

Abstract
ABC transporters form a superfamily of integral membrane proteins involved in translocation of substrates across the membrane driven by ATP hydrolysis. Despite available crystal structures and extensive biochemical data, many open questions regarding their transport mechanisms remain. Therefore, there is a need to explore spectroscopic techniques such as solid state NMR in order to bridge the gap between structural and mechanistic data. In this study, we investigate the feasibility of using E. coli MsbA as a model ABC transporter for solid state NMR studies. We show that optimised solubilisation and reconstitution procedures enable preparing stable and homogenous protein samples. Depending on the solubilisation times, MsbA can be obtained in either an apo- or in a native lipid-A bound form. Building onto these optimizations, the first promising MAS-NMR spectra with narrow lines have been recorded. However, further sensitivity improvements are required so that complex NMR experiments can be recorded within a reasonable amount of time. We therefore demonstrate the usability of paramagnetic doping for rapid data acquisition and explore dynamic nuclear polarisation as a method for general signal enhancement. Our results demonstrate that solid state NMR provides an opportunity to address important biological questions related to complex mechanisms of ABC transporters.
Keywords: ABC transporter, DNP, Lipid A, MAS-NMR, MsbA, PRE
Introduction
ABC (ATP Binding Cassette) transporters are a superfamily of membrane proteins that use ATP as a source of energy to translocate a variety of substrates across the cell membrane. Mammalian transporters such as P-glycoprotein are of great physiological importance and belong generally to the exporters subclass (George & Jones, 2012; Higgins & Linton, 2004). In prokaryotes, ABC transporters act as nutrient importers, -drug exporters and lipid flippases. All ABC transporters show a similar modular topology with two highly conserved nucleotide binding domains (NBDs) for ATP hydrolysis and two transmembrane domains (TMDs), which are specialised towards binding and translocating a range of substrates. The nucleotide binding domain consists of the canonical signature motif LSGGQ, also known as the C-loop, and the conserved Walker A and B domains along with the D-, Q- and H- loops (Jones & George, 2013). These modules are either fused into one single polypeptide chain or assemble separately into homo- or heterodimers (Higgins & Linton, 2004).
The apparently similar architecture of ABC transporters implies a unified molecular transport mechanism. There are however contradicting data questioning this assumption at least for exporters. Nucleotide-bound outward facing structures have both NBDs in close contact as found for MsbA and Sav1866 (Dawson & Locher, 2006; Ward et al, 2007), which is in contrast to the apo-state structures of MsbA and PgP (Aller et al, 2009; Ward et al, 2007) in which a V-shape with well separated NBDs is observed. The latter contradicts biochemical studies from which models were derived in which the NBDs are permanently in contact with each other (George & Jones, 2012). Interestingly, a recent pulsed EPR study on LmrA, a homologue to MsbA from L. lactis, revealed that the apo state covers a large conformational space, which is restricted upon nucleotide binding (Hellmich et al, 2012b). Such an apo state is not likely to be populated in the cell as the ATP concentration is significantly higher than the Km of ATP hydrolysis, but particular sample preparation or crystallisation conditions might favour certain apo state subpopulations.
So far, progress in ABC transporter research has relied on X-ray crystallography combined with biochemical approaches. Furthermore, pulsed EPR spectroscopy, which provides distance measurements via electron dipole-dipole couplings, has become very important for identifying domain movements during the catalytic and transport cycles (Buchaklian & Klug, 2005; Dong et al, 2005; Mishra et al, 2014). In addition, first applications of single molecule methods for imaging as well as for spectroscopy have been reported (Cooper & Altenberg, 2013; Kim et al, 2015). However, compared to other families of membrane proteins, many spectroscopic techniques remain underrepresented although unique mechanistic data could be obtained to complement crystallographic approaches. Especially NMR spectroscopy could provide site-resolved data at atomic resolution, which would be helpful in understanding details of ATP hydrolysis, substrate binding and release mechanism, NBD-TMD cross talk and general structural dynamics.
In general, NMR spectroscopy on membrane proteins is challenging. Membrane-mimicking environments suitable for different NMR approaches are illustrated in Figure 1. For liquid-state NMR, the chosen protein/detergent complex has to be small enough to ensure fast isotropic tumbling while preserving at the same time functional integrity. One possible disadvantage is the high curvature of detergent micelles creating an unnatural environment for membrane proteins. A potential solution is offered by the use of isotropic bicelles or nanodiscs, which are larger than micelles, but still soluble. All of these three systems have been used successfully for liquid-state NMR (Raschle et al, 2010), but proteins of the size of full length ABC transporters pose a general problem due to their molecular weight.
Figure 1:
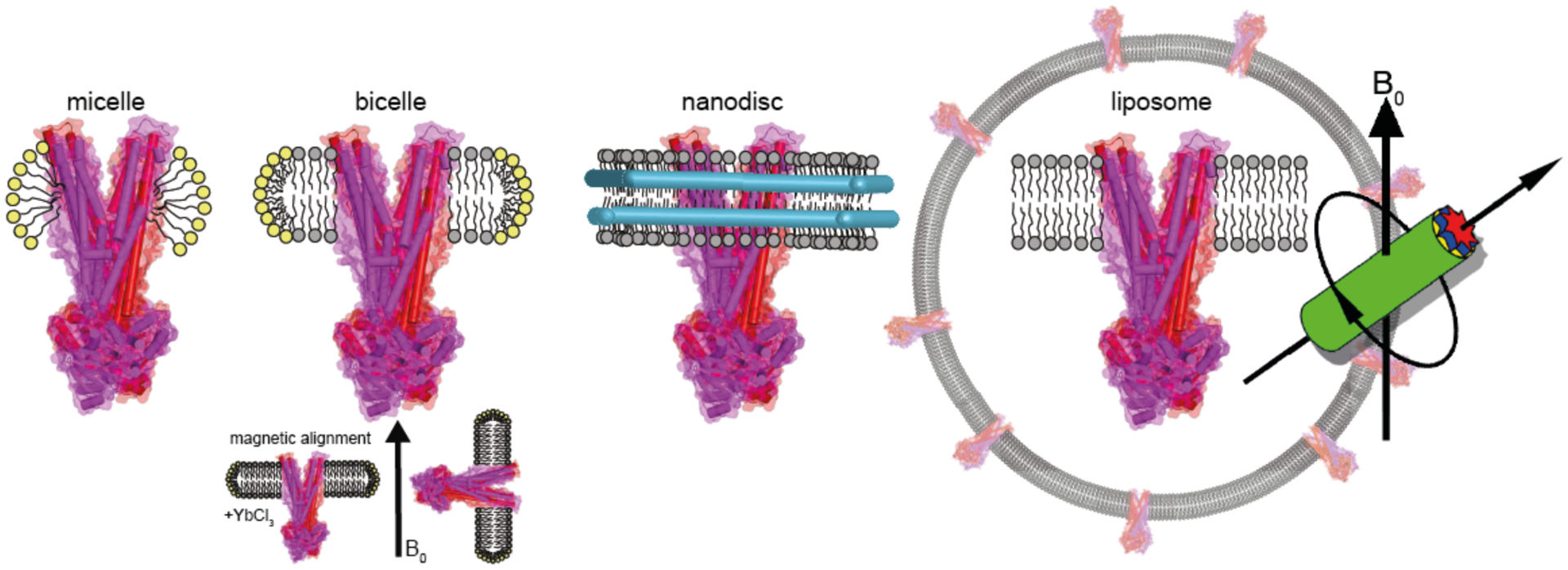
Membrane mimicking environments for investigating membrane proteins by NMR spectroscopy. Micelles, bicelles and nanodiscs are used to keep the protein soluble, which would make them suitable to solution state NMR but their total molecular weight when incorporating proteins such as ABC transporters limits this approach. Oriented solid state NMR exploits the spontaneous alignment of bicelles in magnetic fields. The most universal approach is based on MAS-NMR, which enables experiments directly on proteoliposomes as demonstrated in this paper for the ABC exporter MsbA.
Solid state NMR does not suffer from line broadening due to increased molecular weight and allows working under even more native-like conditions using lipid bilayers. Oriented solid state NMR (O-SSNMR) relies on macroscopically ordered samples, which are either prepared on glass plates as solid support or more elegantly by utilising the spontaneous alignment of anisotropic bicelles within the magnetic field, which can be controlled by the addition of lanthanides (De Angelis & Opella, 2007; Prosser et al, 1996) (Figure 1). However, most applications rely on magic angle sample spinning for which proteoliposomes can be used directly. Fast sample rotation about an axis inclined to the magnetic field at 54.7° (the “magic angle”), averages all anisotropic interactions leading to well-resolved spectra (Figure 1). This approach has been extensively used for hypothesis-driven studies or for obtaining 3D structure models of membrane embedded proteins (Ader et al, 2009; Mao et al, 2014; Park et al, 2012; Wang et al, 2013).
Due to their size and often challenging biochemical properties, ABC transporters are especially difficult targets for NMR spectroscopy. Only a few initial studies on full transporters, all of them based on solid state NMR, have been published so far. For L. lactis LmrA, reconstitution conditions, time-resolved 31P - magic angle sample spinning (MAS) NMR (Hellmich et al, 2008) and wideline 2H-solid state NMR (Siarheyeva et al, 2007) have been reported. The studies were later complemented by EPR spectroscopy (Hellmich et al, 2012b) and by solution state NMR on the isolated NBDs (Hellmich et al, 2012a). Optimized reconstitution conditions and first MAS-NMR spectra were also reported for BmrA from B. subtilis (Kunert et al, 2014). The first solid state NMR experiments on an ABC importer were described for ArtMP in 2D-crystals (Akbey et al, 2014; Lange et al, 2010).
Here, we explore MsbA as a potential target for further solid state NMR studies. As mentioned above, MsbA has been in the focus of a number of studies, but additional spectroscopic data can be especially useful towards completing the mechanistic picture. MsbA is a 584 amino acid E. coli ABC exporter, functional as a homodimer of 130 kDa. Absence of MsbA in E. coli leads to cell death, which can partly be attributed to the toxic nature of lipid A accumulating inside the cell, and partly because of reduced integrity of the outer E. coli membrane, which consist of lipopolysaccharide (LPS) with lipid A as the core moiety (Zhou et al, 1998).
An up to 4 fold in-vitro stimulation of ATPase activity of MsbA in the presence of 3-deoxy-D-manno-2-octulosonic-lipid A has been shown (Doerrler & Raetz, 2002). In addition, transport or stimulated ATPase activity has been demonstrated for many other substrates such as Hoechst 33342, daunomycin, daunorubicin, erythromycin, vinblastine and ethidium (Eckford & Sharom, 2008c; Eckford & Sharom, 2010; Siarheyeva & Sharom, 2009).
