

Abstract
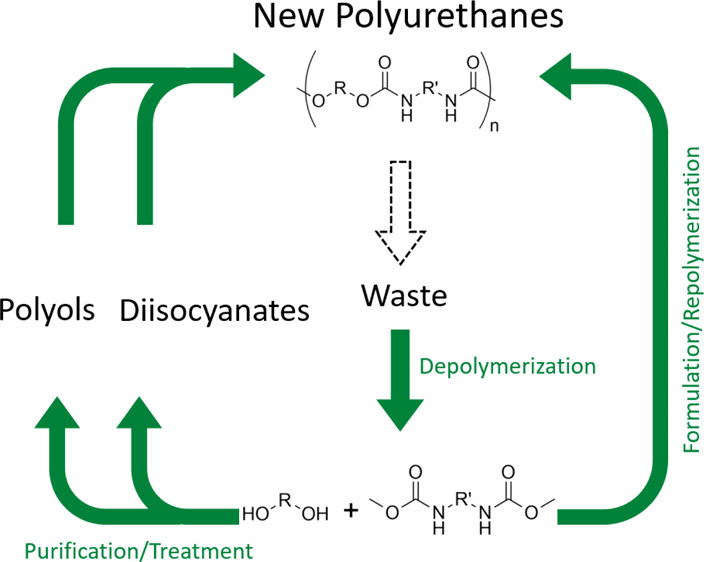
In this paper, we describe a new strategy to recycle polyurethanes (PUs) using base-catalyzed transcarbamoylation. PUs were depolymerized qualitatively in the presence of MeOH (methanol)/tetrahydrofuran as a solvent and tert-butoxide as a base catalyst. The resulting depolymerized mixture constituted by O-dimethylcarbamates and polyols can either be used as the starting material to synthesize new PUs with the transcarbamoylation approach or be purified to recover polyols and diisocyanates. The versatility and easy scaling-up of the experimental procedures and high depolymerization outcomes of the presented method make this strategy very attractive for PU recycling.
Introduction
Polyurethane (PU) is one of the most used polymers all over the world. The global production of PU was 22.9 million tons in 2017 and is increasing by 4.5% per year.1,2 As a result of the increasing quantity of PU demand, a large amount of PU waste, particularly foams, was disposed of by landfilling in the last decades. Postconsumer products as well as scraps from postproduction products reach almost 10% of the total production.3 However, increasing landfill costs, decreasing landfill space, environmental issues, and strengthening of public policies and regulations are forcing consideration of alternative options for the disposal of PU materials. PU recycling has experienced growing attention from the research and industrial world;4 recycling of any kind of plastic to convert it into valuable products is one of the main challenges of today’s society.
PU recycling is an alternative approach to landfilling. There are three kinds of strategies: (i) physical recycling (corresponding to the mechanical transformation of PU foams into flakes, granules, or powder to be used in new materials production); (ii) energy recovery (conversion of PU waste materials into useable heat, electricity, or fuel), and (iii) chemical recycling (consisting of the transformation of polymer chains into valuable chemicals).3 Several processes have been developed to chemically recycle PU foams, such as hydrolysis3,5−7 (the first process developed to recycle PU waste in a chemical way, in particular for flexible PU foams), aminolysis8−10 (the polymer chain is degraded with low molecular-weight amines), phosphorolysis11−19 (a reaction analogous to hydrolysis in which esters of phosphonic or phosphoric acids perform in a similar way to that of water with the formation of a phosphate), and glycolysis20−29 (the PU chain is fragmented by glycols producing polyols). Methanolysis of PU foams was also investigated by several groups,30,31 starting from commercially available PU foams and thermoplastic PU (TPU) elastomer. However, the depolymerization of PU operated at high temperatures (above 200 °C) with supercritical methanol limits its development.
The development of sustainable alternative and environmentally friendly routes to recyclable materials is of great interest, especially when recycling processes are able to obtain the basic valuable chemicals or building blocks which can be reused in the synthesis of chemical material or in the petrochemical industry. In this context, the transcarbamoylation reaction is an interesting reaction, allowing the conversion of a carbamate to another carbamate (Figure 1). Several metal catalysts are known to be able to activate the carbonyl group and catalyze transcarbamoylation,32,33 such as titanium(IV) isopropoxide,34 lanthanum(III) salts,35,36 dihalodistannoxanes,35 zinc acetate36 or bismuth triflate,37 inorganic or organic bases such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD),38 and even without any catalyst with sulfonyl carbamates.39
Figure 1.

