

Abstract
C9orf72 ALS/FTD patients show remarkable clinical heterogeneity, but the complex biology of the repeat expansion mutation has limited our understanding of the disease. BAC transgenic mice were used to better understand the molecular mechanisms and repeat length effects of C9orf72 ALS/FTD. Genetic analyses of these mice demonstrate that the BAC transgene and not integration site effects cause ALS/FTD phenotypes. Transcriptomic changes in cell proliferation, inflammation and neuronal pathways are found late in disease and alternative splicing changes provide early molecular markers that worsen with disease progression. Isogenic sublines of mice with 800, 500 or 50 G4C2 repeats generated from the single-copy C9–500 line show longer repeats result in earlier onset, increased disease penetrance and increased levels of RNA foci and dipeptide RAN protein aggregates. These data demonstrate G4C2 repeat length is an important driver of disease and identify alternative splicing changes as early biomarkers of C9orf72 ALS/FTD.
Introduction
The intronic C9orf72 G4C2 expansion mutation is the most common known genetic cause of both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (1,2). Proposed molecular mechanisms include C9orf72 protein loss-of-function, RNA gain-of-function and repeat-associated non-ATG (RAN) protein toxicity (1–12). Numerous mouse models have been developed to better understand the relative contributions of loss-of-function and gain-of-function mechanisms in disease (13–23). While C9orf72 protein levels measured in autopsy samples are lower in C9 patients (24), C9orf72 knockout mice develop peripheral immune phenotypes but not ALS/FTD-related phenotypes (15,16,18,20), making it unlikely that C9orf72 loss-of-function alone is a major driver of disease. In contrast, Drosophila and mouse models that overexpress specific RAN proteins develop neurodegenerative and motor phenotypes (13–23,25), indicating RAN proteins can be toxic and may play a role in disease. RNA gain-of-function effects may cause RNA processing abnormalities that contribute to disease through the sequestration of RNA-binding proteins by the repeat-containing sense and antisense transcripts. Studies of C9 iPSC-derived neurons (iPSNs) and C9-ALS autopsy tissue have reported transcriptomic changes (26–28). Additionally, a number of RNA-binding proteins (RNA-BPs) that interact with short stretches of G4C2 repeats have been identified through unbiased interactome screens, including Pur-α, ADARB2, hnRNPH, hnRNPA1, hnRNPA2/B1, ALYREF, nucleolin and RanGAP1 (26,29–36). Crosslinking immunoprecipitation (CLIP) analyses using autopsy material from the frontal cortex of C9-ALS patients shows that hnRNPH binds to G4C2 transcripts with short repeats (37). Since there is little consensus on which RNA-binding proteins are sequestered by the G4C2 repeats, the relative contribution of RNA gain-of-function mechanisms in C9orf72 ALS/FTD remains unclear.
There is remarkable clinical heterogeneity among C9orf72 expansion carriers with clinical presentations ranging from muscle wasting characteristic of ALS in some patients, to disinhibition and cognitive deficits characteristic of FTD in others. While some expansion carriers remain asymptomatic into their 90s, the frequency of reduced penetrance is not yet clear. The C9orf72 expansion mutation is found in approximately 7% of sporadic ALS cases, in which there is no family history of the disease (38). Because asymptomatic expansion-positive relatives and asymptomatic expansion carriers in general are unlikely to be tested, the frequency of the C9orf72 expansion mutation is not yet clear (39,40). Repeat length and somatic repeat instability, which are known to contribute to Huntington disease and other repeat expansion disorders (41–43), may contribute to the reduced disease penetrance of C9orf72 ALS/FTD, differences in age of onset and the wide-ranging clinical effects of the C9orf72 expansion mutation (4,40). However, because of ascertainment bias, technical difficulties in measuring repeat length and somatic instability in patients, it is challenging to study the effects of repeat length as a modifier of C9orf72 ALS/FTD (44,45).
To better understand the molecular mechanisms of disease, we and others generated bacterial artificial chromosome (BAC) transgenic mouse models that show molecular phenotypes of the disease including sense and antisense RNA foci and RAN protein aggregates, although the relative levels of these molecular phenotypes have not been directly compared (18–21). In the C9-BAC transgenic mice developed at the University of Florida, mice from several independent lines established on the FVB background (19) developed both the molecular and behavioral features of ALS/FTD including the accumulation of sense and antisense RNA foci and RAN proteins, movement abnormalities, motor neuron loss and decreased survival (19,46,47).
Here, we describe the transgene integration sites of these C9-BAC lines (19), further establishing that the ALS/FTD phenotypes in these lines occur independent of integration effects. RNAseq analyses, using the most penetrant single copy C9–500 line, show transcriptomic profiles consistent with neuronal loss, microglia and oligodendrocyte changes that are distinct at different stages of disease. Additionally, alternative splicing abnormalities are prevalent prior to onset of overt disease features, suggesting their potential utility as early biomarkers of ALS/FTD. Using the single copy C9–500 line, we generated an allelic series of mice containing 800, 500 or 50 repeats and demonstrate that longer repeat tracts in an isogenic background increase disease penetrance and decrease age of onset and survival. These data demonstrate that the ALS/FTD phenotypes in FVB C9-BAC mice are driven by gain-of-function effects of the expansion mutation; these effects occur independent of integration site and repeat length is a major driver of disease.
Results
Phenotypes in C9-BAC mice independent of integration sites
We previously reported the development of a BAC transgenic model of C9orf72 ALS/FTD on the FVB/NJ background (19). Four independent lines were generated by pronuclear injection of a circularized BAC containing a 98.3 kb human DNA insert, including a large G4C2 repeat-expansion mutation (Fig. 1A). To further characterize these mice, we performed whole genome sequencing to determine transgene break points, genomic integration sites and number of transgene copies for each of the four BAC transgenic lines. Transgene break and genomic integration sites were identified by computational analyses of discordant read-pairs from transgenic DNA compared to mouse and human reference genomes. Transgene copy number for each of the lines was determined by comparing the regional coverage depth of the BAC with the average coverage depth of the mouse reference genome. These data show that the transgenes in all four BAC lines were inserted into distinct single integration sites. Transgene copy number for each of the lines was consistent with previous estimates based on Southern blot and quantitative real time polymerase chain reaction (qRT-PCR) analyses (19).
Figure 1.
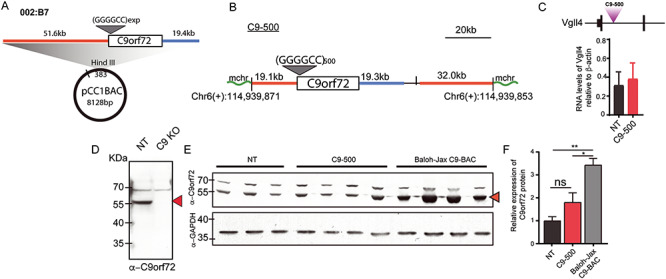
Characterization of transgene integration site, and C9orf72 protein levels in C9-BAC mice. (A) BAC construct containing human C9orf72 gene and 51.6 kb and 19.4 kb of upstream (orange) and downstream (blue) flanking sequence, respectively. (B) Map showing breakpoint and integration of the BAC on mouse chromosome 6. (C) Map showing insertion of C9orf72 transgene in Vgll4 gene and qPCR showing expression of Vgll4 in NT and C9–500 mice. (D, E) Protein blots showing NT, C9–500 and Baloh-Jax mice probed with α-C9orf72 antibody. (F) Protein blot quantitation. Data information: Statistical analyses for panel F were performed using one-way ANOVA with Bonferroni correction for multiple comparisons with mean +/− SEM shown; not significant (ns) = P > 0.05.
In the single-copy C9–500 line, the BAC transgene inserted between Chr 6(+): 114939871 and Chr 6(+): 114939853 in the mouse genome within the first intron of the annotated tumor suppressor gene vestigial like 4 (Vgll4) (Fig. 1B and C). The breakpoint on the circular BAC transgene occurred 19,097 bp upstream of C9orf72. At the site of integration, the C9–500 line contains the full-length C9orf72 gene containing ~500 G4C2 • G2C4 repeats with ~19.1 kb of 5′ human flanking sequence and ~19.4 kb of 3′ human flanking sequence. The remaining sequence includes the pCC1BAC backbone followed by 32.0 kb of human DNA originally 5′ of the breakpoint on the BAC. qRT-PCR using primers targeting Vgll4 shows no significant differences in expression of Vgll4 in C9–500 mice compared to controls (Fig. 1C). In summary, the BAC transgene in the C9–500 line has a single integration site containing a full-length copy of the C9orf72 gene, substantial human flanking sequence and ~500 G4C2 • G2C4 repeats.
In the C9–500/32 line, two copies of the transgene were integrated between mouse chromosome 18 (−) 17 919 900 and the flipped chromosome 18 (+) strand at position 18 526 504 (Supplementary Material, Fig. S1A). No annotated genes were found in this region and no decrease in coverage between the breakpoints was observed, indicating a small mouse chromosomal rearrangement but not a mouse chromosomal deletion occurred in this region (Supplementary Material, Fig. S1A). In the C9–500/32 line, the first copy of the integrated human transgene contains 46.6 kb of upstream and 19.4 kb of downstream flanking sequences and the full-length copy of the C9orf72 gene. The second transgene copy contains the full 51.6 kb upstream flanking region and a small portion of C9orf72 which terminates 3′ of the repeat within the first intron. Southern blot analyses show that the larger repeat expansion is located in the full-length C9orf72 copy and the shorter expansion in the second copy containing the truncated C9orf72 gene (Supplementary Material, Fig. S1B and C).
The C9–36/29 line has three copies of the full-length C9orf72 gene, and one truncated copy inserted into the second intron of metallophosphoesterase 1 (Mppe1) (Supplementary Material, Fig. S2A). Although the deletion of exons 3–4 of Mppe1 is predicted to lead to the expression of a truncated protein lacking the N-terminal region and a portion of the metallophosphoesterase domain, qRT-PCR and RNA sequencing detected no significant differences in transcript levels including over exons 3 and 4 (Supplementary Material, Fig. S2B and C). While it is possible that a truncated metallophosphoesterase protein is expressed and could cause some type of deleterious effect, no overt phenotypic differences were found in this line compared to the other phenotypic lines.
The C9–37 line has a single transgene insertion site on chromosome 4 at a position with no annotated genes. This insertion contains a partial copy of C9orf72 extending from exon 1 into intron 9 plus 19.5 kb of endogenous human upstream flanking sequence. Due to the position of the transgene break, the pCC1BAC backbone plus an additional 17.3 kb of 3′ sequence is integrated further upstream of C9orf72 (Supplementary Material, Fig. S2D). The C9–37 line, which lacks the 3′ end of C9orf72, is the only line that does not contain a full-length copy of the transgene, and the only line that does not develop overt ALS/FTD phenotypes.
