

Abstract
From a life-course perspective, genetic and environmental factors driving childhood obesity may have a lasting influence on health later in life. However, how obesity trajectories vary throughout the life-course remains unknown. Recently, Richardson et al. created powerful early life and adult gene scores for body mass index (BMI) in a comprehensive attempt to separate childhood and adult obesity. The childhood score was derived using questionnaire-based data administered to adults aged 40–69 regarding their relative body size at age 10, making it prone to recall and misclassification bias. We therefore attempted to validate the childhood and adult scores using measured BMI data in adolescence and adulthood among 66 963 individuals from the HUNT Study in Norway from 1963 to 2019. The predictive performance of the childhood score was better in adolescence and early adulthood, whereas the predictive performance of the adult score was better in adulthood. In the age group 12–15.9 years, the variance explained by the childhood polygenic risk score (PRS) was 6.7% versus 2.4% for the adult PRS. In the age group 24–29.9 years, the variance explained by the adult PRS was 3.9% versus 3.6% for the childhood PRS. Our findings support that genetic factors driving BMI differ at young age and in adulthood. Within the framework of multivariable Mendelian randomization, the validated childhood gene score can now be used to determine the consequence of childhood obesity on later disease.
Introduction
Although obesity has tripled among adults, childhood obesity has increased more than 8-fold worldwide since 1975 (1). This growing pandemic is cause for major public health concern (2). From a life-course perspective, genetic and environmental factors driving childhood obesity may have lasting influence on health later in life (3). Several epidemiological studies suggest that childhood obesity is associated with morbidity such as type 2 diabetes and cardiometabolic disease (4,5). However, isolating the impact of childhood obesity from that mediated by obesity later in life is difficult. Many children with obesity carry their excess weight into adulthood (6), and we lack knowledge on the genetic factors underpinning the obesity trajectories (3).
Recently, Richardson et al. created a childhood polygenic risk score (PRS) in an unprecedented attempt to separate the effects of child-onset and adult obesity. The childhood score predicts body mass index (BMI) better at age 10, whereas the adult score is a stronger predictor of adult BMI. Although there is considerable overlap in the genetic variants associated with BMI at each age, the strength of their effect seems to vary across the lifespan (7). Within the framework of multivariable Mendelian randomization, combining childhood and adult scores could distinguish if childhood obesity has a direct effect on disease risk or whether the risk is conferred through adult obesity (7–11).
The aim of our study is to validate the childhood PRS using measured BMI data of individuals in both adolescence and adulthood from the HUNT Study in Norway. Further, we aim to identify the ages at which predictive performance of the early life and adult scores cross over. Richardson et al.’s childhood score was derived using questionnaire-based data administered to adults aged 40–69 regarding their relative body size at age 10, making this variable prone to recall and misclassification bias (12). The validation of Richardson et al.’s recent work in a study sample with measured BMI demonstrates the use of genetics to better understand childhood and adult obesity.
Results
The study sample for this validation study consists of 66 963 participants aged 12–70 years with a total of 185 078 BMI measurements, keeping only the most recent observation per age category left 97 879 observations to be included in analyses of explained variance (Fig. 1).
Figure 1.
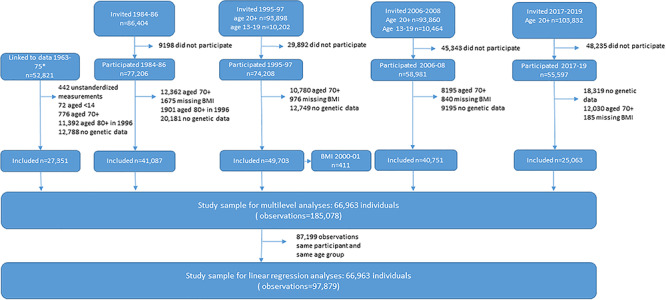
Flowchart of study participants and criteria for inclusion in study sample. *Linkage to data from the tuberculosis screening program 1963–75 required participation in any part of the Trøndelag Health Study.