The Xray crystallographic data of MsbA from E. coli (pdb id: 3B5W; 5.3Å), S. typhimurium (pdb id: 3B5X; 5.5Å) and V. cholerae (pdb id: 3B5Y, 3B6O; 3.7Å) have shown an inward facing V-shaped structure with separated NBDs in the apo-state and an outward facing conformation upon nucleotide binding (Ward et al, 2007). It has also been observed by Cryo EM studies that MsbA in complex with different nucleotides (AMP.PNP, ADP-Vi, ADP-AlFx) forms a unique lattice under same crystallization conditions (Ward et al, 2009). Hints towards association and dissociation of NBDs by ATP have also been provided by cross linking studies (Doshi et al, 2010) and an intermediate closed state was suggested (Doshi et al, 2013). MsbA was also subject to rather extensive and detailed cw- and pulsed EPR studies, which has not only provided evidence for conformational changes but has also given a first impression of the extent of conformational dynamics within defined catalytic states of MsbA. Large amplitude domain movements between an open apo into the closed nucleotide-bound state have been reported in both micelles and proteoliposomes. It was demonstrated that lipid A binds at the surface of helix 6 and global conformational changes were observed for adenosine-5′-(β,γ-imido)triphosphate (AMP.PNP) binding and for the ADP.Vi bound high energy state (Borbat et al, 2007; Mishra et al, 2014; Smriti et al, 2009).
Further in this paper, we present first solid state NMR data on MsbA, which could be obtained through optimisation of key steps during sample preparation. Special emphasis has been given to the reconstitution step, which is crucial for obtaining a homogenous stable preparation and for keeping the membrane proteins stable in vitro (Geertsma et al, 2008; Rigaud et al, 1995; Rigaud & Levy, 2003; Sanders & Landis, 1995). The small sample volume used for MAS NMR (10 – 40 μL) presents an additional constraint, which requires a small lipid to protein ratio. We also evaluate concepts to improve sensitivity based on Gd3+-doping with rapid data acquisition as well as dynamic nuclear polarisation. Finally, first data of lipid A in complex with MsbA will be presented and discussed.
Results and Discussion
(a). Preparing MsbA for solid state NMR
In order to prepare isotope-labelled samples, MsbA was overexpressed in M9 minimal medium resulting in a reproducible yield of approximately 10 mg per litre of culture. The yield even increased to up to 35 mg when amino acids were added to the medium for reverse labelling. It was found that MsbA in complex with lipid A could be purified from E. coli membranes solubilised with 1.25% n-Dodecyl β-D-Maltopyranoside (DDM) for 1 h, while a duration of 12 h resulted in apo-state MsbA (see below). Sample homogeneity and stability were verified by size exclusion chromatography (0.015% DDM) directly after purification and after storage at room temperature for 24 h (Figure 2a). Purity of the preparation was determined by SDS-PAGE (Figure 2b) and confirmed by Matrix Assisted Laser Desorption Ionisation Mass Spectrometry (MALDI-MS) (Figure 2e). The MALDI spectrum clearly shows single, double and triple charged peaks of the MsbA monomer corresponding to its molecular weight including the His-tag with its peptide linker. A western blot of the preparation using Anti-His AP-conjugate was run for confirmation of sample purity (Figure 2d). Long-term stability was further checked by SDS PAGE on MsbA reconstituted in 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) / 1,2-dimyristoyl-sn-glycero-3-phosphate (DMPA) (9:1) (see below for choice of lipid mixture) stored at room temperature. No degradation was observed for up to 30 days (Figure 2c).
Figure 2:
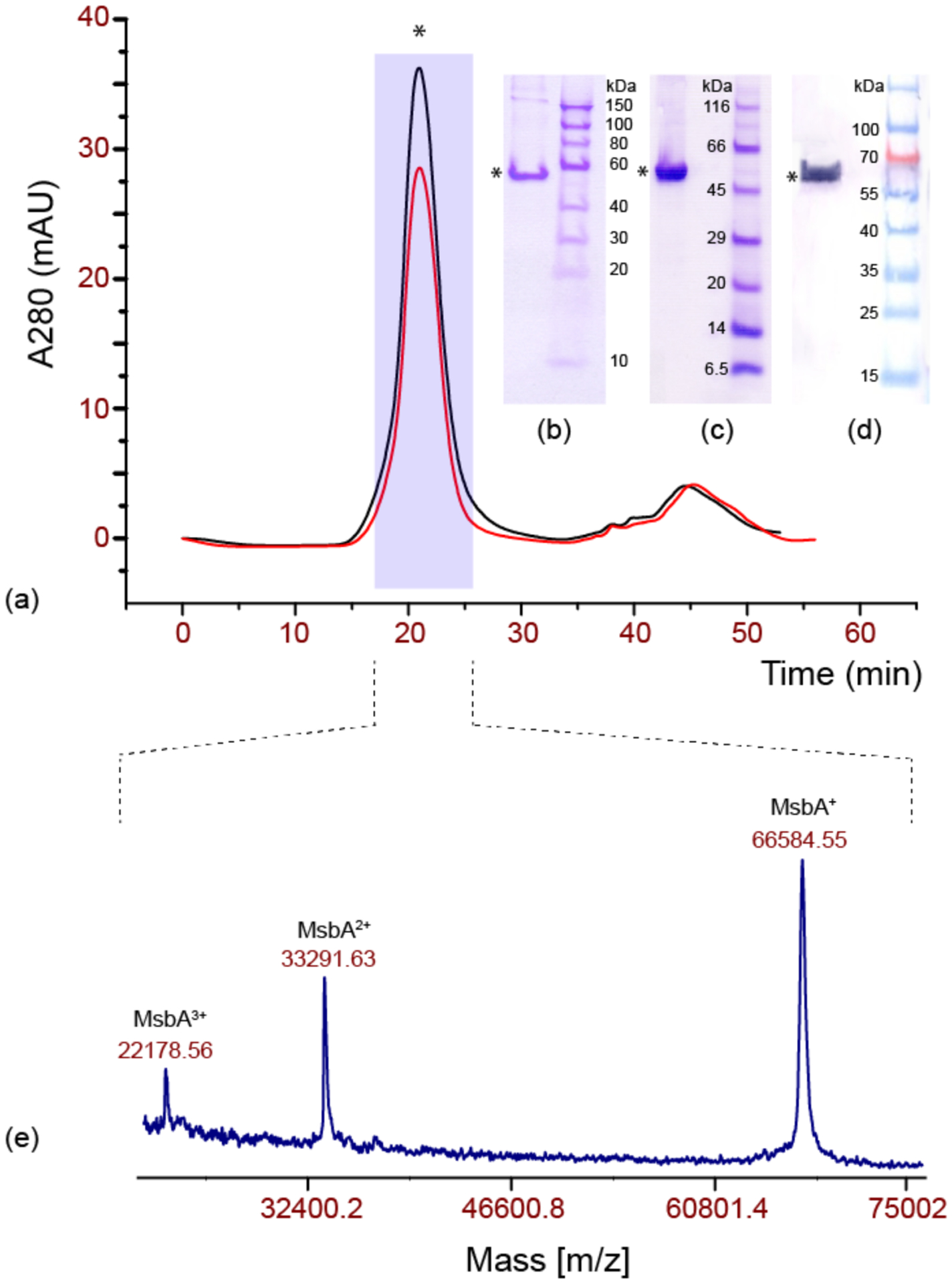
Purification and stability of MsbA. (a) Size exclusion chromatogram of MsbA in DDM, freshly eluted (black) and after 24 h at room temperature (red). The intensity differences are due the different amounts of protein used. (b) SDS PAGE of MsbA in DDM after NiNTA purification. (c) SDS PAGE of MsbA reconstituted in DMPC:DMPA (9:1) mol/mol) at LPR = 75 mol/mol) after storage for 30 days at room temperature. (d) Western blot analysis of MsbA using an anti His-conjugate antibody. (e) MALDI mass spectra of MsbA in detergent.
The reconstitution of MsbA into liposomes has been tested for different lipids and for different reconstitution conditions. Unilamellar vesicles were prepared by extrusion followed by detergent destabilisation to aid homogeneous insertion of MsbA. As detergent is added, liposomes start swelling resulting in a change of turbidity of the liposomal solution, which can be followed by monitoring the optical density at 540 nm as shown in Figure 3a for DMPC/DMPA (9:1). The liposomes are fully saturated at the detergent concentration RSAT but dissolve completely at RSOL resulting in a clear solution. It has been described that homogeneity and reconstitution efficiency of membrane proteins at RSAT is low but improves at a slightly higher concentration further referred to as ROPT (Geertsma et al, 2008; Rigaud & Levy, 2003). In order to find ROPT, we have reconstituted MsbA into DMPC/DMPA liposomes destabilised by increasing DDM concentrations in the range between RSAT and RSOL. Detergent was completely removed by Bio-Beads SM-2 and the resulting proteoliposomes were analysed with respect to the amount of incorporated MsbA and with respect to ATPase activity dependent on homogeneity of incorporation. The degree of MsbA insertion, as determined using a Lowry assay after re-solubilising the proteoliposomes, was found to be almost complete and independent of the level of liposome destabilisation. In contrast, the ATPase activity of MsbA proteoliposomes showed a strong dependence on the DDM concentration, which means that the homogeneity of MsbA orientation, or in other words the NBD accessibility, varies. The highest activity implies most uniform orientation and is observed for ROPT = 3 mM (Figure 3a). This reconstitution procedure allowed preparing samples with a molar lipid-to-protein ratio (LPR) of approx. 50. Furthermore, a clear effect on sample homogeneity due to the size of the unilamellar vesicles, which depends on the pore diameter of the extrusion membranes, was seen (Figure 3b). The most homogeneous samples were obtained for vesicles with an average diameter of 0.1 μm.
Figure 3.
