

Abstract
Genomic instability contributes to a variety of potentially damaging conditions, including DNA-based rearrangements. Breakage in the form of double strand breaks (DSBs) increases the likelihood of DNA damage, mutations and translocations. Certain human DNA regions are known to be involved in recurrent translocations, such as the palindrome-mediated rearrangements that have been identified at the breakpoints of several recurrent constitutional translocations: t(11;22)(q23;q11), t(17;22)(q11;q11) and t(8;22) (q24;q11). These breakpoints occur at the center of palindromic AT-rich repeats (PATRRs), which suggests that the structure of the DNA may play a contributory role, potentially through the formation of secondary cruciform structures. The current study analyzed the DSB propensity of these PATRR regions in both lymphoblastoid (mitotic) and spermatogenic cells (meiotic). Initial results found an increased association of sister chromatid exchanges (SCEs) at PATRR regions in experiments that used SCEs to assay DSBs, combining SCE staining with fluorescence in situ hybridization (FISH). Additional experiments used chromatin immunoprecipitation (ChIP) with antibodies for either markers of DSBs or proteins involved in DSB repair along with quantitative polymerase chain reaction to quantify the frequency of DSBs occurring at PATRR regions. The results indicate an increased rate of DSBs at PATRR regions. Additional ChIP experiments with the cruciform binding 2D3 antibody indicate an increased rate of cruciform structures at PATRR regions in both mitotic and meiotic samples. Overall, these experiments demonstrate an elevated rate of DSBs at PATRR regions, an indication that the structure of PATRR containing DNA may lead to increased breakage in multiple cellular environments.
Introduction
The most common recurrent non-Robertsonian chromosomal translocation in humans is t(11;22)(q23;q11.21). Balanced carriers exhibit no clinical symptoms; however, they are at an increased risk for 3:1 meiotic malsegregation resulting in severely affected offspring with supernumerary-der(22)t(11;22) syndrome (Emanuel Syndrome) (1–4). While the t(11;22) is the most common PATRR-mediated translocation, the PATRR on chromosome 22 has been shown to translocate with PATRR regions on other chromosomes as well, including t(17;22)(q11;q11.21) and t(8;22)(q24.13;q11.21) with translocation rates dependent on the length and symmetry of the PATRR regions (5–11).
Studies on the recurrent t(11;22)(q23;q11.21) have historically suggested that the translocation occurs during meiosis or gametogenesis since de novo translocations are detected in the sperm of healthy males at a frequency of 1.24–9.46×10−5, but not detected in somatic cells (12). Other PATRR-mediated translocations have also been detected in the sperm of healthy males, such as the de novo t(8;22)(q24.13;q11.21) which is detected at a frequency ranging from less than 6.38×10−7 to 1×10−5 (13). The t(17;22)(q11;q11.21) has not been detected in sperm, indicating it has a translocation frequency less than 7×10−7 (5). However recent evidence indicates that this translocation can occur during mitosis in rare situations (14). Collectively, this suggests that while these translocations may occur during both mitosis and meiosis, the cellular environment during meiosis/gametogenesis may increase the likelihood of having a PATRR-mediated translocation occurs.
The breakpoints for the PATRR-mediated translocations occur at the center of PATRR sequence (15–17). These palindromic sequences have identical 5′-3′ sequences on opposing strands, which causes the formation of an inverted repeats on a single strand with the potential to form complex hairpin or cruciform DNA structures (18–20). The presence of these structures could contribute to genomic instability, resulting in increased susceptibility to DSBs and translocations (21). Cruciform structures of PATRR sequences have been previously predicted in silico via the M-fold program (22) and in vitro through atomic force microscopy of PATRR sequences inserted in plasmids (19). This led us to question whether the susceptibility of PATRR DNA sequences to form cruciform structures resulting in genomic instability was maintained in vivo. To begin to address this question, we tested for elevated DNA strand breakage events at PATRR sequences as a marker of increased genomic instability.
Our initial approach took advantage of SCEs, which are a mechanism of efficient recovery of DNA breakage during mitosis and thereby an indirect measure of underlying instability (23–27). This technique involves visualizing metaphase chromosome spreads using a fluorescent microscope with the addition of labeled probes located at PATRR regions. Upon visual inspection, SCE and translocation events can be counted. Through the use of SCE detection coupled with fluorescence in situ hybridization (FISH) using probes specific to the regions containing PATRR DNA, we were able to test if the translocation breakpoint regions demonstrate an increased frequency of SCE events due to a genomic organization that renders them prone to DSBs. Determination of SCE frequency at 22q11, 11q23 and 8q24 was performed using metaphase spreads of lymphoblastoid lines derived from both normal individuals and balanced translocation carriers.
A second approach also tested for DSB susceptibility at the PATRR regions in lymphoblastoid cells, where ChIP experiments were performed using antibodies for the DSB-associated proteins γH2AX, Rad51 and NBS1. The most studied marker of DSBs is γH2AX, which is the phosphorylated form of the H2A histone variant H2AX that is present in 10–15% of mammalian nucleosomes, and it plays an important role in the recruitment and retention of DSB repair proteins (28–32). Spreading of γH2AX is bidirectional and can reach hundreds of kilobases (33,34). Rad51 is a homolog of bacterial RecA that binds to single-strand DNA where it functions as a strand exchange protein. It is involved in DSB repair in both mitotic and meiotic cells (35–37) and can be detected in ChIP experiments at a distance of ~2–4 kb (38). NBS1 is a member of the MRE11-RAD50-NBS1 complex that is required for the efficient detection, processing, and repair of DSBs and binds within 200–500 bp of the DSB (39–42).
Additional ChIP experiments were performed using the 2D3 antibody which binds to cruciform DNA (43). Previous work with this antibody has shown that it binds to a variety of cruciform structures (44,45) including PATRR regions when they were inserted in plasmids (19). This led us to ask if the PATRRs maintained their propensity to form cruciform structures within the cell in both mitotic and meiotic environments.
The main questions were to address whether PATRR sequences within a human cell environment were more susceptible to DSB’s and cruciform structures when compared to non-PATRR DNA sequences. In addition, given that PATRR-mediated recurrent constitutional translocations are more likely to occur during meiosis, was there an increased rate of PATRR DSB’s in a meiotic environment compared to a mitotic environment.
Results
Increased SCEs at PATRR regions in mitotic cells
SCEs at the PATRR regions on 8q24, 11q23 and 22q11 were examined to determine whether these regions exhibited a tendency to undergo SCE events as a result of a genomic organization that renders them prone to DSBs. This was accomplished by coupling SCE analysis with FISH using unique probes for the PATRR regions on metaphase spreads of peripheral blood lymphocytes. Figure 1 shows a diagram and representative images of how the SCEs at PATRR regions were visualized for enumeration. The percentage of SCEs at the PATRR regions were compared to a control region on chromosome 6q26, which is approximately the same distance from the telomere of 6q as 11q23 is from the telomere of 11q. If PATRR structure does not increase the frequency of DSBs, a similar number of SCE events would be expected for 6q26 as the PATRR-containing regions. As shown in Table 1, in normal lymphocytes, the percentage of SCEs at both PATRR regions 11q23 and 8q24 is significantly higher than the background on 6q26. In cultured peripheral blood samples, 11q23 (2.12–2.2%) and 8q24 (2.2–2.5%) are both significantly higher than 6q26 (1.2–1.5%); statistical significance was determined using McNemar test of equality of proportions (1-sided): 11q23—replicate A: P1=0.028 and replicate B: p2=0.004; and 8q24—replicate A: P1=0.008 and replicate B: P2=0.003. The rate of SCE’s at 11q23 as compared to 6q26 was even greater in a normal lymphoblastoid cell line, with SCEs at 11q23 occurring >2.5x more frequently than at 6q26 (4% and 1.4%, respectively; McNemar test of equality of proportions, one tail, P<0.000001). It should be noted that attempts were made to determine the SCE frequency at the PATRR on 22q relative to a background of 21q, however, due to the small chromosome sizes, SCEs could not be accurately counted (data not shown).
