

Summary
Neurodegenerative diseases are characterized by the formation and propagation of protein aggregates especially amyloid fibrils. However, what normally suppresses protein misfolding and aggregation in metazoan cells remains incompletely understood. Here we show that TRIM11, a member of metazoan tripartite motif (TRIM) family, both prevents formation of protein aggregates and dissolves pre-existing protein deposits, including amyloid fibrils. These molecular chaperone and disaggregase activities are ATP independent. They enhance folding and solubility of normal proteins and cooperate with TRIM11 SUMO ligase activity to degrade aberrant proteins. TRIM11 abrogates α-synuclein fibrillization and restores viability in cell models of Parkinson’s disease (PD). Intracranial adeno-associated viral delivery of TRIM11 mitigates α-synuclein-mediated pathology, neurodegeneration, and motor impairments in a PD mouse model. Other TRIMs can also function as ATP-independent molecular chaperones and disaggregases. Thus, we define TRIMs as a potent and multifunctional protein quality control system in metazoa, which might be applied to treat neurodegenerative diseases.
Introduction
Neurodegenerative diseases are associated with misfolding and aggregation of specific polypeptides and the ensuing loss of neurons (Chiti and Dobson, 2006). While some of these diseases are familial due to germline mutations that result in the production of defective proteins, the vast majority of diseases are sporadic and are caused by aggregation of normal proteins expressed at physiological levels. The predominant form of aggregates is amyloid fibril, which consists of β-strands that align perpendicularly to the fibril axis (cross-β structure) (Eisenberg and Jucker, 2012; Knowles et al., 2014). These highly ordered assemblies can emerge from de novo or seeded aggregation of endogenous soluble proteins. The focal formation of amyloid fibrils and their self-templating spread among functionally-connected brain regions underlie the initiation and progression of lesions in human patients (Brettschneider et al., 2015; Jucker and Walker, 2013).
Among these diseases, Parkinson’s disease (PD) is the most common neurodegenerative movement disorder. PD is pathologically characterized by Lewy bodies (LBs) and Lewy neurites (LNs), which contain amyloid fibrils of α-synuclein (α-Syn), a normal protein abundantly expressed in neurons (Poewe et al., 2017; Surmeier et al., 2017). The emergence of α-Syn pathology is followed by progressive degeneration of dopaminergic neurons and motor symptoms including bradykinesia, rigidity, and resting tremor. In addition to PD, α-Syn fibrillization is found in Lewy body dementia (LBD) and multiple system atrophy (MSA), collectively known as synucleinopathies (Goedert et al., 2013). While fatal and increasingly prevalent, PD and many other neurodegenerative diseases remain incurable, underscoring the need for a better knowledge of how metazoan cells including human cells normally prevent and reverse pathological protein fibrillization.
To maintain proteins in their functional soluble state, organisms in all kingdoms of life have evolved protein quality control (PQC) systems. These systems include molecular chaperones that prevent protein misfolding and aggregation, disaggregases that dissolve pre-existing protein deposits, and degradation pathways that recycle proteins by proteolysis (Doyle et al., 2013; Kim et al., 2013; Mack and Shorter, 2016; Rubinsztein, 2006; Saibil, 2013; Tyedmers et al., 2010; Wolff et al., 2014). Molecular chaperones and disaggregases enable normal proteins to attain or regain their native conformations, while degradation pathways eliminate defective or terminally misfolded proteins. The repertoires of these systems in metazoans, as well as their functional coordination, remain incompletely understood. Most known chaperones and disaggregases are driven by energy derived from ATP hydrolysis and are either conserved from prokaryotes to eukaryotes (e. g., Hsp60, Hsp70, and Hsp90 families of molecular chaperones) or absent from metazoans (e.g., the Hsp104 disaggregase). Yet, compared to other organisms, metazoans contain proteins that have become substantially longer and structurally more intricate, and they depend on post-mitotic cells such as neurons and cardiomyocytes that are irreplaceable. This complexity raises an important question as to whether metazoans employ additional specialized PQC machineries to meet the special demand of their life cycle.
Here, we present evidence that tripartite motif (TRIM) proteins may be a metazoan innovation with molecular chaperone and disaggregase activities that are mechanistically distinctive and functionally effective. TRIM proteins are defined by the TRIM/RBCC motif consisting of a RING domain, one or two B-boxes, and a coiled-coil region. These proteins are found only in metazoans and exist as a large number of distinct variants, with ~20 in the nematode Caenorhabditis elegans and over 70 in mice and humans (Hatakeyama, 2011; Ozato et al., 2008). We previously established that TRIM19 (also known as PML) and TRIM11 can mediate proteasomal degradation of defective proteins linked to familial neurodegenerative diseases, such as ataxin-1 with an expanded polyglutamine region (Atxn1 82Q) (Chen et al., 2017; Guo et al., 2014). In the case of TRIM19, defective proteins are marked with poly-SUMO2/3 chains via its SUMO ligase activity, permitting their ubiquitination by SUMO-targeted ubiquitin ligases (STUbLs) and subsequent degradation by the proteasome (Guo et al., 2014). In this study, we show that TRIM11 and other TRIMs can act as ‘stand-alone’ chaperones and disaggregases that are independent of ATP. These TRIM activities suppress de novo and seeded fibrillization of normal proteins including α-Syn in vitro and in cells, and can functionally cooperate with the SUMO ligase activity in the clearance of defective proteins including Atxn1 82Q. Using adeno-associated virus (AAV)-mediated gene transfer, we find that TRIM11 mitigates protein aggregation, neurodegeneration, and motor impairments in a PD mouse model. These findings suggest that TRIMs may comprise a multifunctional PQC system in metazoans with utility in treating various neurodegenerative diseases.
Results
TRIM11 inhibits formation of amorphous aggregates
The role of TRIMs in the proteolysis of defective proteins (Chen et al., 2017; Guo et al., 2014) prompted us to evaluate whether they have other functions in PQC. We primarily focused on human TRIM11 because of its potent effects (Chen et al., 2017). To test whether TRIM11 possesses molecular chaperone activity, we purified a glutathione S-transferase (GST) fusion of TRIM11 from bacteria (Figure S1A). When the model chaperone substrate firefly luciferase was treated at an elevated temperature (42 °C), it rapidly misfolded and lost its enzymatic activity (Figures 1A and 1B), and generated amorphous aggregates detectable by light scattering (Figure 1C). GST-TRIM11, but not GST, protected luciferase from the thermal inactivation over time and in a dose-dependent manner, similar to Hsp70 (Figures 1A and 1B). GST-TRIM11, but not GST, also reduced luciferase aggregation by more than half at an equimolar ratio to luciferase and nearly abolished it at higher (2:1 and 4:1) molar ratios (Figures 1C, S1B, and S1C). Likewise, GST-TRIM11, but not GST, protected citrate synthase from heat-induced aggregation, reducing it by ~70% at a 2:1 molar ratio to citrate synthase (Figure 1D).
Figure 1. TRIM11 prevents the formation of amorphous and fibrillar aggregates.

(A and B) Inactivation of luciferase (10 nM) at 45 °C in the presence of GST, GST-TRIM11, or Hsp70 at 200 nM for the indicated time (A), or at the indicated concentration for 2 min (B). Samples with Hsp70 also contained 5 mM ATP and an ATP regeneration system (same below).
(C and D) Aggregation of luciferase (200 nM, C) and citrate synthase (400 nM, D) at 42 °C and 40 °C, respectively, in presence of GST or GST-TRIM11 at indicated molar ratios.
(E to G) Luciferase was transfected into HCT116 cells that were treated with control or TRIM11 siRNA (E), or stably expressed mCherry or mCherry-TRIM11 (F and G). Cells were heat shock (HS) and recovered at 37 °C. Shown are luciferase activity and protein expression with recombinant GST-TRIM11 and Hsp70 as protein standards.
(H and I) ThT-binding (H, left), sedimentation (H, right), and electron microscopy (I; Scale bar, 100 nm) assays of fibrilization of α-Syn monomers (70 μM) in the absence or presence of GST or GST-TRIM11 at indicated concentrations (H) or at 2 μM (I).
(J) ThT-binding assay of α-Syn (70 μM) fibrillization in the absence or presence of GST (1 μM), GST-TRIM11 (0.5 μM), Hsp70 (0.5 μM), and/or Hsp40 (0.25 μM).
(K) RT-QuIC assay of PFFs (133 nM)-seeded α-Syn (13.3 μM) aggregation in the presence of GST-TRIM11 (0.94, 1.88, 3.75, 7.5, and 15 μM, respectively). Results are representatives of three independent experiments done in 4 technical repeats. Blank, buffer only. Negative control (Neg Con), monomeric α-Syn.
(L) Atxn1 82Q (0.1 μM) fibrillization in the presence of indicates amounts of 6xHis-TRIM11 for 24 h.
(M) Suppresses Atxn1 82Q aggregation by GST-TRIM11. The three parts of SR were from the same exposure of the same blot. Relative amounts of Atxn1 82Q in each fraction are shown.
(N) Binding of 6xHis-TRIM11 and Hsp70 with agarose beads conjugated with ATP via different moiety. Molecular Weight markers (in KDa) are shown.
Data are mean ± SD (n = 3 biological replicates unless otherwise indicated). *P < 0.05, **P < 0.01, ***P < 0.001.
To corroborate these results, we purified GST-TRIM11 from insect Sf9 cells and observed that it protected both luciferase and citrate synthase from heat-induced inactivation and aggregation (Figures S1D to S1F). We also generated 6xHis-tagged TRIM11 from bacteria. To ensure purification of the full-length protein, TRIM11 was fused with maltose-binding protein (MBP) at the N-terminus and the 6xHis tag at the C-terminus, and purified sequentially with amylose and Ni-NTA resins with the MBP moiety being cleaved off of TRIM11 in between (Figures S1G and S1H). The purified 6xHis-TRIM11 protein protected luciferase against heat inactivation, similar to Hsp70 plus its co-chaperone Hsp40 (Figures S1I). Together, these results indicate that the chaperone activity in vitro is intrinsic to TRIM11.
To evaluate the effect of TRIM11 in cells, we treated human HCT116 cells expressing exogenous luciferase at an elevated temperature and then allowed them to recover at 37 °C, under the condition where new protein synthesis was blocked by cycloheximide (CHX). Knockdown of endogenous TRIM11 by means of small interfering RNA (siRNA) resulted in lower luciferase activity upon heat shock and slower luciferase reactivation during recovery (Figure 1E). Conversely, stable overexpression of mCherry-TRIM11, but not mCherry, protected luciferase activity and promoted its reactivation (Figures 1F and S1J). Intracellular TRIM11 protein levels, even upon overexpression, were substantially lower than those of Hsp70 (Figure 1G). These results show that TRIM11 can function as a molecular chaperone in cells.
TRIM11 prevents formation of fibrillar aggregates
While amorphous aggregation often occurs upon acute proteotoxic stresses, accumulation of amyloid fibrils is a pathological hallmark of various chronic neurodegenerative diseases. To investigate whether TRIM11 can inhibit fibrillization of disease-associated proteins, we used α-Syn as a substrate. In vitro, soluble α-Syn monomers spontaneously assembled into amyloid fibrils, as indicated by a binding-assay with the amyloid-specific dye thioflavin-T (ThT) (Figure 1H, left) and negative-stain electron microscopy (EM) (Figure 1I). GST-TRIM11, but not GST, suppressed de novo α-Syn fibrillization in a dose-dependent manner, reducing ThT fluorescence and increasing soluble α-Syn (Figure 1H). EM analysis confirmed that TRIM11 blocked the formation of mature fibrils (Figure 1I). Notably, a small amount of TRIM11 (0.1-0.5 μM) was able to prevent aggregation of α-Syn monomers at over 140-fold in excess (70 μM) (Figures 1H–1J).
Reflecting the self-templating assembly of amyloid fibrils, fibrillization of soluble α-Syn monomers was accelerated by α-Syn preformed fibrils (PFFs) in a real-time quaking-induced conversion (RT-QuIC) assay (Figure 1K). GST-TRIM11 inhibited the PFFs-seeded fibrillization in a dose-dependent manner and near-completely blocked it at a higher dose (Figure 1K). 6xHis-TRIM11 also abrogated both spontaneous and seeded fibrillization of soluble α-Syn monomers (see below).
To extend these analyses, we used Atxn1 82Q, which causes spinocerebellar ataxia type 1 (SCA1) (Orr and Zoghbi, 2007). Like α-Syn, recombinant Atxn1 82Q protein formed ThT-reactive amyloid fibrils upon incubation (Figure 1L). 6xHis-TRIM11 prevented Atxn1 82Q fibrillization in a dose-dependent manner and reduced it by ~80% at a 4:1 molar ratio to Atxn1 82Q (Figure 1L). We also performed a sedimentation assay and analyzed the pellet fraction by dot blot to detect the SDS-resistant (SR) fibrillar aggregates and by Western blot to detect the SDS-soluble (PE) amorphous aggregates (Wanker et al., 1999) (Figure 1M). GST-TRIM11, but not GST, blocked the formation of both SR and PE aggregates, keeping virtually all Atxn1 82Q molecules soluble in the supernatant (SN) even after a prolonged incubation (12 h; Figure 1M). Similarly, 6xHis-TRIM11 prevented the formation of these Atxn1 82Q aggregates. Collectively, these results indicate that TRIM11 suppresses fibrillization of disease-associated proteins.
In contrast to canonical chaperones such as Hsp70, TRIM11 prevented protein aggregation in the absence of ATP (Figures 1 and S1). TRIM11, like other TRIMs, does not contain a recognizable ATP-binding domain. In an in vitro assay, TRIM11 was unable to bind to ATP (Figure 1N).
TRIM11 dissolves pre-existing amorphous aggregates
Compared to prevention of protein aggregates, dissolution of pre-existing protein deposits is a more challenging process. Besides energy derived from ATP hydrolysis, it often requires the coordinated action of multiple components (Doyle et al., 2013; Saibil, 2013). The best characterized disaggregase is perhaps the bi-chaperone system consisting of Hsp70/Hsp40 and the Hsp104 AAA+-ATPase (Glover and Lindquist, 1998). This system is present in bacteria, fungi, and plants but absent in the cytoplasm and nucleus of animal cells (Doyle et al., 2013; Saibil, 2013). Mammalian Hsp110 family (an atypical Hsp70 family) can act together with the Hsp70 and Hsp40 families for disaggregation, although a combination of a specific member(s) from each family at a specific molar ratio(s) appears to be required for such activity (Gao et al., 2015; Nillegoda et al., 2015; Shorter, 2011).
