

Abstract
While the highly-conserved FOXO transcription factors have been studied in Drosophila melanogaster for decades, the ability to accurately control and measure their tissue-specific expression is often cumbersome due to a lack of reagents and to limited, nonhomogeneous samples. The need for quantitation within a distinct cell type is particularly important because transcription factors must be expressed in specific amounts to perform their functions properly. However, the inherent heterogeneity of many samples can make evaluating cell-specific FOXO and/or FOXO load difficult. Here, we describe an extremely sensitive fluorescence in situ hybridization (FISH) approach for visualizing and quantifying multiple mRNAs with single-cell resolution in adult Drosophila cardiomyocytes. The procedure relies upon branched DNA technology, which allows several fluorescent molecules to label an individual transcript, drastically increasing the signal-to-noise ratio compared to other FISH assays. This protocol can be modified for use in various small animal models, tissue types, and for assorted nucleic acids.
Keywords: Fluorescence in situ hybridization, FISH, Drosophila melanogaster, Heart tube, Dorsal vessel, Branched DNA, bDNA, ViewRNA, RNAscope
1. Introduction
Forkhead transcription factors, characterized by a conserved “Forkhead box (Fox)” or “winged helix” DNA-binding domain, were first identified in Drosophila melanogaster [1-3]. Since their initial discovery, 19 subfamilies, designated A–S, have been classified. The “O” subfamily is involved in numerous physiological and pathological processes, including aging, growth, cancer, and neurological and cardiovascular diseases [4-9]. Several features of Drosophila have rendered it essential in helping discern the biological roles of the FOXO subfamily since the beginning of the 21st century [6, 10, 11]. Flies possess a single FOXO gene, dFOXO, which helps simplify research [12]. The longest protein product of this gene shares 89% and 88% Forkhead box domain identity with FOXO3 and FOXO1, respectively [13, 14], the two most abundant and highly studied FOXO proteins in mammals [15]. Generally, use of fly models affords researchers the benefits of easy, tissue-restricted manipulation of gene expression, a short life cycle, conserved aging and disease processes, and the capability of studying large, isogenous populations of offspring [16-18].
FOXO proteins control the expression of a vast array of genes, which have been studied in a variety of cell types [19, 20]. As with other transcription factors [21], subtle changes in FOXO expression or FOXO activity can result in context-dependent, beneficial or detrimental, cellular responses [22, 23]. In skeletal and heart muscle, for example, FOXO-mediated changes in proteostasis can have either adaptive or maladaptive functional consequences that seemingly correlate with the transcription factor’s dose [22-26]. Therefore, precisely quantifying these slight yet critical differences in expression is essential for understanding downstream gene regulation. Accurate quantitation, however, is frequently hampered by low expression levels characteristic of many transcription factors [27, 28], non-optimal reagents or techniques, and heterogeneity of the tissue- or cell-types in the samples being studied.
Different approaches for assessing protein and RNA levels are well-established. Relative protein load can be determined via quantitative western blotting [29]. While valuable, this method can be accompanied by impediments. Monoclonal antibodies are the gold-standard in protein identification experiments, but their production is expensive and laborious [30, 31], and availability is often limited to human, murine, or other vertebrate proteins. Polyclonal antibodies have a higher probability of nonspecific cross-reactivity, which complicates data interpretation, and reproducibility is influenced by greater lot-to-lot variation [29]. To circumvent some of the drawbacks of antibody use and/or to complement protein analysis, methods such as quantitative reverse transcription polymerase chain reaction (qRT-PCR) have been developed and optimized to similarly examine mRNA abundance in cells and tissue. qRT-PCR is a common and sensitive means to determine RNA levels and transcriptional differences in tissue samples under diverse conditions [32]. A key obstacle that regularly faces both total protein and mRNA quantitation, however, is tissue heterogeneity within samples, potentially leading to contamination by undesired cells together with the cells of interest [33]. Moreover, both western blotting and qRT-PCR become increasingly difficult, and results more variable, when the amount of tissue is limited. These problems can be bypassed by alternative approaches that allow direct identification of mRNA exclusively in chosen cells. Highly sensitive in situ detection of RNA allows evaluation of multiple transcripts within nonhomogeneous samples without the need to first isolate specific cell types or extract, purify, and amplify an mRNA of interest.
Early iterations of in situ hybridization (ISH) detected specific DNA and RNA sequences using radiolabeled, nucleic acid-based probes [34-37]. Over the past few decades, protocols for probe design and in situ visualization of RNA have been developed [38-42] and adapted for use in the majority of model systems, including Drosophila. Techniques have been improved upon, including the use of fluorescent probes (fluorescence in situ hybridization, FISH) [43, 44], to significantly increase safety and to maximize the probe signal while minimizing background signals (see Note 1). However, these approaches are adversely influenced by nonspecific primary probe binding, which causes an increase in background signal and a reduction of contrast. High signal-to-noise ratios are particularly vital when investigating modestly expressed transcripts or subtle changes in transcription [28].
Here, we outline two similar FISH approaches for analyzing transcript levels in individual cells within an entire organ using branched DNA (bDNA) technology [40, 45]. bDNA signal amplification assays eliminate most background and allow for high-specificity detection of mRNAs (Fig. 1). Specifically, we tested several probes for different messages in Drosophila cardiac tissue, which has not been especially amenable to conventional RNA labeling methods. The Drosophila heart is a complex organ made up of many cell types, including ostia and valve cells [46-48], and is intimately associated with pericardial, fat, and non-cardiac muscle cells. The difficulty of working with such a small organ (~80 cells) paired with a lack of antibodies makes standard protein quantitation approaches rather elusive. Thus, our modified FISH protocols, based on the commercially available ViewRNA (ThermoFisher Scientific) and RNAscope (Advanced Cell Diagnostics) kits, allow mRNA analysis of modestly-expressed transcription factors, including FOXO, in distinct cells, such as individual cardiomyocytes, while disregarding undesired cells. The bDNA approach comprises multiple amplification steps, in addition to the initial complementary probe hybridization, to produce fluorescent signals with exceptionally high contrast (Figs. 1A and 2) [40, 45]. The FISH methods described here are practical as an independent means for quantifying cellular mRNA in heterogeneous samples and as supplements to the other techniques discussed above.