Figure 1.
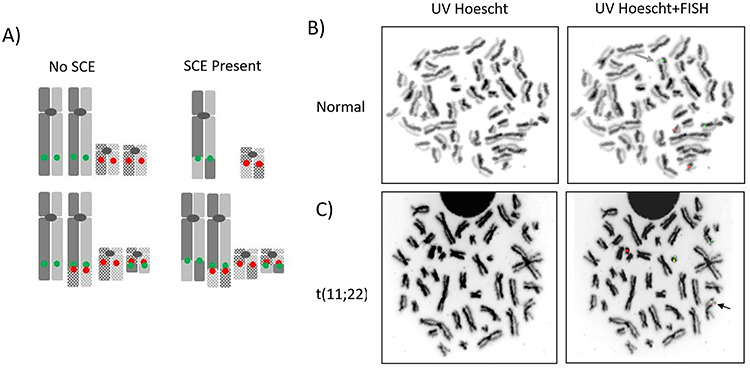
Detection of SCEs at PATRR sites in lymphocytes using UV-Hoechst exposure coupled with FISH probes. (A) Model depicting an SCE event at the PATRR regions on chromosomes 11 (solid) and 22 (stippled) on both normal and t(11;22) chromosomes with FISH probes for PATRR 11 (green) and PATRR22 (red). (B) Representative metaphase spreads derived from normal peripheral blood lymphocytes visualized by UV-Hoescht plus Giemsa and UV-Hoescht plus FISH probes for PATRR 8 (green) and PATRR 22 (red). The green arrow marks an SCE event at PATRR8. (C) Representative metaphase spreads derived from lymphocytes of a t(11;22) balanced translocation carrier. Images are visualized either by UV-Hoescht plus Giemsa or by UV-Hoescht plus FISH probes for PATRR11 (green) and PATRR22 (red). The black arrows identify an SCE event at PATRR11 on the der11 chromosome. In (B) and (C), black and white images of DAPI counterstained chromosomes enable visualization of SCEs with greater contrast.
Table 1.
Percentage of total SCEs in lymphoblastoid cells
| Sample | 11q23 SCE’s total no/(%) | 8q24 SCE’s total no/(%) | 6q26 SCE’s total no/(%) | Total SCE no (all chr.) | Mean SCE (±SD) | Total metaphase |
|---|---|---|---|---|---|---|
| Normal PB—A | 65/(2.2%) | 71/(2.5%) | 44/(1.5%) | 2970 | 5.5 ± 0.74 | 540 |
| Normal PB—B | 62/(2.12%) | 63/(2.2%) | 35/(1.2%) | 2912 | 5.3 ± 0.48 | 540 |
| Normal LCL | 274/(4%) | – | 95/(1.4%) | 6864 | 6.0 ± 0.60 | 1144 |
| t(11;22) LCL—A | 22/(0.6%) | – | 39/(1.1%) | 3460 | 5.5 ± 0.46 | 629 |
| t(11;22) LCL—B | 28/(0.85%) | – | 65/(2.0%) | 3258 | 6.8 ± 0.75 | 479 |
Peripheral blood (PB) lymphocytes and lymphoblastoid cell lines (LCL) were used to assay SCEs at PATRR locations11q23 and 8q24 as well as at control region 6q26. The letters A and B represent separate replicate experiments. For the t(11;22) experiments, the replicates were two different female balanced translocation carriers. Percentage of SCEs was determined by dividing the site-specific SCEs by the total number of SCEs for each sample.
To further confirm the PATRR-SCE association, we next looked at two replicates of lymphoblastoid cell lines containing a balanced t(11;22) to assess if the disruption of one copy of the PATRR at 11q23 would result in a decrease in SCE frequency. These cells had a comparable total number of SCEs compared to normal cells, with 5.5 ± 0.46 SCE/cell (sample A) and 6.8 ± 0.75 SCE/cell (sample B). Interestingly, not only did the frequency decrease in the t(11;22) cells, but it actually dropped below the levels of the control region on 6q26. The frequency of SCEs at 11q23 (0.6–0.85%) is significantly lower than that of 6q26 (1.1–2.0%) (Table 1), with P-values for sample A and B of P = 0.036 and P = 0.0002 respectively (McNemar test of equality of proportions, two-tail). Also of interest was that the SCEs observed at 11q23 were approximately evenly distributed between the intact PATRR on the normal chromosome 11 and the der(11) chromosome (sample A had 11 each, and sample B had 13 der(11) versus 15 normal).
Increased DSBs at PATRR regions in mitotic cells
To confirm whether the increased rate of SCEs is indicative of an increased rate of breakage as well as to increase the specificity of the detection, chromatin immunoprecipitation (IP) assays were performed using peripheral blood lymphocytes with antibodies specific to DSB related proteins: γH2AX, Rad51 and NBS1. The DNA was collected and tested for an increase of PATRR-DNA sequences using quantitative polymerase chain reaction (qPCR) with primers located near the PATRR regions (Supplementary Material, Fig. S8). Levels of PATRR-DNA located on chromosomes 8, 11, 17 and 22 were compared to the same background region on 6q26 as was used in the SCE experiments. Initial results showed an increased association to PATRR-DNA with each DSB-associated antibody tested, with the range for individual PATRRs between 1.03 and 1.58 fold increase over the control region. When combining the data from the four PATRR regions, they demonstrate a significant fold increase of 1.20 (γH2AX), 1.15 (Rad51) and 1.21 (NBS1) with P-values of 0.0002, 0.0005 and 0.0086, respectively, using the one-sample t-test (Table 2 and Supplementary Material, Fig. S1).
Table 2.
Fold increase data of PATRR DNA compared to background 6q26 in ChIP assays using DSB-associated antibodies γH2AX, Rad52 and NBS1 in peripheral blood lymphocytes
| γH2AX | Rad51 | NBS1 | |
|---|---|---|---|
| PATRR17 | 1.58 | 1.41 | 1.36 |
| PATRR8 | 1.20 | 1.03 | 1.33 |
| PATRR11 | 1.10 | 1.07 | 1.05 |
| PATRR22 | 1.25 | 1.16 | 1.21 |
| PATRR median | 1.20 | 1.15 | 1.21 |
| PATRR mean | 1.51 | 1.16 | 1.24 |
| P-value | 0.0002 | 0.0005 | 0.0086 |
Median values are reported for individual PATRRs. Data from all four PATRRs are combined to generate the total PATRR median and mean values for a given antibody. P-values are calculated for the total PATRR value for each antibody using the one-sample t-test comparing the total PATRR to the mean value of 1 using GraphPad Software at www.graphpad.com.
