synthesis could account for ssDNA gaps in the lagging strand, the presence of leading strand gaps suggested that the replisome is able to reinitiate DNA synthesis downstream of a leading strand lesion. Pioneering work in bacteria showed that the DnaG primase can indeed mediate de novo primer synthesis (or repriming) downstream of leading strand lesions, allowing resumption of DNA synthesis (Heller and Marians 2006). These findings were extended to budding yeast, in which the polymerase α-primase (Polα-primase) complex is able to mediate de novo primer synthesis downstream of different lesions induced by DNA-damaging agents (Lopes et al. 2006; Daigaku et al. 2010; Karras and Jentsch 2010; Fumasoni et al. 2015; Branzei and Psakhye 2016; Wong et al. 2020).
Repriming mechanisms are conserved in vertebrate and human cells. However, differently from budding yeast, they rely on a newly discovered protein called PRIMPOL instead of using the main Polα-primase complex (Bianchi et al. 2013; Garcia-Gomez et al. 2013; Mouron et al. 2013; Wan et al. 2013). PRIMPOL possesses both primase and polymerase activities, and information on the roles of its enzymatic activities during the replication stress response is just beginning to emerge. Here, we review our current understanding of the roles of PRIMPOL and its dual enzymatic activities in DDT. We also discuss how PRIMPOL is recruited to stalled replication forks and how changes in its expression levels affect genome stability, cell survival, and cancer cell response to chemotherapy.
Structure.
Encoded by the gene CCDC111, the Primase and DNA-directed Polymerase (PRIMPOL) is a 560 amino acid protein conserved in vertebrates, plants, and lower eukaryotes (Iyer et al. 2005; Bianchi et al. 2013; Garcia-Gomez et al. 2013). Orthologs are also present in species of fungi, algae, and protists but are absent in budding yeast. PRIMPOL was first identified as a member of the archaeo-eukaryotic primase (AEP) superfamily – a family of primases associated with DNA damage response in bacteria (Iyer et al. 2005). The catalytic AEP core of PRIMPOL spans over 300 residues and consists of two highly conserved modules: ModN (residues 35-105) and ModC (residues 108-200; 261-348), which regulate PRIMPOL nucleotidyltransferase activity, required for its primase and polymerase functions (Rechkoblit et al. 2016). ModN directly interacts with the DNA template strand, whereas ModC interacts with incoming nucleotides (Rechkoblit et al. 2016) (Figure 1A). Modifications of key residues in ModN reduce DNA binding affinity, leading to decreased enzymatic activity (Keen, Bailey, et al. 2014; Keen, Jozwiakowski, et al. 2014). Furthermore, modifications in the catalytic residues Asp114, Glu116, or Asp280 of ModC ablate both primase and polymerase functions of PRIMPOL (Rechkoblit et al. 2016). These residues act as activating metal ligands, using divalent cations such as Mn2+ and Mg2+ as catalytic cofactors for enzymatic function (Bianchi et al. 2013; Garcia-Gomez et al. 2013; Wan et al. 2013; Calvo et al. 2019) (Figure 1B). Alteration of these residues, which prevents interaction with metal cofactors, renders PRIMPOL catalytically inactive (Garcia-Gomez et al. 2013; Mouron et al. 2013; Calvo et al. 2019). In addition to the AEP core, PRIMPOL contains an N-terminal helix, a C-terminal Replication Protein A (RPA) binding domain, and a zinc finger (ZnF) domain that stabilizes PRIMPOL’s interaction with ssDNA (Keen, Jozwiakowski, et al. 2014; Rechkoblit et al. 2016) (Figure 1A).
Figure 1. Domain architecture and catalytic residues of PRIMPOL.

A) Domain architecture of PRIMPOL. PRIMPOL catalytic archaeo-eukaryotic primase (AEP) core contains two conserved modules, ModN (yellow) and ModC (red). ModN interacts with ssDNA and the templating base, while ModC interacts with incoming nucleotides. PRIMPOL catalytic residues (grey stars) are essential for both primase and polymerase activity. PRIMPOL domains also include an N-terminal helix (green), a zinc finger (ZnF) domain (purple) critical for stabilization of PRIMPOL on ssDNA and stabilization of the 5’ nucleotide during repriming, and a C-terminal RPA binding domain (blue). A flexible linker between ModC and the ZnF domain regulates the nucleotide incorporation of PRIMPOL. B) Catalytic residues of PRIMPOL. PRIMPOL’s catalytic residues include Asp114, Glu116, and Asp280. A divalent metal cation (Me2+), such as Mn2+ or Mg2+, is shown here coordinated by the catalytic residues Asp114 and Glu116 and the three phosphate groups of an incoming dATP, which facilitates PRIMPOL enzymatic activity (Image from the RCSB PDB (rcsb.org) of PDB ID: 5L2X) (Rechkoblit et al. 2016).
PRIMPOL polymerase activity.
PRIMPOL possesses both primase and polymerase activities in vitro (Bianchi et al. 2013; Garcia-Gomez et al. 2013; Mouron et al. 2013; Wan et al. 2013) (Figure 2). PRIMPOL synthesizes DNA with limited processivity, rarely incorporating more than four nucleotides on an undamaged template (Keen, Jozwiakowski, et al. 2014). This low processivity is, in part, due to a self-regulatory mechanism mediated by the ZnF domain of PRIMPOL, which is connected to the catalytic core via a ~140 amino acid flexible linker (Guilliam and Doherty 2017) (Figure 1 A) and limits the extent of nucleotide incorporation (Keen, Jozwiakowski, et al. 2014; Guilliam and Doherty 2017) (Figure 3). Specifically, the ZnF domain stably binds ssDNA upstream of the catalytic core of PRIMPOL and limits the distance that the core can synthesize DNA away from the ZnF domain to the length of the flexible linker (Rechkoblit et al. 2016) (Figure 3). PRIMPOL processivity is robustly enhanced by the presence of the metal cofactors Mg2+ and Mn2+, which also increase PRIMPOL affinity for DNA templates (Zafar et al. 2014). Both the processivity and affinity for DNA are more highly enhanced by Mn2+ relative to Mg2+. Specifically, PRIMPOL binds DNA with 34-fold higher affinity and incorporates the correct dNTP with 100-fold higher efficiency in the presence of Mn2+ relative to Mg2+ (Tokarsky et al. 2017). Whether both cations are required to enhance PRIMPOL activity in vivo remains a subject of debate (Calvo et al. 2019).
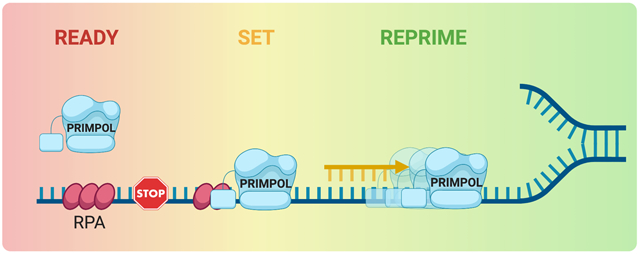
Figure 2. PRIMPOL translesion synthesis and repriming activities.