To investigate whether TRIM11 can dissolve protein aggregates, we used amorphous aggregates formed by thermally denatured luciferase. GST-TRIM11, but not GST, purified from bacteria was able to dissolve misfolded luciferase molecules trapped in aggregates and restore their enzymatic activity in a dose- and time-dependent manner (Figures 2A and 2B). We also employed amorphous aggregates generated by heat-denatured green fluorescence protein (GFP) (DeSantis et al., 2012; Shorter, 2011). GST-TRIM11 dissolved GFP molecules trapped in aggregates and enabled them to reemit fluorescence (Figures 2C and 2D).
Figure 2. TRIM11 dissolves preformed amorphous and fibrillar aggregates.

(A to D) Solubilization and reactivation of preformed luciferase (10 nM, A and B) and GFP (0.45 μM, C and D) aggregates in the presence of GST or GST-TRIM11 at indicated concentrations (A and C) or 1 μM (B and D), or Hsp70/Hsp40-Hsp104 at 0.5 μM.
(E) Reactivation of preformed citrate synthase aggregates (0.15 μM) by 6xHis-TRIM11 and Hsp70/Hsp40-Hsp104.
(F to H) ThT-binding (F), sedimentation (G), and EM (H) assays of α-Syn PFFs (1 μM) treated with buffer, GST, GST-TRIM11, or Hsp70/Hsp40-Hsp104A503S at the indicated concentrations (F and G) or 1 μM (H). Scale bar, 0.5 μm
(I) ThT-binding assay of Atxn1 82Q aggregates (0.2 μM) incubated with 6xHis-TRIM11.
(J and K) Sedimentation assay of Atxn1 82Q aggregates at 50 nM (J) or 200 nM (K) incubated with lysozyme, GST, or TRIM11 at increasing concentrations (10, 50, and 200 nM) (J) or 0.5 μM (K), or with Hsp70 at 1 μM (K).
Data are mean ± SD (n = 3 biological replicates). **P < 0.01, ***P < 0.001.
Similarly, GST-TRIM11 purified from insect Sf9 cells could dissolve and reactivate luciferase aggregates (Figures S2A and S2B). 6xHis-TRIM11 was also able to solubilize aggregated luciferase, GFP (see below), and citrate synthase (Figure 2E). A GFP fusion of TRIM11 (GFP-TRIM11) purified from human embryonic kidney 293T (HEK293T) cells could dissolve luciferase aggregates as well (Figures S2C and S2D).
TRIM11 dissolves preformed amyloid fibrils
To test whether TRIM11 can disassemble amyloid fibrils, we examined its activity on α-Syn PFFs. As a positive control, Hsp70/Hsp40 plus Hsp104A503S, an enhanced version of HSP104 (Jackrel et al., 2014), disaggregated α-Syn PFFs in the presence of ATP (Figures 2F–2H). Interestingly, GST-TRIM11, but not GST, was also capable of disaggregating α-Syn PFFs, reducing their binding to ThT in a dose-dependent manner (Figure 2F) and converting them to the soluble form (Figure 2G). This effect was verified by EM analysis (Figure 2H). Likewise, 6xHis-TRIM11 was able to dissolve α-Syn PFFs (Figures S2E–S2G).
6xHis-TRIM11 also disassembled Atxn1 82Q fibrils in a dose-dependent manner leading to near-complete dissolution at a molar ratio of 5:1 or higher to Atxn1 82Q (Figure 2I). In addition, GST-TRIM11, but not GST, disaggregated SR and PE aggregates of Atxn1 82Q (Figure 2J). Under conditions where virtually all Atxn1 82Q proteins coalesced into SR aggregates, GST-TRIM11 was still able to solubilize the majority of them (Figure 2K, lane 3). In comparison, Hsp70/Hsp40 turned SR aggregates only into the PE form (lane 4), while Hsp104 showed no activity on its own (lane 5). To convert SR aggregates all the way to the supernatant, both Hsp70/Hsp40 and Hsp104 were required (lane 6). TRIM11-mediated disaggregation occurred in the absence of ATP (Figures 2 and S2A to S2G), and was not affected upon treatment with apyrase to degrade ATP (Figure S2G).
Atxn1 82Q formed nuclear inclusions when overexpressed in mammalian cells (Guo et al., 2014; Skinner et al., 1997). To evaluate the effect of TRIM11 in cells, we first expressed Atxn1 82Q in human HeLa cells and then introduced recombinant TRIM11 protein by using HIV TAT peptide-mediated protein transduction (Nagahara et al., 1998) (Figure S2H). The TAT-TRIM11 fusion protein entered cells in a time- and concentration-dependent manner (Figures S2I–S2K). It not only prevented further accumulation of Atxn1 82Q aggregates but also largely reduced pre-existing Atxn1 82Q inclusions (Figures S2L and S2M), suggesting that TRIM11 can function as both a molecular chaperone and a disaggregase for disease-associated proteins in the cellular milieu.
Structural determinants of TRIM11 chaperone and disaggregase activities
To determine which domains are required for TRIM11’s chaperone and disaggregase activities, we generated a panel of deletion mutants that either lacked the RING, B-Box, or coiled-coil domain (∆R, ∆B, and ∆CC, respectively), or contained only the N-terminal RBCC (N) or the C-terminal PRY-SPRY/B30.2 (C) region (Figure 3A). These mutants were purified as 6xHis-tagged recombinant proteins (Figure S3A), as for the wild-type TRIM11 (Figure S1G). Among them, only TRIM11∆R retained molecular chaperone activity. Specifically, TRIM11∆R suppressed heat-induced aggregation of luciferase (Figure S3B), as well as de novo and PFFs-induced fibrilization of α-Syn (Figures 3B and 3C). TRIM11∆R also prevented aggregation of Atxn1 82Q into ThT-reactive fibrils (Figures 3D and 3E) and PE/SR aggregates (Figure 3F). In contrast, none of the other mutants (∆B, ∆CC, N, and C) showed molecular chaperone activity (Figures 3B–3F and S3B).
Figure 3. Structural determinants of TRIM11’s chaperone and disaggregase activities.

(A) Schematic presentation of TRIM11 and its mutations.
(B) ThT-binding assay of α-Syn monomers (70 μM) incubated with GST or 6xHis-TRIM11 (1 μM) for 48 h.
(C) RT-QuIC assay of PFFs (100 nM)-seeded aggregation of α-Syn monomers (20 μM) in the presence of 6xHis-TRIM11 proteins (2 μM).
(D to F) ThT-binding (D and E) and sedimentation (F) assays of Atxn1 82Q (100 nM) incubated with 6xHis-TRIM11 proteins at 400 nM (D and F) or the indicated concentrations (E), at 37 °C for 24 h. Soluble Atxn1 82Q without incubation (37 °C, −) was used as a control.
(G and H) Solubilization and reactivation of preformed luciferase (10 nM, G) and GFP (0.45 μM, H) aggregates by 6xHis-TRIM11 proteins at 200 nM (G) or 100 nM (H).
(I to M) ThT binding (I, K, and L) and sedimentation (J and M) assays of preformed α-Syn (1 μM) (I and J) or Atxn1 82Q (0.2 μM) (K to M) aggregates treated with GST or 6xHis-TRIM11 proteins at increasing concentrations (K) or 1 μM (I, J, L, and M).
Data are mean ± SD (n = 3 biological replicates). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n.s., not significant.
Similarly, TRIM11∆R retained disaggregase activity. TRIM11∆R solubilized luciferase and GFP aggregates (Figures 3G and 3H), α-Syn PFFs (Figure 3I and 3J), and Atxn1 82Q fibrils and PE/SR aggregates (Figures 3K to 3M). None of the other mutants displayed disaggregase activity (Figures 3G–M). Therefore, with the exception of the RING domain, the other regions of TRIM11 are required for the chaperone and disaggregase activities.
An analysis of TRIM11 amino acid sequence revealed that it contains more leucine than any other amino acid (~13%; Figures S3C and S3D). We mutated 44 leucine residues outside the RING domain to alanine (TRIM11LA, Figure S3C). TRIM11LA neither prevented nor reversed protein aggregation (Figures S3E–S3G). Thus, the compositional bias for leucine residue appears to be important for the function of TRIM11, although TRIM11LA might not acquire the native conformation.
TRIM11 recognizes misfolded proteins and responds to proteotoxic stress
The molecular chaperone and disaggregase activities of TRIM11 prompted us to test whether it can recognize misfolded proteins. When expressed alone in cells, TRIM11 localized predominantly to the cytoplasm (Chen et al., 2017) (Figure S4A). However, in the presence of Atxn1 82Q, TRIM11 translocated to the nucleus and accumulated in Atxn1 82Q inclusions (Figures 4A and S4A), parallel to the movement of Hsp70 (Figure S4B). Also similar to Hsp70, both endogenous and exogenous TRIM11 interacted with Atxn1 82Q in cells (Figures 4B and 4C). In vitro, TRIM11 bound directly with ataxin 1 proteins and preferentially with the pathogenic form (Atxn1 82Q) over a non-pathogenic form (Atxn1 30Q) (Figures 4D and S4C). TRIM11 also bound to urea-denatured, but not native, luciferase (Figure 4E). Therefore, TRIM11 can distinguish misfolded states from the native state of the same protein.
Figure 4. TRIM11 recognizes misfolded proteins and responds to heat shock, and other TRIMs can also prevent and reverse protein aggregation.

(A) Co-localization of GFP-Atxn1 82Q and HA-TRIM11 in HEK293T cells. Scale bar, 5 μm
(B and C) Interactions of Atxn1 82Q with endogenous TRIM11 or Hsp70 (B), or exogenous TRIM11, TRIM112EA, or Hsp70 (C), in HEK293T cells.
(D, E, and L) Preferential binding of TRIM11 (D and E) and TRIM21 (L) to Atxn1 82Q over Atxn1 30Q (D), or to denatured (d) over native (n) luciferase (E and L).
(F and G) Localization (F; scale bar, 10 pm) and interaction (G) of GFP-Atxn1 82Q and Flag-TRIM11 proteins in HEK293T cells.
(H and I) Increase in TRIM11 protein and mRNA levels in HCT116 cells (H) and mouse primary HC neurons (I) that were heat shocked at 42 °C for 1 (H) or 0.5 (I) hr and recovered at 37 °C for the indicated times (H) or 3 h (I).
(J) Activity of luciferase (10 nM) when heated at 45 °C for 1 min in the presence of GST or GST-TRIM21 (200 nM).
(K and M) Solubilization and reactivation of preformed luciferase (K, 10 nM) and GFP (M, 0.45 μM) aggregates by TRIM21 (K) and TRIM19 (isoform VI) for 90 and 60 min, respectively.
(N and O) A nucleus-localized luciferase was transfected alone in control or TRIM19-knockdown HCT116 cells (N), or together with control vector or TRIM19 in HCT116 cells (O). Luciferase activity during heat shock at 42 °C for 60 min (O) or 45 °C for 30 min (N) and recovery at 37 °C was assayed.
(P) Localization of GFP-Atxn1 82Q and mCherry-TRIM19 (isoform VI) in HeLa cells that were untreated or extracted with cytoskeleton stripping buffer.
Data are mean ± SD (n = 3 biological replicates). *P < 0.05, **P < 0.01, ***P < 0.001.
TRIM11∆R, like TRIM11, co-localized and interacted with Atxn1 82Q in cells, while the other mutants (∆B, ∆CC, N, and C) did not (Figures 4F and 4G). TRIM11LA, which showed a cytoplasmic localization pattern (Figure S4D), also failed to co-localize or interact with Atxn1 82Q (Figures S4E and S4F). Thus, the regions of TRIM11 beyond the RING domain are required for the interaction with misfolded proteins.
Levels of molecular chaperones and disaggregases often increase in response to proteotoxic stresses. Upon heat shock, endogenous TRIM11 mRNA and protein, along with those of Hsp70, were up-regulated in several human cell lines including HCT116, HeLa, and A549 (Figures 4H, S4G, and S4H) as well as in mouse primary cortical and hippocampal (HC) neurons (Figures 4I and S4I). In contrast, levels of exogenous TRIM11 stably expressed from a heterologous promoter remained unchanged in HCT116 cells (Figure S4J). Thus, endogenous TRIM11 is responsive to proteotoxic stresses, underscoring another parallel with canonical molecular chaperones and disaggregases.
Chaperone and disaggregase activities of other TRIM proteins
TRIM proteins constitute a large family in metazoans (Hatakeyama, 2011; Ozato et al., 2008). To evaluate whether other TRIMs can function similarly to TRIM11, we analyzed human TRIM21 and TRIM19, both of which can mediate the degradation of Atxn1 82Q (Guo et al., 2014) (Figure S4K). TRIM21 is a cytoplasmic receptor for antibodies (Keeble et al., 2008) and, similar to TRIM11, contains a PRY-SPRY/B30.2 region (Ozato et al., 2008). Purified recombinant TRIM21 protein prevented heat inactivation of luciferase (Figure 4J), and solubilized and reactivated previously denatured luciferase (Figure 4K). TRIM21 bound to urea-denatured luciferase more strongly than native luciferase (Figure 4L), indicating its ability to discern misfolded proteins.
TRIM19 is a nuclear protein implicated in diverse cellular processes (Bernardi and Pandolfi, 2007) and, unlike TRIM11 and TRIM21, contains no recognizable C-terminal domain (Ozato et al., 2008). Still, purified recombinant TRIM19 protein re-activated thermally denatured GFP in vitro (Figures 4M and S4L). In cells, knockdown of TRIM19 accelerated the inactivation of a nucleus-localized luciferase upon heat shock and delayed its reactivation during the recovery (Figure 4N and S4M), whereas forced expression of predominantly protected luciferase and promoted its reactivation (Figure 4O). TRIM19 interacts with misfolded proteins including Atxn1 82Q (Guo et al., 2014) and forms proteinaceous nuclear bodies (Bernardi and Pandolfi, 2007) that partially co-localize with Atxn1 82Q (Guo et al., 2014) (Figure 4P, top panels). Interestingly, Atxn1 82Q molecules within TRIM19 nuclear bodies were soluble and could be extracted by a non-denaturing detergent at low concentrations (Figure 4P, bottom panels), suggesting that TRIM19 can function as a disaggregase in cells. Similar to TRIM11, both TRIM21 and TRIM19 prevented and reversed protein aggregation in an ATP-independent manner (Figures 4J, 4K, and 4M).
