

Abstract
Chrysanthemum rhombifolium (Ling et C. Shih), an endemic plant that is extremely well-adapted to harsh environments. However, little is known about its molecular biology of the plant's resistant traits against stress, or even its molecular biology of overall plant. To investigate the molecular biology of C. rhombifolium and mechanism of stress adaptation, we performed transcriptome sequencing of its leaves using an Illumina platform. A total of 130,891 unigenes were obtained, and 97,496 (~74.5%) unigenes were annotated in the public protein database. The similarity search indicated that 40,878 and 74,084 unigenes showed significant similarities to known proteins from NCBI non-redundant and Swissprot protein databases, respectively. Of these, 56,213 and 42,005 unigenes were assigned to the Gene Ontology (GO) database and Cluster of Orthologous Groups (COG), respectively, and 38,918 unigenes were mapped into five main categories, including 18 KEGG pathways. Metabolism was the largest category (23,128, 59.4%) among the main KEGG categories, suggesting active metabolic processes in C. rhombifolium. About 2,459 unigenes were annotated to have a role in defense mechanism or stress tolerance. Transcriptome analysis of C. rhombifolium revealed the presence of 12,925 microsatellites in 10,524 unigenes and mono, trip, and dinucleotides having higher polymorphism rates. The phylogenetic analysis based on GME gene among related species confirmed the reliability of the transcriptomic data. This work is the first genetic study of C. rhombifolium as a new plant resource of stress-tolerant genes. This large number of transcriptome sequences enabled us to comprehensively understand the basic genetics of C. rhombifolium and discover novel genes that will be helpful in the molecular improvement of chrysanthemums.
Keywords: Asteraceae, stress tolerance, ornamental plant, RNA-seq, SSR
Introduction
Chrysanthemum (Chrysanthemum morifolium (Ramat.)Tzvel.; Asteraceae) is among the most popular flowers in China, and the most important cut flowers in the world, having a great ornamental and economical value (Song et al., 2018; Su et al., 2019). However, the long-term artificial domestication of chrysanthemums often causes declines in their resistance to environmental stressors and adaptability (Da Silva, 2003; Chen et al., 2011, 2012; Song et al., 2014), thereby limiting their use in landscaping and industrial production. Therefore, the development of Chrysanthemum cultivars with increased environmental tolerance has always been a goal of breeders (Su et al., 2019). Many stress resistance traits, and corresponding stress resistance gene resources identified in the wild chrysanthemum species (Zhao et al., 2009; Lu et al., 2010; Li et al., 2013), have a great significance for the genetic improvement of chrysanthemum cultivars.
RNA sequencing (RNA-Seq) is a powerful tool for quantifying and analyzing different types of RNA molecules using deep-sequencing technologies (Wang et al., 2009). It provides us large-scale transcript data with high throughput, accuracy, sensitivity and reproducibility which enabled us to generate an unprecedented global view of the transcriptome of the species (Angeloni et al., 2011; Jain, 2011). RNA-seq has been widely used in plants, especially for some non-model species and some large and complex genomes, greatly accelerating the discovery of novel genes, understanding the complex tissue-specific expression patterns, and regulation networks in higher plants (Li and Dewey, 2011; Wang et al., 2014, 2017; Wu et al., 2016).
Chrysanthemum rhombifolium Ling et Shih is a perennial herb endemic to Wushan, Chongqing in China (Shih and Fu, 1983; Bremer and Humphries, 1993) and has a high ornamental value. It has diamond-shaped leaves with dense abaxial pubescence and semi-lignified stems and branches (Figure 1). The species is well-adapted to environments characterized by high temperatures, low soil fertility, and drought (Zhao et al., 2009, 2010). However, few studies performed on C. rhombifolium except using as a sample in molecular phylogeny of Chrysanthemum (Masuda et al., 2009; Zhao et al., 2010; Li et al., 2014; Ma et al., 2020) or in geographical distribution of Chrysanthemum (Zhao et al., 2009; Li et al., 2013). Here, little is known about its molecular biology of overall plant or the plant's resistant traits against stress. This prompted us to characterize its leaf transcriptome using high-throughput RNA sequencing and de novo assembly to provide a comprehensive resource for understanding the biology of C. rhombifolium in general, and gain insights in improving the breeding of chrysanthemums and other related crops.