Fig. 1. Overview of the branched DNA approach.

(A) In bDNA assays, pairs of primary probes (ZZ) identify and hybridize to a specific gene product of interest. Preamplifiers and amplifiers then bind the probe pairs to form tree-like structures. Fluorescent label probes attach to respective amplifier “branches” to give up to 100× higher signals than approaches illustrated in (B) under equivalent imaging conditions. Both ViewRNA and RNAscope follow the same general cascade of hybridization events. Label probes for ViewRNA are predetermined at the time of probe set design. Label probe combinations for RNAscope are determined at the time of the experiment according to Table 1. (B) Alternative FISH approaches include the direct hybridization of several short oligonucleotide probes with a single fluorophore, which require long transcripts to ensure high fluorescence intensity for detection [38, 41]. While direct fluorophore conjugation does not allow for signal-to-noise ratios as high as those afforded by bDNA assays, as described by Titlow et al., a benefit of this method is the capability of viewing protein localization in parallel with RNA localization [38]. (C) An advantage of using a bDNA approach for RNA in situ hybridization is the high specificity of probe pairs compared to individual oligonucleotide probes. If only one member of a probe pair hybridizes to an off-target RNA sequence, the preamplifier cannot bind, and therefore no fluorescent signal will result.
Fig. 2. Characterization of GAL4 expression patterns in DMef2-GAL4 and tinCΔ4-Gal4 using RNAscope and ViewRNA.
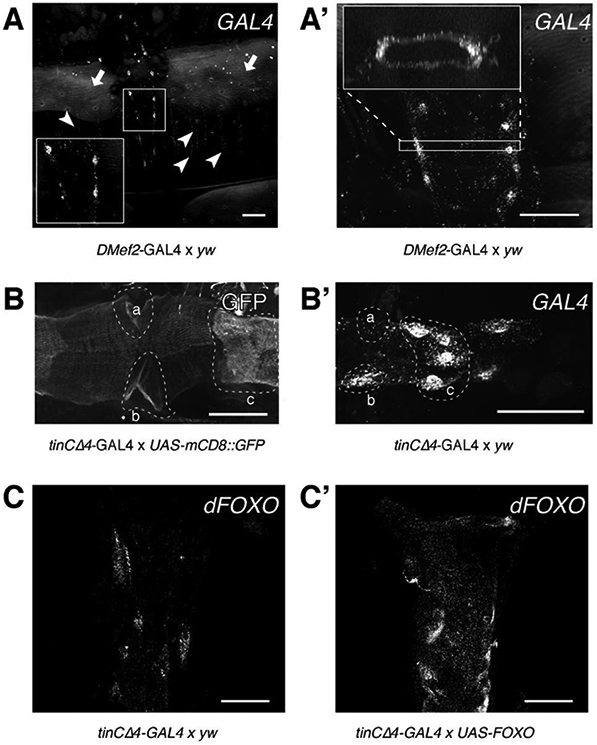
(A) Adult Drosophila abdominal carcass treated with a probe against GAL4 shows expression of GAL4 in abdominal body wall muscles (arrowheads) and in the cardiomyocytes of the heart (inset). Note the background fluorescence from the cuticle (arrows). (A’) RNA molecules can be detected throughout the heart tube (cross-sectional view). (B) Activity of GAL4 assayed via mCD8::GFP reporter gene expression. GFP can be detected throughout the entire heart tube, but shows stronger fluorescence in the ostia cells (a, b) and valve cells (c). (B’) GFP intensity is mirrored by high levels of GAL4 transcripts in the same cell types (a, b, and c, compare to B). (C) Endogenous dFOXO transcripts from control hearts (tinCΔ4-GAL4 × yw) were probed using ViewRNA. (C’) tinCΔ4-GAL4 drives UAS-dFOXO expression to a visibly greater extent than the endogenous dFOXO promoter in control hearts. All scale bars (white) represent 50 μm. (C and C’) were adapted for use with permission from [23].
2. Materials
2.1. Manipulation of Gene Expression in the Drosophila Heart
Anesthetizing pad connected to CO2 tank or triethylamine (FlyNap).
tinCΔ4-GAL4 fly line (cardiac-specific driver line)[49].
DMef2-GAL4 fly line (ubiquitous muscle driver line, Bloomington Drosophila Stock Center, BDSC, BL-27390).
UAS-FOXO wt.m3-1 fly line (inducible dFOXO overexpression line) [50].
yw fly line (control background line).
UAS-mCD8::GFP (membranous GFP-reporter line, BDSC, BL-32184).
Standard Drosophila media: deionized water, molasses (9.10% v/v), agar (12.40 g/L), yeast (17.05 g/L), yellow cornmeal (68.50 g/L), tegosept (p-hydroxybenzoic acid methyl ester, 0.92% v/v), propionic acid (0.275% v/v), and phosphoric acid (0.033% v/v) to desired volume.
Vials for mating and collecting flies.
2.2. Adult Drosophila Heart Dissections (see Note 2)
A list of all required materials is available in [51].
First generation (F1) offspring from fly crosses described below in Subheadings 3.1 and 3.2.
Triethylamine (FlyNap).
Petri dishes (35 × 10 mm).
Petroleum jelly.
Curved microdissection spring scissors and microforceps.
Adult Drosophila hemolymph (AH): 108 mM NaCl, 5 mM KCl, 2 mM CaCl2, 8 mM MgCl2, 15 mM Hepes pH 7.1, 1 mM NaH2PO4, 4 mM NaHCO3, 10 mM sucrose, and 5 mM trehalose. Adjust to pH 7.1, and add sugars immediately before use [51].