PRIMPOL has both TLS and repriming activities. TLS activity has been shown using synthetic DNA substrates containing different types of lesions including 8-oxoguanines, abasic sites, and UV-induced lesions, such as cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs) (green box). The question mark next to 6-4PP reflects the fact that there are conflicting data regarding the ability of PRIPMOL to bypass 6-4PPs, which more prominently distort the DNA helix relative to CPDs. PRIMPOL repriming activity is needed to reinitiate DNA synthesis downstream of chain-terminating-nucleosides (ddNTPs), CPDs, and 6-4PPs in vitro, and in response to UV-C lesions, G-quadruplexes, R-loops, intra- and inter-strand crosslinks, and dNTP depletion in a cellular context (yellow box).
Figure 3. Mechanisms of PRIMPOL recruitment and regulation.

ATR positively regulates PRIMPOL expression, whereas WRNIP1-mediated proteosomal degradation negatively regulates PRIMPOL protein levels. PolDIP2, which interacts with PCNA, has a mechanistically ill-defined role in modulating PRIMPOL activity. In addition, PRIMPOL activity is self-regulated by the ZnF domain's flexible linker, which restricts the distance of nucleotide incorporation by the catalytic core to the length of the linker. RPA facilitates the interaction of PRIMPOL with ssDNA. However, high RPA levels limit PRIMPOL ssDNA binding, thereby decreasing PRIMPOL activity. In the mitochondria, PRIMPOL activity is enhanced by TWINKLE, while it is inhibited by mtSSB. Positive regulators of PRIMPOL expression/function are included in the green boxes, negative regulators are in red boxes, and a factor whose function is still debated is shown in the yellow box.
Similar to other TLS enzymes, PRIMPOL lacks exonuclease activity and is a highly error-prone polymerase (~10−4 errors per nucleotide incorporated, as opposed to ~10−5 for the TLS Pol ζ, and 10−5–10−6 for the replicative polymerases Pol δ and Pol ε) (Shcherbakova et al. 2003; Fortune et al. 2005; Zhong et al. 2006; Zafar et al. 2014; Guilliam et al. 2015). PRIMPOL polymerase activity mainly generates insertions and deletions (also known as “indels”). Although other error-prone polymerases can generate “indels” (Eckert et al. 2002; Baptiste and Eckert 2012; Ananda et al. 2014), PRIMPOL maintains a unique error signature across coding regions, with a proportion of insertion errors higher than any other DNA polymerase. In particular, PRIMPOL generates insertions and deletions of single nucleotides at a similar rate, primarily on short homopolymeric sequences (Guilliam et al. 2015; Martinez-Jimenez et al. 2015). Although speculative, PRIMPOL’s error profile might be due to its ability to realign its primer terminus to a location downstream of a lesion during polymerization (Chi and Lam 2008; Bikard et al. 2010; Garcia-Gomez et al. 2013; Du et al. 2014; Martinez-Jimenez et al. 2015). This process loops out the template DNA and can occur between short, direct nucleotide repeats (Martinez-Jimenez et al. 2015). This peculiar activity of PRIMPOL has been termed “pseudo-TLS” (Mouron et al. 2013) and leads to the synthesis of shorter DNA products than would be generated without looping out or “scrunching” the template DNA.
In vitro, PRIMPOL exerts its TLS activity on synthetic DNA templates carrying different types of damages, including 8-oxoguanine (8-oxo-G), abasic sites (although with low proficiency) and UV-C-induced lesions, such as cyclobutane pyrimidine dimers (CPD) and pyrimidine (6-4) pyrimidone (6-4PP) ((Garcia-Gomez et al. 2013; Mouron et al. 2013), reviewed in (Guilliam and Doherty 2017)) (Figure 2, left panel). 6-4PPs cause a more pronounced distortion of the DNA double helix compared to CPDs, and whether PRIMPOL can bypass these lesions using its polymerase activity remains controversial. Initial in vitro studies suggested that PRIMPOL could bypass DNA templates containing both types of UV-C-induced photoproducts (Bianchi et al. 2013). However, these findings are at odds with recent structural studies showing that the active site of PRIMPOL cannot accommodate bulky 6-4PP lesions (Rechkoblit et al. 2016). In agreement with the structural data, Mouron et al. reported that PRIMPOL primarily uses its primase activity to reinitiate DNA synthesis downstream of 6-4PP lesions (Mouron et al. 2013).
All studies on PRIMPOL TLS activity have been performed using synthetic DNA substrates and purified recombinant protein, raising the question of whether PRIMPOL TLS activity has a physiologically relevant function in vivo. Generating a polymerase-dead / primase-proficient mutant will be crucial to conclusively demonstrate that the polymerase activity of PRIMPOL is required to bypass these lesions in vivo. However, the generation of separation of function PRIMPOL mutants selectively lacking polymerase activity has been hampered by the fact that point mutations in key catalytic residues of PRIMPOL negatively affect both the primase and polymerase activities in vitro (Garcia-Gomez et al. 2013; Mouron et al. 2013; Keen, Bailey, et al. 2014; Keen, Jozwiakowski, et al. 2014; Calvo et al. 2019).
PRIMPOL primase activity.
In addition to its TLS activity, PRIMPOL synthesizes both dNTP and NTP primers on synthetic DNA substrates (Bianchi et al. 2013; Garcia-Gomez et al. 2013; Mouron et al. 2013). Interestingly, PRIMPOL preferentially incorporates dNTPs, whereas the replicative polymerase Polα-primase complex only synthesizes NTP primers during lagging strand DNA synthesis (Pellegrini 2012; Williams and Kunkel 2014). NTPs synthesized by this complex are removed during Okazaki fragment maturation, while NTPs incorporated by replicative polymerases are removed by the ribonucleotide excision repair machinery to avoid distortions in the DNA helix and genomic instability (Meroni et al. 2017; Sassa et al. 2019; Nava et al. 2020). Conversely, DNA primers inserted by PRIMPOL require no further processing, which may explain the unique role of PRIMPOL primase activity in reinitiating DNA synthesis downstream of leading strand lesions, as opposed to the Polα-primase complex. Structurally, PRIMPOL’s unique preference for dNTPs is likely a consequence of the close positioning of the dNTP and the main-chain carbonyl of Asn289 within its active site. The NTP hydroxyl group would sterically clash with Asn289 and require significant rearrangement of residues 289-291 to be incorporated (Rechkoblit et al. 2016). However, a single point mutation of Tyr100 to His is sufficient to disable this steric gate, reverting PRIMPOL’s sugar specificity and leading to similar levels of NTP and dNTP incorporation in vitro (Diaz-Talavera et al. 2019). Remarkably, structural data also reveal an almost complete lack of contact between DNA primer strands and PRIMPOL (Rechkoblit et al. 2016). However, the crystal structure of PRIMPOL lacks the C-terminus ZnF domain (Rechkoblit et al. 2016), which is crucial to stabilize the 5‘ nucleoside triphosphate to initiate and elongate primer synthesis (Martinez-Jimenez et al. 2018).
In vitro, PRIMPOL reprimes downstream of chain-terminating-nucleosides and UV-C-induced CPDs and 6-4PPs (Mouron et al. 2013; Kobayashi et al. 2016) (Figure 2, right panel). The primers generated on synthetic DNA substrates by PRIMPOL can subsequently be elongated by the leading strand replicative polymerase Pol ε (Garcia-Gomez et al. 2013).