Functional cooperation of TRIM11 chaperone/disaggregase and SUMO ligase activities in the removal of defective proteins
TRIM19 and some other TRIMs possess SUMO ligase (E3) activity (Chu and Yang, 2011). Moreover, TRIM19 mediates degradation of defective proteins such as Atxn1 82Q through sequential TRIM19-mediated SUMOylation and STUbL-mediated ubiquitination (Guo et al., 2014). Therefore, we examined whether TRIM11 has SUMO ligase activity and, if so, whether the chaperone/disaggregase activities of TRIM11 functionally cooperate with the SUMO E3 activity to remove defective proteins. In cells, TRIM11 promoted SUMOylation of Atxn1 82Q in the presence or absence of exogenous SUMO2 (Figures 5A to 5C), with an activity that appeared to be stronger than that of TRIM19 (Figure 5B). In vitro, purified recombinant TRIM11 enhanced SUMOylation of Atxn1 82Q (Figure S5A). In contrast, TRIM11 did not promote the ubiquitination of soluble α-Syn stably expressed in HEK293T cells (Figure 5D). Atxn1 82Q is an aberrant protein that easily misfolds and aggregates in cells and in vitro, whereas α-Syn is a normal protein. These results suggest that TRIM11 possesses SUMO ligase activity that may be dependent on the folding state of the substrate.
Figure 5. SUMO ligase activity of TRIM11, and functional cooperation of TRIM11’s discrete activities in the clearance of Atxn1 82Q.
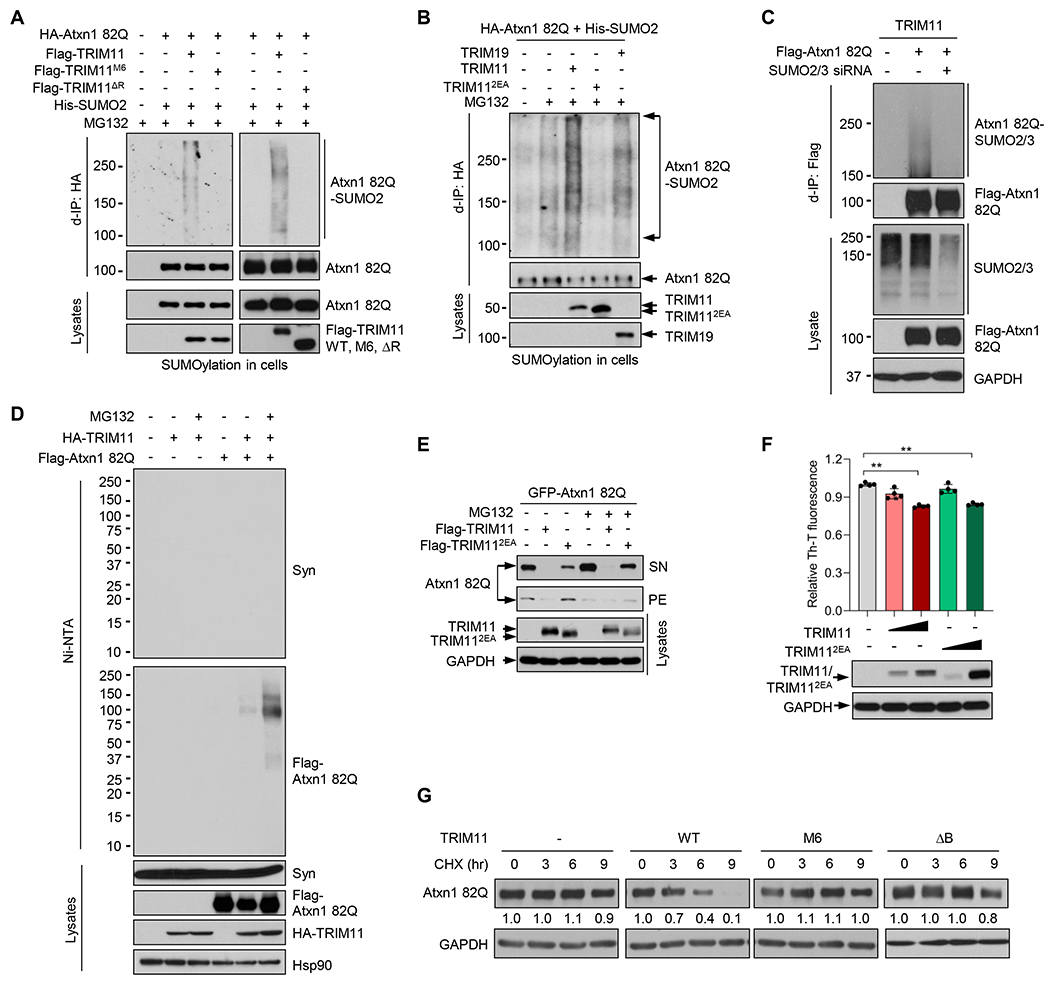
(A and B) SUMOylation of HA-Atxn1 82Q by Flag-TRIM proteins in the presence of 6xHis-SUMO2 in HEK293T cells.
(C) SUMOylation of Flag-Atxn1 82Q by HA-TRIM11 in HEK293T cells treated with control or SUMO2/3 siRNA.
(D) SUMOylation of Flag-Atxnl 82Q, but not α-Syn, by HA-TRIM11 in the presence of 6xHis-SUMO2 in HEK293T cells stably expressing α-Syn.
(E) Levels of GFP-Atxn1 82Q when expressed alone or together with Flag-TRIM11/TRIM112EA in HCT116 cells.
(F) Amyloid contents in HCT116 cells transfected with TRIM11 or TRIM112EA
(G) Half-life of Flag-Atxn1 82Q in HEK293T cells when co-expressed with the indicated TRIM11 proteins. Relative Atxn1 82Q/GAPDH ratios are shown. The exposure of different blots was adjusted so that band intensities at time 0 were comparable.
Data are mean ± SD (n = 4 biological replicates). **P < 0.01.
Given that the in vitro chaperone/disaggregase assays were performed in the absence of ATP or any other SUMOylation reaction components (Figures 1-3 and S1-S3), these activities of TRIM11 occur independently of its SUMO ligase activity. Nevertheless, in cells these activities may cooperate with SUMO ligase activity to remove defective proteins, which in the aggregated state cannot be effectively recycled by the proteasome (Bence et al., 2001). To test this notion, we generated missense mutants of TRIM11 that were defective in SUMO ligase activity, but retained chaperone and disaggregase activities. We replaced six conserved zinc-chelating His or Cys residues in the RBCC region with Ala (Chu and Yang, 2011), or two Glu residues proximal to the RING domain with Ala (Li et al., 2014), TRIM11M6 and TRIM112EA, respectively (Figure 3A). Both mutants failed to SUMOylate Atxn1 82Q (Figures 5A, 5B, and S5A). Nevertheless, TRIM11M6 was as active as TRIM11 in preventing and dissolving amorphous aggregates formed by luciferase and GFP, as well as fibrillar aggregates formed by α-Syn and Atxn1 82Q (Figures 3B to 3M, and S5B). TRIM11M6 also co-localized and interacted with Atxn1 82Q in cells (Figures 4F and 4G). Similarly, TRIM112EA was fully active as a molecular chaperone and disaggregase (Figures S5C to S5E). TRIM112EA also interacted with Atxn1 82Q in cells and in vitro (Figures 4C and S5F) and bound to denatured, but not native, luciferase in vitro (Figure S5G). Therefore, the RING domain of TRIM11, which is dispensable for molecular chaperone and disaggregase activities (Figures 3 and S3), is essential for SUMO ligase activity. Consistently, TRIM11∆R, which retained chaperone/disaggregase activities, failed to SUMOylate Atxn1 82Q (Figure 5A). Therefore, TRIM11’s chaperone and disaggregase activities are structurally separable from its SUMO ligase activity.
We compared TRIM11 and TRIM112EA for the ability to remove defective proteins in cells. When Atxn1 82Q was transiently or stably overexpressed, it readily generated amorphous/fibrillar aggregates in cell lysates (Figures 5E, S5H, and S5I) in addition to nuclear inclusions in cells (Figure 4A), indicating that intracellular levels of Atxn1 82Q far exceeded its solubility limit. TRIM11 strongly reduced levels of both overall and aggregated Atxn1 82Q (Figures 5E, S5H, and S5I). TRIM112EA displayed a weaker inhibitory effect (Figures 5E, S5H, and S5I). Amyloid fibril is a generic conformation that can be adopted by many proteins regardless of primary sequence (Eisenberg and Jucker, 2012; Knowles et al., 2014). TRIM11 reduced levels of intracellular amyloid aggregates (Figure 5F). TRIM112EA could elicit a similar effect, but only when it was expressed at substantially higher levels (Figure 5F). Moreover, while wild-type TRIM11 accelerated Atxn1 82Q degradation, neither TRIM11M6 (which lacked SUMO ligase activity) nor TRIM11∆B (which lacked chaperone/disaggregase activities) showed such an effect (Figure 5G). Together, these results suggest that both chaperone/disaggregase and SUMO ligase activities are required for the effective clearance of defective proteins.
TRIM11 abrogates α-Syn pathology and restores cell viability
While Atxn1 82Q is associated with an inherited disorder, most neurodegenerative diseases are sporadic in which normal proteins expressed at physiological levels assemble into self-templating fibrillar aggregates. To evaluate the role of TRIM11 in suppressing α-Syn fibrillization in cells, we used a PD model where a small amount of recombinant α-Syn PFFs taken up by cells can convert soluble endogenous α-Syn into LB/LN-like inclusions (Luk et al., 2009; Volpicelli-Daley et al., 2011). We first evaluated whether TRIM11 suppresses the seeding capability of recombinant α-Syn. Recombinant α-Syn pre-incubated alone or with GST or GST-TRIM11 (Figures 1H to 1J) was used to treat primary HC neurons isolated from wild-type mice. Upon incubation alone or with GST, recombinant α-Syn, which formed amyloid fibrils (Figures 1H to 1J), strongly induced α-Syn aggregation in primary HC neurons. This aggregation was reflected by the appearance of LB/LN-like inclusions in the cytosol containing α-Syn phosphorylated at Ser129 (pSyn; Figures 6A and 6B), a highly specific marker of α-Syn pathology (Fujiwara et al., 2002), and by the formation of various α-Syn species in cell lysates that were insoluble in the non-denaturing detergent Triton-X100 (Figure 6C). In contrast, recombinant α-Syn acquired minimal activity to induce α-Syn pathology upon incubation with TRIM11 (Figures 6A–6C), which prevented its assembly into amyloid fibrils (Figures 1H to 1J). The pathogenic species of α-Syn are not fully defined, but short α-Syn fibrils elicit the most robust PD phenotypes in mice (Froula et al., 2019). Our results indicate that TRIM11 effectively eliminates the α-Syn species responsible for corrupting its soluble endogenous counterpart.
Figure 6. TRIM11 abrogates α-Syn fibrillation and restores cell viability in PD cell models through its chaperone/disaggregase activities.

(A-C) Representative images of pSyn immunostaining (A), quantification of pSyn pathology density (B), and soluble and insoluble α-Syn in lysates (C) of primary mouse HC neurons incubated with PBS, or α-Syn monomer (200 μM) that were pre-treated with or without GST or GST-TRIM11 (5 μM). Scale bar, 50 μm
(D and E) Representative images of pSyn immunostaining (D) and quantification of pSyn pathology (E, n = five or more randomly chosen fields) in control (EV), TRIM11-expressing, and TRIM112EA-expressing QBI-Syn cells that were incubated with or without α-SynS87N PFFs.
(F and G) Soluble and insoluble α-Syn proteins in (F), and viability (G) of, SH-SY5Y-Syn cells expressing control vector (EV), TRIM11, and TRIM11 mutants and treated with or without α-SynS87N PFFs.
(H) ThT staining (top) and protein expression (bottom) of SH-SY5Y cells transfected with Ctrl siRNA, TRIM11 siRNA (the #2 siRNA targeted 3’UTR), and/or Flag-TRIM11.
(I) Viability (left) and TRIM11 protein levels (right) of mouse primary HC neurons treated with control or TRIM11 siRNA and incubated with α-SynS87N PFFs.
Data are mean ± SD, n = 5 biological replicates for (I) and 3 for the rest, unless otherwise indicated. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To assess whether TRIM11 also affords protection within the cell, we used PFFs of a human α-Syn variant (α-SynS87N), which displays an increased pathogenicity (Luk et al., 2016), to treat QBI-293 cells stably expressing exogenous α-Syn in a soluble state (QBI-Syn cells) (Luk et al., 2009). SynS87N PFFs induced pSyn-containing LB/LN-like inclusions in QBI-Syn cells (Figures 6D and 6E). Forced expression of TRIM11 dramatically reduced SynS87N PFFs-induced pathology (Figures 6D, 6E, and S6A). To extend this analysis, we generated human neuroblastoma SH-SY5Y cells stably expressing α-Syn (SH-SY5Y-Syn) (Figure S6B), in which α-Syn was soluble (Figure 6F, lane 1) and elicited no apparent detrimental effects (Figure S6C). α-SynS87N PFFs induced aggregation of intracellular α-Syn in SH-SY5Y-Syn cells, as shown by the appearance of pSyn and various insoluble α-Syn species, as well as a reduction of soluble α-Syn, in cell lysates (Figures 6F, lane 2, and S6D). This process was accompanied by a decrease in cell viability (Figures 6G and S6E). Forced expression of TRIM11 had no effect on the proliferation of SH-SY5Y-Syn cells in the absence of PFFs (Figures S6F and S6G). However, it inhibited PFFs-induced α-Syn aggregation (Figures 6F, lane 3, and S6D) and restored cell viability (Figures 6G and S6E). Despite the potent effect on α-Syn aggregation, TRIM11 did not alter overall levels of intracellular α-Syn in QBI-Syn or SH-SY5Y-Syn cells (Figures S6A and S6F), suggesting that TRIM11 suppresses α-Syn fibrillization without targeting it for degradation.