Transcarbamoylation reaction of a carbamate (R, R′ = aliphatic or aromatic group and R″ = aliphatic group).
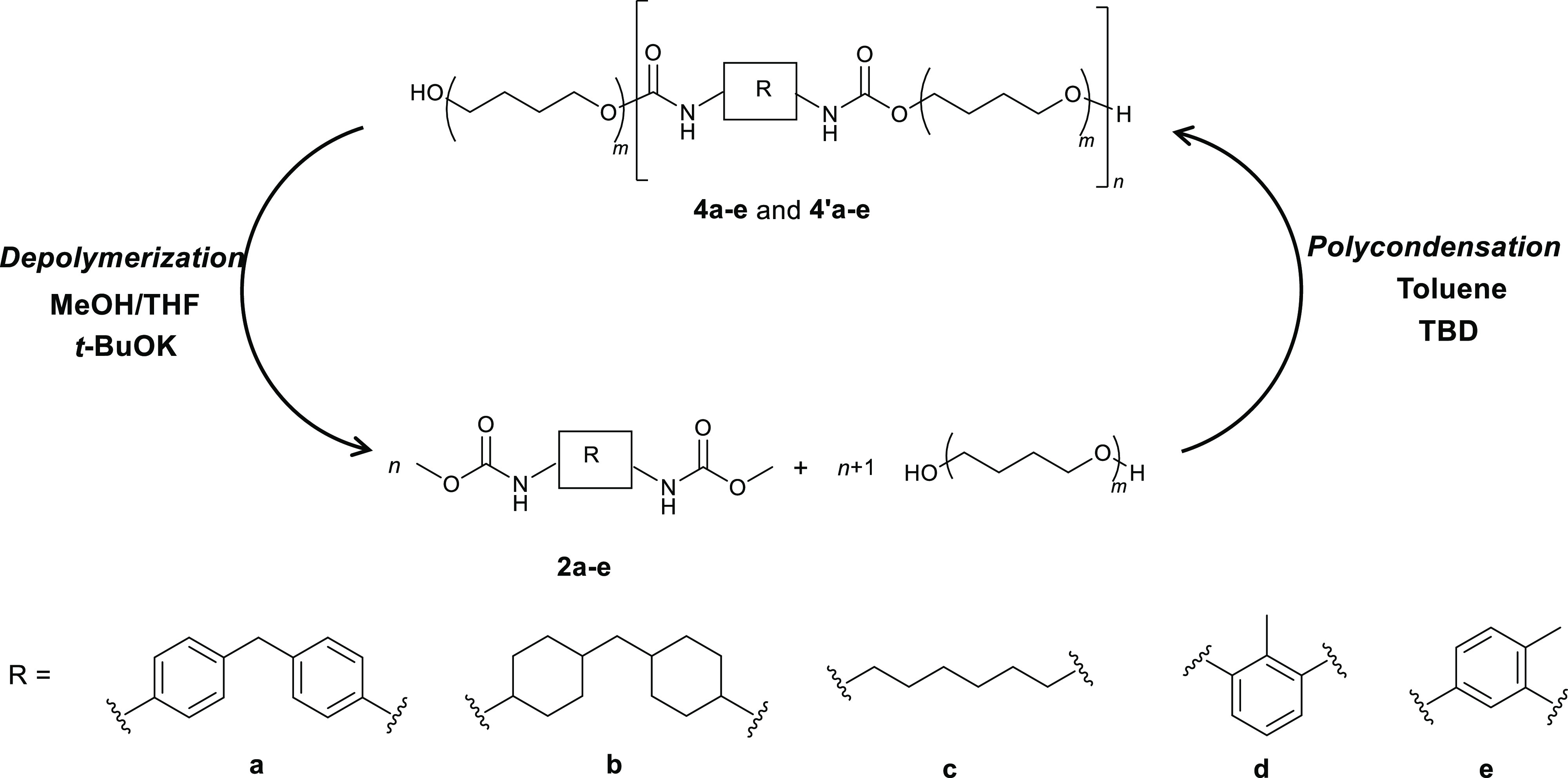
Transcarbamoylation can be used as well to synthesize PUs to avoid the use of toxic isocyanates.40−43 Recent works have shown that transcarbamoylation is an efficient reaction and can be operated in mild conditions with a soft base at low temperatures (<70 °C) offering promising perspectives in the field of PU recycling.38 Here, we investigate a recycling procedure involving the depolymerization of PU wastes producing dicarbamates and polyols coupled to the repolymerization of these intermediates to regenerate new PUs both using transcarbamoylation reaction in mild conditions (Figure 2).38 The key intermediate in this strategy is the O-dimethylcarbamate obtained from the complete depolymerization of PU through transcarbamoylation in the presence of methanol (namely methanolysis). This O-dimethylcarbamate is stable to be reformulated or isolated and reactive enough to form a new urethane bond with the elimination of MeOH allowing the regeneration of PU.
Figure 2.

Recycling of PUs through transcarbamoylation.
Materials and Methods
All solvents used were dry solvents purchased from Sigma-Aldrich. All reagents were purchased from Sigma-Aldrich and used without further purification. 1H NMR spectra were recorded with a Bruker Avance III (400 MHz). DMSO-d6 or CDCl3 was used as the solvent and tetramethylsilane as the internal standard. Chemical shifts (δ) and coupling constants (J) are given in parts per million and in hertz, respectively. High-performance liquid chromatography (HPLC) was monitored using an HP Agilent 1050 series HPLC with a diode array detector. HPLC analysis was performed on a Vydac 218TP column (C18, 5 μm, 4.6 mm i.d. × 250 mm) by using a linear gradient of A [0.1% trifluoroacetic acid (TFA) in H2O] and B (MeCN containing 0.1% TFA) at a flow rate of 1.0 mL/min with UV detection at 254 nm. All solvents were HPLC grade. The volume of the sample injected was set at 5 μL. The molecular weights (Mw) and molecular weight distributions of polymers (polydispersity index) were evaluated by size exclusion chromatography (SEC) using an Agilent 1260 Infinity Series GPC (ResiPore 3 μm, 300 × 7.5 mm), 1.0 mL/min, UV (250 nm), and refractive index (PLGPC 220) detector. All measurements were performed with tetrahydrofuran (THF) as the eluent at a flow rate of 1.0 mL/min at 35 °C. Monodisperse poly(styrene) polymers were used as calibration standards. Attenuated total reflection–IR was used to characterize the infrared absorption spectra of PU samples. The infrared spectra were recorded on a Nicolet Magna 550 spectrometer equipped with a diamond probe.
Synthesis of Model O-Dibutylcarbamates 1a and 1b
Diisocyanate [methylene diphenyl 4,4′-diisocyanate (MDI) or 1,6-diisocyanatohexane (HMDI)] (16 mmol) was added to 40 mL of dry 1-butanol at ambient temperature slowly. Dibutyltin dilaurate (10 μL, 0.016 mmol) was added, and the reaction mixture was stirred at 50 °C for 3 h. The 1-butanol was evaporated under reduced pressure to give a white solid. The residue was purified by silica column chromatography eluting with hexane/ethyl acetate EtOAc (75/25). The fractions were concentrated under reduced pressure to give the product 1 (Figure S1). O-Dibutylcarbamate 1a: white solid (5.8 g, 91% yield). 1H NMR (400 MHz, chloroform-d): δ 7.31–7.26 (m, 4H), 7.12–7.07 (m, 4H), 6.59–6.51 (m, 2H), 4.15 (t, J = 6.7 Hz, 4H), 3.88 (s, 2H), 1.69–1.62 (m, 4H), 1.46–1.35 (m, 4H), and 0.95 (t, J = 7.4 Hz, 6H). O-Dibutylcarbamate 1b: white solid (6.2 g, 95% yield). 1H NMR (400 MHz, DMSO-d6): δ 6.98–6.86 (m, 2H), 3.97–3.83 (m, 4H), 3.50–3.41 (m, 2H), 1.69 (dd, J = 39.9, 12.7 Hz, 4H), 1.56–1.19 (m, 20H), 1.19–1.03 (m, 4H), and 0.89–0.84 (m, 6H).