In human cells, PRIMPOL reinitiates de novo DNA synthesis upon replication fork stalling induced by UV-C lesions, G-quadruplexes, bulky adducts such as those generated upon treatment with benzo[a]pyrene-diol-epoxide (BPDE) or 4-nitroquinoline 1-oxide (4-NQO), and platinum crosslinking agents (Mouron et al. 2013; Schiavone et al. 2016; Piberger et al. 2020; Quinet et al. 2020). Recent studies also show that PRIMPOL repriming is required to “traverse” interstrand crosslinks (ICLs) (González-Acosta et al. 2020). ICLs were initially considered absolute blocks to DNA replication (Zhang et al. 2015). However, ICLs can also be “traversed” in a reaction mediated by the FANCM/MHF DNA translocase (Huang et al. 2013). Gonzalez-Acosta et al. demonstrated that PRIMPOL plays a key role in this reaction, leaving the ICL behind the fork to be repaired post-replicatively (González-Acosta et al. 2020). Moreover, PRIMPOL-mediated repriming can circumvent transcriptional conflicts caused by DNA:RNA hybrids, or R-loops, in avian DT40 cells (Svikovic et al. 2019) (Figure 2, right panel).
A primase-dead, polymerase-proficient mutant of PRIMPOL can be generated by introducing a double mutation in the ZnF element present in its C-terminal domain (C419G/H426Y, CH variant), which abolishes primase activity, while preserving polymerase function (Mouron et al. 2013). Of note, inactivation of the ZnF domain also increases the average number of nucleotides inserted by the polymerase activity of PRIMPOL in vitro (Keen, Jozwiakowski, et al. 2014). This primase-dead CH mutant, as well as other ZnF truncation mutants, were used to confirm the important role of PRIMPOL primase activity in human cells treated with UV-C and cisplatin (Mouron et al. 2013; Quinet et al. 2020). Moreover, similar results were obtained using the same primase-dead CH mutant in avian DT40 cells treated with various DNA-damaging agents, including UV-C radiation, methyl methanesulfonate (MMS), and cisplatin (Keen, Jozwiakowski, et al. 2014; Kobayashi et al. 2016). Collectively, these studies support the notion that the primase activity of PRIMPOL is essential for many of the biologically relevant functions of PRIMPOL in the nucleus (Mouron et al. 2013), as we also discuss in the last section of this review.
PRIMPOL recruitment and regulation.
PRIMPOL protein levels are approximately constant throughout the cell cycle (Mouron et al. 2013). However, PRIMPOL binding to chromatin increases after treatment with different genotoxic agents, such as hydroxyurea and UV-C (Mouron et al. 2013). Recent work in human cells suggests that the ATPase Werner Helicase Interacting Protein 1 (WRNIP1) negatively regulates PRIMPOL expression by promoting proteasomal degradation of PRIMPOL (Yoshimura et al. 2019). Furthermore, studies from our lab suggest a transcriptional role of ATR in regulating PRIMPOL expression (discussed below, see (Quinet et al. 2020)), adding an additional layer of complexity to the mechanisms that control PRIMPOL expression and chromatin recruitment in mammalian cells. Interestingly, treatment with an anti-HIV drug, tenovofir, has been shown to downregulate Chk1, a checkpoint kinase downstream of ATR signaling, and to concomitantly decrease PRIMPOL protein levels, supporting the notion that there is a tight connection between ATR activity and PRIMPOL expression (Fang and Beland 2009; Smith et al. 2010; Duong et al. 2020). However, further studies will be necessary to assess the effect of ATR-dependent post-translational modifications on PRIMPOL activity.
Upon DNA damage, canonical TLS polymerases are recruited to chromatin through interaction with PCNA (Kannouche and Lehmann 2004; Kannouche et al. 2004; Hendel et al. 2011). In contrast, PRIMPOL does not seem to interact with PCNA, and the presence of PCNA does not affect the enzymatic activity of PRIMPOL in vitro (Guilliam et al. 2015). However, recent studies have shown that PRIMPOL interacts with polymerase δ-interacting protein 2 (PolDIP2 or PDIP38), a binding partner of the p50 subunit of Pol δ, as well as of PCNA (Liu et al. 2003; Guilliam et al. 2016) (Figure 3). PolDIP2 also interacts with various TLS polymerases including Pol η, Pol ζ, and REV1 (Tissier et al. 2010). These data leave open the possibility that PRIMPOL might be able to indirectly interact with PCNA and that the PCNA-PolDIP2 complex might be required for PRIMPOL recruitment to replication forks. Interestingly, PRIMPOL interaction with PolDIP2 alone enhances PRIMPOL’s affinity for DNA as well as its processivity in response to UV-C damage (Guilliam et al. 2016). However, Guilliam et al. showed that the complex formed by PCNA and PolDIP2 has an inhibitory effect on PRIMPOL polymerase activity, complicating our understanding of how the PCNA-PolDIP2-PRIMPOL interaction network regulates PRIMPOL activity (Guilliam et al. 2016).
Recruitment of PRIMPOL to chromatin seems to be primarily mediated by interaction with the ssDNA binding protein RPA (Mouron et al. 2013; Wan et al. 2013; Guilliam et al. 2015). RPA is a heterotrimeric complex formed by RPA70, RPA32, and RPA14 (Pokhrel et al. 2019). The interaction between PRIMPOL and RPA is mediated by the C-terminal RPA binding domain of PRIMPOL (Wan et al. 2013; Guilliam et al. 2017) (Figure 1A), which binds to the N-terminus of RPA70, the largest subunit of the RPA heterotrimer (Wold 1997). Interestingly, in vitro studies suggest that the effect of RPA on PRIMPOL recruitment depends on the extent of RPA molecules coating DNA (Guilliam et al. 2015; Guilliam et al. 2017; Martinez-Jimenez et al. 2017). PRIMPOL is unable to displace RPA from ssDNA fragments because RPA has a higher affinity for ssDNA than PRIMPOL (Kim et al. 1994; Martinez-Jimenez et al. 2017). Accordingly, PRIMPOL binds synthetic ssDNA templates more effectively when RPA is not at high concentrations, which can otherwise limit PRIMPOL interaction with ssDNA (Martinez-Jimenez et al., 2017; Guilliam et al., 2017) (Figure 3). In particular, in vitro experiments using circular M13 ssDNA templates show that non-saturating concentrations of RPA stimulate PRIMPOL primase and polymerase activities, whereas high concentrations of RPA are catalytically restrictive (Guilliam et al. 2017; Martinez-Jimenez et al. 2017). On the basis of these findings, Guilliam and Doherty propose that RPA might act as regulator of PRIMPOL catalytic activity in order to limit mutagenic DNA synthesis (Guilliam and Doherty 2017).
In addition to RPA, PRIMPOL interacts with the mitochondrial single-stranded DNA binding protein, mtSSB (Guilliam et al. 2015). Mitochondrial DNA replication relies on DNA polymerase Pol γ along with mtSSB, the mitochondrial replicative helicase Twinkle, and the mitochondrial RNA polymerase (Young and Copeland 2016). In vitro work revealed that mtSSB negatively regulates the primase activity of PRIMPOL and also inhibits PRIMPOL polymerase activity (Guilliam et al. 2015; Stojkovic et al. 2016) (Figure 3). Interestingly, while mtSSB inhibits PRIMPOL function, Twinkle enhances the polymerase activity of PRIMPOL in vitro on DNA templates that are either undamaged or that carry oxidative lesions, such as 8-oxoguanines or abasic sites (Stojkovic et al. 2016) (Figure 3).
PRIMPOL and cell survival.
