

Abstract
Mutations in the lamin A/C gene (LMNA), which encodes A-type lamins, cause several diseases called laminopathies, the most common of which is dilated cardiomyopathy with muscular dystrophy. The role of Ca2+ regulation in these diseases remain poorly understood. We now show biochemical remodeling of the ryanodine receptor (RyR)/intracellular Ca2+ release channel in heart samples from human subjects with LMNA mutations, including protein kinase A-catalyzed phosphorylation, oxidation and depletion of the stabilizing subunit calstabin. In the LmnaH222P/H222P murine model of Emery-Dreifuss muscular dystrophy caused by LMNA mutation, we demonstrate an age-dependent biochemical remodeling of RyR2 in the heart and RyR1 in skeletal muscle. This RyR remodeling is associated with heart and skeletal muscle dysfunction. Defective heart and muscle function are ameliorated by treatment with a novel Rycal small molecule drug (S107) that fixes ‘leaky’ RyRs. SMAD3 phosphorylation is increased in hearts and diaphragms of LmnaH222P/H222P mice, which enhances NADPH oxidase binding to RyR channels, contributing to their oxidation. There is also increased generalized protein oxidation, increased calcium/calmodulin-dependent protein kinase II-catalyzed phosphorylation of RyRs and increased protein kinase A activity in these tissues. Our data show that RyR remodeling plays a role in cardiomyopathy and skeletal muscle dysfunction caused by LMNA mutation and identify these Ca2+ channels as a potential therapeutic target.
Introduction
The lamin A/C gene (LMNA) encodes the A-type nuclear lamins, components of the nuclear lamina, a meshwork of atypical intermediate filaments primarily associated with the inner nuclear membrane of metazoan cells (1–7). Mutations in LMNA cause a range of rare diseases often referred to as ‘laminopathies’ (8,9). Most prevalent among these rare diseases are those affecting striated muscle. Autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD) is the first disease linked to LMNA mutations (10). The classical clinical features of EDMD are early contractures of the elbows, Achilles tendons and postcervical muscles, slowly progressive muscle wasting and weakness with a humeroperoneal distribution, and dilated cardiomyopathy with conduction system defects. LMNA mutations can also cause dilated cardiomyopathy with minimal skeletal muscle involvement or with the affected muscle groups different than those of classical EDMD (11–15).
Dysfunctional ryanodine receptor (RyR)-mediated Ca2+ handling has been implicated in the pathogenesis of heart failure and skeletal myopathies (16,17). RyR channels provide the primary pathway for sarcoplasmic reticulum (SR) Ca2+ release during excitation-contraction coupling in both cardiac and skeletal muscle. The channel is a homotetrameric macromolecular complex comprised of four RyR monomers (565 kDa each), and regulatory subunits/modulators including a Ca2+ channel stabilizing binding protein (calstabin; also known as FK506 binding protein), the catalytic subunit of protein kinase A (PKA), the regulatory subunit of PKA, protein phosphatases 1 and 2A and an A-kinase anchoring protein (18–22). SR Ca2+ release via RyR2 in cardiomyocytes is triggered by Ca2+ entering via cardiac L-type calcium channel during depolarization, whereas RyR1 in skeletal muscle is activated by interaction with the plasma membrane L-type Ca2+ channel. Calstabin stabilizes the channel in the closed state and is required for coupled gating between individual RyR channels (18,23–25). The binding of calstabin to RyR is inhibited by PKA-catalyzed phosphorylation (26–28). S-nitrosylation and oxidation of RyRs also decreases the binding affinity of calstabin for the channel (29–32).
Oxidation, protein kinase A-catalyzed phosphorylation and S-nitrosylation of RyR1 cause depletion of calstabin1 from the channel complex in skeletal muscle of mdx and β-sarcoglycan-deficient mice (33,34). The same occurs for RyR2 and calstabin2 in the ventricular cardiac muscle of mdx mice (35). This RyR remodeling is induced by oxidative stress and leads to further reactive oxygen species generation as part of a vicious cycle of SR Ca2+ leak and mitochondrial Ca2+ overload and ROS production (36,37). As a result of remodeling, RyRs become ‘leaky’ and Ca2+ is released from the SR into the cytoplasm, where it activates downstream pathological processes which combined with SR Ca2+ depletion can weaken muscle contraction (38). Drugs known as RyR calcium release channel stabilizers (Rycals) stabilize leaky channels by preventing calstabin depletion from the oxidized and phosphorylated RyR and have beneficial effects on cardiac and skeletal muscle function in various pathological conditions (33,34,39–42).
We hypothesize that RyR remodeling occurs in striated muscles with pathogenic alterations in A-type lamins. We further hypothesize that this remodeling is not only an ‘end-stage’ event but occurs prior to or at the onset of muscle dysfunction, contributing to the early stages of pathogenesis. To test this hypothesis, we examined RyR2 remodeling in available tissues from humans with cardiomyopathy caused by LMNA mutations and in hearts of LmnaH222P/H222P mice, a murine model that recapitulates the dilated cardiomyopathy and muscular dystrophy in humans (43). We also examined RyR1 remodeling in the skeletal muscle of LmnaH222P/H222P mice. We then investigated the effects of a Rycal on RyR remodeling and cardiac and skeletal muscle function in LmnaH222P/H222Pmice.
Results
RyR remodeling in human subjects with LMNA mutations and LmnaH222P/H222P mice
To examine RyR2 remodeling in cardiomyopathy caused by LMNA mutations, we obtained heart tissue samples from two patients who had received transplants and two controls who died of non-cardiac causes. Immunoblotting of RyR2 immunoprecipitated from tissue lysates demonstrated PKA-catalyzed phosphorylation, channel oxidation as well as calstabin2 depletion from the channel (Fig. 1A and B). Given these findings in human heart samples, we initiated an analysis of RyR remodeling in LmnaH222P/H222P mice (43). Male LmnaH222P/H222P mice develop signs and symptoms of dilated cardiomyopathy starting at approximately 8 to 10 weeks of age. By approximately 16 weeks of age, they have left ventricular dilatation and an approximate 30% decrease in cardiac ejection fraction (EF). The median survival of male LmnaH222P/H222P mice is approximately 28 weeks. Female LmnaH222P/H222P mice have the same cardiac abnormalities but with a later onset, starting at about 16 weeks of age, and a median survival of approximately 36 weeks. We, therefore, analyzed hearts from male LmnaH222P/H222P mice for evidence of RyR2 remodeling at 4, 8, 16 and 20 weeks of age. Immunoblotting of RyR2 immunoprecipitated from hearts of these mice showed an age-dependent increase in RyR2 nitrosylation, channel oxidation and calstabin2 dissociation in the heart initially detectable at about 8 weeks of age (Fig. 1C and D). Hearts of female LmnaH222P/H222P mice showed a similar pattern of RyR2 modification at comparatively older ages (Fig. 1E and F).
Figure 1.
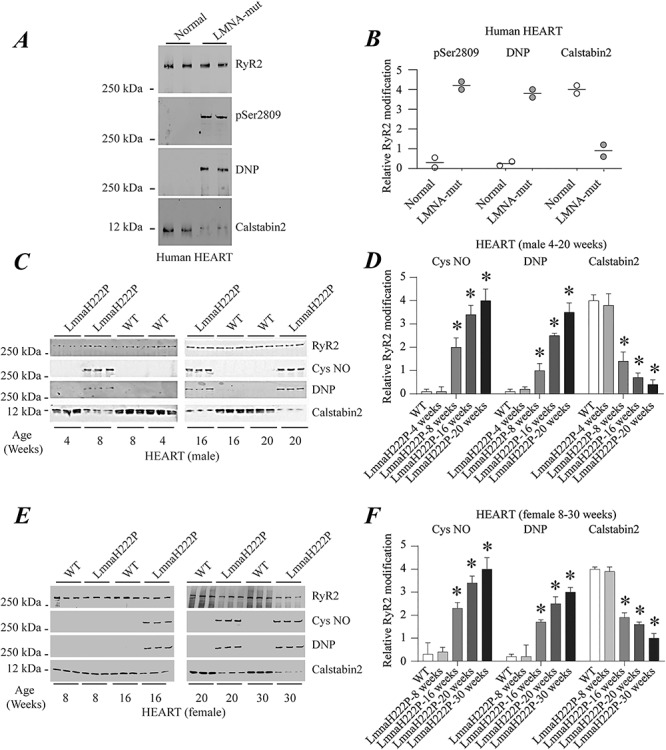
RyR2 remodeling in the hearts of human subjects with cardiomyopathy and LMNA mutations and LmnaH222P/H222P mice. (A) Immunoblot showing RyR2 immunoprecipitated from protein extracts of human hearts and PKA-catalyzed phosphorylation (pSer2809), oxidation (DNP) and calstabin2 depletion. The two samples at the left are from normal controls and the two samples at the right are from human subjects with LMNA mutation and cardiomyopathy. (B) Quantifications of results from scanning immunoblots (values normalized to RyR2). Individual values are shown (n = 2). (C) Immunoblots showing RyR2 immunoprecipitated from protein extracts of male WT and LmnaH222P/H222P (LmmaH222P) mouse hearts and S-nitrosylation (Cys NO), channel oxidation (DNP) and calstabin2 depletion. Three samples from LmnaH222P and WT mice are shown for each time point. (D) Quantifications of results from scanning immunoblots (values normalized to RyR2). Data are means ± SEM (n = 3); *P < 0.05 WT versus LmnaH222P at 8, 16 and 20 weeks of age. (E) Immunoblots showing RyR2 immunoprecipitated from protein extracts of female WT and LmnaH222P/H222P (LmnaH222P) mouse hearts and S-nitrosylation (Cys NO), oxidation (DNP) and calstabin2 depletion. Three samples from LmnaH222P and WT mice are shown for each time point. (F) Quantifications of results from scanning immunoblots (values normalized to RyR2). Data are means ± SEM (n = 3); *P < 0.05 WT versus LmnaH222P at 16, 20 and 30 weeks of age. Migrations of molecular mass standards are indicated to the left of the blots in (A, C andE).