Figure 1.

The plant of C. rhombifolium. (A) The habitat; (B) leaf, up, abaxially, down, adaxially. Scale bars = 2 cm.
Materials and Methods
Plant Materials
We collected C. rhombifolium plants from Wushan of Chongqing in China and planted them in the Nurse Garden of the Northeastern University, China. Fresh, mature leaves were washed with sterile water, immediately frozen in liquid nitrogen, and stored at −80°C.
RNA Isolation and cDNA Library Construction
Total RNA was isolated from the leaves using TRIzol reagent (InvitrogenTM Life Technologies, CA, USA) following the manufacturer's instructions. The RNA quality was assessed using formaldehyde denaturing gel electrophoresis (28S:18S>2), a NanoPhotometer®spectrophotometer (IMPLEN, CA, USA), and RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). For RNA-Seq analysis, three biological replicates were used. Sequencing libraries were generated with 1 μg RNA sample using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer's recommendations. The mRNA was purified from total RNA using beads with Oligo (dT), and cut into short fragments with fragmentation buffer. First-strand cDNA was synthesized using random hexamer primers and M-MuLV Reverse Transcriptase (NEB, USA), and second-strand cDNA was synthesized using buffer, dNTPs, RNase H, and DNA polymerase I. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure was ligated to prepare for hybridization. cDNA fragments, preferentially 250–300 bp in length, were selected by purifying the library fragments with AMPure XP system (Beckman Coulter, Beverly, USA). The size-selected, adaptor-ligated cDNA fragments were incubated with 3 μl USER Enzyme (NEB, USA) at 37°C for 15 min, followed by 5 min at 95°C before PCR. PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. The PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system.
Sequencing and de novo Assembly
We sequenced the transcriptome library using the Illumina HiSeq 2500 platform, and generated paired-end reads. We filtered the raw data using the Filterfq program (BGI, Shenzhen, China) to remove adaptor sequences, reads in which unknown nucleotides (N) were >5%, and low-quality sequences with >20% low-quality bases (quality value ≦10) to generate clean data. The raw data were deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) with the Bioproject accession: PRJNA674029 and BioSample accessions:SAMN16633381- SAMN16633383.
We then used the Trinity software (v2.8.0; http://trinityrnaseq.sourceforge.net/) with default settings for de novo transcriptome assembly (Grabherr et al., 2011). Two contigs thus obtained were connected into a single scaffold to generate unigenes. These unigenes were further spliced to generate longer complete consensus sequences and to remove redundant sequences with TGICL (v 2.1; http://www.tigr.org/tdb/tgi/) (Pertea et al., 2003).
Functional Annotation and Classification of Unigenes
We annotated the obtained unigenes using the NCBI Nr (non-redundant protein database), NCBI Nt (non-redundant nucleotide sequences), Swiss-Prot, Gene ontology terms (GO), and Protein family (Pfam) using BLAST 2 with an E-value cut-off of 10−5 to obtain information on protein function annotation. We also performed functional annotation using Clusters of Orthologous Groups of proteins (KOG/COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases to classify possible COG functions and KEGG pathways and predict possible functional classifications and molecular pathways, respectively (Conesa et al., 2005; Ye et al., 2006; Kanehisa et al., 2008).
Phylogenetic Analysis
GDP-D-mannose 3′, 5′- epimerase (GME), regulates cell wall biosynthesis and ascorbate accumulation, playing an important role in plant development and abiotic stress tolerance (Tao et al., 2018). We extracted annotated GME unigene sequences, aligned them with other GME homologs retrieved from Genbank (http://www.ncbi.nlm.nih.gov/entyez/query.fcgi). Multiple alignments were made using MUSCLE (Edgar, 2004) in Geneious v.8.1.2 (http://www.geneious.com/; Kearse et al., 2012) and adjusted manually. We contructed the phylogenetic tree by the neighbor-joining (NJ) method with 1,000 bootstrap replicates using MEGA 7 (Kumar et al., 2016).
Simple Sequence Repeats (SSRs) Prediction
We predicted SSR regions among all the assembled unigenes using MIcroSAtellite (MISA, http://pgrc.ipk-gatersleben.de/misa/; Zalapa et al., 2012). We detected the SSR motifs of mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides with a minimum of twelve, six, five, five, four, and four repeats, respectively. For other parameters, default settings were used.