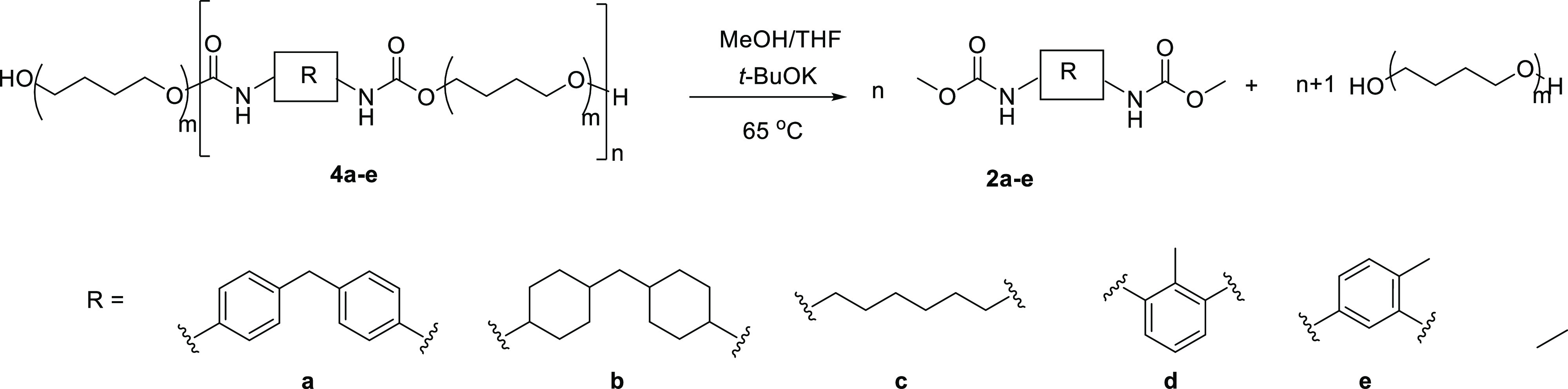
Synthesis of Model O-Dimethylcarbamates 2a–e
Diisocyanate [MDI, hexamethylene diisocyanate (HDI), HMDI, 2,6-tolylene diisocyanate (2,6-TDI), or 2,4-tolylene diisocyanate (2,4-TDI)] (5 mmol) was added to 10 mL of dry methanol at ambient temperature slowly. Dibutyltin dilaurate (5 μL, 0.008 mmol) was added, and the reaction mixture was stirred at 50 °C for 3 h. Methanol was evaporated under reduced pressure to give a white solid. The residue was purified by silica column chromatography eluting with hexane/EtOAc (75/25). The fractions were concentrated under reduced pressure to give the corresponding desired product 2a–e (Figure S2). O-Dimethylcarbamate 2a: white solid (1.52 g, 97% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.52 (s, 2H), 7.33 (d, J = 8.2 Hz, 4H), 7.12–7.04 (m, 4H), 3.77 (s, 2H), and 3.62 (s, 6H). O-Dimethylcarbamate 2b: white solid (1.45 g, 89% yield). 1H NMR (400 MHz, DMSO-d6): δ 7.05–6.96 (m, 2H), 3.05–3.45 (m, 8H), 1.71 (dd, J = 40.5, 12.8 Hz, 4H), 1.54–1.38 (m, 8H), and 1.30–1.05 (m, 8H). O-Dimethylcarbamate 2c: white solid (1.04 g, 90% yield). 1H NMR (400 MHz, DMSO-d6): δ 7.09–7.01 (m, 2H), 3.49 (s, 6H), 2.95–2.90 (m, 4H), 1.38–1.31 (m, 4H), and 1.23–1.19 (m, 4H). O-Dimethylcarbamate 2d: white solid (1.14 g, 96% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.91 (s, 2H), 7.13 (d, J = 1.6 Hz, 3H), 3.64 (s, 6H), and 2.04 (s, 3H). O-Dimethylcarbamate 2e white solid (1.15 g, 97% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.53 (s, 1H), 8.79 (s, 1H), 7.48 (d, J = 2.2 Hz, 1H), 7.18–7.13 (m, 1H), 7.07–7.03 (m, 1H), 3.63 (d, J = 1.8 Hz, 6H), and 2.10 (s, 3H).
Methanolysis of O-Dibutyldicarbamate 1
The urethane 1 (0.224 mmol) was dissolved in methanol (2 mL). A base [NaOH, KOH, LiOH, NaH, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), TBD, tert-butoxide (t-BuOK), 1.0–2.0 equiv per urethane group] was added, and the reaction mixture was heated at 55 or 65 °C with continuous agitation for 20 h. The solvent was evaporated, and water (15 mL) was added and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried on magnesium sulfate, and the solvent was evaporated. The residue was purified by silica column chromatography eluting with hexane/EtOAc (75/25). The fractions were concentrated under reduced pressure to give the product 2.
General Procedure for the Depolymerization of PU 4a–e
PU 4a–e (300 mg) was suspended in THF (2 mL) and methanol (2 mL), 2.0–2.7 equiv per carbamate group of t-BuOK was added to the solution, and the solution was kept at continuous agitation under 65 °C for 20 h. The solvent was then evaporated, and water (20 mL) was added to the reaction mixture and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried on magnesium sulfate, and the solvent was evaporated. The residue was purified by silica column chromatography with hexane/EtOAc (75/25). The fractions were concentrated under reduced pressure to give the product 2a–e.
Optimization of Transcarbamoylation-Based Polycondensation 2a–e with PTMO
O-Dimethylcarbamate 2 (0.235 mmol), PTMO 2000 (450 mg, 0.224 mmol), and TBD (10 mg, 0.07 mmol) were added to 10 mL of dry toluene. The temperature was progressively increased to 130 °C under nitrogen flow (to remove MeOH) and stirring. Then, the polymerization reaction was conducted over 16 h. Finally, after it was cooled to room temperature, the obtained PU 4a–e was solubilized in THF and precipitated in cooled methanol.
General Procedure to Recycle TPU (Pellethane 2363-80AE)
Pellethane 2363-80AE (300 mg) was suspended in THF (3 mL) and methanol (3 mL), t-BuOK (60 mg, 0.54 mmol) was added to the solution, with continuous agitation under 65 °C for 20 h; the solvent was evaporated, and water (20 mL) was added to the reaction mixture and extracted with EtOAc (3×). The combined organic layers were washed with brine and dried on magnesium sulfate, and the solvent was evaporated. The residue was dissolved in dry toluene (20 mL), with a nitrogen inlet, monomer 2 (9 mg, 0.03 mmol), and TBD (10 mg, 0.07 mmol) added to the solution; the temperature was progressively increased to 130 °C under nitrogen flow (to remove MeOH) and stirring. Then, the polymerization reaction was conducted over 16 h. Finally, after it was cooled to room temperature, the obtained PU was solubilized in THF and precipitated in methanol to get regenerated PU (220 mg, yield: 74%).