Glass capillaries (100 μL) pulled to a maximum diameter of 40 μm.
Plastic tubing (1/16”).
Vacuum or suction source.
Stereo dissecting microscope with light source and a dark base, ideally.
2.3. Tissue Fixation and Hybridization
Incubator set to exactly 40°C (see Note 3).
10 mM EGTA in AH.
Phosphate buffered saline (1 × PBS): 137 mM NaCl, 20 mM phosphate, 2.7 mM KCl, pH 7.4.
1 × PBST (PBS + 0.1% Tween-20).
4% formaldehyde in 1 × PBS.
Flat-bottom 96-well plate, white.
Foil or other material (such as a second, opaque 96-well plate placed on top) to protect fluorophores from light.
- FISH assay kit: both kits include wash buffer, protease solution(s), reagents for amplification of the primary probes, color probes, and DAPI (Fig. 1A).
- ViewRNA ISH cell assay kit (ThermoFisher Scientific, QVC0001): in addition to reagents listed above, this kit includes 10 × PBS buffer solution, detergent solution, and diluent solutions.
- RNAscope® Fluorescent Multiplex Reagent Kit (Advanced Cell Diagnostics, Inc., 320850).
2.4. Mounting, imaging, and quantitation
Glass slides and ultra-thin cover slips (22 × 22 mm).
ProLong Gold with DAPI or other acceptable mounting media.
Clear nail polish for sealing cover slips and slides.
Epifluorescent or confocal microscope with filters required for the chosen color probe types (see Note 4).
ImageJ software for quantifying punctae (transcripts) (see Note 5).
3. Methods
3.1. Manipulation of Gene Expression in the Drosophila Heart
3.1.1. Cardiac-Specific Overexpression of dFOXO
Collect virgin females and young males for all crosses.
When using the UAS-GAL4 bipartite system for gene manipulation, typically mate virgin females expressing GAL4 driven by a tissue-specific enhancer with young males homozygous for an allele encoding the desired UAS-construct.
Place virgin tinCΔ4-GAL4 females in vials containing molasses-cornmeal media with either young transgenic (e.g., UAS-FOXO wt.m3-1) or control (e.g., yw) males at 25 °C.
After three days, remove P1 (parental generation) adults from the vials.
10 days after mating was initiated, collect adult progeny (F1), place on fresh media, and maintain at 25 °C.
3.1.2. Muscle-Specific Expression of GAL4 and Membranous GFP
Follow steps 1 – 3 listed in Subheading 3.1.1 using DMef2-GAL4 virgin females and UAS-mCD8::GFP males.
Perform steps 4 and 5 in Subheading 3.1.1.
3.2. Probe Design
bDNA probes are specifically designed to detect the transcript(s) of interest. The target region should cover ~1000 bases, which gives the maximum number of primary probe pairs per mRNA necessary to achieve the strongest signal (see Note 6). Since probe design is a proprietary process, the RefSeq number of the target sequence must be submitted to the probe vendor. They will assess if probe design is feasible, if the probes will be specific for the given transcript, and if alternate splice variants of the gene product may be detected. A number of primary probe sets are readily available, including those targeting glyceraldehyde 3-phosphate dehydrogenase (GAPDH1) as a positive control and yeast-GAL4 as either a positive control in the GAL4-background or a negative control in the absence of GAL4. Probe sets targeting human or other vertebrate RNA may serve as a generic negative control for Drosophila tissue. Positive control(s) can help validate successful FISH, and negative control(s) can assist in determining the degree of background or nonspecific binding. Additional thought must be given to how these probes will be combined, since each probe set of a specific type cannot be combined with a different probe set of the same type in a multiplex experiment (e.g., RNAscope C1 probes can only be used together with a C2 and a C3 probe, but not another C1 probe (Table 1), and ViewRNA probe type 1 can only be used together with type 4 and 6, but not another type 1) (see Notes 7 and 8 for detailed examples of ViewRNA and RNAscope probe set and combination strategies, respectively).
Table 1.
Color probe combinations available for use with RNAscope.
| Probe Channel ID | Amp 4-AltA | Amp 4-AltB | Amp 4-AltC |
|---|---|---|---|
| C1 | Alexa 488 | Atto 550 | Atto 550 |
| C2 | Atto 550 | Alexa 488 | Atto 647 |
| C3 | Atto 647 | Atto 647 | Alexa 488 |
3.3. Adult Drosophila Heart Dissections
A video depicting the protocol for semi-intact adult Drosophila heart preparations is available in [51].
Anesthetize flies using FlyNap.
Coat a thin layer of petroleum jelly on the bottom of a small petri dish. Immobilize ~six anesthetized flies dorsal side down by separating their wings and sticking them into the petroleum jelly.
For each fly, using curved microdissection spring scissors, remove the head and ventral portion of the thorax in one motion by placing the scissors at an angle under the fly’s legs. Leave the dorsal portion of the thorax and wings to keep the preparation firmly stuck to the petroleum jelly. This cut should also remove a small segment of the anterior end of the abdomen.
Fill the petri dish with enough fresh AH to cover the preparations.
Again, for each fly, use the spring scissors and fine-tipped forceps to remove the posterior tip of the abdomen (segment eight). Make two cuts, one up each side of the abdominal cuticle, and remove the ventral cuticle, exposing the abdominal organs.
Carefully remove the internal organs, leaving the beating heart tube untouched and intact against the dorsal cuticle.
Use pulled microcapillaries connected to a suction source to carefully remove fat surrounding the heart tube to expose as much of the tissue as possible. Ensure the heart is not disturbed, as it is easily damaged.
3.4. Fixation
Replace the AH with enough 10 mM EGTA in AH to cover the preparations to arrest hearts within 1 min.