This result used only one background region on chromosome 6q26, which prompted the careful examination of this region and led to the question of whether the same trend would be observed for other background regions. The background region of 6q26 contains multiple elements such as DNAse I hypersensitivity regions and repeat elements that could lead to a higher than average susceptibility to DSB’s (46–49). In particular, the presence of a 5 kb LINE element (Supplementary Material, Fig. S2) in the region could potentially cause above-average levels of γH2AX on the background chromosome. To get a more accurate representation of background levels across the genome, the qPCR experiments were repeated using the same ChIP assays comparing the PATRR-DNA to multiple other backgrounds including 9q34 and 10q26.1. Both of these additional backgrounds are approximately the same distance from the long arm telomere as PATRR-11 is from the telomere of 11q. The presence of repeat elements in the alternative backgrounds is also shown (Supplementary Material, Fig. S2) with each background having a different density and distribution of repeat patterns in order to establish whether PATRRs are enriched in DSBs independent of these backgrounds. The majority of the ChIP assays reveal an increased association of DSB markers at the PATRR regions regardless of the background chromosomal region to which it is compared. When compared to 9q34, all four PATRR regions had individual fold increase levels ranging from 1.17 to 1.65 over the background with the combined PATRR values having significant fold increases of 1.26 (γH2AX), 1.39 (Rad51) and 1.48 (NBS1) with P-values of 0.0016, 0.0001 and 0.0001, respectively. When compared to the 10q26.1 background, the same trend was observed with all but one of the individual combinations assayed indicating an increased propensity for binding DSB proteins and total PATRR values demonstrating significant fold increases of 1.08 (γH2AX), 1.37 (Rad51) and 1.17 (NBS1) with P-values of 0.011, 0.0001 and 0.0002, respectively (Table 3 and Supplementary Material, Fig. S3).
Table 3.
Fold increase data of PATRR DNA compared to backgrounds 9q34 and 10q26.1 for lymphoblastoid cells
| 9q34 | 10q26.1 | |||||
|---|---|---|---|---|---|---|
| γH2AX | Rad51 | NBS1 | γH2AX | Rad51 | NBS1 | |
| PATRR17 | 1.29 | 1.63 | 1.56 | 1.30 | 1.36 | 1.39 |
| PATRR8 | 1.17 | 1.25 | 1.35 | 0.96 | 1.37 | 1.17 |
| PATRR11 | 1.19 | 1.40 | 1.52 | 1.30 | 1.12 | 1.05 |
| PATRR22 | 1.51 | 1.55 | 1.65 | 1.21 | 1.52 | 1.34 |
| PATRR Median | 1.26 | 1.39 | 1.48 | 1.08 | 1.37 | 1.17 |
| PATRR Mean | 1.42 | 1.68 | 1.52 | 1.29 | 1.55 | 1.21 |
| P-value | 0.0016 | 0.0001 | 0.0001 | 0.011 | 0.0001 | 0.0002 |
Data from ChIP assays using DSB-associated antibodies γH2AX, Rad51 and NBS1. Median values are reported for individual PATRRs. Data from all four PATRRs are combined to generate the total PATRR median and mean values for a given antibody. P-values are calculated for the total PATRR value for each antibody using the one-sample t-test comparing the total PATRR to the mean value of 1 using GraphPad Software at www.graphpad.com.
As was done for the SCE experiments, the experiment was repeated using a t(11;22) lymphoblastoid cell line to determine if the disruption of the PATRR sequence in one chromosome would reduce the overall propensity for DSBs at the PATRR regions on chromosomes 11 and 22. The median fold increase data for the t(11;22) samples ranges from 0.78 to 1.23 for all of the PATRR-antibody-background combinations tested (Supplementary Material, Table S1 and Supplementary Material, Fig. S4). This range was lower than that of PATRR 11 and 22 for normal lymphoblasts, 1.05–1.65 (Tables 2 and 3), and about a third of the t(11;22) samples had median fold increase values lower than that of background. Upon normalization of the backgrounds, the combined values from PATRR 11 and 22 had significantly lower mean fold increases in the t(11;22) samples than the normal lymphoblastoid cells for all three antibodies, (P=0.0001; Table 4). As a control, several qPCR plates were set up with PATRR8 primers to test if PATRR8 sites (with 2 intact PATRR regions) maintained an increased fold value in the t(11;22) lines even when PATRR11 and PATRR22 (with only 1 intact PATRR) were decreased. Indeed this was shown to be the case for the majority of the samples, with representatives results shown in Supplementary Material, Fig. S4.
Table 4.
Mean fold increase comparison between t(11;22) and normal lymphoblastoid cells
| Combined values | γH2AX | Rad51 | NBS1 |
|---|---|---|---|
| t(11;22) | 1.12 | 1.12 | 0.99 |
| Normal DNA | 1.82 | 1.69 | 1.45 |
| P-value | 0.0001 | 0.0001 | 0.0001 |
Combined values were generated by first normalizing all three backgrounds for both PATRR 11 and 22 and then generating the mean value shown in the table. P-values were generated using the normalized data for t(11;22) DNA and normal DNA in an unpaired t-test to compare two means at www.graphpad.com.
Increased DSB in meiotic cells
Since the recurrent PATRR-mediated translocations have only been observed in meiotic or germ cells, the question of whether there would be an even greater association of the DSB markers with PATRR regions in a meiotic cellular environment compared to what was observed in the mitotic lymphoblastoid cells was asked. The initial approach involved digesting testicular tissue and performing the same ChIP- qPCR assay as used for the lymphoblastoid cells. The data from the testis samples showed an increase in DSB markers associated with PATRR regions compared to the 6q26 background for all combinations tested, with a range from 1.04 to 1.79 fold increase for individual PATRRs. When combining all PATRR data for a given antibody there remains a significant increase in association above background with 1.49 fold for γH2AX, 1.62 for Rad51 and 1.18 for NBS1 with P-values of 0.0036, 0.0055 and 0.0037, respectively (Supplementary Material, Table S2). When comparing the median fold increase of testis to normal lymphoblastoid results, only one marker (Rad51) showed a consistent increase in meiotic cells compared to mitotic cells across all the PATRR regions, while the other two markers (γH2AX and NBS1) had similar results between the two cell types (Supplementary Material, Fig. S5).
There were concerns that during digestion of testicular tissue a mixed population of cells, not all of which would have the ability to undergo meiosis, would be produced. In order to have a germline cell population with a greater enrichment in meiotic cells, purified spermatogenic cells were used, specifically WA01 (H1) human embryonic stem cells (hESCs) that had been differentiated into spermatogenic cells as previously described (50). These cells showed a significant median fold increase for all antibodies and PATRR combinations when compared to the 6q26 background as well as for the majority of the 9q34 and 10q26.1 background combinations. Median values for all PATRRs within a particular antibody-background combination ranged from 1.04 to 1.26 fold increase with P-values from 0.0001 to 0.0283 (Table 5). Box plots showing the full range of data used to calculate the median are shown in Figure 2 (6q26 background) and Supplementary Material, Figure S6 (9q34 and 10q26.1 backgrounds) with the majority of the replicates demonstrating an increased association of DSB-associated markers at PATRR regions. While an increase of DSBs at PATRR regions over background is indicated by this data, there is no striking difference between the degree of increase for the spermatogenic cells derived in vitro and the normal lymphoblastoid samples.
Table 5.