To further compare these transgenic lines, we measured the levels of exon1a-containing sense RNA transcripts by qRT-PCR and polyGP RAN protein levels using a meso scale discovery (MSD) assay (Supplementary Material, Fig. S3). As previously described (19), qRT-PCR shows that the levels of sense expansion-containing RNA transcripts in the brain from each of the four C9-BAC lines correlate with the number of transgene copies (Supplementary Material, Fig. S3A). In contrast, the levels of polyGP RAN proteins are the highest in the most penetrant C9–500 line in both the cortex and cerebellum (Supplementary Material, Fig. S3B).
In summary, three independent C9orf72 BAC lines show similar ALS/FTD phenotypes (19), strongly supporting the hypothesis that these phenotypes are caused by the repeat expansion mutation and not gene disruptions or other changes at the various integration sites. Of the two lines with relatively short repeats (C9–36/29 vs C9–37), only the C9–36/29 line containing 4 copies of transgenes showed significant differences in survival from NT mice (19). This difference could result from the higher expression of the relatively small G4C2 expansions in the phenotypic C9–36/29 line (4 transgene copies) compared with the C9–37 line (1 transgene copy), the accumulation of GA RAN protein aggregates (19) and higher GP RAN protein levels in some C9–36/29 compared to C9–37 animals (Supplementary Material, Fig. S3B) or the truncation of the C9orf72 gene found in the C9–37 line. The C9–500 line has one copy of the full-length human transgene, no evidence of integration effects on phenotypes, the most penetrant phenotype among the lines (19) and the highest expression of RAN proteins, making the C9–500 mice an attractive model for therapeutic development. We focused our subsequent analyses on mice from the C9–500 line.
C9orf72 protein overexpression not associated with disease in C9–500 mice
Similar to the C9–500 mice developed at UF (19), O’Rourke et al. (20) developed an independent C9-BAC mouse model (Baloh-Jax) using a different BAC construct with the full-length C9orf72 gene and additional flanking sequence on the C57BL/6 background. In contrast to our model, these mice carry multiple copies of the BAC transgene containing ~100–1000 G4C2 repeats and do not develop behavioral or neurodegenerative features of C9orf72 ALS/FTD. To study if the integration of the transgene results in an upregulation of C9orf72 protein levels that might contribute to phenotypes seen in the C9–500 line, we compared the levels of the C9orf72 protein in 20-week-old female C9–500 mice with age-matched NT mice and Baloh-Jax transgenic mice. Splice variants of the C9orf72 mRNA generate two isoforms of the protein with predicted sizes of ~55 kDa and ~35 kDa (48). Commercially available antibodies detect the long isoform. We used the C9orf72 Genetex antibody that detects both human and mouse C9orf72 protein (49) and confirmed antibody specificity by showing that the 55 kDa protein is detected in the control but not C9orf72 KO brain lysates (generously donated by Dr Robert. H. Baloh) (Fig. 1D). Cortical brain lysates from the Baloh-Jax mice show a 3.4-fold elevation of C9orf72 protein compared to NT controls (P < 0.003). In contrast, no significant upregulation in C9orf72 protein was found in C9–500 compared to NT mice (Fig. 1E and F) and the Baloh-Jax mice had a 1.6-fold elevation of protein compared to the C9–500 mice (P < 0.019).
In summary, elevated levels of C9orf72 protein found in the asymptomatic Baloh-Jax mice, but not the phenotypic C9–500 mice, suggest any modest elevations of the C9orf72 protein found in the UF C9–500 line are unlikely to contribute to the ALS/FTD phenotypes.
Neuroinflammatory transcriptome changes predominate at end-stage
RNA dysregulation is thought to play a role in C9orf72 ALS/FTD (27,50,51) but little is known about how the transcriptome is affected early in the course of disease or how these changes progress during the course of disease. In our initial description of these mice (19), we showed that a subset of C9(+) females develop acute phenotypes that progress rapidly, while other mice develop phenotypes later in life and generally have a more slowly progressive disease course. To understand transcriptomic changes that occur in early versus later stages of disease, we performed RNA sequencing on frontal cortex samples from C9–500 mice with similar ages but at different stages of disease compared to NT controls. These groups included 10 female C9–500 mice at 20 weeks of age with no overt cage behavior abnormalities (C9+ presymptomatic), four C9+ animals that had developed acute rapidly progressive phenotypes (20–22 weeks old) (Acute) and three NT controls.
Sample to sample correlation based on differential gene expression measured using DESeq2 shows that the Acute cohort of C9–500 mice have unique gene expression profiles. Additionally, global gene expression changes in the NT animals are similar to the C9(+) animals but Acute animals were significantly different from both the NT and C9(+) presymptomatic animals (Fig. 2A). In the Acute mice, we identified 2514 upregulated and 2921 downregulated genes compared to NT animals, with false discovery rate (FDR) < 0.05 (Fig. 2A). A heat map of the top 50 significant genes in the Acute vs NT mice is shown in Supplementary Material, Figure S4. Gene ontology (GO) analyses in the Acute vs NT mice shows that the upregulated pathways include negative regulation of cell proliferation, inflammatory response and actin cytoskeleton organization (P-values indicated on the left) (Fig. 2B). Significantly downregulated pathways include brain development, synaptic organization and neuron projection development (Fig. 2B). Since the Acute C9-BAC mice mimic the neuropathology seen in end-stage C9-ALS patients, we compared the gene expression profiles of the cortex from Acute mice and C9-ALS patients. RNA sequencing data obtained from the Prudencio et al. (27) study were reanalyzed using STAR (52) for alignment and Kallisto (53) to obtain transcript per million (TPM) values. Using these parameters, we identified 36 genes that were consistently dysregulated between C9-ALS and unaffected individuals in this dataset. Of these, 15 of the 36 were also dysregulated in our Acute mice, an ~2-fold greater overlap than expected by chance (P < 0.04, hypergeometric test), including SerpinH1 (Supplementary Material, Fig. S5). This gene belongs to the serine protease inhibitor family, and several members of this gene family, including SerpinA3 and SerpinA1 are hypothesized to disrupt neuronal function and have been found to be differentially expressed in C9orf72 ALS patient autopsy tissue (27,28). In contrast, no significant differences in overall gene expression were observed between NT and C9(+) presymptomatic mice.
Figure 2.
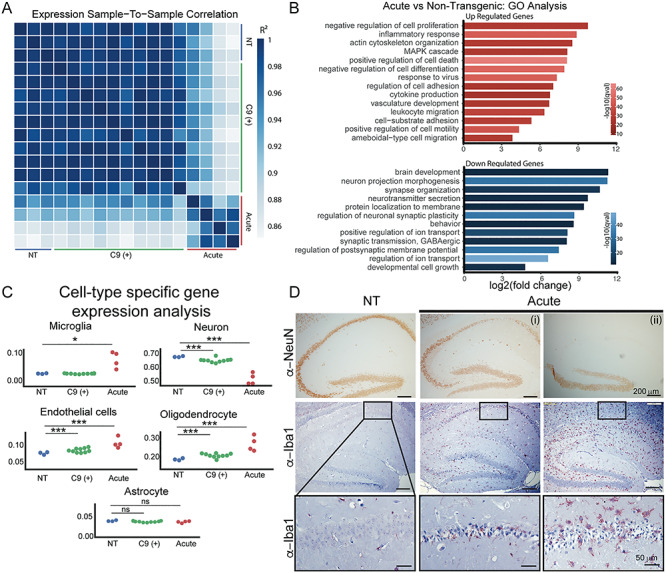
Prominent neurodegenerative and neuroinflammatory changes in acute end-stage C9-BAC mice (A) Sample-to-sample correlation plot shows increased correlation in gene expression changes among Acute mice and C9(+) presymptomatic mice but not between groups. (B) Gene ontology analyses of gene expression changes in Acute vs NT mice. (C) Cell-type enrichment analyses in NT, C9(+) presymptomatic and Acute mice. Statistical tests and significance are shown in Table 1. (D) Immunohistochemistry of Acute C9-BAC mice stained for neuronal (NeuN) and microglial (Iba1) markers. Inset shows zoom-in of microglial staining. Acute (i) and (ii) show variations in pathology that are seen in two different Acutely affected end-stage C9-BAC mice.
Since a large number of neuroinflammatory and neurodegenerative gene expression changes in the Acute cohort may have resulted from inflammatory and neurodegenerative processes that cause loss of neurons or increases in inflammatory cells, we estimated cell type changes in each sample. For this analysis, we used publicly available datasets that had characterized gene expression in seven different cell types (neurons, microglia, astrocytes, endothelial cells, oligodendrocyte precursor cells, myelinating oligodendrocytes and newly formed oligodendrocytes) in the mouse brain (55). Because there is a considerable overlap between the expression profiles of oligodendrocyte precursor cells, myelinating oligodendrocytes and newly formed oligodendrocytes, we combined these cell types into a single category called ‘oligodendrocytes’. We observed an increase in the estimated proportion of gene expression changes specific for microglial, oligodendrocyte and endothelial cells in Acute vs NT mice, and a decrease in estimated proportion of gene expression changes specific for neurons in Acute vs NT mice (Fig. 2C, Table 1). Consistent with these findings, Acute mice IHC showed overt loss of immunoreactivity to the neuronal marker NeuN and increased staining of the microglial marker Iba1 in the Acute mice compared to NT mice (Fig. 2D). Additionally, significant albeit small differences in gene expression were found between C9(+) presymptomatic and NT mice in estimated changes specific to endothelial, oligodendrocyte and neuronal cells, but not microglia (Fig. 2C, Table 1) and consistent with these more subtle changes, no overt pathology was found in the hippocampus (Supplementary Material, Fig. S6) of C9(+) presymptomatic mice compared to NT mice.
Table 1.