Cardiomyopathy caused by LMNA mutations frequently occurs with variable skeletal muscular dystrophy. As LmnaH222P/H222P mice age, they develop a progressive skeletal muscle pathology that mimics human EDMD. Skeletal muscle pathology begins to manifest at about 16 weeks of age in male LmnaH222P/H222P mice (43). We, therefore, analyzed skeletal muscle for evidence of RyR1 remodeling from male LmnaH222P/H222P mice at 4, 8, 16 and 20 weeks of age. Quadriceps muscle from these mice showed an age-dependent increase in RyR1 nitrosylation, channel oxidation and calstabin1 dissociation, which was prominent by 16 weeks of age (Fig. 2A and B). Skeletal muscle of female LmnaH222P/H222P mice showed a similar pattern of RyR1 modification, with a shift toward older ages (Fig. 2C and D).
Figure 2.
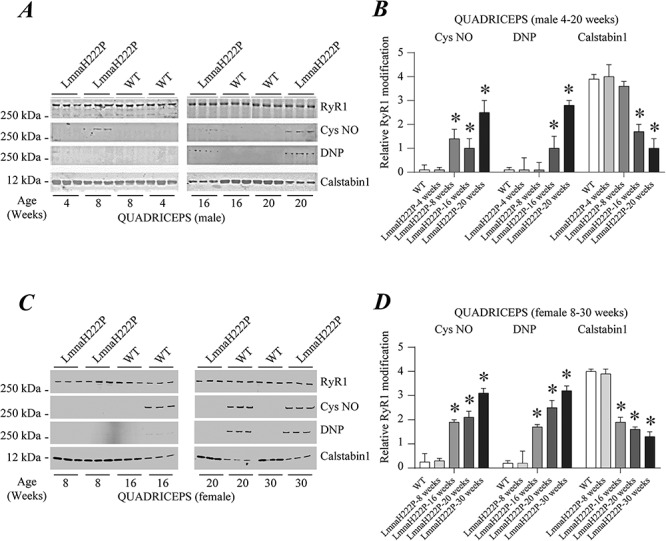
RyR1 remodeling in skeletal muscle of LmnaH222P/H222P mice. (A) Immunoblots showing RyR1 immunoprecipitated from protein extracts of male WT and LmnaH222P/H222P (LmnaH222P) mouse quadriceps and S-nitrosylation (Cys NO), channel oxidation (DNP) and calstabin1 depletion. Three samples from LmnaH222P and WT mice are shown for each time point. (B) Quantifications of results from scanning immunoblots (values normalized to RyR1). Data are means ± SEM (n = 3); *P < 0.05 WT versus LmnaH222P at 8, 16 and 20 weeks of age. (C) Immunoblots showing RyR1 immunoprecipitated from protein extracts of female WT and LmnaH222P/H222P (LmnaH222P) mouse quadriceps and S-nitrosylation (Cys NO), oxidation (DNP) and calstabin1 depletion. Three samples from LmnaH222P and WT mice are shown for each time point. (D) Quantifications of results from scanning immunoblots (values normalized to RyR1). Data are means ± SEM (n = 3); *P < 0.05 WT versus LmnaH222P at 16, 20 and 30 weeks of age. Migrations of molecular mass standards are indicated to the left of the blots in (A) and (C).
Effect of a Rycal on RyR2 remodeling and cardiac function in LmnaH222P/H222P mice
To determine if RyR2 remodeling contributes to cardiac pathology in LmnaH222P/H222P mice, we examined the effects of Rycal S107 which fixes the channel leak. S107 prevents calstabin dissociation from the RyR macromolecular complex without effects on the oxidation and phosphorylation of the channels (38,44). We treated male LmnaH222P/H222P mice with S107 (50 mg/kg/day administered in drinking water) or placebo starting at 14 weeks of age. At 20 weeks of age, we performed echocardiography and then immediately sacrificed the mice to obtain heart tissue to analyze RyR2 remodeling. We performed immunoblotting of RyR2 immunoprecipitated from cardiac lysates of male wild type (WT) mice and LmnaH222P/H222P mice treated with either placebo or S107 (Fig. 3A). Treatment with S107 prevented calstabin2 depletion from RyR2 in LmnaH222P/H222P mice, with associated calstabin2 levels similar to those in WT mice (Fig. 3B). Channel oxidation and PKA-catalyzed phosphorylation of RyR2 were similar in hearts of mice treated with placebo or S107, with both greater than in WT controls (Fig. 3C and D). When analyzed using echocardiography male LmnaH222P/H222P mice treated with S107 had significantly decreased left ventricular end diastolic and end systolic diameters compared to those receiving placebo (Fig. 3E and F). Treatment with S107 resulted in a significant increase in left ventricular fractional shortening (FS) and left ventricular EF (Fig. 3G and H). Taken together these are echocardiographic indicators of improved left ventricular function. Heart rates were similar in all mice (Supplementary Material, Fig. S1A).
Figure 3.
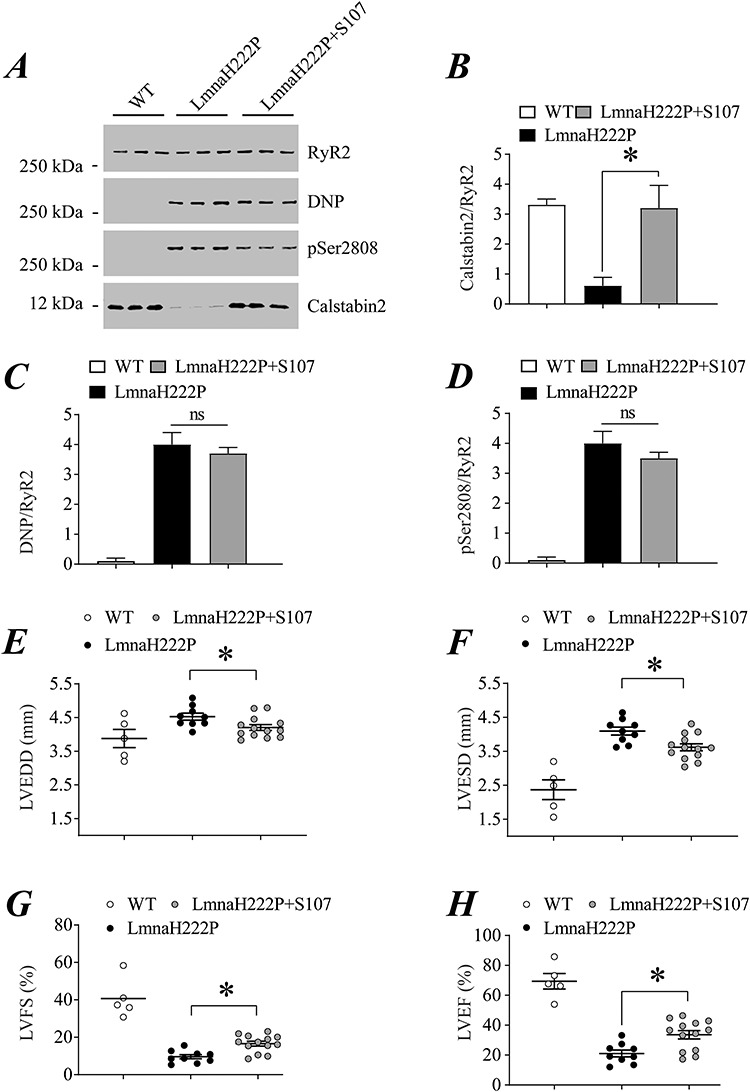
Effect of S107 on RyR2 remodeling and echocardiographic parameters in male LmnaH222P/H222P mice. (A) Immunoblot showing RyR2 immunoprecipitated from protein extracts of hearts of male WT mice and LmnaH222P/H222P mice treated with either placebo (LmnaH222P) or S107 (LmnaH222P + S107) and oxidation (DNP), PKA-catalyzed phosphorylation (pSer2808) and calstabin2 depletion. Samples are from mice at 20 weeks of age; treated mice received placebo or drug for 6 weeks. Migration of molecular mass standards is indicated to the left of the blot. (B–D) Quantification of associated calstabin2 (B), oxidized (C) and phosphorylated (D) RyR2 normalized to total RyR2 from WT mice and LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). (E–H) Left ventricular end diastolic diameter (LVEDD) (E), left ventricular end systolic diameter (F), left ventricular FS (G), and left ventricular EF (H) determined by echocardiography in WT mice and LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). Values are means ± SEM (Control n = 5, LmnaH222P n = 9, LmnaH222P + S107 n = 13); *P < 0.05 LmnaH222P versus LmnaH222P + S107.
We also examined the effects of S107 on RyR2 remodeling and echocardiographic parameters in female LmnaH222P/H222P mice. Because female mice develop cardiac dysfunction at older ages than male littermates, we started treatment with placebo or S107 at 22 weeks of age and examined their hearts at 30 weeks. We performed immunoblotting of RyR2 immunoprecipitated from protein extracts of hearts of female WT mice and LmnaH222P/H222P mice treated with either placebo or S107 (Fig. 4A). Treatment with S107 prevented calstabin2 depletion from RyR2 in hearts of female LmnaH222P/H222P mice, with associated calstabin2 levels similar to those in WT mice (Fig. 4B). Channel oxidation and PKA-catalyzed phosphorylation of RyR2 were similar in hearts of mice treated with placebo or S107, with both greater than in WT controls (Fig. 4C and D). Similar to their male littermates but at older ages, female LmnaH222P/H222P mice treated with S107 had significantly decreased left ventricular end diastolic and end systolic diameters compared to those receiving placebo (Fig. 4E and F). Indices of left ventricular systolic function were preserved in treated mice compared to those receiving placebo (Fig. 4G and H). Heart rates were similar in all mice (Supplementary Material, Fig. S1B).