Fold increase of PATRR DNA compared to backgrounds 6q26, 9q34 and 10q26.1 for the hESC-derived spermatogenic cells
| 6q26 | 9q34 | 10q26.1 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| γH2AX | Rad51 | NBS1 | γH2AX | Rad51 | NBS1 | γH2AX | Rad51 | NBS1 | |
| PATRR17 | 1.30 | 1.26 | 1.36 | 1.29 | 1.13 | 1.20 | 1.27 | 1.06 | 1.04 |
| PATRR8 | 1.06 | 1.06 | 1.21 | 1.01 | 0.98 | 0.95 | 1.07 | 1.07 | 0.87 |
| PATRR11 | 1.45 | 1.16 | 1.21 | 1.37 | 1.06 | 1.07 | 1.32 | 1.07 | 1.18 |
| PATRR22 | 1.46 | 1.39 | 1.19 | 1.69 | 1.50 | 1.18 | 1.43 | 1.54 | 1.97 |
| PATRR Median | 1.23 | 1.18 | 1.22 | 1.22 | 1.08 | 1.07 | 1.26 | 1.15 | 1.04 |
| PATRR Mean | 1.43 | 1.25 | 1.38 | 1.40 | 1.25 | 1.20 | 1.44 | 1.32 | 1.18 |
| P-value | 0.0011 | 0.0005 | 0.0039 | 0.0001 | 0.0172 | 0.0044 | 0.0007 | 0.0287 | 0.0283 |
Data from ChIP assays using DSB-associated antibodies γH2AX, Rad51 and NBS1. Median values are reported for individual PATRRs. Data from all four PATRRs are combined to generate the total PATRR median and mean values for a given antibody. P-values are calculated for the total PATRR value for each antibody using the one-sample t-test comparing the total PATRR to the mean value of 1 using GraphPad Software at www.graphpad.com.
Figure 2.
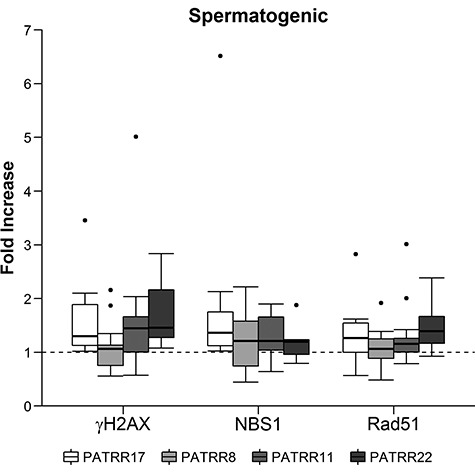
Median fold increase box plots for hESC-derived spermatogenic cells over the background 6q26. Data are from multiple ChIP assays using DSB-associated antibodies for γH2AX, Rad51 and NBS1 and testing for their association at PATRR regions on chromosomes 17, 8, 11 and 22.
Detection of cruciform structure at PATRR regions
To examine if the PATRR regions in both mitotic and meiotic cells had an increased tendency to form cruciform structures compared to the background regions, the same ChIP-qPCR assay was performed using the cruciform binding antibody 2D3. Combinations of PATRR/background were tested for both the normal lymphoblastoid and in vitro-derived spermatogenic cells, and 22 of the 24 individual combinations showed a significant fold increase of the 2D3 at PATRR regions. Lymphoblastoid sample medians ranged from 0.90 to 1.94 fold increase and spermatogenic cell median’s ranging from 0.98 to 1.38. Even more striking was the total PATRR mean fold increase values, combining the data for all the PATRRs within each background and cell type; all values were found to be highly significant over the background with P-values ranging from 0.0001 to 0.0171 (Table 6). Box plots showing the full range of the data from multiple ChIP experiments are shown in Figure 3 (6q26 background) and Supplementary Material, Figure S7 (9q34 and 10q26.1 backgrounds). While both lymphoblastoid (mitotic) and spermatogenic (meiotic) cells have an increased association of 2D3 at the PATRR regions, there is no significant difference in the degree of the increase between the two cell types.
Table 6.
Fold increase of PATRR DNA compared to backgrounds 6q26, 9q34 and 10q26.1 for normal lymphoblastoid cells (lymph) and hESC-derived spermatogenic cells (sperm)
| 6q26 | 9q34 | 10q26.1 | ||||
|---|---|---|---|---|---|---|
| Lymph | Sperm | Lymph | Sperm | Lymph | Sperm | |
| PATRR17 | 1.68 | 1.31 | 1.70 | 1.30 | 1.36 | 1.17 |
| PATRR8 | 1.30 | 1.07 | 1.50 | 0.98 | 1.03 | 1.14 |
| PATRR11 | 1.12 | 1.17 | 1.23 | 1.15 | 0.90 | 1.14 |
| PATRR22 | 1.35 | 1.38 | 1.94 | 1.26 | 1.30 | 1.36 |
| PATRR median | 1.29 | 1.17 | 1.54 | 1.16 | 1.10 | 1.14 |
| PATRR Mean | 1.29 | 1.37 | 1.49 | 1.17 | 1.12 | 1.23 |
| P-value | 0.0001 | 0.0001 | 0.0001 | 0.0004 | 0.0171 | 0.0001 |
Data from ChIP assays using the 2D3 antibody. Median values are reported for individual PATRRs. Data from all four PATRRs are combined to generate the total PATRR median and mean values for a given antibody. P-values are calculated for the total PATRR value for each antibody using the one-sample t-test comparing the total PATRR to the mean value of 1 using GraphPad Software at www.graphpad.com.
Figure 3.
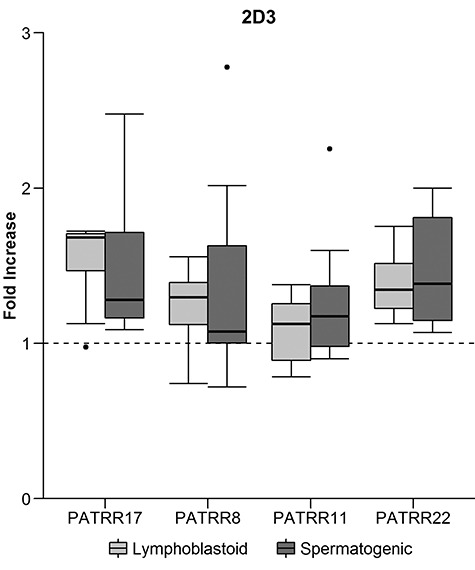
Median fold increase box plots for normal lymphoblastoid and hESC-derived spermatogenic cells over the 6q26 background. Data are from multiple ChIP assays using the cruciform binding antibody 2D3.
Discussion
Previous studies have shown that the breakpoints of the recurrent t(11;22)(q23;q11.2), t(8;22)(q24;q11.2) and t(17;22)(q11;q11.2) constitutional translocations fall within the center of the palindromic sequences between two AT-rich inverted repeats. It has been proposed that their unique organization makes them more susceptible to DSBs with evidence suggesting that the PATRR sequences extrude cruciform DNA (6,15,19,20,22,51–53). While the actual translocation is thought to occur along the meiosis-gametogenesis pathway, likely involving non-homologous end joining (NHEJ) (5,52,54), it is possible that a propensity for DSBs in these regions might predispose them to breakage in a mitotic environment as well, which could be resolved through homologous repair and sister chromatid exchanges (SCEs) (55,56). The SCE results presented here support the idea that DNA with the ability to form cruciform structures is more susceptible to SCEs and that the distribution of SCEs on chromosomes is not random. If SCEs occurred at random, a similar number would be expected at all three 8q24, 11q23 and 6q26 chromosomal locations based on their position with respect to the telomere, whereas an increase at the PATRR locations on chromosomes 8 and 11 was observed. This observation is further supported by the ChIP data which has demonstrated an increase in DSB-associated markers and proteins at the PATRR regions in mitotic cells. The results in this paper provide evidence that a susceptibility to DSBs at PATRR regions exists in both mitotic and meiotic cells. However, we know from previous studies that the translocation itself does not occur in mitotic cells (52,54,57). This may indicate that the PATRR sequence and resulting conformation contributes to the initial DSB but that other cellular factors specific to the meiotic—gametogenesis pathway are required for the translocations to take place.