Cell type analysis on transcriptome data from NT, C9(+) presymptomatic and Acute mice
| Samples | Mean 1 | Mean 2 | Statistics | P-value | P adj | |
|---|---|---|---|---|---|---|
| Endothelial cells | ||||||
| NT vs C9(+) | 0.075 | 0.084 | −13.735 | <0.0001 | <0.0001 | |
| NT vs Acute | 0.075 | 0.106 | −31.664 | <0.0001 | <0.0001 | |
| C9(+) vs Acute | 0.084 | 0.106 | −27.118 | <0.0001 | <0.0001 | |
| Microglia | ||||||
| NT vs C9(+) | 0.025 | 0.025 | −2.22 | 0.02 | 1 | |
| NT vs Acute | 0.025 | 0.075 | −52.523 | <0.0001 | <0.0001 | |
| C9(+) vs Acute | 0.025 | 0.075 | −53.598 | <0.0001 | <0.0001 | |
| Astrocyte | ||||||
| NT vs C9(+) | 0.037 | 0.035 | 0.651 | 0.5 | 1 | |
| NT vs Acute | 0.037 | 0.034 | 3.575 | <0.0001 | 0.7 | |
| C9(+) vs Acute | 0.035 | 0.034 | 4.256 | <0.0001 | 0.04 | |
| Neuron | ||||||
| NT vs C9(+) | 0.68 | 0.65 | 25.62 | <0.0001 | <0.0001 | |
| NT vs Acute | 0.68 | 0.52 | 53.21 | <0.0001 | <0.0001 | |
| C9(+) vs Acute | 0.65 | 0.52 | 54.08 | <0.0001 | <0.0001 | |
| Oligodendrocytes | ||||||
| NT vs C9(+) | 0.186 | 0.207 | −18.742 | <0.0001 | <0.0001 | |
| NT vs Acute | 0.186 | 0.269 | −46.016 | <0.0001 | <0.0001 | |
| C9(+) vs Acute | 0.207 | 0.269 | −44.485 | <0.0001 | <0.0001 | |
Taken together, robust changes in gene expression were seen in Acute mice compared to NT littermates. These changes are consistent with the neuronal loss and increased numbers of microglia in Acute compared to NT mice. Additionally, both Acute and presymptomatic C9(+) animals showed changes in cell type-specific genes associated with neurons, endothelial cells and oligodendrocytes compared to NT mice, and these changes increased with increased disease severity.
Alternative splicing changes characterize disease states in ALS/FTD
Changes in alternative splicing and alternative polyadenylation were previously reported in autopsy brains from C9-positive ALS but not C9-negative sporadic ALS cases (27). We examined alternative splicing variants in C9–500 mice using MISO analyses and show that 240 and 539 genes showed alternative splicing changes in C9(+) presymptomatic vs NT and C9(+) Acute vs NT cohorts, respectively (Fig. 3A). These alternative splicing abnormalities provide a robust molecular signature of C9orf72 ALS/FTD. A total of 83 of these alternative splicing events (intersection events) were significantly different in both the Acute and C9(+) presymptomatic cohorts (Fig. 3A and B) compared to NT mice, and the psi values of these 83 shared splicing events was higher in animals with acute disease compared to the C9(+) presymptomatic cohort. These data indicate that splicing changes occur in C9(+) animals prior to the onset of overt phenotypes and suggest that these changes worsen with disease progression. For example, Elavl2, an RNA-binding protein enriched in the neurons that affect neuronal excitability (56), was found to be alternatively spliced in both C9(+) presymptomatic animals and Acute animals and the psi values were highest in the acutely affected animals (Fig. 3C). A summary of the alternatively spliced events found in both C9(+) presymptomatic and Acute animals is shown in Figure 3D. Although beyond the scope of this study, these genes may be useful as biomarkers to monitor disease progression in C9-ALS/FTD patients (Fig. 3D, Table 2).
Figure 3.
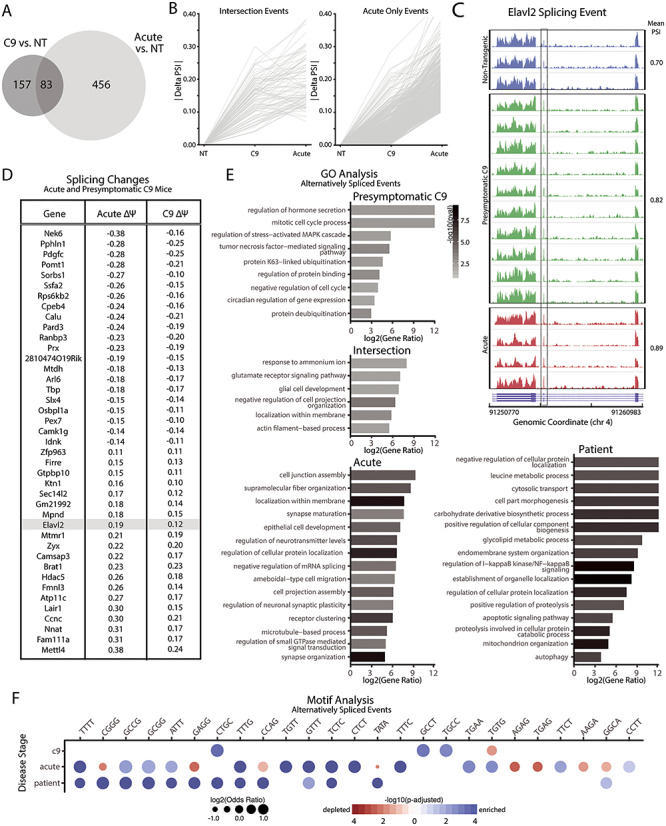
Abundant alternative splicing changes in C9-BAC mice. (A) Venn diagram showing number of alternative splicing changes in Acute and presymptomatic mice compared to NT mice. (B) Alternative splicing events found in both presymptomatic and Acute animals show delta psi values for a group of markers increase with disease severity. (C) Elavl2 is alternatively spliced in both the C9(+) presymptomatic [C9(+)] and Acute mice. (D) Gene IDs and corresponding Δpsi values in C9(+) presymptomatic and Acute animals. Additional information on alternative splicing events is provided in Table 2. (E) Gene Ontology categories are shown for alternatively spliced events found only in C9(+) presymptomatic mice, both presymptomatic and Acute animals (intersection), only Acute animals and C9-ALS patients. (F) Motif analysis of significant alternative splicing changes in C9(+) presymptomatic mice, acute mice and C9-ALS patients shows no enrichment of single motif or unique signature of sequestration of single RNA-binding protein.
Table 2.
Common alternative splicing events in C9(+) presymptomatic and Acute mice
| #Event | symb | Acute ∆psi | Acute P-value | C9(+) ∆psi | C9 (+) P-value |
|---|---|---|---|---|---|
| chr2:38514581:38514658:+@chr2:38514733:38514864:+@chr2:38550553:38550671:+ | Nek6 | -0.38 | 0.03 | -0.16 | 0.02 |
| chr15:93410692:93410783:+@chr15:93420284:93420490:+@chr15:93424014:93424075:+ | Pphln1 | -0.29 | 0.03 | -0.25 | 0.02 |
| chr3:81036416:81037554:+@chr3:81131018:81131076:+@chr3:81141405:81141600:+ | Pdgfc | -0.29 | 0.03 | -0.25 | 0.01 |
| chr2:32244234:32244389:+@chr2:32244786:32244950:+@chr2:32245547:32245677:+ | Pomt1 | -0.28 | 0.03 | -0.21 | 0.01 |
| chr19:40344356:40344439:-@chr19:40340045:40340146:-@chr19:40336986:40337035:- | Sorbs1 | -0.27 | 0.03 | -0.10 | 0.01 |
| chr2:79657147:79658349:+@chr2:79658670:79658846:+@chr2:79660233:79660310:+ | Ssfa2 | -0.26 | 0.03 | -0.15 | 0.02 |
| chr19:4161068:4161215:-@chr19:4159566:4159705:-@chr19:4159403:4159460:- | Rps6kb2 | -0.26 | 0.03 | -0.16 | 0.01 |
| chrX:74018130:74018366:-@chrX:74017901:74018017:-@chrX:74017130:74017511:- | Irak1 | -0.14 | 0.03 | -0.15 | 0.02 |
| chr11:31908825:31908875:+@chr11:31918801:31918824:+@chr11:31920409:31920582:+ | Cpeb4 | -0.24 | 0.03 | -0.16 | 0.03 |
| chr6:29361294:29361487:+@chr6:29361560:29361753:+@chr6:29366040:29366206:+ | Calu | -0.24 | 0.03 | -0.21 | 0.02 |
| chr5:5556065:5556156:-@chr5:5555238:5555406:-@chr5:5546353:5546405:- | Gtpbp10 | 0.11 | 0.03 | 0.12 | 0.03 |
| chr8:127409556:127409707:+@chr8:127410729:127410773:+@chr8:127415570:127415797:+ | Pard3 | -0.24 | 0.03 | -0.19 | 0.01 |
| chr17:56686006:56686061:+@chr17:56692521:56692721:+@chr17:56696602:56696638:+ | Ranbp3 | -0.23 | 0.03 | -0.20 | 0.03 |
| chr7:27507947:27508103:+@chr7:27515251:27515447:+@chr7:27516040:27520041:+ | Prx | -0.23 | 0.03 | -0.19 | 0.03 |
| chr6:112644588:112644747:-@chr6:112628462:112628524:-@chr6:112619851:112620904:- | Rad18 | -0.20 | 0.03 | -0.20 | 0.03 |
| chr6:149310322:149310377:+@chr6:149324732:149324800:+@chr6:149325405:149329784:+ | 2810474O19Rik | -0.20 | 0.03 | -0.15 | 0.04 |
| chr15:34117880:34118110:+@chr15:34121423:34121512:+@chr15:34123668:34123766:+ | Mtdh | -0.18 | 0.03 | -0.13 | 0.02 |
| chr2:124298575:124298655:+@chr2:124400243:124400365:+@chr2:124489985:124490012:+ | Sema6d | 0.12 | 0.03 | 0.12 | 0.03 |
| chr16:59618795:59618850:-@chr16:59618666:59618678:-@chr16:59613321:59613844:- | Arl6 | -0.18 | 0.03 | -0.17 | 0.03 |
| chr17:15499888:15500024:+@chr17:15501154:15501202:+@chr17:15502962:15503128:+ | Tbp | -0.18 | 0.03 | -0.17 | 0.03 |
| chr16:3984991:3987176:-@chr16:3982979:3983242:-@chr16:3981585:3981687:- | Slx4 | -0.15 | 0.03 | -0.14 | 0.01 |
| chr2:22957018:22957118:-@chr2:22950184:22950270:-@chr2:22946576:22946761:- | Abi1 | 0.16 | 0.03 | 0.17 | 0.03 |
| chr18:12862107:12862713:-@chr18:12841184:12841346:-@chr18:12819505:12819661:- | Osbpl1a | -0.15 | 0.03 | -0.11 | 0.01 |
| chr10:19904563:19904713:-@chr10:19894725:19894802:-@chr10:19894250:19894358:- | Pex7 | -0.14 | 0.03 | -0.10 | 0.03 |
| chr1:193362013:193362133:-@chr1:193361580:193361667:-@chr1:193359719:193359847:- | Camk1g | -0.14 | 0.03 | -0.14 | 0.02 |