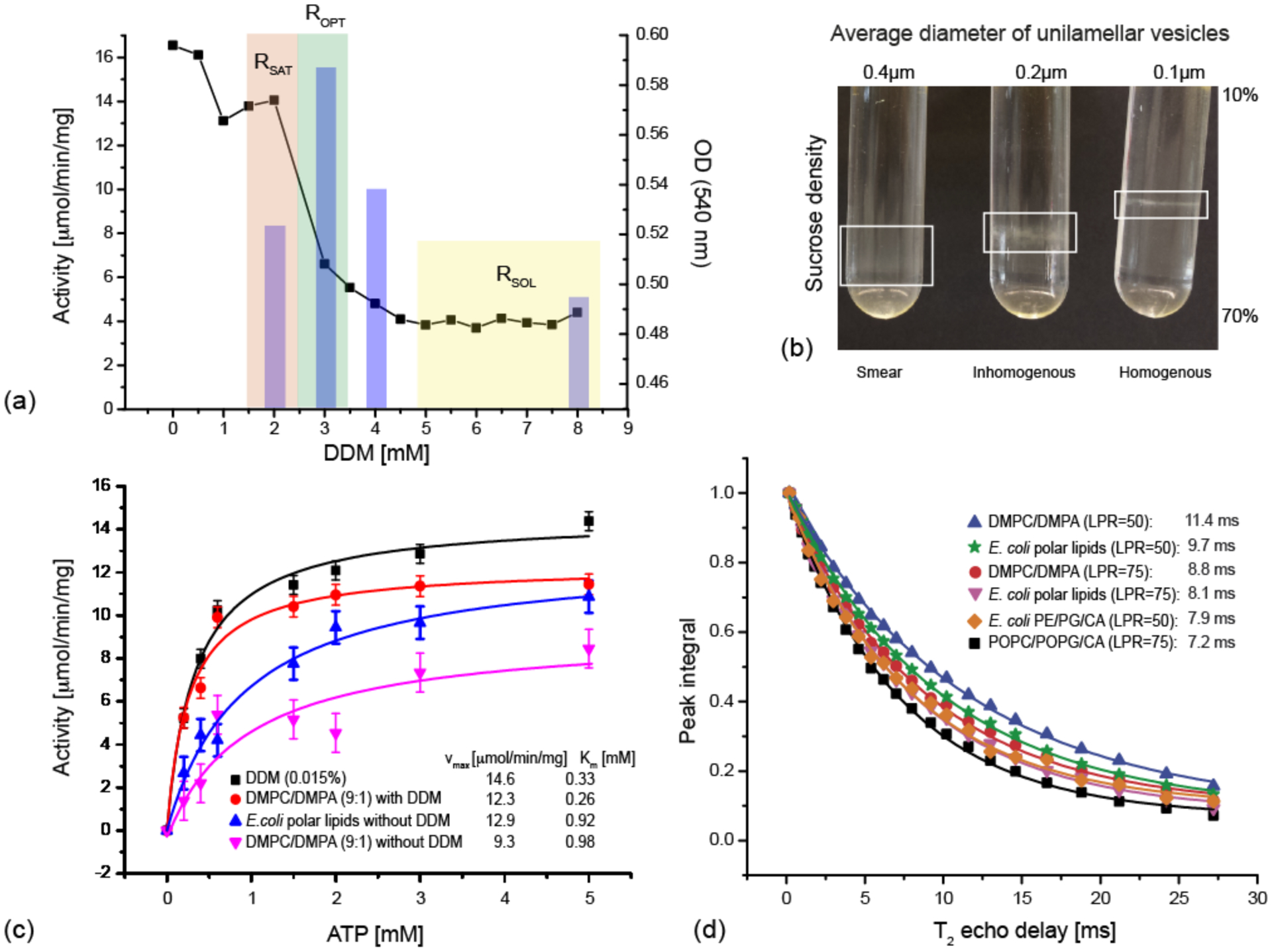
Reconstitution of MsbA. (a) Optimised DMPC/DMPA (9:1) liposome destabilisation for homogenous MsbA proteoliposome preparation. Destabilisation was followed by changes in optical density at 540 nm (black squares). Liposomes are fully saturated at RSAT and fully solubilised at RSOL. The ATPase activity determined from proteoliposomes after detergent removal (bars) shows a maximum at ROPT. Since the amount of MsbA within the proteoliposomes is the same for all destabilisation conditions starting from ROPT, ATPase activity reports on NBD accessibility / homogeneity of MsbA orientation. ATPase activities were determined at an ATP concentration of 6mM. (b) Effect of extrusion of lipids through membranes of different pore size used to prepare unilamellar vesicles (MsbA in DMPC/DMPA (9:1), LPR=50 mol/mol) on sample homogeneity as seen by sucrose density gradients (10–70% w/v). (c) Characterisation of basal ATPase activity using the colorimetric molybdate blue assay for MsbA in 0.015% DDM (black), reconstituted in destabilised DMPC/DMPA (9:1) (red), reconstituted in E. coli polar lipids (blue), reconstituted in DMPC/DMPA (9:1) (magenta). A LPR of 75 mol/mol was used in all cases. (d) Determination of the 15N transversal relaxation time T2’ of [U-15N]-MsbA reconstituted into different lipid mixtures. The largest value is obtained for DMPC/DMPA (9:1) with a LPR of 50 mol/mol, which corresponds to the smallest homogeneous 15N linewidth (27 Hz) indicating good spectral resolution.
The ATPase activity in different proteoliposomes is compared to detergent solubilised MsbA in Figure 3c. Very similar values were observed. The slightly higher activity in DDM could be caused by some inaccessible NBDs in proteoliposomes. The activity in E. coli polar lipids and in DMPC/DMPA is almost identical and their values are consistent with previously reported data (Zou & McHaourab, 2009).
Our data show that active and homogeneous MsbA proteoliposomes with a lipid to protein ratio (LPR) compatible with the requirements of MAS-NMR can be prepared. One of the most important criteria for NMR spectroscopy is spectral resolution. Therefore, [U-15N]-MsbA has been reconstituted into proteoliposomes containing different lipid compositions (see Figure 3d). We have carried out 15N spin-echo experiments to determine the 15N transverse relaxation time T2’ (De Paepe et al, 2003). Spin echo decays are shown in Figure 3d. Their values clearly depend on LPR and/or on the lipid composition and are found between 7.2 ms and 11.4 ms corresponding to homogeneous linewidths of 45 and 27 Hz. MsbA in DMPC/DMPA (9.1) at an LPR of 50 in mol/mol has been used for all further experiments as it shows the longest T2’ and also the same activity as in E. coli polar lipids.
(b). 2D-MAS NMR spectra on MsbA containing proteoliposomes
Based on these optimised conditions, uniform- and extensive selective labelled samples were prepared. Temperature dependent 13C - cross polarisation (CP) - MAS NMR spectra were recorded to probe the effect of the lipid and water phase on resolution and signal intensity (Figure 4a). The overall signal intensity is almost the same in frozen samples (255 K) compared to the lipid gel phase (273 K), but resolution improves in the non-frozen case. In the liquid-crystalline phase at 313 K, CP signal intensities decreased compared to 273 K due to increased mobility on the intermediate timescale (μs) interfering with cross polarisation and proton decoupling. On the other hand, the directly measured averaged C-H dipole couplings in MsbA are close to the rigid limit of 21.5 kHz (order parameter SCH=0.9) and are not significantly reduced when comparing all three cases (Figure 4c). This means that fast and large amplitude motions do not play a major role, at least not for the residues, which can be cross-polarised. The temperature dependence of all resonances as seen by direct polarisation is shown in Figure 4b. In principle, the same trend as for cross polarisation is observed. Usually, highly mobile region undergoing fast, large-amplitude fluctuations will be suppressed in CP experiments, but become visible under direct polarisation. As this is not a dominating phenomenon here, MsbA must be relatively rigid and well structured.
Figure 4.
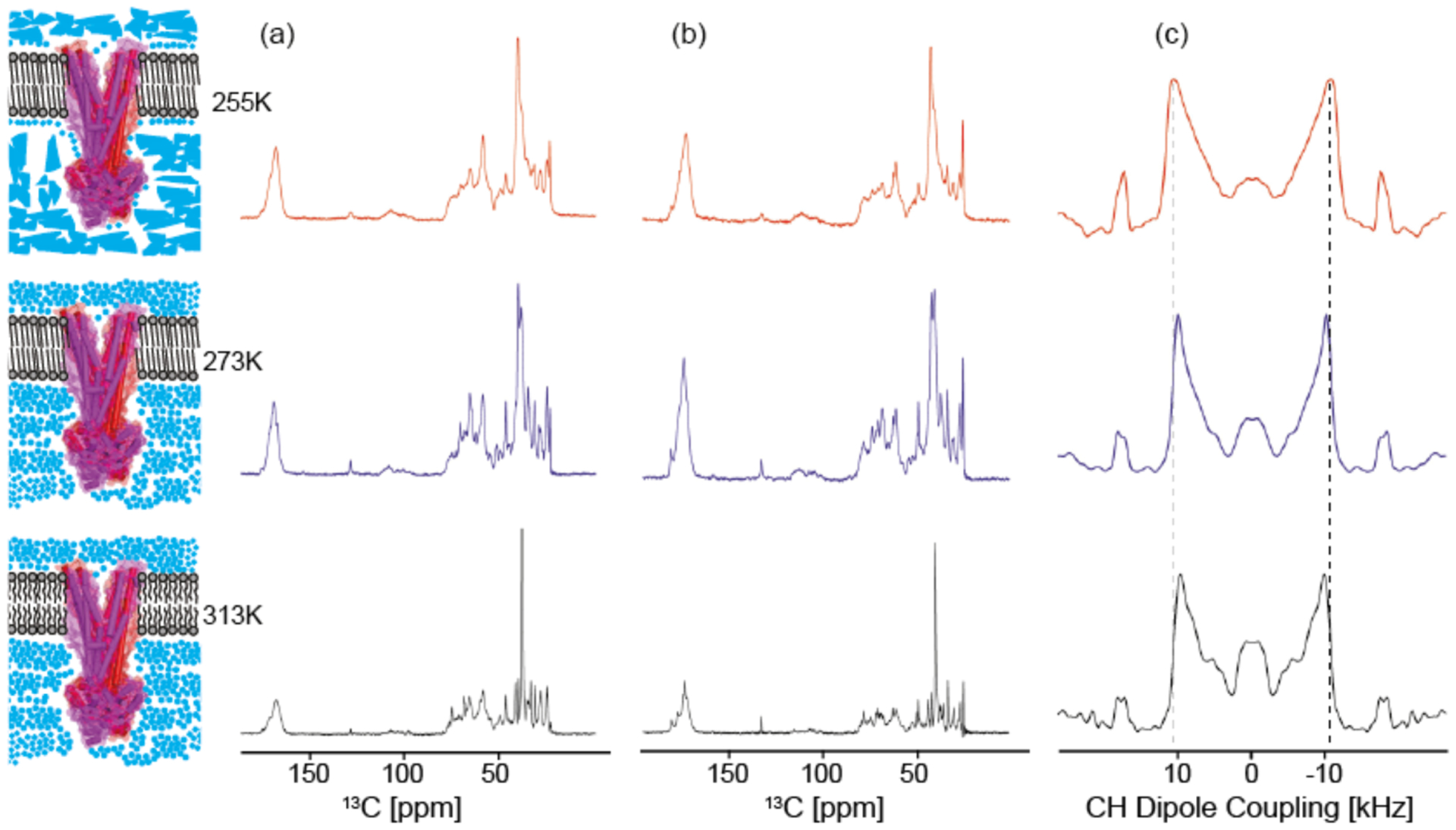
(a) 13C-CP spectra, (b) directly polarised (DP) spectra and (c) C-H dipolar spectra of MsbA in DMPC/DMPA in the gel phase and below the freezing point of water (255 K), in the gel phase above the freezing point of water (273 K) and in the liquid-crystalline phase (313 K). Resolution in both (a) and (b) is better compared to frozen samples. Sensitivity slightly decreases with increasing temperature, but there is no direct dependence on the lipid phase transition. DP spectra visualize all regions of the protein while CP filters mainly regions, which are less flexible. This is also seen in (c), which displays the average C-H dipole coupling in MsbA from cross-polarised segments. The splittings are close to the rigid limit of 21.5 kHz indicating that these parts of the protein are not involved in large amplitude fluctuations.