Figure 4.
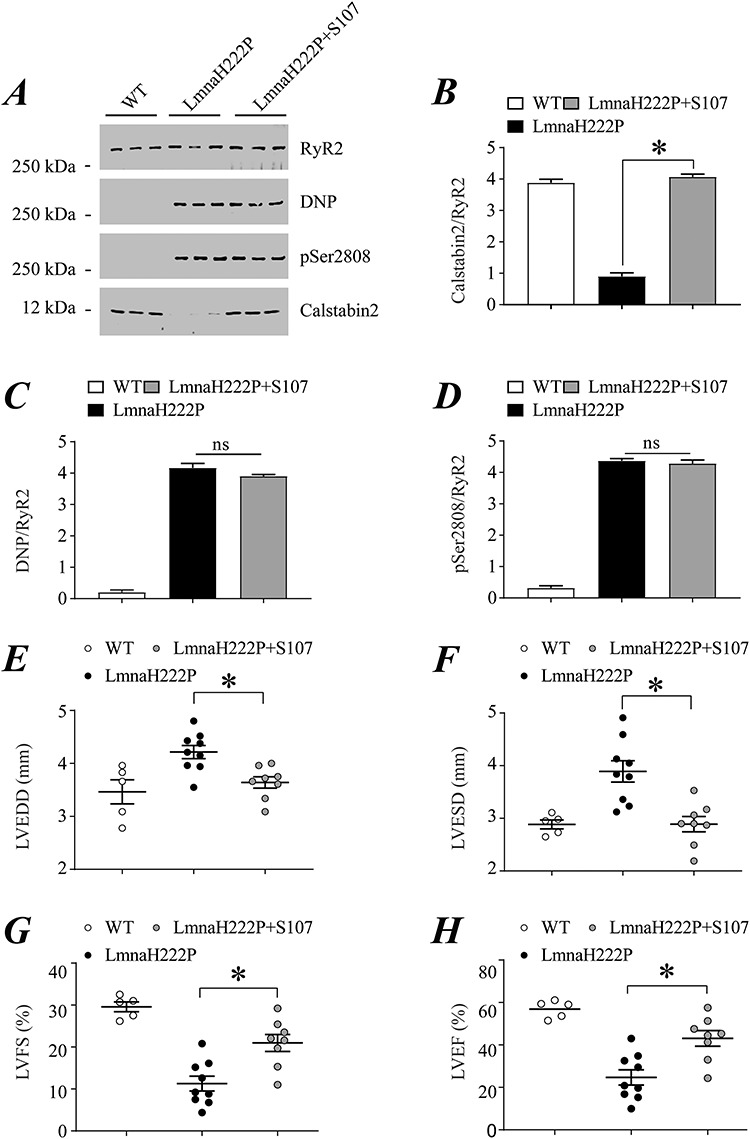
Effect of S107 on RyR2 remodeling and echocardiographic parameters in female LmnaH222P/H222P mice. (A) Immunoblot showing RyR2 immunoprecipitated from extracts of hearts of female WT mice and LmnaH222P/H222P mice treated with either placebo (LmnaH222P) or S107 (LmnaH222P + S107) and oxidation (DNP), PKA-catalyzed phosphorylation (pSer2808) and calstabin2 depletion. Samples are from mice at 30 weeks of age; treated mice received placebo or drug for 8 weeks. Migration of molecular mass standards are indicated to the left of the blot. (B–D) Quantification of associated calstabin2 (B), oxidized (C) and phosphorylated (D) RyR2 normalized to total RyR2 from WT mice and LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). (E–H) Left ventricular end diastolic diameter (LVEDD) (E), left ventricular end systolic diameter (F), left ventricular FS and left ventricular EF (H) determined by echocardiography in WT mice and LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). Values are means ± SEM (Control n = 5, LmnaH222P n = 9, LmnaH222P + S107 n = 8); *P < 0.05 LmnaH222P versus LmnaH222P + S107.
Effect of a Rycal on RyR1 remodeling and skeletal muscle function in LmnaH222P/H222P mice
Both male and female LmnaH222P/H222P mice develop skeletal myopathy. At older ages, severe histopathological abnormalities occur in the diaphragm and soleus muscle and more moderate pathology in gastrocnemius, quadriceps, triceps and tibialis anterior muscles (43). We, therefore, examined RyR1 remodeling and the effects of S107 on diaphragm and soleus function in LmnaH222P/H222P mice. We performed immunoblotting of RyR1 immunoprecipitated from protein extracts of diaphragm muscles of male WT mice and LmnaH222P/H222P mice at 20 weeks of age, after 6 weeks of treatment with either placebo or S107 (Fig. 5A). Treatment with S107 prevented calstabin1 depletion from RyR1 in diaphragm muscle of male LmnaH222P/H222P mice, with associated calstabin1 levels similar to those in WT mice (Fig. 5B). Channel oxidation and PKA-catalyzed phosphorylation of RyR1 were similar in diaphragms of mice treated with placebo or S107, with both greater than in WT controls (Fig. 5C and D). We also assessed the isometric contractile properties of diaphragm muscle ex vivo by measuring diaphragm-specific force production measured at incremental frequencies (10–120 Hz) under isometric conditions in male WT mice, LmnaH222P/H222P mice treated with placebo or S107 (Fig. 5E–G). Although the diaphragmatic force production was not significantly different when the muscles were stimulated at low frequencies (10, 20 and 30 Hz), male LmnaH222P/H222P mice treated with placebo had a significant decrease in diaphragm force production with medium and high stimulation frequencies (50–120 Hz). This measure of muscle function was identical in S107-treated LmnaH222P/H222P mice and WT controls (Fig. 5H). Similar results were observed in the soleus muscle of male LmnaH222P/H222P mice (Supplementary Material, Fig. S2).
Figure 5.
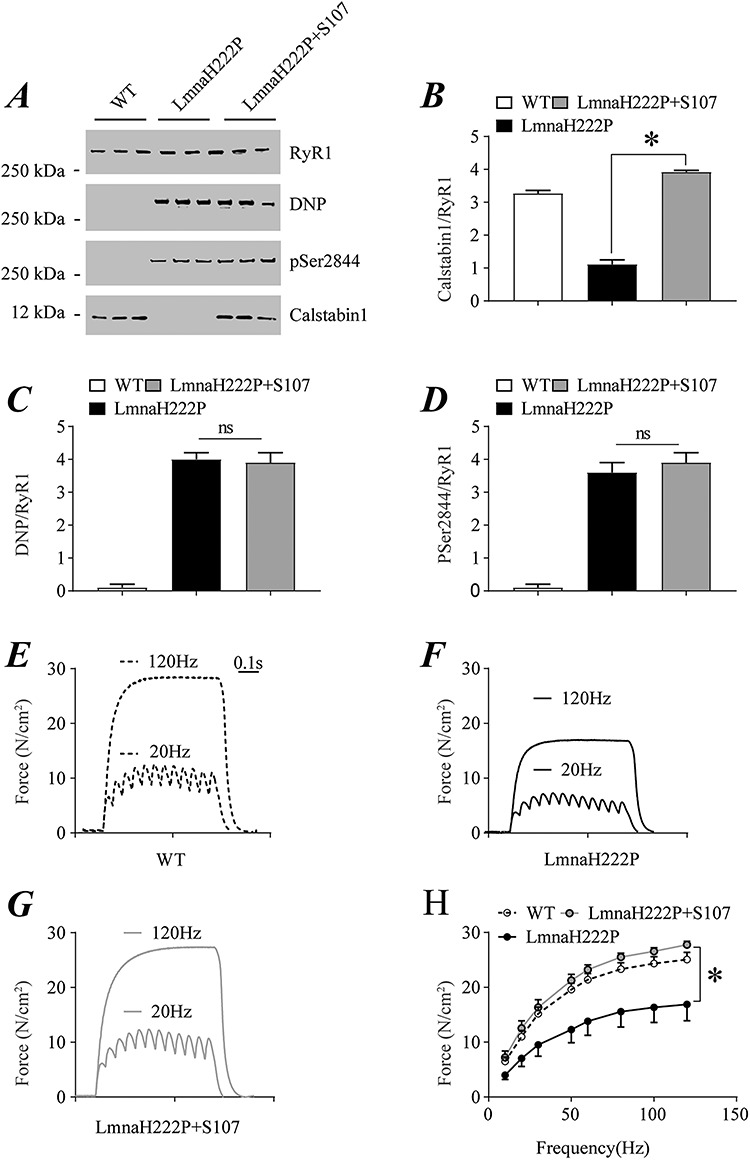
Effect of S107 on RyR1 remodeling and isometric contractile properties of diaphragm from male LmnaH222P/H222P mice. (A) Immunoblot showing RyR1 immunoprecipitated from extracts of diaphragms of male WT mice and LmnaH222P/H222P mice treated with either placebo (LmnaH222P) or S107 (LmnaH222P + S107) and oxidation (DNP), PKA-catalyzed phosphorylation (pSer2844) and calstabin1 depletion. Samples are from mice 20 weeks of age; treated mice received placebo or drug for 6 weeks. Migration of molecular mass standards is indicated to the left of the blot. (B–D) Quantification of associated calstabin1 (B), oxidized (C) and phosphorylated (D) RyR1 normalized to total RyR1 from WT mice and LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). Values are means ± SEM (Control n = 3, LmnaH222P n = 3, LmnaH222P + S107 n = 3); *P < 0.05; ns not significant. (E–G) Representative records of diaphragmatic specific force production measured ex vivo at 20 and 120 Hz in muscle bundles under isometric conditions of WT (E), LmnaH222P/H222P mice treated with placebo (LmnaH222P) (F) or S107 (LmnaH222P + S107) (G). (H) Average force-frequency relationship recorded in WT, LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). Values are means ± SEM (Control n = 5, LmnaH222P n = 4, LmnaH222P + S107 n = 5); *P < 0.05, LmnaH222P versus LmnaH222P + S107.