The childhood and adult PRSs were only moderately correlated in our dataset, with a correlation coefficient of 0.34. In the age group 12–15.9 years, the variance explained by the childhood PRS was 6.7% versus 2.4% for the adult PRS (Table 1). In the age group 24–29.9 years, the variance explained by the adult PRS was 3.9% versus 3.6% for the childhood PRS. Results were similar in analyses excluding the proxy single nucleotide polymorphism (SNPs) (Supplementary Material, Fig. S1, Supplementary Material, Table S1). Thus, the crossover in terms of explained variance occurs in adolescence and early adulthood. This finding holds true for all years combined and when studying 1995–97 separately, yet is more uncertain for earlier times (Supplementary Material, Figs S2 and S3). The results among older adults in 1960–70 should be interpreted with caution as they require survival to and participation in genetic testing in the 1990 or 2000 (Supplementary Material, Table S1). Correspondingly, the receiver operating characteristic (ROC) curve analyses indicate that the childhood score is superior to the adult score in predicting overweight in the age group 12–15.9, whereas there is no difference between the two scores in the age group 16–17.9 (Fig. 2, Supplementary Material, Fig. S4, Supplementary Material, Tables S2 and S3). Interestingly, the marginal effect of the childhood score on BMI, i.e. how much BMI increases per standard deviation of the gene score, is relatively constant throughout the life-course, whereas the marginal effect of the adult score on BMI increases with age (Fig. 3). The marginal effects of the two scores cross in early adulthood yet their confidence intervals overlap in adolescence and early adulthood. This implies that neither score is better at predicting BMI in this age range. Redoing the analyses with FaT mass and Obesity-associated protein SNP (FTO) alone showed an increasing explained variance with age, however, results for the 1960 and 1970 were more uncertain (Supplementary Material, Table S4).
Table 1.
Additional variance explained by the childhood and adult gene scores after accounting for age, sex, genetic principal components, genotyping batch and time in all years combined as well as in 1963–75 and 1995–97. The gene scores include proxies for most of the missing SNPs
| Year | Age | Incremental R2 adult grs (%) | P-value | Confidence interval (%) | Incremental R2 childhood grs (%) | P-value | Confidence interval (%) | Difference in R2 adult grs vs. childhood grs (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| All years | 12–15.9 | 2.4 | <0.001 | 1.4 | 3.5 | 6.7 | 0 | 5.0 | 8.3 | -4.3 |
| 16–17.9 | 3.1 | <0.001 | 1.9 | 4.4 | 4.0 | 0 | 2.7 | 5.3 | -0.9 | |
| 18–23.9 | 3.1 | <0.001 | 2.6 | 3.7 | 3.9 | 0 | 3.2 | 4.5 | -0.8 | |
| 24–29.9 | 3.9 | <0.001 | 3.4 | 4.4 | 3.6 | 0 | 3.1 | 4.1 | 0.3 | |
| 30–69.9 | 3.6 | <0.001 | 3.3 | 3.8 | 1.9 | 0 | 1.7 | 2.1 | 1.7 | |
| 1963–75 | 12–15.9 | 2.7 | <0.001 | 1.4 | 4.1 | 6.5 | 0 | 4.6 | 8.4 | -3.8 |
| 16–17.9 | 3.3 | <0.001 | 1.8 | 4.8 | 4.7 | 0 | 3.0 | 6.3 | -1.4 | |
| 18–23.9 | 3.1 | <0.001 | 2.2 | 4.1 | 4.0 | 0 | 2.9 | 5.1 | -0.9 | |
| 24–29.9 | 4.2 | <0.001 | 3.1 | 5.3 | 4.3 | 0 | 3.2 | 5.4 | -0.1 | |
| 30–69.9 | 3.4 | <0.001 | 2.8 | 4.0 | 3.5 | 0 | 2.9 | 4.1 | -0.1 | |
| 1995–97 | 12–15.9 | 2.0 | 0.017 | 0.4 | 3.7 | 7.0 | 0 | 3.9 | 10.1 | -5.0 |
| 16–17.9 | 2.7 | 0.023 | 0.4 | 5.1 | 2.2 | 0.066 | -0.1 | 4.6 | 0.5 | |
| 18–23.9 | 3.5 | <0.001 | 2.3 | 4.8 | 4.2 | 0 | 2.8 | 5.5 | -0.7 | |
| 24–29.9 | 5.1 | <0.001 | 3.9 | 6.3 | 4.3 | 0 | 3.2 | 5.5 | 0.8 | |
| 30–69.9 | 3.6 | <0.001 | 3.3 | 4.0 | 2.6 | 0 | 2.3 | 2.9 | 1.0 | |
Figure 2.
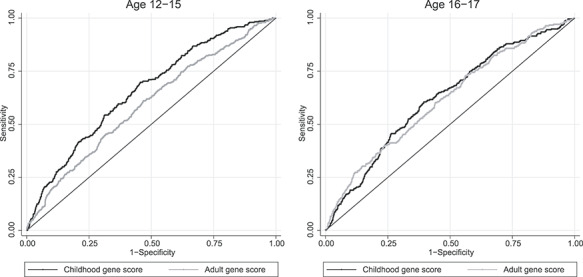
Receiver operator characteristics curves to compare the predictive ability of the childhood and the adult gene scores for overweight at age 12–15 and age 16–17 in the HUNT Study.
Figure 3.