Two-dimensional 13C-13C Proton Driven Spin Diffusion (PDSD) spectra were recorded to assess spectral resolution (Figure 5a). In order to avoid spectral overcrowding in this initial analysis due to the large size of the MsbA dimer, we have used extensive reverse labelling in order to simplify our spectra. In the resulting protein, only eight types of amino acids (D, E, Q, G, H, K, T, S) are 13C- and 15N labelled corresponding to 35% of all residues. In order to minimize isotope scrambling, the amino acids used for reverse labelling were carefully chosen. This choice was based on the work of Bellstedt and co-workers who performed individual amino acid-selective unlabeling experiments in E. coli BL21(DE3) (Bellstedt et al, 2013). Their data indicate residual carbon scrambling for Ala but not for Arg, Cys, Ile, Leu, Met, Phe, Pro, Tyr and Val. Indeed, unwanted signals in the Ala Cα-Cβ region of our PDSD spectrum are observed (Figure 5a). Overall, good spectral dispersion is obtained and resolution and sensitivity is good enough to identify individual cross peaks with linewidth between 0.5 and 0.7 ppm (Figs. 5b and c). These values are slightly larger than the homogenous linewidth (Figure 5b), which is due to 13C-13C and 13C-15N J-couplings as well as due to remaining sample heterogeneities. The resolution of individual cross-peaks is further illustrated for an MsbA sample in which only lysines are 15N and 13C labelled. Based on the secondary structure dependent chemical shift index (Wang & Jardetzky, 2002), the NCA spectrum of [13C-15N-K]-MsbA can be divided into regions originating from α-helices, β-sheets and random coils (Figure 5c). MsbA contains 22 lysines (plus one in the linker between His-tag and protein). Based on the crystal structure, 15 lysines are located in the N-terminal TMDs (9 x α-helix, 6 x turns/loops/end-residues of helices) and 7 are found in the C-terminal NBDs (4x α-helix, 1x β-sheet, 2 x turns/loops/end-residues of helices) including K382 in the Walker A motif. Therefore, the β-sheet resonances in Figure 5c can be mainly assigned to the β-sheet regions found in the NBDs around residue K365.
Figure 5.
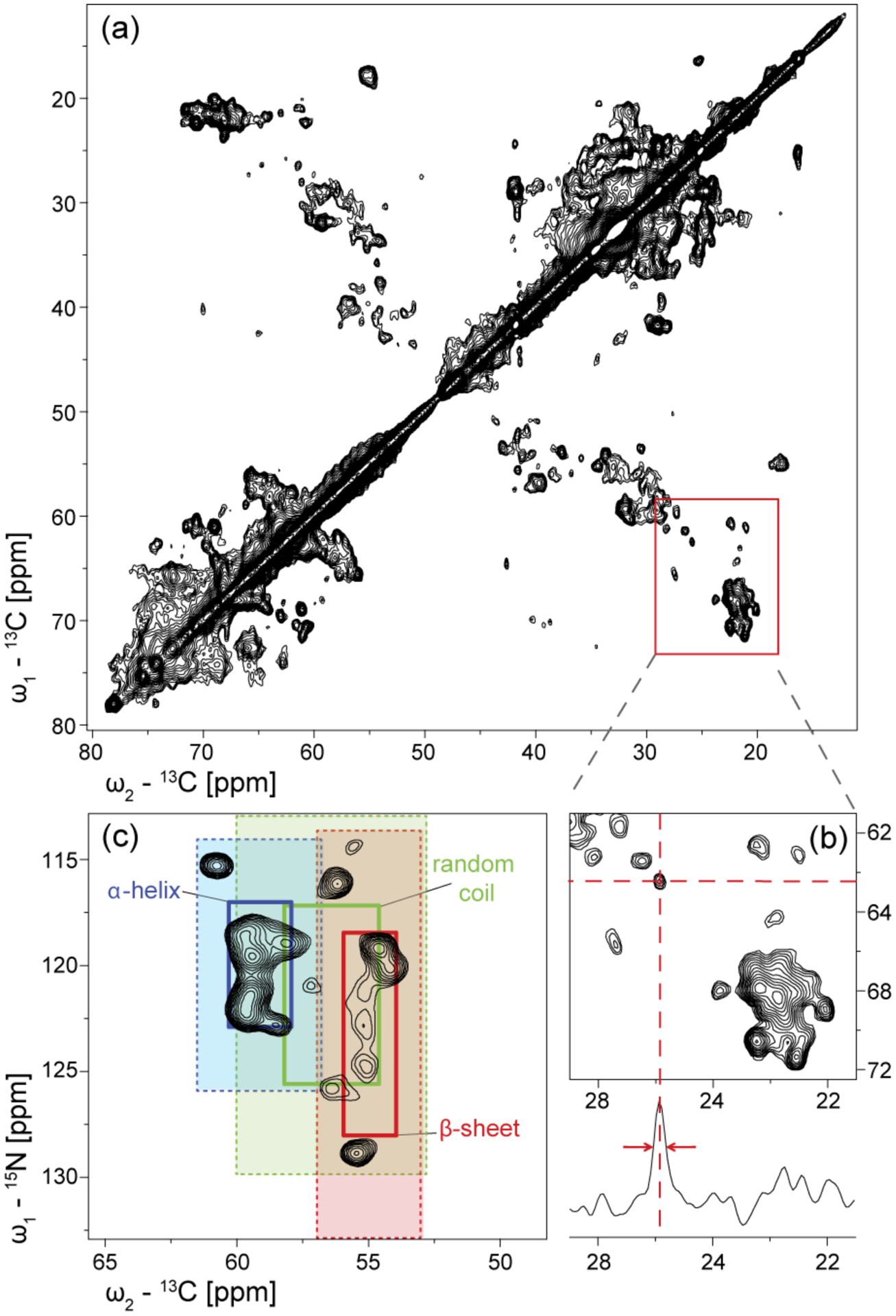
(a) Aliphatic region of the 13C-13C PDSD spectrum (20 ms mixing time) of [13C,15N-DEQGHKTS]-MsbA reconstituted in DMPC/DMPA (9:1) at LPR=50 mol/mol. Labelling was achieved using extensive reverse labelling (see materials and methods). The homogeneous sample preparation results in a number of individually resolved cross peaks. (b) A 1D trace taken at 62.5 ppm shows a 0.5 ppm linewidth (106 Hz) at half height for an isolated peak. (c) 15N-13C NCA spectrum of [13C, 15N-K]-MsbA with secondary structure regions highlighted (1- and 2-times standard deviation) (Wang & Jardetzky, 2002). MsbA contains 22 lysines.
(c). Options to improve NMR sensitivity
Although the spectra described above demonstrate that site-resolved data can be successfully obtained from MsbA, further improvements in detection sensitivity would be helpful in order to carry out a resonance assignment and/or to obtain site-resolved structural details. Here, two different approaches have been evaluated with respect to their compatibility with this ABC exporter. In the following part, improving the signal-to-noise ratio (SNR) per unit time by rapid data acquisition based on paramagnetic doping is described. This is followed by a comparison with the signal enhancements achieved by dynamic nuclear polarisation.
Generally, an experiment requires long recycle delays (seconds) between successive scans for the recovery of spin polarization, which depends on the size of the spin lattice relaxation time T1. For quantitative spectra, a recycle delay time of 5 × T1 is required, while the optimum between magnetisation recovery and data acquisition per unit time results in 1.3 × T1. One possibility to improve the SNR per unit time is therefore given by reducing T1 through the addition of suitable paramagnetic relaxation agents to the protein sample so that more spectra can be accumulated within the same period of time. It has been successfully demonstrated that 1H-T1, which determines the recycle delay time in cross polarisation experiments, is significantly reduced in a homogeneous manner in microcrystalline samples by doping with Cu2+-EDTA. In combination with very fast MAS rates, low power decoupling schemes, perdeuterated samples and short recycle delays (0.2 – 0.3 s), a significant improvement in SNR per unit time has been achieved (Linser et al, 2007; Wickramasinghe et al, 2009). We could show for green proteorhodopsin (GPR) that doping with Gd3+-DOTA results in dramatically reduced 1H-T1 at much lower dopant concentrations compared to Cu2+-EDTA (Ullrich et al, 2014). Rapid data acquisition at moderate MAS rates (10 kHz) was achieved by utilising MAS probes with a reduced electric field component of the applied RF during decoupling, which reduces sample heating.
Due to its very high relaxivity and its successful demonstration on GPR we have applied Gd3+-DOTA doping to [U-15N]-MsbA containing proteoliposomes in order to improve SNR per unit time. In Figure 6a, the averaged amide proton spin lattice relaxation time (1H-T1) of MsbA as a function of dopant concentration is shown. It changes from 650 ms for the diamagnetic sample to 80 ms in presence of 1mM Gd3+-DOTA, which is a reduction by almost 90%. By reducing the recycle delay accordingly, the theoretical gain in SNR per unit time compared to the non-doped sample could be 3-fold as shown before for GPR. However, such an improvement is hardly possible considering the limited duty cycle of conventional MAS probes, but even with longer recycle delays of 1 s, a 20% better SNR is achieved upon doping (Figure 6a).
Figure 6.