We similarly performed immunoblotting of immunoprecipitated RyR1 from lysates of diaphragm muscle of female WT mice and LmnaH222P/H222P mice at 30 weeks of age, after 8 weeks of treatment with either placebo or S107 (Fig. 6A). As in male mice, treatment with S107 prevented calstabin1 depletion from RyR1 in the diaphragm muscle of female LmnaH222P/H222P mice, with associated calstabin1 levels similar to those in WT mice (Fig. 6B). Channel oxidation and PKA-catalyzed phosphorylation of RyR1 were similar in diaphragms of mice treated with placebo or S107, with both greater than in WT controls (Fig. 6C and D). We also assessed the isometric contractile properties of diaphragm muscle ex vivo by measuring diaphragm-specific force production measured at incremental frequencies (10–120 Hz) under isometric conditions in WT mice, LmnaH222P/H222P mice treated with placebo and LmnaH222P/H222P mice treated with S107 (Fig. 6E–G). Whereas female LmnaH222P/H222P mice treated with placebo had a significant decrease in diaphragm force production, S107-treated LmnaH222P/H222P mouse diaphragm generated the same force as that from WT controls (Fig. 6H). Similar results were observed in the soleus muscle of female LmnaH222P/H222P mice (Supplementary Figure S3).
Figure 6.
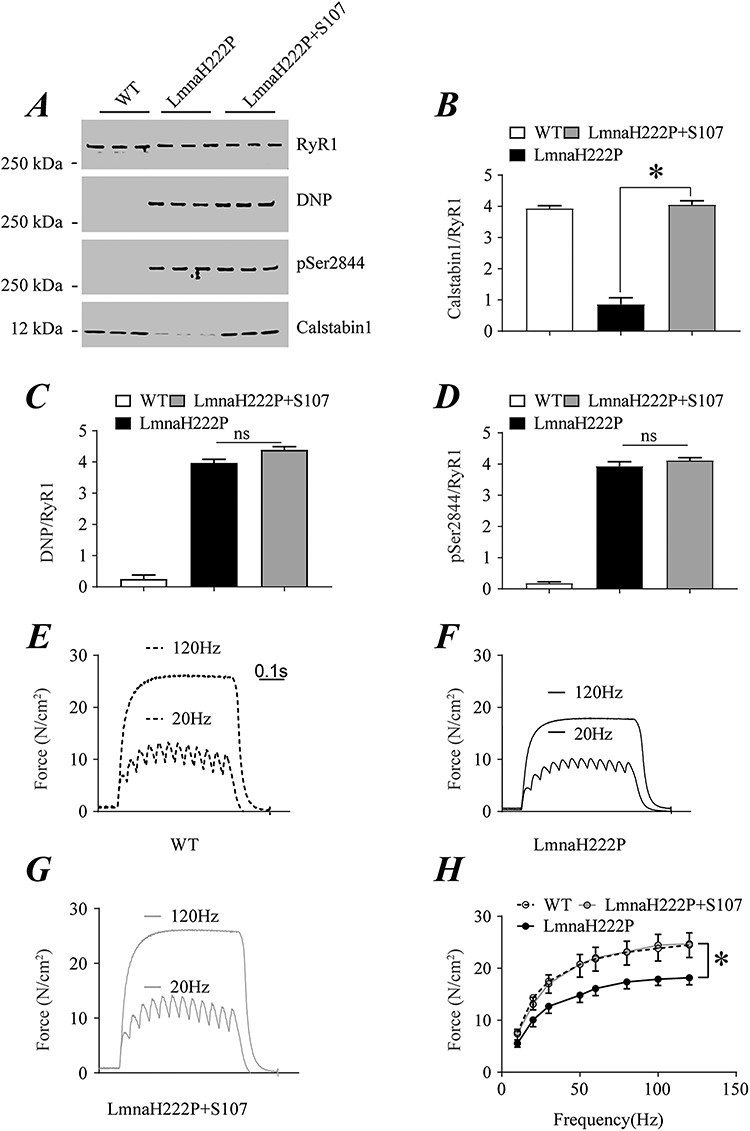
Effect of S107 on RyR1 remodeling and isometric contractile properties of diaphragm from female LmnaH222P/H222P mice. (A) Immunoblot showing RyR1 immunoprecipitated from extracts of diaphragms of female WT mice and LmnaH222P/H222P mice treated with either placebo (LmnaH222P) or S107 (LmnaH222P + S107) and oxidation (DNP), PKA-catalyzed phosphorylation (pSer2844) and calstabin1 depletion. Samples are from mice at 30 weeks of age; treated mice received placebo or drug for 8 weeks. Migration of molecular mass standards are indicated to the left of the blot. (B–D) Quantification of associated calstabin1 (B), oxidized (C) and phosphorylated (D) RyR1 normalized to total RyR1 from WT mice and LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). Values are means ± SEM (Control n = 3, LmnaH222P n = 3, LmnaH222P + S107 n = 3); *P < 0.05; ns not significant. (E–G) Representative records of diaphragmatic specific force production measured ex-vivo at 20 and 120 Hz in muscle bundles under isometric conditions of WT (E), LmnaH222P/H222P mice treated with placebo (LmnaH222P) (F) or S107 (LmnaH222P + S107) (G). (H) Average force-frequency relationship recorded in WT, LmnaH222P/H222P mice treated with placebo (LmnaH222P) or S107 (LmnaH222P + S107). Values are means ± SEM (Control n = 5, LmnaH222P n = 7, LmnaH222P + S107 n = 7); *P < 0.05, LmnaH222P versus LmnaH222P + S107.
After 6 weeks of treatment with the Rycal S107, 20-week-old male LmnaH222P/H222P mice demonstrated significantly increased grip strength compared to those treated with placebo (Fig. 7A). Similarly, after 8 weeks of treatment with S107, 30-week-old female LmnaH222P/H222P mice had significantly increased grip strength compared to those that received a placebo (Fig. 7B).
Figure 7.
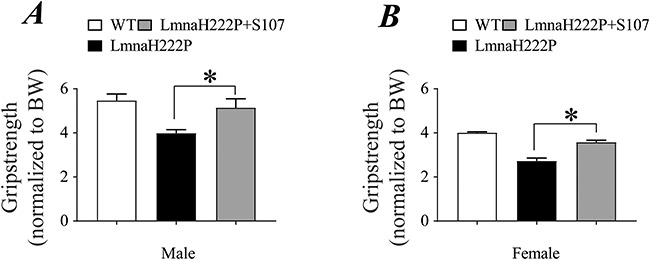
Effect of S107 treatment on grip strength in LmnaH222P/H222P mice. (A) Grip strength of male WT mice, LmnaH222P/H222P mice treated with placebo (LmnaH222P) and LmnaH222P/H222P mice treated with S107 (LmnaH222P + S107). Values are means ± SEM (WT n = 5, LmnaH222P n = 4, LmnaH222P + S107 n = 5); *P < 0.05, LmnaH222P versus LmnaH222P + S107. (B) Grip strength of female WT mice, LmnaH222P/H222P mice treated with placebo (LmnaH222P) and LmnaH222P/H222P mice treated with S107 (LmnaH222P + S107). Values are means ± SEM (WT n = 5, LmnaH222P n = 9, LmnaH222P + S107 n = 9); *P < 0.05 LmnaH222P versus LmnaH222P + S107.
Mechanisms responsible for RyR remodeling
To identify the mechanisms responsible for RyR1 and RyR2 oxidation in LmnaH222P/H222P mouse striated muscle, we performed immunoblotting of RyR2 and RyR1 immunoprecipitated from heart and diaphragm lysates of male WT and LmnaH222P/H222P mice at 20 weeks of age. We found an increase of cardiac NADPH oxidase 2 (Nox2) and diaphragmatic NADPH oxidase 4 (Nox4) binding to RyR2 and RyR1 channels, respectively (Fig. 8A and B). This may account for the increased channels oxidation as previously reported (45–47). Endogenous S-nitrosylation of RyR channels by a nitric oxide donor such as nitric oxide synthase (iNOS) results in depletion of calstabin from the channels (33). We found increased iNOS binding to RyR2 and RyR1, respectively, in hearts and diaphragms of LmnaH222P/H222P mice (Fig. 8A and B). SMAD3 phosphorylation, an upstream mediator of Nox expression, was also increased in both hearts and diaphragms of LmnaH222P/H222P mice (Fig. 8C and D). Furthermore, several other proteins were oxidized in both hearts and diaphragms of LmnaH222P/H222P mice compared to the controls, an indicator of high levels of oxidative stress in these tissues (Fig. 8E and F). Calcium/calmodulin-dependent protein kinase II (CaMKII) and PKA are tethered to RyRs and phosphorylates the channels in heart failure (26,48) and muscular dystrophies (33,34). CaMKII is activated by Ca2+-calmodulin and phosphorylates cardiac RyR2 channels on Ser2815. We found increased RyR2 phosphorylation by CaMKII on Ser2815 in hearts of LmnaH222P/H222P mice compared to controls (Fig. 8G). Adrenaline and noradrenaline bind to β-adrenergic receptors and activate adenylyl cyclase, which produces cAMP, resulting in downstream activation of PKA causing chronic RyRs hyper-phosphorylation. We also found increased PKA activity in both hearts and diaphragms from LmnaH222P/H222P mice, indicating increased circulating catecholamines levels (Fig. 8H and I).
Figure 8.
