

Abstract
The four contiguous all-carbon quaternary centers of waihoensene, coupled with the absence of any traditional reactive functional groups other than a single alkene, renders it a particularly challenging synthetic target among angular triquinane natural products. Here, we show that its polycyclic frame can be assembled concisely by using a strategically selected quaternary center to guide the formation of the other three through judiciously selected C–C bond formation reactions. Those events, which included a unique Conia-ene cyclization and a challenging Pauson–Khand reaction, afforded a 17-step synthesis of the molecule in enantioenriched form.
Keywords: Conia-ene, Pauson-Khand, waihoensene, total synthesis, triquinanes
Graphical Abstract
Despite the power of modern chemical synthesis, the preparation of unique, non-functionalized terpenes remains challenging. Here, we show how the strategic use of a minimal number of functional groups, coupled with retrosynthetic planning based on the use of a guiding all-carbon quaternary center to forge three additional such centers as fueled by some unique C-C bond forming steps, can afford an expeditious, 17-step total synthesis of the unique angular triquinane natural product (+)-waihoensene.
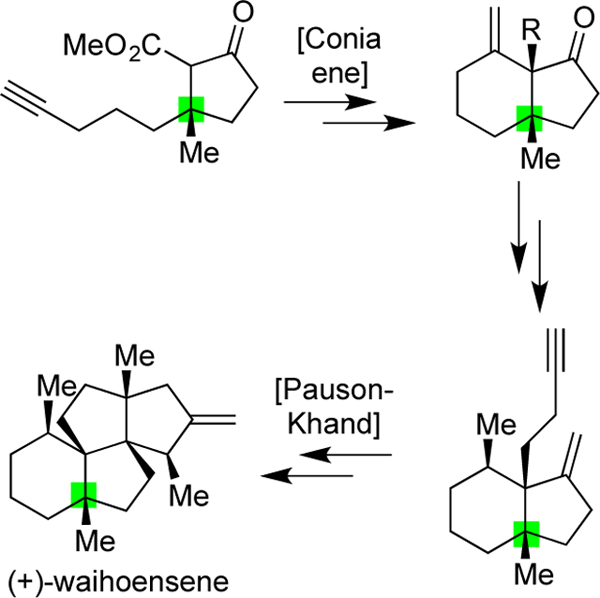
The structures of angular triquinane natural products have long fascinated synthetic chemists, not only due to the challenges presented by their diverse fused ring systems and congested frames, but also because of their overall stereochemical complexity and density.[1] Indeed, as shown in Scheme 1, many of these natural products (such as 1 and 2) possess three all-carbon quaternary centers,[2] often contiguous (colored here in blue). Some, such as laurenene (3),[3] possess a fourth (denoted here in orange), though to the best of our knowledge, only the natural product waihoensene (4),[4] isolated in 1997 by the Weavers group from Podocarpus totara var. waihoensis,[5] has four such centers arrayed in a row. Within some, quaternary centers comprise as much as 20% of their carbon atoms. Further, many natural products in this class, particularly as exemplified by 3 and 4, are devoid of typically reactive functional groups; both of these molecules possess only a lone alkene outside of their alkane-based frames. Over the past few years, our group has engaged in a research program seeking to develop strategies and tactics to efficiently synthesize similarly unique terpenes (such as 5–7) lacking in traditional functional groups.[6] Key lessons learned from those endeavors include: 1) using a bare modicum of added functional groups for critical C–C bond formations and strategically using those groups for multiple purposes, and 2) viewing the quaternary centers within the targets as key components to include in retrosynthetic planning, specifically in thinking about how the presence of one (such as the green colored center in 7) could assist in the formation of others. Herein, we show how the application of these ideas has led to a concise 17-step asymmetric total synthesis of waihoensene (4).
Scheme 1.

Angular-triquinane natural products (1-4) possessing numerous all-carbon quarternary stereocenters, many of which are contiguous and the development of a new approach to access waihoensene (4).