To further evaluate the role of different TRIM11 activities, we examined various TRIM11 mutants. Like TRIM11, TRIM112EA strongly inhibited PFFs-induced α-Syn aggregation in QBI-Syn and SH-SY5Y-Syn cells and restored their viability (Figures 6D, 6E, S6D, and S6E), without altering levels of intracellular α-Syn (Figures S6A and S6F). Likewise, the other two SUMO ligase-defective mutants, TRIM11M6 and TRIM11∆R, suppressed α-Syn aggregation and restored cell viability (Figures 6F and 6G). In contrast, the chaperone/disaggregase-defective TRIM11∆B and TRIM11∆CC showed virtually no effect (Figures 6F and 6G). Collectively, these results indicate that the effect of TRIM11 on α-Syn pathology and cytotoxicity is largely due to its chaperone activity, disaggregase activity, or both.
To evaluate the role of endogenous TRIM11, we knocked it down in SH-SY5Y cells using independent small interfering RNAs (siRNAs, Figures 6H and S6H). This led to ~20% increase in intracellular amyloid-like aggregates (Figure 6H). Expression of an siRNA-resistant form of TRIM11 not only reversed the increase of amyloid-like aggregates in TRIM11-knockdown cells, but also further reduced these aggregates to levels ~30% below those in control cells (Figure 6H). Knocking down TRIM11 in HCT116 cells led to a similar increase in amyloid-like aggregates, which again could be reversed and further reduced by the siRNA-resistant TRIM11 (Figure S6I). Moreover, knocking down TRIM11 in mouse primary HC neurons decreased cell viability in the absence of α-Syn PFFs and dramatically sensitized these cells to PFFs-mediated cytotoxicity (Figure 6I). Collectively, these results indicate that TRIM11 affords protection to neurons and other cells under unstressed conditions and especially under proteotoxic stress conditions.
TRIM11 suppresses α-Syn pathology, neurodegeneration, and motor impairments in a mouse PD model
To evaluate the effect of TRIM11 in mammals, we employed a PD mouse model in which a single stereotaxic injection of α-Syn PFFs targeting the striatum induces LB/LN-like aggregates at the injection site as well as brain regions that are connected to the striatum, including piriform cortex (Pir) and frontal cortex (Fc). The appearance of α-Syn pathology is followed by progressive loss of dopaminergic neurons in the substantial nigra (SN) (Luk et al., 2012). Thus, this model recapitulates the two cardinal features of PD: LB/LN-like aggregates and neurodegeneration. We cloned TRIM11 and a control protein (GFP) in adeno-associated virus 9 (AAV9), a serotype that efficiently targets cells in the central nervous system (CNS) (Foust et al., 2009). We injected PFFs generated from recombinant mouse α-Syn protein, together with AAV9-TRIM11 or AAV9-GFP, unilaterally into dorsal striatum of young wild-type mice (C57BL6/C3H) (Figure 7A). At 120 days post injection (dpi), expression of GFP and TRIM11 was detected in the injected side of striatum and the ipsilateral Pir and Fc regions (Figures 7B and S7A). By then, LB/LN-like α-Syn aggregates were evident at the injection site and especially in the ipsilateral Pir and Fc regions in mice injected with AAV9-GFP, as assayed by both immunofluorescence (Figures 7B and S7A) and immunoperoxidase staining (Figures 7C, 7D, and S7B). Interestingly, mice injected with AAV9-TRIM11 displayed a strong reduction in α-Syn pathology (Figures 7B–7D, S7A, and S7B), with ~80% and ~65% decrease in the Pir and Fc regions, respectively (Figure 7D). Moreover, while α-Syn pathology was commonly present in GFP-expressing neurons, it was rarely detectable in TRIM11-expressing neurons (Figures 7B and S7A), further indicating that TRIM11 inhibits α-Syn aggregation.
Figure 7. TRIM11 mitigates α-Syn pathology, neurodegeneration, and behavioral defects in a PD mouse model.

(A) Schematic representation of stereotaxic injections (top) and experimental timeline (bottom).
(B) Representative immunofluorescence images of pSyn and TRIM11-HA/GFP in frontal cortex and piriform cortex of AAV9-GFP- and AAV9-TRIM11-injected mice. Scale bar, 100 μm.
(C and D) Representative IHC images of pSyn in cortical regions (C) and percentage of the total area occupied by pSyn inclusions (D; mean + SD, n ≥ 4 mice).
(E and F) Representative IHC images of TH-positive neurons in the injected ipsilateral and the non-injected contralateral regions (Scale bar, 190 μm) (E) and quantification of TH-positive neuron loss relative to the contralateral side of the same brain (mean + SEM, n ≥ 4 mice) (F).
(G) Distance travelled in each quadrant by AAV9-GFP and AAV9-TRIM11 mice during the open-field test. Data are the mean ± SEM (n ≥ 11 mice).
(H and I) Total distance travelled by (H), and total freezing time of (I), AAV9-GFP and AAV9-TRIM11 mice during the open-field test (mean ± SEM, n ≥ 11 mice).
(J) A model for the multiple roles of TRIMs in protein quality control. TRIMs prevent protein misfolding (a) and amorphous and fibrillar aggregation (b). They also dissolve pre-existing aggregates (b’) and promote their refolding (a’). For defective or terminally misfolded proteins, TRIMs may mark them with poly-SUMO2/3 chains (c), enabling them to be ubiquitinated by STUbLs (d) and subsequently degraded in the proteasome (e).
*P < 0.05, **P < 0.01.
Dopaminergic neurons, which project from the SN to the striatum, degenerate during the progression of PD (Poewe et al., 2017; Surmeier et al., 2017). As previously observed (Luk et al., 2012), inoculation of PFFs at the striatum led to formation of pSyn-containing inclusions inside SN dopaminergic neurons (Figure S7C). At 180 dpi, mice co-injected with AAV9-GFP exhibited significant loss of dopaminergic neurons, as shown by immunohistochemistry (IHC) analysis of tyrosine hydroxylase (TH) (Figures 7E and 7F), the rate-limiting enzyme in dopamine synthesis. The neuronal loss was restricted to the SN ipsilateral to the injection site, due to the exclusive unilaterality of the nigrostriatal pathway. TRIM11 noticeably protected TH-positive neurons, reducing their loss from ~47% to < 30% (Figures 7E and 7F).
To evaluate the effect of TRIM11 on motor functions, we performed an open-field test (Brooks and Dunnett, 2009). Animals in the AAV9-TRIM11 group showed greater locomotor activity and less anxiety compared to those in the AAV9-GFP group, with ~50% longer distance travelled around the center or the sides (Figure 7G) or in the whole field (Figure 7H). Sudden and transient loss of movement is a characteristic of PD patients. This loss of movement was also observed in GFP-injected mice, as shown by their relatively long freezing time (Figure 7I). In comparison, TRIM11-injected mice showed ~60% reduction in freezing time (Figure 7I). These data indicate that TRIM11 rescues α-Syn aggregation-mediated motor impairments.
Intracranial delivered AAV9-TRIM11 did not affect overall levels of α-Syn (Figure S7D). Moreover, when introduced into cultured hippocampus and cortical neurons, AAV-TRIM11 did not alter the abundance of endogenous α-Syn (Figure S7E). Collectively, these results indicate that TRIM11 inhibits the seeding and cell-to-cell transmission of α-Syn fibrils and protects animals against neurodegeneration and motor impairments, likely through its molecular chaperone activity, disaggregase activity, or both.
DISCUSSION
The late onset of PD and other neurodegenerative diseases suggest the existence of a metazoan system(s) that can suppress protein fibrilization at a younger age. Our studies suggest that metazoan TRIM proteins may comprise a distinctive and multifunctional PQC system that is neuroprotective (Figure 7J). Individual TRIMs such as TRIM11, TRIM19, and TRIM21 can function as molecular chaperones, impeding amorphous aggregation of model substrates and fibrillar aggregation of proteins associated with neurodegenerative diseases. This chaperone activity is independent of ATP, distinguishing TRIMs from canonical chaperones. This activity is highly effective, as TRIM11 prevents fibrillization of α-Syn monomers that are ~140-fold higher in molar concentration. The underlying mechanisms remain to be determined. TRIMs recognize misfolded, but not native, proteins (Guo et al., 2014) (Figures 4 and S4). In the case of TRIM19, this specificity is mediated by at least two substrate recognition sites, each discerning a different feature on misfolded proteins (Guo et al., 2014). We envision a scenario in which TRIMs might provide a platform that recruits an unfolded or partially folded protein, enables it to attain the native conformation, and then releases the folded protein. This mechanism would be similar to that used by a small ATP-independent chaperone, Spy, from E. coll (Stull et al., 2016), although the function of Spy in PQC appears to be limited as a molecular chaperone.
Individual TRIMs can also dissolve amorphous aggregates and amyloid fibrils without the energy derived from ATP hydrolysis. TRIM11 operates at a concentration close to their client proteins and thus may function as a “dissolvase”. This would be analogous to nuclear-import receptors (NIRs) such as karyopherin-β2 (Kapβ2), which reverse fibrillization of RNA-binding proteins (RBPs) in an ATP-independent manner (Guo et al., 2018; Hofweber et al., 2018; Yoshizawa et al., 2018). Kapβ2 stably binds to the nuclear localization signal of an RBP, which permits additional weak and dynamic interactions involving other regions of both proteins leading to the dissolution of higher-order RBP assemblies (Yoshizawa et al., 2018). The stable interaction of a TRIM with a misfolded protein might also enable similar weak and dynamic interactions for disaggregation. Nevertheless, in contrast to NIRs, TRIMs likely have a broad repertoire of client proteins. It is possible that TRIMs employ a similar mechanism to prevent and reverse protein fibrillization. In this scenario, TRIMs might selectively act on the small amounts of oligomers or amyloid fibrils that are initially generated and dissolve them, hence blocking the rate-limiting nucleation step.
TRIM11 displays a potent activity in suppressing α-Syn fibrillization in cell culture and animal models of PD. This potent activity appears to be in contrast to the Hsp70 disaggregase system consisting of members of Hsp70, Hsp40, and Hsp110 families, which can fragment large fibrils into smaller fibrils that seed further fibrillization (Tittelmeier et al., 2020). The extent to which TRIM11 chaperone or disaggregase activity contributes to the in vivo effect is unclear. Molecules within amyloid fibrils can exchange with those in solution in a biologically-relevant time scale (Carulla et al., 2005), which could blur the functional demarcation of these activities in cells. Nevertheless, given their large number, potent activities, and responsiveness to proteotoxic stresses, TRIM proteins likely constitute an important family of molecular chaperones and disaggregases that maintain metazoan proteins in their functional soluble states.
The molecular chaperone and disaggregase activities of TRIMs may aid in the recycling of aberrant proteins, which in their aggregated form cannot be effectively degraded by the proteasome, and instead may impair proteasome function (Bence et al., 2001). These activities may keep aberrant proteins soluble so that they can be SUMOylated via the ligase activity, ubiquitinated by STUbLs, and subsequently degraded in the proteasome (Figure 7J). TRIMs appear to play a broad role in maintaining protein hemostasis. For example, TRIM11 stimulates proteasome function by counteracting the proteasome-associated deubiquitinase USP14 (Chen et al., 2018), and TRIM25 maintains protein quality in the ER compartment by activating the antioxidant transcription factor Nrf2 (Liu et al., 2020). The multifunctionality of individual TRIMs could be highly advantageous. The ability to chaperone and disaggregate clients and promote their degradation provides several safeguards that may combine or synergize to buffer proteotoxicity.
The potency and multifunctionality of TRIM11 and other TRIMs likely make them valuable for treating neurogenerative diseases. While small compounds that bolster the TRIM system can be beneficial, direct expression of individual TRIMs may offer an attractive alternative. Gene transfer mediated by AAVs has become an important approach for treating CNS disorders (Deverman et al., 2018), and its potential is emphasized by recent positive outcomes in clinical trials (Mendell et al., 2017). Our study provides a proof-of-concept for using TRIMs as transgenes to treat neurodegenerative diseases. By suppressing α-Syn fibrillization, a common player in the disease pathogenesis of synucleinopathies, TRIM11 is likely suitable for treating both familial and sporadic diseases of different etiologies. A recent study revealed that single nucleotide polymorphisms (SNPs) in the TRIM11 gene are associated with progressive supranuclear palsy (PSP), the most common form of atypical parkinsonism characterized by the accumulation of fibrillary tau aggregates (Jabbari et al., 2018). This finding, along with the effects of TRIM11 on diverse substrates, indicates a potential link between TRIM11 and other diseases. Moreover, the very large number of human TRIMs likely allows for the identification of an optimal TRIM(s) for any given misfolded protein associated with neurodegenerative diseases.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiaolu Yang (xyang@pennmedicine.upenn.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
cell culture
HEK293T, HeLa cells, HCT16, A549, and SH-SY5Y cells were purchased from ATCC, QBI-293 cells were purchased from QBiogene. QBI-293 cells stably expressing wild-type human α-Syn (QBI-WT-Syn, called QBI-Syn in the current study) were previously described (Luk et al., 2009). To generate SH-SY5Y cells stably expressing human α-Syn without any tag (SH-SY5Y-Syn) and HCT116 cells stably expressing Atxn1 82Q, pTRPE-α-Syn and pTRPE-GFP-Atxn1 82Q, respectively, were transfected into HEK293T together with the packing plasmids Gag, VSVG, and REV to generate lentiviral vectors expressing α-Syn. 48 h after transfection, the vector-containing medium were harvested, centrifuged at 1,200 rpm for 5 min, filtered through 0.45 μm filters (Millipore), and concentrated by centrifugation at 10,000 rpm for 20 h at °C. SH-SY5Y and HCT116 cells were then transduced with the concentrated lentiviral vectors in the presence of 8 μg/ml polybrene (Sigma-Aldrich).