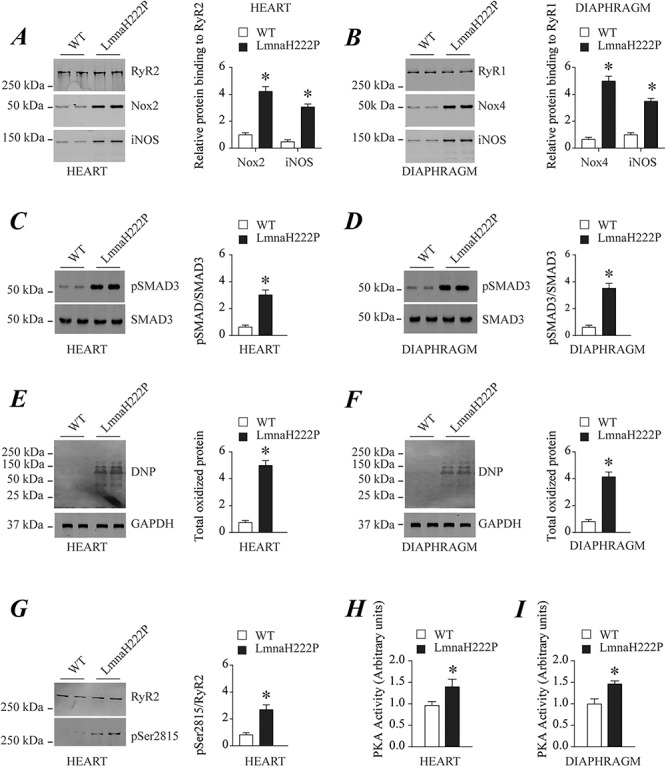
Mechanisms responsible for RyR2 and RyR1 remodeling in hearts and diaphragms of LmnaH222P/H222P mice. (A) Immunoblot and quantification showing RyR2 immunoprecipitated from protein extracts of WT and LmnaH222P/H222P (LmnaH222P) mouse hearts and Nox2 and iNOS binding to the channel. (B) Immunoblot and quantification showing RyR1 immunoprecipitated from protein extracts of WT and LmnaH222P mouse diaphragms and Nox4 and iNOS binding to the channel. (C) Immunoblot and quantification showing levels of SMAD3 phosphorylation from protein extracts of WT and LmnaH222P mouse hearts. (D) Immunoblot and quantification showing levels of SMAD3 phosphorylation from protein extracts of WT and LmnaH222P mouse diaphragms. (E) Immunoblot and quantification showing total protein oxidation from protein extracts of WT and LmnaH222P mouse hearts. (F) Immunoblot and quantification showing total protein oxidation from protein extracts of WT and LmnaH222P mouse diaphragms. (G) Immunoblot and quantification showing levels of RyR2 phosphorylation by CaMKII from protein extracts of WT and LmnaH222P mouse hearts. In (A) through (G), migrations of molecular mass standards are indicated to the left of the blots; values are means ± SEM (WT n = 4, LmnaH222P n = 4); *P < 0.05 WT versus LmnaH222P. (H) Bar graphs showing PKA activity (normalized to the WT levels) from protein extracts of WT LmnaH222P mouse hearts. (I) Bar graphs showing PKA activity (normalized to the WT levels) from protein extracts of WT and LmnaH222P diaphragms. In (E) and (F), values are means ± SEM (WT n = 3, LmnaH222P n = 3); *P < 0.05 WT versus LmnaH222P.
Discussion
The molecular basis of how LMNA mutations induce cardiomyopathy and muscular dystrophy is not well understood. Questions regarding the role of Ca2+ signaling in the heart and skeletal muscle remain unanswered. Here we report a mechanism responsible for defective Ca2+ regulation in striated muscle induced by pathogenic LMNA mutation. RyR Ca2+ channels are PKA-phosphorylated, oxidized, nitrosylated and depleted of the stabilizing subunit calstabin in heart and skeletal muscle of LmnaH222P/H222P mouse model of the disease. PKA activity is increased in both heart and diaphragm of LmnaH222P/H222P mice, mirroring an increase in circulating catecholamines levels. Transforming growth factor-β (TGF-β) signaling, an upstream mediator of RyR channel oxidation via Nox enzymes (49,50), is also increased. This is in accordance with previous studies showing increased SMAD2/3 and 4 phosphorylation and nuclear translocation in hearts of LmnaH222P/H222P (43,51). Indeed, Nox2 and Nox4 binding to RyRs channels is significantly increased, thereby contributing to channel oxidation. Furthermore, iNOS association with RyRs was enhanced, which may account for increased RyRs S-nitrosylation. This biochemical remodeling of RyRs channel is known as the ‘leaky signature of RyR’ leading to an increase of the SR Ca2+ leak which is a major contributor to cardiomyopathy, arrhythmias and muscle weakness. We observed similar biochemical modification of RyR2 in the heart tissue of human patients with cardiomyopathy-causing LMNA mutations and dilated cardiomyopathy.
In mice, RyR2 remodeling in the heart is associated with dilated cardiomyopathy and reduced cardiac function. In skeletal muscle, RyR1 post-translational modification is associated with diaphragmatic dysfunction and limb muscle weakness. S107 treatment of both male and female LmnaH222P/H222P mice prevented calstabin dissociation from RyR channels and improved cardiac and skeletal muscle function despite the persistence of the channels’ post-translational modifications. RyR1/2 channels remodeling may also occur in non-muscle tissues such as the brain and pancreatic β-cells and has been associated with cognitive impairment (41,52) and defective insulin release (53). However, abnormalities in those organs have never been reported in LmnaH222P/H222P mice or humans with cardiomyopathy-causing LMNA mutations.
A few previous studies reported evidence of Ca2+ dysregulation without clear mechanisms in the case of LMNA mutations. RNA sequence analysis throughout the course of disease in hearts of Lmna−/− mice showed downregulation of genes involved in Ca2+ signaling such as RyR2, troponin I3 and tropomyosin 4 (54). CaMKII was upregulated, mirroring an increase in cytosolic Ca2+ (54). CaMKII is initially activated by Ca2+-calmodulin, remains active owing to auto-phosphorylation, tethers to RyR2 and phosphorylates the channel on Ser2815 (38,55). We found an increase of RyR2 phosphorylation at Ser2815 in hearts from LmnaH222P/H222P mice, which may be associated with an increase in the cytosolic Ca2+ concentrations. Mutant human induced pluripotent stem cell (iPSC)-derived immature cardiomyocytes carrying the most prevalent Finnish founder LMNA mutation, p.S143P, have altered Ca2+ dynamics (56). At baseline, Ca2+ levels and transient rise time and decay time are elevated in these immature cardiomyocytes consistent with possible SR Ca2+ depletion and impaired reuptake, both of which could be due to SR Ca2+ leak via remodeled RyR channels (56). Monolayers of electrically paced cardiomyocytes derived from iPSCs of patients with LMNA haploinsufficiency mutations also have abnormal Ca2+ flux associated with upregulation of CACNA1 encoding the pore-forming subunit of L-type calcium channels (57). Furthermore, Lee et al. reported RyR2 phosphorylation by CaMKII in iPSC-derived cardiomyocytes from patients with an LMNA mutation and dilated cardiomyopathy, which is consistent with our findings (58). A recent study has also demonstrated elevated levels of sarcolipin and an alteration of Ca2+ cycling in cardiomyocytes isolated from LmnaH222P/H222P mice (59). These results and ours are consistent with Ca2+ dyshomeostasis observed in failing human hearts and animal models of heart failure in which RyR2 plays a major role in cardiac Ca2+ dysregulation (26,60).
PKA-catalyzed phosphorylation of RyR2 dissociates the stabilizing subunit calstabin2, leading to channel instability and increasing Ca2+ ‘leak’ through the channel. Chronic RyR2 Ca2+ ‘leak’ depletes SR Ca2+ stores and reduces the levels available for sarcomere cross bridging formations during systole, thereby leading to decreased heart contractility and failure (38). This is in line with previous studies reporting progressive heart failure observed in knock-in mice with aspartic acid replacing the RyR2 PKA phosphorylation site, serine 2808 (61). Our data show evidence of RyR2 remodeling as early as at 8 weeks of age and 16 weeks of age, respectively, in male and female LmnaH222P/H222P mice. The resulting SR Ca2+ leak may contribute to the progressive heart failure and predisposition to lethal arrhythmias characteristic of human subjects with dilated cardiomyopathy caused by LMNA mutations (62).
Human subjects with cardiomyopathy-causing LMNA mutations often have associated variable forms of muscular dystrophy, most frequently in an EDMD pattern (10–15). We similarly observed RyR1 remodeling in the skeletal muscle of LmnaH222P/H222P mice. This finding is comparable with RyR1 remodeling previously reported in mouse models of Duchenne muscular dystrophy (33) and β-sarcoglycanopathy (34). Overall, remodeled RyR1 appears to be a common defect in multiple forms of muscular dystrophy, including those caused by LMNA mutation.
In contrast to LmnaH222P/H222P mice, most pediatric and adult human patients with EDMD or other forms of muscular dystrophy caused by LMNA mutations do not have respiratory muscle involvement. However, a small subset of patients with LMNA mutations presents early in childhood with congenital muscular dystrophy (15,63–65), some of whom suffer from severe respiratory insufficiency and nocturnal hypoventilation requiring ventilatory support. While the contribution of possible diaphragmatic dysfunction to respiratory insufficiency in these human cases has not been ascertained, it is possible that S107 may be beneficial given its effects on diaphragm contractile function in LmnaH222P/H222Pmice.