The evidence for the increased occurrence of DSBs at the PATRR regions is strengthened by the fact that this association is reduced in the t(11;22) lymphoblastoid samples for both SCE and ChIP experiments where there is only 1 intact PATRR at 22q11 and 11q23. It is interesting to note that even with one intact PATRR at each site, these cells demonstrated a decrease in site-specific DSBs that was greater than expected, with levels dropping below that of background at times. This was the case for both SCE replicates as well as for 6 of the 18 comparisons in the ChIP experiments (Tables 1, Supplementary Material, Table S1). This could reflect either a lack of sensitivity in these experiments or it may suggest that the removal of some of the PATRR sequences is enough to decrease the underlying genomic instability at these regions.
It is known from previous work that there is a correlation between PATRR length and translocation frequency with longer PATRRs more frequently involved in balanced translocations. The t(11;22), which involves the two longest PATRRs, is the most frequent of the PATRR-mediated translocations. The plots reported here display the PATRRs in order of the length of their most common variants, with PATRR17 (~200 bp), PATRR8 (300–350 bp), PATRR11 (450 bp) and PATRR22 (600 bp) (5,7,8,10,11,13,58). Although tempting to speculate that this association might translate to an increased rate of DSBs at the longer PATRRs, the results suggest that there is no correlation between PATRR length and DSB frequency. This may suggest that other factors that promote the subsequent steps of the translocation are more influenced by length than the rate of DSBs. Another possibility based on recent studies of fragile sites (59) is that the DSBs may be occurring at a higher frequency at the longer PATRRs but the binding of the repair proteins that we are using to detect DSBs could be impaired due to the structure at these regions. However, it should also be noted that for many of the experiments described here, PATRR8 and PATRR11 have slightly lower averages than PATRR17 and PATRR22 while at the same time PATRR8 and PATRR11 have primers that, due to sequence constraints, are located farther from the center of the PATRR and expected breakpoint, particularly in the pre-PATRR regions (Supplementary Material, Fig. S8). This difference in distance from the PATRR combined with slight batch differences in shearing fragment length may partially disguise any length association with DSB frequency.
Previous work has shown that the PATRR sequences have the ability to form cruciform when PATRR sequences are inserted into plasmids (19–21,60). With the use of the 2D3 antibody, that work has been expanded here to test 2D3’s association at PATRR regions in both mitotic and meiotic cells. Indeed, the ChIP results reported in Figures 3 and Supplementary Material, Figure S7 as well as Table 6 indicate that 2D3 binds to PATRR regions at a rate above that of background levels. This would suggest that in both mitotic and meiotic environments the PATRR regions have at least some ability to form cruciform structures. The cruciform structure results correlate with the increased DSB frequency results in the ChIP experiments. Collectively this supports the hypothesis that the underlying secondary structure of the PATRR regions renders them more susceptible to DSBs. The fact that relatively similar levels of both DSBs and cruciform structures are detected in populations that are solely mitotic compared to populations that are enriched in meiotic cells suggests that additional keys to understanding PATRR-mediated translocations might need to focus on steps and factors that take place after the DSB is generated.
Materials and Methods
Cell lines and testicular tissue
Lymphoblastoid cells lines were transformed by Epstein–Barr virus and established from peripheral blood samples of either a normal adult (CEPH) or an adult with a balanced t(11;22). The cell lines were cultured at 37°C, 5% CO2, in RPMI-1640 media supplemented with 15% fetal bovine serum, 100 u/ml penicillin, 100 μg/ml streptomycin and 1 mM L-glutamine.
Testicular tissue was acquired through the human cooperative tissue network (https://chtn.org) with IRB approval from normal white males. The testicular samples used in this paper were shipped fresh in RPMI media on ice and were between 1.00–4.65 g from donors ranging from 58–79 years old. Immediately upon receipt samples were processed through mechanical and enzymatic digestion to form a single cell suspension. The sample was placed in HBBS with magnesium and calcium media (approximately 5 ml media per gram of tissue) plus the addition of Pen/Strep (Gibco #15140–122) to a final concentration of 100 units/ml, collagenase F at a final concentration of 0.1 units/ml, and Dispase II to 1.88 mg/mL. The testis samples were gently teased apart and filtered through a 40 μm Nylon cell strainer to achieve a single cell suspension. The suspension was centrifuged at 2000 rcf for 10 min to pellet the cells that were then washed twice in PBS and finally resuspended in PBS (about 1 ml per gram of testis) and 1x protease inhibitor added.
In vitro-derived spermatogenic cells
In vitro-derived spermatocytes were obtained by differentiating NIH-registry approved H1 (WA01) hESCs into spermatogenic lineages as previously described (50). Briefly, H1 hESCs were cultured for 10 days in mouse spermatogonial stem cell (SSC) medium containing the following: MEMalpha, 0.2% Bovine Serum Albumin, 5 μg/ml insulin, 10 μg/ml transferrin, 60 μM putrescine, 2 mM L-glutamine, 50 μM β-mercaptoethanol, 1 ng/ml hbFGF (human basic fibroblast growth factor), 20 ng/ml GDNF (glial-derived neurotrophic factor), 30 nM sodium selenite, 2.36 μM palmitic acid, 0.21 μM palmitoleic acid, 0.88 μM stearic acid, 1.02 μM oleic acid, 2.71 μM linoleic acid, 0.43 μM linolenic acid, 10 mM HEPES and 0.5X penicillin/streptomycin. After 10 days, all spermatogenic cells were collected and cell pellets including undifferentiated and differentiating spermatogonia, primary and secondary spermatocytes, and haploid spermatids were used for ChIP experiments.
SCE frequency by FISH
Lymphoblastoid cell lines were incubated with 20 μM 5-Bromo-2-Deoxyuridine (BrdU) for 2 days followed by treatment with 10 μg/ml colcemid for 30 min. Cells were then harvested, exposed to hypotonic treatment (0.04 mM KCl, 20 mM HEPES, 0.5 mM EDTA), and fixed in 3:1 methanol/acetic acid using standard procedures (61). The slides with metaphase spreads were aged by air drying at room temperature for 2 days. Visualization and determination of SCE frequency used a fluorescence plus Giemsa approach as previously described (62–65), where aged slides were immersed in 2.0 μg/ml Hoechst solution and exposed to a UV light source (G.E. Black Light F15T8.PL) for 4 h. Slides were then placed in 0.5 × SSC for 1 h followed by staining in 5% Giemsa for 5 min. Slides were prepared in triplicate, with only cells that underwent two rounds of BrdU, indicated by the characteristic bifiliary chromatid pattern, selected for analysis.
