

To the Editor:
Zebrafish (Danio rerio) has become a well-established and powerful in vivo model to study the molecular basis of T-cell acute lymphoblastic leukemia (T-ALL) [1]. Langenau et al. [2] were the first to generate a cancer model using transgenesis. Expression of the mouse Myc oncogene under the D. rerio lymphoblast-specific rag2 promoter resulted in rapid onset of leukemia [2]. However, since the development of this Myc model, the number of other oncogenedriven T-ALL models in zebrafish has remained limited.
The TLX1-driven T-ALL subtype encompasses 5–10% of pediatric and 30% of adult T-ALL cases, and is caused by a translocation that juxtaposes the TLX1 transcription factor near the T-cell receptor α or β enhancer region [3]. TLX1 acts as an oncogene in T-ALL, mainly by binding DNA regulatory regions to repress a network of T-ALL tumor suppressor genes including BCL11B, RUNX1, WT1, TET1, EZH2, FBXW7, PTEN, PTPN2 and mitotic checkpoint genes, leading to genomic instability and aneuploidy [4, 5]. Notably, 43.5% of TLX1/3-positive T-ALL harbor mutations in PHF6 [6] when compared to 9.5% in TLX1/3-negative cases [6], suggesting a functional cooperation between these genetic perturbations might drive leukemogenesis. PHF6 is amongst the most frequently affected genes in T-ALL due to loss-of-function mutations or deletions [6]. However, the exact functionality of PHF6 in TALL remains largely unclear. Nevertheless, recent studies in mice support a role of PHF6 in the regulation of leukemia stem cell activity [7, 8]. Interestingly, PHF6 has also been detected as a tumor promoting factor in B-ALL [9–11], suggesting a high degree of context specificity regarding the role of PHF6 in leukemia biology.
Here, we used zebrafish to model TLX1-driven leukemia in vivo and investigate its putative cooperation with PHF6 deficiency in T-ALL leukemogenesis. We generated a stable Tg(rag2:hTLX1;rag2:EFGP) zebrafish line (henceforth called hTLX1 fish) by over-expressing human TLX1 and EGFP in developing lymphocytes (Supplementary Fig. 1a, b). Fish was screened by fluorescence microscopy for T-ALL development every 2 weeks for a total of 18 months. In total, 161 hTLX1 zebrafish were monitored; 2 fish (1.2%) developed T-ALL (samples named “T”) at 13 and 14 months of age (Fig. 1a). To assess functional cooperation with PHF6 loss during T-ALL formation, the hTLX1 stable line was crossed to a phf6 mutant line that harbors a 10 bp deletion in exon 2, leading to a frameshift that creates a premature stop codon (Supplementary Fig. 1c). No T-ALL tumors could be detected in the phf6+/− mutant (n = 104) only, suggesting the need for additional driver events. In contrast, hTLX1;pkf6+/− zebrafish developed T-ALL with slightly higher penetrance (nonsignificant; log rank three group comparison: p = 0.0563; log-rank comparison hTLXl versus hTLX1;phf6+/−: p= 0.1483) of 3.7% (6/162, samples named “PT”) and average latency of 11 months (range 5–18 months) (Fig. 1a, b). Although penetrance was low in both models, these results demonstrate that human TLX1 can drive T-ALL formation in zebrafish.
Fig. 1. Phenotypic and molecular analysis of TLX1-driven zebrafish T-ALL.