Exchange the EGTA solution with 4% formaldehyde in 1 × PBS, and incubate with gentle rocking at room temperature for 20 min (see Note 9).
Wash 3× in 1 × PBST at room temperature for 10 min each, and replace with PBS.
While the fixed samples remain in PBS, carefully cut off most of the remaining thoracic material, and trim the sides of each abdominal cuticle.
Delicately transfer the preparations to appropriately marked well(s) of a 96-well plate filled with 150 μL 1 × PBS, up to six specimens per well. Use as many wells as needed for a given experiment.
The samples may be stored in 1 × PBS at 4 °C overnight (see Note 10).
3.5. In Situ Hybridization
3.5.1. ViewRNA
Ensure the incubator is set to and maintains 40 °C.
Prepare wash buffer by combining wash buffer component 1 with water, and then add component 2 according to the manufacturer’s protocol.
Warm all diluent buffers to 40 °C for 30 min before use.
Carefully aspirate the 1 × PBS from each well containing specimens (see Note 11), and replace it immediately with 100 μL detergent solution included in the ViewRNA ISH Cell Assay kit. Cover the 96-well plate, and incubate at room temperature for 5 min.
Draw out the detergent solution, and rinse heart tube preparations 2× with 150 μL 1 ×PBS at room temperature for 2 min each to remove all residual detergent solution.
Replace PBS with 100 μL protease solution in each well (1:4000 in 1 × PBS, see Note 12) to expose the mRNA for hybridization. Cover the 96-well plate, and incubate at room temperature for 10 min with gentle agitation.
Remove the protease solution, and rinse 3× with 150 μL 1 × PBS at room temperature for 2 min each to remove residual protease.
Substitute PBS with 100 μL working probe set solution (ViewRNA probes diluted 1:100 in probe set diluent buffer). This solution contains all probe sets used in the assay (Fig. 1a, ZZ probe pairs). Cover the 96-well plate, and incubate at 40 °C for 3 h.
Aspirate the working probe set solution, and wash 3× with 150 μL wash buffer at room temperature for 2 min each to remove residual probes. IMPORTANT: Do not soak samples in wash buffer longer than 30 min.
Exchange wash buffer with 100 μL working pre-amplifier mix solution (pre-amplifier mix diluted 1:25 in amplifier diluent buffer) (Fig. 1a, dashed lines). Cover the 96-well plate, and incubate at 40 °C for 30 min.
Remove the working pre-amplifier mix solution, and wash 3× with 150 μL wash buffer at room temperature for 2 min each to remove remaining pre-amplifier molecules. IMPORTANT: Do not soak samples in wash buffer longer than 30 min.
Swap wash buffer with 100 μL working amplifier mix solution (amplifier mix diluted 1:25 in amplifier diluent buffer) (Fig. 1a, dotted lines). Cover the 96-well plate, and incubate at 40 °C for 30 min.
Extract the working amplifier mix solution from the wells, and wash 3× with 150 μL wash buffer at room temperature for 2 min each to remove unbound amplifier molecules. IMPORTANT: Do not soak samples in wash buffer longer than 30 min.
Protect samples from light from now on, as the label probes contain fluorophores that are photolabile.
Replace wash buffer with 100 μL working label probe mix solution (label probe mix diluted 1:25 in label probe diluent buffer, protect from light) (Fig. 1a, colored circles). Cover the 96-well plate, and incubate at 40 °C for 30 min.
Draw out the working label probe mix solution, and wash 2× with 150 μL wash buffer at room temperature for 2 min each and 1× for 10 min. IMPORTANT: Do not soak samples in wash buffer longer than 30 min.
Remove wash buffer. Stain the samples with a 1:100 DAPI solution in 1 × PBS at room temperature for 1 min if DNA counterstaining is necessary or if you are not using mounting medium that contains DAPI. Rinse once with 1 × PBS at room temperature for 2 min, and store in 1 × PBS until mounted (see Note 10).
3.5.2. RNAscope (See Note 13)
Ensure the incubator is set to and maintains 40 °C.
Bring all reagents to room temperature.
Prepare 100 mL of 1 × wash buffer from 50 × stock solution.
Protect samples from light for the entirety of the procedure.
If using C2 and/or C3 probes, dilute each probe 1:50 in C1 probe. 50 – 100 μL of probe solution is sufficient for a 96-well plate well.
Transfer the probe solution into as many empty wells as are appropriate for the given experiment. Heat the probes to 40 °C for 10 min.
In parallel to step 6, replace 1 × PBS in the specimen-containing wells (from Subheading 3.3, step 5) with 3-4 drops of Protease III, and incubate at 40 °C for 10 min (see Note 12).
Carefully remove the Protease III, and immediately rinse samples 3× with 1 × PBS at room temperature for 2 min each to eliminate remaining protease (see Note 11).
Aspirate the PBS, and transfer 50 μL of the pre-warmed probe solution to the heart samples (Fig. 1a, ZZ probe pairs). Incubate at 40 °C for 2 h or overnight.
Draw off the probe solution, and wash 3× with 1 × wash buffer at room temperature for 5 min with gentle agitation to remove unbound probes.
Replace wash buffer with 2 drops AMP 1-FL solution (Fig. 1a, dashed lines), and incubate at 40 °C for 30 min.
Extract the AMP 1-FL solution, and wash 3× with 1 × wash buffer at room temperature for 5 min each with gentle agitation to remove residual AMP 1-FL solution.
Replace wash buffer with 2 drops AMP 2-FL solution (Fig. 1a, dotted lines), and incubate at 40 °C for 15 min.
Remove the AMP 2-FL solution, and wash 3× with 1 × wash buffer at room temperature for 5 min each with gentle agitation to remove remaining AMP 2-FL solution.
Replace wash buffer with 2 drops AMP 3-FL solution, and incubate at 40 °C for 30 min.