Result and Discussion
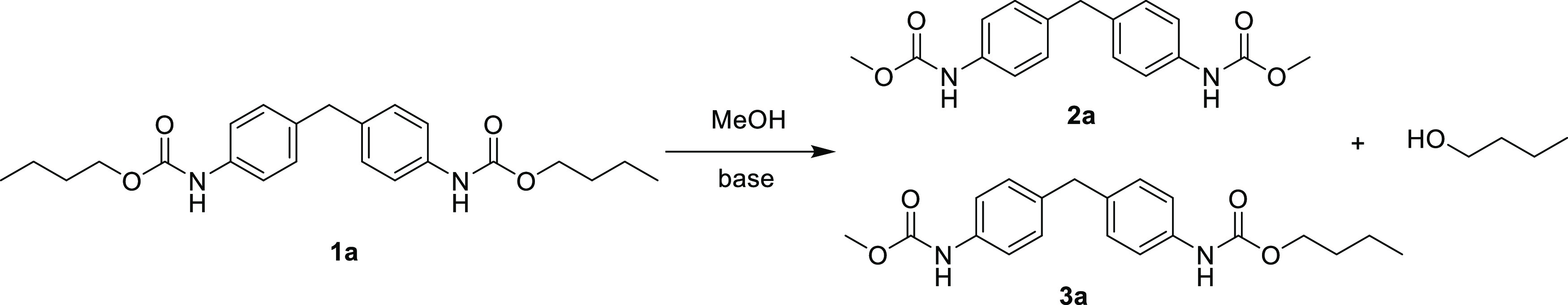
We first investigated the reactivity of transcarbamoylation. Dibutyl(methylenebis(4,1-phenylene))dicarbamate 1a was chosen as the model substrate for aromatic carbamates to optimize reaction conditions including bases, temperatures, and reaction times. The results are summarized in Table 1. According to the previous work carried out by our group,38 we first used 2 equiv per urethane group of potassium hydroxide (KOH) as the base at 65 °C in the presence of methanol (Table 1, entry 1). The reaction afforded a mixture of compounds 1a, 2a, and 3a (detected by HPLC) conducting a yield of 45% in disubstituted dicarbamate 2a. In order to increase the yield of the compound 2a, other inorganic bases such as sodium hydroxide (NaOH), lithium hydroxide (LiOH), and sodium hydride (NaH) as well as organic bases such as potassium t-BuOK; TBD, triethylamine (Et3N), and DBU (Table 1, entries 2–8) were screened. Most of the bases evaluated (NaOH, KOH, LiOH, NaH, DBU, TBD, and t-BuOK) afforded dimethylcarbamates with good conversions. It was shown that the reaction is base-dependent, t-BuOK and TBD providing the highest yield (80 and 79% respectively). The impact of the reaction time (Table 1, entries 5, 9, and 10), equivalent of base (Table 1, entry 5, and entries 11–12), and reaction temperature (Table 1, entry 5, and entry 13) was studied. The yield of 2a decreased with the reduction of reaction time or the amount of base. The best condition is 2 equiv of potassium t-BuOK as the base at 65 °C reacted for 20 h (Table 1, entry 5) and was kept for the rest of the study. Furthermore, the reactivity of aliphatic diurethane with the use of the dicarbamate 1b as model substrate of aliphatic PU was also investigated resulting in the same observations (see the Supporting Information, Table S1 for details). It is noteworthy that in our trials no byproducts such as aromatic amines were detected by liquid chromatography/mass spectrometry (LC/MS) due to the mild reaction conditions used. The excellent reactivity of aromatic and aliphatic dicarbamates subjected to transcarbamoylation reaction with methanol is consequently particularly interesting to develop depolymerization strategies of PU in mild conditions.
Table 1. Optimization of the Transcarbamoylation Reaction Conditionsa.
| entry | base | equivb | temp. (°C) | conversionc | 2a/3ac | isolated yieldd (%) |
|---|---|---|---|---|---|---|
| 1 | KOH | 2.0 | 65 | 66% (20 h) | 76/24 | 45 |
| 2 | NaOH | 2.0 | 65 | 78% (20 h) | 90/10 | 73 |
| 3 | LiOH | 2.0 | 65 | 68% (20 h) | 80/20 | 57 |
| 4 | NaH | 2.0 | 65 | 79% (20 h) | 90/10 | 70 |
| 5 | t-BuOK | 2.0 | 65 | 85% (20 h) | 97/3 | 80 |
| 6 | TBD | 2.0 | 65 | 86% (20 h) | 97/3 | 79 |
| 7 | Et3N | 2.0 | 65 | 1% (20 h) | ND | trace |
| 8 | DBU | 2.0 | 65 | 65% (20 h) | 85/15 | 60 |
| 9 | t-BuOK | 2.0 | 65 | 79% (14 h) | 88/12 | 75 |
| 10 | t-BuOK | 2.0 | 65 | 61% (8 h) | 71/29 | ND |
| 11 | t-BuOK | 1.5 | 65 | 76% (20 h) | 86/24 | 75 |
| 12 | t-BuOK | 1.0 | 65 | 48% (20 h) | 60/40 | ND |
| 13 | t-BuOK | 2.0 | 55 | 38% (20 h) | 50/50 | ND |
Reactions were run with 0.224 mmol of 1 in 2 mL of MeOH with 0.448, 0.672, or 0.896 mmol of base for 8–20 h at 55 or 65 °C.
Equivalent per urethane group.
Conversion of compound 2a group determined by LC/MS analysis of the crude products.
Isolated yield (by column chromatography) of compound 2a. ND: not determined.
After the optimization of the transcarbamoylation reaction with the model dicarbamates, several PUs were synthesized to perform transcarbamoylation-based depolymerizations. PUs obtained from different diisocyanates (see the Supporting Information) were reacted with t-BuOK using methanol (MeOH) and THF as solvents at 65 °C to produce the corresponding O-dimethylcarbamate and polytetrahydrofuran (PTMO). The results are shown in Table 2. Two equivalents of a base were used as the standard, and 5 different PUs (4a–4e) which are widely used in PU industry were chosen (Table 2, entries 1–5) to investigate the depolymerization by the described process. For all those PUs, depolymerization was observed affording the corresponding O-dimethylcarbamates (2a–2e) regardless of the chemical structure of the PUs (Table 2, entry 1–5) and their molecular weights (conversion ranging from 77–81%; Table 2, entries 1 and 6–8). It is worth noting that with the increase of the amount of the base, the conversion rate and yield were improved, leading to the obtention of monomer 2a with high yields. We first optimized the base catalysis conditions (Table 2, entries 7 and 9–11). The best amount of t-BuOK was found to be 2.3 equiv (Table 2, entry 9). A remarkable fact is that THF can be substituted by MeOH by just increasing reaction times (THF being a good swelling solvent): for example, PU 4a (Mw = 6500 g/mol, 300 mg) reacted in 4 mL of MeOH at 65 °C for 20 h leads to 2a with a 73% yield.