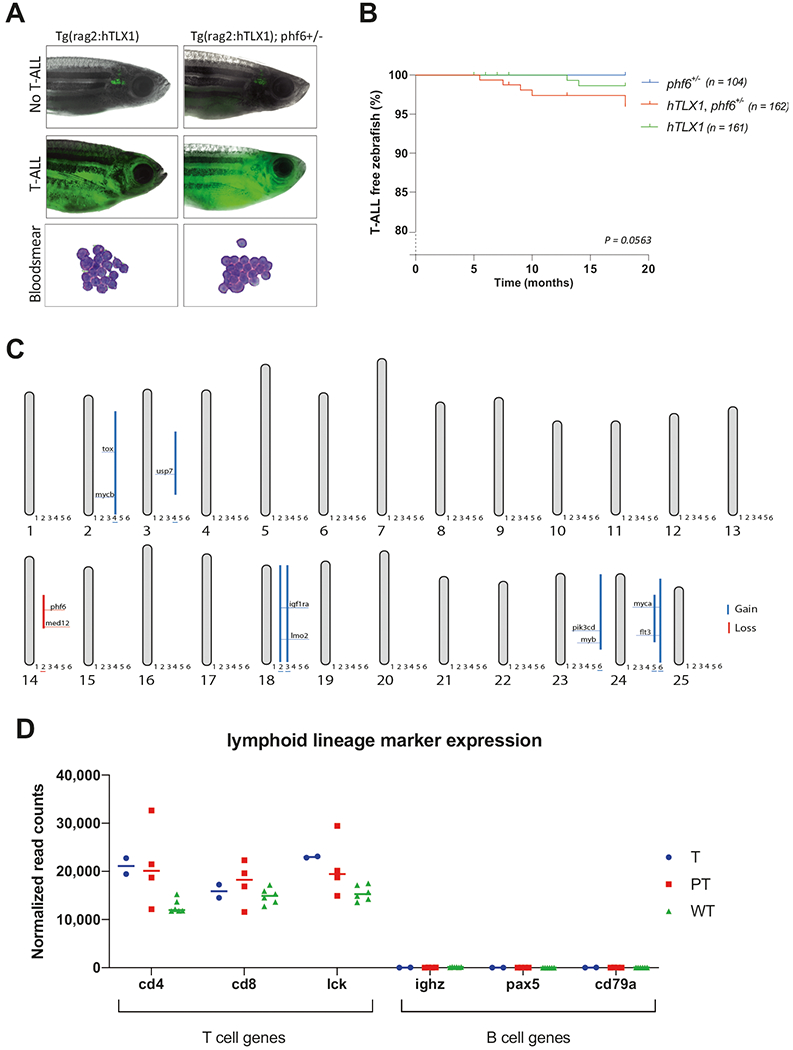
a Representative images of EGFP positive T-ALL bearing fish for both hTLX1 (left) as hTLX1; phf6+/− (right) transgenic zebrafish lines. The hematoxylin–eosin stained blood smears from leukemic fish show a high presence of malignant lymphoblasts (bottom panels). b Kaplan–Meier incidence curves depicting the percentage of leukemia-free animals over time (months), log-rank comparison for the three groups: p = 0.0563. c Schematic overview representing all the chromosomes and the detected copy number variations of six analyzed T-ALLs, with gains indicated by blue lines and losses by red lines. Sample annotation: 1 = T1, 2 = T2, 3 = PT1, 4 = PT2, 5 = PT3, 6 = PT4. d Normalized RNA-seq read counts showing high expression of T-cell marker (cd4, cd8, and lck) and low expression of B-cell marker (ighz, pax5, and cd79a) in leukemic and thymocyte samples. Horizontal lines indicate the median of each group. “T” refers to samples from hTLX1 fish, “PT” refers to samples from hTLX1;phf6+/− fish, and “WT” refers to normal wild-type thymocytes.
To confirm transformation and assess the molecular pathways driving T-ALL formation, malignant cells were collected from the fish when 90% of the animal was overtaken by EGFP+ cells. Transplantation assays confirmed the malignant transformation (Supplementary Table 1), with blood smears demonstrating a strong abundance of lymphoblasts with high nuclear:cytoplasmic ratio, characteristic for ALL (Fig. 1a). RNA (GSE151816) and DNA copy number variation (CNV) sequencing was performed to examine leukemic genomes. CNV-seq revealed that five of six T-ALLs analyzed contained large chromosomal duplications, containing known T-ALL oncogenes such as tox, usp7, lmo2, myb, myca, and mycb (Fig. 1c). Moreover, a large deletion containing the phf6 gene could be identified in one TLX1-only T-ALL sample (T2) (Supplementary Fig. 2a), suggestive for a selective advantage of PHF6 alterations in TLX1-driven T-ALL. This deletion corresponded with a 50% downregulation of pfh6 expression in the T2 T-ALL compared to wild-type thymocytes (Supplementary Fig. 2b). Since the rag2 promoter regulates the hTLXl transgene, and rag2 is expressed by both B and T lymphoblasts, leukemias could conceivably be T-, B- or mixed-lineage ALL [12, 13]. Gene expression analysis of T and B cell-specific genes confirmed the T-cell origin of these obtained leukemia (Fig. 1d). For further transcriptome profiling, additional T-ALLs from transplantation assays were also analyzed (Supplementary Table 1), with hTLX1 and phf6+/−;hLTX1 T-ALLs treated as one cohort, regardless of their phf6 mutation status and further referred to as phf6/hTLX1 T-ALL. Expression of the TCRβ variable regions revealed that the T-ALL samples were mono- or oligo-clonal (Supplementary Fig. 2c). Gene Set Enrichment Analysis (GSEA) of differentially expressed genes in phf6/hTLX1 T-ALL samples compared to wild-type thymocytes revealed downregulation of mitotic