Extract the AMP 3-FL solution, and wash 3× with 1 × wash buffer at room temperature for 5 min each with gentle agitation to remove residual AMP 3-FL solution.
Replace wash buffer with 2 drops AMP 4-FL solution, and incubate at 40°C for 15 min. Refer to Table 1 for the desired color combination (see Note 4).
Draw out the AMP 4-FL solution, and wash 3 × with 1 × wash buffer at room temperature for 5 min each with gentle agitation to remove any lingering AMP 4-FL solution.
Replace wash buffer with 2 drops DAPI solution if DNA counterstaining is needed and if the mounting medium does not already contain DAPI.
Rinse with 1 × PBS and ideally mount the samples immediately in antifade reagent. Alternatively, samples can be stored at 4 °C but for no longer than 8-12 h, as the signal will start to fade in 1 × PBS (see Note 10).
3.6. Mounting the Adult Drosophila Heart Tube Preparations
A video file that explains a suitable approach for mounting adult Drosophila heart preparations is available in [52].
Place two small drops (~15 μL) of mounting media centered ~1 cm apart on a glass slide, and place two cover slips on the drops.
Place a 10 – 20 μL drop of mounting medium on a third cover slip.
Remove the hybridized heart preparations gently from the 96-well plate using fine-tipped forceps. Handle specimens at the edge of the leftover cuticle. Do not make contact with the heart tube. Place the preparations heart side-down on the drop of mounting medium on the third coverslip. Up to six flies can be placed per drop.
Carefully but quickly, invert the cover slip, and place it on top of the glass slide such that the adult heart preparations lie between the two already placed cover slips. This forms a bridge that protects the heart samples from becoming disturbed.
Ensure that all hearts are now facing up and are unobscured by residual cuticle.
Use mounting medium to fill in any air pockets present between the glass slide and cover slip.
Seal the edges with clear nail polish.
Keep slides protected from light until ready to image.
3.7. Imaging of Hearts via Confocal Microscopy (See Note 7)
Choose appropriate beam splitter, emission filters, laser power, and camera gain, and ideally use the same settings throughout the imaging of samples. The fluorophores conjugated to the label probes used by ViewRNA (DAPI, FITC, Cy3, Cy5, and Cy7) are commonly employed and pre-set emission settings are available with most microscope software.
To faithfully capture all fluorescent dots, take care to acquire a sufficient amount of data in the x, y, and z optical planes. For a typical setup, a 40× immersion objective is sufficient to image close to the Nyquist criterion. However, this must be determined for each instrument used. This is equally important for imaging along the z-axis, in which undersampling can easily cause erroneous data representation due to missed slices.
Owing to the rounded and varying morphology of the adult Drosophila heart tube between the anterior-most conical chamber and the posterior end, the number of z-sections required to image the whole tissue varies along the heart’s length.
Capturing the highest dynamic range is also critical since the intensities can vary between the detected mRNAs. Individual transcripts appear as distinct punctae with mostly uniform intensities, however lower intensity dots should be disregarded (see Subheading 3.8). A negative control probe is crucial to establish the amount of background punctae per cell, which should be corrected for in the final analysis.
3.8. Image Analysis and Quantitation (See Note 8)
3.8.1. Basic Image Analysis and Quantitation of mRNA Particles Using ImageJ Particle Counter
Open an image that was generated in Subheading 3.7 in the microscope program. Separate emission channels for each label probe and/or DAPI. Stacked images or individual z-sections may be converted to grayscale and exported as TIFFs to ImageJ (Fig. 3A).
For a particular cardiac region, open all emission channel files in ImageJ, and convert each image to 8-bit (Image > Type > 8-bit).
Change the threshold (Image > Adjust > Threshold) such that the upper and lower limits adequately incorporate punctae that represent transcripts but exclude those that may signify off-target binding or background. Maintain the threshold settings throughout analysis of all other images. Here, we used an arbitrary upper limit of 150 and a lower limit of 55 (Fig. 3B, see Note 14)[23].
Because the images include signals from tissue other than the heart, use the freehand selection tool to outline a region of interest (ROI) that includes only cardiomyocytes and no adjacent adipose tissue, pericardial cells, or retractors of tergite muscles (Fig 3C, yellow outline). Copy the ROI tracing to all other channels from the same heart section (Ctrl+Shift+e on PC or Cmd+Shift+e on Mac, see Note 15).
Count the number of particles in the ROI, which corresponds to the number of transcripts in each channel (Analyze > Analyze particles).
For individual specimens in which multiple cardiomyocytes were imaged separately, combine the total number of dFOXO particles (or mRNA particles under investigation) and the total number of GAPDH particles determined for each channel, from each cell per heart.
Normalize the total number of dFOXO particles to GAPDH mRNA particles per heart (Fig. 3D).
Fig. 3. Quantitation of dFOXO mRNA particles from a ViewRNA FISH experiment in a semi-intact Drosophila heart tube preparation.

(A) Representative images of a tinCΔ4-GAL4 > UAS-dFOXO heart, probed for dFOXO and GAPDH transcripts, converted to grayscale for analysis using ImageJ. Inset displays a region of interest (cardiomyocyte) prior to threshold modification. (B) After conversion to 8-bit, the threshold of each image is arbitrarily changed to a range between 55 and 150 to help eliminate background signals and off-target probe binding from subsequent particle analysis. (C) Representative images, post-threshold adjustment, with the inset displaying the same cardiomyocyte data as in (A). Larger regions of interest, which exclude non-cardiac tissue, can be selected using the free-hand tool in ImageJ (yellow line). (D) Values from tinCΔ4-GAL4 × yw control vs. tinCΔ4-GAL4 > UAS-dFOXO hearts (Fig. 2C and C’) were obtained using the ImageJ particle analysis tool. dFOXO/GAPDH was calculated for each heart or region of interest. The graph shows a small representation of the difference calculated in normalized dFOXO transcripts between the hearts of the two genotypes. The use of ViewRNA to detect more subtle changes in cardiac dFOXO transcript levels was previously published [23].