In order to identify SCEs at the PATRR breakpoints, the Hoechst-UV-exposed slides were hybridized with site-specific FISH probes followed by a DAPI counterstain. For the 22q11 breakpoint two cosmid probes were used which flanked the region (68a1 and c87f9 from the LL22NCO3 cosmid library), while other breakpoints were spanned by BAC probes: RP11-158 k1 for 8q24 breakpoint and RP11-442e11 for the 11q23 breakpoint (2,6,66). The control of 6q26 used the BAC probe RP11-480a20 and was selected due to its location that is roughly the same distance from the telomere as 11q23 is from the 11q telomere (51). BAC probes were selected from the UCSC database (http://genome.ucsc.edu/) on the basis of their location and were purchased from BACPAC resources (http://bacpac.chori.org/). Labeling of 1 μg of each of the five non-commercial probes was carried out following standard nick-translation procedures with either biotin-16-dUTP or digoxigenin-11-dUTP (Roche, USA) (67). The slides, after exposure to the 4 h UV light source, underwent a serial ethanol dehydration [70, 90, 100% (v/v)] and co-hybridized at 74°C for 3 min with biotin-and digoxigenin-labeled probes. After an overnight incubation at 37°C, slides were washed in SSC (1 × SSC for 3 × 5 min and 0.5 × SSC for 2 × 5min), treated with blocking solution (3%BSA and 4 × SSC) for 30 min and incubated with detection solution (FITC-Avidin antibody (biotin-probe) or Cy3-conjugated IgG Anti-digoxin (digoxigenin-probe) (Vectashield, Vector Laboratories, USA) in 1% BSA and 4x SSC). The slides were embedded with Vectashield mounting medium containing DAPI counterstain, and visualization of the hybridization signals and differential SCE pattern was accomplished using a Zeiss Fluorescence microscope equipped with a CCD camera, Chroma Technology filters and Applied Imaging software analysis.
qPCR-ChIP
ChIP experiments were performed essentially as described in (68) with a few modifications. Crosslinking reactions were performed in 10 mL PBS using 1 × 107 cells. ChIP-IT® Fixation Buffer (Active Motif #53038) was used to prepare Complete Fixation Solution according to manufacturer’s directions (for 10 ml media, combine 785 μl water, 90 μl ChIP-IT® Fixation Buffer, and 375 μl 37% formaldehyde) and 1.25 ml was added to the crosslinking reaction. The reaction gently rocked at room temperature for 15 min, quenched with glycine to a final concentration of 125 mM and incubated on ice for 2 min. Cells were pelleted by centrifugation (5 min, 1000 rpm) and washed twice in chilled PBS. Cell pellets were resuspended in 800 μl ChIP Wash Buffer (0.25% Triton X-100, 50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM EDTA, 10 mM sodium butyrate, 1x protease inhibitor) followed by centrifugation at 2000 rcf for 5 min. Cells were then resuspended in 800 μl ChIP IP Buffer (150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.5% NP-40, 50 mM Tris, pH 7.5, 0.25% deoxycholate, 1x protease inhibitor). Samples were again centrifuged at 2000 rcf for 5 min and dried cell pellets stored in −80° freezer until ready to shear.
Frozen crosslinked cell pellets were thawed and resuspended in 120 μl RIPA Cell Lysis Buffer (Amresco #N653-100 mL) plus 1x protease inhibitor. Chromatin shearing was performed using a Covaris® S220 Focused Ultrasonicator at the following conditions: frequency sweeping mode, 5% duty cycle, intensity 3, peak incident power of 105 watts, 200 cycles per burst, 55 s treatment time, bath temperature 4–5°C, continuous degassing mode, in 120 μl volume snap cap tube (Covaris #520045). Samples are then centrifuged at 10000 rcf for 5 min and soluble sheared material collected for further analysis. To test quality of shearing, 15 μl of soluble sheared is run on agarose gel after treatment with 0.5 mg/ml RNase A and 0.7 mg/ml Protinase K at 65°C for 2 h.
IPs were set up using 36 μl of soluble sheared material plus 3 μg antibody, adjusted to 600 μl total volume with RIPA buffer. The following antibodies were used: 2D3 (Abcam, ab150343), γH2AX (Millipore, #05-636), NBS1 (Thermo Scientific, MA1-23263), Rad51 (Novus Biologicals, NB100-148), and normal mouse IgG (Santa Cruz, sc-2025). Samples were rocked overnight at 4°C. The following day, 35 μl Salmon Sperm DNA/Protein A Agarose beads (Millipore, #16-157) and rocked at 4°C for 4 h. The samples were then washed once with 1 ml RIPA buffer and twice with TE, each time rocking samples for 5 min at room temperature and centrifuged at 1000 g for 5 min. Samples were harvested by adding 125 μl 10% Chelex bead slurry, incubating at 95°C for 10 min, treatment with RNase and Protinase K, and centrifugation at 1000 g for 5 min. The supernatant was collected, quantified by nanodrop and analyzed byqPCR.
The qPCR was analyzed on Applied Biosystems 7500 real-time PCR system, using the fast setting for 96 wells, comparative CT for the quantitation setting and SYBR Green for the reagents setting. Run conditions were as follows: 20 s hold at 95°C, 40 cycles of 3 s at 95°C plus 30 s at 60°C, followed by the melt curve program. Each reaction was run in triplicate on a 96-well plate and contained 10 μl of reaction mixture with final concentrations of 1X Fast SYBR Green Master Mix (Applied Biosystems, #4385612), 0.2 μM primers, and 12.5 ng DNA. The qPCR analysis used multiple primers located immediately adjacent to the PATRR regions (Supplementary Material, Fig. S8) as well as control primers on chromosomes 6, 9, 10, and 12 that were approximately the same distance from the telomere as PATRR11 is from the chromosome 11 telomere. Primer efficiency (E) was calculated for each primer pair using the above reaction conditions but with a gradient of eight DNA concentrations: 100, 50, 25, 12.5, 6.25, 3.12 and 1.56 ng. The CT data were exported to excel where the slope was calculated using the following excel function: =slope(CT, log10). The E value was then calculated using the slope: E =10(−1/slope). E values along with primer sequences are reported in Supplementary Material, Table S3.
Negative controls (no DNA) and normal mouse IgG IPs were used as controls against contamination and to ensure target antibody IPs worked. For successful IPs, the input and the target antibody IP samples were used in subsequent fold increase analysis. The averaged triplicate CT values were used to first calculate percent input, for both PATRR-associated primers and the background primers, using the primer E value in this equation: % input = 100 × E(CTinput-CTsample). The fold increase was then determined dividing the percent input for the PATRR primers by the percent input for the background primers.
Supplementary Material
Acknowledgments
The authors thank Daphne Koumbi for her work on the SCE protocol and Marcella Devoto for assistance with the SCE statistics. We also thank Lindsay Horvath for her assistance with the ChIP protocol and Sabina Spigner and Myrna Yousuf for their experimental assistance. We are grateful to the NAPcore center at CHOP for their help and for providing access to the Covaris shearing equipment. This work was supported by National Cancer Institute CA39926 and the Charles E.H. Upham chair in pediatrics (B.S.E) and NIH K22ES025418 (C.A.E).
Conflict of Interest statement. None declared.