spindle genes (Fig. 2a). This is in keeping with previous reports pointing towards a critical role for TLX1 in G2–M cell cycle checkpoint [4, 14–16], disruption of the mitotic spindle and the observed genomic instability detected in this study and the previously reported TLX1 mouse model [4]. Evidence for implication of myc comes with the observed duplication of the myc locus in three samples and enriched Myc target genes in zebrafish T-ALL versus wild-type T cells (Fig. 2a and Supplementary Table 2). Each corresponding T-ALL sample showed an enhanced Myc activity either by the upregulation of myca or the upregulation mycb compared to wild-type thymus cells (Fig. 2a and Supplementary Fig. 2d), highlighting the importance of Myc in zebrafish T-ALL. In addition to Myc, other known T-ALL oncogenes and tumor suppressor genes were dysregulated in the zebrafish T-ALLs compared to wild-type thymocytes. This includes significantly enhanced expression of notch1a, usp7, and nras and a significant reduction in ikzf1, rb1, gata3, fat1a, tet1, and bcl11ba, all tumor suppressor genes known to be regulated by TLX1 in mice and human [4, 5] (Fig. 2b). The important role of Bcl11b has been previously established as a key suppressor gene in human and mouse T-ALL and directly repressed by TLX1 [4]. In addition, downregulation of cish, a Jak/Stat target, was also detected, suggesting reduced Jak/Stat activity in our T-ALL model (Fig. 2b).
Fig. 2. Comparative of gene expression analysis the phf6/hTLX1, hulk, hMYC, otg, and shrek T-ALL models.
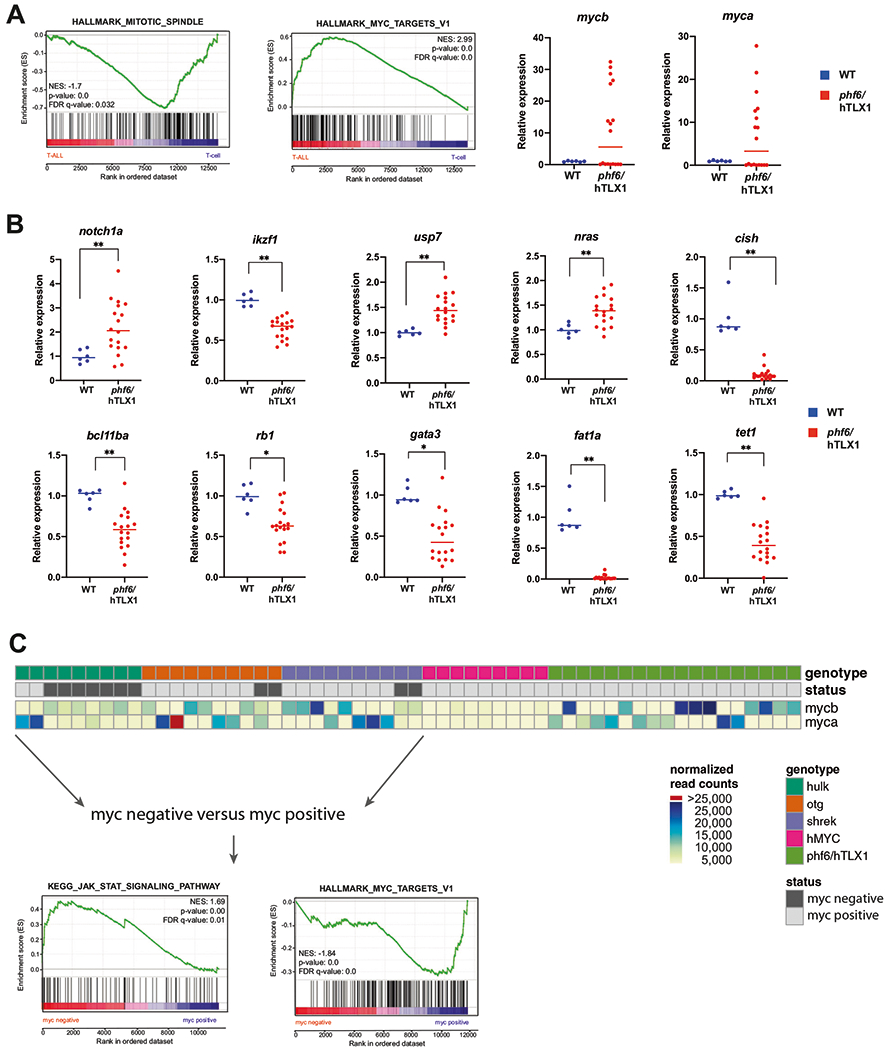
a (left) GSEA of differentially expressed genes in phf6/hTLX1 T-ALL compared to WT thymocytes. (right) Relative expression for myca and mycb in T-ALL samples (red) versus WT thymocytes (blue). The median of each group is indicated by a horizontal line. b Relative expression of notch1a, ikzf1, usp7, nras, cish, bcl11ba, rb1, gata3, fat1a, and tet1 in phf6/hTLX1 T-ALL samples (red) versus WT thymocytes (blue) (*p < 0.05; **p < 0.001). The median of each group is indicated by a horizontal line. c Schematic overview of all T-ALL samples with respect to their activation Myc, as determined by their overexpression of myca or mycb compared to wild-type thymocytes or the introduction of the hMYC transgene. Heatmap indicates normalized read counts of myca and mycb. GSEA analysis of myc negative samples compared to myc-positive samples of the hulk, otg, shrek T-ALLs reveals a positive enrichment for JAK/STAT targets, while a negative enrichment for MYC targets is detected.
To gain a comprehensive perspective on how our novel phf6/hTLX1 T-ALL model relates to existing zebrafish T-ALL models, we compared their expression profiles with four other T-ALL models (hulk, shrek, otg [17] (GSE151816) and Tg(rag2:hMYC;lck:GFP) (GSE119173) [13]) processed and sequenced in the Frazer lab. The hulk, shrek, and otg zebrafish lines were generated by chemical mutagenesis, and their underlying germline mutations are unknown [17]. In a first step, Principle Component Analysis (PCA) was performed (Supplementary Fig. 3), with T-ALL samples and thymocyte samples mainly separated by the first principal component (x-axis). The second principal component is mainly defined by lab to lab sequencing variability, clearly separating samples sequenced in the Frazer lab and the samples sequenced in the Speleman lab.