While it has been proposed that waihoensene (4) can arise biosynthetically from laurenene (3), the only synthesis achieved at the time our work commenced was a non-biomimetic, but highly creative approach from the Lee group which delivered racemic material.[7] Their effort, shown in a condensed format in Scheme 1, illustrates the tenets of quaternary center-guided synthesis in that the quaternary center present within 8 assisted in the formation of the other three. Of note, the two additional centers within 9, formed concurrently via a cascade process, proceeded with 3.3:1 selectivity for 9 and other isomers, while installation of the remaining two methyl groups, leading to the final all-carbon quaternary center, was stereospecific. Our retrosynthetic analysis of the target as guided by its four contiguous quaternary centers suggested that the same initial quaternary center used by the Lee group would be the ideal starting point, but not in the form of a six-membered ring. Rather, it was the neighboring five-membered ring of the final target as expressed by 12, where our approach sought to use its lone quaternary center to introduce each additional quaternary center individually in a stereocontrolled fashion. Thus, from 12, we anticipated that a Conia-ene reaction could lead to a second ring and quaternary center as expressed in 11, which, if elaborated to 10, could then enable the formation of the third center found in 9 through a Pauson–Khand reaction.[8] In this way, the original ester, alkyne, and ketone functional groups of 12 would serve as the only functional group lynchpins needed to achieve all bond formations, chain homologations, and other elements of tailoring required to reach the final target. As validation for the power of this approach, as well as interest in waihoensene as a target in general, a synthesis of (+)-waihoensene by Yang, Huang, and co-workers was published while this paper was under review using similar key bond constructions, albeit in unique ways,[9] as was an additional model study from Tu and Wang.[10]
Our efforts began with the preparation of a chiral and protected version of 12 in the form of 19 as shown in Scheme 2. Following the reaction of Grignard reagent 14[11] with 13 to afford functionalized α,β-unsaturated ketone 15, use of Hoveyda’s asymmetric conjugate addition chemistry,[12] as guided by N-heterocyclic carbene complex 16, generated the all-carbon quaternary center of 17 in excellent yield (81–92%) and high enantioselectivity (82–92% ee), variable based on the scale of the reaction. From here, a partially regioselective addition of a methyl ester could be achieved via deprotonation with NaHMDS and subsequent trapping with Mander’s reagent, affording a 1.8:1 mixture of 19 and 18 in 88% combined yield. Extensive screening revealed that no other conditions were superior in terms of throughput, noting, however, that LiHMDS did give higher selectivity in terms of desired enolate formation, but a lower numerical yield of 19. We attribute the potential regioselectivity observed for all bases to be due to a weak directing effect from the alkyne.[6c,13] While this step did not proceed with ideal levels of selectivity, the undesired regioisomer (18) could be recycled to 17 through a Krapcho decarboxylation.[14] Of note, efforts to forge the critical quaternary carbon in an enantioselective manner with the ester present were unsuccessful. While the key precursor (21) could be prepared using a method developed by our group starting from lactone 20,[15] subsequent attempts at asymmetric methyl addition were uniformly unsuccessful (see SI for details); the racemic addition reaction, by contrast, worked quite well when promoted with CuBr•Me2S.
Scheme 2.

Asymmetric introduction of the guiding quaternary carbon: (a) (5-bromo-1-pentynyl)trimethylsilane (1.0 equiv), Mg (2.0 equiv), I2 (trace), THF, reflux, 4 h; 13, 0 to 23 °C, 10 h, 54%; (b) 16 (3.75 mol %), Cu(OTf)2 (7.5 mol %), THF, 23 °C, 10 min, then AlMe3 (3.0 equiv), 15, −78 °C, 12 h, 1 g scale: 81%, 92% ee; 5 g scale: 92%, 82% ee; (c) NaHMDS (2.3 equiv), THF, 0 °C, 2 h, then Mander’s reagent (1.6 equiv), −78 °C, 3 h, 32% 18; 56% 19 (d) LiCl (2 equiv), H2O (5 equiv), DMSO, 150 °C, 3 h, 76%; (e) MeNHOMe•HCl (1.3 equiv), pyridine (5.0 equiv), CH2Cl2, 23 °C, 6 h; (f) NaH (1.3 equiv), THF, 0 °C, 30 min, then 14 (1.3 equiv), −78 to 0 °C, 1 h then MeOH, 50 °C, 1 h, 54% overall; (g) CuBr•Me2S (1.2 equiv), MeMgBr (2.4 equiv), THF, −40 °C, 30 min, then 21, 2 h, 77%.