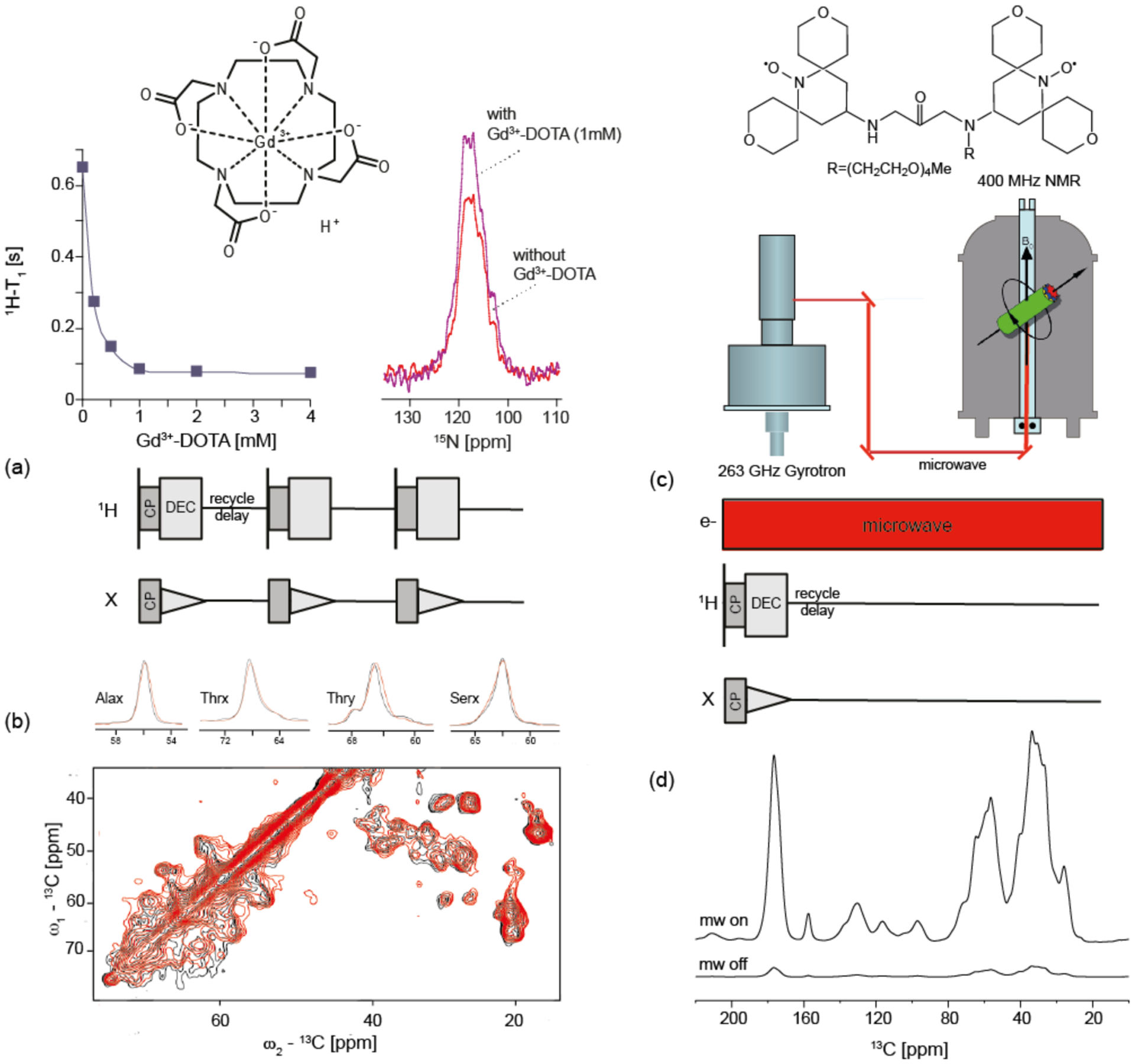
Approaches to improve the detection sensitivity for MAS-NMR on MsbA. (a) Sample doping with Gd3+-DOTA reduces the averaged amide 1H-T1 in 15N-MsbA significantly. In presence of 1 mM Gd3+-DOTA, 1H-T1 was reduced by almost 90%. A comparison of 15N-CP MAS spectra of dia- and paramagnetic MsbA samples recorded each with a recycle delay of 1 s shows a 20%-fold better SNR upon doping. (b) Superposition of 13C-13C PDSD spectra of diamagnetic (black) and paramagnetic (red) MsbA samples recorded with recycle delays of 3 and 1 sec, respectively. The number of scans and the amount of sample was the same in both spectra. The spectrum of the doped sample was recorded three times faster. Cross sections along ω2 for Cα-Cβ cross peaks show no doping induced line broadening and no relevant peak shifts. (c) Basic setup for dynamic nuclear polarisation (DNP). Samples are doped with suitable biradicals such as AmuPOL. NMR experiments are conducted under continuous microwave irradiation resulting in a large sensitivity enhancement. (d) 1D 13C-CP spectra of MsbA with and without microwave irradiation demonstrate a 20-fold signal increase.
The effect of paramagnetic doping on multidimensional spectra of MsbA was then examined in order to test whether the relaxation enhancement spreads homogeneously through the sample and whether additional line broadening occurs. In Figure 6b 13C-13C PDSD spectra of [U-13C,15N]-MsbA with and without doping are compared. Recycle delays of 3 s and 1 s, respectively, were chosen to obtain quantitative spectra. Under these conditions the spectrum of the doped sample was recorded three times faster compared to the non-doped sample. Neither significant peak shifts nor line broadening were observed and, as already observed for GPR (Ullrich et al, 2014), the paramagnetic relaxation enhancement occurs within the whole protein. These results confirm the great advantage of using Gd3+-DOTA as paramagnetic doping to enhance sensitivity through a better SNR per time unit in solid state NMR spectroscopy of membrane proteins.
A more general approach is given by dynamic nuclear polarisation (DNP), which allows transfer of the large electron polarization to nuclei (γe/γn ~ 660) (review see e.g. (Maly et al, 2008)). The most commonly used mechanism for DNP enhanced solid state NMR relies on the cross effect using allowed transitions in a three spin system consisting of two dipolar coupled electron spins and a nearby proton from which magnetization spreads throughout the sample. The polarization transfer from electrons to the nuclear spins is most efficient when using bi-radicals with strongly dipolar coupled paramagnetic centres such as in AMUPol (Sauvee et al, 2013) (Figure 6c). The basic experimental scheme is illustrated in Figure 6c: A high power microwave source (gyrotron) is coupled to a NMR spectrometer. The microwaves are directed directly at the rotating sample through a waveguide. The microwave frequency has to match the electron Larmor frequency in the NMR magnet (e.g. 263 GHz vs. 400 MHz at 9.4 T). Using this setup, NMR experiments are conducted as usual but under continuous microwave irradiation using an additional channel (Figure 6d). For these experiments, low temperatures are needed (~100 K) to reach sufficiently long electron relaxation times of the polarizing agents added to the sample. In addition, it is also required to immerse water-containing samples in a glass-forming matrix acting as cryo-protectant and at the same time preventing radical aggregation (Zagdoun et al, 2013). Here, MsbA containing proteoliposomes prepared as described above were incubated with D8-glycerol/H2O/D2O mixture containing the radical AMUPol (20 mM). Performing DNP experiments at 263 GHz/400 MHz at 100 K on these samples resulted in 20-fold signal enhancement (Figure 6d), which enables a 400-times faster data acquisition. Comparable enhancements on other membrane proteins using the same experimental setup but different polarizing agents have been reported from our group (Mao et al, 2013; Mao et al, 2014; Ong et al, 2013).
Upon comparing both approaches, it seems obvious that Dynamic Nuclear Polarization (DNP) clearly outperforms PRE in terms of sensitivity. However, the best choice of methods depends on a number of boundary conditions, which need to be considered. Paramagnetic Relaxation Enhancement (PRE) experiments can be performed at high fields and non-frozen samples ensuring best spectral resolution especially in case of extensively labelled samples. Fast data acquisition bears the risk of sample heating due to high power proton decoupling. However, this approach will become fully accessible once MAS probes with reduced electric fields and higher duty cycles become available, which are currently being developed. In contrast, DNP works best at lower fields and especially requires performing such experiments under cryogenic conditions. This requirement is often associated with freezing-induced heterogeneity resulting in line-broadening as seen in Figs. 6d and 7d. Therefore, this approach is best applied to cases with reduced spectra complexity. Furthermore, working at low temperatures is especially useful when particular states need to be trapped or unfavourable dynamics has to be quenched (Bajaj et al, 2009; Ong et al, 2013).
Figure 7.
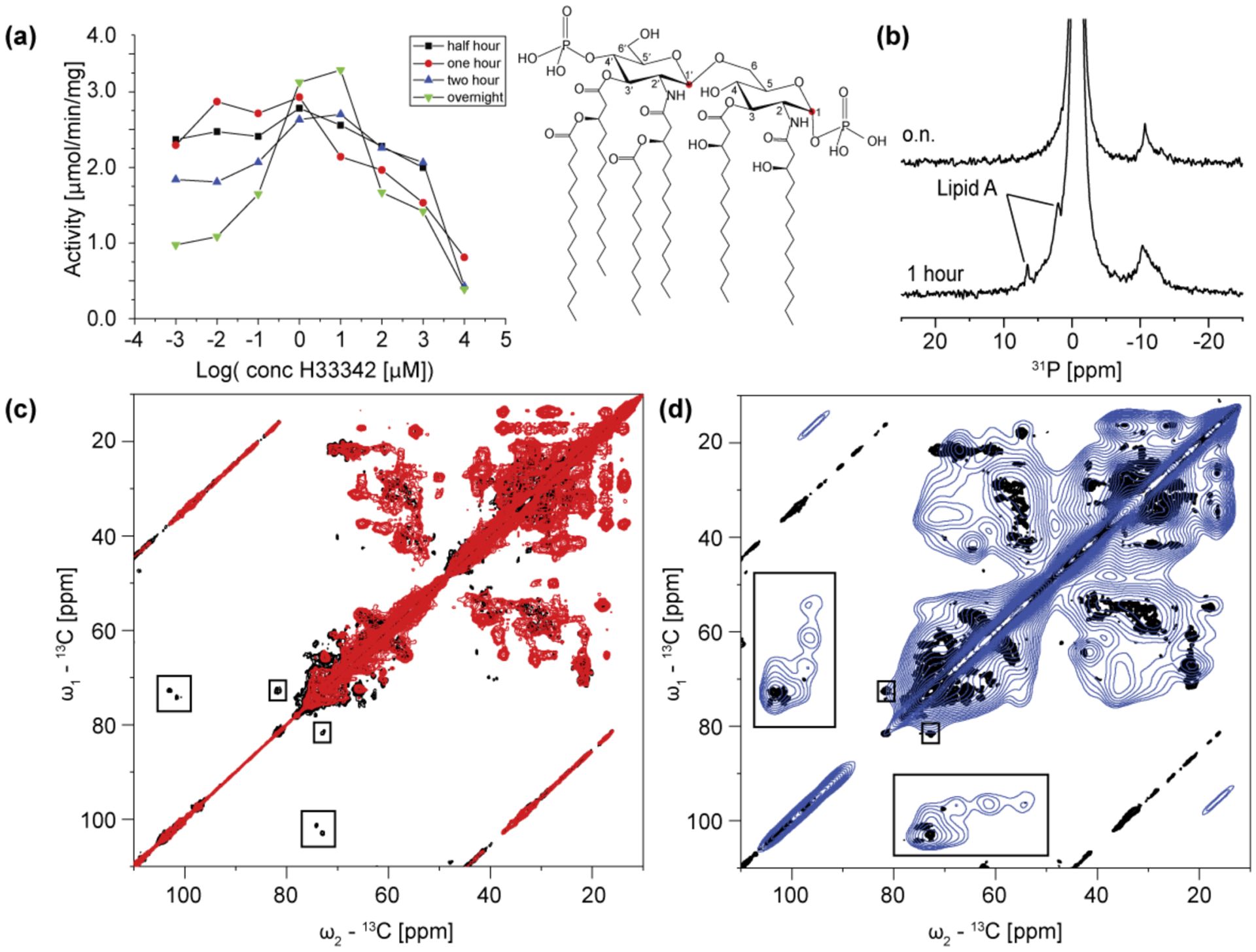
(a) Stimulation of MsbA ATPase activity by Hoechst-33342 depending on the duration of detergent solubilisation prior to purification. The longer the sample has been solubilised the more pronounced is substrate-stimulated ATPase activity. (b) 31P-CP spectra of MsbA reconstituted in DMPC/DMPA lipid bilayers after one hour and after overnight solubilisation from E. coli membranes. The sample exposed for only one hour to detergent shows two additional 31P signals, which are tentatively assigned to the phosphate groups of Lipid A within the binding pocket of MsbA. Shown here is the structure of the most abundant and toxic form of E. coli Lipid A. (c) 13C-13C PDSD spectra (20ms mixing time) of MsbA samples prepared using overnight (red) and one hour (black) solubilisation. The latter spectrum (black) shows signals typical for sugar molecules (box). (d) 13C-13C PDSD spectra (100ms mixing time) of MsbA samples exposed to one hour solubilisation at 270K (black) and 110K under DNP-conditions (blue). In the spectrum recorded under DNP condition additional signals from the sugar backbone of Lipid A can be clearly assigned.