Pathway analyses using Enrichr, David, and iRegulon revealed that, as observed in phf6/hTLX1 T-ALL, myc-regulated genes were enriched in the shrek, otg, and hMYC models compared to wild-type thymocytes. Almost all shrek and otg samples showed upregulation of either myca or mycb (Fig. 2c), underscoring the important rol of Myc in T-ALL pathogenesis. Notably, Myc targets were not significantly enriched in hulk samples, however, three hulk T-ALL showed upregulation of myca or mycb. Interestingly, in the PCA analysis, these three hulk T-ALLs show highest PC1 values compared to the other hulk samples, and thus more similar to other T-ALL groups on PC1 (Supplementary Fig. 3). Further comparison between T-ALLs found that hulk T-ALLs showed enrichment of Jak/Stat pathway genes through Enrichr analysis, as compared to hMYC T-ALL. Indeed, the canonical Jak/Stat target gene cish was activated in hulk T-ALL but was low expressed in hMYC (Supplementary Fig. 4a). Of note, some hulk samples showed levels of cish that were comparable to the low expression seen in hMYC T-ALL. Interestingly, these “Jak/Stat low” samples expressed high levels of myca (Supplementary Fig. 4a). High cish expression was also observed in otg and shrek samples without myca or mycb activation. This suggests that Jak/Stat signaling could alternatively drive T-ALL leukemogenesis. To explore this further, we divided the hulk, otg, and shrek samples into two groups based on the activation of myca/mycb. Samples where myca or mycb expression was higher than all the wild-type thymocytes were classified as “myc positive,” while samples where myca and mycb expression levels were lower or equal to wild-type thymocytes were classified as “myc negative.” GSEA revealed that the myc-positive samples indeed showed the MYC target signature while Jak/Stat activation showing a positive enrichment in myc-negative samples. The latter was evident through a significant enrichment for following gene sets: “KEGG_JAK_ STAT_SIGNALING_PATHWAY,” “HALLMARK_IL2_STAT5_SIGNALING,” “HALLMARK_IL6_JAK_STAT3_SIGNALING” (Fig. 2c and Supplementary Table 3). Detailed analysis of key JAK/STAT target genes [18] revealed a higher expression in the myc-negative group compared to the myc-positive and the hMYC T-ALL cases (Supplementary Fig. 4b). In addition, all phf6/hTLX1 T-ALL showed Myc activation and low levels of key Jak/Stat target genes compared to wild-type thymocytes (Supplementary Fig. 4c), with a negative enrichment for the gene set “HALLMARK_IL2_STAT5” in the phf6/hTLX1 T-ALL compared to wild-type thymocytes (Supplementary Table 2).