With one ring and quaternary center forged effectively, our focus next turned to generating a second ring and quaternary center through a Conia-ene reaction. As shown in Scheme 3, that step, following TBAF-mediated removal of the TMS group from the alkyne, proceeded smoothly and quantitatively with only 0.96 mol % each of Ph3PAuCl and AgOTf to afford the desired bicyclo[4.3.0] framework of 11.[16] Critically, this reaction proved highly scalable, even when using gram quantities of starting material, with no decrease in overall efficiency; to our knowledge, this Conia-ene reaction is the first leading to the formation of a six-membered ring containing two vicinal quaternary carbons starting from a five-membered ring precursor.[17]
Scheme 3.

Total synthesis of waihoensene (4) by strategic formations of three quaternary carbons from a starting quaternary center: (a) TBAF (1.6 equiv), THF, 0 to 23 °C, 2 h, 94%; (b) Ph3PAuCl (0.96 mol %), AgOTf (0.96 mol %), CH2Cl2, 23 °C, 4 h, quantitative; (c) H2 (balloon pressure), PtO2 (4.6 mol %), CH2Cl2 23 °C, 2 h, quantitative, 3.2 : 1 dr. (d) t-BuOK (9.0 equiv), Ph3PCH3Br (10.0 equiv), toluene, reflux, 1 h, then 22, 12 h; (e) DIBAL-H (2.0 equiv), CH2Cl2 −78 °C, 1 h, then t-BuOH (30 equiv), NaHC03 (10.0 equiv), Dess-Martin periodinane (8.0 equiv), 23 °C, 1 h, 55% overall; (f) DIBAL-H (2.5 equiv), CH2Cl2 −78 °C, 1 h, 67% overall; (g) NaHCO3 (10.7 equiv), Dess-Martin periodinane (1.9 equiv), CH2Cl2, 0 to 23 °C, 2 h, 94%; (h) diethyl cyanomethylphosphonate (4.6 equiv), t-BuOK (5.0 equiv), THF, 23 °C, 1 h, then 26; reflux, 18 h, 93%, cis : trans =1.7 :1; (i) Mg (30 equiv), MeOH (0.02 M), 0 to 23 °C, 3 h, 72%; (j) DIBAL-H (1.8 equiv), CH2Cl2, 0 °C, 1 h; (k) Ohira-Bestmann reagent (1.5 equiv), K2CO3 (2.5 equiv), MeOH, 30 °C, 12 h, 55% overall; (I) Co2(CO)8(1.1 equiv), mesitylene, 23 °C, 2 h, then CO (balloon pressure), 160 °C, 24 h, 50%; (m) CuCN (3.0 equiv), MeLi (6.0 equiv), THF, −78 to 23 °C, 10 min, then BF3•Et2O (2.4 equiv), then 9, −78 to −55 °C, 2 h, 54%; (n) LiHMDS (2.0 equiv), THF, 0 °C, 2 h, then Mel (5.0 equiv), 0 to 23 °C, 12 h, 81%; (o) t-BuOK (9.0 equiv), Ph3PCH3Br (10.0 equiv), toluene, reflux, 1 h, then 29, 1 h, 83%.
Now that the alkyne had served its role in generating the alkene of 11, we sought to convert it into the respective methyl group of the final target through a facially selective reduction. That event would formally require the addition of hydrogen from the concave face. Pleasingly, however, conventional hydrogenation using Pd/C (Table 1, Entry 1) led to the desired product (22) as the predominant diastereomer as confirmed by X-ray crystallographic analysis, albeit in low yield (20%). Further screening revealed that hydrogenation with PtO2 (Entry 2) in CH2Cl2 gave much better yield and selectivity (76% 22, 3.2:1 dr). Given that a hydrogen atom transfer (HAT)-type hydrogenation, as developed by Shenvi,[18] demonstrated the same trend in diastereoselectivity (2.5:1, Entry 3), these collective outcomes suggest that 22 is both kinetically and thermodynamically favored as the hydrogenation product. Intriguingly, a reversal in stereoselectivity was observed when Crabtree’s catalyst was employed (Entry 4), possibly due to directing effects by the neighboring ester motif.[19] As such, these results could provide guidance on stereodivergent applications of such intermediates for the synthesis of other molecules.