Results and Discussion
Illumina Paired-End Sequencing and Assembly
The 151,037,024 raw sequencing reads obtained from the Illumina sequencing were cleaned by removing low-quality data and adaptors, yielding 147,842,128 clean reads with Q20 bases at 97.9%, and a GC content of 44.44%. Using the overlapping information of high-quality reads from the Trinity software, 254,853 transcripts with an average length of 921 bp and N50 of 1,301 bp were generated. After that 130,891 unigenes with an average length of 807 bp and N50 of 1,034 bp were obtained (Table 1). The number and average length of the unigenes we obtained was larger and longer than the transcriptomes of the related species, C. nankingense (45,789) and C. lavandulifolium (108,737) (Wang et al., 2013, 2014), indicating the high quality of sequencing.
Table 1.
Summary of sequence assembling of C. rhombifolium transcriptome.
| Category | Transcripts | Unigene | |
|---|---|---|---|
| Number | 300–500 bp | 102,216 | 59,296 |
| 500-1 Kbp | 74,883 | 41,353 | |
| 1 K-2 Kbp | 53,959 | 21,862 | |
| >2 Kbp | 23,795 | 8,380 | |
| Total number | 254,853 | 130,891 | |
| Mean length | 921 | 807 | |
| N50 | 1,301 | 1,034 | |
| Total nucleotides | 234,824,746 | 105,639,077 | |
Functional Annotation and Classification of All Non-redundant Unigenes
We used the Nr, Nt, Pfam, KOG, Swiss-prot, GO, and KEGG databases to annotate the assembled unigenes. Among the 130,891 unigenes obtained, at least 97,496 unigenes (74.48%) could be annotated with the searched databases −40,878 (Nr), 55,831 (Nt), 37,488 (KO), 74,084 (Swiss-prot), 56,213 (Pfam), 56,213 (GO), and 42,005 (KOG/COG), suggesting that this project generated a substantial fraction of the expressed genes in this study (Table 2). The unigenes annotated with the Nr database mainly comprised Quercus suber L. (51%), Helianthus annuus L. (12.9%), Lactuca sativa Linn. (10%), Cynara cardunculus L., Sp. Pl. (5.8%), and Hordeum vulgare Linn. (2.3%) sequences (Figure 2). The highest similarity observed was with Q. suber, a species with resistance to wind, drought, and barren environments (Pereira-Leal et al., 2014).
Table 2.
Summary of functional annotation of assembled unigenes in C. rhombifolium.
| Database | Number of unigenes annotated | Percentage (%) |
|---|---|---|
| Annotated in NR | 40,878 | 31.23 |
| Annotated in NT | 55,831 | 42.65 |
| Annotated in KO | 37,488 | 28.64 |
| Annotated in SwissProt | 74,084 | 56.59 |
| Annotated in PFAM | 56,213 | 42.94 |
| Annotated in GO | 56,213 | 42.94 |
| Annotated in KOG | 42,005 | 32.09 |
| Annotated in all databases | 7,286 | 5.56 |
| Annotated in at least one database | 97,496 | 74.48 |
| Total unigenes | 130,891 | 100 |
Figure 2.
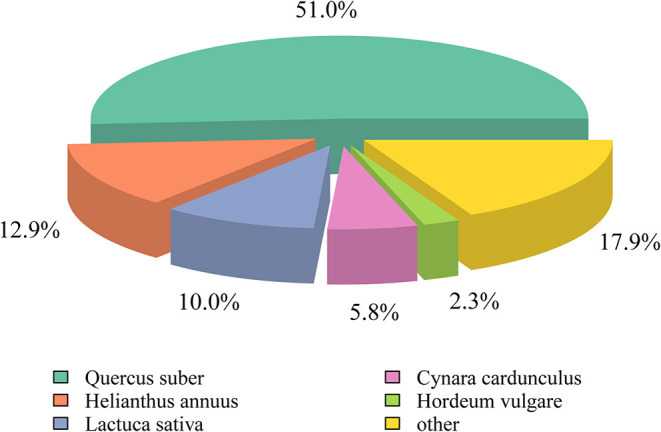
The percentage (%) of C. rhombifolium unigenes hits the species in the Nr database.