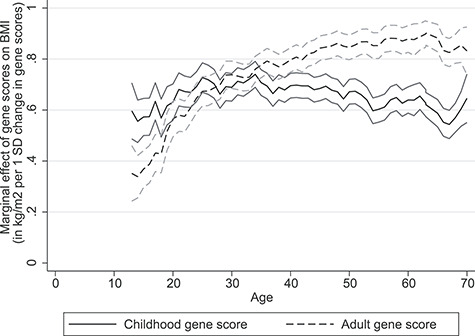
Marginal effects with 95% confidence intervals of the childhood and adult gene scores on BMI as a function of age.
Discussion
In this study, we validated the new British childhood and adult gene scores for BMI with repeated BMI measurements of a large Norwegian population aged 12–70 over six decades. Our study confirmed that both PRSs are valid instruments. Comparable to the British study (7), the age of crossover in terms of strength of prediction for the early life score to the adult score occurred in adolescence and young adulthood.
Strength and limitations of this study
The British and Norwegian study populations are well matched in terms of ethnicity and have comparable cohorts that were children and middle aged in the same decades. The British score for childhood BMI is unprecedented in statistical power yet this comes at a cost. Although the adult score is derived from measured BMI in middle-aged adults of the UK Biobank (UKBB), the childhood score is derived from the same adults’ self-reported adiposity at age 10 making it prone to recall and misclassification bias (7). Appropriately, the validation of both scores with standardized BMI measurements of adolescents and middle-aged adults in the Norwegian HUNT Study population can help address this limitation. Our findings should be taken together with results from the extensive simulation analyses conducted by Richardson et al. to investigate measurement error in the self-reported childhood score. Their results suggest that misclassification would only mask an effect of the adult score if it were to influence a disease outcome in the same direction as the childhood score.
Our study widens the age range for assessment of the scores (7) and is able to identify at what ages the crossover in terms of strength of prediction from the early life score to the adult score occurs. Limited by the current knowledge of genetic variation underlying each trait, we acknowledge that this may in part contribute to the observed difference between the two scores. The generalizability of the British childhood score could also be questioned being derived from data of older cohorts that were children approximately half a century ago (7,13). Our dataset contains both genetic material and repeated BMI measurements for a large sample of individuals from 12 to 70 years of age over six decades. Hence, being able to show that the British childhood gene score is still associated with childhood BMI in younger cohorts from the HUNT Study is a defining strength of our study. We validate that Richardson et al.’s childhood PRS for BMI is a predictor of childhood obesity in the past and the present. Notably, the differences in the crossover age for these two groups could potentially be explained by how comparable they are to the original UK Biobank population, which the scores were derived from.
Comparison with other studies
The factors driving the various obesity trajectories throughout the life-course remain unknown. Genetic studies show promising attempts in separating childhood and adult obesity. Although some studies justify the adult gene score as a valid instrument for life-long BMI (14–16), others support the use of a more age appropriate gene score in children (17–22). Although largely limited by statistical power, previous childhood gene scores uncovered novel variants associated with infant and childhood adiposity (3,17,18) and showed that many variants represent age-related differences in strength of association with BMI (17–19,21).
Richardson et al.’s PRS for childhood BMI is unprecedented in terms of its statistical power. It includes 295 genetic variants associated with a measure of childhood BMI reported by 453 169 participants of the UK Biobank (7,13). Our validation study could only include 289 of these 295 genetic variants, marginally reducing its strength (Supplementary Material, Table S5). Regardless, the new childhood score explains an additional 7% of variance in BMI for individuals aged 12–16 when applied to our dataset, which is roughly triple that of comparable childhood scores (17). There is large overlap in genetic variants associated with obesity in children and adults. However, only the independent SNPs most significantly associated with measures of BMI were included in the gene scores. FTO, one of the SNPs with the strongest effect size, shows an increasing explained variance with age. However, trends for 1960 and 1970 were more uncertain (Supplementary Material, Table S4). Several genetic variants appear to have a stronger influence on BMI at different ages suggesting that the genetic background for child-onset and adult obesity can be separated (7). Our study supports this notion as the variance explained by the childhood score was greater in adolescence and early adulthood, whereas the variance explained by the adult score was greater in adults (Fig. 3, Supplementary Material, Fig. S2, Table 1). Similarly, there was a distinct difference in the ROC plots for obesity in adulthood and for overweight in the younger age categories (Fig. 2, Supplementary Material, Figs S5 and S6). The corresponding estimated area under the curves is presented in Supplementary Material, Table S2. The ROC plots for overweight from age 18 to 70 appear to show an equal effect of both scores on BMI (Supplementary Material, Figs S7 and S8). This is somewhat misleading and is rather a reflection of the high prevalence of overweight in the HUNT Study population. Interestingly, the childhood score is a better predictor of BMI in adolescence in both the HUNT Study and the Avon Longitudinal Study of Parents and Children (ALSPAC) cohorts (7). Correspondingly, the adult score is a better predictor of BMI in adulthood, likely in part reflecting the effects of puberty. That two large studies confirm a similar age range as a cross-point separating the genetics of childhood and adult obesity is novel and could lay groundwork for future research. Richardson et al. used multivariable Mendelian Randomization to show that childhood body size does not increase risk of coronary artery disease and type 2 diabetes after taking into account adulthood body size. In light of these findings, we have proposed a window in the life course concerning the critical age where the negative impact of obesity from early life no longer can be mitigated with respect to later life disease. The effect of the childhood score is relatively constant throughout the life-course, whereas the effect of the adult score on BMI increases with age (Fig. 3). Although we acknowledge the difference in normal range of BMI in children and adults, this does not change the interpretation of our findings. Comparatively, the genome-wide polygenic score for BMI is vastly more powerful than both the childhood and adult scores created by Richardson et al. Although its effect on weight also emerges in early life, this effect increases into adulthood making it a better predictor of BMI in adults (23).