Lmna H222P/H222P mice treated with S107 have superior cardiac and skeletal muscle function compared to those treated with placebo. S107 is a member of a class of drugs called Rycals that stabilize RyR1 and RyR2 channels by preventing the dissociation of calstabin. This in turn reduces SR Ca2+ leak. Rycals may therefore provide benefits for both the cardiomyopathy and skeletal myopathy that often occur together in patients with LMNA mutations. Another Rycal is currently under investigation in a clinical trial for myopathy caused by RYR1 mutations (https://clinicaltrials.gov/ct2/show/NCT04141670).
Besides defective RyR regulation reported in the current study, cardiomyopathy caused by LMNA mutation is associated with several cell signaling pathways abnormalities. These include mitogen-activated protein (MAP) kinases, protein kinase B and mammalian target of rapamycin (mTOR) (66–68). Treatment of LmnaH222P/H222P mice with inhibitors of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), which blocks activity of the MAP kinases extracellular signal-regulated kinases 1 and 2, has beneficial effects on left ventricular diameters and left ventricular EF, significantly reduces cardiac fibrosis and prolongs survival (67,69–71). Treatment with temsirolimus, an mTOR inhibitor, reduces heart size and preserves FS in LmnaH222P/H222P mice (67). Combination treatment with a Rycal and either a MEK1/2 or mTOR inhibitor may therefore be worthy of preclinical investigation in LmnaH222P/H222Pmice.
Materials and Methods
Human heart samples
Tissue bank samples of explanted hearts from human subjects with LMNA mutations were obtained without identifiers from Myobank-AFM de l’lnstitut de Myologie. One subject had a LMNA E33D mutation with a left ventricular EF of 40% prior to transplantation; the other had a LMNA delK261 mutation and a left ventricular EF of 25% prior to transplantation. Myobank-AFM received approval from the French Ministry of Health and from the Committee for Protection of Patients to share tissues and cells of human origin for scientific purposes, ensuring the donors’ anonymity, respect of their volition and consent according to the legislation. Control human heart samples were obtained from the National Disease Research Interchange from subjects who died from causes other than the cardiac disease; information regarding donor confidentiality and consent can be found at https://www.ndriresource.org.
Mice
The Institutional Animal Care and Use Committee at Columbia University Irving Medical Center approved the use of animals and the study protocol. LmnaH222P/H222P mice have been described previously (43). These mice and their WT counterparts had the 129S1 genetic background. Mice were bread and genotyped as described previously (69,70). Mice were fed a chow diet and housed in a barrier facility with 12 h/12 h light/dark cycles. S107 was added to acidified water, in a blinded fashion, at a concentration of 0.25 mg/ml. The feeding method for S107 medicated water or acidified water (placebo) was ad libitum. The average of mouse water intake was 5 ml/day (72) and body weight was about 25 g. The consumption of S107 by each individual mouse was approximately 50 mg/kg/day. There is no significant water intake difference between male and female mice (72). Mice were euthanized by CO2 overdose followed by cervical dislocation.
Immunoprecipitation and immunoblotting
RyR2 and RyR1 were immunoprecipitated extracts of heart, diaphragm and soleus muscle each containing 100 μg of total protein using anti-RyR2 or anti-RyR1-specific antibodies (2 μg) in 0.5 ml of a modified radioimmune precipitation assay buffer (50 mm Tris–HCl, pH 7.2, 0.9% NaCl, 5.0 mm NaF, 1.0 mm Na3VO4, 1% Triton X-100 and protease inhibitors). Antibodies were incubated with extracts overnight at 4°C. RyR1-specific antibody was RyR1-1327, an affinity-purified rabbit polyclonal antibody raised against a KLH-conjugated peptide with the amino acid sequence CAEPDTDYENLRRS, corresponding to residues 1327-1339 of mouse skeletal RyR1, with an additional cysteine residue added to the amino terminus, and affinity-purified with the unconjugated peptide (44). RyR2-specific antibody was an affinity-purified polyclonal rabbit antibody using the peptide CKPEFNNHKDYAQEK corresponding to amino acids 1367–1380 of mouse RyR2 with a cysteine residue added to the amino terminus (73). The immune complexes were incubated with protein A-Sepharose beads (Sigma-Aldrich) at 4°C for 1 h and the beads were washed three times with the modified radio-immunoprecipitation assay buffer. The immunoprecipitated proteins were size-fractionated on SDS-polyacrylamide gels (4–20% for RyR1/RyR2 and calstabin) and transferred to nitrocellulose membranes for 2 h at a current of 200 mA. Immunoblots were probed with the following primary antibodies: anti-RyR1/2 (Affinity Bioreagents, 1:2000 dilution), anti-Cys-NO (Sigma-Aldrich, 1:1000 dilution), anti-calstabin (FKBP12 C-19, Santa Cruz Biotechnology, 1:1000 dilution), anti-SMAD3 (Abcam, 1:1000 dilution), anti-phospho-SMAD3 (Abcam, 1:1000 dilution), anti-Nox2 (Abcam, 1:10000 dilution), anti-Nox4 (Abcam, 1:10000 dilution) and anti-iNOS (Abcam, 1:10000 dilution). Some blots were also probed with antibodies that recognize specific phosphorylated residues in RyR. The RyR2-Ser2809 phospho-epitope specific anti-RyR2 antibody (1:1000 dilution) is a custom made affinity-purified polyclonal rabbit antibody (Zymed Laboratories) that recognizes the peptide CRTRRI-(pS)-QTSQ corresponding to RyR2 PKA phosphorylated at Ser2809. This antibody recognizes pSer2808 (mouse), 2809 (human) and 2844 sites (74). The RyR2-pSer2815 phospho-epitope specific anti-RyR2 antibody (1:1000 dilution) is a custom made affinity-purified polyclonal rabbit antibody generated using the peptide CSQTSQV-(pS)-VD corresponding to RyR2 CaMKII phosphorylated at Ser2815 (75). To determine channel oxidation, the carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenol (DNP) by reaction with 2,4-dinitrophenylhydrazine. The DNP signal associated with the total oxidized protein or with RyR was determined using a specific anti-DNP antibody according to the manufacturer’s instructions (Millipore). All immunoblots were developed using an Odyssey system (LI-COR Biosciences), with infrared-labeled anti-mouse or anti-rabbit IgG (Abcam, 1:10000 dilution) secondary antibodies. The post-translational modification of RyR1/2, including phosphorylation, oxidation, nitrosylation and calstabin1/2, NOX2/4, iNOS binding were normalized to total immunoprecipitated RyR1/2. Phosphorylation of SMAD3 was normalized to total SMAD3 expression and protein oxidation (DNP) were normalized to GAPDH.
PKA activity assay
Samples were thawed on ice, PKA activity was determined by using a colorimetric assay (Abcam, ab139435) according to manufacturer’s instruction and as previously described (44).
Echocardiography
Mice were anesthetized (1–3% isoflurane, 100% oxygen) and examined by transthoracic echocardiography using a high-resolution ultrasound system (Vevo 3100, Visualsonics), as described in (76). Animals were placed on a heating table in a supine position to be maintained at 37°C. A single-lead electrocardiogram was monitored throughout, and recorded during echocardiography, to ensure heart rates were similar between groups (>350 bpm). Two-dimensional and M-mode images were recorded at the left ventricular parasternal short axis position at the level of papillary muscle to assess internal diameters, allowing the calculation of the FS and EF by the Teicholz method. The analysis was performed in Vevo Lab (Visualsonics).
Isometric contractile properties of skeletal muscle
After euthanasia, the entire diaphragm and soleus were surgically excised. Isometric contractile properties were assessed, as described elsewhere (44,77). The excised diaphragm strip and soleus were mounted into jacketed tissue bath chambers filled with equilibrated and oxygenated Krebs solution. The muscles were supramaximally stimulated using square wave pulses (Model S48; Grass Instruments). The force–frequency relationship was determined by sequentially stimulating the muscles for 600 ms at 10, 20, 30, 50, 60, 80, 100 and 120 Hz with 1 min between each stimulation train (44,77). After measurement of contractile properties, muscles were measured at Lo (the length at which the muscle produced maximal isometric tension), dried and weighed. For comparative purposes, muscle force production was normalized for total muscle strip cross-sectional area and expressed in N/cm2. The total muscle strip cross-sectional area was determined by dividing muscle mass by its length and tissue density (1.056 g/cm3) (78).
Grip strength
A Grip Strength Meter (GPM-100; Melquest) was used to measure forelimb grip strength. As a mouse grasped the bar, the peak pull force in grams was recorded on a digital force transducer. In the conventional test, a mouse was allowed to grasp the bar mounted on the force gauge. The gauge was reset to 0 g after stabilization, and the mouse’s tail was slowly pulled back by an inspector, as previously described (79,80).
Statistics
Student’s t-test was used when comparing the differences between two groups. Values in experiments with more than two experimental groups were compared using one-way ANOVA. To validate ANOVA results, a nonparametric (Mann–Whitney) test was performed and concordance checked. For muscle function, groups were compared by two-way ANOVA.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Heart, Lung and Blood Institute of the National Institutes of Health under award number R01HL142903 to H.J.W. and A.R.M. R.M.O. and L.S. were supported by National Institutes of Health Diversity Supplements. J.K. was supported by the New York Academy of Medicine Glorney-Raisbeck Junior Faculty Research Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest statement. H.J.W.—scientific advisory board and equity owner AlloMek Therapeutics; consultant Eiger BioPharmaceuticals; sponsored research funding Navitor Pharmaceuticals and Sarepta Therapeutics. A.R.M.—scientific advisory board, board of directors and equity owner ARMGO, Inc. Columbia University also owns equity in ARMGO, Inc.