The COG analysis enabled the functional classification of 42,005 unigenes (Figure 3). The most frequently identified classes were “Translation, ribosomal structure and biogenesis” (7,147; 17%), followed by “Posttranslational modification, protein turnover, chaperones” (6,016; 14.3%), “Energy production and conversion” (4,830; 11.5 %), “General function prediction only” (4,706; 11.2%), “Amino acid transport and dynamics” (2407; 5.7%), “Intracellular trafficking, secretion, and vesicular transport (2218; 5.3%), “signal transduction” (2,145; 5.1%), “Lipid transport and metabolism” (2065, 4.9%) and “Carbohydrate transport and metabolism (2057; 4.9%). The least frequently identified groups were “Nuclear structure” (183; 0.4%), “Extracellular structures” (36; 0.09%), and “Cell motility” (24; 0.06%). Similar patterns have been reported in some angiosperms, such as Chrysanthemum nankingense (Wang et al., 2013) and Camelina sativa (Liang et al., 2013). We found 205 unigenes belonging to “Defense mechanism,” which indicated the existence of stress resistance genes in C. rhombifolium.
Figure 3.

COG classifications of unigenes in the transcriptome of C. rhombifolium unigenes.
Based on the Nr annotation, 42,005 unigenes were assigned to three ontologies and classified into 48 functional GO categories using the Blast2GO software. Of these, 1050 (68.5%), 163 (10.6%), and 320 (20.9%) GO terms were related to cellular components, biological processes, and molecular functions, respectively (Figure 4). The assignment of GO terms in C. rhombifolium in this study focused on “cellular processes,” “metabolic processes,” “single-organism processes,” “cell,” “cell parts,” “macromolecular complex,” “membrane part,” “organelles,” and “binding and catalytic activity,” which reflected the functional gene expression characteristics during its normal growth. This result was similar to those GO terms in some drought- resistance species, e.g., bread wheat, oak, Boea hygrometrica, Boea hygrometrica, and so on (Gupta et al., 2003; Durand et al., 2010; Xiao et al., 2015; Zhu et al., 2015), which mainly due to the selective gene expression caused by various environments and physiological states.
Figure 4.

Functional classification of C. rhombifolium unigenes based on Gene Ontology (GO) categorization. The results are summarized in three main GO categories: biological process (shown in red color), cellular component (shown in green color) and molecular function (shown in blue color). The x-axis indicates the subcategories, and the y-axis indicates the numbers related to the total number of GO terms present.
Based on the sequence homology searches against the KEGG database, 56,213 unigenes were assigned to five ontologies and classified into 18 functional KEGG pathways (Figure 5). Among these pathways, the “translation pathway” (6,912; 12.3% of KEGG unigenes) was the largest category in “metabolism” followed by “carbohydrate metabolism” (4,876), “overview” (4,496), “amino acid metabolites” (3,306), “folding, sorting, and degradation” (3,112), “energy metabolism” (2,663), “transport and catabolism” (2,297), and “lipid metabolism” (2,168). In this study, we highlight the pathways enriched for the interaction between plants and their environment, including: “metabolism of terpenoids and polyketides” (752), “signal transduction” (592), “environmental adaptation” (719), and “replication and repair” (396). Our results are consistent with those from other studies identifying plant genes and gene products with important roles in drought-resistant plants (Gechev et al., 2012; Xiao et al., 2015). More than 74% of the unigenes from C. rhombifolium were mapped in the known databases, which is higher than that reported for C. nankingense (64%) (Wang et al., 2013).
Figure 5.

Pathway assignment based on the Kyoto Encyclopedia of Genes and Genomes (KEGG). (A) Classification based on cellular process categories, (B) classification based on environmental information processing categories, (C) classification based on genetic information processing categories, (D) classification based on metabolism categories, and (E) classification based on organismal systems categories.