PRIMPOL is not essential for cell survival, as PRIMPOL-knockout (PRIMPOL-KO) mice and human cell lines are viable (Bianchi et al. 2013; Mouron et al. 2013; Bailey et al. 2019; González-Acosta et al. 2020; Quinet et al. 2020). However, loss of PRIMPOL is associated with slower proliferation and generation of RPA and yH2AX foci, suggesting that PRIMPOL might play an important, albeit mechanistically ill-defined, role at replication forks in unperturbed conditions (Mouron et al. 2013; Wan et al. 2013).
The notion that PRIMPOL-mediated repriming is an important mechanism to skip DNA lesions and continue DNA synthesis is supported by different studies showing that human cells lacking PRIMPOL are characterized by defects in replication fork progression and restart, increased mutagenesis, increased sister chromatid exchanges, and micronuclei formation following UV-C radiation and treatment with ICL-inducing agents (Mouron et al. 2013; Bailey et al. 2019; González-Acosta et al. 2020). Moreover, it is possible that the replication defects associated with PRIMPOL loss are partially mitigated by alternative DDT pathways that either employ other TLS polymerases or a different mechanism of lesion bypass. For example, loss of PRIMPOL significantly affects proliferation and viability in avian DT40 cells only when PRIMPOL is depleted in combination with the TLS polymerases Pol η and Pol ζ, suggesting that these polymerases partially compensate for the loss of PRIMPOL repriming, at least in avian cells (Kobayashi et al. 2016). Along the same lines, loss of PRIMPOL does not seem to sensitize human cells to UV-C, unless the TLS enzyme Pol η is co-depleted (Bailey et al. 2019), supporting the notion that Pol η and PRIMPOL might have complementary roles in response to UV damage.
Further studies have shown how loss of PRIMPOL leads to increased cellular sensitivity to different DNA-damaging agents in avian DT40 and human cells, including UV-C radiation, cisplatin, 4-NQO, BPDE, MMS, chain-terminating nucleoside analogs, mitomycin C (MMC), and trimethyl psoralen activated with UV-A (TMP-UVA) (Bianchi et al. 2013; Keen, Bailey, et al. 2014; Kobayashi et al. 2016; Olivieri et al. 2020). In addition, treatment with ICL-inducing agents such as MMC and TMP-UVA increases the frequency of metaphase chromosomal abnormalities in PRIMPOL-KO human cells, in agreement with the recent finding that repriming by PRIMPOL is required for ICL traverse (González-Acosta et al. 2020). Similar to human cells, PRIMPOL-KO mice are hypersensitive to MMC and display a significant impairment of bone marrow cell proliferation when treated with this drug (González-Acosta et al. 2020). Collectively, these studies demonstrate that PRIMPOL mediates the cellular response to an array of DNA-damaging agents that perturb replication fork progression. Conversely, treatment with agents that generate DNA breaks, such as camptothecin, ICRF193, and γ-rays, does not affect viability in PRIMPOL-deficient DT40 cells, suggesting that PRIMPOL is not involved in DNA break repair (Kobayashi et al. 2016).
In summary, these studies demonstrate that PRIMPOL repriming is paramount to preserve genomic stability and allow replication forks to cope with a different array of DNA lesions. The fact that loss of PRIMPOL leads to increased sister chromatid exchanges and mutagenesis also underscores a possible link between changes in PRIMPOL expression and cancer development. Moreover, these findings suggest that PRIMPOL repriming might represent a novel target to increase cancer cell sensitivity to DNA-damaging chemotherapy.
Biological functions of PRIMPOL.
PRIMPOL repriming operates in both the mitochondria and the nucleus. Biochemical fractionation experiments indicated that, at least in HeLa cells, PRIMPOL is almost twice as abundant in the mitochondria than in the nucleus (Garcia-Gomez et al. 2013). Mutation of Tyr 89 to Asp (PRIMPOL-Y89D) in the PRIMPOL coding gene, CCDC111, is associated with development of chronic progressive external ophthalmoplegia (CPEO), one of the most common mitochondrial disorders characterized by weakness of the extraocular and facial muscle groups (Kasamo et al. 2019). However, a conclusive association between the Y89D mutation and CPEO requires further investigation of the pathogenicity and the inheritance pattern of this PRIMPOL variant (Finsterer 2020). Moreover, this mutation has been associated with the development of another ophthalmologic disorder, high myopia (Zhao et al. 2013; Keen, Bailey, et al. 2014; Yuan et al. 2020), although this also remains controversial as studies using a larger cohort of patients suggested that the PRIMPOL-Y89D mutation is a common polymorphism present also in healthy individuals (Keen et al. 2015; Li J and Zhang 2015). Current models for the function of PRIMPOL in the mitochondria suggest that PRIMPOL helps the mitochondrial genome (mtDNA) tolerate high levels of oxidative damage, such as 8-oxo-G and abasic lesions, that severely stall the mitochondrial polymerase, Pol γ (Garcia-Gomez et al. 2013; Zafar et al. 2014; Stojkovic et al. 2016; Bailey et al. 2019). In agreement with this model, PRIMPOL-KO mice and cell lines have clear defects in mitochondrial DNA (mtDNA) replication (Garcia-Gomez et al. 2013; Bailey et al. 2019).
As previously discussed, in vitro studies showed that PRIMPOL TLS catalytic activity allows bypass of the most common type of mitochondrial oxidative lesions, 8-oxo-G (Garcia-Gomez et al. 2013). However, the mitochondrial mutation spectrum in both humans and mice suggest that 8-oxo-G lesions are often mispaired with adenines during bypass, as dATP presents the most thermodynamically favorable pairing across from this damage (Irimia et al. 2009). This misincorporation ultimately results in an increase of G>T and C>A transversions after lesions are repaired by base excision repair (David et al. 2007; Ameur et al. 2011; Kennedy et al. 2013). However, studies using synthetic templates containing 8-oxo-G lesions showed that PRIMPOL preferentially misincorporates nucleotides that would not give rise to G>T and C>A transversions, and that PRIMPOL inserts dATP across 8-oxo-G lesions less frequently than Pol γ (Stojkovic et al. 2016). The discrepancy between PRIMPOL enzymatic activity and the observed mutational signature suggests that the TLS activity of PRIMPOL might not be required to overcome 8-oxo-G lesions in the mitochondrial compartment. Along the same lines, PRIMPOL has a very limited abasic site bypass activity compared to other canonical TLS polymerases, such as Pol η, Pol ι, and REV1, suggesting that PRIMPOL TLS activity might not be required to bypass abasic lesions in the mitochondria (Choi et al. 2010; Garcia-Gomez et al. 2013; Keen, Jozwiakowski, et al. 2014; Zafar et al. 2014).
Of note, mtDNA is rich in guanines, providing a favorable environment for the formation of G-quadruplexes, which also perturb replication fork progression and cause fork stalling (Bedrat et al. 2016; Falabella et al. 2019). Moreover, mtDNA is rich in R-loops (Holt 2019), which hamper DNA replication, repair, and transcription (Huertas and Aguilera 2003; Li X and Manley 2005; Tuduri et al. 2009; Gan et al. 2011; Wahba et al. 2011; Groh and Gromak 2014). Recent in vitro studies demonstrated that PRIMPOL can bypass stable G-quadruplex templates via its TLS activity in the presence of Pif1, a G-quadruplex resolvase that localizes to the mitochondria (Butler et al. 2020). Error-prone bypass of these structures by PRIMPOL could account for increased mutagenesis at stable G-quadruplex regions in the mitochondria (Butler et al. 2020). However, studies in avian DT40 cells showed that PRIMPOL is unable to directly replicate through G-quadruplexes or R-loops (Schiavone et al. 2016; Svikovic et al. 2019). Instead, PRIMPOL can reprime downstream of these structures (Schiavone et al. 2016; Svikovic et al. 2019). Thus, we speculate that the repriming activity of PRIMPOL is required to restart DNA synthesis downstream of G-quadruplex or R-loop structures present in the mtDNA in vivo.