Validation of separate gene scores for adult and childhood BMI enables us to study childhood obesity and its relation to later health. The question of whether childhood obesity has a direct effect on disease risk or if the risk is conferred through adult obesity is baffling and has led to conflicting results (24). Previous observational studies and phenome-wide Mendelian randomization studies have found associations with high BMI in early life and increased risk for morbidity (25,26) including coronary artery disease (4,26), type 2 diabetes (5,26) and several types of cancer such as breast cancer (27,28). Other studies imply that high BMI in childhood does not have a direct effect on risk for later disease unless it is sustained throughout adulthood (29,30). This argument is supported clinically as severely obese adolescents have shown reversal of type 2 diabetes and improvements in cardiovascular risk factors after surgical weight loss (31).
Richardson et al.’s (7) attempt to distinguish childhood obesity’s relation to later disease is the most comprehensive to date. Using the childhood and adult PRSs as separate genetic instruments, they distinguish the causal role of childhood obesity within the framework of multivariable Mendelian randomization (8,9). After validating the childhood and adult PRSs for BMI with the HUNT population, Richardson et al.’s analytical approach can now be used to test a multitude of disease outcomes. Most previous studies have focused on the impact of childhood obesity on the risk for disease later in life; however, this approach should also consider the impact of childhood obesity on pediatric outcomes. Previous Mendelian randomization studies have found causal associations of childhood obesity on type 1 diabetes (20) and childhood asthma (21). The results of these and other Mendelian randomization studies should now be reevaluated using the more powerful childhood gene score in a multivariable framework.
Generalizability of the findings
Genetic risk is likely to differ among child populations throughout the world as genetic variants associated with childhood BMI may vary between ethnicities (32). Regardless, all child populations are likely to have age-related differences in strength of association between genetic variants and BMI. Furthermore, the HUNT Study with BMI measurements in adolescents and adults over six decades is an appropriate dataset for validating increasingly powerful childhood gene scores for BMI in the future.
Conclusion and implications
Our findings support that genetic factors driving BMI differ at young age and in adulthood. This creates ample opportunity to study the life-course trajectories and consequences of obesity. Within the framework of multivariable Mendelian randomization, the validated childhood PRS can now be used to determine causality (7). It could resolve whether childhood obesity is an early metabolic derangement where most risk evolves in adulthood (30) or whether childhood obesity impacts risk for later disease directly. Guided by Richardson et al.’s recent results for type 2 diabetes and coronary artery disease, it is plausible that obesity only presents a risk for somatic diseases after adolescence and early adulthood. It seems, however, likely that childhood obesity has lasting effects on later well-being and psychiatric disease. This validation study confirms the essence of the timing of obesity in a life-course. Both the UK Biobank and the HUNT Study identify adolescence as a critical age that separates the genetics of childhood and adult obesity. This new knowledge could be an important clue in uncovering the mechanisms underlying the global disease. Although efforts to alleviate obesity should be pursued at all ages, using human genetics to disentangle the contribution of childhood and adult BMI to disease risk can be an attractive and cost-effective approach to help improve prevention strategies.
Materials and Methods
Our findings are based on 66 963 individuals of European descent aged 12–70 years. The study population consists of participants from the HUNT Study (1984–2019) linked to previous height and weight measurements in the tuberculosis screening program (1963–75). Established in 1943, the tuberculosis screening program contributed to the surveillance of tuberculosis in the general Norwegian population (33). The HUNT Study is a large population-based study with data based on clinical examinations, self-reported health characteristics, assays of biological samples and genotyping. The entire adult population of the former Nord-Trøndelag county was invited to participate, and the HUNT Study was conducted in four waves: HUNT1 (1984–86), HUNT2 (1995–97), HUNT3 (2006–08) and HUNT4 (2017–19). Similarly, the Young-HUNT Study was conducted in 1995–97, 2000–01, 2006–08 and 2017–19 and recruited all teenagers aged 13–19 from the same region. The HUNT Study is considered representative of the Norwegian population despite participation decline from 88% in HUNT1 to 70% in HUNT2 and subsequently 54% in HUNT3 and HUNT4 (34).