Table 2. Depolymerization of PUsa.
| entry | 4 | n | m | Mw | t-BuOK (equiv)b | conversionc (%) | isolated yieldd (%) |
|---|---|---|---|---|---|---|---|
| 1 | 4a | 5 | 14 | 6500 | 2.0 | 79 | 75 |
| 2 | 4b | 5 | 14 | 5250 | 2.0 | 65 | 53 |
| 3 | 4c | 6 | 14 | 8200 | 2.0 | 71 | 63 |
| 4 | 4d | 3 | 14 | 4700 | 2.0 | 78 | 74 |
| 5 | 4e | 4 | 14 | 5900 | 2.0 | 78 | 65 |
| 6 | 4a | 8 | 27 | 20,000 | 2.0 | 77 | 73 |
| 7 | 4a | 11 | 27 | 28,000 | 2.0 | 81 | 78 |
| 8 | 4a | 10 | 40 | 33,600 | 2.0 | 78 | 70 |
| 9 | 4a | 11 | 27 | 28,000 | 2.3 | 90 | 85 |
| 10 | 4a | 11 | 27 | 28,000 | 2.5 | 83 | 83 |
| 11 | 4a | 11 | 27 | 28,000 | 2.7 | 85 | 81 |
300 mg of PU was suspended in 4 mL of solvent (MeOH/THF, 1/1), stirred at 65 °C, and t-BuOK was added then reacted for 20 h.
Equivalent per urethane group.
Determined by 1H NMR analysis of the crude products.
Isolated yield (by column chromatography) of dicarbamate 2.
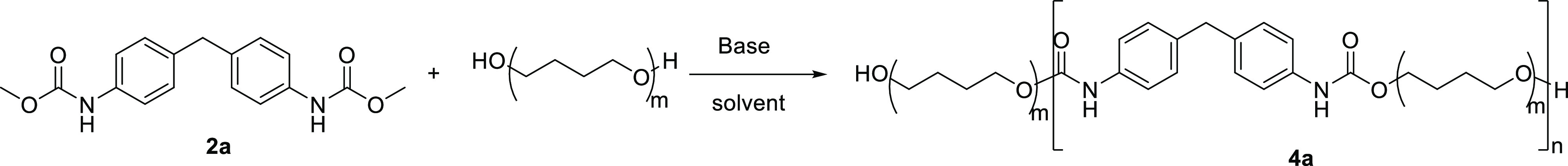
One important advantage of methanolysis is the production of O-methylcarbamate, a useful reactive intermediate that can be used to regenerate PUs through transcarbamoylation associated to the release of MeOH which can be easily eliminated by evaporation during the reaction.40−42 We tried to achieve repolymerization to prepare PU by using the dicarbamate 2a and PTMO (2000); they were subjected to transcarbamoylation, with an organic base TBD to catalyze the reaction.43 The results are shown in Table 3. We first tried a solvent-free condition; however, only a trace amount of PU was observed (Table 3, entry 1) possibly due to its poor solubility in liquid PTMO (2000). A solvent is required to observe the formation of PU, and toluene was therefore chosen as the solvent for the rest of the study (Table 3, entries 2–10). The effect of the quantity of base (TBD) to catalyze the reaction was then studied. When 0.15 and 0.25 equiv per urethane group of base were used (Table 3, entries 3 and 4), the molecular weights and yields increased. As expected, the increase of the reaction time improved the polymerization yield (from 33 to 54%) (Table 3, entries 3 and 5), while there was no noticeable improvement under a higher reaction temperature (Table 3, entry 6). The use of an excess of dicarbamate improves polycondensation: 1.1 and 1.2 equiv have been tested, leading fortunately to PU with molecular weight of 15,800 g/mol (Table 3, entries 7 and 8). Attempts to use longer reaction time or some other base (t-BuOK) or solvent (THF) failed to improve the reactivity of transcarbamoylation (Table 3, entries 9–11).
Table 3. Optimization of Transcarbamoylation-Based Polycondensationa.
| entry | 2a (equiv) | base (equiv) | temp. (°C) | reaction times (h) | solvent | Mw (g/mol)b | Mw/Mnb | yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.0 | TBD (0.05) | 130 | 8 | 6600 | 1.11 | trace | |
| 2 | 1.0 | TBD (0.05) | 130 | 8 | toluene | 8900 | 1.21 | 35 |
| 3 | 1.0 | TBD (0.15) | 130 | 8 | toluene | 11,200 | 1.31 | 33 |
| 4 | 1.0 | TBD (0.25) | 130 | 8 | toluene | 11,200 | 1.51 | 34 |
| 5 | 1.0 | TBD (0.15) | 130 | 16 | toluene | 11,200 | 1.38 | 54 |
| 6 | 1.0 | TBD (0.15) | 150 | 16 | toluene | 11,200 | 1.55 | 56 |
| 7 | 1.1 | TBD (0.15) | 130 | 16 | toluene | 15,800 | 1.35 | 55 |
| 8 | 1.2 | TBD (0.15) | 130 | 16 | toluene | 15,800 | 1.41 | 51 |
| 9 | 1.1 | TBD (0.15) | 130 | 20 | toluene | 13,500 | 1.53 | 57 |
| 10 | 1.1 | t-BuOK (0.15) | 130 | 16 | toluene | 8900 | 1.45 | 52 |
| 11 | 1.1 | TBD (0.05) | reflux | 16 | THF | 4300 | 1.08 | trace |
Reactions were performed under nitrogen flow with 0.224, 0.235, or 0.269 mmol diurethane 2a, 0.05–0.25 equiv per urethane group of TBD, and 0.224 mmol PTMO in 10 mL of anhydrous toluene or THF with corresponding base at appropriate reaction time and temperature.
Determined by SEC.
Based on these optimal reaction conditions, we turned our attention to validate the cycle depolymerization/repolymerization on different PU structures (Table 4, entries 1–5). Five PUs 4a–4e (see the Supporting Information) were synthesized as starting material to screen the applicability of our chemical recycling processes. Depolymerization was performed according to the procedure described in Table 2, and after extraction, the products of depolymerization were used in the repolymerization reaction. Polycondensation was conducted at 130 °C under catalysis of TBD (0.15 equiv per urethane group) in toluene, with 1.1 equiv of monomer 2. Five different structures of PUs (scaffold corresponding to MDI, 2,6-TDI, 2,4-TDI, HMDI, and HDI) have been evaluated. Because of the high reactivity of the structure 2a (Table 4, entry 1), the Mw of the repolymerized PU (4′a) provided the best results in our system (Mw = 18,100 g/mol); all structures were able to achieve the original PU structure, although other compounds 4′b–4′e were not as remarkable as 4′a (the Mw are 8900–13,000 g/mol).