Table 1.
Screening of conditions for the stereospecific reduction of 11.
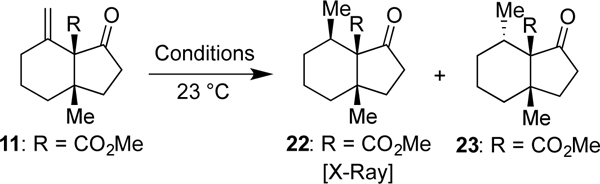 | ||||
|---|---|---|---|---|
| Entry | Conditions | 22 [%][a] | 23 [%][a] | dr |
| 1 | H2, 5% Pd/C (50 wt%). EtOH | 20 | 8 | 2 5:1.0 |
| 2[b] | H2, ptO2(5 mol %), CH2Cl2 | 76 | 24 | 3.2:1.0 |
| 3 | Mn(dpm)3, t-BuOOH, PhSiH3, i-PrOH | 47 | 19 | 2.5: 1.0 |
| 4 | H2, Crabtree’s cat. (10 mol %), CH2Cl2 | 11 | 65 | 1.0 : 5.9 |
Isolated yields, reactions ran on 0.05–0.10 mmol scale unless otherwise specified
Reaction ran on 5.0 mmol scale.
Given that this additional chiral center had now been secured, the ketone within 22 was then olefinated with excess Ph3P=CH2, using KOt-Bu in toluene at reflux to generate the ylide and then reacting it with the substrate for 12 h.[20] No other condition set afforded the desired product effectively, with the need for elevated temperature, prolonged reaction time, and large excess in reagents likely reflecting the neighboring steric bulk of the vicinal quaternary carbons. Next, with an eye towards performing the designed Pauson–Khand reaction, the ester was converted into aldehyde 26 through a one-pot procedure involving sequential DIBAL-H reduction, t-BuOH quench, and Dess–Martin periodinane-mediated oxidation as developed during our total synthesis of 6 (cf. Scheme 1);[6b] a slightly higher yield was obtained by performing the reduction and oxidation steps separately. However, despite the facility of this operation, subsequent conversion of the aldehyde within 26 to the chain homologated alkyne needed for the Pauson–Khand step (i.e. 10) proved quite challenging. Many efforts proved fruitless, either due to an inability to forge the alkyne of 10 or to afford the natural product using a differentially functionalized version of 10.[21] Ultimately, the desired intermediate was accessed through a 4-step sequence involving an initial Horner–Wadsworth–Emmons homologation, producing an inconsequential 1.7:1 mixture of alkene isomers that were subsequently erased by chemoselective single electron transfer (SET)-based reduction using magnesium metal in MeOH.[22] These operations produced 27 in 67% overall yield. Then, following DIBAL-H reduction of the nitrile to the respective aldehyde, a highly silica gel-unstable material (potentially due to a Prins-type cyclization, although unverified), immediate treatment of this crude product with the Ohira–Bestmann reagent afforded 10 in 55% yield.[23]
With this compound in hand, we were delighted to find that the designed Pauson–Khand reaction worked to forge the third quaternary center of the target as well as generate the full angular triquinane framework of waihoensene. That event, achieved by stirring 10 with stoichiometric Co2(CO)8 and then heating the resultant cobalt complex at 160 °C in mesitylene under a CO atmosphere, delivered 9 in 50% yield. The uncommonly high temperature used for this reaction[24] was critical to its success, likely due to the increased reaction rate overcoming the energy-barrier needed for quaternary center formation.[25] Pleasingly, our synthetic 9 matched all reported spectral data obtained by the Lee group.[7a] We then followed the same general sequence utilized in that synthesis to access waihoensene (4), optimizing the final step by using the Wittig homologation procedure deployed earlier for the conversion of 22 to 24. That event proceeded in 83% yield, while the Lee procedure using the Petasis reagent[26] led to a 32% yield. All spectral data for synthetic 4, including the sign and general magnitude of its optical rotation, matched that reported by Weavers,[5] confirming its absolute configuration. We also obtained a single-crystal of compound (±)-28, further verifying the integrity of our sequence. In total, we have prepared ~50 mg of the natural product across all runs of our sequence.