In summary, we show that TLX1 is able to drive T-ALL formation in zebrafish, at least partially, by disrupting normal mitosis leading to the occurrence of CNVs in line with previously in vitro and mouse data [4, 16]. This points at a strongly conserved TLX1-driven mechanism of leukemia formation. While multiple T-ALL drivers have been discovered the past decades, so far in vivo studies and modeling in zebrafish remained limited. The long latency and low penetrance that is seen in the Tg(rag2-hTLX1) model is reminiscent to the findings in the mouse model, indicating the importance of additional genetic events to fully establish leukemia. Also in healthy elderly humans, driver mutations in bona fide oncogenes or tumor suppressors are frequently observed also supporting the multistep process of leukemia formation [19]. Given the accessibility of zebrafish for large-scale screens, our model provides an excellent opportunity for further acceleration studies towards identifying additional cooperating events in TLX1-driven T-ALL. Our comparative transcriptome analysis demonstrates the crucial role of Myc as a T-ALL driver. In human T-ALL, MYC is upregulated, amongst others, through oncogenic activation of NOTCH1 which occurs in >50% of the cases [3]. Zebrafish transgenic Myc models develop T-ALL with short latency with nearly 100% penetrance, while other T-ALL models (TLX1, hulk, shrek, otg) activate endogenous myc in a high percentage of cases. Furthermore, our data suggest that the Jak/Stat pathway provides an alternative driver pathway for T-ALL, as also frequently seen in human T-ALL, e.g., through mutations causing IL7R/JAK/STAT pathway activation in as much as 20–30% of human T-ALLs [3]. In contrast to human T-ALL and mouse models in which STAT5 was shown to drive MYC expression through binding to its downstream enhancer [20, 21], in zebrafish T-ALL, Myc activation appears to be driven by other mechanisms, which could include copy number alterations as shown in this study.
Of further interest, our data support the relevance and need to develop JAK/STAT-driven zebrafish models which may be particularly useful to emulate this subtype of human T-ALL, since not all human T-ALL are driven by MYC hyperactivity.
Supplementary Material
Acknowledgements
We would like to thank our zebrafish caretaker Karen Vermeulen for all her nurturing work. In addition, we would like to thank following agencies for funding reagents, zebrafish facility costs, and personnel expenses: FWO Vlaanderen (Fund for scientific research Flanders) (post)doctoral grants for SL, SV, and KD, FWO research project GO37918N; GOA: BOF16/GOA/023; BOF: 01J15119; Villa Joep; Kinderkankerfonds; Kom op tegen kanker: Emmanuel van der Schueren grant for LD and SL; Stichting tegen kanker project number: F/2014/373; the computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, FWO and the Flemish Government—department EWI. DML is funded by NIH (R01CA211734) and the MGH Research Scholars Program. JKF and his laboratory were supported by the OUHSC Jimmy Everest Section of Pediatric Hematology-Oncology and grants from Hyundai Hope On Wheels, the Oklahoma Center for the Advancement of Science and Technology (HRP-067), and an INBRE pilot project award from the National Institute of General Medical Sciences (P20 GM103447). JKF holds the Children’s Hospital Foundation E.L. and Thelma Gaylord Endowed Chair in Pediatric Hematology-Oncology. Research reported in this publication was supported in part by the National Cancer Institute Cancer Center Support Grant P30CA225520 awarded to the University of Oklahoma Stephenson Cancer Center and used the Molecular Biology Shared Resource. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Supplementary information The online version of this article (https://doi.org/10.1038/s41375-020-0938-2) contains supplementary material, which is available to authorized users.