| chr13:58160152:58160238:+@chr13:58162838:58162881:+@chr13:58163433:58164693:+ | Idnk | -0.14 | 0.03 | -0.11 | 0.03 |
| chr8:69744461:69744587:-@chr8:69744192:69744252:-@chr8:69742859:69743269:- | Zfp963 | 0.11 | 0.03 | 0.11 | 0.02 |
| chrX:50573224:50573351:-@chrX:50573015:50573141:-@chrX:50569277:50570291:- | Firre | 0.15 | 0.03 | 0.13 | 0.02 |
| chr5:5557213:5557406:-@chr5:5555238:5555382:-@chr5:5547927:5548000:- | Gtpbp10 | 0.15 | 0.03 | 0.11 | 0.04 |
| chr16:87914154:87914404:-@chr16:87896809:87896895:-@chr16:87895897:87896351:- | Grik1 | -0.18 | 0.03 | -0.20 | 0.03 |
| chr14:47704187:47704277:+@chr14:47704420:47704488:+@chr14:47705763:47705800:+ | Ktn1 | 0.16 | 0.03 | 0.10 | 0.03 |
| chr10:14760289:14760710:+@chr10:14766677:14767025:+@chr10:14770155:14770556:+ | Nmbr | -0.12 | 0.03 | -0.17 | 0.02 |
| chr11:4118625:4118729:-@chr11:4116681:4116756:-@chr11:4111894:4111937:- | Sec14l2 | 0.17 | 0.03 | 0.12 | 0.03 |
| chr19:4811172:4811634:-@chr19:4793926:4794009:-@chr19:4792398:4792820:- | Gm21992 | 0.18 | 0.03 | 0.14 | 0.04 |
| chr17:56011866:56011962:+@chr17:56012271:56012492:+@chr17:56014941:56015109:+ | Mpnd | 0.18 | 0.03 | 0.15 | 0.03 |
| chr4:91260761:91260986:-@chr4:91254141:91254179:-@chr4:91250767:91253570:- | Elavl2 | 0.19 | 0.03 | 0.12 | 0.03 |
| chrX:71376887:71376910:+@chrX:71379014:71379064:+@chrX:71383200:71383275:+ | Mtmr1 | 0.21 | 0.03 | 0.19 | 0.03 |
| chr17:85615594:85615743:-@chr17:85614648:85614727:-@chr17:85609090:85611748:- | Six3os1 | -0.17 | 0.03 | -0.25 | 0.01 |
| chr7:96694770:96695025:+@chr7:96702642:96702740:+@chr7:96703926:96704161:+ | Tenm4 | 0.17 | 0.03 | 0.19 | 0.01 |
| chr6:42350946:42351148:+@chr6:42351270:42351362:+@chr6:42351475:42351969:+ | Zyx | 0.22 | 0.03 | 0.20 | 0.01 |
| chr8:3606267:3606406:+@chr8:3606504:3606820:+@chr8:3607791:3607890:+ | Camsap3 | 0.22 | 0.03 | 0.17 | 0.03 |
| chr5:140713053:140713172:+@chr5:140714293:140714384:+@chr5:140714567:140714685:+ | Brat1 | 0.23 | 0.03 | 0.23 | 0.01 |
| chr11:102224734:102224955:-@chr11:102219145:102219281:-@chr11:102218416:102218781:- | Hdac5 | 0.26 | 0.03 | 0.18 | 0.03 |
| chr15:99319131:99319254:-@chr15:99318918:99319017:-@chr15:99317223:99318372:- | Fmnl3 | 0.26 | 0.03 | 0.14 | 0.04 |
| chr2:124298575:124298655:+@chr2:124400243:124400364:+@chr2:124489985:124490012:+ | Sema6d | 0.11 | 0.03 | 0.18 | 0.02 |
| chr5:32785549:32785626:-@chr5:32774396:32774466:-@chr5:32764773:32764948:- | Pisd | -0.12 | 0.03 | -0.17 | 0.04 |
| chrX:60295189:60295292:-@chrX:60293607:60293657:-@chrX:60292319:60292383:- | Atp11c | 0.27 | 0.03 | 0.17 | 0.04 |
| chr7:4010466:4010503:-@chr7:4010251:4010300:-@chr7:4007073:4009834:- | Lair1 | 0.30 | 0.03 | 0.15 | 0.03 |
| chr4:21746656:21746774:+@chr4:21747483:21747576:+@chr4:21748137:21750546:+ | Ccnc | 0.30 | 0.03 | 0.21 | 0.02 |
| chr2:157560448:157560535:+@chr2:157561213:157561293:+@chr2:157561542:157562519:+ | Nnat | 0.31 | 0.03 | 0.17 | 0.02 |
| chr19:12577090:12577170:+@chr19:12583923:12584122:+@chr19:12586835:12589696:+ | Fam111a | 0.31 | 0.03 | 0.17 | 0.04 |
| chr17:94747617:94748417:-@chr17:94743932:94743994:-@chr17:94740386:94740755:- | Mettl4 | 0.38 | 0.03 | 0.24 | 0.01 |
| chr12:101082451:101084025:+@chr12:101084284:101084499:+@chr12:101088042:101088927:+ | D130020L05Rik | -0.17 | 0.03 | -0.30 | 0.01 |
| chr6:145211170:145211244:+@chr6:145214522:145214784:+@chr6:145215184:145215271:+ | Etfrf1 | 0.37 | 0.03 | 0.26 | 0.03 |
| chr7:73514090:73514151:-@chr7:73507806:73507913:-@chr7:73504988:73505128:- | Chd2 | -0.23 | 0.03 | -0.15 | 0.03 |
| chr10:128645721:128645993:-@chr10:128643875:128643968:-@chr10:128642629:128642733:- | Ikzf4 | -0.31 | 0.03 | -0.16 | 0.04 |
| chr8:85841002:85841103:+@chr8:85843021:85843112:+@chr8:85875634:85875723:+ | Phkb | -0.15 | 0.03 | -0.16 | 0.02 |
| chr18:59062381:59062562:+@chr18:59065835:59065885:+@chr18:59072193:59072417:+ | A730017C20Rik | -0.20 | 0.03 | -0.20 | 0.03 |
| chr7:123179717:123179859:+@chr7:123180249:123180395:+@chr7:123180540:123180677:+ | Tnrc6a | -0.34 | 0.03 | -0.19 | 0.04 |
| chr11:95046921:95047046:-@chr11:95046449:95046590:-@chr11:95044482:95045763:- | Itga3 | 0.26 | 0.03 | 0.22 | 0.03 |
| chrX:135751945:135752017:+@chrX:135796142:135796336:+@chrX:135797553:135797629:+ | Gprasp1 | -0.24 | 0.03 | -0.08 | 0.04 |
| chr4:118234959:118235103:-@chr4:118231486:118231791:-@chr4:118230695:118230888:- | Ptprf | 0.18 | 0.03 | 0.05 | 0.04 |
| chr16:45408856:45409053:-@chr16:45400222:45400303:-@chr16:45396969:45397295:- | Cd200 | -0.15 | 0.03 | -0.04 | 0.02 |
| chr2:71228600:71228690:+@chr2:71233649:71233708:+@chr2:71235915:71236030:+ | Dync1i2 | -0.15 | 0.03 | -0.06 | 0.03 |
| chrX:151835224:151835292:+@chrX:151836222:151836329:+@chrX:151840056:151840154:+ | Huwe1 | -0.13 | 0.03 | -0.04 | 0.03 |
| chr9:31157695:31157847:-@chr9:31156190:31156225:-@chr9:31153414:31153499:- | Aplp2 | -0.12 | 0.03 | -0.04 | 0.03 |
| chr3:146458743:146458840:+@chr3:146459679:146459840:+@chr3:146463899:146465771:+ | Ctbs | 0.14 | 0.03 | 0.06 | 0.04 |
| chr11:70609166:70609254:+@chr11:70609408:70609431:+@chr11:70609554:70609717:+ | Mink1 | -0.15 | 0.03 | -0.07 | 0.02 |
| chr4:11287475:11287633:-@chr4:11285078:11285173:-@chr4:11281004:11281124:- | Dpy19l4 | -0.13 | 0.03 | -0.07 | 0.03 |
| chr11:28434081:28434157:-@chr11:28396046:28396165:-@chr11:28385692:28388948:- | Ccdc85a | 0.15 | 0.03 | 0.09 | 0.04 |
| chr4:9635901:9635969:-@chr4:9624360:9624401:-@chr4:9610843:9610938:- | Asph | -0.12 | 0.03 | -0.06 | 0.04 |
| chr8:77155202:77155318:+@chr8:77187520:77187664:+@chr8:77210644:77210774:+ | Nr3c2 | -0.13 | 0.03 | -0.07 | 0.03 |
| chr13:107022012:107022114:-@chr13:106996747:106997129:-@chr13:106993848:106993942:- | Kif2a | 0.12 | 0.03 | 0.06 | 0.04 |
| chr7:109960074:109960422:-@chr7:109935482:109935553:-@chr7:109934693:109934802:- | Dennd5a | 0.14 | 0.03 | 0.08 | 0.04 |
| chr10:81314992:81315118:+@chr10:81317326:81317403:+@chr10:81317766:81319974:+ | Pip5k1c | -0.14 | 0.03 | -0.08 | 0.04 |
| chrX:74337911:74338054:+@chrX:74338134:74338368:+@chrX:74338459:74338601:+ | Plxna3 | 0.12 | 0.03 | 0.08 | 0.02 |
| chrX:95166424:95166486:-@chrX:95165593:95165970:-@chrX:95130836:95131015:- | Arhgef9 | -0.11 | 0.03 | -0.07 | 0.01 |
| chrX:152178964:152179090:+@chrX:152180997:152181092:+@chrX:152202908:152203169:+ | Iqsec2 | 0.11 | 0.03 | 0.09 | 0.03 |
| chr16:32898878:32898939:+@chr16:32900633:32900707:+@chr16:32902419:32902545:+ | Fyttd1 | -0.10 | 0.03 | -0.09 | 0.03 |
| chr11:75349669:75349814:+@chr11:75382250:75382394:+@chr11:75387411:75387500:+ | Smyd4 | -0.08 | 0.03 | -0.10 | 0.03 |
| chr4:129600983:129601134:+@chr4:129601407:129601533:+@chr4:129602195:129602261:+ | Tmem234 | 0.09 | 0.03 | 0.12 | 0.03 |
| chr2:31950529:31950590:+@chr2:31962215:31962281:+@chr2:31969630:31969792:+ | Aif1l | -0.07 | 0.03 | -0.13 | 0.04 |
| chr5:81771584:81771680:+@chr5:81792361:81792412:+@chr5:81794264:81795347:+ | Adgrl3 | 0.10 | 0.03 | 0.17 | 0.01 |
GO analyses were used to better understand the categories of alternatively spliced genes that show changes in presymptomatic and Acute mice and changes common to presymptomatic and acutely affected animals (Fig. 3E). MISO was also performed to detect alternative splicing changes in previously published RNAseq data from human autopsy tissue (Fig. 3E). Splicing changes occurring in both acutely affected mice and end-stage C9-ALS patients were enriched for several similar GO categories including genes with alternative splicing abnormalities that are normally involved in neuronal death, oxidative stress, cytoskeletal pathways and inflammation. In contrast, pathways dysregulated in presymptomatic mice include synaptic transmission and membrane localization.