Table 4. Recycling PUs through Depolymerization and Polycondensationa.
| depolymerization
of PU into monomers |
repolymerized
PU |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | 4 | R | Mw/Mnb,f | Mwb | yield (%)c | 4′ | Mwd,f | Mw/Mnd,f | yield (%)e |
| 1 | 4a | 2a | 1.21 | 28,000 | 85 | 4′a | 18,100 | 1.34 | 51 |
| 2 | 4b | 2b | 1.24 | 28,000 | 73 | 4′b | 10,800 | 1.30 | 31 |
| 3 | 4c | 2c | 1.24 | 22,000 | 89 | 4′c | 13,000 | 1.35 | 65 |
| 4 | 4d | 2d | 1.38 | 14,000 | 91 | 4′d | 8900 | 1.42 | 56 |
| 5 | 4e | 2e | 1.33 | 20,000 | 81 | 4′e | 10,000 | 1.33 | 41 |
Depolymerization conditions as described in Table 2. Polymerization: the same process as described in Table 3 (with extra 0.1 equiv of monomer 2).
PU synthesized by diisocyanate approach.
Isolated yield (monomer 2, by column chromatography) of depolymerization, calculated for the urethane group.
PU synthesized by transcarbamoylation.
Yield of polymerization.
Determined by SEC.
Encouraged by these results, we have extended our investigation to recycle commercial TPU (Pellethane 2363-80AE, Mw = 110,331 g/mol, Mw/Mn = 2.10). Pellethane was first depolymerized to afford the O-dimethylcarbamate 2a and PTMO (Figure 3A), which can be identified clearly by NMR in the spectrum of the crude of the depolymerization reaction [Figure 3B, 1H NMR spectra of raw Pellethane (black trace), depolymerized Pellethane (green trace), O-dimethylcarbamate 2a (blue trace)]. This mixture was then incubated with TBD to regenerate PU successfully [Figure 3B, crude of the regenerated PU (red trace)] which was isolated by precipitation (Mw = 19,500 g/mol, Mw/Mn = 1.40). Alternatively to this route, the O-dimethylcarbamate 2a can be isolated due to its stability and possibly converted subsequently into MDI44,45 offering new perspectives to regenerate diisocyanates to feed back into the classical PU industry.
Figure 3.

(A) Depolymerization of commercial TPU (Pellethane 2363-80AE) and regeneration of PU. (B) 1H NMR spectra of raw Pellethane 2363-80AE (black), depolymerized Pellethane (green), the isolated O-dimethylcarbamate 2a (blue), and the regenerated PU (red).
With the aim to recycle PU scraps, we then turned our attention to the PU foams coming from waste electronic and electrical equipment (WEEE; foams from fridge/freezer) as well as construction and demolition waste (foams of insulating panels). Following this strategy, foams were first depolymerized using transcarbamoylation (see the Supporting Information). We can clearly observe in the NMR spectra of the crudes of depolymerization, shown in Figure 4A, the characteristic signals of O-methylurethane at 3.62 ppm as well as aromatic signals at 7.1 and 7.3 ppm corresponding to mainly O-dimethylcarbamate 2a originating from 4,4′-MDI-based PU (a major constituent of rigid foam) as well as other aromatic signals associated probably to different possible MDI derivatives (isomers, polymeric forms) or aromatic polyols in the case of scrap from insulating panels. Unfortunately, because of the complex composition of the waste foams (e.g., mixtures of polyols, undefined additives: brominated flame retardant, etc.), carbamate 2a is the only constituent identified by 1H NMR.
Figure 4.

(A) Methanolysis of waste PU. 1H NMR analysis of depolymerized foam (insulating panel, black; fridge/freezer, blue; monomer 2a, red); (B) IR of foam from fridge/freezer (blue) and corresponding regenerated PU (red).
The mixture resulting from depolymerization containing carbamates and polyols can then be used to regenerate new PUs. Reconstituted PU was prepared by polycondensation using TBD, starting from depolymerized foams from WEEE and removing unpolymerized materials and unincorporated substances after precipitation, PU was obtained, with characteristic signals in IR spectrum (Figure 4B).
Alternatively in the case of a more complicated mixture of depolymerization (foams of insulating panel), because of too much uncertain compositions in some rigid foams46−48 (e.g., polymeric MDI, undefined PUs; mixture of polyols, brominated flame retardant; catalysts; salts...), purification is preferred by the isolation of the carbamate 2a with a yield of 16% (wt %) that could be further reformulated or further transformed into diisocyanates, associated to the classical recovery of polyols done in glycolysis for example.
Conclusions
In this report, a new chemical recycling process of PU (TPU and PU foams) is presented based on the transcarbamoylation reaction. PUs are depolymerized under mild and efficient conditions using a base and THF/MeOH as the solvent at 65 °C to provide quantitatively polyols and O-dimethylcarbamates making this strategy simple and easy to scale up. It is also noteworthy that THF can be substituted by MeOH offering great prospects. The obtention of O-dimethylcarbamates makes this strategy also very attractive since this intermediate is able to react with polyols to regenerate PUs using transcarbamoylation releasing MeOH. The reactivity of this intermediate also offers huge versatility in the outputs of this strategy since the mixture obtained from depolymerization can be used directly to regenerate new PUs through polycondensation or by modifying the formulation (e.g., by adding polyols) to rectify or confer new properties or performances to the regenerated PU.49 Alternatively, each constituent resulting from depolymerization can be isolated, particularly the O-dimethylcarbamates which due to their stability can be further reacted to synthesize new PUs (nonisocyanate routes)49 or converted to diisocyanates to be fed back into the classical PU industry to generate virgin PUs. The last route is of interest in the case of the composition of PUs that is not suitable for the market or does not satisfy new regulations due to the presence of toxic substances (e.g., brominated flame retardant).
Acknowledgments
L.Z. thanks the China Scholarship Council for the financial support. The authors are grateful to the Institut de Recherche de Chimie Paris (IRCP), the Centre National de la Recherche Scientifique (CNRS), and Chimie ParisTech for the facilities and the administrative support. Xavier Lantoinette and Marianne Fleury from ecosystem (French take-back scheme) are acknowledged for providing PU foam wastes from appliances as well as helpful discussion. The authors also thank Chaire Mines Urbaines and Fondation ParisTech for their support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04855.