The derivation of the study sample is shown in Supplementary Material, Figure S1. Nearly all participants with BMI measured before the age of 18 also had at least one additional BMI measured later in adulthood (Supplementary Material, Table S6).
BMI assessment
BMI was calculated as weight in kilograms per height in meters squared. Weight was measured to the nearest half kilogram with the participants wearing light clothes and no shoes and height was measured to the nearest centimeter (35). The World Health Organization defines overweight as BMI greater than or equal to 25 and obesity as BMI greater than or equal to 30 (36). BMI strongly relates to longitudinal growth. To define corresponding cut-offs for obesity and overweight for participants younger than 18 years, we calculated their BMI z-score using age- and sex-specific references from the International Obesity Task Force (37).
Genotyping and computation of the child and adult PRSs
Genetic analyses were performed on blood samples collected from adults participating in HUNT2 and HUNT3 (34). Genotyping was carried out with one of three different Illumina HumanCoreExome arrays (HumanCoreExome12 v1.0, HumanCoreExome12 v1.1, and UM HUNT Biobank v1.0, Illumina, CA), as described previously (38,39). Imputation was performed using Minimac3 from a panel combined from the Haplotype Reference Consortium and 2202 HUNT low-pass sequenced individuals with indel calling.
Using summary statistics from the genome-wide association study in the UK Biobank (7), we constructed both childhood and adult gene scores for BMI with data from the HUNT Study participants. For the childhood and adult scores respectively, we first multiplied the number of risk alleles for each of the common variants with the estimated effect size of that particular variant on BMI published by Richardson et al. (7), and then summarized overall common variants in respective scores to create a weighted PRS (40). We accepted proxy SNPs with an R2 ≥ 0.8 as well as a D′ ≥0.95 using publicly available reference haplotypes of a European British in UK and Scotland population from Phase 3 (Version 5) of the 1000 Genomes Project. We were unable to locate proxy SNPs for 11 of 39 missing SNPs from the adult score and 6 of 17 SNPs from the childhood score (Supplementary Material, Table S5). After ascertainment that the forward strand was analyzed in both the UKBB and the HUNT Study, we included all palindromic SNPs. For comparison, we generated scores excluding the proxy SNPs as well as imputed SNPs with an r2 < 0.8 (nine adult score SNPs and one childhood score SNP).
Richardson et al.’s childhood and adult PRSs include 295 and 557 common variants identified to be associated with childhood and adult BMI, respectively. We included 289 of the 295 common variants in the childhood score, whereof 11 were proxies for 17 of the common variants excluded due to lacking information in the HUNT dataset. Correspondingly, we included 546 of the 557 common variants in the adult score, whereof 28 were proxies for 39 of the common variants excluded due to lacking information (Supplementary Material, Table S5).
Statistical analyses
We used the gene scores including palindromic SNPs in the main analyses. First, in order to check the overlap in genetic predisposition to obesity as defined by the two scores, we calculated the Pearson’s correlation coefficient between the childhood and adult gene scores for all ages combined, using only one observation per individual. The main validation analyses involved linear regression between measures of BMI with both the childhood and adult scores adjusted for age (included as a continuous variable), sex, time of measurement (as indicator variables for each time period), 20 genetic principal components and genotyping batch. We performed these analyses separately by age groups 12–15.9, 16–17.9, 18–23.9, 24–29.9 and 30–70, and we included only the most recent observation for each individual per age group. We calculated the difference in explained variance by comparing the variance explained by models with and without a gene score, and performed bootstrapping with 1000 replications to have confidence intervals for the differences in explained variance. We performed analyses with BMI measured in the HUNT Study as well as in the tuberculosis screening program in the 1960 and 1970 both separately and over all times combined to evaluate the ability of both scores to predict the BMI both overall and at specific time points. To describe the age of crossover in strength of association between each score and BMI, we included all available BMI measurements and performed linear mixed models with observations nested in individuals. Adjustment models were similar to the linear models described earlier, but rather than analyzing separately over age groups, we included interaction terms between gene scores (as continuous variables) and age groups (as an indicator variable in 3-year bands). We subsequently estimated the marginal effects of gene scores on BMI over age, using the user written spost13 package for Stata. In additional analyses, we also included interaction terms between gene scores and time of measurement (as an indicator variable) and between age groups (as indicator variable) and time point (as an indicator variable). We estimated the marginal effects of gene scores on BMI over both age and time of measurement. We then generated ROC plots as undertaken in Richardson et al.’s study (7) to investigate the ability of both scores to predict overweight and obesity in different age categories. Because obesity was rare among adolescents in our sample, we present ROC plots for overweight in the main results. We redid the analyses using only the FTO SNP to explore its role over six decades. We performed additional analyses using scores without proxy SNPs and excluding SNPs with poorer imputation quality to confirm consistent results. The gene scores were generated using R version 3.6.2 and all subsequent analyses were performed using Stata16.