3.8.2. Alternative ImageJ Protocol [53]
Install the Bioformats plugin, which allows for seamless import of most scientific file formats into ImageJ.
Load the image stack acquired in Subheading 3.7 into ImageJ. Create a Maximum Intensity projection of the stack (Image>Stacks>Z-project>Max.Intensity).
Adjust image brightness (Ctrl+Shift+C on PC or Cmd+Shift+C on Mac), and open the ROI Manager (Analyze>Tools>ROI Manager).
Create a cell outline with the freehand selection tool (see Note 15), and add the ROI to the ROI manager by pressing ‘t’. Repeat this for all cells you want to analyze.
Select all ROIs (check ‘Show all’). Flatten the image, and save it to document an overview of all ROIs in the open stack.
Activate the full stack image, and select the first ROI from the ROI Manager. Duplicate this ROI (Ctrl+Shift+D on PC or Cmd+Shift+D on Mac).
Discard image data outside the ROI (Edit>Clear Outside).
Scroll through this stack, and select the image range covering all slices of interest.
Reduce dimensionality to use only the desired range (e.g. 1-37). To avoid potential double-counting of punctae present in adjacent slices select only even slices, Image>Stack>Tolls>Make Substack. Enter the image range followed by −2 to select every other image (e.g. 1-37-2).
Split this stack into separate Colors (Image>Color>Split Channels), and continue with thresholding.
Run the thresholding command (Ctrl+Shift+T on PC or Cmd+Shift+T on Mac) on the first channel. Use MaxEntropy with Red and Dark background. Calculate threshold for each image, check ‘Dark background’, and apply.
Use ImageJ’s built-in ‘Analyze Particles’ function to count all punctae of a specific size range automatically. First, determine the typical puncta size by measuring the area of several particles (typically 0.2-2 μm2). Set circularity to 0.2-1, show ‘Overlay’, and check ‘Display results’.
Repeat steps 11 - 12 for the remaining channels.
Repeat steps 6 - 13 for the remaining ROIs of the full stack.
Acknowledgements:
We thank Holly Howarth for excellent technical assistance. This work was supported by R01HL124091 (to A.C.) and Cabrini University Academic Affairs funds (to A.C.B.).
Footnotes
Protocols for the design and in situ detection of single or multiple RNA probes using both chromogenic and fluorescent labels have been developed and adapted for a majority of model systems. A variety of techniques have also been established to amplify the signal through enzymatic reactions, including alkaline phosphatase or horseradish-peroxidase that cause local precipitation of a specific compound, such as nitro-blue tetrazolium, 3,3’ diaminobenzidin or TSA, or more recently by hybridization chain reaction [54]. However, each of these approaches, like early versions of FISH, will be directly affected by nonspecific binding of the probe (Fig. 1B).
Gene overexpression or knockdown [55] can be achieved in different tissues in Drosophila and in other small model organisms, although the procedure for exposure will differ depending on the tissue being investigated.
These procedures were successfully tested with several incubators, including a Robbins scientific model 400, an Eppendorf Thermomixer C for 96-well plates with heated lid, and a Shel-Lab-2350-T water-jacketed incubator.
The microscopes used in 3.7 were a Leica TCS SPE RGBV confocal microscope with a 40× oil immersion lens for ViewRNA experiments and a Zeiss Imager.Z1 with Apotome.2 and a 40× water immersion lens for RNAscope experiments. Alternatively, an epifluorescence microscope may be used. A limitation of epifluorescence versus confocal microscopy is the inability to scan at a specific focal depth without interfering, out-of-focus signals. However, because bDNA FISH methods produce distinct punctae, or dots, when hybridized to individual transcripts, background signal from other focal planes should not interfere significantly with interpretation of results after analysis.
Additional suitable FISH image acquisition and analysis methods are described in detail in [38].
While a target mRNA of 1000 bases is ideal, the target region may be as short as 300 bases; however, signal reduction occurs due to fewer binding probes. Probe design can also be optimized for localization and quantitation of miRNAs, viral RNA, and lncRNA in specific cells in addition to mRNA transcripts. One miRNA can be identified at a time as well as two mRNA targets. miRNA probes are detected using Fast Red fluorescent dye.
The following probes were designed for ViewRNA: the dFOXO probe set was designed against the longest mRNA product of the dFOXO gene, NM_001275628 (type 1, 550 nm excitation, ThermoFisher Scientific, catalog number VF1-18189-VC). The internal control, GAPDH1, probe set was designed against the mRNA sequence NM_080369 (type 6, 650 nm excitation, ThermoFisher Scientific, catalog number VF6-18191-VC). Probe set type 4 (488 nm excitation) was not used due to potential autofluorescence of adipose tissue surrounding the heart [56]. A probe set against a nonendogenous, bacterial mRNA sequence, dapB (type 1, 550 nm excitation) ThermoFisher Scientific, catalog number VF1-11712-VC), was used as a negative control. Up to four different probe types can be combined without overlapping, but they must be predetermined at the probe design step.
The following probes were ordered for RNAscope: Drosophila melanogaster GAPDH1 (NM_080369.3, Advanced Cell Diagnostics, 470801-C1), and yeast GAL4 (NM_001184062.1, Advanced Cell Diagnostics, 428501-C1). Up to three probe types can be combined, and final fluorophore combinations are determined at the time of the experiment as illustrated in Table 1.
When tissue is fixed for longer than 30 minutes, ssRNA specifically becomes degraded or modified by crosslinking [57, 58].
For ViewRNA assays, it was determined that samples should not remain in PBS at 4 °C for more than 16 h before performing in situ hybridization. However, in the case of RNAscope, samples remaining at 4 °C for 1 week after fixation yielded acceptable results. The critical step is to mount hearts immediately after the final color probe step since bDNA probes are not stable long after hybridization. Once mounted, samples are stable for several weeks.