In summary, we show that alternative splicing abnormalities increase with disease severity in C9-BAC mice and hence may provide useful biomarkers and tools to understand disease progression.
Motif analyses of intronic regions flanking exons dysregulated in C9 ALS/FTD
In myotonic dystrophy, a well-established RNA gain-of-function disease, CUG or CCUG expansion RNAs sequester MBNL proteins into intranuclear foci, thus preventing their normal function in regulating posttranscriptional processing including alternative splicing and polyadenylation (57). As expected, sequence analyses of abnormally spliced exons from DM1 skeletal muscle are most significantly enriched for MBNL YGCY binding motifs (Supplementary Material, Fig. S7).
In contrast to DM1, sequence analyses of abnormal splicing events in C9(+) presymptomatic mice and Acute mice shows the enrichment of a larger number and more diverse set of tetramer motifs, including AT and multiple types of GC-rich repeat motifs (Fig. 3F). A diverse set of repeat motifs are also enriched in the C9-ALS patient splicing data (Fig. 3F). In contrast to the prominent involvement of a single category of RBP in DM1, the multiple types of RNA-binding motifs found in the dysregulated genes in C9 ALS/FTD are consistent with published literature and indicate that C9orf72 RNA dysregulation may involve a larger group of RNA-binding proteins, or may reflect concurrent processes that prevent unambiguous identification of a single RBP driver of disease (Fig. 3F).
In contrast to DM1, motif enrichment analyses show that the alternative splicing changes found in our C9-BAC mice are unlikely to be caused by sequestration of a single category of RBP.
Repeat length increases penetrance and decreases survival in allelic series of C9-BAC mice
Repeat length is a known modifier of disease severity in multiple repeat expansion diseases including Huntington disease, DM1 and multiple spinocerebellar ataxias (55,59). Somatic instability and technical difficulties measuring G4C2 repeat length in human C9orf72 patients have made the contribution of repeat length to age of onset and disease severity in C9orf72 ALS/FTD difficult to determine. To test the hypothesis that the length of the G4C2 repeat is an important modifier of age of onset and disease risk, we established an allelic series of mice from our most penetrant BAC transgenic line (C9–500). Taking advantage of intergenerational repeat instability observed during the maintenance of our colony (19), we selected and bred C9–500 animals that had repeat contractions or expansions and established sublines with 50 (C9–50) or 800 (C9–800) repeats (Fig. 4A and B). Limited somatic repeat instability was observed between tail and brain DNA from the 50 and 800 lines (Fig. 4B). The limited somatic instability of the G4C2• G2C4 expansion, the single-copy of the C9orf72 transgene and the identical insertion site shared by the C9–800, C9–500 and C9–50 sublines make these mice an ideal resource to test the effects of repeat length on age of onset and disease penetrance.
Figure 4.
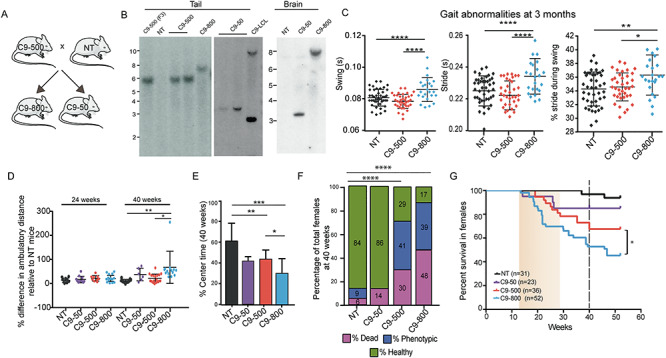
Earlier onset and increased penetrance in isogenic C9 sublines with longer G4C2 expansions. (A) Breeding strategy of expansion and contraction lines established from the C9–500 animals with spontaneous intergenerational instability. (B) Southern blots of tail and brain DNA from F3 C9–500 mice and C9–800 and C9–50 sublines. C9-LCL is a lymphoblastoid cell line obtained from a C9-ALS patient. (C) Gait abnormalities in C9–800 mice at 3 months of age. NT n = 44, C9–500 n = 38, C9–800 n = 24. All parameters listed in Table 3. (D) Open field analyses comparing the percentage change in ambulatory distance in C9–50, C9–500 and C9–800 mice compared to NT mice at 24 weeks (NT n = 9, C9–50 = 10, C9–500 = 9, C9–800 = 13) and 40 weeks (NT n = 9, C9–50 = 6, C9–500 = 18, C9–800 = 10). (E) Open field analyses showing percentage time spent in center of chamber for NT, C9–50, C9–500, C9–800 cohorts (NT n = 9, C9–50 = 6, C9–500 = 18, C9–800 = 10). (F) Population census with percentage of female mice that are sick, healthy and phenotypic using multifactorial scoring criteria. (G) Kaplan–Meier survival curves of female mice from NT (n = 31), C9–50 (n = 23), C9–500 (n = 36) and C9–800 (n = 52) cohorts. Data information: Statistical analyses for panels D and E were performed using one-way ANOVA and Tukey’s multiple comparison test with mean +/− SEM shown; not significant (ns) = P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. In (F), Chi square test was used, ****P < 0.0001. In (G) Gehan–Breslow–Wilcoxon test, *P < 0.05.
To understand the effects of repeat length on disease, we performed a series of experiments on female mice from this allelic series. First, we performed DigiGait analyses at an early timepoint to test if phenotypes in the C9–800 mice were detected earlier than in the C9–500 cohort. At 12 weeks of age, C9–800 mice showed abnormalities in 9 of 42 DigiGait parameters while the C9–500 mice showed six differences compared to NT mice (Table 3). Additionally, 12 parameters were different between C9–800 and C9–500 mice (Fig. 4C) including three key parameters typically involved in ALS: swing time, stride time and time of a stride when paw is in swing motion (60,61). These data show gait abnormalities are found earlier in mice with longer repeats.
Table 3.
DigiGait parameters of 12-week-old females
| Parameters (41 in total) | Unit | NT vs C9–500 | NT vs C9–800 | C9–500 vs C9–800 |
|---|---|---|---|---|
| Significant Parameters (Desired FDR = 5%) | (real#) | 6 | 9 | 12 |
| Swing | (s) | ns | * * * * P < 0.0001 | * * * * P < 0.0001 |
| Swing/Stride | (%) | ns | * * P = 0.0052 | * P = 0.0176 |
| Brake | (s) | ns | ns | ns |
| Brake/Stride | (%) | ns | ns | ns |
| Propel | (s) | ns | ns | * P = 0.0255 |
| Propel/Stride | (%) | ns | ns | ns |
| Stance | (s) | * P = 0.0444 | ns | * * P = 0.0020 |
| Stance/Stride | (%) | ns | * * P = 0.0052 | * P = 0.0174 |
| Stride | (s) | ns | * * * * P < 0.0001 | * * * * P < 0.0001 |
| Brake/Stance | (%) | ns | ns | ns |
| Propel/Stance | (%) | ns | ns | ns |
| Stance/Swing | (real#) | ns | * P = 0.0134 | ns |
| Stride Length | (cm) | ns | * * * * P < 0.0001 | * * * * P < 0.0001 |
| Stride Frequency | (steps/s) | * P = 0.0292 | * * * P = 0.0001 | * * * * P < 0.0001 |
| Paw Angle | (deg) | ns | ns | ns |
| Absolute Paw Angle | (deg) | ns | ns | ns |
| Paw Angle Variability | (deg) | ns | ns | ns |
| Stance Width | (cm) | * P = 0.0292 | ns | ns |
| Step Angle | (deg) | ns | ns | ns |
| Stride Length Variability | (cm) | ns | ns | ns |
| Stance Width Variability | (cm) | * * P = 0.059 | ns | ns |
| Step angle Variability | (deg) | ns | ns | * P = 0.0283 |
| #Steps | (real#) | ns | ns | ns |
| Stride Length CV | (CV%) | * * * * P < 0.0001 | * * * * P < 0.0001 | ns |
| Stance Width CV | (CV%) | * * * P = 0.0006 | ns | * P = 0.0243 |
| Step Angle CV | (CV%) | ns | ns | * P = 0.0202 |
| Swing Duration CV | (CV%) | ns | ns | ns |
| Paw Area at Peak Stance | (cm2) | ns | ns | ns |
| Paw Area Variability at Peak Stance | (cm2) | ns | ns | ns |
| Hind Limb Shared Stance Time | (s) | ns | ns | ns |
| Shared/Stance | (%) | ns | ns | ns |
| Stance Factor | (real#) | ns | ns | ns |
| Gait Symmetry | (real#) | ns | * * * P = 0.0001 | * * * * P < 0.0001 |
| MAX dA/dT | (cm2/s) | ns | ns | ns |
| MIN dA/dT | (cm2/s) | ns | ns | ns |
| Tau—Propulsion | (real#) | ns | ns | ns |
| Overlap Distance | (cm) | ns | ns | ns |
| Paw Placement Positioning | (cm) | ns | ns | ns |
| Ataxia Coefficient | (real#) | ns | ns | ns |
| Midline Distance | (cm) | ns | ns | ns |
| Paw Drag | (mm2) | ns | ns | ns |
#, number; CV, coefficient of variation; ns, not significant in one-way ANOVA; desired FDR (false discovery rate) set to 5%; NT n = 44; C9–500 n = 38; C9–800 n = 24.
Open field studies previously showed both slowed movement and hyperactivity in older C9–500 mice (12–18 months) (19). To test if increased repeat length causes earlier open field abnormalities, we examined the allelic series of mice earlier in disease at 24 and 40 weeks of age. At 24 weeks there were no differences in ambulatory distance between any of the C9–50, C9–500 and C9–800 sublines compared to NT animals. At 40 weeks the C9–800 group showed a significant increase in ambulatory distance compared to both the C9–500 and NT cohorts (Fig. 4D). In contrast, no significant differences were found between the C9–500 or C9–50 animals and NT controls at this age. Open field analyses were also used to measure decreases in center time, a phenotype associate with anxiety-like behavior. At 40 weeks, both the C9–500 and C9–800 animals showed decreased center time compared to NT controls. Additionally, center time in the C9–800 animals was decreased compared to C9–500 mice (Fig. 4E). Taken together, these data show open field abnormalities are found earlier with increased repeat length.
Phenotypic mice show abnormal cage behavior including kyphosis, inactivity, severe dehydration and hind-limb paralysis. Population census and cage behavior analyses (19) to compare the percentages of dead, phenotypic and apparently healthy animals at 40 weeks showed significant differences between the C9–500 and C9–800 cohorts compared to NT controls. In contrast, although several animals in the C9–50 line died, including one animal that showed acute phenotypes and paralysis, the overall survival of animals in the C9–50 line was not significantly different from NT animals (Fig. 4F). These data demonstrate that repeats as short as 50 can cause C9orf72 ALS/FTD phenotypes, but with reduced penetrance compared to the C9–500 and C9–800 lines. As previously reported (19), death in the C9–500 line begins at ~20 weeks of age. In contrast, death of animals in the C9–800 line begins earlier, at approximately 12 weeks of age. Kaplan–Meier analysis shows a significant decrease in survival in the C9–800 line compared to C9–500 animals by 52 weeks (Fig. 4G). Taken together, these data demonstrate that increased repeat length leads to increased penetrance and earlier disease onset.