Supplementary Material
Acknowledgments
The Trøndelag Health Study (The HUNT Study) is a collaboration between HUNT Research Centre, (Faculty of Medicine and Health Sciences, NTNU, Norwegian University of Science and Technology), Nord-Trøndelag County Council, Central Norway Regional Health Authority and the Norwegian Institute of Public Health. The genotyping in HUNT was financed by the National Institutes of Health; University of Michigan; The Research Council of Norway; The Liaison Committee for Education, Research and Innovation in Central Norway; and the Joint Research Committee between St. Olavs hospital and the Faculty of Medicine and Health Sciences, NTNU. The genotype quality control and imputation has been conducted by the K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, NTNU, Norwegian University of Science and Technology.
Conflicts of Interest statement. Dr Vie and Dr Bjørngaard report grants from The Research Council of Norway during the conduct of the study. Dr Brandkvist reports grants from The Liaison Committee for Education, Research and Innovation in Central Norway during the conduct of the study. All authors have no financial relationships with any organizations that might have an interest in the submitted work in the previous 5 years; no other relationships or activities that could appear to have influenced the submitted work.
Contributor Information
Maria Brandkvist, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Trondheim, Norway; Children’s Clinic, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway; Obesity Centre, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway.
Johan Håkon Bjørngaard, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Trondheim, Norway; Faculty of Nursing and Health Sciences, Nord University, Levanger, Norway.
Rønnaug Astri Ødegård, Children’s Clinic, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway; Obesity Centre, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway; Department of Clinical and Molecular Medicine, NTNU, Norwegian University of Science and Technology, Trondheim, Norway.
Bjørn Olav Åsvold, K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Trondheim, Norway; Department of Endocrinology, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway; HUNT Research Centre, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Levanger, Norway.
George Davey Smith, Medical Research Council Integrative Epidemiology Unit, University of Bristol, Bristol, UK; Population Health Sciences, Bristol Medical School, University of Bristol, Barley House, Oakfield Grove, Bristol, UK.
Ben Brumpton, K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Trondheim, Norway; Medical Research Council Integrative Epidemiology Unit, University of Bristol, Bristol, UK; Clinic of Thoracic and Occupational Medicine, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway.
Kristian Hveem, K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Trondheim, Norway; Department of Research, Innovation and Education, St. Olavs hospital, Trondheim University Hospital, Trondheim, Norway.
Tom G Richardson, Medical Research Council Integrative Epidemiology Unit, University of Bristol, Bristol, UK.
Gunnhild Åberge Vie, Department of Public Health and Nursing, NTNU, Norwegian University of Science and Technology, Trondheim, Norway; Obesity Centre, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway.
Data Sharing
Data from the HUNT Study used in research projects will when reasonably requested by others be made available upon request to the HUNT Data Access Committee (hunt@medisin.ntnu.no). The HUNT data access information (available here: http://www.ntnu.edu/hunt/data) describes in detail the policy regarding data availability. The Norwegian Institute of Public Health will consider applications for data from the Tuberculosis screening program (https://www.fhi.no/en/op/data-access-from-health-registries-health-studies-and-biobanks/).
Funding
The Liaison Committee for Education, Research and Innovation in Central Norway (project number 90057601 to M.B) and the Norwegian Research Council (250335 to G.Å.V, 295989 to JHB); a research unit funded by Stiftelsen Kristian Gerhard Jebsen; Faculty of Medicine and Health Sciences, NTNU; The Liaison Committee for Education, Research and Innovation in Central Norway; and the Joint Research Committee between St. Olavs Hospital and the Faculty of Medicine and Health Sciences, NTNU and the Medical Research Council Integrative Epidemiology Unit at the University of Bristol, which is supported by the Medical Research Council (MC_UU_00011/1) and the University of Bristol (T.G.R., B.B., G.D.S.). T.G.R. is a UKRI research fellow (MR/S003886/1 to T.G.R.).