HEK293T and HeLa cells were cultured in DMEM medium, HCT116 cells in McCoy’s 5A medium, A549 cells in RPMI1640 medium, SH-SY5Y and SH-SY5Y-Syn cells in DMEM:F12 (1:1 mix) medium plus GlutaMAX, and QBI-Syn in DMEM medium supplemented with G418 (1000 g/mL; Gibco), penicillin/streptomycin, and L-glutamine. All medium contained 10% FBS. Cells were cultured at 37 °C in a humidified 5% CO2 atmosphere unless otherwise indicated.
Mouse primary cortical and hippocampal neurons were obtained from Penn Neuron Culture Service Center or isolated in our own lab and were cultured as previously described (Volpicelli-Daley et al., 2014).
Mice
Wild-type male C3H/HeJ (8-10 weeks of age) were purchased from Jackson Laboratories and housed at 22 - 24 °C on a 12 hr light/dark cycle with ad libitum access to water and a standard diet (LabDiet 5010). All procedures involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
METHOD DETAILS
Plasmids
TRIM11 mutations were: ∆R (a. a. 1-55 deleted), ∆B (a. a. 88-127 deleted), ∆CC (a. a. 128-207 deleted), N (a. a. 1-286 retained), C (a. a. 287-468 retained), M6 (six zinc-chelating residues within the RBCC region, or two in each of the RING, B1-box, and B2-box domains, were replaced: C16R, C19R, C92A, H95A, C111A, and C114A), 2EA (two conserved residues proximal to the RING domain were mutated: E12A and E13A), and LA (44 Leu residues were mutated to Ala, Figure S3C). These mutants were generated by PCR expect for LA, which was synthesized by Gene Universal Inc. (Newark, DE).
For transient expression of protein in mammalian cells, Flag-TRIM11 and Flag-TRIM11 mutants (each with the FLAG epitope at the N-terminus) were cloned into pcDNA3.1. GFP-Hsp70, GFP-TRIM11, GFP-Flag-TRIM11, and GFP-TRIM11LA were made in pEGFP-C1 (Clontech). mCherry-TRIM19 (isoform VI) was cloned in pRK5. For stable expression in mammalian cells, TRIM11, TRIM112EA, and α-Syn were cloned into lentiviral vector GFP-T2A-mCherry/pTRPE (Posey et al., 2016) (kindly provided by J. L. Riley).
For expression in bacteria, GST-TRIM11 and GST-TRIM11 mutants were constructed in pGEX-1ZT, a derivative of pGEX-1λT with additional cloning sites. To generate proteins fused with maltose-binding protein (MBP) and 6xHis, TRIM11 and its mutants were inserted into pMAL-c2X (which contains an N-terminal MBP, a gift of Dr. Paul Riggs, Addgene plasmid #75286), with the 6xHis tag introduced to C-terminus through a PCR reaction. TAT-TRIM11 was cloned into pTAT-HA, a gift from S. Dowdy (Addgene plasmid #35612), in which TRIM11 was N-terminally fused sequentially with the 6xHis tag, an HIV-TAT peptide (YGRKKRRQRRR), and the HA epitope.
For expression in Sf9 cells, TRIM11 was cloned into pFastBac vector containing an N-terminal GST tag. All constructs made for this study were confirmed by DNA sequencing.
The following plasmids (all in pRK5) were previously described: Flag-TRIM19 (isoform VI) (Chu and Yang, 2011); GFP-Atxn1 82Q, Flag-Atxn1 82Q, HA-Atxn1 82Q-Flag, Flag-Atxn1 30Q, 6xHis-SUMO2, Flag-Luciferase, and Flag-NLS-Luciferase (Guo et al., 2014); pTRPE-mCherry and pTRPE-mCherry-TRIM11 (Chen et al., 2017); and pPROEX HTb-6xHis-TEV-GFP (Jackrel and Shorter, 2014). Luciferase-6xHis (a Photinus pyralis luciferase variant) was a gift from P. Goloubinoff (Sharma et al., 2010).
DNA and siRNA transfection
DNA plasmids were transfected using Lipofectamine 2000 (Invitrogen). For transfecting TRIM11 siRNA, mouse hippocampal neuron cells were plated in 96-well plates at 15,000 cells/well with plating medium (combined Neurobasal medium containing B27, GlutaMax, penicillin/streptomycin and 10% FBS) and cultured for 18 h, and then cultured in neuronal medium (combined Neurobasal medium containing B27, GlutaMax, and penicillin/streptomycin) for an additional day (Luna et al., 2018). TRIM11 siRNA (1 pmol) was transfected into cells by Lipofectamine RNAiMAX. Mouse TRIM11 siRNA was purchased from Santa Cruz Biotech (sc-76735). Human TRIM11 siRNAs and negative control siRNA were synthesized by Integrated DNA Technologies (IDT) (Table S1).
Lentiviral and retroviral transduction
Cells stably expressing TRIM11 and α-Syn were generated by lentiviral transduction using the 3rd generation lentiviral packaging system. HEK293T cells were transfected with each lentiviral vector together with the helper plasmids Gag, Rev and VSVG. High titer viral vectors were obtained by centrifugation at 10,000 rpm for 18-20h) and used to achieve high efficiency of transduction without the need of drug selection.
Semi-quantitative PCR
Total RNA was isolated from cells by TRIzol reagent (Invitrogen) and 1-2 µg RNA of each sample was reversed to cDNA by the First-strand cDNA Synthesis System (Marligen Biosciences). The primers were synthesized by IDT (Table S1).
Immunofluorescence
Cells plated on coverslips were fixed with 4% paraformaldehyde for 10 min at room temperature and permeabilized with methanol for 10 min at −20 °C. Cells were washed with PBS and blocked in PBS with 2% bovine serum albumin (BSA) for 30 min at room temperature. Cells were incubated with the indicated primary antibodies overnight at 4 °C, and fluorescence-labeled secondary antibodies for 1-2 h at room temperature. The coverslips were mounted to glass slides in medium containing 4’,6-diamidino-2-phenylindole (DAPI). Images were acquired using a conventional or a ZEISS 710 confocal microscope. For testing the effect of TRIM19 on Atxn1 82Q in cells, GFP-Atxn1 82Q and mCherry-TRIM19 VI were co-expressed in HeLa cells. Cells were untreated or treated with cytoskeleton stripping (CT) buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2 1% Tween-20, and 0.25% sodium deoxycholate). Fixation and fluorescence analysis were performed as described above.
Cell lysate fractionation, immunoblotting, and dot blot assay
Cells were lysed with the lysis buffer [50 mM Tris, pH 8.8, 100 mM NaCl, 5 mM MgCl2, 0.5% IGEPAL CA-630 (chemically indistinguishable from Nonidet P-40), 1 mM DTT, 250 IU/ml benzonase, 1 mM PMSF, and 1x complete protease inhibitor cocktail] for 30 min on ice. Lysates were centrifuged for 15-20 min at 13,000 rpm and 4 °C. The NP-40-soluble supernatants were designated as SN fraction, and protein concentrations were measured using Bradford assay (Bio-Rad Labs). The NP-40-insoluble pellets were re-suspended in the pellet buffer (20 mM Tris, pH 8.0, 15 mM MgCl2, 1 mM DTT, 250 IU/ml benzonase, 1 mM PMSF, and 1X complete protease inhibitor cocktail) for 30 min on ice. Aggregated proteins in pellets that could be solubilized by SDS were designated as the SDS-soluble pellet fraction (PE). Both the SN and PE fractions were boiled in buffer containing 2% SDS and analyzed by SDS-PAGE and immunoblotting.
Aggregated proteins in the pellets that were SDS-resistant, called the SR fraction, were analyzed by filter retardation (dot blot) assay as previously described (Guo et al., 2014; Wanker et al., 1999). Briefly, the pellets were boiled in 2% SDS loading buffer (under reducing conditions) at 95 °C for 10 min and then applied onto 0.2 μm cellulose acetate membrane (Sterlitech) that was clamped onto a Bio-dot vacuum apparatus (Bio-Rad). The SDS-resistant aggregates on the membrane were detected with the indicated antibodies and the ECL system.
Atxn1 82Q fused to GFP, HA, and/or FLAG were detected by the antibodies corresponding to the protein or epitopes. Luciferase (untagged or 6xHis tagged), GFP (untagged), and ±-synuclein (untagged) were detected by antibodies against these proteins. The intensity of the signals in Western and dot blots was quantified by ImageJ program.
Protein purification
To purify GST, GST-TRIM11, GST-TRIM21, and GST-TRIM11 mutants from bacteria, BL21 DE3 cells containing the corresponding plasmids were grown at 37 °C to A600 nm = 0.6 - 0.8, and induced for protein expression with 0.1 mM IPTG at 30 °C for ~4 h. Cells were suspended in a buffer containing 50 mM Tris-Cl, pH 7.4, 500 mM NaCl, 200 mM KCl, 10% glycerol, complete protease inhibitor cocktail, 1 mM phenylmethylsulphonyl fluoride, 1 mM DTT, and 1 mg/ml lysozyme. To purify GST-TRIM11 from insect cells, Sf9 cells infected with baculoviruses expressing GST-TRIM11 were generated and cultured by the Molecular Screening & Protein Expression Facility at the Wistar Institute. Cells were lysed with lysis buffer (50 mM Tris-Cl, pH 7.4, 300 mM NaCl, 0.05% Triton-X100, 1 mM DTT, protease inhibitor cocktail, and benzonase nuclease) on ice for 30 min.
Bacterial and insect cell lysates were sonicated and then centrifuged at 13,000 rpm and 4 °C for 30 min. The supernatants were applied to a column (34694, Qiagen) packed with glutathione beads and incubated at 4 °C for 2 h. The column was washed extensively with the 1xPBS buffer containing 0.5 M NaCl, followed by 1xPBS buffer. The bound proteins were eluted in the elution buffer containing 20 mM glutathione and further purified on Superdex 200 Increase 10/300GL gel filtration column (GE Healthcare) driven by an NGC™ Liquid Chromatography Systems (BIORAD). The column was washed with two column volumes of gel filtration buffer (10 mM Tris-Cl pH 7.5). 500 μl samples were injected and eluted with 24 ml gel filtration buffer at 0.9 ml/min. Fractions were collected at 0.5 ml each, and fractions containing GST or GST fusions were concentrated with centrifugal filters (Millipore, cat#: UFC800308).
To purify 6xHis-TRIM11 from bacteria, MBP- and 6xHis-fused TRIM11 and TRIM11 mutants were expressed BL21 DE3 cells. Cells were grown at 37 °C to A600 nm = 0.6 - 0.8 in LB medium plus 0.2% glucose. Protein expression was induced with IPTG (0.2 mM) at 30 °C for 4 h. Bacteria were collected by centrifugation, resuspended in Column Buffer (20 mM Tris-Cl, pH 7.4, 200 mM NaCl, 1 mM EDTA, plus 1x protease inhibitor cocktail and 1 mM PMSF), and lysed by sonication. After centrifugation at 13,000 rpm and 4 °C for 30 min, the supernatants were loaded onto Amylose resin column. After extensive washing, protein-bound amylose resins were treated with TEV protease at 4 °C overnight in a buffer containing 50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 0.5 mM EDTA and 1 mM DTT, to cleave MBP tag and release 6xHis-TRIM11 proteins from the resins. Cleaved proteins were diluted with native lysis buffer (20 mM Hepes, 100 mM NaCl, pH 7.2) and incubated with Ni-NTA Agarose (Qiagen) at 4 °C for 4 h. Beads were washed extensively with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM Imidazole, pH 8.0). Fusion proteins were eluted 2x with 500 μl of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 400 mM Imidazole, pH 8.0), desalted on PD 10 columns (Genesee) and concentrated with centrifugal filters (Millipore). Proteins were aliquoted and stored at −80 °C.
To purify GFP-Flag-TRIM11, HA-Atxn1 82Q-Flag, and Flag-TRIM19 (isoform VI) from mammalian cells, HEK293T cells were transiently transfected with the corresponding plasmids, and proteins were purified using anti-FLAG mAb M2 beads as previously described (Guo et al., 2014). Briefly, cells were lysed with a lysis buffer (50 mM Tris-Cl at pH 7.4, 200 mM NaCl, 0.2% Triton, 10% glycerol, 1 mM DTT, 1 mM PMSF and complete protease inhibitor cocktail). The supernatants were incubated with anti-FLAG M2 beads at 4 °C overnight. The beads were sequentially washed once each with lysis buffer containing KCl at 0, 0.5, or 1 M, respectively, and with PBS for three times. The bound proteins were eluted with 3xFLAG peptides.
For purifying His-luciferase, BL21 DE3 cells transformed with the corresponding plasmids were induced for protein expression by 0.5 mM IPTG for 3 h at 37 °C. Proteins were purified by Ni-NTA Agaroses previously described (Sharma et al., 2010).
For purifying TAT-TRIM11, BL21 DE3 cells transformed with pTAT-HA-TRIM11 were cultured in one liter of medium containing 0.1 mM IPTG and 0.1 mM ZnCl2 (Sigma-Aldrich). Cell pellets were lysed in 10 ml of native lysis buffer (20 mM Hepes, 100 mM NaCl, pH 7.2) or denaturing lysis buffer (8 M urea, 100 mM NaCl, 20 mM Hepes, pH 8.0). Cell lysates were centrifuged, supernatant was incubated with Ni-NTA beads (Qiagen) at 4 °C for 1 h. Beads were loaded onto columns, washed twice with 2.5 ml wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM Imidazole, pH 8.0). Fusion proteins were eluted 4 times with 500 μl of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM Imidazole, pH 8.0), desalted on PD 10 columns (Sigma, cat# 17-0851) into 1xPBS, flash-frozen in 10% glycerol, and stored at −80 °C. The concentration of all purified proteins was determined by SDS-PAGE and Coomassie blue staining with BSA as the standard.
Recombinant Hsp104 and Hsp104A503S (Jackrel et al., 2014), human α-Syn and α-SynS87N (Volpicelli-Daley et al., 2014), and GFP (Jackrel and Shorter, 2014) were purified as previously described. Eluted GFP protein was further purified by gel filtration as described above.