The model that the primase, rather than the polymerase, activity of PRIMPOL is required to deal with fork-stalling lesions in the mitochondria is supported by studies performed in mouse embryonic fibroblasts (Torregrosa-Munumer et al. 2017) and by a recent study suggesting that the primase activity of PRIMPOL is essential to protect human cells from tenofovir-induced mitochondrial toxicity (Duong et al. 2020). Tenofovir is a nucleoside reverse transcriptase inhibitor (NRTI) used in the antiretroviral therapy for HIV patients. This drug is toxic for mtDNA replication because the incorporation of the NRTI by Pol γ causes replication fork stalling (Duong et al. 2020). Duong et al. identified a mutation in the catalytic site of PRIMPOL (D114N) that increases tenofovir-induced mitochondrial toxicity. The D114N mutation targets a key residue required for metal binding and abolishes the primer synthesis activity of PRIMPOL, as well as its ability to bind DNA (Duong et al. 2020). On the basis of these findings, Duong et al. proposed that PRIMPOL alleviates tenofovir-induced mitochondrial toxicity by repriming downstream of chain-terminated nucleotides, thereby rescuing stalled replication forks (Duong et al. 2020).
While our understanding of PRIMPOL repriming in the mitochondria is still in its infancy, several studies documented a key role for the primase activity of PRIMPOL during DNA replication in the nucleus (Mouron et al. 2013; Wan et al. 2013; Keen, Jozwiakowski, et al. 2014; Kobayashi et al. 2016; Bailey et al. 2019; Blanco et al. 2019; González-Acosta et al. 2020; Piberger et al. 2020; Quinet et al. 2020). As already discussed, loss of the primase activity of PRIMPOL leads to increased sensitivity to a variety of genotoxic agents, including cisplatin, UV-C irradiation, and MMS in avian DT40 cells (Keen, Jozwiakowski, et al. 2014; Kobayashi et al. 2016). Moreover, expression of a polymerase-proficient, primase-dead PRIMPOL mutant fails to rescue UV-C-induced replication fork stalling in human cells (Mouron et al. 2013).
In addition, recent studies suggested that hyperactivation of PRIMPOL-mediated repriming has a protective role under specific genetic backgrounds, such as cells defective for Breast Cancer Susceptibility Proteins, BRCA1 and BRCA2 (Quinet et al. 2020). Aside from their well-defined role in DNA double-stranded break repair via homologous recombination, BRCA proteins protect reversed replication forks from degradation by nucleases (Schlacher et al. 2011; Schlacher et al. 2012; Ying et al. 2012; Ray Chaudhuri et al. 2016; Quinet et al. 2017). Fork reversal is a physiologically relevant mechanism that allows replication forks to cope with DNA damage encountered ahead of their path by reversing their course (Leon-Ortiz et al. 2014; Zeman and Cimprich 2014; Neelsen and Lopes 2015; Berti and Vindigni 2016; Quinet et al. 2017; Berti et al. 2020). However, fork reversal can also lead to the pathological degradation of replication intermediates when reversed forks are not adequately protected by BRCA proteins (Kolinjivadi et al. 2017; Lemacon et al. 2017; Mijic et al. 2017; Pasero and Vindigni 2017; Taglialatela et al. 2017). We recently discovered that treatment with multiple cisplatin doses suppresses replication fork reversal and promotes repriming by PRIMPOL in BRCA1-deficient cells (Quinet et al. 2020). These studies suggest that in the absence of BRCA proteins, cells adapt to treatment with multiple cisplatin doses by suppressing fork reversal and upregulating PRIMPOL-mediated repriming as an alternative strategy to cope with cisplatin-induced DNA lesions and prevent pathological reversed fork degradation. Furthermore, we found that PRIMPOL-mediated repriming can be more generally activated also in BRCA-proficient cells under conditions of impaired fork reversal, such as loss of the SMARCAL1 SWI/SNF translocase, or upon PRIMPOL overexpression. In agreement with our results, recent work by Bai et al. showed that treatment with hydroxyurea leads to the activation of PRIMPOL repriming in human cells lacking HLTF, which is another member of the SWI/SNF translocase family required for replication fork reversal (Bai et al. 2020). Collectively, these studies suggest fork repriming and reversal are two alternative mechanisms by which cells deal with different forms of replication stress. The balance between these two pathways can be tilted toward fork repriming either by increasing PRIMPOL expression levels, as we observed upon treatment with multiple cisplatin doses (Quinet et al. 2020), or by depleting fork reversal factors (Bai et al. 2020; Quinet et al. 2020). Of note, these studies also point to the PRIMPOL pathway as a novel target to increase chemotherapy response. For example, preclinical studies show that ATR inhibition synergizes with cisplatin treatment (Huntoon et al. 2013), and combinatorial treatments with ATR inhibitors are currently in clinical trials (NCI-2016-00355). The ATR pathway plays a central role in the control of replication fork stability (Saldivar et al. 2017) and is also a key regulator of the PRIMPOL-dependent adaptive response to cisplatin treatment (Quinet et al. 2020). Accordingly, overexpression of PRIMPOL in BRCA1-deficient human cells leads to increased resistance to co-treatment of cisplatin and ATR inhibitor suggesting that the PRIMPOL pathway is an important modulator of chemotherapy response and that monitoring PRIMPOL levels in tumor samples may represent a novel predictive parameter to stratify patients in personalized cancer therapy (Quinet et al. 2020). In this regard, the point mutant PRIMPOL-Y100H that alters PRIMPOL’s unique preference for dNTPs (Diaz-Talavera et al. 2019) has been identified in lung carcinoma, as reported in the COSMIC database (http://cancer.sanger.ac.uk/cosmic) (Bamford et al. 2004), suggesting that mutations in the PRIMPOL gene may also be used as a predictive parameter in cancer therapy.
Conclusions and Future Directions.
Altogether, a growing body of evidence highlights the complex relationship between PRIMPOL expression levels, cell survival and fitness, DNA damage tolerance, and cancer cell response to chemotherapeutic treatment. As we discussed, loss of PRIMPOL increases cellular sensitivity to different replication inhibitors and DNA-damaging agents. At the same time, hyperactivation of PRIMPOL repriming decreases sensitivity to replication stress (Bai et al. 2020) and enables cells to adapt to multiple rounds of drug treatment under genetic backgrounds that prevent reversed fork formation (Berti and Vindigni 2016; Quinet et al. 2017; Berti et al. 2020; Quinet et al. 2020) or that compromise replication fork stability (Schlacher et al. 2011; Schlacher et al. 2012; Ying et al. 2012; Ray Chaudhuri et al. 2016; Quinet et al. 2020).