References
- 1.White R, Rose K, Zon L. Zebrafish cancer: the state of the art and the path forward. Nat Rev Cancer. 2013;13:624–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299:887–90. [DOI] [PubMed] [Google Scholar]
- 3.Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood Am Soc Hematol. 2017;129:1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med. 2010;16:1321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Durinck K, Van Loocke W, Van der Meulen J, Van de Walle I, Ongenaert M, Rondou P, et al. Characterization of the genomewide TLX1 binding profile in T-cell acute lymphoblastic leukemia. Leukemia. 2015;29:2317–27. [DOI] [PubMed] [Google Scholar]
- 6.Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42:338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wendorff AA, Quinn SA, Rashkovan M, Madubata CJ, Ambesi-Impiombato A, Litzow MR, et al. Phf6 loss enhances HSC self-renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Disco. 2019;9:436–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu YC, Chen TC, Lin CC, Yuan CT, Hsu CL, Hou HA, et al. Phf6-null hematopoietic stem cells have enhanced self-renewal capacity and oncogenic potentials. Blood Adv. 2019;3:2355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meacham CE, Lawton LN, Soto-Feliciano YM, Pritchard JR, Joughin BA, Ehrenberger T, et al. A genome-scale in vivo loss-of-function screen identifies phf6 as a lineage-specific regulator of leukemia cell growth. Genes Dev. 2015;29:483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soto-Feliciano YM, Bartlebaugh JME, Liu Y, Sánchez-Rivera FJ, Bhutkar A, Weintraub AS, et al. PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes Dev. 2017;31:973–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thakral D, Kaur G, Gupta R, Benard-Slagter A, Savola S, Kumar I, et al. Rapid identification of key copy number alterations in B-and T-cell acute lymphoblastic leukemia by digital multiplex ligation-dependent probe amplification. Front Oncol. 2019;9:871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia EG, Iyer S, Garcia SP, Loontiens S, Sadreyev RI, Speleman F, et al. Cell of origin dictates aggression and stem cell number in acute lymphoblastic leukemia. Leukemia. 2018;32: 1860–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borga C, Park G, Foster C, Burroughs-Garcia J, Marchesin M, Ahmed ST, et al. Simultaneous B and T cell acute lymphoblastic leukemias in zebrafish driven by transgenic MYC: implications for oncogenesis and lymphopoiesis. Leukemia. 2019;33:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Keersmaecker K, Ferrando AA. TLX1-induced T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2011;17:6381–6. [DOI] [PubMed] [Google Scholar]
- 15.Chen E, Lim MS, Rosic-Kablar S, Liu J, Jolicoeur P, Dubé ID, et al. Dysregulated expression of mitotic regulators is associated with B-cell lymphomagenesis in HOX11-transgenic mice. Oncogene. 2006;25:2575–87. [DOI] [PubMed] [Google Scholar]
- 16.Kawabe T, Muslin AJ, Korsmeyer SJ. HOX11 interacts with protein phosphatases PP2A and PP1 and disrupts a G2/M cell-cycle checkpoint. Nature. 1997;385:454–8. [DOI] [PubMed] [Google Scholar]
- 17.Frazer JK, Meeker ND, Rudner L, Bradley DF, Smith ACH, Demarest B, et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia. 2009;23:1825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribeiro D, Melão A, van Boxtel R, Santos CI, Silva A, Silva MC, et al. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018;2:2199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinz S, Unser S, Rascle A. Signal transducer and activator of transcription STAT5 is recruited to c-Myc super-enhancer. BMC Mol Biol. 2016;17. 10.1186/s12867-016-0063-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bempt MV, Demeyer S, Broux M, Bie JD, Bornschein S,Mentens N, et al. Cooperative enhancer activation by TLX1 and STAT5 drives development of NUP214-ABL1/TLX1-positive T cell acute lymphoblastic leukemia. Cancer Cell. 2018;34:271–85.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.