Prevention of protein misfolding and aggregation
For prevention of luciferase misfolding, native luciferase (10 nM) was mixed with recombinant GST, GST-TRIM11, GST-TRIM112EA or Hsp70 protein at 45 °C for the indicated durations. Luciferase activity was measured with the Luciferase Assay System (Promega). Prevention of luciferase and citrate synthase aggregation was measured by light scattering (Nillegoda et al., 2015). Luciferase (200 nM) was mixed with GST or GST-TRIM11 in a HEPES buffer (50 mM HEPES-KOH, pH7.4, 50 mM KCl, 5 mM MgCl2, 2 mM DTT). Samples were heated at 42 °C, and aggregation was monitored by A 600nm on a microplate reader (Synergy HT-Gen5, BioTek). Citrate synthase (400 nM) was mixed with GST/GST-TRIM11 in Tris buffer (50 mM Tris-Cl, pH 8.0), heated at 40 °C, and monitored by A360 nm on the microplate reader.
For prevention of Atxn1 82Q aggregation, purified HA-Atxn1 82Q-Flag (50 nM) was incubated alone or in the presence of 400 nM GST or GST-TRIM11 at 37 °C for indicated durations. Atxn1 82Q fibrillization was analyzed by ThT-binding. Atxn1 82Q aggregation was also examined by sedimentation assay. After centrifugation at 13,000rpm for 20 min., the pellet fraction was analyzed by Western blot to detect SDS-soluble (PE) aggregates, and by dot blot to detect relatively large SDS-resistant (SR) aggregates.
For prevention of α-Syn fibrillation, human α-Syn monomer (70 μM) was incubated with GST, GST-TRIM11, Hsp70 (ADI-NSP-555, Enzo), Hsp40 (ADI-SPP-400, Enzo), or Hsp104 (Jackrel et al., 2014) at the indicated concentrations at 37 °C for 5 days with continuous shaking at 1,000 rpm. Formation of α-Syn fibrils was analyzed by ThT-binding and electron microscopy.
For samples containing Hsp70 or Hsp70/Hsp40, ATP (5 or 10 mM) and an ATP regeneration system (10 mM creatine phosphate and 0.5 μM creatine kinase) were also provided.
Real time quaking induced conversion (RT-QuIC) assay
TRIM11-mediated avoidance of α-Syn aggregation was analyzed with RT-QuIC assay as described previously (Harischandra et al., 2019). Briefly, α-Syn PFFs used as seeds for the RT-QuIC assay were created by incubating 1 mg/ml recombinant human α-Syn for 7 days at 37 °C with 1,000 rpm shaking. α-Syn PFFs (2 ng) were incubated with an increasing amount of recombinant TRIM11 protein (1.25, 2.5, 5, 10, and 20 μg) in a buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, and μM ThT in the presence of 20 μg soluble recombinant human α-Syn protein in Nunc™ MicroWell™ 96-well optical-bottom plates. The assay also included blank wells containing PBS only, negative control wells containing soluble α-Syn and no PFFs, and positive control wells containing α-Syn PFFs. Each sample was run in 4 replicates at the same time, and the average fluorescence intensity is shown. The results are representative of three independent experiments. The reaction was carried out using Cytation3 multimode microplate reader, incubated at 37 °C, and shaken intermittently (1-min shake-1-min rest cycle) at 807 cpm in a double orbital configuration for 95 h. ThT fluorescence was recorded every 15 min throughout the experiment.
Disaggregation of luciferase, GFP, and citrate synthase
Luciferase reactivation was described previously (Jackrel et al., 2014). Firefly luciferase (100 nM) in a luciferase refolding buffer (LRB; 25 mM HEPES-KOH [pH 7.4], 150 mM KOAc, 10 mM Mg(OAc)2, mM DTT) was heated at 45 °C for 8-10 min. Denatured luciferase (10 nM, monomer concentration) was treated with the indicated concentrations of TRIM11 or other proteins at 25 °C for 90 min. GFP disaggregation was carried out as described (DeSantis et al., 2012). GFP (4.5 μM) in buffer A [20 mM Tris-HCl, pH 7.5, 100 mM KCl, 20 mM MgCl2, 5 mM DTT, 0.1 mM EDTA, and 10% (v/v) glycerol] was incubated at 85 °C for 15 min. GFP aggregates (0.45 μM) were incubated with the indicated proteins at 25 °C for 60 min. Disaggregation of GFP aggregates was detected by measuring fluorescence at 510 nm upon excitation at 395 nm (Infinite M200 pro) and sedimentation assay. To generate citrate synthase aggregates, citrate synthase (0.15 μM) in buffer (HEPES-KOH pH 7.5, 0.5 mM DTT) was incubated at 43 °C for 45 min as described previously (Buchner et al., 1998; Haslbeck et al., 2005). For reactivation of citrate synthase aggregates, aggregates were shifted to 25 °C and the stabilizing ligand oxaloacetic acid (1 mM) was added. The Hsp70/Hsp40-Hsp104 system and TRIM11 at different concentrations were equilibrated in the buffer for 15 min on ice prior to addition to the reaction. After incubation for 60 min, citrate synthase activity was measured. For samples containing the Hsp104-Hsp70/Hsp40 bi-chaperone system, ATP (10 mM) and an ATP regeneration system (10 mM creatine phosphate and 0.5 μM creatine kinase) were provided.
Disaggregation of Atxn1 82Q aggregates and α-Syn fibrils
To facilitate the formation of Atxn1 82Q aggregates, purified HA-Atxn1 82Q-Flag (1 μM) was kept at −80 °C for 1-2 month or undergone multiple freeze-thaw cycles, followed by incubation at 37 °C for 24 h with shaking at 1,000 rpm. Atxn1 82Q aggregates (0.2 μM) were incubated without or with GST, GST-TRIM11, Hsp70/Hsp40 or Hsp104/Hsp70/Hsp40 at the indicated concentrations (with the concentration of Hsp40 being half of that of Hsp70 or Hsp104) at 30 °C for 3 h. Disaggregation was assessed by sedimentation analysis or ThT-binding assay. Disaggregation of preformed α-Syn fibrils was performed as described (Jackrel et al., 2014). α-Syn monomer (80 μM) was assembled into fibrils by incubation in buffer containing 40 mM HEPES-KOH, pH 7.4, 150 mM KCl, 20 mM MgCl2, and 1 mM DTT for 48 h at 37 °C with constant shaking at 1,200 rpm. Preformed α-Syn fibrils (1 μM) were incubated without or with GST, GST-TRIM11, or Hsp70/Hsp40-Hsp104A503S (0.2 and 1 μM) for 3 h at 30 °C. In sample containing Hsp proteins, ATP (10 mM) and an ATP regeneration system (10 mM creatine phosphate and 0.5 μM creatine kinase) were also provided. To better observe disaggregation, preformed α-Syn fibrils were sometimes kept at 4 °C for 2 days prior to the disaggregation assay. Disaggregation was assessed by ThT-binding, sedimentation, and electron microscopy assays (Shorter and Lindquist, 2004).
ThT staining
To measure amyloid fibrils formed by purified recombinant proteins, 100 μl of freshly made 20 μM ThT solution in PBS were added to reaction mixtures for 15 min prior to the measurement of fluorescence. To measure amyloid fibrils in cell lysates, cells were seeded in 6-well plates at 1x106 cells per well and treated as indicated. Cells were lysed, and the supernatant fraction was used for protein quantification. Pellet fraction that contain fibrils was re-suspended in PBS by brief sonication and added with ThT (20 μM) (Tang et al., 2015). Fluorescence was measured at Ex450nm/Em480nm by a plate reader (Infinite M200 pro). Each sample was performed in triplicate.
Negative-stain electron microscopy
α-Syn fibrils (3 μl) in PBS buffer were adsorbed on glow discharged grids coated with Formvar-Carbon membrane for 1 min and stained twice with phosphotungstic acid (PTA) for 10 seconds and 1 min, respectively. Samples were viewed using JEOL1010 transmission electron microscope operated at 80KV. Images were captured by a Hamamatsu digital camera and AMT Advantage Image Capture Software at the Electron Microscopy Resource Laboratory of University of Pennsylvania Perelman School of Medicine.
ATP-binding
Binding to ATP was assayed using an ATP Affinity Test Kit (AK-102, Jena Bioscience GmbH), which contained blank agarose beads, and agarose beads that were conjugated with ATP via the phosphate moiety (aminophenyl-ATP-agarose, or AP-ATP-agarose, C10-spacer), the ribose moiety (2′/3′-EDA-ATP-agarose, or EDA-ATP agarose), or the adenine base at different positions (8-[(6-Amino)hexyl]-amino-ATP-agarose or 8AHATP-agarose) and (N6-(6-Amino)hexyl-ATP-agarose, or 6AH-ATP agarose). Beads (50 μl slurry) were equilibrated 3x with wash buffer and then incubated with 6xHis-TRIM11 or Hsp70 (1 μg each) at 4 °C for 3 h with slight agitation. The beads were then washed 3x with wash buffer, and the bound proteins were eluted with elution buffer. Input and bound proteins were analyzed by Western blot.
Luciferase protection and reactivation in cells
For in vivo luciferase protection and reactivation, a cytoplasm-localized luciferase (pRK5-Flag-Luciferase) was transiently transfected into HCT116 cells that stably expressing mCherry or mCherry-TRIM11 (Chen et al., 2017), or into HCT116 cells pre-treated with control or TRIM11 siRNA. Also, a nucleus-localized luciferase (pRK5-Flag-NLS-Luciferase) was co-transfected with or without pRK5-Flag-TRIM19 (VI) into HCT116 cells or transfected into HCT116 cells pre-treated with control or TRIM19 siRNA. These cells were seeded in 96-well plates, and transfection efficiency was ~60-70%. 24 h after transfection, cycloheximide (20 μg/ml) was added to culture medium to inhibit new protein synthesis (Zhai et al., 2008). Cells were heated at 42 °C for 1 h or 45 °C for 30 min, and then cultured at 37 °C for another 1.5 or 3 h. Luciferase activity was measured with the Luciferase Assay System (Promega).
Transduction of TAT-TRIM11 into cells
For visualization of transduction of recombinant TAT-TRIM11, HeLa cells plated on cover slips were cultured with medium containing pTAT-TRIM11 or control eluents from untransformed or TAT-HA-transformed bacteria for various concentrations and time points. At desired time point, cells were rapidly washed three times with PBS and either immediately fixed in 4% paraformaldehyde on ice for 15 min, or trypsinized, spun down and flash frozen for SDS-PAGE analysis. Fixed cells were permeabilized with 0.2% Triton X-100 for 15 min, blocked with 1% BSA, and incubated with anti-HA primary antibody (1:500). Cells were then washed and incubated with Texas Red-conjugated anti-rabbit secondary (1:1000) (Vector Labs). Cells were mounted with medium containing DAPI (Vector Labs) and visualized using a fluorescent microscope.
For testing the effect of pTAT-TRIM11 on preformed Atxn1 82Q-GFP aggregates, HeLa cells were maintained in standard culture conditions and seeded at 40% density overnight. Atxn1 82Q-GFP was transiently transfected into cells. At 12 h post-transfection, upon visualization of noticeable aggregates, cell culture medium was removed and replaced with medium containing 50 μM native-purified pTAT-TRIM11 or control eluates from BL21 transformed with empty vector or untransformed BL21. Cells were incubated overnight in standard conditions. Cells were then were washed, fixed in 4% paraformaldehyde, and visualized using a fluorescent microscope. For quantification of Atxn1 82Q-GFP signal, cells from ten or more randomly selected fields were examined. Aggregate size parameters were set using ImageJ and the total number of aggregates was measured for Atxn1 82Q transfected cells. The statistical significance was calculated through one-way ANOVA.
Co-immunoprecipitation
HEK293 cells transfected with or without indicated plasmids were lysed in lysis buffer (50 mM Tris, pH 7.4, 200 mM NaCl, 0.2% Triton, 1 mM DTT, 1 mM PMSF, and 1x complete protease inhibitor cocktail) for 20 min on ice. Cell lysates were centrifuged, and supernatants were incubated with anti-FLAG M2 beads at 4 °C overnight. The beads were washed three times with the lysis buffer. Immunoprecipitates and whole cell lysates were subjected to SDS-PAGE and immunoblotting.
In vitro pull-down
To testing interaction of TRIM11 with Atxn1 proteins, Flag-Atxn1 82Q and Flag-Atxn1 30Q were expressed in HEK293T cells and purified using anti-FLAG M2 beads. These beads were incubated with GST or GST-TRIM11 purified from bacteria at 4 °C for 4 h or overnight. For testing the interaction of TRIM11 with native and denatured luciferase, His-Luciferase bound to Ni-NTA Agarose were treated with or without 8 M urea for 5 min. Bead were washed with binding buffer (50 mM Tris-HCl at pH 7.4, 200 mM NaCl, 0.2% Triton, 10% glycerol, 1 mM DTT) and incubated with GST, GST-TRIM11, or GST-TRIM112EA purified from bacteria at 4 °C for 4 h or overnight. Beads were washed three times with the binding buffer. The pull-down samples and input proteins were analyzed by immunoblotting and/or Coomassie blue staining.
In vivo and in vitro SUMOylation
For in vivo SUMOylation assays, cells were transfected with the indicated plasmids and treated with or without the proteasome inhibitor MG132 (10 μM) for 4-6 h. After 48 h, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Triton, 1 mM DTT, 1 mM PMSF, and 1x complete protease inhibitor cocktail) supplemented with 2% SDS and 50 mM DTT. Cell lysates were boiled at 95 °C for 10 min. One aliquot was saved for input. The rest of the cell lysates were diluted 10-fold in a lysis buffer containing no SDS and incubated with anti-HA beads, anti-Flag mAb M2 beads, or Ni-NTA beads at 4 °C overnight. Beads were washed extensively. Immunoprecipitated proteins and cell lysates were analyzed by immunoblotting with the indicated antibodies. In vitro SUMOylation assays were performed using SUMO E1, SUMO E2 (UbcH9), SUMO-2, and purified recombinant TRIM11, TRIM112EA, TRIM19, and/or Atxn1 82Q as previously described (Guo et al., 2014).
Half-life of Atxn1 82Q
HEK293T cells were transfected with Flag-Atxn1 82Q together with wild type TRIM11 or the indicated TRIM11 mutants. After 12 h, cycloheximide (CHX, 150 μg/ml) was added to cell culture medium. Cells were harvested at different times after CHX treatment and snap-frozen at −80 °C. Cells were lysed with lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.5% Trition-X100, 1 mM DTT and complete protease inhibitor cocktail) and boiled with SDS-loading buffer for Western blot analysis.