An important outcome of PRIMPOL activity is that it leads to accumulation of ssDNA gaps on the replicating DNA because it restarts DNA synthesis a few nucleotides downstream of a replication blocking lesion. Interestingly, an emerging theme in the replication field is that aberrant accumulation of ssDNA gaps might represent a major and previously unappreciated determinant of vulnerability to targeted cancer therapy (Panzarino et al. 2019; Nayak et al. 2020; Piberger et al. 2020). For example, a recent study suggested that ssDNA gaps generated by PRIMPOL repriming over bulky BPDE lesions are prone to sister chromatid exchanges (Piberger et al. 2020). This could explain the mutagenesis and genomic rearrangements in cells treated with BPDE. While Piberger et al. showed that siRNA-mediated PRIMPOL depletion did not alter cell survival upon BPDE treatment (Piberger et al. 2020), a recent CRISPR-Cas9 knockout screening identified that loss of PRIMPOL sensitizes cells to BPDE (Olivieri et al. 2020). This discrepancy might be attributable to knockdown efficiency or cell line-specific differences, and future studies should aim to clarify the potential link between PRIMPOL-mediated ssDNA gap formation, genomic rearrangements and mutagenesis, and sensitivity to genotoxic agents. Furthermore, Nayak et al showed how ssDNA gaps induced by oncogenes reduce cellular fitness, and can be offset by TLS, perhaps as a means of gap suppression (Nayak et al. 2020). Follow-up studies by the same group proposed that ssDNA gaps are a major driver of BRCA-deficient cell sensitivity to genotoxic stress, and that suppression of ssDNA gap formation confers resistance to cisplatin (Panzarino et al. 2019). Along the same lines, depletion of the deubiquitinase USP1 in BRCA1-deficient cells leads to ssDNA gap accumulation and decreased cell viability, which is again rescued upon gap suppression (Lim et al. 2018). Collectively, these studies point to a link between ssDNA gap accumulation and cancer cell response to chemotherapy. However, future studies are required to demonstrate that the ssDNA gaps that form under these different conditions are indeed PRIMPOL-dependent.
Another important avenue for future research will be to determine how the ssDNA gaps formed by PRIMPOL-mediated repriming are eventually filled post-replicatively and whether defects in these ssDNA gap filling mechanisms lead to increased sensitivity to targeted cancer therapy. For example, in the context of BPDE treatment, gap filling by RAD51-mediated recombination leads to genomic rearrangements (Piberger et al. 2020), whereas ssDNA gap filling via TLS could lead to mutagenesis (Diamant et al. 2012; Sale 2013; Quinet et al. 2016; Quinet et al. 2018). Notably, one study found that PRIMPOL-deficient invasive breast cancers from The Cancer Genome Atlas (TCGA) have higher global mutation loads relative to PRIMPOL-proficient tumors (Pilzecker et al. 2016). In PRIMPOL-deficient breast cancers, TpC dinucleotides, which are preferentially deaminated by APOBEC3B, undergo transversions more frequently than in PRIMPOL-proficient tumors (Pilzecker et al. 2016). Pilzecker et al. suggest that PRIMPOL acts to limit mutagenesis by repriming beyond abasic sites generated by APOBEC3B in the leading DNA strand and that a high-fidelity mechanism, such as template switching, is utilized to fill PRIMPOL-mediated ssDNA gaps, thereby limiting mutation burden (Pilzecker et al. 2016). However, this gap filling pathway remains to be elucidated. Future work should focus on defining the molecular mechanisms of ssDNA gap filling upon PRIMPOL-dependent repriming, as well as determining how the choice between PRIMPOL-mediated repriming and other DDT mechanisms is regulated. These studies are mandatory to define the molecular contexts in which PRIMPOL-mediated repriming activity can be targeted or modulated to improve cancer cell response to chemotherapeutics.
Acknowledgements
We thank J. Mendez for his careful reading of the manuscript and insightful comments. Figures were created with BioRender.com.
Disclosure of interests
The authors report no conflict of interests. This work was supported by the NCI under grant number R01CA237263.
References
- Ameur A, Stewart JB, Freyer C, Hagstrom E, Ingman M, Larsson NG, Gyllensten U. 2011. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet. 7(3):e1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananda G, Hile SE, Breski A, Wang Y, Kelkar Y, Makova KD, Eckert KA. 2014. Microsatellite interruptions stabilize primate genomes and exist as population-specific single nucleotide polymorphisms within individual human genomes. PLoS Genet. 10(7):e1004498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, Cimprich KA. 2020. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey LJ, Bianchi J, Doherty AJ. 2019. PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res. 47(8):4026–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR et al. 2004. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 91(2):355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptiste BA, Eckert KA. 2012. DNA polymerase kappa microsatellite synthesis: two distinct mechanisms of slippage-mediated errors. Environ Mol Mutagen. 53(9):787–796. [DOI] [PubMed] [Google Scholar]
- Bedrat A, Lacroix L, Mergny JL. 2016. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 44(4):1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Cortez D, Lopes M. 2020. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Berti M, Vindigni A. 2016. Replication stress: getting back on track. Nat Struct Mol Biol. 23(2):103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD et al. 2013. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell. 52(4):566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Loot C, Baharoglu Z, Mazel D. 2010. Folded DNA in action: hairpin formation and biological functions in prokaryotes. Microbiol Mol Biol Rev. 74(4):570–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco L, Calvo PA, Diaz-Talavera A, Carvalho G, Calero N, Martinez-Carron A, Velazquez-Ruiz C, Villadangos S, Guerra S, Martinez-Jimenez MI. 2019. Mechanism of DNA primer synthesis by human PrimPol. Enzymes. 45:289–310. [DOI] [PubMed] [Google Scholar]
- Branzei D, Psakhye I. 2016. DNA damage tolerance. Curr Opin Cell Biol. 40:137–144. [DOI] [PubMed] [Google Scholar]
- Branzei D, Szakal B. 2016. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst). 44:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TJ, Estep KN, Sommers JA, Maul RW, Moore AZ, Bandinelli S, Cucca F, Tuke MA, Wood AR, Bharti SK et al. 2020. Mitochondrial genetic variation is enriched in G-quadruplex regions that stall DNA synthesis in vitro. Hum Mol Genet. 29(8):1292–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo PA, Sastre-Moreno G, Perpina C, Guerra S, Martinez-Jimenez MI, Blanco L. 2019. The invariant glutamate of human PrimPol DxE motif is critical for its Mn(2+)-dependent distinctive activities. DNA Repair (Amst). 77:65–75. [DOI] [PubMed] [Google Scholar]
- Chang DJ, Cimprich KA. 2009. DNA damage tolerance: when it's OK to make mistakes. Nat Chem Biol. 5(2):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Walker GC. 2017. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 58(5):235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Ong K, Giaever G, Nislow C. 2013. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 20(5):648–659. [DOI] [PubMed] [Google Scholar]
- Chi LM, Lam SL. 2008. Nuclear magnetic resonance investigation of primer--template models: formation of a pyrimidine bulge upon misincorporation. Biochemistry. 47(15):4469–4476. [DOI] [PubMed] [Google Scholar]
- Choi JY, Lim S, Kim EJ, Jo A, Guengerich FP. 2010. Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J Mol Biol. 404(1):34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD. 2010. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 465(7300):951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David SS, O'Shea VL, Kundu S. 2007. Base-excision repair of oxidative DNA damage. Nature. 447(7147):941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, Livneh Z. 2012. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 40(1):170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Talavera A, Calvo PA, Gonzalez-Acosta D, Diaz M, Sastre-Moreno G, Blanco-Franco L, Guerra S, Martinez-Jimenez MI, Mendez J, Blanco L. 2019. A cancer-associated point mutation disables the steric gate of human PrimPol. Sci Rep. 9(1):1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Gertz EM, Wojtowicz D, Zhabinskaya D, Levens D, Benham CJ, Schaffer AA, Przytycka TM. 2014. Potential non-B DNA regions in the human genome are associated with higher rates of nucleotide mutation and expression variation. Nucleic Acids Res. 42(20):12367–12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong VN, Zhou L, Martinez-Jimenez MI, He L, Cosme M, Blanco L, Paintsil E, Anderson KS. 2020. Identifying the role of PrimPol in TDF-induced toxicity and implications of its loss of function mutation in an HIV+ patient. Sci Rep. 10(1):9343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert KA, Mowery A, Hile SE. 2002. Misalignment-mediated DNA polymerase beta mutations: comparison of microsatellite and frame-shift error rates using a forward mutation assay. Biochemistry. 41(33):10490–10498. [DOI] [PubMed] [Google Scholar]