Frequency and Distribution of SSRs
In total, 12,925 SSR regions were identified in 10,524 unigenes. Among the identified SSRs, 128 different motifs were identified, the distribution and frequencies of which are shown in Figure 6. Mononucleotide motifs were the most abundant, and A/T were the largest subset (6,328). Overall, 6,429 mononucleotide, 2,463 di-repeats, 3,694 tri-repeats, 199 tetra-repeats, 56 penta-repeats, and 84 hexa-repeats were found in the C. rhombifolium leaf transcriptome. Among the unigenes containing SSRs, 941 SSRs presented compound formation, and 1,874 contained more than one SSR. On average, one SSR was found every 8.17 kb. The observed number of SSR sequences in our study was higher than EST-SSR ever reports in Chrysanthemum (Wang et al., 2013). The SSR sequences may gain or loss of repeats at a locus in their rapid evolution for adaptation to various environments (King et al., 1997; Trifonov, 2004). The mass EST-SSR loci in C. rhombifolium may be caused by its harsh habitats. These ESTs will provide a valuable repository of abundant information for future functional SSR studies.
Figure 6.

Simple sequence repeat (SSR) types present in the C. rhombifolium transcriptome.
Phylogenetic Analysis of GME
Using the annotated sequence of GME in C. rhombifolium and other GME homologs, we constructed a phylogenetic tree among related species. All of the GME sequences from the same taxa were clustered together and GME in C. rhombifolium were grouped into a single clade with the sequences of Helianthus annuus and other Asteraceae species (Figure 7), this result revealed a close relationship of C. rhombifolium and other Asteraceas species, which consistent with the taxonomy based on morphology.
Figure 7.

Phylogenetic analysis of the GME in plants. Helianthus annuus XM_022174384.1; Lactuca sativa (XM_023912155.1); Cynara cardunculus var. scolymus (XM_025140401.1), Chrysanthemum rhombifolium (R|Cluster-20886.25690); Camelina sativa (XM_010456910.1); Capsella rubella (XM_006287892.2); Nelumbo nucifera (XM_010275940.2); Morus notabilis (XM_024169380.1); Vitis riparia (XM_034830226.1); Vitis vinifera (XM_002283862.4); Ziziphus jujuba (XM_016019918.2); Gossypium arboretum (XM_017764784.1); Gossypium hirsutum (XM_016869694.1); Durio zibethinus (XM_022891507.1); PREDICTED: Herrania umbratica (XM_021427500.1); Theobroma cacao (XM_018122066.1); Pistacia vera (XM_031412928.1); Citrus sinensis (XM_006471610.2); Citrus unshiu (HQ224947.1).
Conclusions
We obtained 130,891 unigenes from the leaf of C. rhombifolium by NGS transcriptomics, of which 97,496 (~74.5%) unigenes were successfully annotated in the public protein database. A total of 12,925 SSRs were detected in 10,524 unigenes. This is the first genetic study of C. rhombifolium as a plant resource of stress-tolerant genes. These large numbers of transcriptome sequences have enabled us to comprehensively understand the basic genetics of C. rhombifolium and discover novel genes that will be helpful in the molecular improvement of chrysanthemums.
Data Availability Statement
The datasets presented in this study can be found in GenBank. The accession numbers can be found in the article.
Author Contributions
YM conceived and designed the experiments. LZ collected the plants. WZ, HX, XD, JL, and JH performed the experiments and analyzed the data. YM, WZ, and LZ wrote the paper. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This project was supported by the National Nature Science Foundation of China (Nos. 31470699, 31872710, and 31770200).