References
- 1. Gerace, L., Blum, A. and Blobel, G. (1978) Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J. Cell Biol., 79, 546–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McKeon, F.D., Kirschner, M.W. and Caput, D. (1986) Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature, 319, 463–468. [DOI] [PubMed] [Google Scholar]
- 3. Aebi, U., Cohn, J., Buhle, L. and Gerace, L. (1986) The nuclear lamina is a meshwork of intermediate-type filaments. Nature, 323, 560–564. [DOI] [PubMed] [Google Scholar]
- 4. Fisher, D.Z., Chaudhary, N. and Blobel, G. (1986) cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA, 83, 6450–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldman, A.E., Maul, G., Steinert, P.M., Yang, H.Y. and Goldman, R.D. (1986) Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc. Natl. Acad. Sci. USA, 83, 3839–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin, F. and Worman, H.J. (1993) Structural organization of the human gene encoding nuclear Lamin a and nuclear Lamin C. J. Biol. Chem., 268, 16321–16326. [PubMed] [Google Scholar]
- 7. Turgay, Y., Eibauer, M., Goldman, A.E., Shimi, T., Khayat, M., Ben-Harush, K., Dubrovsky-Gaupp, A., Sapra, K.T., Goldman, R.D. and Medalia, O. (2017) The molecular architecture of lamins in somatic cells. Nature, 543, 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Worman, H.J. and Bonne, G. (2007) "Laminopathies": a wide spectrum of human diseases. Exp. Cell Res., 313, 2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guillin-Amarelle, C., Fernandez-Pombo, A., Sanchez-Iglesias, S. and Araujo-Vilar, D. (2018) Lipodystrophic laminopathies: diagnostic clues. Nucleus, 9, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bonne, G., Di Barletta, M.R., Varnous, S., Becane, H.M., Hammouda, E.H., Merlini, L., Muntoni, F., Greenberg, C.R., Gary, F., Urtizberea, J.A. et al. (1999) Mutations in the gene encoding Lamin a/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet., 21, 285–288. [DOI] [PubMed] [Google Scholar]
- 11. Fatkin, D., MacRae, C., Sasaki, T., Wolff, M.R., Porcu, M., Frenneaux, M., Atherton, J., Vidaillet, H.J., Jr., Spudich, S., De Girolami, U. et al. (1999) Missense mutations in the rod domain of the Lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med., 341, 1715–1724. [DOI] [PubMed] [Google Scholar]
- 12. Bonne, G., Mercuri, E., Muchir, A., Urtizberea, A., Becane, H.M., Recan, D., Merlini, L., Wehnert, M., Boor, R., Reuner, U. et al. (2000) Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the Lamin A/C gene. Ann. Neurol., 48, 170–180. [PubMed] [Google Scholar]
- 13. Muchir, A., Bonne, G., van der Kooi, A.J., van Meegen, M., Baas, F., Bolhuis, P.A., de Visser, M. and Schwartz, K. (2000) Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet., 9, 1453–1459. [DOI] [PubMed] [Google Scholar]
- 14. Brodsky, G.L., Muntoni, F., Miocic, S., Sinagra, G., Sewry, C. and Mestroni, L. (2000) Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation, 101, 473–476. [DOI] [PubMed] [Google Scholar]
- 15. Quijano-Roy, S., Mbieleu, B., Bonnemann, C.G., Jeannet, P.Y., Colomer, J., Clarke, N.F., Cuisset, J.M., Roper, H., De Meirleir, L., D'Amico, A. et al. (2008) De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol., 64, 177–186. [DOI] [PubMed] [Google Scholar]
- 16. Kushnir, A., Wajsberg, B. and Marks, A.R. (2018) Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res., 1865, 1687–1697. [DOI] [PubMed] [Google Scholar]
- 17. Lawal, T.A., Todd, J.J. and Meilleur, K.G. (2018) Ryanodine receptor 1-related myopathies: diagnostic and therapeutic approaches. Neurotherapeutics, 15, 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brillantes, A.B., Ondrias, K., Scott, A., Kobrinsky, E., Ondriasova, E., Moschella, M.C., Jayaraman, T., Landers, M., Ehrlich, B.E. and Marks, A.R. (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell, 77, 513–523. [DOI] [PubMed] [Google Scholar]
- 19. Marx, S.O., Reiken, S., Hisamatsu, Y., Gaburjakova, M., Gaburjakova, J., Yang, Y.M., Rosemblit, N. and Marks, A.R. (2001) Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J. Cell Biol., 153, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zalk, R., Clarke, O.B., des Georges, A., Grassucci, R.A., Reiken, S., Mancia, F., Hendrickson, W.A., Frank, J. and Marks, A.R. (2015) Structure of a mammalian ryanodine receptor. Nature, 517, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zalk, R., Lehnart, S.E. and Marks, A.R. (2007) Modulation of the ryanodine receptor and intracellular calcium. Annu. Rev. Biochem., 76, 367–385. [DOI] [PubMed] [Google Scholar]
- 22. Denniss, A., Dulhunty, A.F. and Beard, N.A. (2018) Ryanodine receptor Ca(2+) release channel post-translational modification: central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol., 101, 49–53. [DOI] [PubMed] [Google Scholar]
- 23. Marx, S.O., Ondrias, K. and Marks, A.R. (1998) Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science, 281, 818–821. [DOI] [PubMed] [Google Scholar]
- 24. Marx, S.O., Gaburjakova, J., Gaburjakova, M., Henrikson, C., Ondrias, K. and Marks, A.R. (2001) Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res., 88, 1151–1158. [DOI] [PubMed] [Google Scholar]
- 25. Kaftan, E., Marks, A.R. and Ehrlich, B.E. (1996) Effects of rapamycin on ryanodine receptor/ca(2+)-release channels from cardiac muscle. Circ. Res., 78, 990–997. [DOI] [PubMed] [Google Scholar]
- 26. Marx, S.O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N. and Marks, A.R. (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell, 101, 365–376. [DOI] [PubMed] [Google Scholar]
- 27. Ward, C.W., Reiken, S., Marks, A.R., Marty, I., Vassort, G. and Lacampagne, A. (2003) Defects in ryanodine receptor calcium release in skeletal muscle from post-myocardial infarct rats. FASEB J., 17, 1517–1519. [DOI] [PubMed] [Google Scholar]
- 28. Reiken, S., Lacampagne, A., Zhou, H., Kherani, A., Lehnart, S.E., Ward, C., Huang, F., Gaburjakova, M., Gaburjakova, J., Rosemblit, N. et al. (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J. Cell Biol., 160, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chelu, M.G., Danila, C.I., Gilman, C.P. and Hamilton, S.L. (2004) Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc. Med., 14, 227–234. [DOI] [PubMed] [Google Scholar]
- 30. Sun, J., Xin, C., Eu, J.P., Stamler, J.S. and Meissner, G. (2001) Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci. USA, 98, 11158–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aracena, P., Sanchez, G., Donoso, P., Hamilton, S.L. and Hidalgo, C. (2003) S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J. Biol. Chem., 278, 42927–42935. [DOI] [PubMed] [Google Scholar]
- 32. Durham, W.J., Aracena-Parks, P., Long, C., Rossi, A.E., Goonasekera, S.A., Boncompagni, S., Galvan, D.L., Gilman, C.P., Baker, M.R., Shirokova, N. et al. (2008) RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell, 133, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bellinger, A.M., Reiken, S., Carlson, C., Mongillo, M., Liu, X., Rothman, L., Matecki, S., Lacampagne, A. and Marks, A.R. (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med., 15, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andersson, D.C., Meli, A.C., Reiken, S., Betzenhauser, M.J., Umanskaya, A., Shiomi, T., D'Armiento, J. and Marks, A.R. (2012) Leaky ryanodine receptors in beta-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet. Muscle, 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fauconnier, J., Thireau, J., Reiken, S., Cassan, C., Richard, S., Matecki, S., Marks, A.R. and Lacampagne, A. (2010) Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA, 107, 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andersson, D.C., Betzenhauser, M.J., Reiken, S., Meli, A.C., Umanskaya, A., Xie, W., Shiomi, T., Zalk, R., Lacampagne, A. and Marks, A.R. (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab., 14, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zima, A.V. and Mazurek, S.R. (2016) Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol., 171, 39–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dridi, H., Kushnir, A., Zalk, R., Yuan, Q., Melville, Z. and Marks, A.R. (2020) Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat. Rev. Cardiol., 17, 732–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wehrens, X.H., Lehnart, S.E., Reiken, S.R., Deng, S.X., Vest, J.A., Cervantes, D., Coromilas, J., Landry, D.W. and Marks, A.R. (2004) Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science, 304, 292–296. [DOI] [PubMed] [Google Scholar]
- 40. Bellinger, A.M., Reiken, S., Dura, M., Murphy, P.W., Deng, S.X., Landry, D.W., Nieman, D., Lehnart, S.E., Samaru, M., LaCampagne, A. et al. (2008) Remodeling of ryanodine receptor complex causes "leaky" channels: a molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA, 105, 2198–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dridi, H., Liu, X., Yuan, Q., Reiken, S., Mohamad, Y., Sittenfeld, L.R., Apostolou, P., Buron, J., Sicard, P., Matecki, S. et al. (2020) Role of defective calcium regulation in cardiorespiratory dysfunction in Huntington's disease. JCI Insight, 5, e140614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dridi, H., Jung, B., Yehya, M., Daurat, A., Reiken, S., Moreau, J., Marks, A.R., Matecki, S., Lacampagne, A. and Jaber, S. (2020) Late ventilator-induced diaphragmatic dysfunction after extubation. Crit. Care Med., 48, e1300–e1305. [DOI] [PubMed] [Google Scholar]
- 43. Arimura, T., Helbling-Leclerc, A., Massart, C., Varnous, S., Niel, F., Lacene, E., Fromes, Y., Toussaint, M., Mura, A.M., Keller, D.I. et al. (2005) Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet., 14, 155–169. [DOI] [PubMed] [Google Scholar]