RNA foci and RAN protein aggregates increase with increased repeat length in C9-BAC mice
Next, we performed experiments to understand the molecular changes associated with increases in repeat length. Since the BAC transgene in the C9–500 mice is inserted into the first intron of the Vgll4 gene (Fig. 1C), we evaluated the expression of Vgll4 in the sublines derived from C9–500 mice. We observed no significant differences in expression of Vgll4 in C9–50, C9–500 and C9–800 mice compared to NT mice (Supplementary Material, Fig. S8A). At 20 weeks of age, there was a significant increase in sense and antisense foci in the C9–800 dentate gyrus compared to mice from the C9–500 line (Fig. 5A and B). Similar to our previously published data on mice with shorter repeat tracts (29–37 repeats) (19), sense and antisense RNA foci were not detected in animals in the C9–50 line (Fig. 5A and B). Next, we compared RAN protein aggregates in the retrosplenial cortex by immunohistochemistry (IHC) or immunofluorescence (IF) using human α-GA1 and α-GP1 antibodies (46) (Supplementary Material, Figs 5C and S8B). At 40 weeks of age, there was a significant increase in the percentage of cells with polyGA aggregates (71% vs 40%) and polyGP aggregates (30% vs 15%) detected by IHC or IF in the C9–800 compared to the C9–500 mice (Fig. 5C–E). The levels of soluble GP protein measured by MSD showed similar trends at 40 weeks (Supplementary Material, Fig. S8C). Similarly, IF studies performed at an earlier timepoint (20 weeks) showed higher levels of aggregates in groups of mice with longer repeat lengths including C9–800 and C9–500 mice. Infrequent aggregates were found in mice from the C9–50 subline but these were not significantly different from NT mice (Supplementary Material, Fig. S8D and E).
Figure 5.
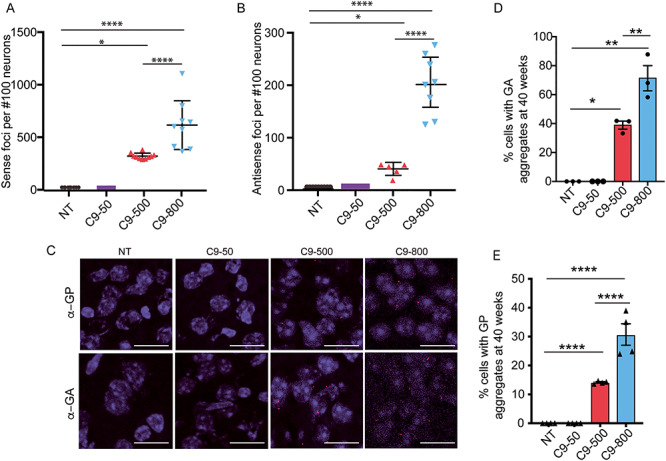
Molecular features of C9orf72 ALS/FTD in C9-BAC mice increase with repeat length. (A, B) Quantification of fluorescence in situ hybridization (FISH) detection of sense (A) and antisense (B) RNA foci in the dentate gyrus of the hippocampus. (C) Immunofluorescence (IF) showing representative images of GA and GP RAN protein aggregates in the retrosplenial cortex of 40-week-old female NT, C9–50, C9–500 and C9–800 mice. Scale bar corresponds to 20 μm. (D) Quantification of GA and GP RAN protein aggregates was done in a blinded fashion and is shown as the percentage of neurons with aggregates in NT, C9–50, C9–500 and C9–800 mouse cohorts. Data information: Statistical analyses for (A), (B) and (D) done using a one-way ANOVA with Bonferroni correction for multiple comparisons. Mean +/− SEM, *P < 0.05, **P < 0.01, ****P < 0.0001.
In summary, our data demonstrate that repeat length is a modifier of disease in our C9-BAC transgenic mouse model. Mice with longer G4C2 repeat tracts show increased disease penetrance and earlier ages of onset and are characterized by increased levels of RNA foci and RAN protein aggregates.
Discussion
To understand the complex molecular mechanisms of C9orf72 ALS/FTD, more than a dozen mouse models have been developed (13–17,19–23,62). Among these, we reported multiple lines of C9orf72 ALS/FTD BAC transgenic mice that develop the behavioral, neuropathological and molecular features of disease (19). To further understand disease in this mouse model, we performed whole genome sequencing and show four independent lines of these BAC mice have unique integration sites. RNA sequencing identified alternative splicing changes affecting RNA processing and degradation pathways as an early molecular signature of disease, which worsens with disease progression. Similar to C9orf72 ALS/FTD patients (27,50,51), gene expression changes in neuroinflammatory and neurodegenerative pathways predominate in severely affected end-stage animals. Finally, isogenic sub-lines of C9-BAC mice containing 50, 500 or 800 G4C2 repeats show that animals with longer repeat lengths have increased levels of RNA foci and RAN protein aggregates as well as earlier ages of onset and increased disease penetrance.
Patients with C9orf72 expansion mutations show remarkable heterogeneity in clinical presentation (9,44) and age of onset (1,2,4,5,7,8,10,44,58,63), but the potential role of repeat length as a disease modifier has been difficult to assess for a number of reasons. First, there is substantial somatic repeat instability and repeat lengths in blood are likely to be substantially shorter than those found in affected brain tissue (64,65). Additionally, Southern blotting is the most common way to measure repeat length but somatic instability and the variation of repeat lengths within a single individual make comparisons of repeat length and age of onset in patients inaccurate. Finally, GC-rich expansion mutations are difficult to amplify by PCR and to sequence making small-pool PCR strategies to accurately measure the distributions of repeat lengths challenging (66,67).
Our generation of an isogenic series of C9-BAC mice with 800, 500 or 50 repeats has allowed us to directly demonstrate that longer repeat tracts result in earlier onset of behavioral abnormalities and decreased survival in mice compared to shorter alleles. These results, combined with the somatic instability seen in patients, suggest that targeting DNA repair pathways may be a viable approach for mitigating disease in C9orf72 ALS/FTD (41–43). The paralysis phenotype found in a single C9–50 animal, combined with previously published phenotypes in animals with four copies of the transgene containing 36 and 29 repeats (19), suggest additional human studies are needed to understand the relative risks of shorter repeat expansion mutations and the potential role of somatic instability to exacerbate disease. This may be particularly relevant for families with sporadic cases of C9orf72 ALS/FTD.
Ectopic cytoplasmic localization of mutant TDP43, FUS and MATR3 or overexpression of wildtype TDP43, FUS and MATR3 protein has been shown to be associated with ALS phenotypes in patients and model systems, respectively (68–72). C9orf72, a DENN (differentially expressed in normal and neoplastic cells) domain containing protein, has been shown to play a role in autophagy and immune-regulatory functions (73–75). Recently, it has been shown that a dose-dependent increase in motor deficits was observed when single copy C9–500 mice were crossed with C9orf72 heterozygous and homozygous knockout mice suggesting both loss-of-function and gain-of-function effects may contribute to C9orf72 ALS/FTD (76). Additionally, it has been proposed that overexpression of C9orf72 protein may lead to toxicity in mouse models of disease (77). Our data, showing that the asymptomatic Baloh-Jax mice have higher levels of C9orf72 protein levels compared to our C9–500 mice combined with our allelic series data, indicate that repeat length and not C9orf72 overexpression is the primary driver of the ALS/FTD phenotypes found in our mice.
While several BAC transgenic models have been reported for C9orf72 ALS/FTD, only the mice developed by Liu et al. (19) and Jiang et al. (18) show neuropathological and behavioral phenotypes. Several reasons could contribute to the differences in phenotypes observed in these different mouse models including repeat length, differences in transgene expression, regulatory elements or sequence differences within human transgenes used to generate the models, differences in sense and antisense RNA foci and DPR protein accumulation and mouse background effects. A systematic comparison across different BAC mouse models would provide an understanding of the relative contribution of the parameters listed above to disease.
Substantial data suggest RNA gain-of-function effects contribute to C9orf72 ALS/FTD and a large number of RNA-binding proteins have been proposed to be sequestered by the repeats. Circular dichroism studies performed on short stretches of sense repeats G4C2 show that the repeat can adopt G quadruplex, R loop or hairpin conformations and the antisense repeats likely adopt a hairpin conformation (25,31,78). Since several of these predicted RNA-binding proteins also contain low complexity domains, it is possible that multiple RNA-binding proteins interact with repeat RNAs and form dynamic liquid droplet like structures that may be complicated to resolve (79–81). The variability and complexity in secondary structures formed by the repeats make it more likely that multiple RNA-binding proteins are sequestered by these repeat motifs (29), a possibility consistent with our data showing that genes with abnormal splicing events have a variety of RNA recognition motifs (RRMs).
Transcriptomic data from our acute C9-BAC mice are consistent with gene expression changes caused by ongoing apoptotic processes involved in cell death and inflammation found at end-stage disease. In contrast, transcriptomic profiles of presymptomatic mice showed minimal changes in gene expression but abundant changes in alternative splicing. Robust alternative splicing changes are also detected in neurons differentiated from C9orf72 patient iPSCs (iPSN), and similarly showed few gene expression differences compared to C9orf72 patient autopsy tissue (26). In our mice, 240 genes are mis-spliced in C9(+) presymptomatic animals, and the psi values of 83 of these genes increase further in mice with acute end-stage disease. These genes include Elavl2, which has an RRM and has been predicted to play a role in ALS because of its similarity to TDP-43, FUS (81) and Pard3, which regulates neuronal polarity, has also been shown to be mis-spliced in ALS patient autopsy tissue (83). These results, combined with the 82 other alternatively spliced genes whose psi values increase with disease progression, highlight alternative splicing changes as a novel and early molecular signature of C9orf72 ALS/FTD.
A role of RAN proteins in disrupting alternative splicing has also been proposed. In cell culture experiments, ~5000 mis-spliced events were observed when astrocytes were treated with PR RAN protein (84). Additionally, it has been shown that in cell culture systems, GR and PR RAN proteins associate with low complexity domains of RNA-binding proteins (79,80,85) and can cause mis-splicing in a U2snRNP-dependent manner (83). In cell culture, these low complexity domain proteins are typically involved in the formation of membrane-less organelles such as stress granules and neuronal speckles (38,86). Since splicing factors normally localize to the neuronal speckles, it is possible that RAN proteins interact with RNA-binding proteins containing low complexity domains and disrupt splicing globally. Global splicing deficits would be likely to cause an increase in overall alternative spliced changes rather than the disruption of specific set of events mediated by the sequestration of a single family of RNA-binding protein as is seen in myotonic dystrophy (57). These data, combined with our data showing phenotypic C9–500 mice have more RAN protein aggregates compared to asymptomatic C9–500 (19), suggest that RAN proteins could contribute to the early RNA splicing abnormalities found in our mice.