References
- Angeloni F., Wagemaker C. A. M., Jetten M. S. M., Camp H. J. M. O. D., Janssen-Megens E. M., Francoijs K. J., et al. (2011). De novo transcriptome characterization and development of genomic tools for Scabiosa columbaria L. using next generation sequencing techniques. Mol. Ecol. Resour. 11, 662–674 10.1111/j.1755-0998.2011.02990.x [DOI] [PubMed] [Google Scholar]
- Bremer K., Humphries C. J. (1993). Generic monograph of the Asteraceae-Anthemideae. Bull. Nat. Hist. Mus. Lond. 23, 71–177. [Google Scholar]
- Chen L., Chen Y., Jiang J., Chen S., Chen F., Guan Z., et al. (2012). The constitutive expression of Chrysanthemum dichrum ICE1 in Chrysanthemum grandiflorum improves the level of low temperature, salinity and drought tolerance. Plant Cell Rep. 31, 1747–1758. 10.1007/s00299-012-1288-y [DOI] [PubMed] [Google Scholar]
- Chen S., Cui X., Chen Y., Gu C., Miao H., Gao H., et al. (2011). CgDREBa transgenic chrysanthemum confers drought and salinity tolerance. Environ. Exp. Bot. 74, 255–260 10.1016/j.envexpbot.2011.06.007 [DOI] [Google Scholar]
- Conesa A., Gotz S., Garcia-Gomez J. M., Terol J., Talon M., Robles M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 10.1093/bioinformatics/bti610 [DOI] [PubMed] [Google Scholar]
- Da Silva J. A. T. (2003). Chrysanthemum: advances in tissue culture, cryopreservation, postharvest technology, genetics and transgenic biotechnology. Biotechnol. Adv. 21, 715–766. 10.1016/S0734-9750(03)00117-4 [DOI] [PubMed] [Google Scholar]
- Durand J., Bodénès C., Chancerel E., Frigerio J. M., Vendramin G., Sebastiani F., et al. (2010). A fast and cost-effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genomics 11:570. 10.1186/1471-2164-11-570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gechev T. S., Dinakar C., Benina M., Toneva V., Bartels D. (2012). Molecular mechanisms of desiccation tolerance in resurrection plants. Cell. Mol. Life Sci. 69, 3175–3186. 10.1007/s00018-012-1088-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P. K., Rustgi S., Sharma S., Singh R., Kumar N., Balyan H. S. (2003). Transferable ESTSSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genomics 270, 315–323 10.1007/s00438-003-0921-4 [DOI] [PubMed] [Google Scholar]
- Jain M. (2011). Next-generation sequencing technologies for gene expression profiling in plants. Brief. Funct. Genomics. 11, 63–70. 10.1093/bfgp/elr038 [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. 10.1093/nar/gkm882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. G., Soller M., Kashi Y. (1997). Evolutionary tuning knobs. Endeavour 21, 36–40. 10.1016/S0160-9327(97)01005-3 [DOI] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Wan Q., Guo Y. P., Abbott R. J., Rao G. Y. (2014). Should I stay or should I go: biogeographic and evolutionary history of a polyploid complex (Chrysanthemum indicum complex) in response to Pleistocene climate change in China. New Phytol. 201, 1031–1044. 10.1111/nph.12585 [DOI] [PubMed] [Google Scholar]
- Li J., Wan Q., Richard J., Abbott R. J., Rao G. Y. (2013). Geographical distribution of cytotypes in the Chrysanthemum indicum complex as evidenced by ploidy level and genome-size variation. J. Syst. Evol. 51, 196–204. 10.1111/j.1674-4918.2012.00241.x [DOI] [Google Scholar]
- Liang C., Liu X., Yiu S. M., Lim B. L. (2013). De novo assembly and characterization of Camelina sativa transcriptome by paired-end sequencing. BMC Genomics 14:146. 10.1186/1471-2164-14-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Zhang Q. X., Mo G. Z., Chen X. J. (2010). Collection and analysis of drought-resistant wild resources. Heilongjiang Agric. Sci. 7, 84–89. 10.3969/j.issn.1002-2767.2010.07.028 [DOI] [Google Scholar]
- Ma Y. P., Zhao L., Zhang W. J., Zhang Y. H., Xing X., Duan X.-X., et al. (2020). Origins of cultivars of Chrysanthemum - evidence from the chloroplast genome and nuclear LFY gene. J. Syst. Evol. 58, 925–944. 10.1111/jse.12682 [DOI] [Google Scholar]
- Masuda Y., Yukawa T., Kondo K. (2009). Molecular phylogenetic analysis of members of Chrysanthemum and its related genera in the tribe Anthemideae, the Asteraceae in East Asia on the basis of the internal transcribed spacer (ITS) region and the external transcribed spacer (ETS) region of nrDNA. Chromosome Bot. 4, 25–36. 10.3199/iscb.4.25 [DOI] [Google Scholar]