(d). MsbA in complex with lipid A
Membrane proteins are usually extracted from their native membrane using buffers with high (1–1.5%) detergent concentration and solubilisation times of 30 min to 1 h. MsbA extracted with DDM as detergent, applied in a concentration of 1.25% w/v for 1 h showed a high basal activity and its activity could not be stimulated with Hoechst 33342 (see Figure 7a). We tested the effect of different solubilisation times on the activity of MsbA in detergent micelles. The basal activity of MsbA was elevated at short solubilisation times (2.8 μmol/mg/min after 1 h) and it decreased as the solubilisation time was increased (1.43 μmol/mg/min after overnight). A clear stimulation in the activity of protein by Hoechst 33342 could only be observed for the sample prepared by overnight solubilisation. Practically no stimulation was seen for shorter solubilisation times. We explain this phenomenon with the presence of the native substrate, Lipid A, in the binding pocket of MsbA, which could only be effectively removed when the protein was exposed to detergent for 12 h or longer.
Lipid A is the hydrophobic membrane anchor of the LPS from the outer membrane of Gram-negative bacteria. It is an endotoxin, responsible for the activation of the host innate immune system (Zhou et al, 1998). Lipid A is structurally heterogeneous. It consists of a conserved diglucosamine disaccharide backbone with fatty acids attached as ester- or amide linked substituents. Phosphate groups are also attached to 4´ and/or 1 positions. Variations in its structure can usually occur in the number, position and type of fatty acids and also in number of phosphate groups to which additional monosaccharides might be attached. The structural variation of lipid A can also vary from bacteria to bacteria or/and depend on environmental factors such as the presence of divalent cations, temperature and other growth conditions (Erridge et al, 2002; Raetz & Whitfield, 2002; Rebeil et al, 2004). The standard and most abundant structure of lipid A from E. coli is depicted in Figure 7a. However, many different variants have been isolated so far from different E. coli strains (Baltzer & Mattsbybaltzer, 1986; El Hamidi et al, 2005; Henderson et al, 2013; Jones et al, 2008).
We used solid state NMR in order to demonstrate the presence of lipid A in our preparations. In the 31P MAS-NMR spectrum of MsbA solubilised for only one hour, two signals could be seen at 6.57 and 2.06 ppm, which were not present when the protein was solubilised overnight (Fig 7b). Baltzer and Mattsby-Baltzer (Baltzer & Mattsbybaltzer, 1986) measured the 31P NMR spectra of seven different lipid A fractions isolated from E. coli strain 0111. Their 31P chemical shifts were ranging between 5.5 and 1.7 ppm. Taking into account the different experimental conditions (solution state NMR at 45°C of isolated lipid A molecules in chloroform/methanol/water mixture vs. solid state NMR at 3°C of lipid A bound to MsbA embedded within liposomes at pH 7.4) these results correspond well to our 31P NMR data and support the assignment of the two 31P resonances as lipid A signals.
13C-13C PDSD spectra of [13C,15N-DEQGHKTS]-MsbA prepared by one hour solubilisation recorded with 20 and 100 ms mixing time (Figure 7c and d, black) also showed additional cross peaks as compared with the spectrum of an overnight solubilised sample (Figure 7c, red). The 1D chemical shifts of these signals at 81.5, 101.2 and 103.0 ppm and two additional resonances in the spectrum recorded with longer mixing time at 97.2 and 104.0 ppm are typical for cyclic sugars. Ribeiro et al. assigned all 13C chemical shifts of standard lipid A from E. coli by multidimensional solution NMR techniques. Based on their results the signal at 97.2 ppm can be assigned to C-1 from the second glucosamine unit and those with a chemical shift at around 100 ppm should arise from the C-1` of the first glucosamine monomer. These carbon atoms are highlighted in red on Figure 7a. The observation of three peaks with chemical shifts of approx. 100 ppm indicates that three structurally different types of lipid A are present. Additional cross-peaks could be detected between C-1 and the other carbons from the glucosamine moiety at 68.5, 61.7 and 55.0 ppm by DNP-enhanced MAS-NMR at low temperature. These 13C spectra show that the co-purified Lipid A was at least partially 13C labelled using the expression protocol described here.
Based on these observations, it can be concluded that MsbA in DDM can only be prepared in a pure apo-state by overnight solubilisation. Other types of detergents (e.g. DM, LDAO) were used successfully also with one hour of solubilisation and the activity of the obtained protein showed clear stimulation with Hoechst 33342 (Doerrler & Raetz, 2002; Eckford & Sharom, 2008b; Eckford & Sharom, 2008c; Siarheyeva & Sharom, 2009).
Conclusions and Perspective
We have shown that milligram quantities of isotope labelled, pure, stable and mono-disperse MsbA can be produced. Reconstitution into DMPC/DMPA liposomes represents the optimum in terms of homogeneity, dense packing, ATPase activity and spectral resolution. First 2D-MAS NMR experiments of extensively and selectively labelled samples show that individual sites can be resolved. We have successfully demonstrated the applicability of Gd3+-doping to improve signal-to-noise per unit time, which is especially useful for studies at high field and at non-frozen samples. Furthermore, we have demonstrated a large signal enhancement by DNP, which will become essential for freeze-trapping functional states on selectively labelled MsbA. These approaches allowed us to identify 13C-labelled lipid A in complex with labelled MsbA by utilising the duration of detergent solubilisation. These initial experiments confirm that MsbA is a promising target for further in-depth solid state NMR studies especially with respect to nucleotide and lipid interactions, which are currently in progress and will be reported elsewhere.
Materials and methods
(a). Reagents
Luria broth, Tris and HEPES were obtained from Carl Roth GmbH & Co. Ni-NTA beads were purchased from Qiagen and Biobeads were ordered from Bio-Rad. ATP and sodium meta-arsenite were purchased from Sigma-Aldrich. Complete EDTA free protease inhibitor cocktail tablets were bought from Roche and Run Blue precast SDS-PAGE gels from Expedeon. Lipids were obtained from Avanti Polar Lipids. All other compounds were from PanReac AppliChem reagents. For size exclusion chromatography Sephadex 200 10/300GL and PD10 column for buffer exchange were from GE Healthcare Life Sciences. Labelled glucose (13C) and other amino acids were ordered from Cambridge Isotope Laboratories (CIL) while ammonium chloride (15N) was obtained from Cortecnet. The Gd3+-DOTA complex was bought from BOC sciences.
(b). Expression and Purification of MsbA
MsbA was cloned into a pET 19b vector containing an N-terminal His10 tag connected via an 11 amino acid peptide linker (Borbat et al, 2007). The plasmid was transformed into C43(DE3) cells for protein expression. For obtaining a high yield of isotope labelled MsbA, cells were grown initially in LB-medium, collected by centrifugation and transferred to M9 minimal medium (Marley et al, 2001). Expression was started by adding 10 ml of preculture to one liter of LB at 37 °C and 220 rpm. When the OD600nm reached 0.6 – 0.7, cells were harvested and then resuspended into 500 ml of M9 minimal medium. The cells were further incubated at 37 °C, 220 rpm for one hour for adaptation. The protein expression was induced with 1mM IPTG at 20 °C and 260 rpm once the OD600nm has reached a value between 1.7 and 2.1 After 17 h of expression, the cells were harvested and resuspended in buffer A (10 mM Tris, 250 mM Sucrose, 150 mM NaCl, 2.5 mM MgSO4, pH 7.5) with protease inhibitor, 0.5 mM dithiothreitol (DTT) and DNAase. Membranes were prepared by passing the resuspended cells through a cell disrupter at a pressure of 1.5–1.7 kbar (2–3 times), followed by centrifugation at 8000 rpm for 10 min to remove cell debris and a final ultracentrifugation step at 55000 rpm for one hour. Membranes were solubilised in buffer B (50 mM HEPES, 300 mM NaCl, 5 mM MgCl2, 10% Glycerol, pH 7.5) with 1.25% DDM, 0.5mM DTT and 10 mM imidazole at 4°C either overnight (apo sample) or for one hour (lipid A bound sample). The insoluble fraction was removed by ultracentrifugation at 55000 rpm for 1 h. The remaining supernatant was loaded onto Ni-NTA prewashed with buffer B containing 50 mM imidazole. After 1.5 −2 h binding, elution was carried out using buffer B containing 0.015% DDM and 400 mM imidazole. SDS - PAGE, size exclusion chromatography using Sephadex 200 column and MALDI-MS were done to confirm purity and homogeneity of the protein. Using this procedure, [U-15N]-MsbA, [U-13C,15N]-MsbA, [13C,15N-K]-MsbA and [13C,15N-DEQGHKTS]-MsbA were produced. Samples were prepared by supplemented M9 with 13C-glucose (2 g/l M9 minimal media) and 15NH4Cl (1 g/l M9 minimal media). For [13C,15N-DEQGHKTS]-MsbA, unlabelled amino acids Ala (0.5 g/l), Cys (0.05 g/l), Val (0.23 g/l), Leu (0.23 g/l), Ile (0.23 g/l), Met (0.25 g/l), Pro (0.1 g/l), Phe (0.13 g/l), Tyr (0.17 g/l), Trp (0.05 g/l), Asn (0.4 g/l), Arg (0.4 g/l) were added for extensive reverse labelling. In case of [13C,15N-K]-MsbA, 13C-15N-Lys together with all other unlabelled amino acids was added to M9 minimal medium.
(c). Reconstitution of MsbA
The following lipid mixtures were used for reconstitution: DMPC:DMPA (9:1), E. coli polar lipids, E. coli PE:PG:CA (65 : 25 : 10) and POPC:POPG:CA (65 : 25 : 10) at molar lipid to protein ratios of 75 : 1 and 50 : 1. Each lipid mixture was solubilised in CHCl3:CH3OH (2:1) and drying under a stream of nitrogen gas followed by vacuum rotor evaporation. The dried lipids were resuspended in buffer C (50 mM HEPES, 50 mM NaCl) and extruded 11 times through membranes with pore diameters of 0.1, 0.2 and 0.4 μm. Liposomes were destabilised with 3 mM DDM. After dropwise addition of protein in the DDM/liposome solution, the mixture was allowed to incubate at room temperature for 30 min. Detergent was removed by biobeads (80 mg/ml), which were first added for 12 h at 4 °C and then twice for one hour at room temperature. Sample homogeneity was verified by a discontinuous sucrose gradient prepared by layering 1 ml of 10, 30, 50 and 70% (w/v) sucrose in 50 mM HEPES, 50 mM NaCl. About 400 μL of sample was layered on top of the sucrose solution and centrifuged at 28,000 rpm for 16 h at 4 °C.
(d). ATPase activity
The activity of MsbA was determining by monitoring the release of inorganic phosphate. Each reaction mixture containing MsbA (solubilized in detergent or reconstituted into liposomes) and ATP (0.2 – 5 mM) in buffer (50 mM HEPES, 50 mM NaCl, 10 mM MgCl2) was incubated at 37 °C for 20 min. Control samples for each reaction were kept on ice. The reaction was stopped by adding 12% SDS. The release of inorganic phosphate (Pi) was measured using the molybdenium blue method (Chifflet et al, 1988; González-Romo et al, 1992). Stimulated activity was observed by determining the release of Pi upon adding Hoechst 33342 at increasing concentrations of 0.001, 0.01, 0.1, 1, 10, 100, 1000 and 10000 μM in the presence of 0.2 mM ATP (Eckford & Sharom, 2008a).
(e). Sample preparation for NMR
For MAS-NMR, proteoliposomes were pelleted and centrifuged into 3.2 or 4 mm MAS rotors. Usually, an amount of 10 – 15 mg of MsbA was used. For paramagnetic relaxation enhancement, samples were doped with the appropriate amount of gadolinium- 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (Gd3+-DOTA) (Ullrich et al, 2014). The Gd3+-DOTA powder was dissolved in NMR buffer and added to the sample at the desired concentration. The sample was incubated for 15 minutes at 4 °C followed by ultracentrifugation and transfer into a 4 mm MAS rotor.
For DNP, reconstituted samples were incubated overnight at 4 °C with 20 mM AMUPOL in a buffer containing 10 % H2O, 30 % D8-glycerol and 60 % D2O. The solution was removed carefully and the pellets were transferred to a 3.2 mm zirconium oxide rotor by centrifugation.
(f). Solid state NMR
For all NMR experiments, standard settings for cross polarisation (CP) and decoupling were used. A typical 1H 90° pulse had a duration of 3 μs, the CP contact time was chosen between 0.8 and 1 ms and high power proton decoupling of 70–100 kHz was applied using SPINAL64 (Fung et al, 2000). Spectra were processed using TOPSPIN 3.2. An exponential window function was applied to 1D data and a shifted cos2 function was used for 2D spectra. Depending on the experiment, between 10 and 20 mg of MsbA were used.
15N-T2’ measurements (Figure 3d):
15N-spin echo experiments on [U-15N]-MsbA were recorded at 270K at 10kHz sample spinning using a Bruker 600WB Avance I spectrometer with a 4 mm DVT-HCN E-free MAS probe. The rotor-synchronized spin echo was acquired after a CP step under high power proton decoupling with a 3 s recycle delay. The full integral intensity of the amide region (109 – 121 ppm) was used for data analysis.
Temperature-dependent spectra (Figure 4):
The temperature-dependant experiments on [U-13C]-MsbA were recorded at 10 kHz sample spinning using a Bruker 600WB Avance I spectrometer with a 4 mm DVT-HCN E-free MAS probe. Order parameters were derived from 13C-1H dipolar couplings measured from PISEMA experiments (Dvinskikh et al, 2003; Wu et al, 1994). A 50 kHz RF field was used during the Lee–Goldberg spin exchange at the magic angle.
13C-13C and 13C-15N correlation spectra (Figure 5):
The 13C-13C through space correlation spectra on [13C,15N-DEQGHKTS]-MsbA were recorded using a proton-driven spin diffusion experiment (PDSD) (Szeverenyi et al, 1982) at 14 kHz sample spinning and 270K using a Bruker 850WB Avance III spectrometer equipped with a 3.2 mm DVT-HXY MAS probe. Spectra were recorded with 800 increments and 128 scans each, with a 20 ms mixing time and a 3 s recycle delay. Spectral widths were 80 kHz in ω2 and 52 kHz in ω1. The NCA spectrum of [15N-Lys]-MsbA was recorded at 11.4 kHz sample spinning at 270 K using a Bruker 600WB Avance I spectrometer with a 4 mm DVT-HCN E-free MAS probe. For the NC cross polarisation step, a mixing time of 6 ms was applied. A constant 15N lock field of 28.5 kHz (2.5 × νr) and a linearly ramped (90–100) 13C field around 1.5 × νr was used. The CW decoupling during this step was set to 100 kHz. The spectrum was acquired with 200 increments with 512 scans each and 3 s recycle delay. Spectral widths were 60 kHz in ω2 and 5.5 kHz ω1.
Gd3+-DOTA doping (Figs. 6a,b):
All experiments were carried out using a Bruker 600WB Avance I spectrometer with a 4 mm DVT-HCN E-free MAS probe. Amide proton T1 values (Figure 6a) were obtained by saturation recovery experiments following a 15N CP step. Relaxation times were determined by analysing the full intensity of the 15N amide resonances (109 – 121 ppm). The 13C-13C PDSD spectrum (Figure 6b) was recorded with a 20 ms mixing time and 512 increments with 144 scans each. Spectral widths were 60 kHz in ω2 and 40 kHz in ω1. Upon Gd3+-DOTA doping, the recycle delay was reduced from 3 to 1 s.
DNP-enhanced MAS-NMR (Figs. 6d, 7a):
The DNP enhanced MAS NMR spectra were recorded using a Bruker DNP system consisting of a 400 MHz WB Avance II spectrometer, a 263 GHz Gyrotron as a microwave source and a 3.2 mm HCN-DNP-MAS probe. The temperature was set 105K and a sample-spinning rate of 8 kHz was used. The 13C-13C PDSD spectrum was acquired with a 100 ms mixing time and 256 increments with 32 scans each. Spectral widths were 40 kHz in ω2 and 25 kHz in ω1. A recycle delay of 2.5 s was used.
MsbA in complex with Lipid A (Figure 7):
The 13C-13C PDSD spectrum of [13C,15N-DEQGHKTS]-MsbA were acquired at 14kHz sample spinning and 270 K using a Bruker 850WB Avance III spectrometer equipped with a 3.2 mm DVT-HXY MAS probe. Mixing times of 20 and 100 ms were applied and spectra were recorded with 800 increments with 128 scans each. Spectral widths were 80 kHz in ω2 and 52 kHz in ω1. A recycle delay of 3 s was used. 31P MAS NMR spectra (Figure 7b) were recorded at 10kHz sample spinning and 270K using a Bruker 600WB Avance I spectrometer with a 4 mm DVT-HX MAS probe. A CP contact time of 3 ms was applied followed by high power proton decoupling.
Acknowledgements
The work was supported by a DFG research grant through SFB 807 ‘Transport and communication across membranes’ and by a DFG equipment grant (GL 307/4-1) to C.G.. H. M. acknowledges support by NIH grant U54-GM087519. MALDI-MS data were kindly provided by Dr. Ute Bahr (Institute for Pharmaceutical Chemistry, Goethe University Frankfurt)
References
- Ader C, Schneider R, Hornig S, Velisetty P, Vardanyan V, Giller K, Ohmert I, Becker S, Pongs O, Baldus M (2009). Coupling of activation and inactivation gate in a K+-channel: potassium and ligand sensitivity. EMBO J. 28, 2825–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbey U, Nieuwkoop AJ, Wegner S, Voreck A, Kunert B, Bandara P, Engelke F, Nielsen NC, Oschkinat H (2014). Quadruple-resonance magic-angle spinning NMR spectroscopy of deuterated solid proteins. Angew. Chem. Int. Ed. Engl 53, 2438–2442. [DOI] [PubMed] [Google Scholar]
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG (2009). Functional and shunt states of bacteriorhodopsin resolved by 250 GHz dynamic nuclear polarization-enhanced solid-state NMR. Proc Natl Acad Sci U S A 106, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltzer LH, Mattsbybaltzer I (1986). Heterogeneity of Lipid-a - Structural Determination by C-13 and P-31 Nmr of Lipid-a Fractions from Lipopolysaccharide of Escherichia-Coli 0111. Biochemistry 25, 3570–3575. [DOI] [PubMed] [Google Scholar]
- Bellstedt P, Seiboth T, Haefner S, Kutscha H, Ramachandran R, Goerlach M (2013). Resonance assignment for a particularly challenging protein based on systematic unlabeling of amino acids to complement incomplete NMR data sets. J. Biomol. NMR 57, 65–72. [DOI] [PubMed] [Google Scholar]
- Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, McHaourab HS (2007). Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 5, 2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchaklian AH, Klug CS (2005). Characterization of the Walker A motif of MsbA using site-directed spin labeling electron paramagnetic resonance spectroscopy. Biochemistry 44, 5503–5509. [DOI] [PubMed] [Google Scholar]
- Chifflet S, Torriglia A, Chiesa R, Tolosa S (1988). A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal. Biochem 168, 1–4. [DOI] [PubMed] [Google Scholar]
- Cooper RS, Altenberg GA (2013). Association/Dissociation of the Nucleotide-binding Domains of the ATP-binding Cassette Protein MsbA Measured during Continuous Hydrolysis. J. Biol. Chem 288, 20785–20796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson RJ, Locher KP (2006). Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185. [DOI] [PubMed] [Google Scholar]
- De Angelis AA, Opella SJ (2007). Bicelle samples for solid-state NMR of membrane proteins. Nature Protocols 2, 2332–2338. [DOI] [PubMed] [Google Scholar]
- De Paepe G, Giraud N, Lesage A, Hodgkinson P, Bockmann A, Emsley L (2003). Transverse dephasing optimized solid-state NMR spectroscopy. J. Am. Chem. Soc 125, 13938–13939. [DOI] [PubMed] [Google Scholar]
- Doerrler WT, Raetz CRH (2002). Lipid A stimulates the ATPase activity of purified, reconstituted MsbA, an essential, Escherichia coli ABC transporter. FASEB J. 16, A893–A894. [Google Scholar]
- Dong J, Yang G, McHaourab HS (2005). Structural basis of energy transduction in the transport cycle of MsbA. Science 308, 1023–1028. [DOI] [PubMed] [Google Scholar]
- Doshi R, Ali A, Shi W, Freeman EV, Fagg LA, van Veen HW (2013). Molecular Disruption of the Power Stroke in the ATP-binding Cassette Transport Protein MsbA. J. Biol. Chem 288, 6801–6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi R, Woebking B, van Veen HW (2010). Dissection of the conformational cycle of the multidrug/lipidA ABC exporter MsbA. Proteins-Structure Function and Bioinformatics 78, 2867–2872. [DOI] [PubMed] [Google Scholar]
- Dvinskikh SV, Zimmermann H, Maliniak A, Sandström D (2003). Heteronuclear dipolar recoupling in liquid crystals and solids by PISEMA-type pulse sequences. J. Magn. Reson 164, 165–170. [DOI] [PubMed] [Google Scholar]
- Eckford PD, Sharom FJ (2008a). Functional Characterization of Escherichia coli MsbA interaction with nucleotides and substrates. J. Biol. Chem 283, 12840–12850. [DOI] [PubMed] [Google Scholar]
- Eckford PD, Sharom FJ (2008b). Interaction of the P-Glycoprotein Multidrug Efflux Pump with Cholesterol: Effects on ATPase Activity, Drug Binding and Transport†. Biochemistry 47, 13686–13698. [DOI] [PubMed] [Google Scholar]
- Eckford PDW, Sharom FJ (2008c). Functional characterization of Escherichia coli MsbA - Interaction with nucleotides and substrates. J. Biol. Chem 283, 12840–12850. [DOI] [PubMed] [Google Scholar]
- Eckford PDW, Sharom FJ (2010). The reconstituted Escherichia coli MsbA protein displays lipid flippase activity. Biochem. J 429, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hamidi A, Tirsoaga A, Novikov A, Hussein A, Caroff M (2005). Microextraction of bacterial lipid A: easy and rapid method for mass spectrometric characterization. J. Lipid Res 46, 1773–1778. [DOI] [PubMed] [Google Scholar]
- Erridge C, Bennett-Guerrero E, Poxton IR (2002). Structure and function of lipopolysaccharides. Microb. Infect 4, 837–851. [DOI] [PubMed] [Google Scholar]
- Fung B, Khitrin A, Ermolaev K (2000). An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson 142, 97–101. [DOI] [PubMed] [Google Scholar]
- Geertsma ER, Mahmood NABN, Schuurman-Wolters GK, Poolman B (2008). Membrane reconstitution of ABC transporters and assays of translocator function. Nature Protocols 3, 256–266. [DOI] [PubMed] [Google Scholar]
- George AM, Jones PM (2012). Perspectives on the structure-function of ABC transporters: The Switch and Constant Contact Models. Progress in Biophysics & Molecular Biology 109, 95–107. [DOI] [PubMed] [Google Scholar]
- González-Romo P, Sánchez-Nieto S, Gavilanes-Ruíz M (1992). A modified colorimetric method for the determination of orthophosphate in the presence of high ATP concentrations. Anal. Biochem 200, 235–238. [DOI] [PubMed] [Google Scholar]
- Hellmich UA, Duchardt-Ferner E, Glaubitz C, Woehnert J (2012a). Backbone NMR resonance assignments of the nucleotide binding domain of the ABC multidrug transporter LmrA from Lactococcus lactis in its ADP-bound state. Biomolecular NMR assignments 6, 69–73. [DOI] [PubMed] [Google Scholar]
- Hellmich UA, Haase W, Velamakanni S, van Veen HW, Glaubitz C (2008). Caught in the Act: ATP hydrolysis of an ABC-multidrug transporter followed by real-time magic angle spinning NMR. FEBS Lett. 582, 3557–3562. [DOI] [PubMed] [Google Scholar]
- Hellmich UA, Lyubenova S, Kaltenborn E, Doshi R, van Veen HW, Prisner TF, Glaubitz C (2012b). Probing the ATP hydrolysis cycle of the ABC multidrug transporter LmrA by pulsed EPR spectroscopy. J. Am. Chem. Soc 134, 5857–5862. [DOI] [PubMed] [Google Scholar]
- Henderson JC, O’Brien JP, Brodbelt JS, Trent MS (2013). Isolation and chemical characterization of lipid A from gram-negative bacteria. Journal of visualized experiments : JoVE, e50623–e50623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CF, Linton KJ (2004). The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol 11, 918–926. [DOI] [PubMed] [Google Scholar]
- Jones JW, Shaffer SA, Ernst RK, Goodlett DR, Turecek F (2008). Determination of pyrophosphorylated forms of lipid A in Gram-negative bacteria using a multivaried mass spectrometric approach. Proc Natl Acad Sci U S A 105, 12742–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PM, George AM (2013). Mechanism of the ABC transporter ATPase domains: catalytic models and the biochemical and biophysical record. Crit. Rev. Biochem. Mol. Biol 48, 39–50. [DOI] [PubMed] [Google Scholar]
- Kim J, Wu S, Tomasiak TM, Mergel C, Winter MB, Stiller SB, Robles-Colmanares Y, Stroud RM, Tampe R, Craik CS, Cheng Y (2015). Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature 517, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert B, Gardiennet C, Lacabanne D, Calles-Garcia D, Falson P, Jault J-M, Meier BH, Penin F, Böckmann A (2014). Efficient and stable reconstitution of the ABC transporter BmrA for solid-state NMR studies. Frontiers in Molecular Biosciences 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange V, Becker-Baldus J, Kunert B, van Rossum B-J, Casagrande F, Engel A, Roske Y, Scheffel FM, Schneider E, Oschkinat H (2010). A MAS NMR Study of the Bacterial ABC Transporter ArtMP. ChemBioChem 11, 547–555. [DOI] [PubMed] [Google Scholar]
- Linser R, Chevelkov V, Diehl A, Reif B (2007). Sensitivity enhancement using paramagnetic relaxation in MAS solid-state NMR of perdeuterated proteins. J. Magn. Reson 189, 209–216. [DOI] [PubMed] [Google Scholar]
- Maly T, Debelouchina GT, Bajaj VS, Hu KN, Joo CG, Mak-Jurkauskas ML, Sirigiri JR, van der Wel PCA, Herzfeld J, Temkin RJ, Griffin RG (2008). Dynamic nuclear polarization at high magnetic fields. J. Chem. Phys 128, 052211–052219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Akhmetzyanov D, Ouari O, Denysenkov V, Corzilius B, Plackmeyer J, Tordo P, Prisner TF, Glaubitz C (2013). Host-guest complexes as water-soluble high-performance DNP polarizing agents. J. Am. Chem. Soc 135, 19275–19281. [DOI] [PubMed] [Google Scholar]
- Mao J, Do NN, Scholz F, Reggie L, Mehler M, Lakatos A, Ong YS, Ullrich SJ, Brown LJ, Brown RC, Becker-Baldus J, Wachtveitl J, Glaubitz C (2014). Structural Basis of the Green-Blue Color Switching in Proteorhodopsin as Determined by NMR Spectroscopy. J. Am. Chem. Soc [DOI] [PubMed] [Google Scholar]
- Marley J, Lu M, Bracken C (2001). A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75. [DOI] [PubMed] [Google Scholar]
- Mishra S, Verhalen B, Stein RA, Wen P-C, Tajkhorshid E, McHaourab HS (2014). Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. eLife 3, e02740–e02740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong YS, Lakatos A, Becker-Baldus J, Pos KM, Glaubitz C (2013). Detecting substrates bound to the secondary multidrug efflux pump EmrE by DNP-enhanced solid-state NMR. J. Am. Chem. Soc 135, 15754–15762. [DOI] [PubMed] [Google Scholar]
- Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, Opella SJ (2012). Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 491, 779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RS, Hunt SA, DiNatale JA, Vold RR (1996). Magnetically aligned membrane model systems with positive order parameter: Switching the sign of S-zz with paramagnetic ions. J. Am. Chem. Soc 118, 269–270. [Google Scholar]
- Raetz CRH, Whitfield C (2002). Lipopolysaccharide endotoxins. Annu. Rev. Biochem 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Etzkorn M, Wagner G (2010). Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr. Opin. Struct. Biol 20, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ (2004). Variation in lipid A structure in the pathogenic yersiniae. Mol. Microbiol 52, 1363–1373. [DOI] [PubMed] [Google Scholar]
- Rigaud J-L, Pitard B, Levy D (1995). Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim. Biophys. Acta 1231, 223–246. [DOI] [PubMed] [Google Scholar]
- Rigaud JL, Levy D (2003). Reconstitution of membrane proteins into liposomes. Liposomes, Pt B 372, 65–86. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Landis GC (1995). Reconstitution of membrane-proteins into lipid-rich bilayered mixed micelles for NMR-studies. Biochemistry 34, 4030–4040. [DOI] [PubMed] [Google Scholar]
- Sauvee C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P (2013). Highly efficient, water-soluble polarizing agents for dynamic nuclear polarization at high frequency. Angew. Chem. Int. Ed. Engl 52, 10858–10861. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A, Lopez JJ, Lehner I, Hellmich UA, van Veen HW, Glaubitz C (2007). Probing the molecular dynamics of the ABC multidrug transporter LmrA by deuterium solid-state nuclear magnetic resonance. Biochemistry 46, 3075–3083. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A, Sharom FJ (2009). The ABC transporter MsbA interacts with lipid A and amphipathic drugs at different sites. Biochem. J 419, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smriti Zou, P., McHaourab HS (2009). Mapping Daunorubicin-binding Sites in the ATP-binding Cassette Transporter MsbA Using Site-specific Quenching by Spin Labels. J. Biol. Chem 284, 13904–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeverenyi NM, Sullivan MJ, Maciel GE (1982). Observation of spin exchange by two-dimensional fourier transform 13C cross polarization-magic-angle spinning. Journal of Magnetic Resonance (1969) 47, 462–475. [Google Scholar]
- Ullrich SJ, Holper S, Glaubitz C (2014). Paramagnetic doping of a 7TM membrane protein in lipid bilayers by Gd(3)(+)-complexes for solid-state NMR spectroscopy. J. Biomol. NMR 58, 27–35. [DOI] [PubMed] [Google Scholar]
- Wang S, Munro RA, Shi L, Kawamura I, Okitsu T, Wada A, Kim SY, Jung KH, Brown LS, Ladizhansky V (2013). Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat. Methods 10, 1007–1012. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Jardetzky O (2002). Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11, 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Mulligan S, Carragher B, Chang G, Milligan RA (2009). Nucleotide dependent packing differences in helical crystals of the ABC transporter MsbA. Journal of Structural Biology 165, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Reyes CL, Yu J, Roth CB, Chang G (2007). Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe NP, Parthasarathy S, Jones CR, Bhardwaj C, Long F, Kotecha M, Mehboob S, Fung LW, Past J, Samoson A, Ishii Y (2009). Nanomole-scale protein solid-state NMR by breaking intrinsic 1HT1 boundaries. Nat. Methods 6, 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Ramamoorthy A, Opella SJ (1994). High-resolution heteronuclear dipolar solid-state NMR-spectroscopy. J. Magn. Reson., Ser A 109, 270–272. [Google Scholar]
- Zagdoun A, Casano G, Ouari O, Schwarzwalder M, Rossini AJ, Aussenac F, Yulikov M, Jeschke G, Coperet C, Lesage A, Tordo P, Emsley L (2013). Large Molecular Weight Nitroxide Biradicals Providing Efficient Dynamic Nuclear Polarization at Temperatures up to 200 K. J. Am. Chem. Soc 135, 12790–12797. [DOI] [PubMed] [Google Scholar]
- Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CRH (1998). Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. FASEB J. 12, A1284–A1284. [DOI] [PubMed] [Google Scholar]
- Zou P, McHaourab HS (2009). Alternating Access of the Putative Substrate-Binding Chamber in the ABC Transporter MsbA. J. Mol. Biol 393, 574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]