Longer repeat lengths result in the expression of both longer expansion RNAs and increased RAN protein aggregates. Since both RNA GOF and protein toxicity are closely connected to repeat length, additional work will be needed to clarify the relative roles of RNA gain-of-function effects and RAN proteins in disease. Our mouse model is an excellent platform to address the contribution of specific disease mechanisms and for the development of potential therapies. These C9-BAC mice were recently used in two separate studies that highlight the contributions of RAN proteins to C9orf72 ALS/FTD. The first study showed that a human antibody targeting GA RAN proteins is sufficient to rescue behavior and survival phenotypes, without changing the levels of their corresponding expansion RNAs (46). The second study showed reducing RAN translation by targeting the protein kinase R (PKR) pathway using a dominant negative form of the protein (PKR-K296R) or metformin mitigated disease (47). These preclinical data highlight the therapeutic potential of targeting RAN proteins.
Since the discovery of the C9orf72 expansion mutation, research has focused on understanding disease mechanisms and identifying therapeutic targets. Transgenic mouse models have contributed to these efforts, but ectopic overexpression and transgene integration can affect the resulting phenotypes and suitability for testing potential therapies. This issue is not restricted to C9-ALS as this issue also occurs in the widely used R6/2 mouse model of Huntington disease (87). Multiple features make our C9–500 BAC transgenic mice a valuable tool both for understanding the biology and time course of the disease and also for testing novel therapeutic strategies. First, the phenotypes in our mice, which contain the full-length C9orf72 gene and substantial flanking sequences, are not affected by integration site. Second, the C9–500 line carries one copy of the transgene expressed at levels comparable to the endogenous C9orf72 ortholog in mice. Third, we have identified a group of alternative splicing as a molecular signature of disease that worsen with disease progression and may be useful biomarkers of disease. Finally, we demonstrate that repeat length increases disease penetrance and that the phenotypes in our mice are caused by a gain of function of the repeat expansion. Taken together, our data provide mechanistic insight into C9orf72 ALS/FTD and demonstrate that our mice are a robust tool to study the disease and to test therapeutic strategies for disease intervention.
Materials and Methods
Mouse generation and maintenance
C9-BAC transgenic mice were mated with FVB-N/J mice obtained from Jackson Laboratory. Pups were genotyped based on genomic DNA (gDNA) extracted from the tail using the primers C9-GT F and R (Table 3) and the protocol previously described by Liu et al. (19) NT littermates from each of these lines were used as controls in the study. The repeat size of each mouse in the study was determined using Southern blotting described below. Behavioral tests were performed at 12, 24 and 40 weeks of age. DigiGait and open field analyses were performed based on the manufacturer’s protocol. For molecular and pathological staining, mice were perfused transcardially using 1x PBS and tissues were embedded in 10% formalin or OCT frozen in cold 2-methylbutane for further analyses. As originally reported in the Liu et al. (19) more severe phenotypes were found in female mice, therefore in this study female mice were used to characterize the effect of repeat length (Figs 4 and 5) and transcriptomic abnormalities (Figs 2 and 3).
Phenotypic characterization of C9-BAC mice
Cage behavior of animals was regularly monitored, and changes were recorded on a weekly basis. At 40 weeks of age, two blinded independent investigators scored a cohort of mice based on appearance (normal, scruffy and hunched); behavior (normal, hyperactive, delayed or absent righting response); neurological changes (clasping, seizures, excessive grooming, paralysis of one or both limbs) and hydration (normal, mild or significant). A scoring system was established based on these phenotypes and animals were euthanized if they suffered >20% weight loss or as recommended by the veterinarian. For additional details please see Matters Arising Article by Nguyen et al. (88).
Mate pair preparation and whole genome sequencing
Input DNA was received and quantified using a fluorometric-based method specific for double-stranded DNA. The quality of the gDNA was evaluated using capillary electrophoresis-based technology (Fragment Analyzer, AATI). Four micrograms of good quality DNA were processed into Illumina-compatible libraries using Nextera Mate Pair Sample Preparation Kit (Illumina) following the Gel-Plus procedure with the following modifications. The fragmented DNA was size-selected for a target range of 5–8 kb fragments using an automated DNA size-selection technology, BluePippin (Sage Science) and the adapter-ligated mate pair fragments were enriched using 10 amplification cycles. Final libraries were quantified using the KAPA Library Quantification Kit (KAPA Biosystem), Qubit Fluorometer (Life Technologies) and 2100 BioAnalyzer (Agilent). Libraries were sequenced on an Illumina HiSeq2500 sequencer using 2 × 125 bp cycles.
Bioinformatics analysis for transgene integration
All read data were processed prior to alignment. Processing included: (1) ab initio duplicate removal using in-house scripts; (2) adapter identification/clipping and low-quality end trimming (Q = 20) using Cutadapt 1.8.1 (89) and (3) phiX decontamination by mapping with GEM mapper (90) against the phiX reference (acc. NC_001422.1). Read pairs were further screened for the presence of the Nextera ligation adapter, which was also trimmed. After determination of the library insert size and mate orientation by mapping a subset of the reads against the mouse reference genome (mm10) with the GEM mapper, we decided to consider for downstream analysis the entire set of processed mate-pairs, given that the overall percentage of reads mapping in forward-reverse orientation (representing paired-end contamination) was low (~1%). Processed read data were then mapped, using BWA-MEM (91), against a combined index that included, apart from the full mouse reference genome (mm10), the human C9orf72 sequence and the pCC1 vector used in transfection. Despite the fact that most duplicate sequences have been removed in the initial processing of the data, additional duplicates were identified and marked by the MarkDuplicates tool from Picard (92) (from Broad Institute). Integration sites were identified by manual inspection of discordant read-pairs in which one end mapped to the mouse reference genome and the other end to the C9orf72 or pCC1 sequences. Analysis of soft-clipped reads at the integration sites allowed determining, with base-pair resolution, the exact breakpoint location in both the mouse genome and the C9orf72 sequence. Further analysis on structure of the integration was based on read-pairing information and in copy-number analysis of the C9orf72 and pCC1 sequences. For the latter, both the C9orf72 and the pCC1 sequences were segmented based on integration breakpoints and deletion site boundaries and, for each segment, average depth of coverage was computed using Depth of coverage from the Genome Analysis Toolkit (GATK) (93) The obtained values were then compared to the average depth of coverage over the mouse reference genome, estimated from a set of intervals overlapping genic regions. For calling variants over both the C9orf72 and pCC1 sequences, alignments from all four samples were combined into a single BAM file and GATK’s HaplotypeCaller was used to produce a gVCF file. Variants were then emitted by GATK’s GenotypeGVCFs.
Transcriptome sequencing
Animals were perfused transcardially with sterile 1X PBS and the frontal cortex was harvested for RNA isolation. RNA was isolated using the Direct-Zol RNA miniprep kit (Zymo Research). RNA-seq libraries were prepared from total RNA (500–700 ng) using the directional RNA-seq kit with NEB Ultra Ribominus (New England Labs) following the manufacturer’s protocol. The RNA was then sequenced using the Illumina NextSeq500 machine at the Center for NeuroGenetics, University of Florida.
RNA sequencing data analysis
Reads obtained after sequencing were aligned using STAR (52) to either the human genome (hg19) or mouse genome (mm10). Quality analysis of the reads was performed using RSeQC. We used Kallisto (53) to obtain TPM values for some analyses. GENCODE was used for gene annotations from STAR alignment wand RefSeq was used for Kaliisto gene annotations. DEseq2 was used to measure gene expression differences, run using the Bioconductor package for R. We used MISO based on MISO annotations version 2.0 to identify changes in alternative splicing (Katz et al.) (54). Only events with Bayes factor ≥ 5 for at least one pair-wise comparison were considered. Events were filtered with |ΔΨ| ≥ 0.10 compared to the NT mice and the Venn diagrams were plotted using Biovenn. To identify enriched biological pathways, gene ontology analysis was performed using Metascape using gene sets from GO biological processes and was confirmed using DAVID annotation using default statistical thresholds (P-value < 0.01, maximum enrichment >1.5). Graphs were plotted on R using ggplot2. C9-ALS patient data were obtained from Prudencio et al. (27). Data from frontal cortex on autopsy tissue samples were filtered based on RIN scores and samples used were: C9(+)-ALS—SRR1927020, SRR1927022, SRR1927024, SRR1927026, SRR1927028, SRR1927030, SRR1927032 and SRR1927034; Control—SRR1927056, SRR1927058, SRR1927060, SRR1927062, SRR1927064, SRR1927066, SRR1927068, SRR1927070 and SRR1927071.
Cell-type enrichment analysis
The index for relative expression of genes in different cell types was set up using publicly available data on cell type enrichment in the mouse cortex (55). Since the differences between the oligodendrocyte cell types were subtle, we combined myelinating oligodendrocytes, oligodendrocyte precursor cells and mature oligodendrocytes into one classification ‘oligodendrocyte’. For a gene to belong to a particular cell type based on the developed index, expression had to be >20FPKM and absolute logFC between any sample and mean of all other samples >2. Bayesian statistical model was used to calculate the proportions of cell types within the current dataset. The python package Pymc3 was used to perform these analyses and Matplotlib was used to generate the graphs.
Motif analysis
To analyze the number of enriched motifs, we used the monotonically changing cassette exons from alternatively spliced events generated from MISO with a Bayes factor > 5 and |ΔΨ| ≥ 0.10 in NT and Acute animals. Fisher’s exact test and Bonferroni correction were performed and the P adjusted < 0.05 was set as the significance threshold. The regions 250 bp upstream, exon, 250 bp downstream were used to count for all possible combination of 4-mers. We used Fisher’s exact test to identify motifs that are enriched on depleted specifically in Acute mice when compared to NT mice.
Splicing analysis
For splicing analyses, mm10 annotated events were run on MISO to identify alternatively spliced events to identify skipped exon (SE) events. Delta PSI values for NT vs C9 mean PSIs and NT vs Acute mean PSIs were calculated and reported in Table 2. Wilcoxon rank-sum test was also run for both comparisons. Delta PSI cutoff was set at 0.1 (or − 0.1) and a rank sum P-value < 0.05 to establish significance. GO analysis was run with metascape.org.