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The researchers were independent from funders, and all authors had full access to all of the data (including statistical reports and tables) in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1. NCD Risk Factor Collaboration (NCD-RisC) (2017) Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet, 390, 2627–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ayton, A. and Ibrahim, A. (2019) Obesity is a public health emergency. BMJ, 366, l5463. [DOI] [PubMed] [Google Scholar]
- 3. Couto Alves, A., De Silva, N.M.G., Karhunen, V., Sovio, U., Das, S., Taal, H.R., Warrington, N.M., Lewin, A.M., Kaakinen, M., Cousminer, D.L. et al. (2019) GWAS on longitudinal growth traits reveals different genetic factors influencing infant, child, and adult BMI. Sci. Adv., 5, eaaw3095–eaaw3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bibbins-Domingo, K., Coxson, P., Pletcher, M.J., Lightwood, J. and Goldman, L. (2007) Adolescent overweight and future adult coronary heart disease. N. Engl. J. Med., 357, 2371–2379. [DOI] [PubMed] [Google Scholar]
- 5. Hannon, T.S., Rao, G. and Arslanian, S.A. (2005) Childhood obesity and type 2 diabetes mellitus. Pediatrics, 116, 473–480. [DOI] [PubMed] [Google Scholar]
- 6. Ogden, C.L., Carroll, M.D., Curtin, L.R., Lamb, M.M. and Flegal, K.M. (2010) Prevalence of high body mass index in US children and adolescents, 2007-2008. JAMA, 303, 242–249. [DOI] [PubMed] [Google Scholar]
- 7. Richardson, T.G., Sanderson, E., Elsworth, B., Tilling, K. and Davey Smith, G. (2020) Use of genetic variation to separate the effects of early and later life adiposity on disease risk: Mendelian randomisation study. BMJ, 369, m1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burgess, S. and Thompson, S.G. (2015) Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol., 181, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sanderson, E., Davey Smith, G., Windmeijer, F. and Bowden, J. (2019) An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol., 48, 713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith, G.D. and Ebrahim, S. (2003) 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol., 32, 1–22. [DOI] [PubMed] [Google Scholar]
- 11. Sanderson, E. (2020) Multivariable Mendelian randomization and mediation. Cold Spring Harb. Perspect. Med., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grimes, D.A. and Schulz, K.F. (2002) Bias and causal associations in observational research. Lancet, 359, 248–252. [DOI] [PubMed] [Google Scholar]
- 13. Sudlow, C., Gallacher, J., Allen, N., Beral, V., Burton, P., Danesh, J., Downey, P., Elliott, P., Green, J., Landray, M. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reed, Z.E., Micali, N., Bulik, C.M., Davey Smith, G. and Wade, K.H. (2017) Assessing the causal role of adiposity on disordered eating in childhood, adolescence, and adulthood: a Mendelian randomization analysis. Am. J. Clin. Nutr., 106, 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Millard, L.A., Davies, N.M., Timpson, N.J., Tilling, K., Flach, P.A. and Davey Smith, G. (2015) MR-PheWAS: hypothesis prioritization among potential causal effects of body mass index on many outcomes, using Mendelian randomization. Sci. Rep., 5, 16645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buscot, M.J., Wu, F., Juonala, M., Lehtimäki, T., Pitkänen, N., Sabin, M.A., Viikari, J.S.A., Raitakari, O.T. and Magnussen, C.G. (2020) Longitudinal association of a body mass index (BMI) genetic risk score with growth and BMI changes across the life course: the cardiovascular risk in young Finns study. Int. J. Obes. (Lond), 44, 1733–1742 [DOI] [PubMed] [Google Scholar]
- 17. Felix, J.F., Bradfield, J.P., Monnereau, C., van der Valk, R.J., Stergiakouli, E., Chesi, A., Gaillard, R., Feenstra, B., Thiering, E., Kreiner-Moller, E. et al. (2016) Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum. Mol. Genet., 25, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bradfield, J.P., Taal, H.R., Timpson, N.J., Scherag, A., Lecoeur, C., Warrington, N.M., Hypponen, E., Holst, C., Valcarcel, B., Thiering, E. et al. (2012) A genome-wide association meta-analysis identifies new childhood obesity loci. Nat. Genet., 44, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hohenadel, M.G., Baier, L.J., Piaggi, P., Muller, Y.L., Hanson, R.L., Krakoff, J. and Thearle, M.S. (2016) The impact of genetic variants on BMI increase during childhood versus adulthood. Int. J. Obes. (Lond), 40, 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Censin, J.C., Nowak, C., Cooper, N., Bergsten, P., Todd, J.A. and Fall, T. (2017) Childhood adiposity and risk of type 1 diabetes: a Mendelian randomization study. PLoS Med., 14, e1002362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen, Y.C., Fan, H.Y., Huang, Y.T., Huang, S.Y., Liou, T.H. and Lee, Y.L. (2019) Causal relationships between adiposity and childhood asthma: bi-directional Mendelian randomization analysis. Int. J. Obes. (Lond), 43, 73–81. [DOI] [PubMed] [Google Scholar]