α-Synuclein pathology and cytotoxicity in cell models of PD
For testing α-Syn-seeded pathology in primary neurons, hippocampal neurons isolated from wild type mice were plated in poly (D-lysine)-coated 96-well plates. α-Syn monomers previously incubated with buffer, GST, or GST-TRIM11 were added to these neuronal cells. After two weeks, cells in 96-well plates were fixed with 4% paraformaldehyde/4% sucrose for 15 min at room temperature and then permeabilized with 0.3% Triton for 15 min. Cells were blocked in PBS with 3% BSA/3% FBS, and incubated with anti-phospho-α-Syn S129 (pSyn) (1:1000) and anti-NeuN (1:1000) at 4 °C overnight. Cells were then incubated with fluorescence-labeled secondary antibodies and counterstained with DAPI. The plates were processed using an IN Cell Analyzer 2200. Cell numbers and pathology density were calculated using In Cell Developer Toolbox 1.9.2 software as previously described (Luna et al., 2018).
For α-Syn PFFs-seeded pathology in QBI-Syn cells, cells were plated in 60-mm tissue culture plates, grown to ~80—90% confluence, and changed to serum-free media 1 h before transduction. α-Syn fibrils were diluted with PBS and sonicated in a water bath sonicator for 10 min (10s on and 10s off). For each plate, 4.5 εg of sonicated α-Syn S87N PFFs were added with cationic-liposomal transfection reagent (Lipofectamine 2000) with a protein:reagent ratio of 7.5:1. Cells were then further incubated for 4 h, washed twice with PBS and 0.5% trypsin/EDTA to remove extracellular α-Syn fibrils, and transferred to 6-well tissue culture plates or poly (D-lysine)-coated glass coverslips. To observe α-Syn PFF-induced pathology, cells were maintained for 5-7 days and stained with an antibody for phosphorylated α-Syn S129 (pSyn) as previously described (Luk et al., 2009). Fluorescence intensity was calculated using ImageJ software.
For α-Syn PFFs-induced pathology and toxicity in SH-SY5Y cells, sonicated human α-Syn fibrils (final concentration, 2 μg/ml) were transfected into cells seeded in 6-well plates by mixing with Lipofectamine 2000 (1 μg fibrils with 3 μL Lipofectamine) and incubating for 20 min. Cells were cultured for two weeks, and analyzed for protein expression by Western blot and viability by CellTiter-Glo Luminescent Cell Viability Assay.
To test the effect of TRIM11 knock down, mouse hippocampal neurons were plated in poly (D-lysine)-coated 96-well plates. Control siRNA or mouse TRIM11 siRNA was introduced into cells. At 24 h post-transfection, sonicated mouse α-Syn PFFs (final concentration, 2 μg/ml) were added into cells. After cultured for 8 days, cells were analyzed for protein expression by Western blot and viability by CellTiter-Glo Luminescent Cell Viability Assay as described previously (Volpicelli-Daley et al., 2014).
Cell viability assays
Cell viability was analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or CellTiter-Glo Luminescent Cell Viability Assay (Promega). Cells were seeded into 96-well plates at least in triplicate. CellTiter-Glo Luminescent Cell Viability Assay was performed according to manufacturer’s instruction. For MTT assay, 20 μL MTT (5 mg/ml) was added into each well. The reaction was performed at 37 °C for 4 h, and then the culture media were discarded. Dimethyl sulfoxide (150 μl) was added into the wells, and the plate was subjected to vibration for 5-10 min until purple precipitates were dissolved. The absorbance was measured at 490 nm.
rAAV9-TRIM11 and rAAV9-GFP production
To generate the plasmid pENN.AAV9.CB7.CI.hTRIM11-HA.WPRE.rBG (rAAV9-TRIM11) (in which TRIM11 expression is driven by CB7, a chicken β-actin promoter with cytomegalovirus enhancer elements), human TRIM11 tagged with a C-terminal HA was PCR amplified by using Phusion® High-Fidelity PCR Kit (NEB) and subsequently cloned into pENN.AAV9.CB7.CI.WPRE.RBG backbone at EcoRI and HindIII sites. rAAV9-GFP was constructed in the same vector. DNA plasmids were prepared by using endotoxin-free mega-prep kit (Qiagen) and characterized by structure and sequence analysis. AAV packaging was performed by the Vector Core of the University of Pennsylvania’s Gene Therapy Program as described previously (Lock et al., 2010).
Stereotaxic injection
For injections involving PFFs, full-length wild type mouse α-Syn was purified, and allowed to form fibrils at 5 mg/ml concentration as previously described (Luk et al., 2016; Luk et al., 2012). Prior to intrastriatal injection, PFFs were diluted in sterile PBS and sonicated for 10-cycles (30 second on, 30 second off; high setting) using Diagenode Bioruptor 300 (Qsonica, LLC). Mice were anaesthetized using ketamine hydrochloride (100 mg/kg, intraperitoneal [i.p.]) and xylazine (10 mg/kg, i.p.). The Angle 2 stereotaxic instrument (Leica Biosystems) was used with a 10 μl Hamilton syringe for injection. PFFs (5 μg) were injected together with rAAV9-TRIM11 or rAAV9-GFP (~9 × 1012 total viral particles each) into the striatum of one hemisphere at the following stereotaxic coordinates in relation to Bregma (mm): 0.5 AP, −1.88 ML, −3.56 DV. Animals were monitored regularly and provided supportive care first 10 days following stereotaxic injection till they fully recovered from surgery.
Mice were sacrificed on 120 or 180 days post injection (dpi) by overdose with ketamine/xylazine. For histological studies, mice were transcardially perfused with 1xPBS and 4% PFA followed by 48 h post-fixation of extracted brains in 4% PFA before being processed and embedded in paraffin. All procedures involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
Immunohistochemistry
For immunohistochemistry, slides were baked at 60 °C for 30 min followed by deparaffinization with Xylenes and rehydration through ethanol gradient. For antigen retrieval, sections were heated at 90 °C in citrate buffer (10 mM sodium citrate, pH 8.5) for 30 min and cooled down to room temperature overnight. Sections were then washed with PBS and permeabilized with blocking buffer (10% goat serum, 2% BSA, 0.1% Triton X-100 and 0.05% Tween 20 in PBS) for 1 h at room temperature. Antibodies directed against pSyn (clone 81A, 1:10,000), HA (1:500, Cell Signaling, 3724S), and GFP (1:500; Invitrogen, A11122) were incubated with the sections overnight at 4 °C and processed as described before (Panicker et al., 2015). Hoechst stain was added to the sections for 5 min at room temperature to label nuclei. Sections were then dehydrated, cleared in xylene and mounted on slides using DPX mounting medium (Electron Microscopy Science). Samples were visualized and photomicrographs of sections were captured using an inverted fluorescence microscope (Revolve, Echo Laboratories).
DAB (3,3′-diaminobenzidine) staining was performed on SN and striatal sections, as described previously (Ghosh et al., 2016; Gordon et al., 2016). Briefly, tissue sections were incubated with anti-TH (1:1,500, Millipore, 657012) or anti-pSyn antibody followed by biotinylated anti-rabbit or mouse secondary antibody. Extent of dopaminergic axonal degeneration was semi-quantified by optical density analysis (NIH ImageJ software) after transforming the color images into 8-bit greyscale images and calculated as the difference between right and left striatal/SN optical density as described previously (Ip et al., 2017; Kneynsberg et al., 2016). Based on DAB images in coronal sections of the ipsilateral side, α-Syn pathology was also spotted against the mouse brain map.
Behavioral measurements
PFFs and AAV9-GFP or AAV9-TRIM11 co-injected mice were analyzed for exploratory locomotor activity at 180 dpi, using the multiple unit open field maze (SD Instruments) consisting of four activity chambers with each chamber measuring 50 cm (length) × 50 cm (width) x 38 cm (height) made from white high density and non-porous plastic. Mice were placed in the center of the chamber at the start of the test. After a 2 min accustomization period, mice were monitored for total distance traveled, total movement time, and time spent in each of the designated quadrants of the chamber for 10 minutes using a video-camera coupled to automated tracking software (ANY-maze, SD Instruments).
Quantification and statistical analysis
A two-tailed Student’s t-test was used to evaluate the statistical significance in the mean value between two populations unless otherwise indicated. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TRIM11 | Millipore | Cat# ABC926; RRID: AB_2861378 |
| Anti-α-synuclein | Cell Signaling | Cat# 2642; RRID: AB_2192679 |
| Anti-α-synuclein phopho-Ser129 | Abcam | Cat# ab168381; RRID: AB_2728613 |
| Anti-α-synuclein phopho-Ser129 | Biolegend | Cat# 825701; RRID: AB_2564891 |
| Anti-SUMO2/3 | Abgent | Cat# AP1224a; RRID: AB_2198423 |
| Anti-GFP mouse mAb | Clontech | Cat# 632380; RRID: AB_10013427 |
| Anti-GFP rabbit pAb | Invitrogen | Cat#: A11122; RRID:AB_221569 |
| Anti-HA rabbit pAb | Santa Cruz | Cat# sc-805; RRID: AB_631618 |
| Anti-HA rabbit mAb (C29F4) | Cell Signaling | Cat# 3724; RRID: AB_1549585 |
| Anti-HA Affinity Matrix Rat mAb | Sigma-Aldrich | Cat# 11815016001; RRID: AB_390914 |
| Anti-GAPDH | Santa Cruz | Cat# sc-32233; RRID: AB_627679 |
| Anti-GST | GE Healthcare Life Sciences | Cat# 27457701; RRID: AB_771432 |
| Anti-β-actin | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Anti-α-tubulin | Sigma-Aldrich | Cat# T9026; RRID: AB_477953 |
| Anti-Flag rabbit mAb | Cell signaling | Cat# 14793; RRID: AB_2572291 |
| Anti-Flag mouse mAb (M2) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Anti-FLAG M2 agarose beads | Sigma-Aldrich | Cat# A1205, RRID: AB_2687448 |
| Anti-Hsp70 | Enzo Life Sciences | Cat# ADI-SPA-810-D; RRID: AB_2039260 |
| Anti-Hsp90 | Cell Signaling | Cat# 4874; RRID: AB_2121214 |
| Anti-NeuN | Millipore | Cat# MAB377; RRID: AB_2298772 |
| Anti-Tyrosine Hydroxylase | Millipore | Cat# 657012; RRID:AB_696697 |
| Bacterial and Virus Strains | ||
| BL21(DE3) | ThermoFisher | Cat# C600003 |
| DH5α Competent Cells | ThermoFisher | Cat# 18258012 |
| TRIM11 lentiviral vector | This paper | N/A |
| TRIM11 2EA lentiviral vector | This paper | N/A |
| mCherry-TRIM11 lentiviral vector | (Chen et al., 2017) | N/A |
| mCherry lentiviral vector | (Chen et al., 2017) | N/A |
| α-Syn Lentiviral vector | This paper | N/A |
| TRIM11 AAV9 vector | This paper | N/A |
| GFP AAV9 vector | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SUMO E1 | Boston Biochem | Cat# E-315 |
| SUMO E2 (UbcH9) | Boston Biochem | Cat# E2-465 |
| 6xHis-SUMO2 | Boston Biochem | Cat# UL-753 |
| Human α-synuclein | The Michael J. Fox Foundation | N/A |
| Human α-synuclein S87N | (Luna et al., 2018) | N/A |
| Mouse α-synuclein | (Luna et al., 2018) | N/A |
| GFP | This paper | N/A |
| GST | This paper | N/A |
| GST-TRIM11 | This paper | N/A |
| GST-TRIM11 2EA | This paper | N/A |
| 6xHis-TRIM11 | This paper | N/A |
| 6xHis-TRIM11 ∆R | This paper | N/A |
| 6xHis-TRIM11 ∆B | This paper | N/A |
| 6xHis-TRIM11 ∆CC | This paper | N/A |
| 6xHis-TRIM11 N | This paper | N/A |
| 6xHis-TRIM11 C | This paper | N/A |
| 6xHis-TRIM11 M6 | This paper | N/A |
| 6xHis-TRIM11 LA | This paper | N/A |
| Flag-Atxn1 82Q-HA | This paper | N/A |
| GST-TRIM21 | This paper | N/A |
| Flag-PML (isoform VI) | This paper | N/A |
| Hsp40 | Enzo Life Sciences | Cat# ADI-SPP-400 |
| Hsp70 | Enzo Life Sciences | Cat# ADI-NSP-555 |
| Hsp104 | (Jackrel et al., 2014) | N/A |
| Hsp104 A503S | (Jackrel et al., 2014) | N/A |
| Luciferase | Sigma-Aldrich | Cat# L9506 |
| Citrate Synthase | Sigma-Aldrich | Cat# C3260 |
| Mg2+-ATP | Sigma-Aldrich | Cat# A9187 |
| Phosphocreatine | Sigma-Aldrich | Cat# P1937 |
| Creatine kinase | Sigma-Aldrich | Cat# C3755 |
| FLAG Peptide | Sigma-Aldrich | Cat# F3290 |
| MG132 | Sigma-Aldrich | Cat# C2211 |
| Benzonase | Sigma-Aldrich | Cat# E1014 |
| TEV protease | Sigma-Aldrich | Cat# T4455 |
| Isopropyl-1-thio-D-galactopyranoside (IPTG) | Sigma-Aldrich | Cat# I6758 |
| Complete protease inhibitor Cocktail | Sigma-Aldrich | Cat# 11697498001 |
| L-Glutathione reduced | Sigma-Aldrich | Cat# G4251 |
| Ni-NTA Agarose | Qiagen | Cat# 30230 |
| Glutathione Superflow Agarose | Thermo Fisher | Cat# 25236 |
| TRIzol Reagent | Invitrogen | Cat# 15596 |
| Amylose resin | New England BioLabs | Cat# E8021 |
| 4’,6-diamidino-2-phenylindole (DAPI) | Vector Laboratories | H-1200 |
| Critical Commercial Assays | ||
| ATP Affinity Test Kit | Jena Bioscience GmbH | Cat# AK-102 |
| MTT assay | Sigma-Aldrich | Cat# M2128 |
| Phusion® High-Fidelity PCR Kit | NEB | Cat# E0553 |
| First-strand cDNA Synthesis System | Marligen Biosciences | Cat# 11801 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7570 |
| Super Sensitive™ IHC Detection System | BioGenex | Cat# QD400 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | Cat# CCL-247; RRID: CVCL_0291 |
| A549 | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| SH-SY5Y | ATCC | Cat# CRL-2266; RRID: CVCL_0019 |
| HeLa | ATCC | Cat# CCL-2; RRID: CVCL_0030 |
| HEK293T | ATCC | Cat# CRL-11268; RRID: CVCL_0063 |
| QBI-293 | QBiogene | N/A |
| Mouse primary neurons | Penn Neuron Culture Service Center and our own lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: male C3H/HeJ | Jackson Laboratories | Stock# 000659 |
| Oligonucleotides | ||
| siRNAs target mouse TRIM11 | Santa Cruz | Cat# sc-76735 |
| siRNA target human TRIM19 | Santa Cruz | Cat# sc-36284 |
| siRNAs target Human TRIM11 | IDT | Table S1 |
| Negative control siRNA | IDT | Table S1 |
| RT-PCR primers for human and/or mouse TRIM11, Hsp70, Hsp90, and GAPDH | IDT | Table S1 |
| Recombinant DNA | ||
| pcDNA3.1-Flag-TRIM11 | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 ∆R | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 ∆B | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 ∆CC | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 N | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 C | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 M6 | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 2EA | This paper | N/A |
| pcDNA3.1-Flag-TRIM11 LA | This paper | N/A |
| pEGFP-C1-TRIM11 | This paper | N/A |
| pEGFP-C1-Flag-TRIM11 | This paper | N/A |
| pEGFP-C1-TRIM11 LA | This paper | N/A |
| pEGFP-C1-Hsp70 | This paper | N/A |
| pRK5-Flag-TRIM19 (VI) | (Guo et al., 2014) | N/A |
| pRK5-mCherry-TRIM19 (VI) | This paper | N/A |