In conclusion, we have completed an enantioselective total synthesis of (+)-waihoensene in 17-steps from commercially available starting materials. This overall length compares favorably to past efforts. That outcome is due, in large part, to utilizing a modicum of functional groups, none of which were extraneous, to repeatedly effect key C–C bond formations and overall tailoring coupled with overarching retrosynthetic design orchestrated by viewing the quaternary centers as a central organizing feature. Efforts to extend these lessons, and some of the unique elements of the Conia-ene and Pauson–Khand steps, to other targets are the subject of current investigations.
Supplementary Material
Acknowledgements
We thank Dr. Antoni Jurkiewicz, Dr. Josh Kurutz, and Dr. C. Jin Qin for assistance with NMR and mass spectrometry, respectively. We also thank Dr. Alexander Filatov and Mr. Andrew McNeece for X-ray crystallographic analysis of our intermediates. Financial support for this work came from the University of Chicago and the National Institutes of Health (R01-GM132570).
References
- [1].For a review on syntheses of polyquinane natural products, see: Mehta G, Srikrishna A, Chem. Rev 1997, 97, 671–720. [DOI] [PubMed] [Google Scholar]
- [2].For the isolation of bipolaridide D, see: (a) Liu M, Sun W, Shen L, He Y, Liu J, Wang J, Hu Z, Zhang Y, Angew. Chem. Int. Ed 2019, 58, 12091–12095; for the isolation of crinipellins A and B, see: [DOI] [PubMed] [Google Scholar]; (b) Kupka J, Anke T, Oberwinkler F, Antibiot J. 1979, 32, 130–135; [DOI] [PubMed] [Google Scholar]; (c) Anke T, Heim J, Knoch F, Mocek U, Steffan B, Steglich W, Angew. Chem. Int. Ed. Engl 1985, 24, 709–711; for syntheses of crinipellins A and B, see: [Google Scholar]; (d) Piers E, Renaud J, J. Org. Chem 1993, 58, 11–13; [Google Scholar]; (e) Piers E, Renaud J, Rettig SJ, Synthesis 1998, 590–602; [Google Scholar]; (f) Wender PA, Dore TM, Tetrahedron Lett. 1998, 39, 8589–8592; [Google Scholar]; (g) Kang T, Song SB, Kim W-Y, Kim BG, Lee H-Y, J. Am. Chem. Soc 2014, 136, 10274–10276; [DOI] [PubMed] [Google Scholar]; (h) Huang Z, Huang J, Qu Y, Zhang W, Gong J, Yang Z, Angew. Chem. Int. Ed 2018, 57, 8744–8748. [DOI] [PubMed] [Google Scholar]
- [3].For the isolation of laurenene, see: (a) Corbett RE, Lauren DR, Weavers RT, J. Chem. Soc. Perkin Trans 1979, 1, 1774–1790; [Google Scholar]; (b) Corbett RE, Lauren DR, Weavers RT, J. Chem. Soc. Perkin Trans 1979, 1, 1791–1794; for syntheses of laurenene, see: [Google Scholar]; (c) Tsunoda T, Amaike M, Tambunan USF, Fujise Y, Itô S, Kodama M, Tetrahedron Lett. 1987, 28, 2537–2540; [Google Scholar]; (d) Crimmins MT, Gould LD, J. Am. Chem. Soc 1987, 109, 6199–6200; [Google Scholar]; (e) Paquette LA, Okazaki ME, Caille J-C, J. Org. Chem 1988, 53, 477–481; [Google Scholar]; (f) Wender P, von Geldern TW, Levine BH, J. Am. Chem. Soc 1988, 110, 4858–4860; [Google Scholar]; (g) Mehta G, Rao KS, J. Org. Chem 1988, 53, 425–427. [Google Scholar]