Gel-loading tips may be used to ensure no accidental aspiration of the heart tube preparations. Avoid touching the samples, as they tend to stick to the pipette tips. After aspiration, immediately replace buffers so tissues do not dry out.
Conditions for protease treatment (e.g. time and concentration) must be empirically determined for different tissues. While 10 min is necessary for the extracellular matrix-rich heart tube, it leads to a noticeable thinning of the abdominal wall muscles, which therefore might require shorter incubation times.
While mostly similar to the ViewRNA Cell Assay kit, the RNAscope kit comes with dropper bottles for most reagents, minimizing the handling of the solutions. 1-3 drops per well (25 - 75 μL) are usually sufficient to keep the samples immersed.
The lower limit chosen in ImageJ will minimize punctae that may represent off-target binding of probes from either the ViewRNA or RNAscope assays.
Appropriate ROIs are best determined using an overlay of all channels plus DAPI and pasting the ROI onto the images from individual channels. However, this can introduce an element of human error, as the freehand tool is ultimately used to outline the ROI.
References
- 1.Weigel D, Jürgens G, Küttner F, et al. (1989) The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 57:645–658 . doi: 10.1016/0092-8674(89)90133-5 [DOI] [PubMed] [Google Scholar]
- 2.Tuteja G, Kaestner KH (2007) SnapShot: Forkhead transcription factors II. Cell 131 [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann E, Knöchel W (1996) Five years on the wings of fork head. Mech. Dev 57:3–20 [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Zhou Y, Graves DT (2014) FOXO transcription factors: their clinical significance and regulation. Biomed Res Int 2014:925350 . doi: 10.1155/2014/925350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xin Z, Ma Z, Jiang S, et al. (2017) FOXOs in the impaired heart: New therapeutic targets for cardiac diseases. Biochim. Biophys. Acta - Mol. Basis Dis 1863:486–498 [DOI] [PubMed] [Google Scholar]
- 6.Antikainen H, Driscoll M, Haspel G, Dobrowolski R (2017) TOR-mediated regulation of metabolism in aging. Aging Cell 16:1219–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLaughlin CN, Broihier HT (2017) Keeping neurons young and foxy: FoxOs promote neuronal plasticity. Trends Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornsveld M, Dansen TB, Derksen PW, Burgering BMT (2017) Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol [DOI] [PubMed] [Google Scholar]
- 9.Sun X, Chen WD, Wang YD (2017) DAF-16/FOXO transcription factor in aging and longevity. Front. Pharmacol 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SS, Kennedy S, Tolonen AC, Ruvkun G (2003) DAF-16 target genes that control C. elegans life-span and metabolism. Science (80- ) 300:644–647 . doi: 10.1126/science.1083614 [DOI] [PubMed] [Google Scholar]
- 11.Proshkina EN, Shaposhnikov MV, Sadritdinova AF, et al. (2015) Basic mechanisms of longevity: A case study of Drosophila pro-longevity genes. Ageing Res. Rev 24:218–231 [DOI] [PubMed] [Google Scholar]
- 12.Jünger MA, Rintelen F, Stocker H, et al. (2003) The Drosophila Forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol 2:20 . doi: 10.1186/1475-4924-2-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ronnebaum SM, Patterson C (2010) The FoxO family in cardiac function and dysfunction. Annu Rev Physiol 72:81–94 . doi: 10.1146/annurev-physiol-021909-135931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Flockhart I, Vinayagam A, et al. (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12:357 . doi: 10.1186/1471-2105-12-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salih DA, Brunet A (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol 20:126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perrimon N, Bonini NM, Dhillon P (2016) Fruit flies on the front line: the translational impact of Drosophila. Dis Model Mech 9:229–231 . doi: 10.1242/dmm.024810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bilder D, Irvine KD (2017) Taking stock of the Drosophila research ecosystem. Genetics 206:1227–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–15 . doi: 10.1101/lm.1331809 [DOI] [PubMed] [Google Scholar]
- 19.Murtaza G, Khan AK, Rashid R, Muneer S, Hasan SMF CJ (2017) FOXO transcriptional factors and long-term living. Oxid Med Cell Longev 2017:3494289 . doi: 10.1155/2017/3494289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannenhalli S, Kaestner KH (2009) The evolution of Fox genes and their role in development and disease. Nat Rev Genet 10:233–240 . doi: 10.1038/nrg2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, et al. (2005) Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development 132:2463–2474 . doi: 10.1242/dev.01827 [DOI] [PubMed] [Google Scholar]
- 22.Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27:2276–2288 . doi: 10.1038/onc.2008.21 [DOI] [PubMed] [Google Scholar]
- 23.Blice-Baum AC, Zambon AC, Kaushik G, et al. (2017) Modest overexpression of FOXO maintains cardiac proteostasis and ameliorates age-associated functional decline. Aging Cell 16:93–103 . doi: 10.1111/acel.12543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demontis F, Perrimon N (2010) FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143:813–825 . doi: 10.1016/j.cell.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferdous A, Battiprolu PK, Ni YG, et al. (2010) FoxO, autophagy, and cardiac remodeling. J. Cardiovasc. Transl. Res 3:355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sengupta A, Molkentin JD, Yutzey KE (2009) FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 284:28319–31 . doi: 10.1074/jbc.M109.024406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giambruno R, Grebien F, Stukalov A, et al. (2013) Affinity purification strategies for proteomic analysis of transcription factor complexes. J Proteome Res 12:4018–4027 . doi: 10.1021/pr4003323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banks CAS, Lee ZT, Boanca G, et al. (2014) Controlling for gene expression changes in transcription factor protein networks. Mol Cell Proteomics 13:1510–1522 . doi: 10.1074/mcp.M113.033902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor SC, Posch A (2014) The design of a quantitative western blot experiment. Biomed Res. Int 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipman NS, Jackson LR, Weis-Garcia F, Trudel LJ (2005) Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J 46:258–68 . doi: 10.1093/ilar.46.3.258 [DOI] [PubMed] [Google Scholar]