- 22. Craig, S.J.C., Kenney, A.M., Lin, J., Paul, I.M., Birch, L.L., Savage, J.S., Marini, M.E., Chiaromonte, F., Reimherr, M.L. and Makova, K.D. (2020) Polygenic risk score based on weight gain trajectories is predictive of childhood obesity. bioRxiv in press, 606277. [Google Scholar]
- 23. Khera, A.V., Chaffin, M., Wade, K.H., Zahid, S., Brancale, J., Xia, R., Distefano, M., Senol-Cosar, O., Haas, M.E., Bick, A. et al. (2019) Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell, 177, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Llewellyn, A., Simmonds, M., Owen, C.G. and Woolacott, N. (2016) Childhood obesity as a predictor of morbidity in adulthood: a systematic review and meta-analysis. Obes. Rev., 17, 56–67. [DOI] [PubMed] [Google Scholar]
- 25. Juonala, M., Magnussen, C.G., Berenson, G.S., Venn, A., Burns, T.L., Sabin, M.A., Srinivasan, S.R., Daniels, S.R., Davis, P.H., Chen, W. et al. (2011) Childhood adiposity, adult adiposity, and cardiovascular risk factors. N. Engl. J. Med., 365, 1876–1885. [DOI] [PubMed] [Google Scholar]
- 26. Dong, S.-S., Zhang, K., Guo, Y., Ding, J.-M., Feng, J.-C., Yao, S., Hao, R.-H., Rong, Y., Jiang, F., Chen, J.-B. et al. (2020) Phenome-wide investigation of the causal associations between childhood BMI and adult outcomes: a two-sample Mendelian randomization study. bioRxiv in press, 2020.2006.2001.127530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baer, H.J., Colditz, G.A., Rosner, B., Michels, K.B., Rich-Edwards, J.W., Hunter, D.J. and Willett, W.C. (2005) Body fatness during childhood and adolescence and incidence of breast cancer in premenopausal women: a prospective cohort study. Breast Cancer Res., 7, R314–R325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biro, F.M. and Wien, M. (2010) Childhood obesity and adult morbidities. Am. J. Clin. Nutr., 91, 1499s–1505s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bjerregaard, L.G., Jensen, B.W., Ängquist, L., Osler, M., Sørensen, T.I.A. and Baker, J.L. (2018) Change in overweight from childhood to early adulthood and risk of type 2 diabetes. N. Engl. J. Med., 378, 1302–1312. [DOI] [PubMed] [Google Scholar]
- 30. Franks, P.W., Hanson, R.L., Knowler, W.C., Sievers, M.L., Bennett, P.H. and Looker, H.C. (2010) Childhood obesity, other cardiovascular risk factors, and premature death. N. Engl. J. Med., 362, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inge, T.H., Miyano, G., Bean, J., Helmrath, M., Courcoulas, A., Harmon, C.M., Chen, M.K., Wilson, K., Daniels, S.R., Garcia, V.F. et al. (2009) Reversal of type 2 diabetes mellitus and improvements in cardiovascular risk factors after surgical weight loss in adolescents. Pediatrics, 123, 214–222. [DOI] [PubMed] [Google Scholar]
- 32. Martin, A.R., Kanai, M., Kamatani, Y., Okada, Y., Neale, B.M. and Daly, M.J. (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet., 51, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bjorge, T., Engeland, A., Tverdal, A. and Smith, G.D. (2008) Body mass index in adolescence in relation to cause-specific mortality: a follow-up of 230,000 Norwegian adolescents. Am. J. Epidemiol., 168, 30–37. [DOI] [PubMed] [Google Scholar]
- 34. Hunt Research Center . (2020) HUNT Databank. https://hunt-db.medisin.ntnu.no/hunt-db/#/ (8 April 8 2020, date last accessed).
- 35. Krokstad, S., Langhammer, A., Hveem, K., Holmen, T.L., Midthjell, K., Stene, T.R., Bratberg, G., Heggland, J. and Holmen, J. (2013) Cohort profile: the HUNT Study, Norway. Int. J. Epidemiol., 42, 968–977. [DOI] [PubMed] [Google Scholar]
- 36. World Health Organization . (2020) Obesity and Overweight. http://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (14 October 2020, date last accessed).
- 37. Cole, T.J., Flegal, K.M., Nicholls, D. and Jackson, A.A. (2007) Body mass index cut offs to define thinness in children and adolescents: international survey. BMJ, 335, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brandkvist, M., Bjorngaard, J.H., Odegard, R.A., Asvold, B.O., Sund, E.R. and Vie, G.A. (2019) Quantifying the impact of genes on body mass index during the obesity epidemic: longitudinal findings from the HUNT Study. BMJ, 366, l4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nielsen, J.B., Thorolfsdottir, R.B., Fritsche, L.G., Zhou, W., Skov, M.W., Graham, S.E., Herron, T.J., McCarthy, S., Schmidt, E.M., Sveinbjornsson, G. et al. (2018) Genome-wide association study of 1 million people identifies 111 loci for atrial fibrillation. bioRxiv, in press. [Google Scholar]
- 40. National Cancer Institute, Division of Cancer Epidemiology & Genetics . (2020). https://ldlink.nci.nih.gov/?tab=home (14 October 2020, date last accessed)).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.