- Falabella M, Fernandez RJ, Johnson FB, Kaufman BA. 2019. Potential Roles for G-Quadruplexes in Mitochondria. Curr Med Chem. 26(16):2918–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JL, Beland FA. 2009. Long-term exposure to zidovudine delays cell cycle progression, induces apoptosis, and decreases telomerase activity in human hepatocytes. Toxicol Sci. 111(1):120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J 2020. PRIMPOL variants cause multi-system mitochondrial disorder. Neurosci Res. [DOI] [PubMed] [Google Scholar]
- Fortune JM, Pavlov YI, Welch CM, Johansson E, Burgers PM, Kunkel TA. 2005. Saccharomyces cerevisiae DNA polymerase delta: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem. 280(33):29980–29987. [DOI] [PubMed] [Google Scholar]
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D. 2015. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polalpha/Primase/Ctf4 Complex. Mol Cell. 57(5):812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta MC, Noguchi E. 2017. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes (Basel). 8(3). [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X. 2011. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 25(19):2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ et al. 2013. PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell. 52(4):541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Acosta D, Blanco-Romero E, Mutreja K, Llanos S, Míguez S, García F, Muñoz J, Blanco L, Lopes M, Méndez J. 2020. PrimPol primase mediates replication traverse of DNA interstrand crosslinks. bioRxiv.2020.2005.2019.104729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MF, Woodgate R. 2013. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 5(10):a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Gromak N. 2014. Out of balance: R-loops in human disease. PLoS Genet. 10(9):e1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Bailey LJ, Brissett NC, Doherty AJ. 2016. PolDIP2 interacts with human PrimPol and enhances its DNA polymerase activities. Nucleic Acids Res. 44(7):3317–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Brissett NC, Ehlinger A, Keen BA, Kolesar P, Taylor EM, Bailey LJ, Lindsay HD, Chazin WJ, Doherty AJ. 2017. Molecular basis for PrimPol recruitment to replication forks by RPA. Nat Commun. 8:15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Doherty AJ. 2017. PrimPol-Prime Time to Reprime. Genes (Basel). 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Jozwiakowski SK, Ehlinger A, Barnes RP, Rudd SG, Bailey LJ, Skehel JM, Eckert KA, Chazin WJ, Doherty AJ. 2015. Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 43(2):1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Marians KJ. 2006. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 439(7076):557–562. [DOI] [PubMed] [Google Scholar]
- Hendel A, Krijger PH, Diamant N, Goren Z, Langerak P, Kim J, Reissner T, Lee KY, Geacintov NE, Carell T et al. 2011. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 7(9):e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ. 2019. The mitochondrial R-loop. Nucleic Acids Res. 47(11):5480–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. 2013. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell. 52(3):434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Aguilera A. 2003. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell. 12(3):711–721. [DOI] [PubMed] [Google Scholar]
- Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, Karnitz LM. 2013. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 73(12):3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Eoff RL, Guengerich FP, Egli M. 2009. Structural and functional elucidation of the mechanism promoting error-prone synthesis by human DNA polymerase kappa opposite the 7,8-dihydro-8-oxo-2'-deoxyguanosine adduct. J Biol Chem. 284(33):22467–22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Koonin EV, Leipe DD, Aravind L. 2005. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 33(12):3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche PL, Lehmann AR. 2004. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle. 3(8):1011–1013. [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR. 2004. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 14(4):491–500. [DOI] [PubMed] [Google Scholar]
- Karras GI, Jentsch S. 2010. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 141(2):255–267. [DOI] [PubMed] [Google Scholar]
- Kasamo K, Nakamura M, Daimou Y, Sano A. 2019. A PRIMPOL mutation and variants in multiple genes may contribute to phenotypes in a familial case with chronic progressive external ophthalmoplegia symptoms. Neurosci Res. [DOI] [PubMed] [Google Scholar]
- Keen BA, Bailey LJ, Jozwiakowski SK, Doherty AJ. 2014. Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic Acids Res. 42(19):12102–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen BA, Bailey LJ, Jozwiakowski SK, Doherty AJ. 2015. Author response: PRIMPOL mutation: functional study does not always reveal the truth. Invest Ophthalmol Vis Sci. 56(2):1183. [DOI] [PubMed] [Google Scholar]
- Keen BA, Jozwiakowski SK, Bailey LJ, Bianchi J, Doherty AJ. 2014. Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res. 42(9):5830–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. 2013. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9(9):e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Paulus BF, Wold MS. 1994. Interactions of human replication protein A with oligonucleotides. Biochemistry. 33(47):14197–14206. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Guilliam TA, Tsuda M, Yamamoto J, Bailey LJ, Iwai S, Takeda S, Doherty AJ, Hirota K. 2016. Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle. 15(15):1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L et al. 2017. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell. 67(5):867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, Kirk-Bell S. 1972. Post-Replication Repair of DNA in Ultraviolet-Irradiated Mammalian Cells. European Journal of Biochemistry. 31(3):438–445. [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N et al. 2017. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun. 8(1):860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Ortiz AM, Svendsen J, Boulton SJ. 2014. Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair (Amst). 19:152–162. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang Q. 2015. PRIMPOL mutation: functional study does not always reveal the truth. Invest Ophthalmol Vis Sci. 56(2):1181–1182. [DOI] [PubMed] [Google Scholar]
- Li X, Manley JL. 2005. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 122(3):365–378. [DOI] [PubMed] [Google Scholar]
- Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D et al. 2018. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell. 72(6):925–941 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY. 2003. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem. 278(12):10041–10047. [DOI] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM. 2006. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell. 21(1):15–27. [DOI] [PubMed] [Google Scholar]
- Markkanen E 2017. Not breathing is not an option: How to deal with oxidative DNA damage. DNA Repair (Amst). 59:82–105. [DOI] [PubMed] [Google Scholar]
- Martinez-Jimenez MI, Calvo PA, Garcia-Gomez S, Guerra-Gonzalez S, Blanco L. 2018. The Zn-finger domain of human PrimPol is required to stabilize the initiating nucleotide during DNA priming. Nucleic Acids Res. 46(8):4138–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez MI, Garcia-Gomez S, Bebenek K, Sastre-Moreno G, Calvo PA, Diaz-Talavera A, Kunkel TA, Blanco L. 2015. Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair (Amst). 29:127–138. [DOI] [PubMed] [Google Scholar]
- Martinez-Jimenez MI, Lahera A, Blanco L. 2017. Human PrimPol activity is enhanced by RPA. Sci Rep. 7(1):783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini R 1976. Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochim Biophys Acta. 425(4):419–427. [DOI] [PubMed] [Google Scholar]