- 31.KÖHLER G MILSTEIN C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497 . doi: 10.1038/256495a0 [DOI] [PubMed] [Google Scholar]
- 32.Sarkar G, Sommer SS (1989) Access to a messenger RNA sequence or its protein products is not limited by tissue or species specificity. Science (80- ) 244:331–334 [DOI] [PubMed] [Google Scholar]
- 33.Murphy RM, Lamb GD (2013) Important considerations for protein analyses using antibody based techniques: down-sizing Western blotting up-sizes outcomes. J Physiol 591:5823–5831 . doi: 10.1113/jphysiol.2013.263251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardue ML GJ (1969) Molecular hybridization of radioactive DNA to the DNA of cytological preparations. PNAS 64:600–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connor C (2008) Fluorescence in situ hybridization (FISH). Nat Educ 1–5 . doi: 1(1):171 [Google Scholar]
- 36.Gall JG, Pardue M Lou (1969) Formation and detection of RNA-DNA hybrid molecules in cytological preparations*. Proc Natl Acad Sci U S A 63:378–383 . doi: 10.1073/pnas.63.2.378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison PR, Conkie D, Paul J, Jones K (1973) Localisation of cellular globin messenger RNA by in situ hybridisation to complementary DNA. FEBS Lett 32:109–112 . doi: 10.1016/0014-5793(73)80749-5 [DOI] [PubMed] [Google Scholar]
- 38.Titlow JS, Yang L, Parton RM, et al. (2018) Super-resolution single molecule FISH at the drosophila neuromuscular junction. In: Methods in Molecular Biology. pp 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tautz D, Pfeifle C (1989) A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 98:81–85 . doi: 10.1007/BF00291041 [DOI] [PubMed] [Google Scholar]
- 40.Battich N, Stoeger T, Pelkmans L (2013) Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat Methods 10:1127–1136 . doi: 10.1038/nmeth.2657 [DOI] [PubMed] [Google Scholar]
- 41.Batish M, Raj A, Tyagi S (2011) Single molecule imaging of RNA in situ. Methods Mol Biol 714:3–13 . doi: 10.1007/978-1-61779-005-8_1 [DOI] [PubMed] [Google Scholar]
- 42.Anderson R (2010) Multiplex fluorescence in situ hybridization (M-FISH). In: Methods in molecular biology (Clifton, N.J.). pp 83–97 [DOI] [PubMed] [Google Scholar]
- 43.Rudkin GT, Stollar BD (1977) High resolution detection of DNA-RNA hybrids insitu by indirect immunofluorescence. Nature 265:472–473 [DOI] [PubMed] [Google Scholar]
- 44.Singer RH, Ward DC (1982) Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc Natl Acad Sci U S A 79:7331–5 . doi: 10.1073/pnas.79.23.7331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, Flanagan J, Su N, et al. (2012) RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagnostics 14:22–29 . doi: 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lammers K, Abeln B, Hüsken M, et al. (2017) Formation and function of intracardiac valve cells in the Drosophila heart. J Exp Biol 220:jeb.156265 . doi: 10.1242/jeb.156265 [DOI] [PubMed] [Google Scholar]
- 47.Cammarato A, Ahrens CH, Alayari NN, et al. (2011) A mighty small heart: The cardiac proteome of adult Drosophila melanogaster. PLoS One 6:e18497 . doi: 10.1371/journal.pone.0018497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rugendorff A, Younossi-Hartenstein A, Hartenstein V (1994) Embryonic origin and differentiation of the Drosophila heart. Roux’s Arch Dev Biol 203:266–280 . doi: 10.1007/BF00360522 [DOI] [PubMed] [Google Scholar]
- 49.Yin Z, Xu XL, Frasch M (1997) Regulation of the twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development 124:4971–4982 [DOI] [PubMed] [Google Scholar]
- 50.Wessells RJ, Fitzgerald E, Cypser JR, et al. (2004) Insulin regulation of heart function in aging fruit flies. Nat Genet 36:1275–1281 . doi: 10.1038/ng1476 [DOI] [PubMed] [Google Scholar]
- 51.Vogler G, Ocorr K (2009) Visualizing the beating heart in Drosophila. J Vis Exp 31:6–8 . doi: 10.3791/1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alayari NN, Vogler G, Taghli-Lamallem O, et al. (2009) Fluorescent labeling of Drosophila heart structures. J Vis Exp 1–5 . doi: 10.3791/1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linkert M, Rueden CT, Allan C, et al. (2010) Metadata matters: Access to image data in the real world. J. Cell Biol 189:777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi HMT, Calvert CR, Husain N, et al. (2016) Mapping a multiplexed zoo of mRNA expression. Development 143:3632–3637 . doi: 10.1242/dev.140137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Viswanathan MC, Blice-Baum AC, Sang T-K, Cammarato A (2016) Cardiac-restricted expression of VCP/TER94 RNAi or disease alleles perturbs Drosophila heart structure and impairs function. J Cardiovasc Dev Dis 3:1–20 . doi: 10.3390/jcdd3020019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swatland HJ (1987) Autofluorescence of adipose tissue measured with fibre optics. Meat Sci 19:277–284 . doi: 10.1016/0309-1740(87)90074-X [DOI] [PubMed] [Google Scholar]
- 57.Masuda N, Ohnishi T, Kawamoto S, et al. (1999) Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res 27:4436–4443 . doi: 10.1093/nar/27.22.4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evers LD, Fowler CB, Cunningham BR, et al. (2011) The effect of formaldehyde fixation on RNA: optimization of formaldehyde adduct removal. J Mol Diagnostics 13:282–288 . doi: 10.1016/j.jmoldx.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]