- [4].For a recent review on syntheses of natural products with contiguous all-carbon quaternary stereocenters, see: Büschleb M, Dorich S, Hanessian S, Tao D, Schesnthal KB, Overman LE, Angew. Chem. Int. Ed 2016, 55, 4156–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Clarke DB, Hinkley SFR, Weavers RT, Tetrahedron Lett. 1997, 38, 4297–4300. [Google Scholar]
- [6].(a) Snyder SA, Wespe DA, von Hof JM, J. Am. Chem. Soc 2011, 133, 8850–8853; [DOI] [PubMed] [Google Scholar]; (b) Hu P, Snyder SA, J. Am. Chem. Soc 2017, 139, 5007–5010; [DOI] [PubMed] [Google Scholar]; (c) Hu P, Chi HM, Debacker KC, Gong X, Keim JH, Hsu IT, Snyder SA, Nature 2019, 569, 703–707; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yi H, Hu P, Snyder SA, Angew. Chem. Int. Ed 2020, 59, 2674–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) Lee H, Kang T, Lee H-Y, Angew. Chem. Int. Ed 2017, 56, 8254–8257; for an earlier model study of the tetracyclic framework, see: [DOI] [PubMed] [Google Scholar]; (b) Ergüden J-K, Moore HW, Org. Lett 1999, 1, 375–377. [DOI] [PubMed] [Google Scholar]
- [8].For original reports on the Pauson–Khand reaction, see: (a) Khand IU, Knox GR, Pauson PL, Watts WE, Foreman MI, J. Chem. Soc., Perkin Trans 1973, 1, 977–981; [Google Scholar]; (b) Pauson PL, Tetrahedron 1985, 41, 5855–5860; for examples of Pauson–Khand reactions forming angular triquinane structures, see: [Google Scholar]; (c) Ishizaki M, Iwahara K, Kyoumura K, Hoshino O, Synlett 1999, 5, 587–589; [Google Scholar]; (d) Renaud J-L, Aubert C, Malacria M, Tetrahedron 1999, 55, 5113–5128; [Google Scholar]; (e) Ishizaki M, Iwahara K, Niimi Y, Satoh H, Hoshino O, Tetrahedron 2001, 57, 2729–2738; also worth noting is that Ref. 2h defines an approach towards the linear portion of an angular triquinane. [Google Scholar]
- [9].(a) Snyder SA, Wespe DA, von Hof JM, J. Am. Chem. Soc 2011, 133, 8850–8853; [DOI] [PubMed] [Google Scholar]; (b) Hu P, Snyder SA, J. Am. Chem. Soc 2017, 139, 5007–5010; [DOI] [PubMed] [Google Scholar]; (c) Hu P, Chi HM, Debacker KC, Gong X, Keim JH, Hsu IT, Snyder SA, Nature 2019, 569, 703–707; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yi H, Hu P, Snyder SA, Angew. Chem. Int. Ed 2020, 59, 2674–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].(a) Lee H, Kang T, Lee H-Y, Angew. Chem. Int. Ed 2017, 56, 8254–8257; for an earlier model study of the tetracyclic framework, see: [DOI] [PubMed] [Google Scholar]; (b) Ergüden J-K, Moore HW, Org. Lett 1999, 1, 375–377. [DOI] [PubMed] [Google Scholar]
- [11].Harris MR, Konev MO, Jarvo ER, J. Am. Chem. Soc 2014, 136, 7825–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].(a) Brown MK, May TL, Baxter CA, Hoveyda AH, Angew. Chem. Int. Ed 2007, 46, 1097–1100; [DOI] [PubMed] [Google Scholar]; (b) May TL, Brown MK, Hoveyda AH, Angew. Chem. Int. Ed 2008, 47, 7358–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Posner GH, Lentz CM, J. Am. Chem. Soc 1979, 101, 934–946. [Google Scholar]
- [14].Krapcho AP, Weimaster JF, Eldridge JM, Jahngen EGE, Lovey AJ, Stephens WP, J. Org. Chem 1978, 43, 138–147. [Google Scholar]