Experimental procedures and characterization data, 1H NMR spectra of compounds, and gel permeation chromatography (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cornille A.; Auvergne R.; Figovsky O.; Boutevin B.; Caillol S. A perspective approach to sustainable routes for non-isocyanate polyurethanes. Eur. Polym. J. 2017, 87, 535–552. 10.1016/j.eurpolymj.2016.11.027. [DOI] [Google Scholar]
- Zia K. M.; Bhatti H. N.; Ahmad Bhatti I. Methods for polyurethane and polyurethane composites, recycling and recovery: A review. React. Funct. Polym. 2007, 67, 675–692. 10.1016/j.reactfunctpolym.2007.05.004. [DOI] [Google Scholar]
- Behrendt G.; Naber B. The chemical recycling of polyurethane. J. Univ. Chem. Technol. Metall. 2009, 44, 3–23. [Google Scholar]
- DeGaspari J. From trash to cash. Mech. Eng. 1999, 121, 48–51. 10.1115/1.1999-jun-1. [DOI] [Google Scholar]
- Campbell G. A.; Meluch W. C. Polyurethane foam recycling. Superheated steam hydrolysis. Environ. Sci. Technol. 1976, 10, 182–185. 10.1021/es60113a008. [DOI] [Google Scholar]
- Dai Z.; Hatano B.; Kadokawa J.-I.; Tagaya H. Effect of diaminotoluene on the decomposition of polyurethane foam waste in superheated water. Polym. Degrad. Stab. 2002, 76, 179–184. 10.1016/s0141-3910(02)00010-1. [DOI] [Google Scholar]
- Nikje M. M. A.; Nikrah M.; Mohammadi F. H. A. Microwave-assisted polyurethane bond cleavage via hydroglycolysis process at atmospheric pressure. J. Cell. Plast. 2008, 44, 367–380. 10.1177/0021955x08090279. [DOI] [Google Scholar]
- Xue S.; Omoto M.; Hidai T.; Imai Y. Preparation of epoxy hardeners from waste rigid polyurethane foam and their application. J. Appl. Polym. Sci. 1995, 56, 127–134. 10.1002/app.1995.070560202. [DOI] [Google Scholar]
- Van Der Wal H. R. New chemical recycling process for polyurethanes. J. Reinf. Plast. Compos. 1994, 13, 87–96. 10.1177/073168449401300106. [DOI] [Google Scholar]
- Chuayjuljit S.; Norakankorn C.; Pimpan V. Chemical recycling of rigid polyurethane foam scrap via base catalyzed aminolysis. J. Met., Mater. Miner. 2002, 12, 19–22. [Google Scholar]
- Datta J.; Włoch M.. Recycling of polyurethanes. In Polyurethane Polymers: Blends and Interpenetrating Polymer Networks; Thomas S., Datta J., Reghunathan A., Haponiuk J., Eds.; Elsevier: Amsterdam, 2017; pp 323–358. [Google Scholar]
- Mitova V.; Grancharov G.; Molero C.; Borreguero A. M.; Troev K.; Rodriguez J. F. Chemical degradation of polymers (polyurethanes, polycarbonate and polyamide) by esters of H-phosphonic and phosphoric acids. J. Macromol. Sci., Part A: Pure Appl. Chem. 2013, 50, 774–795. 10.1080/10601325.2013.792667. [DOI] [Google Scholar]
- Troev K.; Tsekova A.; Tsevi R. Chemical degradation of polyurethanes 2. Degradation of flexible polyether foam by dimethyl phosphonate. Polym. Degrad. Stab. 2000, 67, 397–405. 10.1016/s0141-3910(99)00106-8. [DOI] [Google Scholar]
- Troev K.; Grancharov G.; Tsevi R.; Tsekova A. A novel approach to recycling of polyurethanes: chemical degradation of flexible polyurethane foams by triethyl phosphate. Polymer 2000, 41, 7017–7022. 10.1016/s0032-3861(00)00054-9. [DOI] [Google Scholar]
- Troev K.; Grancharov G.; Tsevi R. Chemical degradation of polyurethanes 3. Degradation of microporous polyurethane elastomer by diethyl phosphonate and tris(1-methyl-2-chloroethyl) phosphate. Polym. Degrad. Stab. 2000, 70, 43–48. 10.1016/s0141-3910(00)00086-0. [DOI] [Google Scholar]
- Troev K.; Atanasov V.; Tsevi R.; Grancharov G.; Tsekova A. Chemical degradation of polyurethanes. Degradation of microporous polyurethane elastomer by dimethyl phosphonate. Polym. Degrad. Stab. 2000, 67, 159–165. 10.1016/s0141-3910(99)00105-6. [DOI] [Google Scholar]
- Troev K.; Atanassov V.; Tzevi R. Chemical degradation of polyurethanes. II. Degradation of microporous polyurethane elastomer by phosphoric acid esters. J. Appl. Polym. Sci. 2000, 76, 886–893. . [DOI] [Google Scholar]
- Troev K.; Tsekova A.; Tsevi R. Chemical degradation of polyurethanes: Degradation of flexible polyester polyurethane foam by phosphonic acid dialkyl esters. J. Appl. Polym. Sci. 2000, 78, 2565–2573. . [DOI] [Google Scholar]
- Grancharov G.; Mitova V.; Shenkov S.; Topliyska A.; Gitsov I.; Antonya G. T.; Ivan Troev K. Smart polymer recycling: Synthesis of novel rigid polyurethanes using phosphorus-containing oligomers formed by controlled degradation of microporous polyurethane elastomer. J. Appl. Polym. Sci. 2007, 105, 302–308. 10.1002/app.25676. [DOI] [Google Scholar]
- Gerlock J.; Braslaw J.; Zinbo M. Polyurethane waste recycling. 1. Glycolysis and hydroglycolysis of water-blown foams. Ind. Eng. Chem. Process Des. Dev. 1984, 23, 545–552. 10.1021/i200026a023. [DOI] [Google Scholar]
- Wu C.-H.; Chang C.-Y.; Cheng C.-M.; Huang H.-C. Glycolysis of waste flexible polyurethane foam. Polym. Degrad. Stab. 2003, 80, 103–111. 10.1016/s0141-3910(02)00390-7. [DOI] [Google Scholar]
- Schulzke T.; Iakovleva A.; Cao Q.; Conrad S.; Zabelkin S.; Grachev A. Polyurethane foams produced from pyrolysis oil—Production and possible application. Biomass Bioenergy 2018, 115, 195–202. 10.1016/j.biombioe.2018.04.006. [DOI] [Google Scholar]
- Wu J.; Wang Y.; Wan Y.; Lei H.; Yu F.; Liu Y.; Chen P.; Yang L.; Ruan R. Processing and properties of rigid polyurethane foams based on bio-oils from microwave-assisted pyrolysis of corn stover. Int. J. Agric. Biol. Eng. 2009, 2, 40–50. 10.3965/j.issn.1934-6344.2009.01.040-050. [DOI] [Google Scholar]