- Meroni A, Mentegari E, Crespan E, Muzi-Falconi M, Lazzaro F, Podesta A. 2017. The Incorporation of Ribonucleotides Induces Structural and Conformational Changes in DNA. Biophys J. 113(7):1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P et al. 2017. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun. 8(1):859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. 2007. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 71(1):13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, Mendez J. 2013. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol. 20(12):1383–1389. [DOI] [PubMed] [Google Scholar]
- Nava GM, Grasso L, Sertic S, Pellicioli A, Muzi Falconi M, Lazzaro F. 2020. One, No One, and One Hundred Thousand: The Many Forms of Ribonucleotides in DNA. Int J Mol Sci. 21(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, Cantor SB. 2020. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Science Advances. 6(24):eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M. 2015. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol. 16(4):207–220. [DOI] [PubMed] [Google Scholar]
- Olivieri M, Cho T, Alvarez-Quilon A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H et al. 2020. A Genetic Map of the Response to DNA Damage in Human Cells. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzarino NJ, Krais J, Peng M, Mosqueda M, Nayak S, Bond S, Calvo J, Cong K, Doshi M, Bere M et al. 2019. Replication gaps underlie BRCA-deficiency and therapy response. bioRxiv.781955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Vindigni A. 2017. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu Rev Genet. 51:477–499. [DOI] [PubMed] [Google Scholar]
- Pellegrini L 2012. The Pol alpha-primase complex. Subcell Biochem. 62:157–169. [DOI] [PubMed] [Google Scholar]
- Piberger AL, Bowry A, Kelly RDW, Walker AK, Gonzalez D, Bailey LJ, Doherty AJ, Méndez J, Morris JR, Bryant HE et al. 2020. PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. bioRxiv.773242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilzecker B, Buoninfante OA, Pritchard C, Blomberg OS, Huijbers IJ, van den Berk PC, Jacobs H. 2016. PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res. 44(10):4734–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokhrel N, Caldwell CC, Corless EI, Tillison EA, Tibbs J, Jocic N, Tabei SMA, Wold MS, Spies M, Antony E. 2019. Dynamics and selective remodeling of the DNA-binding domains of RPA. Nat Struct Mol Biol. 26(2):129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Lemacon D, Vindigni A. 2017. Replication Fork Reversal: Players and Guardians. Mol Cell. 68(5):830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Lerner LK, Martins DJ, Menck CFM. 2018. Filling gaps in translesion DNA synthesis in human cells. Mutat Res Genet Toxicol Environ Mutagen. 836(Pt B):127–142. [DOI] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, Menck CF. 2016. Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res. 44(12):5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, Gonzalez-Acosta D, Vessoni AT, Cybulla E, Wood M et al. 2020. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell. 77(3):461–474 e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 535(7612):382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechkoblit O, Gupta YK, Malik R, Rajashankar KR, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2016. Structure and mechanism of human PrimPol, a DNA polymerase with primase activity. Sci Adv. 2(10):e1601317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp WD, Howard-Flanders P. 1968. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol. 31(2):291–304. eng. [DOI] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA. 2017. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 18(10):622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE. 2012. Competition, collaboration and coordination--determining how cells bypass DNA damage. J Cell Sci. 125(Pt 7):1633–1643. [DOI] [PubMed] [Google Scholar]
- Sale JE. 2013. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol. 5(3):a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa A, Fukuda T, Ukai A, Nakamura M, Takabe M, Takamura-Enya T, Honma M, Yasui M. 2019. Comparative study of cytotoxic effects induced by environmental genotoxins using XPC- and CSB-deficient human lymphoblastoid TK6 cells. Genes Environ. 41:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, Doherty AJ. 2016. PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol Cell. 61(1):161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 145(4):529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 22(1):106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI, Chilkova O, Rogozin IB, Johansson E, Kunkel TA. 2003. Unique error signature of the four-subunit yeast DNA polymerase epsilon. J Biol Chem. 278(44):43770–43780. [DOI] [PubMed] [Google Scholar]
- Smith J, Tho LM, Xu N, Gillespie DA. 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 108:73–112. [DOI] [PubMed] [Google Scholar]
- Stojkovic G, Makarova AV, Wanrooij PH, Forslund J, Burgers PM, Wanrooij S. 2016. Oxidative DNA damage stalls the human mitochondrial replisome. Sci Rep. 6:28942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svikovic S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, Sale JE. 2019. R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J. 38(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R et al. 2017. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell. 68(2):414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier A, Janel-Bintz R, Coulon S, Klaile E, Kannouche P, Fuchs RP, Cordonnier AM. 2010. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair (Amst). 9(8):922–928. [DOI] [PubMed] [Google Scholar]
- Tokarsky EJ, Wallenmeyer PC, Phi KK, Suo Z. 2017. Significant impact of divalent metal ions on the fidelity, sugar selectivity, and drug incorporation efficiency of human PrimPol. DNA Repair (Amst). 49:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrosa-Munumer R, Forslund JME, Goffart S, Pfeiffer A, Stojkovic G, Carvalho G, Al-Furoukh N, Blanco L, Wanrooij S, Pohjoismaki JLO. 2017. PrimPol is required for replication reinitiation after mtDNA damage. Proc Natl Acad Sci U S A. 114(43):11398–11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C et al. 2009. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol. 11(11):1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica-Ross M. 2011. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell. 44(6):978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, Huang J. 2013. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 14(12):1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 73(1):134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Kunkel TA. 2014. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst). 19:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold MS. 1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 66:61–92. [DOI] [PubMed] [Google Scholar]
- Wong RP, Garcia-Rodriguez N, Zilio N, Hanulova M, Ulrich HD. 2020. Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol Cell. 77(1):3–16 e14. [DOI] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. 2012. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 72(11):2814–2821. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Oikawa M, Jinbo H, Hasegawa Y, Enomoto T, Seki M. 2019. WRNIP1 Controls the Amount of PrimPol. Biol Pharm Bull. 42(5):764–769. [DOI] [PubMed] [Google Scholar]
- Young MJ, Copeland WC. 2016. Human mitochondrial DNA replication machinery and disease. Curr Opin Genet Dev. 38:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Wang Q, Li Y, Cheng S, Liu J, Liu Y. 2020. Concurrent pathogenic variants in SLC6A1/NOTCH1/PRIMPOL genes in a Chinese patient with myoclonic-atonic epilepsy, mild aortic valve stenosis and high myopia. BMC Med Genet. 21(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafar MK, Ketkar A, Lodeiro MF, Cameron CE, Eoff RL. 2014. Kinetic analysis of human PrimPol DNA polymerase activity reveals a generally error-prone enzyme capable of accurately bypassing 7,8-dihydro-8-oxo-2'-deoxyguanosine. Biochemistry. 53(41):6584–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol. 16(1):2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC. 2015. DNA interstrand cross-link repair requires replication-fork convergence. Nat Struct Mol Biol. 22(3):242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Wu J, Xue A, Su Y, Wang X, Lu X, Zhou Z, Qu J, Zhou X. 2013. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum Genet. 132(8):913–921. [DOI] [PubMed] [Google Scholar]
- Zhong X, Garg P, Stith CM, Nick McElhinny SA, Kissling GE, Burgers PM, Kunkel TA. 2006. The fidelity of DNA synthesis by yeast DNA polymerase zeta alone and with accessory proteins. Nucleic Acids Res. 34(17):4731–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]