- [15].Eagan JM, Hori M, Wu J, Kanyiva KS, Snyder SA, Angew. Chem. Int. Ed 2015, 54, 7842–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].For the original report of the gold-catalyzed Conia-ene reaction of β-keto esters, see:Kennedy-Smith JJ, Staben ST, Toste FD, J. Am. Chem. Soc 2004, 126, 4526–4527; for some selected applications of similar transformations in total syntheses which are exo selective using β-keto esters, see:Tsuji H, Yamagata K-I, Itoh Y, Endo K, Nakamura M, Nakamura E, Angew. Chem. Int. Ed 2007, 46, 8060–8062;Takahashi K, Midori M, Kawano K, Ishihara J. and Hatakeyama S, Angew. Chem. Int. Ed 2008, 47, 6244–6246;Liu X, Lee C-S, Org. Lett 2012, 14, 2886–2889;Urabe F, Nagashima S, Takahashi K, Ishihara J, Hatakeyama S, J. Org. Chem 2013, 78, 3847–3857;Ye Q, Qu P, Snyder SA, J. Am. Chem. Soc 2017, 139, 18428–18431,Huang J, Bao W, Huang S, Yang W, Lizhi Z, Du G, Li C-S, Org. Lett 2018, 20, 7466–7469; for a review on catalytic Conia-ene and related reactions, see:Hack D, Blümel M, Chauhan P, Philipps AR, Enders D, Chem. Soc. Rev 2015, 44, 6059–609.
- [17].There is an example of the reverse process, namely 5-membered ring formation onto a 6-membered to also afford a 6–5 fused ring system within Ref. 14a. The Yang/Huang synthesis of Ref. 9 is a second example.
- [18].(a) Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA, J. Am. Chem. Soc 2014, 136, 1300–1303; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Obradors C, Martinez RM, Shenvi RA, J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Crabtree RH, Davis MW, J. Org. Chem 1986, 51, 2655–2661. [Google Scholar]
- [20].Fitjer L, Quabeck U, Synth. Commun 1985, 15, 855–864. [Google Scholar]
- [21].For example, if an alcohol residue from the aldehyde was present along with the alkyne, the Pauson–Khand step worked, but we were unable to excise that alcohol group.
- [22].Pfaffenbach M, Gaich T, Eur. J. Org. Chem 2015, 3427–3429. [Google Scholar]
- [23].(a) Ohira S, Synth. Commun 1989, 19, 561–564; [Google Scholar]; (b) Müller SG, Liepold B, Roth GJ, Bestmann HJ, Synlett 1996, 521–522; [Google Scholar]; (c) Roth GJ, Liepold B, Müller SG, Bestmann HJ, Synlett 2004, 59–62. [Google Scholar]
- [24].For selected reviews on the Pauson-Khand reaction, see: (a) Geis O, Schmalz H-G, Angew. Chem. Int. Ed 1998, 37, 911–914; [DOI] [PubMed] [Google Scholar]; (b) Brummond KM, Kent JL, Tetrahedron 2000, 56, 3263–3283; [Google Scholar]; (c) Blanco-Urgoiti J, Añorbe L, Pérez-Serrano L, Domínguez G, Pérez-Castells J, Chem. Soc. Rev 2004, 33, 32–42; [DOI] [PubMed] [Google Scholar]; (d) Boñaga LVR, Krafft ME, Tetrahedron 2004, 60, 9795–9833. [Google Scholar]
- [25].In the Yang/Huang synthesis (Ref. 9) a temperature of 85 oC for their Pauson–Khand reaction could be achieved when laughing gas was used.
- [26].(a) Petasis NA, Bzowej EI, J. Am. Chem. Soc 1990, 112, 6392–6394; [Google Scholar]; (b) Csuk R, Glänzer BI, Tetrahedron 1991, 47, 1655–1664. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.