- Al-Salem S. M.; Lettieri P.; Baeyens J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manage. 2009, 29, 2625–2643. 10.1016/j.wasman.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Borda J.; Pásztor G.; Zsuga M. Glycolysis of polyurethane foams and elastomers. Polym. Degrad. Stab. 2000, 68, 419–422. 10.1016/s0141-3910(00)00030-6. [DOI] [Google Scholar]
- Wu C.-H.; Chang C.-Y.; Li J.-K. Glycolysis of rigid polyurethane from waste refrigerators. Polym. Degrad. Stab. 2002, 75, 413–421. 10.1016/s0141-3910(01)00237-3. [DOI] [Google Scholar]
- Molero C.; de Lucas A.; Rodríguez J. F. Recovery of polyols from flexible polyurethane foam by “split-phase” glycolysis with new catalysts. Polym. Degrad. Stab. 2006, 91, 894–901. 10.1016/j.polymdegradstab.2005.06.023. [DOI] [Google Scholar]
- Molero C.; de Lucas A.; Rodríguez J. F. Recovery of polyols from flexible polyurethane foam by “split-phase” glycolysis: Glycol influence. Polym. Degrad. Stab. 2006, 91, 221–228. 10.1016/j.polymdegradstab.2005.05.008. [DOI] [Google Scholar]
- Simón D.; García M. T.; de Lucas A.; Borreguero A. M.; Rodríguez J. F. Glycolysis of flexible polyurethane wastes using stannous octoate as the catalyst: Study on the influence of reaction parameters. Polym. Degrad. Stab. 2013, 98, 144–149. 10.1016/j.polymdegradstab.2012.10.017. [DOI] [Google Scholar]
- Asahi N.; Sakai K.; Kumagai N.; Nakanishi T.; Hata K.; Katoh S.; Moriyoshi T. Methanolysis investigation of commercially available polyurethane foam. Polym. Degrad. Stab. 2004, 86, 147–151. 10.1016/j.polymdegradstab.2004.04.002. [DOI] [Google Scholar]
- Liu L.; Tang L.; Wu Y.; Ni Y.; Zhu Z. Degradation process investigation of thermoplastic polyurethane elastomer in supercritical methanol. Polym. Degrad. Stab. 2013, 98, 2520–2528. 10.1016/j.polymdegradstab.2013.09.010. [DOI] [Google Scholar]
- Deepa P.; Jayakannan M. Solvent-free and nonisocyanate melt transurethane reaction for aliphatic polyurethanes and mechanistic aspects. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2445–2458. 10.1002/pola.22578. [DOI] [Google Scholar]
- Rokicki G.; Piotrowska A. A new route to polyurethanes from ethylene carbonate, diamines and diols. Polymer 2002, 43, 2927–2935. 10.1016/s0032-3861(02)00071-x. [DOI] [Google Scholar]
- Shapiro G.; Marzi M. Facile and selective O-alkyl transesterification of primary carbamates with titanium(IV) alkoxides. J. Org. Chem. 1997, 62, 7096–7097. 10.1021/jo971498z. [DOI] [PubMed] [Google Scholar]
- Jousseaume B.; Laporte C.; Toupance T.; Bernard J. M. Investigations in the catalytic species of the distannoxane-catalyzed transcarbamoylation. Tetrahedron Lett. 2003, 44, 5983–5985. 10.1016/s0040-4039(03)01487-4. [DOI] [Google Scholar]
- Dumrul H.; Yuksel F. Synthesis and characterization of novel symmetrical and asymmetrical substituted Zn(II) phthalocyanines. Polyhedron 2013, 63, 83–90. 10.1016/j.poly.2013.07.015. [DOI] [Google Scholar]
- Jousseaume B.; Laporte C.; Toupance T.; Bernard J.-M. Efficient bismuth catalysts for transcarbamoylation. Tetrahedron Lett. 2002, 43, 6305–6307. 10.1016/s0040-4039(02)01391-6. [DOI] [Google Scholar]
- Rhoné B.; Semetey V. Base-catalyzed transcarbamoylation. Synlett 2017, 28, 2004–2007. 10.1055/s-0036-1588866. [DOI] [Google Scholar]
- Isaksson R.; Kumpiņa I.; Larhed M.; Wannberg J. Rapid and straightforward transesterification of sulfonyl carbamates. Tetrahedron Lett. 2016, 57, 1476–1478. 10.1016/j.tetlet.2016.02.071. [DOI] [Google Scholar]
- Maisonneuve L.; Lamarzelle O.; Rix E.; Grau E.; Cramail H. Isocyanate-free routes to polyurethanes and poly(hydroxy urethane)s. Chem. Rev. 2015, 115, 12407–12439. 10.1021/acs.chemrev.5b00355. [DOI] [PubMed] [Google Scholar]
- Unverferth M.; Kreye O.; Prohammer A.; Meier M. A. R. Renewable non-isocyanate based thermoplastic polyurethanes via polycondensation of dimethyl carbamate monomers with diols. Macromol. Rapid Commun. 2013, 34, 1569–1574. 10.1002/marc.201300503. [DOI] [PubMed] [Google Scholar]
- Firdaus M.; Meier M. A. R. Renewable polyamides and polyurethanes derived from limonene. Green Chem. 2013, 15, 370–380. 10.1039/c2gc36557j. [DOI] [Google Scholar]
- Kébir N.; Nouigues S.; Moranne P.; Burel F. Nonisocyanate thermoplastic polyurethane elastomers based on poly(ethylene glycol) prepared through the transurethanization approach. J. Appl. Polym. Sci. 2017, 134, 44991. 10.1002/app.44991. [DOI] [Google Scholar]
- Uriz P.; Serra M.; Salagre P.; Castillon S.; Claver C.; Fernandez E. A new and efficient catalytic method for synthesizing isocyanates from carbamates. Tetrahedron Lett. 2002, 43, 1673–1676. 10.1016/s0040-4039(02)00094-1. [DOI] [Google Scholar]
- Kreye O.; Mutlu H.; Meier M. A. R. Sustainable routes to polyurethane precursors. Green Chem. 2013, 15, 1431–1455. 10.1039/c3gc40440d. [DOI] [Google Scholar]
- Kairytė A.; Vėjelis S. Evaluation of forming mixture composition impact on properties of water blown rigid polyurethane (PUR) foam from rapeseed oil polyol. Ind. Crops Prod. 2015, 66, 210–215. 10.1016/j.indcrop.2014.12.032. [DOI] [Google Scholar]
- Polyurethane Handbook: Chemistry, Raw Materials, Processing, Application, Properties; Oertel G., Abele L., Eds.; Hanser, Macmillan: USA, 1994. [Google Scholar]
- Fleurent H.; Thijs S. The use of pentanes as blowing agent in rigid polyurethane foam. J. Cell. Plast. 1995, 31, 580–599. 10.1177/0021955x9503100606. [DOI] [Google Scholar]
- Ye S.; Xiang X.; Wang S.; Han D.; Xiao M.; Meng Y. Nonisocyanate CO2-based poly(ester-co-urethane)s with tunable performances: A potential alternative to improve the biodegradability of PBAT. ACS Sustainable Chem. Eng. 2020, 8, 1923–1932. 10.1021/acssuschemeng.9b06294. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.