| pRK5-Flag-TRIM21 | This paper | N/A |
| pRK5-His-SUMO2 | (Guo et al., 2014) | N/A |
| pRK5-GFP-Atxn1 82Q | (Guo et al., 2014) | N/A |
| pRK5-HA-Atxn1 82Q-Flag | (Guo et al., 2014) | N/A |
| pRK5-Flag-Atxn1 82Q | (Guo et al., 2014) | N/A |
| pRK5-Flag-Atxn1 30Q | (Guo et al., 2014) | N/A |
| pRK5-Flag-Luciferase | (Guo et al., 2014) | N/A |
| pRK5-Flag-NLS-Luciferase | (Guo et al., 2014) | N/A |
| pTRPE | (Posey et al., 2016) | N/A |
| pTRPE-TRIM11 | This paper | N/A |
| pTRPE-TRIM11 2EA | This paper | N/A |
| pTRPE-α-synuclein | This paper | N/A |
| pTRPE-GFP-Atxn1 82Q | This paper | N/A |
| pTRPE-mCherry | (Chen et al., 2017) | N/A |
| pTRPE-mCherry-TRIM11 | (Chen et al., 2017) | N/A |
| AAV9-GFP | Penn Gene Therapy Vector Core | N/A |
| AAV9-TRIM11 | Penn Gene Therapy Vector Core | N/A |
| pFastBac-GST-TRIM11 | This paper | N/A |
| pGEX-1ZT-TRIM11 | This paper | N/A |
| pGEX-1ZT-TRIM11 2EA | This paper | N/A |
| pMAL-c2X-TRIM11-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 ∆R-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 ∆B-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 ∆CC-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 N-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 C-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 M6-6xHis | This paper | N/A |
| pMAL-c2X-TRIM11 LA-6xHis | This paper | N/A |
| pTAT-HA | Gift from Steven Dowdy | Addgene Plasmid Cat# 35612 |
| pTAT-HA-TRIM11 | This paper | N/A |
| Luciferase-6xHis | (Sharma et al., 2010) | N/A |
| pPROEX HTb-6xHis-TEV-GFP | (Jackrel and Shorter, 2014) | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | ImageJ.Ink | https://imagej.net/Welcome |
| In Cell Developer Toolbox 1.9.2 software | GE Healthcare | N/A |
| ZEN lite | Carl Zeiss Microscopy | N/A |
Acknowledgments
We thank P. Goloubinoff, A. Kanthasamy, J. L. Marsh, H. T. Orr, J. Riley, and N. Takahashi for plasmids and/or cell lines; the Michael J. Fox Foundation for Parkinson’s Research for recombinant α-Syn protein; D. Brady for help with protein purification; R. Wang, R. Niu, and J. Wang for technical assistance; L. Mu for graphic design; and L. Makinen for help with manuscript preparation. Supported by grants from NSF (DGE-1321851) to K.L.M.; NIH (K12GM081259 and K22NS09131401) to M.P.T.; NIH (R01NS088322) to K.C.L.; NIH (R01GM099836), MDA (MDA277268), the Life Extension Foundation, the Packard Center for ALS Research at Johns Hopkins University, and Target ALS to J.S.; NIH (R01CA182675, R01CA184867, R01CA235760, and R01CA243520) and Wealth Strategy Holding Limited to X.Y.; and the Penn Institute for Translational Medicine and Therapeutics (under NIH grant UL1TR000003) to K.C.L. and X.Y.
Footnotes
Declaration of interests
X.Y. and L.G. are inventors on a patent application owned by the University of Pennsylvania related to TRIM proteins. X.Y. is a founder of Evergreen Therapeutics LLC, which received investments from Wealth Strategy Holding Limited. J.S. is a consultant for Dewpoint Therapeutics.
REFERENCES
- Bence NF, Sampat RM, and Kopito RR (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Bernardi R, and Pandolfi PP (2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8, 1006–1016. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, and Trojanowski JQ (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SP, and Dunnett SB (2009). Tests to assess motor phenotype in mice: a user’s guide. Nat Rev Neurosci 10, 519–529. [DOI] [PubMed] [Google Scholar]
- Buchner J, Grallert H, and Jakob U (1998). Analysis of chaperone function using citrate synthase as nonnative substrate protein. Methods Enzymol 290, 323–338. [DOI] [PubMed] [Google Scholar]
- Carulla N, Caddy GL, Hall DR, Zurdo J, Gairi M, Feliz M, Giralt E, Robinson CV, and Dobson CM (2005). Molecular recycling within amyloid fibrils. Nature 436, 554–558. [DOI] [PubMed] [Google Scholar]
- Chen L, Brewer MD, Guo L, Wang R, Jiang P, and Yang X (2017). Enhanced degradation of misfolded proteins promotes tumorigenesis. Cell Rep 18, 3143–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhu G, Johns EM, and Yang X (2018). TRIM11 activates the proteasome and promotes overall protein degradation by regulating USP14. Nat Commun 9, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, and Dobson CM (2006). Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75, 333–366. [DOI] [PubMed] [Google Scholar]
- Chu Y, and Yang X (2011). SUMO E3 ligase activity of TRIM proteins. Oncogene 30, 1108–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN, and Shorter J (2012). Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151, 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, and Sah DWY (2018). Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov 17, 641–659. [DOI] [PubMed] [Google Scholar]
- Doyle SM, Genest O, and Wickner S (2013). Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol 14, 617–629. [DOI] [PubMed] [Google Scholar]
- Eisenberg D, and Jucker M (2012). The amyloid state of proteins in human diseases. Cell 148, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, and Kaspar BK (2009). Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 27, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froula JM, Castellana-Cruz M, Anabtawi NM, Camino JD, Chen SW, Thrasher DR, Freire J, Yazdi AA, Fleming S, Dobson CM, et al. (2019). Defining alpha-synuclein species responsible for Parkinson’s disease phenotypes in mice. J Biol Chem 294, 10392–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, and Iwatsubo T (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4, 160–164. [DOI] [PubMed] [Google Scholar]
- Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda NB, Szlachcic A, Guilbride DL, Saibil HR, Mayer MP, and Bukau B (2015). Human Hsp70 Disaggregase Reverses Parkinson’s-Linked alpha-Synuclein Amyloid Fibrils. Mol Cell 59, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Langley MR, Harischandra DS, Neal ML, Jin H, Anantharam V, Joseph J, Brenza T, Narasimhan B, Kanthasamy A, et al. (2016). Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson’s Disease. J Neuroimmune Pharmacol 11, 259–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, and Lindquist S (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, and Braak H (2013). 100 years of Lewy pathology. Nat Rev Neurol 9, 13–24. [DOI] [PubMed] [Google Scholar]
- Gordon R, Neal ML, Luo J, Langley MR, Harischandra DS, Panicker N, Charli A, Jin H, Anantharam V, Woodruff TM, et al. (2016). Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nature communications 7, 12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Giasson BI, Glavis-Bloom A, Brewer MD, Shorter J, Gitler AD, and Yang X (2014). A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol Cell 55, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, et al. (2018). Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 173, 677–692 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harischandra DS, Rokad D, Neal ML, Ghaisas S, Manne S, Sarkar S, Panicker N, Zenitsky G, Jin H, Lewis M, et al. (2019). Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of alpha-synuclein. Sci Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Miess A, Stromer T, Walter S, and Buchner J (2005). Disassembling protein aggregates in the yeast cytosol. The cooperation of Hsp26 with Ssa1 and Hsp104. J Biol Chem 280, 23861–23868. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S (2011). TRIM proteins and cancer. Nat Rev Cancer 11, 792–804. [DOI] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, et al. (2018). Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 173, 706–719 e713. [DOI] [PubMed] [Google Scholar]
- Ip CW, Klaus LC, Karikari AA, Visanji NP, Brotchie JM, Lang AE, Volkmann J, and Koprich JB (2017). AAV1/2-induced overexpression of A53T-alpha-synuclein in the substantia nigra results in degeneration of the nigrostriatal system with Lewy-like pathology and motor impairment: a new mouse model for Parkinson’s disease. Acta neuropathologica communications 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbari E, Woodside J, Tan MMX Shoai M, Pittman A, Ferrari R, Mok KY, Zhang D, Reynolds RH, de Silva R, et al. (2018). Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Ann Neurol 84, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, and Shorter J (2014). Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, and Shorter J (2014). Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis Model Mech 7, 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, and Walker LC (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble AH, Khan Z, Forster A, and James LC (2008). TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci U S A 105, 6045–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, and Hartl FU (2013). Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82, 323–355. [DOI] [PubMed] [Google Scholar]
- Kneynsberg A, Collier TJ, Manfredsson FP, and Kanaan NM (2016). Quantitative and semi-quantitative measurements of axonal degeneration in tissue and primary neuron cultures. J Neurosci Methods 266, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, and Dobson CM (2014). The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15, 384–396. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu H, Wu W, Zhuo W, Liu W, Zhang Y, Cheng M, Chen YG, Gao N, Yu H, et al. (2014). Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res 24, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tao S, Liao L, Li Y, Li H, Li Z, Lin L, Wan X, Yang X, and Chen L (2020). TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun 11, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, and Wilson JM (2010). Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 21, 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD, Pitkin RM, Decker SC, Trojanowski JQ, and Lee VM (2016). Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep 16, 3373–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, and Lee VM (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, and Lee VM (2009). Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106, 20051–20056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna E, Decker SC, Riddle DM, Caputo A, Zhang B, Cole T, Caswell C, Xie SX, Lee VMY, and Luk KC (2018). Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol 135, 855–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack KL, and Shorter J (2016). Engineering and Evolution of Molecular Chaperones and Protein Disaggregases with Enhanced Activity. Front Mol Biosci 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, et al. (2017). Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, and Dowdy SF (1998). Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nature medicine 4, 1449–1452. [DOI] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, et al. (2015). Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 524, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, and Zoghbi HY (2007). Trinucleotide repeat disorders. Annu Rev Neurosci 30, 575–621. [DOI] [PubMed] [Google Scholar]
- Ozato K, Shin DM, Chang TH, and Morse HC 3rd (2008). TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 8, 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker N, Saminathan H, Jin H, Neal M, Harischandra DS, Gordon R, Kanthasamy K, Lawana V, Sarkar S, Luo J, et al. (2015). Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J Neurosci 35, 10058–10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, and Lang AE (2017). Parkinson disease. Nat Rev Dis Primers 3, 17013. [DOI] [PubMed] [Google Scholar]
- Posey AD Jr., Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, et al. (2016). Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 44, 1444–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786. [DOI] [PubMed] [Google Scholar]
- Saibil H (2013). Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 14, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, De los Rios P, Christen P, Lustig A, and Goloubinoff P (2010). The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat Chem Biol 6, 914–920. [DOI] [PubMed] [Google Scholar]
- Shorter J (2011). The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 6, e26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, and Lindquist S (2004). Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304, 1793–1797. [DOI] [PubMed] [Google Scholar]
- Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, and Orr HT (1997). Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature 389, 971–974. [DOI] [PubMed] [Google Scholar]
- Stull F, Koldewey P, Humes JR, Radford SE, and Bardwell JCA (2016). Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat Struct Mol Biol 23, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, and Halliday GM (2017). Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Dai S, He Y, Doty RA, Shultz LD, Sampson SB, and Dai C (2015). MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160, 729–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittelmeier J, Sandhof CA, Ries HM, Druffel-Augustin S, Mogk A, Bukau B, and Nussbaum-Krammer C (2020). The HSP110/HSP70 disaggregation system generates spreading-competent toxic alpha-synuclein species. EMBO J 39, e103954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, and Bukau B (2010). Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 11, 777–788. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, and Lee VM (2014). Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9, 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, and Lee VM (2011). Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, and Lehrach H (1999). Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol 309, 375–386. [DOI] [PubMed] [Google Scholar]
- Wolff S, Weissman JS, and Dillin A (2014). Differential scales of protein quality control. Cell 157, 52–64. [DOI] [PubMed] [Google Scholar]
- Yoshizawa T, Ali R, Jiou J, Fung HYJ, Burke KA, Kim SJ, Lin Y, Peeples WB, Saltzberg D, Soniat M, et al. (2018). Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 173, 693–705 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, and Bellen HJ (2008). NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452, 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.