- Pereira-Leal J. B., Abreu I. A., Alabaça C. S., Almeida M. H., Ricardo C. P. P. (2014). A comprehensive assessment of the transcriptome of cork oak (Quercus suber) through EST sequencing. BMC Genomics 15:371. 10.1186/1471-2164-15-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G., Huang X., Liang F., Antonescu V., Sultana R., Karamycheva S., et al. (2003). TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 10.1093/bioinformatics/btg034 [DOI] [PubMed] [Google Scholar]
- Shih C., Fu G. X. (1983). Flora Reipublicae Popularis Sinicae 76. Beijing: Science Press. [Google Scholar]
- Song A., An J., Guan Z., Jiang J., Chen F., Lou W., et al. (2014). The constitutive expression of a two transgene construct enhances the abiotic stress tolerance of chrysanthemum. Plant Physiol. Biochem. 80, 114–120. 10.1016/j.plaphy.2014.03.030 [DOI] [PubMed] [Google Scholar]
- Song C., Liu Y., Song A., Dong G., Zhao H., Sun W., et al. (2018). The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant. 11, 1482–1491. 10.1016/j.molp.2018.10.003 [DOI] [PubMed] [Google Scholar]
- Su J., Jiang J., Zhang F., Liu Y., Chen F. (2019). Current achievements and future prospects in the genetic breeding of chrysanthemum: a review. Horticulture Res. 6, 109–128 10.1038/s41438-019-0193-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J. J., Wu H., Li Z. Y., Huang C. H., Xu X. B. (2018). Molecular evolution of GDP-D-Mannose Epimerase (GME), a key gene in plant ascorbic acid biosynthesis. Front. Plant Sci. 9:1293. 10.3389/fpls.2018.01293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonov E. N. (2004). The tuning function of tandemly repeating sequences: a molecular device for fast adaptation, in Evolutionary Theory and Processes: Modern Horizons (Kluwer Academic Publishers; Springer; ), 115–138. [Google Scholar]
- Wang H., Jiang J., Chen S., Qi X., Peng H., Li P., et al. (2013). Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 8:e62293. 10.1371/journal.pone.0062293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Huang H., Ma Y. P., Fu J. X., Wang L. L., Dai S. L. (2014). Construction and de novo characterization of a transcriptome of Chrysanthemum lavandulifolium: analysis of gene expression patterns in floral bud emergence. Plant Cell Tissue Organ Culture 116, 297–309 10.1007/s11240-013-0404-1 [DOI] [Google Scholar]
- Wang Y., Liu K., Bi D., Zhou S. B., Shao J. W. (2017). Characterization of the transcriptome and EST-SSR development in Boea clarkeana, a desiccation-tolerant plant endemic to China. Peer J. 5:e3422. 10.7717/peerj.3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q. J., Chen Z. D., Sun W. J., Deng T. T., Chen M. J. (2016). De novo Sequencing of the leaf transcriptome reveals complex light-responsive regulatory Networks in Camellia sinensis cv. Baijiguan. Front Plant Sci. 7:332. 10.3389/fpls.2016.00332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L. H., Yang G., Zhang L. C., Yang X. H., Zhao S., et al. (2015). The resurrection genome of Boea hygrometrica: a blueprint for survival of dehydration. Proc. Natl. Acad. Sci. U.S.A. 112, 5833–5837 10.1073/pnas.1505811112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Fang L., Zheng H., Zhang Y., Chen J., Zhang Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34(Suppl. 2), W293–W297. 10.1093/nar/gkl031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalapa J. E., Cuevas H., Zhu H. Y., Steffan S., Senalik D., Zeldin E., et al. (2012). Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in plant sciences. Am. J. Bot. 99, 193–208. 10.3732/ajb.1100394 [DOI] [PubMed] [Google Scholar]
- Zhao H. B., Chen F. D., Chen S. M., Wu G. S., Guo W. M. (2010). Molecular phylogeny of Chrysanthemum, Ajania and its allies (Anthemideae, Asteraceae) as inferred from nuclear ribosomal ITS and chloroplast trnL-F IGS sequences. Plant Syst. Evol. 284, 153–169. 10.1007/s00606-009-0242-0 [DOI] [Google Scholar]
- Zhao H. E., Liu Z. H., Hu X., Yin J. L., Li W., Rao G. Y., et al. (2009). Chrysanthemum genetic resources and related genera of Chrysanthemum collected in China. Genet Resour Crop Ev. 56, 937–946. 10.1007/s10722-009-9412-8 [DOI] [Google Scholar]
- Zhu Y., Wang B., Phillips J., Zhang Z. N., Du H., Xu T., et al. (2015). Global transcriptome analysis reveals acclimation-primed processes was involved in the acquisition of desiccation tolerance in Boea hygrometrica. Plant Cell Physiol. 56, 1429–1441. 10.1093/pcp/pcv059 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets presented in this study can be found in GenBank. The accession numbers can be found in the article.