Southern blot
Tail gDNA extraction and Southern blot experiments were performed following previously published protocols in Liu et al. (19). Briefly, 10 μg of gDNA extracted from the tail or brain was digested with EcoRI and BamHI overnight at 37°C. The digested gDNA samples were then run on a 0.7% agarose gel for ~5–6 h, depurinated (0.2 N HCl), denatured (1.5 M NaCl, 0.5 M NaOH) and neutralized (1.5 M NaCl, 0.5 M Tris HCl) for 15 min each. DNA was transferred overnight by capillary blotting to a positively charged nylon membrane and was UV cross-linked the next morning. For hybridization, the membrane was prehybridized for 1 h using Amersham Rapid-Hyb buffer (GE Healthcare). The probe was labeled using dCTP-P32 using the random primed DNA labeling kit (Invitrogen) and was hybridized to the membrane. After 3 h of hybridization, the membrane was washed with 2x SSC, 0.1%SDS for 20 min at room temperatures and then with 0.2x SSC, 0.1%SDS solution at 65°C two times for 15 min each. Radioactivity was visualized on an X-ray film after 2–3 days of exposure at −80°C.
Quantitative real time PCR
Total RNA was isolated from mouse frontal cortex and spinal cord tissues with TRIzol (Invitrogen). Following DNase treatment (Ambion) using manufacturer’s protocol, cDNA was prepared using the SuperScript III RT kit (Invitrogen) and random-hexamer primers (Applied Biosystems). Two-step quantitative RT-PCR was performed on a MyCycler Thermal Cycler system (Bio-Rad) using SYBR Green PCR Master Mix (Bio-Rad). See primer lists in Table 4.
Table 4.
Primers used for PCR
| Primer name | Sequence |
|---|---|
| Mppe1-F | GGCACCATGTCTGGTTTGTT |
| Mppe1-R | AGACTGGTCGGTGATCTCGT |
| Probe-F | AGAACAGGACAAGTTGCC |
| Probe-R | AACACACACCTCCTAAACC |
| C9GT-F | AGTTGGGTCCATGCTCAACAA |
| C9GT-R | ACTGTTCTAGGTACCGGGCT |
| 1a-F | GCCCACGTAAAAGATGACGC |
| 1a-R | CCTCCTAAACCCACACCTGC |
| AS-F | AGTCGCTAGAGGCGAAAGC |
| AS-R | CGAGTGGGTGAGTGAGGAG |
| C9mRNA-F | TCTCCAGCTGTTGCCAAGAC |
| C9mRNA-R | TCCATTCTCTCTGTGCCTTCT |
| mACT-F | TCGTGCGTGACATCAAAGAG |
| mACT-R | GATCTTCATGGTGCTAGGAG |
| Vgll4-F | CAGGATGCAGACCCTCCC |
| Vgll4-R | TGGTCGTTTTCACAGTCCAAG |
Fluorescent in situ hybridization (FISH)
Frozen sections measuring 10 μm embedded in OCT were cut on the cryostat. Frozen sections were fixed in 4% PFA in PBS for 20 min and incubated in prechilled 70% ethanol for 30 min or longer at 4°C. Following rehydration in 40% formamide in 2× SSC at room temperature for 10 min, the slides were prehybridized with hybridization solution (40% formamide, 2× SSC, 20 μg/ml BSA, 100 mg/ml dextran sulfate, 250 μg/ml yeast tRNA, 2 mM Vanadyl Sulfate Ribonucleosides) for 30 min at 55°C and then incubated with 200 ng/ml of denatured DNA probe (C2G4)3-Cy3 for sense foci and (G4C2)3-Cy3 for antisense foci) in hybridization solution at 55°C. After 3 h of hybridization, the slides were washed three times with 40% formamide in 2× SSC and briefly washed one time in PBS. Autofluorescence of lipofuscin was quenched by 0.25% of Sudan Black B in 70% ethanol. Slides were mounted with ProLong mounting medium containing DAPI (Invitrogen) and imaged on LSM880 Confocal microscope.
Immunohistochemistry
Sections measuring 5 μm were deparaffinized in xylene and rehydrated through gradient ethanol solution. Antigen retrieval was performed by incubating the slides in a steamer with 10 mM citrate buffer (pH 6.0) for 30 min or with 10 mM EDTA (pH 6.4) for 12 min. The slides were cooled down to RT and then washed for 10 min in running tap water. The slides were incubated in 95–100% formic acid for 5 min and subsequently washed for 10 min in running tap water. To eliminate nonspecific binding and excessive background, slides were blocked with a serum-free block or rodent block (Biocare Medical) for 15 min. Primary antibody diluted in 1:10 blocking solution was applied on the slides and incubated overnight at 4°C (see below for dilution information). Slides were washed three times with 1XPBS and incubated with linking reagent (streptavidin or alkaline phosphatase; Covance) or biotinylated rabbit anti-goat IgG (Vector Labs) for 30 min at room temperature. After washing with 1XPBS, these sections were then incubated in 3% hydrogen peroxide (in methanol) for 15 min to eliminate any endogenous peroxidase activity. After washing in running tap water for 15 min, labeling reagent (HRP, Covance; Vectastain ABC-AP kit) was then applied to the slides for 30 min at room temperature. The slides were developed with NovaRed or DAB (Vector Labs) and then were counterstained with hematoxylin (modified Harris, Sigma Aldrich), rehydrated in gradient ethanol solution and coverslipped for visualization. Images were taken on the Olympus BX51 microscope using the cellSense software. For hematoxylin and eosin staining, the slides were deparaffinized in xylene and rehydrated through gradient ethanol solution. The slides were then soaked in hematoxylin (modified Harris, Sigma Aldrich) for 1 min and washed in running distilled water for 10 min. Next, the slides were immersed in Eosin Y (Sigma Aldrich) for 30 s and washed in distilled water for 10 min. The slides were dehydrated and coverslipped before visualization. For cresyl violet staining, slides were deparaffinized in xylene and subsequently rehydrated in gradient ethanol solution. The slides were incubated in 0.25% cresyl violet at 60°C for 8–10 min and differentiated in 95% ethanol for 1–5 min. Slides were then immersed in 100% ethanol and xylene and coverslipped for visualization.
Immunofluorescence
Frozen sections were warmed up at room temperature for 2 h. Slides were fixed with 4% PFA at room temperature for 10 min and immediately permeabilized with ice-cold 1:1 methanol-acetone for 5 min at −20°C. Slides were blocked with background sniper at RT for 30 min to 1 h and incubated overnight with primary antibody (polyGA 1:2000, polyGP 1:1000, generously donated by Neurimmune Inc.) prepared in a 1:10 dilution of background sniper. The slides were washed three times with 1xPBS the next day and incubated with secondary antibody (Cy3-anti-human IgG antibody, 1:5000) for 1–2 h. The slides were washed with 1x PBS and mounted with Prolong containing DAPI. Slides were imaged with a Zeiss LSM880 Confocal microscope.
Western blot
Brain lysates were solubilized with RIPA buffer (1% sodium deoxycholate, 1% Triton X-100, 50 mM Tris pH 7.5, 150 mM NaCl and proteinase inhibitor). A total of 10 μl of lysates was run on a 4–12% Bis-Tris gel and transferred to a nitrocellulose membrane. The membrane was blocked in 5% milk diluted in PBST (1XPBS with 0.05% Tween-20). The membrane was then incubated with the primary antibody (α-C9orf72, 1:1000) overnight at 4°C. The blots were washed three times with 1XPBS the next morning and were incubated with secondary antibody for 1 h at RT. The membrane was developed using ECL prime and was visualized for signal.
Image J quantification
Quantification was performed by two independent blinded investigators using the cell counter plugin in ImageJ (National Institute of Health). Serial sections 20–30 μm apart were stained, imaged and quantified for subsequent analyses.
Behavioral analyses
DigiGait, open field analyses and scoring criteria for cage behavioral assessments were performed as previously described in Liu et al. (19).
Statistics
GraphPad Prism 7 was used to perform the statistical analyses in the manuscript. Significance threshold was set at P < 0.05. The significance values were set as ns = not significant P > 0.05, P < 0.05—‘*’, P < 0.01—‘**’, P < 0.001—‘***’, P < 0.0001—‘****’. Comparisons between groups were performed using mean ± SEM values using one-way analysis of variance (ANOVA) and multiple comparisons. Significance in survival was measured using the Log Mantel-Cox test. DigiGait analyses were performed using multiple comparisons.
Supplementary Material
Acknowledgments
We thank Dr John Cleary, Dr Yuanjing Liu and Dr Maurice Swanson for input and advice on the manuscript, Dr Fabio Montrasio and Dr Jan Grimm for providing human antibodies for polyGA and polyGP detection.
Conflict of Interest statement. The authors declare that they have no conflict of interest.
Contributor Information
Amrutha Pattamatta, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA.
Lien Nguyen, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA.
Hailey R Olafson, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA.
Marina M Scotti, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA.
Lauren A Laboissonniere, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA.
Jared Richardson, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA; McKnight Brain Institute, University of Florida, Gainesville, FL 32610, USA.
J Andrew Berglund, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Biochemistry and Molecular Biology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; RNA Institute and Department of Biological Sciences, University at Albany, Albany, NY 12222, USA.
Tao Zu, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA.
Eric T Wang, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA; McKnight Brain Institute, University of Florida, Gainesville, FL 32610, USA.
Laura P W Ranum, Center for NeuroGenetics, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; Department of Molecular Genetics and Microbiology, Colllege of Medicine, University of Florida, Gainesville, FL 32610, USA; University of Florida Genetics Institute, University of Florida, Gainesville, FL 32610, USA; McKnight Brain Institute, University of Florida, Gainesville, FL 32610, USA; Fixel Institute, University of Florida, Gainesville, FL 32610, USA.
Funding
This work was supported by the National Institutes of Health (R01 NS098819); Target ALS; Amyotrophic Lateral Sclerosis Association; Robert Packard Center for ALS Research; and Muscular Dystrophy Association.
References
- 1. DeJesus-Hernandez, M., Mackenzie, I.R., Boeve, B.F., Boxer, A.L., Baker, M., Rutherford, N.J., Nicholson, A.M., Finch, N.A., Flynn, H., Adamson, J. et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Renton, A.E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J.R., Schymick, J.C., Laaksovirta, H., van Swieten, J.C., Myllykangas, L. et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ash, P.E., Bieniek, K.F., Gendron, T.F., Caulfield, T., Lin, W.L., Dejesus-Hernandez, M., van Blitterswijk, M.M., Jansen-West, K., Paul, J.W., 3rd, Rademakers, R. et al. (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron, 77, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chio, A., Restagno, G., Brunetti, M., Ossola, I., Calvo, A., Canosa, A., Moglia, C., Floris, G., Tacconi, P., Marrosu, F. et al. (2012) ALS/FTD phenotype in two Sardinian families carrying both C9ORF72 and TARDBP mutations. J. Neurol. Neurosurg. Psychiatry, 83, 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., Janssens, J., Bettens, K., Van Cauwenberghe, C., Pereson, S. et al. (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol., 11, 54–65. [DOI] [PubMed] [Google Scholar]
- 6. Mori, K., Weng, S.M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., Schmid, B., