

Abstract
An essential role for cilia in the pathogenesis of congenital heart disease (CHD) has emerged from findings of a large-scale mouse forward genetic screen. High throughput screening with fetal ultrasound imaging followed by whole exome sequencing analysis recovered a preponderance of cilia related genes and cilia transduced cell signaling genes among mutations identified to cause CHD. The perturbation of left-right patterning in CHD pathogenesis is suggested by the association of CHD with heterotaxy, but also by the finding of the co-occurrence of laterality defects with CHD in birth defect registries. Many of the cilia and cilia cell signaling genes recovered were found to be related to Hedgehog signaling. Studies in mice showed cilia transduced hedgehog signaling coordinates left-right patterning with heart looping and differentiation of the heart tube. Cilia transduced Shh signaling also regulates later events in heart development, including outflow tract septation and formation of the atrioventricular septum. More recent work has shown mutations in cilia related genes may also contribute to valve disease that largely manifest in adult life. Overall, these and other findings show cilia play an important role in CHD and also in more common valve diseases.
Keywords: congenital heart disease, cilia, forward genetics, mouse models, mutagenesis, fetal ultrasound imaging
1. Introduction
Congenital heart defects (CHD) are one of the most common structural birth defects, affecting up to 0.5 % of live births[1]. If bicuspid aortic valves are included (see below), the incidence of CHD would more likely fall within the range of 1–3% [2, 3]. CHD is characterized by disturbance in the structure of the heart and can involve abnormalities of the cardiac valves, outflow tract septation defects, or abnormalities in patterning or growth of the cardiac chambers and their inflow/outflow connections. These different anatomical defects can occur separately, causing simple CHD, or in combination, causing complex CHD. They can result in disturbance of unidirectional blood flow or they can cause abnormal mixing of blood flow between the left vs. right sides of the heart, which will compromise the efficient oxygenation of blood.
The human heart is comprised of four chambers, two atria and two ventricles, that have distinct left-right asymmetric inflow and outflow connections allowing the left heart to receive oxygenated blood from the lung and circulate it systemically to and from the body and the right heart to receive and pump deoxygenated blood to the lung for reoxygenation. This anatomical left-right asymmetric four chamber anatomy is an evolutionary adaption that allows for the efficient extraction of oxygen from air. Disturbance of this distinct left-right asymmetric cardiac anatomy can result in failure to oxygenate blood efficiently, either due to mixing of oxygenated/deoxygenated blood or from the inability to route deoxygenated blood to the lungs and oxygenated blood back to the body. Hence it is not surprising that except for simple atrial or ventricular septal defects (ASD, VSD), CHD were previously associated with high morbidity and mortality.
With technical advances in surgical repair patients with even very complex CHD are now surviving their critical structural heart defects, including CHD that were previously uniformly fatal[4, 5]. As a result, currently there are now more adults with CHD then children born annually with CHD[6]. From this ever expanding cohort of CHD adults, there is realization that such surviving patients suffer increased morbidity and mortality, often with clinical sequelae that might include exercise intolerance, poor neurodevelopmental outcomes, heart failure, liver failure, and increased cancer risk[7–9]. As the etiology of these clinical complications are not understood, it has not been possible to identify patients at risk for early intervention. Without knowledge of the pathogenic mechanisms, therapeutic interventions can only be focused on treating symptoms, limiting their effectiveness. While previously such clinical sequelae were largely attributed to hypoxia or surgical trauma, data from multiple clinical studies have shown the causes are largely arising from patient intrinsic factors, suggesting they may arise from the same genetic defects causing their CHD[10]. This would suggest only with insights into the genetic landscape of CHD, can we hope to obtain mechanistic insights into these disease processes, and only then can we hope to develop targeted therapy to address these clinical sequelae that continue to compromise the long term survival and health related quality of life of CHD patients. It is with these needs in mind, we have embarked on the use of systems genetics to interrogate the genetic landscape of CHD using the mouse as a model system.
2.1. Systems Genetics Identifies Role of Cilia in CHD pathogenesis
To investigate the genetic landscape of CHD, we chose mice as our model system. There are multiple important advantages with using mice for these studies, as completely inbred mice are readily available[11]. This overcomes one of the major limitations in human genetic analysis, i.e. the genetic heterogeneity of the human population. Moreover, as the reference genome of inbred mouse strains are readily available, this allows genetic variation or mutation causing a disease phenotype to be easily recovered using whole exome or whole genome sequencing analysis. Most importantly, mice have the same four chamber left-right asymmetric cardiac anatomy as humans, making it possible to model the same simple and complex CHD phenotypes seen in human patients[12]. Hence, together these factors make mice the ideal model system for interrogating the genetic landscape of CHD.
In the setting of the mouse model, we undertook a systems approach with large scale forward genetics to investigate the genetic etiology of CHD. This entailed the use of chemical mutagenesis with ethylnitrosourea (ENU), a mutagen that induces point mutations, to randomly mutagenize the mouse genome. The ENU mutagenized mice were then bred to generate heterozygous G1 and G2 offspring, and these were intercrossed to generate G3 fetuses to allow for recovery of recessive mutations causing CHD[13]. Cardiovascular phenotyping was conducted using fetal echocardiography, an imaging modality that allows visualization of heart structure and function also commonly used clinically to diagnose CHD prenatally[14]. This made it possible to conduct the screen noninvasively, allowing the same litter of fetuses to be queried longitudinally over multiple days of gestation. Using this very high throughput and also very sensitive imaging modality for the diagnosis of CHD, we were able to screen over 100,000 mouse fetuses, and recovered mutants with the majority of CHD observed clinically (Fig.1). This included not only simple CHD such as atrial septal defect and ventricular septal defect, but also more complex lesions such as double outlet right ventricle (DORV), transposition of the great arteries (TGA), persistent truncus arteriosus (PTA), or various aortic arch anomalies or valvular defects (Fig.1). Also recovered from the screen were mutants with hypoplastic left heart syndrome (HLHS; Fig.1), a lesion that was previously suggested to have a hemodynamic origin that would preclude its modeling in mice (given its very short gestation period)[15].
Figure 1. Wide spectrum of congenital heart defect phenotypes observed in mutant mice recovered from the forward genetic screen.
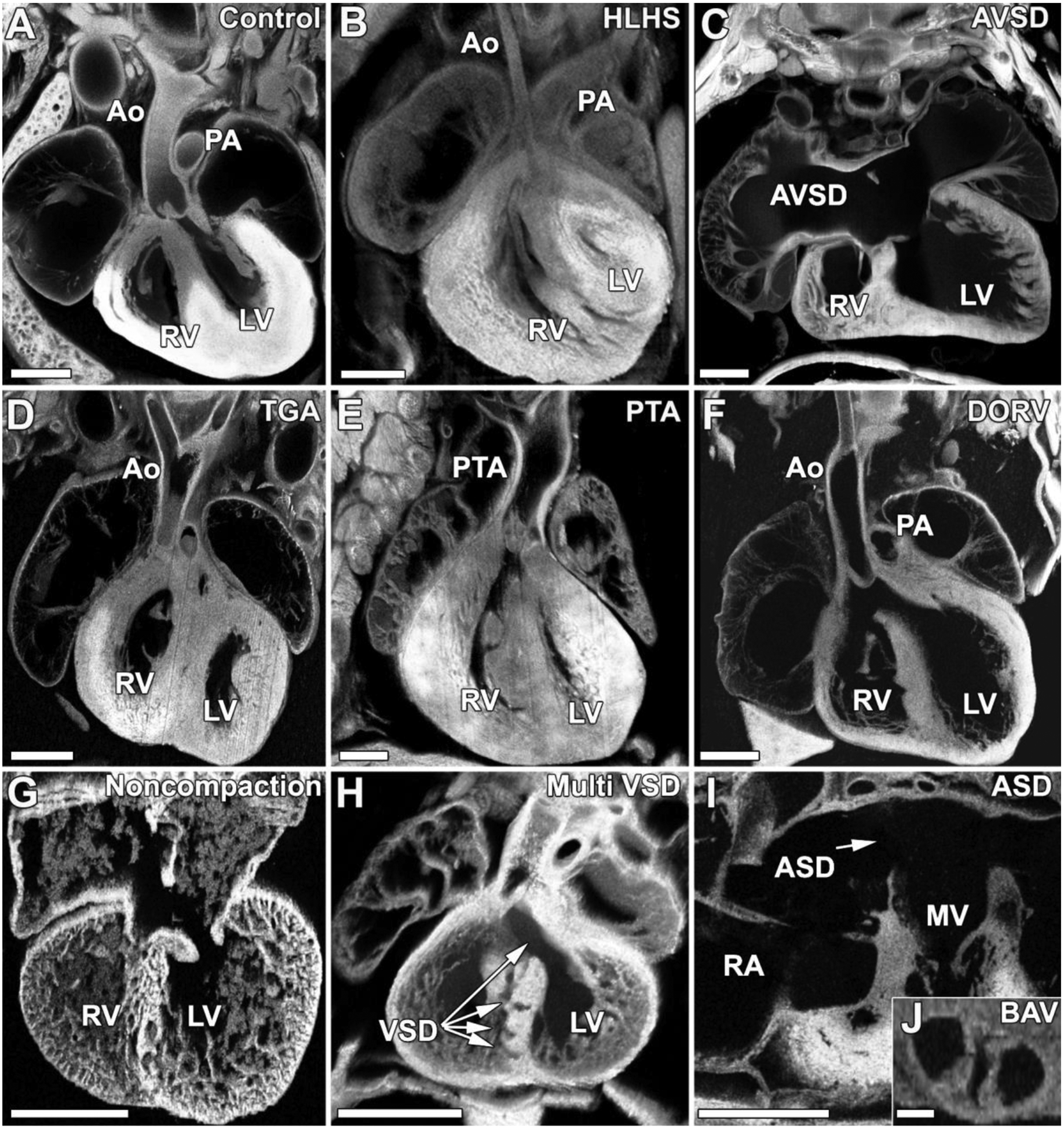
Mutant mice recovered from the mutagenesis screen exhibit a wide range of congenital heart disease phenotypes. For comparison a normal mouse heart is shown in (A). In panels (B-J) are shown a wide range of CHD phenotypes recovered from the screen including hypoplastic left heart syndrome (HLHS) in which left sided heart structures are underdeveloped (B), atrioventricular septal defect in which the septa between the right and left atria and the right and left ventricles fail to form properly (C), transposition of the great arteries (TGA) in which the aorta exits the right ventricle while the pulmonary artery emerges from the left ventricle (D), persistent truncus arteriosis (PTA) in which a single outflow tract arises from the ventricles (E), double outlet right ventricle (DORV) in which both the aorta and pulmonary arteries arises from the right ventricle (F), ventricular noncompaction in which the compact myocardium fails to form appropriately (G), swiss cheese heart in which multiple ventricular septal defects (VSD) are present (H), atrial septal defect (ASD) in which the atrial septum fails to form appropriately (I), or bicuspid aortic valve (BAV) in which the normally tricuspid aortic valve develops with only two cusps (J). Ao, aorta; PA, pulmonary artery; LV, left ventricle; RV, right ventricle; MV, mitral valve.
2.2. Left-Right Patterning Disturbance in the Pathogenesis of Congenital Heart Disease
Interestingly, approximately half of the mutant lines recovered with CHD also exhibited left-right patterning defects, indicating left-right patterning may have an important role in CHD pathogenesis[13]. Normal patterning of organ development includes the specification of left-right organ situs, with situs solitus referring to the normal organ left-right positioning. The heart apex points to the left (levocardia), the stomach and spleen are positioned on the left, and the lung and liver develop distinct left-right asymmetric pattern of lobation (Fig. 2). The bile duct is positioned on the right and the gut undergoes counterclockwise looping around the superior mesenteric artery. In the cardiovascular system, further specification of left-right asymmetry within the cardiac chambers and their left vs. right inflow/outflow connections are essential for establishing the systemic vs. pulmonary circulation needed for efficient oxygenation of blood in the lung and its delivery to the rest of the body. The disturbance of laterality can result in either situs inversus with complete mirror symmetric patterning of organ laterality, or heterotaxy with randomization of organ situs (Fig. 2). Thus, lines with CHD associated with laterality defects can yield mutants with heterotaxy or situs inversus. Importantly, such lines can also yield mutants with situs solitus, though such mice with normal visceral organ patterning typically show no CHD.
Figure 2. Mutations causing laterality defects can result in three situs phenotypes.
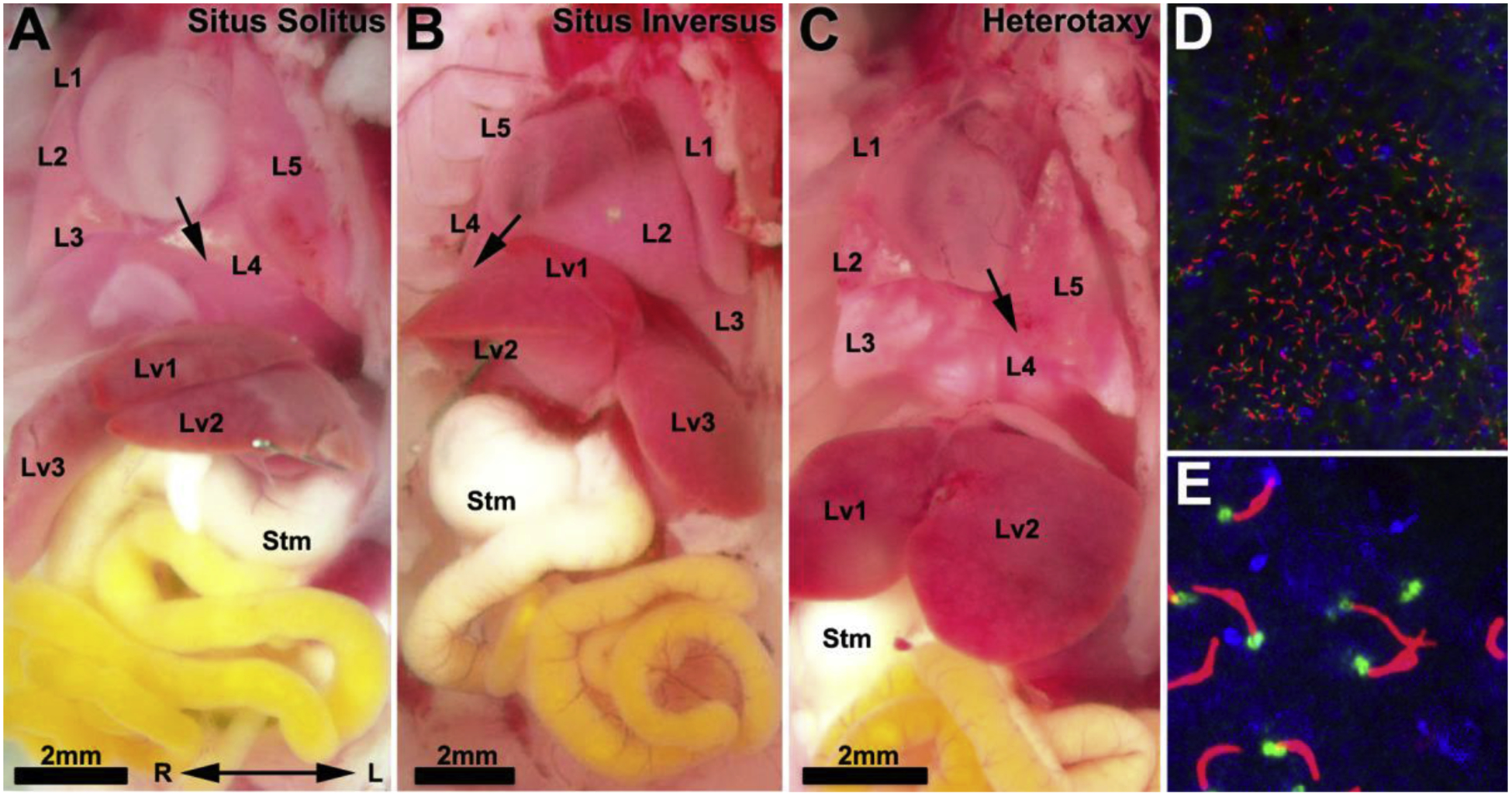
(A–C). Laterality mutants can present with three phenotypes: situs solitus with normal visceral organ patterning in which the heart apex points to the left with four right lung lobes and one left lung lobe, and the dominant liver lobe on the right (A); situs inversus with complete mirror reversal of visceral organ patterning (B); or heterotaxy, in which there is randomization of visceral organ patterning such that the heart apex in this mutant points leftward while the stomach (Stm) is positioned on the right (C). L1-L5, lung lobes 1–5; Lv1-Lv3, liver lobes 1–3; stm, stomach.
(D,E). Motile cilia in the embryonic node are visualized with acetylated tubulin (red) labeing the ciliary axoneme and gamma tubulin (green) labeling the basal body.
The association of complex CHD with heterotaxy is actually well described clinically, pointing to the importance of left-right patterning in CHD pathogenesis[16, 17]. Several epidemiological studies have provided further support for the importance of left-right patterning in human CHD. One such investigation reviewed data from the National Birth Defects Prevention Study (NBDPS) for nonsyndromic cases of situs inversus or heterotaxy cases and determined the incidence of CHD[18]. From 517 such cases, they showed a surprisingly high incidence of CHD, with 77% exhibiting CHD associated with either heterotaxy or situs inversus. The overall incidence of laterality defects in this registry was 1 in 10,000, similar to findings from other studies[18]. Hence, the co-occurrence of CHD and laterality defects is unlikely by chance. The large majority of CHD observed in these cases comprised complex CHD, 67.7% with heterotaxy and 9.3% with situs inversus[18]. This included cooccurrence of a wide spectrum of CHD lesions including single ventricle (SV) lesions, total anomalous pulmonary venous return (TAPVR), double outlet right ventricle (DORV), or atrioventricular septal defect (AVSD; Table 1)[18] with laterality defects. This was most commonly observed in association with heterotaxy (Table 1).
Table 1.
Prevalence of laterality defects among
| Ascertainmenta | SV | TAPVR | DORV | AVSD | PA | HLHS | TOF | TGA | PTA | TA |
|---|---|---|---|---|---|---|---|---|---|---|
| CHDb with Laterality Defects | 52.9% | 56.8% | 34.0% | 31.1% | 19.6% | 18.9% | 14.4% | 13.0% | 14.2% | 4.1% |
| Laterality Defects with CHDc | 11.8% | 23.4% | 20.9% | 36.8% | 4.9% | 2.7% | 3.9% | 3.1% | 0.8% | 1.7% |
| SIT with CHDd | 5.8% | 2.2% | 7.9% | 5.0% | 1.4% | 1.4% | 2.9% | 2.4% | 0% | 0.7% |
First ascertainment was either based on finding CHD or laterality defects. SV:single ventricle, TAPVR: total anomalous pulmonary venous return, DORV: double outlet right ventricle, AVSD: atrioventricular septal defects, PA: pulmonary atresia, HLHS: hypoplastic left heart syndrome, TOF: Tetralogy of Fallot, TGA: transposition of the great arteries; PTA: persistent truncus arteriosus, TA: tricuspid atresia
Based on Pradat et al.2003, CHD subjects found in several birth defects registry were examined for laterality defects (situs inversus, splenic abnormalities, gut malrotation) [19].
Based Lin et al. 2014 with cases having laterality defects comprising heterotaxy or situs inversus examined for CHD.
Based Lin et al. 2014 comprising cases with situs inversus further examined for CHD [18].
These findings are consistent with another epidemiological study primarily focused on CHD that also examined for the presence of other birth defects, including the disturbance of laterality[19]. In this study, large birth defects registries from California, France and Sweden were examined encompassing 4.4 million births[19]. From these births,12,000 infants with CHD were identified. The overall incidence of severe CHD was observed at 1.43 cases per 1000 births[19]. Examination for CHD cases that also had laterality defects indicated by the finding of situs inversus, splenic abnormalities, or gut malrotation showed a surprisingly high cooccurrence of laterality defects. This ranged from a high of 52.9% in patients with SV lesions to 4.1% in patients with tricuspid atresia (Table 1)[19]. As laterality defects are seen with an incidence of 1 in 10,000, their co-occurrence with CHD would indicate a mechanistic link. Laterality defects were highest for SV, TAPVR, DORV, and AVSD (Table 1). Comparison between the laterality vs. CHD focused analyses showed striking differences for SV and TAPVR. The CHD focused analysis showed 52.9% of SV and 56.8% of TAPVR patients also exhibited laterality defects, while the laterality focused analysis demonstrated that among laterality cases with CHD 11.8% had SV and 23.4% had TAPVR phenotypes (Table 1)[18, 19]. These differences may reflect the exclusion of extracardiac anomalies in the laterality focused study, as only nonsyndromic cases were examined. This would suggest the finding of CHD with laterality disturbance may be associated with increased risk for other birth defects. Overall, the same broad spectrum of structural heart defect phenotypes are observed in the CHD focused study, but generally with reduced prevalence compared to the laterality focused analysis (Table 1). It is interesting to note that the only lesion that is equally prevalent in both groups is AVSD, which is observed at 31.1% in the CHD study vs. 36.8% in the laterality focused study[18, 19]. This suggests the possibility that AVSD may be a lesion that is developmentally most intimately tied to the disturbance of laterality. Overall, these epidemiological studies indicate the pathogenesis of CHD is intimately interwoven with the disturbance of left-right patterning.
2.3. Central Role of Cilia in the Pathogenesis of Congenital Heart Disease
The CHD causing mutations in mutants recovered from our screen was carried out using whole exome sequencing analysis, allowing the successful identification of 91 CHD causing pathogenic mutations in 61 genes[13]. The pathogenic mutation in each line was readily identified by genotyping analysis, with the pathogenic mutation being the only mutation that is consistently homozygous in all of the CHD mutants. Interestingly, examination of the 61 genes showed 35 were cilia related (Fig.3)[13]. This enrichment for cilia genes was surprising, given the screen was completely phenotype driven and gene agnostic. Additionally, many of the other CHD causing mutations were in genes involved in hedgehog signaling and other cilia transduced cell signaling pathways[13]. Also observed was enrichment for mutations in genes mediating vesicular/endocytic trafficking, a cell process that is essential for ciliogenesis and in cilia transduced cell signaling[13, 20]. Together, these findings point to the perturbation of cilia playing a central role in the pathogenesis of CHD.
Figure 3. Pathogenic mutations in ciliome genes enriched in mouse ENU mutagenesis screen.
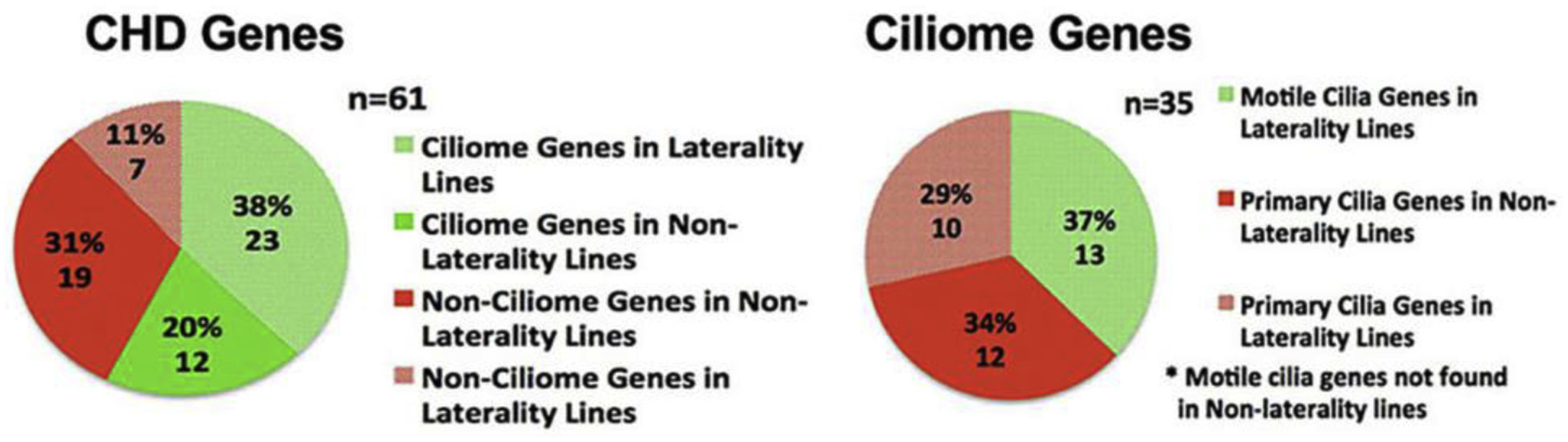
Exome sequencing from 113 CHD mutant mouse lines recovered 91 pathogenic mutations in 61 genes. 35 of these mutations affected cilia genes and caused CHD including 23 in lines exhibiting CHD with laterality defects, and 12 in lines without laterality defects. Note mutations in motile cilia genes were recovered only in mutant lines with laterality defects. Adapted from Li et al. Nature 2015 [13].
Given the known importance of cilia in left-right patterning, the enrichment for cilia genes may reflect the essential role of cilia in left-right patterning. This would be consistent with the epidemiological studies indicating the substantial involvement of left-right patterning defects in a broad spectrum of CHD lesions. Thus, of the 35 cilia-CHD genes recovered, 23 mediate CHD with laterality defects. These are comprised of 13 genes required for motile cilia function, and 10 in primary cilia function, consistent with the known requirement for both motile and primary cilia in the regulation of left-right patterning[13]. Motile cilia at the embryonic node mediate leftward nodal flow[21, 22]. This flow is then sensed by primary cilia at the node periphery leading to downstream activation of the nodal signaling cascade, though the precise mechanisms remain uncertain[23]. However, it is important to note that the role of cilia in CHD pathogenesis is not simply to regulate left-right patterning, as 12 (34%) of the primary cilia-CHD genes do not cause laterality defects. As many of cilia transduced cell signaling pathways are known to play an important role in cardiovascular development, including Shh, Wnt, Pdgf, and Tgfβ-BMP signaling, the disruption of these cell signaling pathways could play a role in CHD pathogenesis[24].
3. Role of Motile Cilia in CHD Pathogenesis
Most of the motile cilia-CHD mutations recovered in our screen are in genes known to cause primary ciliary dyskinesia (PCD), a sinopulmonary disease associated with mucociliary clearance deficits due to immotile or dyskinetic cilia in the airway. All of the motile cilia mutations were observed to cause CHD in the setting of heterotaxy. In fact, PCD is well described to be associated with male infertility and situs inversus, a spectrum of phenotypes previously referred to as Kartagener’s syndrome[25]. However, the link between PCD and CHD was not discovered until more recent studies showed PCD patients can exhibit heterotaxy and in such PCD-heterotaxy patients, CHD is commonly observed[26, 27]. It is interesting to note that while many of our mutations in PCD genes caused immotility or weak flickering dyskinetic ciliary motion in the airway, ciliary motion in the brain ependyma is often preserved, albeit the cilia waveform and beat frequency may be abnormal[13]. In mice with mutations in PCD-cilia genes, hydrocephalus is often observed, indicating cilia generated flow in the brain may play an important role in the maintenance of cerebrospinal fluid (CSF) homeostasis. However, PCD patients rarely exhibit hydrocephalus, with previous studies suggesting only 1.3% of PCD patients have hydrocephalus[28]. Interestingly, we also recovered a CHD causing mutation in FoxJ1, a transcription factor known to regulate the expression of genes required for motile cilia formation[29]. The FoxJ1 mutant mice exhibited CHD associated with heterotaxy, as with mutations in other PCD genes, and they also exhibited hydrocephalus. Importantly, FOXJ1 mutations were previously found to be associated with PCD patients displaying hydrocephalus [30]. Hydrocephalus may arise with FOXJ1 mutation perhaps because of its broad transcriptional effects on regulating motile ciliogenesis, suggesting there may be more ependymal cilia gene related redundancies in human as compared to mice.
4. Primary Cilia and CHD Pathogenesis
Among the 22 primary cilia related gene recovered, we noted four genes, Sufu, Kif7, Jbts17, and Lrp2 are associated with cilia transduced Shh signaling, a pathway known to have important roles in cardiovascular development[31]. Indeed, among non-cilia CHD genes recovered, an enrichment is observed for genes in cilia transduced cell signaling pathways, including Tbc1d32 and Megf8 involved in Shh signaling, Fuz, Ptk7, and Prickle1 in Wnt signaling, and Cfc1, Ltbp1, Pcks5, Tab1, and Smad6 in Tgfβ/BMP signaling[13]. These findings suggest one of the important roles of cilia in heart development and CHD pathogenesis is the modulation of cilia transduced cell signaling.
Also worth noting is the fact that many of the CHD-cilia related genes recovered in our screen are in the ciliogenesis planar cell polarity (CPLANE) protein-protein interaction network[32]. The CPLANE network was recently identified by proteomic analysis, and has been shown to play an important role in the regulation of ciliogenesis and cilia transduced cell signaling[32]. Strikingly, we found three of the five core components of the CPLANE network, Fuz, Jbts17, and Wdpcp, are among the CHD-cilia genes recovered in our screen[13, 32, 33]. These core CPLANE genes are highly conserved in evolution and are found from sea anemone to man[34]. This suggests the ancient function of the CPLANE core proteins have evolved in the context of the cilia to regulate heart development, the first and most critical organ to form in the mammalian embryo.
5. One Mutation Giving Rise to Three Distinct Laterality Phenotypes
The analysis of many mutant lines with CHD associated with heterotaxy have shown that in most lines the same mutation can give rise to three different laterality phenotypes comprising either normal situs solitus, situs inversus totalis, or heterotaxy, with CHD typically only observed with heterotaxy (Figure 2)[35]. This contrasts with the epidemiological studies showing CHD in the human population can be observed with situs inversus albeit at much lower incidence then with heterotaxy (Table 1)[19]. This difference is likely a reflection of the inbred genetic background and monogenic etiology of disease in mice, while disease in the genetically heterogeneous human population is more likely to exhibit oligogenic interactions associated with more complex genetics. We note the finding of three different laterality phenotypes from a single mutation is observed for mutations in all of the motile cilia/PCD related genes and also some of the primary cilia and non-cilia mutations causing CHD with heterotaxy. However, it is notable that mutations in some genes consistently give rise to heterotaxy only, such as Megf8 a negative regulator of Shh signaling, or Mmp21, a matrix metalloprotease[36–40]. The observation that mutations in some genes can yield three different laterality phenotypes, with CHD seen only with heterotaxy, would suggest there are normal individuals with the same disease-causing genotype as individuals with CHD and heterotaxy in the human population. Such individuals with “masked” disease genotypes would confound efforts to recover pathogenic mutations. Given the epidemiological findings showing disturbance of left-right patterning in a broad spectrum of human CHD, this suggests the possibility that some of the well described variable penetrance and variable expressivity seen in human CHD might be related to laterality related masking of disease phenotypes.
6.1. Hedgehog Signaling Coordinates Left-right Patterning with Heart Looping
Cilia transduced hedgehog (Hh) signaling is required for normal establishment of left-right asymmetry, including left-right patterning of the lateral plate mesoderm (LPM), the tissue from which the heart tube is derived. In Smo KO embryos deficient in HH signaling, the left sided activation of Lefty and Nodal in the LPM is not observed, and while the heart tube forms, it does not loop[41, 42]. This heart looping defect is associated with defects in myocardial differentiation which may be due to failure to upregulate expression of the cardiac transcription factor Nkx2.5[41]. Thus, Hh signaling plays a dual role during early development - it is required for establishing normal left-right asymmetry, and it also regulates cardiomyocyte differentiation and looping of the heart tube. This coordinate regulation can help ensure heart looping is appropriately timed relative to specification of the left-right axis and differentiation of the myocardium. While Shh is known to regulate many of the later cardiovascular developmental processes, the looping of the heart tube appears to be orchestrated by Indian Hedgehog (Ihh), as it is the only Hh member expressed in the definitive endoderm directly underlying the LPM containing the cardiac progenitors[42].
Thus, specification of the left-right body axis and right sided looping of the heart tube, are both Hh dependent processes, pointing to the central importance of cilia and left-right patterning in heart development. This is consistent with findings from both our mouse screen and the human epidemiological studies showing the disturbance of left-right patterning is closely linked to the pathogenesis of CHD. We note the negative regulation of Shh may have particular importance, as several negative regulators of Shh signaling recovered from CRISPR screens are known to disrupt normal heart development in conjunction with the disturbance of left-right patterning[38, 43]. Thus, mutations in Megf8 cause heterotaxy with CHD[44]. Interesting to note it causes heterotaxy exclusively, and always with CHD comprising transposition of the great arteries (TGA) with either a D or L-looped heart (Figs.1D and 4) [38, 39]. This suggests the precise level of Hh signaling is critically important and must be fine-tuned, first early in development to direct right sided looping of the heart tube, and then later to direct proper patterning of outflow tract alignment.
Figure 4. Megf8 mutant mice with dextrocardia and transposition of the great arteries.
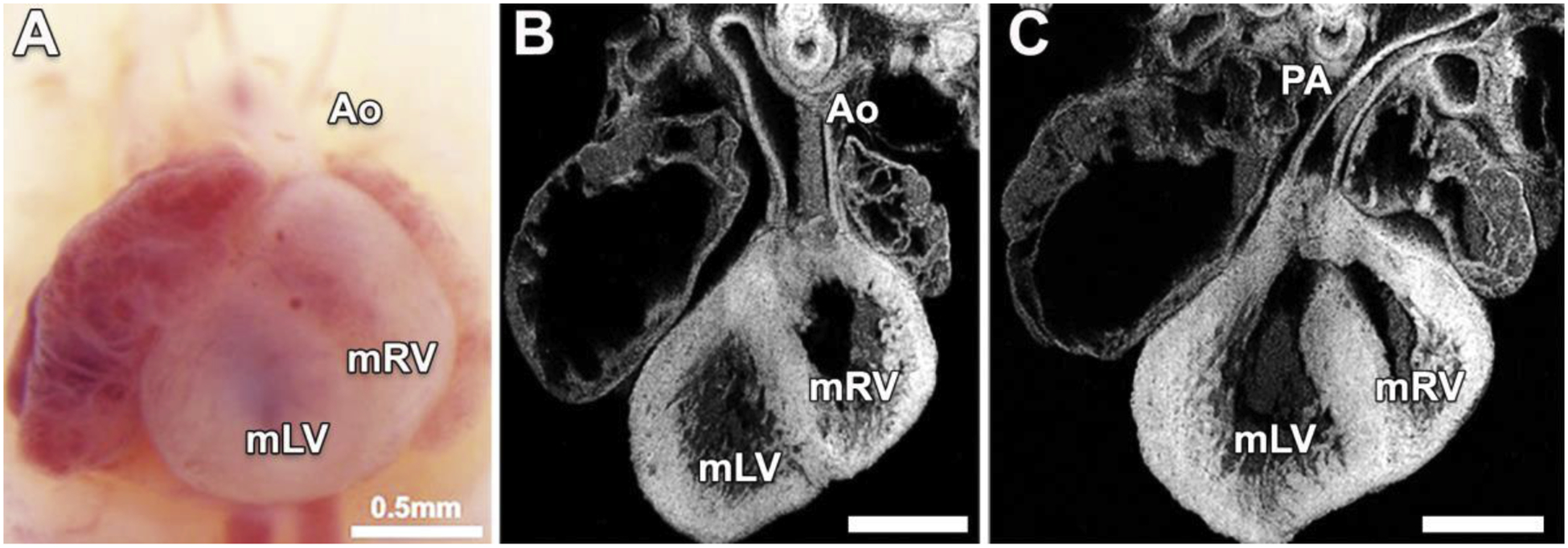
Necropsy image (A) and histological sections of the same heart (B,C) from a Megf8 mutant mouse show dextrocardia with transposition of the great arteries (TGA). Note the heart apex points to the right, with the aorta (Ao) connected to the morphological right ventricle (mRV) located on the anatomical left (B), and the pulmonary artery (PA) connected to the morphological left ventricle (mLV) located on the anatomical right (C).
6.2. Shh Deficiency Causes AVSD and Outflow Tract Septation Defects
Analysis of the Shh KO mice has further shown a critical role for Shh signaling in atrioventricular septation. The atrioventricular septum is largely derived from the dorsal mesenchyme protrusion (DMP), a structure comprising of cells derived from the second heart field. Shh signaling is required for proper migration of the DMP into the atrium to generate the atrioventricular septum. Thus, Shh KO embryos exhibit AVSD, a CHD phenotype that was seen with equally high prevalence in both epidemiological studies examining the cooccurrence of CHD with laterality defects (Table 1)[31, 45, 46]. This suggests the possibility that these second heart field progenitors may be specified at the earliest stages of heart development, perhaps at the time when left-right patterning is first specified at the LPM in the 3–6 somite stages. In addition, the Shh KO embryos also were observed to have an outflow tract septation defect, exhibiting persistent truncus arteriosus (PTA) (Fig.1E)[31]. This is due to a dual requirement for Shh signaling in both cardiac neural crest cells and in cells of the second heart field[31]. We note unlike AVSD, the PTA phenotype was barely detected in the laterality focused human study at 0.8%, while in the CHD focused study, it was seen with 10-fold higher incidence at 14.2% (Table 1)[18, 19]. This suggests outflow tract septation may be less dependent on the very early left-right patterning developmental processes than atrioventricular septation.
7.1. Role of Cilia in Common Valve Defects
Primary cilia are present in the endocardial cushions, which will give rise to the heart valves (Fig.5) [47]. They play important roles in coordinating several signaling pathways known to play an important role in cardiac valve development including Notch, Shh, Wnt, Pdgf, and Tgfb signaling[48]. Additionally, primary cilia in the heart valve primordia may sense and respond to changes in shear stress to initiate proper valve development[49, 50]. Supporting the essential role of cilia in heart valve development are the recent studies showing defects in primary cilia are linked to common valve defects, including bicuspid aortic valve and mitral valve prolapse, both valve defects that usually present later in adult life[51–53]. This suggests that developmental abnormalities may be acted upon and elaborated by changes occurring over the lifetime that may result in disease, such as calcific deposits, abnormal matrix deposition or fibrosis causing stiffening of the valves. Further, the fact that we can detect cilia in adult mouse heart valve tissue indicates that cilia defects may have a further role to play in heart valve homoestasis and disease (Fig.6). Relevant to this are recent studies showing cilia perturbation may contribute to cardiac fibrosis[54].
Figure 5. Cc2d2a mutant mice with laterality defects show AVSD with cilia defects in the AV cushion mesenchyme.
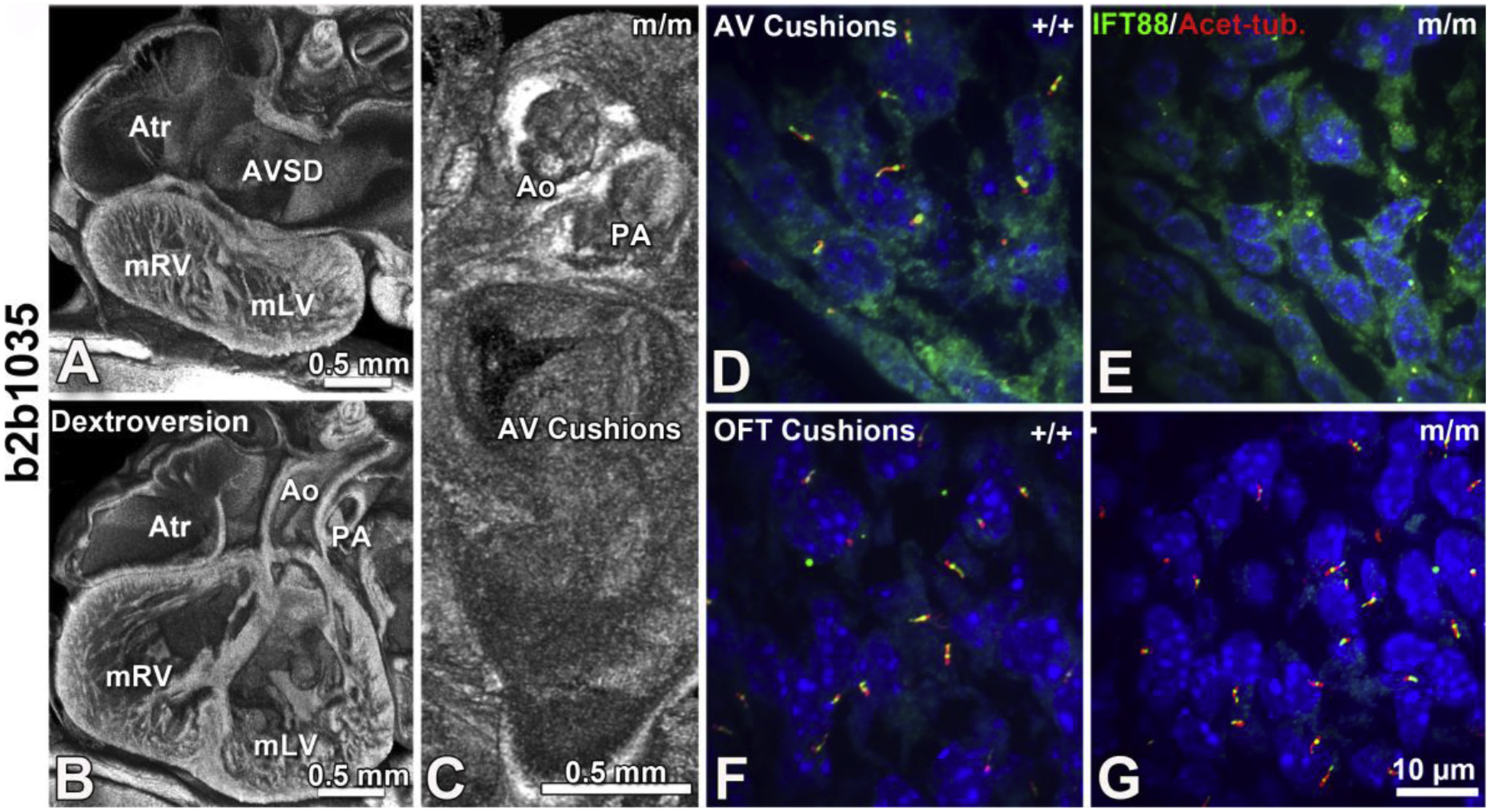
Mice with mutations in Cc2d2a present with laterality defects and congenital heart disease including dextroversion and atrioventricular septal defects (AVSD, A,B) with malformation of the atrioventricular cushions but normal outflow tract patterning (C). Consistent with this Cc2d2a mutant mice also show reduced cilia in the AV cushions compared to control (D vs E), but normal ciliation in the outflow tract cushions (F vs G). Adapted from Li et al. Nature 2015 [13].
Figure 6. Primary cilia in adult mouse aortic valve.
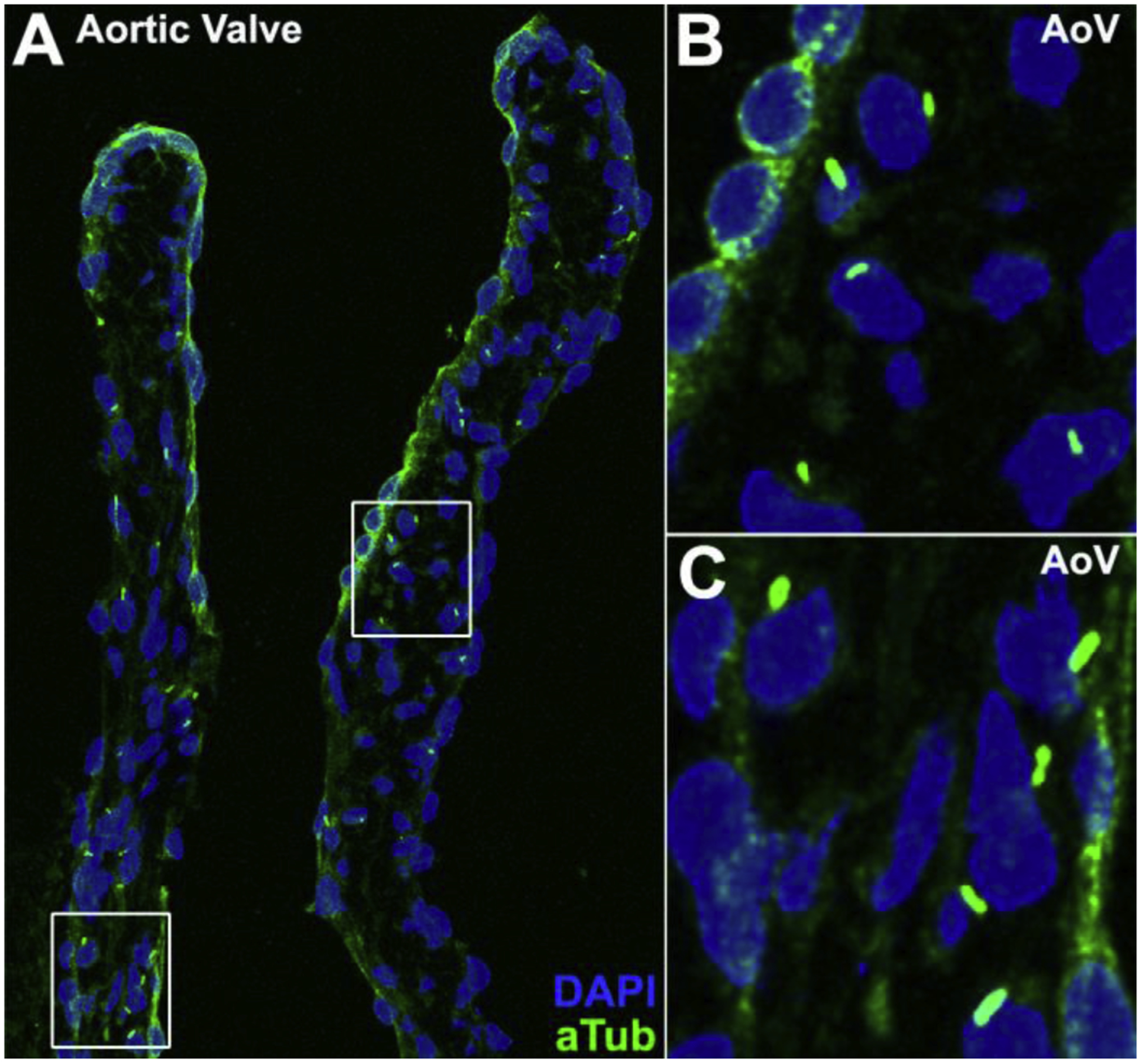
Primary cilia are observed in the aortic valve of a 3-month-old C57BL6/J mouse with immunostaining using an acetylated tubulin antibody (A). Boxed regions in panel (A) are shown in enlarged views in panels (B) and (C).
7.2. Bicuspid Aortic Valve
Bicuspid aortic valve (BAV) is in fact the most common birth defect with an estimated prevalence of 1–3% in the general population[2]. BAV has been reported to be associated with ciliopathies, a class of syndromic diseases associated with cilia defects[55]. This has been confirmed by recent work that has linked BAV with primary cilia defects[56, 57]. In mouse, primary cilia can be found on aortic valve interstitial cells during aortic valve development, where they participate in Shh signaling[56]. Genetic ablation of primary cilia in these cells results in highly penetrant BAV phenotype with aortic stenosis[56]. In humans, a recent genome wide association study identified an association between BAV phenotypes and mutations affecting primary cilia[57]. Specifically, mutations in or near several genes associated with the exocyst, a complex necessary for ciliogenesis, were found to be highly associated with BAV[57]. Further, knock-out of the associated exocyst gene Exoc5 in both mouse and zebrafish resulted in ciliogenesis defects and ciliopathy phenotypes including BAV[57].
7.3. Mitral Valve Prolapse
Mitral valve prolapse is another common cardiac valve disease that affects 2–3% of the population[58]. In this disorder the mitral valve does not close properly, often due to myxomatous valve disease, which can result in mitral regurgitation and necessitate mitral valve surgery[58]. This disease has previously been associated with the ciliopathy polycystic kidney disease, suggesting primary cilia may also play a role in myxomatous valve degeneration and mitral valve prolapse[59]. This is supported by studies identifying cilia mutations causing mitral valve prolapse. The first gene identified to cause non-syndromic mitral valve prolapse was Filamentin-A (FLNA)[60, 61]. FLNA interacts with MKS3 at the primary cilium and is necessary for ciliogenesis and proper orientation of the basal body, the centrosome derived structure forming the base of the cilia [62]. More recent studies have identified the cilia gene Dachsous1 (DCHS1) to cause mitral valve prolapse[63]. DCHS1 is localized to the base of the cilium and play a role in planar cell polarity signaling[64]. Additional studies further revealed mutations in DAZ Interacting Zinc Finger Protein 1 (DZIP1), another cilia gene, to also cause mitral valve prolapse[65]. Mutations in DZIP1 were identified to cause mitral valve prolapse in a multigenerational family with inherited autosomal dominant disease[65]. DZIP1 was previously found to localize to the centrosome where it modulates Shh signaling[66]. Knock-out of Dzip1 in mice caused primary cilia defects with impaired extracellular matrix deposition during heart valve development that resulted in myxomatous valve degeneration and mitral valve prolapse[65]. Together, these studies demonstrate the importance of primary cilia in mitral valve development, and the role of cilia defects in mitral valve prolapse.
8. Conclusions
Our forward genetic screen showed cilia plays an important role in the pathogenesis of CHD. This involves both primary and motile cilia, and cilia transduced cell signaling. The involvement of left-right patterning defects is suggested not only by the association of CHD with heterotaxy, but also by the high incidence seen for co-occurrence of laterality defects with CHD in analysis of data from several human birth defect registries. The recovery of many CPLANE genes among the CHD causing mutations, including the highly conserved CPLANE core components, suggest the ancient function of CPLANE genes have been recruited to regulate development of the heart. This involves a role for cilia in mediating hedgehog signaling and coordinating regulation of left-right patterning with early development of the heart tube. The additional roles of cilia in mediating later events in heart development include the regulation of outflow tract septation, atrioventricular septum formation, and the regulation of valvular morphogenesis. A role for cilia is also observed in valve diseases presenting mostly later in adult life. Overall, these findings indicate the role of cilia in congenital heart disease may extend throughout life. Further investigations are needed to elucidate how disturbances of left-right patterning may contribute to the developmental etiology of congenital heart disease and what might be the role for the CPLANE network in these developmental and disease processes.
Highlights.
Cilia and cilia transduced cell signaling have key roles in congenital heart disease
Disturbance in left-right patterning closely linked to congenital heart disease
Shh signaling coordinates left-right patterning with heart development
Negative regulation of Shh critical for left-right patterning and heart development
Cilia defects also contribute to valve disease prevalent in adults
Funding Sources:
This work was supported by NIH grant 1F30HD097967 (GCG), NIH grants HL142788 and HL132024 (CWL), and DOD grants W81XWH-15-1-0649 and W81XWH-16-1-0613 (CWL).
Abbreviations:
- ASD
atrial septal defect
- AVSD
atrioventricular septal defect
- BAV
bicuspid aortic valve
- CHD
congenital heart disease/defect
- CPLANE
ciliogenesis planar cell polarity
- DMP
dorsal mesenchyme protrusion
- DORV
double outlet right ventricle
- ENU
ethylnitrosourea
- Hh
hedgehog
- HLHS
hypoplastic left heart syndrome
- KO
knock out
- LPM
lateral plate mesoderm
- NBDPS
National Birth Defects Prevention Study
- PCD
primary ciliary dyskinesia
- PTA
persistent truncus arteriosus
- Shh
sonic hedgehog
- TAPVR
total anomalous pulmonary venous return
- TGA
transposition of the great arteries
- VSD
ventricular septal defect
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflict of interest.
References
- [1].Liu Y, Chen S, Zuhlke L, Black GC, Choy MK, Li N, et al. , Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies, Int J Epidemiol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Longobardo L, Jain R, Carerj S, Zito C, Khandheria BK, Bicuspid aortic valve: unlocking the morphogenetic puzzle, The American journal of medicine 129(8) (2016) 796–805. [DOI] [PubMed] [Google Scholar]
- [3].Tutar E, Ekici F, Atalay S, Nacar N, The prevalence of bicuspid aortic valve in newborns by echocardiographic screening, American heart journal 150(3) (2005) 513–515. [DOI] [PubMed] [Google Scholar]
- [4].Gupta A, Amin Z, Popular Hybrid Congenital Heart Procedures without Cardiopulmonary Bypass, Frontiers in surgery 4 (2017) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Holst KA, Said SM, Nelson TJ, Cannon BC, Dearani JA, Current interventional and surgical management of congenital heart disease: specific focus on valvular disease and cardiac arrhythmias, Circulation research 120(6) (2017) 1027–1044. [DOI] [PubMed] [Google Scholar]
- [6].Gilboa SM, Devine OJ, Kucik JE, Oster ME, Riehle-Colarusso T, Nembhard WN, et al. , Congenital heart defects in the United States: estimating the magnitude of the affected population in 2010, Circulation 134(2) (2016) 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cohen S, Gurvitz MZ, Beauséjour-Ladouceur V, Lawler PR, Therrien J, Marelli AJ, Cancer Risk in Congenital Heart Disease–What is The Evidence?, Canadian Journal of Cardiology (2019). [DOI] [PubMed] [Google Scholar]
- [8].Lui GK, Saidi A, Bhatt AB, Burchill LJ, Deen JF, Earing MG, et al. , Diagnosis and management of noncardiac complications in adults with congenital heart disease: a scientific statement from the American Heart Association, Circulation 136(20) (2017) e348–e392. [DOI] [PubMed] [Google Scholar]
- [9].Sanz JH, Berl MM, Armour AC, Wang J, Cheng YI, Donofrio MT, Prevalence and pattern of executive dysfunction in school age children with congenital heart disease, Congenital heart disease 12(2) (2017) 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rollins CK, Newburger JW, Roberts AE, Genetic Contribution to Neurodevelopmental Outcomes in Congenital Heart Disease: Are Some Patients Pre-Determined to Have Developmental Delay?, Current opinion in pediatrics 29(5) (2017) 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rosenthal N, Brown S, The mouse ascending: perspectives for human-disease models, Nature cell biology 9(9) (2007) 993–999. [DOI] [PubMed] [Google Scholar]
- [12].Wessels A, Sedmera D, Developmental anatomy of the heart: a tale of mice and man, Physiological genomics 15(3) (2003) 165–176. [DOI] [PubMed] [Google Scholar]
- [13].Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, et al. , Global genetic analysis in mice unveils central role for cilia in congenital heart disease, Nature 521(7553) (2015) 520–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu X, Francis R, Kim AJ, Ramirez R, Chen G, Subramanian R, et al. , Interrogating congenital heart defects with noninvasive fetal echocardiography in a mouse forward genetic screen, Circulation: Cardiovascular Imaging 7(1) (2014) 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hornberger LK, Sanders SP, Rein AJ, Spevak PJ, Parness IA, Colan SD, Left heart obstructive lesions and left ventricular growth in the midtrimester fetus: a longitudinal study, Circulation 92(6) (1995) 1531–1538. [DOI] [PubMed] [Google Scholar]
- [16].Desgrange A, Le Garrec J-F, Meilhac SM, Left-right asymmetry in heart development and disease: forming the right loop, Development 145(22) (2018). [DOI] [PubMed] [Google Scholar]
- [17].Mishra S, Cardiac and non-cardiac abnormalities in heterotaxy syndrome, The Indian Journal of Pediatrics 82(12) (2015) 1135–1146. [DOI] [PubMed] [Google Scholar]
- [18].Lin AE, Krikov S, Riehle‐Colarusso T, Frías JL, Belmont J, Anderka M, et al. , Laterality defects in the national birth defects prevention study (1998–2007): birth prevalence and descriptive epidemiology, American journal of medical genetics Part A 164(10) (2014) 2581–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pradat P, Francannet C, Harris J, Robert E, The epidemiology of cardiovascular defects, part I: a study based on data from three large registries of congenital malformations, Pediatric cardiology 24(3) (2003) 195–221. [DOI] [PubMed] [Google Scholar]
- [20].Pedersen LB, Mogensen JB, Christensen ST, Endocytic control of cellular signaling at the primary cilium, Trends in biochemical sciences 41(9) (2016) 784–797. [DOI] [PubMed] [Google Scholar]
- [21].Nakamura T, Hamada H, Left-right patterning: conserved and divergent mechanisms, Development 139(18) (2012) 3257–3262. [DOI] [PubMed] [Google Scholar]
- [22].Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, et al. , Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein, Cell 95(6) (1998) 829–837. [DOI] [PubMed] [Google Scholar]
- [23].Shinohara K, Hamada H, Cilia in left–right symmetry breaking, Cold Spring Harbor perspectives in biology 9(10) (2017) a028282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Koefoed K, Veland IR, Pedersen LB, Larsen LA, Christensen ST, Cilia and coordination of signaling networks during heart development, Organogenesis 10(1) (2014) 108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW, Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease, American journal of respiratory and critical care medicine 188(8) (2013) 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harrison MJ, Shapiro AJ, Kennedy MP, Congenital heart disease and primary ciliary dyskinesia, Paediatric respiratory reviews 18 (2016) 25–32. [DOI] [PubMed] [Google Scholar]
- [27].Engesaeth VG, Warner J, Bush A, New associations of primary ciliary dyskinesia syndrome, Pediatric pulmonology 16(1) (1993) 9–12. [DOI] [PubMed] [Google Scholar]
- [28].Behan L, Dimitrov BD, Kuehni CE, Hogg C, Carroll M, Evans HJ, et al. , PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia, European respiratory journal 47(4) (2016) 1103–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yu X, Ng CP, Habacher H, Roy S, Foxj1 transcription factors are master regulators of the motile ciliogenic program, Nature genetics 40(12) (2008) 1445. [DOI] [PubMed] [Google Scholar]
- [30].Wallmeier J, Frank D, Shoemark A, Nöthe-Menchen T, Cindric S, Olbrich H, et al. , De Novo Mutations in FOXJ1 Result in a Motile Ciliopathy with Hydrocephalus and Randomization of Left/Right Body Asymmetry, The American Journal of Human Genetics 105(5) (2019) 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smoak IW, Byrd N, Abu-Issa R, Goddeeris M, Anderson R, Morris J, et al. , Sonic hedgehog is required for cardiac outflow tract and neural crest cell development, Developmental biology 283(2) (2005) 357–372. [DOI] [PubMed] [Google Scholar]
- [32].Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel A-L, et al. , The ciliopathy associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery, Nature genetics 48(6) (2016) 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, et al. , Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton, PLoS biology 11(11) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Adler PN, Wallingford JB, From planar cell polarity to ciliogenesis and back: the curious tale of the PPE and CPLANE proteins, Trends in cell biology 27(5) (2017) 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tan SY, Rosenthal J, Zhao X-Q, Francis RJ, Chatterjee B, Sabol SL, et al. , Heterotaxy and complex structural heart defects in a mutant mouse model of primary ciliary dyskinesia, The Journal of clinical investigation 117(12) (2007) 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Akawi N, McRae J, Ansari M, Balasubramanian M, Blyth M, Brady AF, et al. , Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families, Nature genetics 47(11) (2015) 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guimier A, Gabriel GC, Bajolle F, Tsang M, Liu H, Noll A, et al. , MMP21 is mutated in human heterotaxy and is required for normal left-right asymmetry in vertebrates, Nature genetics 47(11) (2015) 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pusapati GV, Kong JH, Patel BB, Krishnan A, Sagner A, Kinnebrew M, et al. , CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog, Developmental cell 44(1) (2018) 113–129. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang Z, Alpert D, Francis R, Chatterjee B, Yu Q, Tansey T, et al. , Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy, Proceedings of the National Academy of Sciences 106(9) (2009) 3219–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Perles Z, Moon S, Ta-Shma A, Yaacov B, Francescatto L, Edvardson S, et al. , A human laterality disorder caused by a homozygous deleterious mutation in MMP21, Journal of medical genetics 52(12) (2015) 840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang XM, Ramalho-Santos M, McMahon AP, Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node, Cell 105(6) (2001) 781–792. [PubMed] [Google Scholar]
- [42].Tsiairis CD, McMahon AP, An Hh-dependent pathway in lateral plate mesoderm enables the generation of left/right asymmetry, Current Biology 19(22) (2009) 1912–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Breslow DK, Hoogendoorn S, Kopp AR, Morgens DW, Vu BK, Kennedy MC, et al. , A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies, Nature genetics 50(3) (2018) 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Aune CN, Chatterjee B, Zhao X-Q, Francis R, Bracero L, Yu Q, et al. , Mouse model of heterotaxy with single ventricle spectrum of cardiac anomalies, Pediatric research 63(1) (2008) 9–14. [DOI] [PubMed] [Google Scholar]
- [45].Briggs LE, Burns TA, Lockhart MM, Phelps AL, Van den Hoff MJ, Wessels A, Wnt/β‐ catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion, Developmental Dynamics 245(2) (2016) 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Goddeeris MM, Rho S, Petiet A, Davenport CL, Johnson GA, Meyers EN, et al. , Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart, Development 135(10) (2008) 1887–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Slough J, Cooney L, Brueckner M, Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis, Dev Dyn 237(9) (2008) 2304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wheway G, Nazlamova L, Hancock JT, Signaling through the primary cilium, Frontiers in cell and developmental biology 6 (2018) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Van der Heiden K, Groenendijk BC, Hierck BP, Hogers B, Koerten HK, Mommaas AM, et al. , Monocilia on chicken embryonic endocardium in low shear stress areas, Dev Dyn 235(1) (2006) 19–28. [DOI] [PubMed] [Google Scholar]
- [50].Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, et al. , Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition, Circ Res 108(9) (2011) 1093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Avierinos J-F, Gersh BJ, Melton Iii LJ, Bailey KR, Shub C, Nishimura RA, et al. , Natural history of asymptomatic mitral valve prolapse in the community, Circulation 106(11) (2002) 1355–1361. [DOI] [PubMed] [Google Scholar]
- [52].Zuppiroli A, Rinaldi M, Kramer-Fox R, Favilli S, Roman MJ, Devereux RB, Natural history of mitral valve prolapse, The American journal of cardiology 75(15) (1995) 1028–1032. [DOI] [PubMed] [Google Scholar]
- [53].Michelena H, Desjardins V, Avierinos J, Valvulopathies| Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community, in: Circulation, 117 (2008), pp. 2776–2784, et al. , Elsevier Masson, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Villalobos E, Criollo A, Schiattarella GG, Altamirano F, French KM, May HI, et al. , Fibroblast primary cilia are required for cardiac fibrosis, Circulation 139(20) (2019) 2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Karp N, Grosse-Wortmann L, Bowdin S, Severe aortic stenosis, bicuspid aortic valve and atrial septal defect in a child with Joubert Syndrome and Related Disorders (JSRD) - a case report and review of congenital heart defects reported in the human ciliopathies, Eur J Med Genet 55(11) (2012) 605–10. [DOI] [PubMed] [Google Scholar]
- [56].Toomer KA, Fulmer D, Guo L, Drohan A, Peterson N, Swanson P, et al. , A role for primary cilia in aortic valve development and disease, Dev Dyn 246(8) (2017) 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fulmer D, Toomer K, Guo L, Moore K, Glover J, Moore R, et al. , Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis, Circulation 140(16) (2019) 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Levine RA, Hagege AA, Judge DP, Padala M, Dal-Bianco JP, Aikawa E, et al. , Mitral valve disease--morphology and mechanisms, Nat Rev Cardiol 12(12) (2015) 689–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lumiaho A, Ikaheimo R, Miettinen R, Niemitukia L, Laitinen T, Rantala A, et al. , Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1, Am J Kidney Dis 38(6) (2001) 1208–16. [DOI] [PubMed] [Google Scholar]
- [60].Le Tourneau T, Le Scouarnec S, Cueff C, Bernstein D, Aalberts JJJ, Lecointe S, et al. , New insights into mitral valve dystrophy: a Filamin-A genotype-phenotype and outcome study, Eur Heart J 39(15) (2018) 1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kyndt F, Gueffet J-P, Probst V, Jaafar P, Legendre A, Bouffant FL, et al. , Mutations in the Gene Encoding Filamin A as a Cause for Familial Cardiac Valvular Dystrophy, Circulation 115(1) (2007) 40–49. [DOI] [PubMed] [Google Scholar]
- [62].Adams M, Simms RJ, Abdelhamed Z, Dawe HR, Szymanska K, Logan CV, et al. , A meckelin-filamin A interaction mediates ciliogenesis, Hum Mol Genet 21(6) (2012) 1272–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, et al. , Mutations in DCHS1 cause mitral valve prolapse, Nature 525(7567) (2015) 109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dau C, Fliegauf M, Omran H, Schlensog M, Dahl E, van Roeyen CR, et al. , The atypical cadherin Dachsous1 localizes to the base of the ciliary apparatus in airway epithelia, Biochem Biophys Res Commun 473(4) (2016) 1177–1184. [DOI] [PubMed] [Google Scholar]
- [65].Toomer KA, Yu M, Fulmer D, Guo L, Moore KS, Moore R, et al. , Primary cilia defects causing mitral valve prolapse, Sci Transl Med 11(493) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang C, Low WC, Liu A, Wang B, Centrosomal protein DZIP1 regulates Hedgehog signaling by promoting cytoplasmic retention of transcription factor GLI3 and affecting ciliogenesis, J Biol Chem 288(41) (2013) 29518–29. [DOI] [PMC free article] [PubMed] [Google Scholar]