- 44. Matecki, S., Dridi, H., Jung, B., Saint, N., Reiken, S.R., Scheuermann, V., Mrozek, S., Santulli, G., Umanskaya, A., Petrof, B.J. et al. (2016) Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc. Natl. Acad. Sci. USA, 113, 9069–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Byrne, J.A., Grieve, D.J., Bendall, J.K., Li, J.M., Gove, C., Lambeth, J.D., Cave, A.C. and Shah, A.M. (2003) Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res., 93, 802–805. [DOI] [PubMed] [Google Scholar]
- 46. Hidalgo, C., Sanchez, G., Barrientos, G. and Aracena-Parks, P. (2006) A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J. Biol. Chem., 281, 26473–26482. [DOI] [PubMed] [Google Scholar]
- 47. Xia, R., Webb, J.A., Gnall, L.L., Cutler, K. and Abramson, J.J. (2003) Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am. J. Physiol. Cell Physiol., 285, C215–C221. [DOI] [PubMed] [Google Scholar]
- 48. Respress, J.L., van Oort, R.J., Li, N., Rolim, N., Dixit, S.S., deAlmeida, A., Voigt, N., Lawrence, W.S., Skapura, D.G., Skardal, K. et al. (2012) Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res., 110, 1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Waning, D.L., Mohammad, K.S., Reiken, S., Xie, W., Andersson, D.C., John, S., Chiechi, A., Wright, L.E., Umanskaya, A., Niewolna, M. et al. (2015) Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med., 21, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lou, Z., Wang, A.P., Duan, X.M., Hu, G.H., Song, G.L., Zuo, M.L. and Yang, Z.B. (2018) Upregulation of NOX2 and NOX4 mediated by TGF-beta signaling pathway exacerbates cerebral ischemia/reperfusion oxidative stress injury. Cell. Physiol. Biochem., 46, 2103–2113. [DOI] [PubMed] [Google Scholar]
- 51. Chatzifrangkeskou, M., Le Dour, C., Wu, W., Morrow, J.P., Joseph, L.C., Beuvin, M., Sera, F., Homma, S., Vignier, N., Mougenot, N. et al. (2016) ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the Lamin a/C gene. Hum. Mol. Genet., 25, 2220–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lacampagne, A., Liu, X., Reiken, S., Bussiere, R., Meli, A.C., Lauritzen, I., Teich, A.F., Zalk, R., Saint, N., Arancio, O. et al. (2017) Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease-like pathologies and cognitive deficits. Acta Neuropathol., 134, 749–767. [DOI] [PubMed] [Google Scholar]
- 53. Santulli, G., Pagano, G., Sardu, C., Xie, W., Reiken, S., D'Ascia, S.L., Cannone, M., Marziliano, N., Trimarco, B., Guise, T.A. et al. (2015) Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Invest., 125, 1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shao, Z., Koh, W., Ni, Y., Li, W., Agatisa-Boyle, B., Merkurjev, D. and Tang, W.H.W. (2020) RNA sequence analyses throughout the course of mouse cardiac laminopathy identify differentially expressed genes for cell cycle control and mitochondrial function. Sci. Rep., 10, 6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Picht, E., DeSantiago, J., Huke, S., Kaetzel, M.A., Dedman, J.R. and Bers, D.M. (2007) CaMKII inhibition targeted to the sarcoplasmic reticulum inhibits frequency-dependent acceleration of relaxation and Ca2+ current facilitation. J. Mol. Cell. Cardiol., 42, 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shah, D., Virtanen, L., Prajapati, C., Kiamehr, M., Gullmets, J., West, G., Kreutzer, J., Pekkanen-Mattila, M., Helio, T., Kallio, P. et al. (2019) Modeling of LMNA-related dilated cardiomyopathy using human induced pluripotent stem cells. Cell, 8, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bertero, A., Fields, P.A., Smith, A.S.T., Leonard, A., Beussman, K., Sniadecki, N.J., Kim, D.H., Tse, H.F., Pabon, L., Shendure, J. et al. (2019) Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol., 218, 2919–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee, J., Termglinchan, V., Diecke, S., Itzhaki, I., Lam, C.K., Garg, P., Lau, E., Greenhaw, M., Seeger, T., Wu, H. et al. (2019) Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature, 572, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morales Rodriguez, B., Dominguez-Rodriguez, A., Benitah, J.P., Lefebvre, F., Marais, T., Mougenot, N., Beauverger, P., Bonne, G., Briand, V., Gomez, A.M. et al. (2020) Activation of sarcolipin expression and altered calcium cycling in LMNA cardiomyopathy. Biochem. Biophys. Rep., 22, 100767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wehrens, X.H., Lehnart, S.E., Reiken, S., Vest, J.A., Wronska, A. and Marks, A.R. (2006) Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. USA, 103, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shan, J., Betzenhauser, M.J., Kushnir, A., Reiken, S., Meli, A.C., Wronska, A., Dura, M., Chen, B.X. and Marks, A.R. (2010) Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Invest., 120, 4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kumar, S., Baldinger, S.H., Gandjbakhch, E., Maury, P., Sellal, J.M., Androulakis, A.F., Waintraub, X., Charron, P., Rollin, A., Richard, P. et al. (2016) Long-term arrhythmic and nonarrhythmic outcomes of Lamin a/C mutation carriers. J. Am. Coll. Cardiol., 68, 2299–2307. [DOI] [PubMed] [Google Scholar]
- 63. Mercuri, E., Poppe, M., Quinlivan, R., Messina, S., Kinali, M., Demay, L., Bourke, J., Richard, P., Sewry, C., Pike, M. et al. (2004) Extreme variability of phenotype in patients with an identical missense mutation in the Lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch. Neurol., 61, 690–694. [DOI] [PubMed] [Google Scholar]
- 64. Pasqualin, L.M., Reed, U.C., Costa, T.V., Quedas, E., Albuquerque, M.A., Resende, M.B., Rutkowski, A., Chadi, G. and Zanoteli, E. (2014) Congenital muscular dystrophy with dropped head linked to the LMNA gene in a Brazilian cohort. Pediatr. Neurol., 50, 400–406. [DOI] [PubMed] [Google Scholar]
- 65. Karaoglu, P., Quizon, N., Pergande, M., Wang, H., Polat, A.I., Ersen, A., Ozer, E., Willkomm, L., Hiz Kurul, S., Heredia, R. et al. (2017) Dropped head congenital muscular dystrophy caused by de novo mutations in LMNA. Brain and Development, 39, 361–364. [DOI] [PubMed] [Google Scholar]
- 66. Muchir, A., Pavlidis, P., Decostre, V., Herron, A.J., Arimura, T., Bonne, G. and Worman, H.J. (2007) Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest., 117, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Muchir, A., Wu, W., Choi, J.C., Iwata, S., Morrow, J., Homma, S. and Worman, H.J. (2012) Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by Lamin A/C gene mutation. Hum. Mol. Genet., 21, 4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ramos, F.J., Chen, S.C., Garelick, M.G., Dai, D.F., Liao, C.Y., Schreiber, K.H., MacKay, V.L., An, E.H., Strong, R., Ladiges, W.C. et al. (2012) Rapamycin reverses elevated mTORC1 signaling in Lamin a/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med., 4, 144ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Muchir, A., Shan, J., Bonne, G., Lehnart, S.E. and Worman, H.J. (2009) Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet., 18, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu, W., Muchir, A., Shan, J., Bonne, G. and Worman, H.J. (2011) Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in Lamin A/C gene. Circulation, 123, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu, W., Chordia, M.D., Hart, B.P., Kumarasinghe, E.S., Ji, M.K., Bhargava, A., Lawlor, M.W., Shin, J.Y., Sera, F., Homma, S. et al. (2017) Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by Lamin A/C gene mutation. Bioorg. Med. Chem., 25, 1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tordoff, M.G., Bachmanov, A.A. and Reed, D.R. (2007) Forty mouse strain survey of water and sodium intake. Physiol. Behav., 91, 620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bussiere, R., Lacampagne, A., Reiken, S., Liu, X., Scheuerman, V., Zalk, R., Martin, C., Checler, F., Marks, A.R. and Chami, M. (2017) Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem., 292, 10153–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wehrens, X.H., Lehnart, S.E., Huang, F., Vest, J.A., Reiken, S.R., Mohler, P.J., Sun, J., Guatimosim, S., Song, L.S., Rosemblit, N. et al. (2003) FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell, 113, 829–840. [DOI] [PubMed] [Google Scholar]
- 75. Wehrens, X.H., Lehnart, S.E., Reiken, S.R. and Marks, A.R. (2004) Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res., 94, e61–e70. [DOI] [PubMed] [Google Scholar]
- 76. Santulli, G., Cipolletta, E., Sorriento, D., Del Giudice, C., Anastasio, A., Monaco, S., Maione, A.S., Condorelli, G., Puca, A., Trimarco, B. et al. (2012) CaMK4 gene deletion induces hypertension. J. Am. Heart Assoc., 1, e001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dridi, H., Yehya, M., Barsotti, R., Reiken, S., Angebault, C., Jung, B., Jaber, S., Marks, A.R., Lacampagne, A. and Matecki, S. (2020) Mitochondrial oxidative stress induces leaky ryanodine receptor during mechanical ventilation. Free Radic. Biol. Med., 146, 383–391. [DOI] [PubMed] [Google Scholar]
- 78. Umanskaya, A., Santulli, G., Xie, W., Andersson, D.C., Reiken, S.R. and Marks, A.R. (2014) Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. USA, 111, 15250–15255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cabe, P.A., Tilson, H.A., Mitchell, C.L. and Dennis, R. (1978) A simple recording grip strength device. Pharmacol. Biochem. Behav., 8, 101–102. [DOI] [PubMed] [Google Scholar]
- 80. Smith, J.P., Hicks, P.S., Ortiz, L.R., Martinez, M.J. and Mandler, R.N. (1995) Quantitative measurement of muscle strength in the mouse. J. Neurosci. Methods, 62, 15–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.