References
- 1. Zackai, E.H. and Emanuel, B.S. (1980) Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am. J. Med. Genet., 7, 507–521. [DOI] [PubMed] [Google Scholar]
- 2. Shaikh, T.H., Budarf, M.L., Celle, L., Zackai, E.H. and Emanuel, B.S. (1999) Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am. J. Hum. Genet., 65, 1595–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carter, M.T., St Pierre, S.A., Zackai, E.H., Emanuel, B.S. and Boycott, K.M. (2009) Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): clinical features of 63 individuals. Am. J. Med. Genet. A, 149A, 1712–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fraccaro, M., Lindsten, J., Ford, C.E. and Iselius, L. (1980) The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum. Genet., 56, 21–51. [DOI] [PubMed] [Google Scholar]
- 5. Kurahashi, H., Shaikh, T., Takata, M., Toda, T. and Emanuel, B.S. (2003) The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am. J. Hum. Genet., 72, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gotter, A.L., Nimmakayalu, M.A., Jalali, G.R., Hacker, A.M., Vorstman, J., Conforto Duffy, D., Medne, L. and Emanuel, B.S. (2007) A palindrome-driven complex rearrangement of 22q11.2 and 8q24.1 elucidated using novel technologies. Genome Res., 17, 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Inagaki, H., Ohye, T., Kogo, H., Yamada, K., Kowa, H., Shaikh, T.H., Emanuel, B.S. and Kurahashi, H. (2005) Palindromic AT-rich repeat in the NF1 gene is hypervariable in humans and evolutionarily conserved in primates. Hum. Mutat., 26, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mishra, D., Kato, T., Inagaki, H., Kosho, T., Wakui, K., Kido, Y., Sakazume, S., Taniquchi-Ikeda, M., Morisada, N., Iijima, K. et al. (2014) Breakpoint analysis of the recurrent constitutional t(8;22)(q24.13;q11.21) translocation. Mol. Cytogenet., 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kurahashi, H. and Emanuel, B.S. (2001) Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum. Mol. Genet., 10, 2605–2617. [DOI] [PubMed] [Google Scholar]
- 10. Kato, T., Inagaki, H., Yamada, K., Kogo, H., Ohye, T., Kowa, H., Nagaoka, K., Taniguchi, M., Emanuel, B.S. and Kurahashi, H. (2006) Genetic variation affects de novo translocation frequency. Science, 311, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tong, M., Kato, T., Yamada, K., Inagaki, H., Kogo, H., Ohye, T., Tsutsumi, M., Wang, J., Emanuel, B.S. and Kurahashi, H. (2010) Polymorphisms of the 22q11.2 breakpoint region influence the frequency of de novo constitutional t(11;22)s in sperm. Hum. Mol. Genet., 19, 2630–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurahashi, H. and Emanuel, B.S. (2001) Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat. Genet., 29, 139–140. [DOI] [PubMed] [Google Scholar]
- 13. Sheridan, M.B., Kato, T., Haldeman-Englert, C., Jalali, G.R., Milunsky, J.M., Zou, Y., Klaes, R., Gimelli, G., Gimelli, S., Gemmill, R.M. et al. (2010) A palindrome-mediated recurrent translocation with 3:1 meiotic nondisjunction: the t(8;22)(q24.13;q11.21). Am. J. Hum. Genet., 87, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Correll-Tash, S., Conlin, L., Mininger, B.A., Lilley, B., Mennuti, M.T. and Emanuel, B.S. (2018) The recurrent t(11;22)(q23;q11.2) can occur as a post-zygotic event. Cytogenet. Genome Res., 156, 185–190. [DOI] [PubMed] [Google Scholar]
- 15. Edelmann, L., Spiteri, E., Koren, K., Pulijaal, V., Bialer, M.G., Shanske, A., Goldberg, R. and Morrow, B.E. (2001) AT-rich palindromes mediate the constitutional t(11;22) translocation. Am. J. Hum. Genet., 68, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurahashi, H., Shaikh, T.H., Zackai, E.H., Celle, L., Driscoll, D.A., Budarf, M.L. and Emanuel, B.S. (2000) Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22). Am. J. Hum. Genet., 67, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kurahashi, H., Shaikh, T.H. and Emanuel, B.S. (2000) Alu-mediated PCR artifacts and the constitutional t(11;22) breakpoint. Hum. Mol. Genet., 9, 2727–2732. [DOI] [PubMed] [Google Scholar]
- 18. Kurahashi, H., Shaikh, T.H., Hu, P., Roe, B.A., Emanuel, B.S. and Budarf, M.L. (2000) Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22). Hum. Mol. Genet., 9, 1665–1670. [DOI] [PubMed] [Google Scholar]
- 19. Kurahashi, H., Inagaki, H., Yamada, K., Ohye, T., Taniguchi, M., Emanuel, B.S. and Toda, T. (2004) Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocations. J. Biol. Chem., 279, 35377–35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kogo, H., Inagaki, H., Kato, T., Emanuel, B.S. and Kurahashi, H. (2007) Cruciform extrusion propensity of human translocation-mediating palindromic AT-rich repeats. Nucleic Acids Res., 35, 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inagaki, H., Ohye, T., Kogo, H., Kato, T., Bolor, H., Taniguchi, M., Shaikh, T.H., Emanuel, B.S. and Kurahashi, H. (2009) Chromosomal instability mediated by non-B DNA: cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res., 19, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gotter, A.L., Shaikh, T.H., Budarf, M.L., Rhodes, C.H. and Emanuel, B.S. (2004) A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum. Mol. Genet., 13, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carrano, A.V., Thompson, L.H., Lindl, P.A. and Minkler, J.L. (1978) Sister chromatid exchange as an indicator of mutagenesis. Nature, 271, 551–553. [DOI] [PubMed] [Google Scholar]
- 24. Hedner, K., Hӧgstedt, B., Kolnig, A.M., Mark-Vendel, E., Strӧmbeck, B. and Mitelman, F. (1982) Relationship between sister chromatid exchanges and structural chromosome aberrations in lymphocytes of 100 individuals. Hereditas, 97, 237–245. [DOI] [PubMed] [Google Scholar]
- 25. Deen, D.F., Morgan, W.F., Tofilon, P.J. and Barcellos-Hoff, M.H. (1989) Measurement of sister chromatid exchanges and their relationship to DNA damage, repair and cell killing. Pharmacol. Ther., 42, 349–360. [DOI] [PubMed] [Google Scholar]
- 26. Dhillon, V.S., Kler, R.S. and Dhillon, I.K. (1996) Choromosome instabililty and sister chromatid exchange (SCE) studies in patients with carcinoma of cervix uteri. Cancer Genet. Cytogenet., 86, 54–57. [DOI] [PubMed] [Google Scholar]
- 27. Dominguez, I., Daza, P., Natarajan, A.T. and Cortés, F. (1998) A high yield of translocations parallels the high yield of sister chromatid exchanges in the CHO mutant EM9. Mutat. Res., 398, 67–73. [DOI] [PubMed] [Google Scholar]
- 28. Rogakou, E.P., Pilch, D.R., Orr, A.H., Ivanova, V.S. and Bonner, W.M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- 29. Stucki, M. and Jackson, S.P. (2006) gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst), 5, 534–543. [DOI] [PubMed] [Google Scholar]
- 30. Meier, A., Fiegler, H., Muñoz, P., Ellis, P., Rigler, D., Langford, C., Blasco, M.A., Carter, N. and Jackson, S.P. (2007) Spreading of mammalian DNA-damage response factors studied by ChIP-chip at damaged telomeres. EMBO J., 26, 2707–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Savic, V., Yin, B., Maas, N.L., Bredemeyer, A.L., Carpenter, A.C., Helmink, B.A., Yang-Iott, K.S., Sleckman, B.P. and Bassing, C.H. (2009) Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell, 34, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahadevaiah, S.K., Turner, J.M., Baudat, F., Rogakou, E.P., de Boer, P., Blanco-Rodríguez, J., Jasin, M., Keeney, S., Bonner, W.M. and Burgoyne, P.S. (2001) Recombinational DNA double-strand breaks in mice precede synapsis. Nat. Genet., 27, 271–276. [DOI] [PubMed] [Google Scholar]
- 33. Iacovoni, J.S., Caron, P., Lassadi, I., Nicolas, E., Massip, L., Trouche, D. and Legube, G. (2010) High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J., 29, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Price, B.D. and D'Andrea, A.D. (2013) Chromatin remodeling at DNA double-strand breaks. Cell, 152, 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krejci, L., Altmannova, V., Spirek, M. and Zhao, X. (2012) Homologous recombination and its regulation. Nucleic Acids Res., 40, 5795–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheridan, S.D., Yu, X., Roth, R., Heuser, J.E., Sehorn, M.G., Sung, P., Egelman, E.H. and Bishop, D.K. (2008) A comparative analysis of Dmc1 and Rad51 nucleoprotein filaments. Nucleic Acids Res., 36, 4057–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan, Z.M., Huang, Y., Ishiko, T., Nakada, S., Utsugisawa, T., Kharbanda, S., Wang, R., Sung, P., Shinohara, A., Weichselbaum, R. and Kufe, D. (1998) Regulation of Rad51 function by c-Abl in response to DNA damage. J. Biol. Chem., 273, 3799–3802. [DOI] [PubMed] [Google Scholar]
- 38. Aymard, F., Bugler, B., Schmidt, C.K., Guillou, E., Caron, P., Briois, S., Iacovoni, J.S., Daburon, V., Miller, K.M., Jackson, S.P. and Legube, G. (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol., 21, 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berkovich, E., Monnat, R.J., Jr. and Kastan, M.B. (2007) Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol., 9, 683–690. [DOI] [PubMed] [Google Scholar]
- 40. Vago, R., Leva, V., Biamonti, G. and Montecucco, A. (2009) DNA ligase I and Nbs1 proteins associate in a complex and colocalize at replication factories. Cell Cycle, 8, 2600–2607. [DOI] [PubMed] [Google Scholar]
- 41. Beishline, K., Kelly, C.M., Olofsson, B.A., Koduri, S., Emrich, J., Greenberg, R.A. and Azizkhan-Clifford, J. (2012) Sp1 facilitates DNA double-strand break repair through a nontranscriptional mechanism. Mol. Cell. Biol., 32, 3790–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borde, V. (2007) The multiple roles of the Mre11 complex for meiotic recombination. Chromosom. Res., 15, 551–563. [DOI] [PubMed] [Google Scholar]
- 43. Frappier, L., Price, G.B., Martin, R.G. and Zannis-Hadjopoulos, M. (1989) Characterization of the binding specificity of two anticruciform DNA monoclonal antibodies. J. Biol. Chem., 264, 334–341. [PubMed] [Google Scholar]
- 44. Steinmetzer, K., Zannis-Hadjopoulos, M. and Price, G.B. (1995) Anti-cruciform monoclonal antibody and cruciform DNA interaction. J. Mol. Biol., 254, 29–37. [DOI] [PubMed] [Google Scholar]
- 45. Bell, D., Sabloff, M., Zannis-Hadjopoulos, M. and Price, G. (1991) Anti-cruciform DNA affinity purification of active mammalian origins of replication. Biochim. Biophys. Acta, 1089, 299–308. [DOI] [PubMed] [Google Scholar]
- 46. Daga, A., Ansari, A., Rawal, R. and Umrania, V. (2015) Characterization of chromosomal translocation breakpoint sequences in solid tumours: “an in silico analysis”. Open Med Inform J, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu, T.C. and Lichten, M. (1994) Meiosis-induced double-strand break sites determined by yeast chromatin structure. Science, 263, 515–518. [DOI] [PubMed] [Google Scholar]
- 48. Hedges, D.J. and Deininger, P.L. (2007) Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res., 616, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gasior, S.L., Wakeman, T.P., Xu, B. and Deininger, P.L. (2006) The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol., 357, 1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Easley, C.A., 4th., Phillips, B.T., McGuire, M.M., Barringer, J.M., Valli, H., Hermann, B.P., Simerly, C.R., Rajkovic, A., Miki, T., Orwig, K.E. and Schatten, G.P. (2012) Direct differentiation of human pluripotent stem cells into haploid spermatogenic cells. Cell Rep., 2, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ashley, T., Gaeth, A.P., Inagaki, H., Seftel, A., Cohen, M.M., Anderson, L.K., Kurahashi, H. and Emanuel, B.S. (2006) Meiotic recombination and spatial proximity in the etiology of the recurrent t(11;22). Am. J. Hum. Genet., 79, 524–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kurahashi, H., Inagaki, H., Ohye, T., Kogo, H., Kato, T. and Emanuel, B.S. (2006) Palindrome-mediated chromosomal translocations in humans. DNA Repair (Amst), 5, 1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kurahashi, H., Inagaki, H., Ohye, T., Kogo, H., Kato, T. and Emanuel, B.S. (2006) Chromosomal translocations mediated by palindromic DNA. Cell Cycle, 5, 1297–1303. [DOI] [PubMed] [Google Scholar]
- 54. Ohye, T., Inagaki, H., Kogo, H., Tsutsumi, M., Kato, T., Tong, M., Macville, M.V., Medne, L., Zackai, E.H., Emanuel, B.S. and Kurahashi, H. (2010) Paternal origin of the de novo constitutional t(11;22)(q23;q11). Eur. J. Hum. Genet., 18, 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilson, D.M., III and Thompson, L.H. (2007) Molecular mechanisms of sister-chromatid exchange. Mutat. Res., 616, 11–23. [DOI] [PubMed] [Google Scholar]
- 56. Helleday, T. (2003) Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res., 532, 103–115. [DOI] [PubMed] [Google Scholar]
- 57. Kato, T., Kurahashi, H. and Emanuel, B.S. (2012) Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev., 22, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kato, T., Inagaki, H., Kogo, H., Ohye, T., Yamada, K., Emanuel, B.S. and Kurahashi, H. (2008) Two different forms of palindrome resolution in the human genome: deletion or translocation. Hum. Mol. Genet., 17, 1184–1191. [DOI] [PubMed] [Google Scholar]
- 59. Kaushal, S., Wollmuth, C.E., Das, K., Hile, S.E., Regan, S.B., Barnes, R.P., Haouzi, A., Lee, S.M., House, N.C.M., Guyumdzhyan, M. et al. (2019) Sequence and nuclease requirements for breakage and healing of a structure-forming (AT)n sequence within fragile site FRA16D. Cell Rep., 27, 1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Inagaki, H., Ohye, T., Kogo, H., Tsutsumi, M., Kato, T., Tong, M., Emanuel, B.S. and Kurahashi, H. (2013) Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat. Commun., 4, 1592. [DOI] [PubMed] [Google Scholar]
- 61. Trask, B.J. (1999) Fluorescence in situ hybridization. In Birren, B., Green, E.D., Hieter, P., Klapholz, S., Myers, R.M., Riethman, H. and Roskams, J. (eds), Genome Analysis: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 303–413. [Google Scholar]
- 62. Latt, S.A. (1974) Localization of sister chromatid exchanges in human chromosomes. Science, 185, 74–76. [DOI] [PubMed] [Google Scholar]
- 63. Perry, P. and Wolff, S. (1974) New Giemsa method for the differential staining of sister chromatids. Nature, 251, 156–158. [DOI] [PubMed] [Google Scholar]
- 64. Wolff, S. and Perry, P. (1974) Differential Giemsa staining of sister chromatids and the study of chromatid exchanges without autoradiography. Chromosoma, 48, 341–353. [DOI] [PubMed] [Google Scholar]
- 65. Emanuel, B.S. (1978) Compound lateral asymmetry in human chromosome 6: BrdU-dye studies of 6q12→6q14. Am. J. Hum. Genet., 30, 153–159. [PMC free article] [PubMed] [Google Scholar]
- 66. Shaikh, T.H., Kurahashi, H., Saitta, S.C., O'Hare, A.M., Hu, P., Roe, B.A., Driscoll, D.A., McDonald-McGinn, D.M., Zackai, E.H., Budarf, M.L. and Emanuel, B.S. (2000) Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum. Mol. Genet., 9, 489–501. [DOI] [PubMed] [Google Scholar]
- 67. Rigby, P.W., Dieckmann, M., Rhodes, C. and Berg, P. (1977) Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase I. J. Mol. Biol., 113, 237–251. [DOI] [PubMed] [Google Scholar]
- 68. Nelson, J.D., Denisenko, O. and Bomsztyk, K. (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc., 1, 179–185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.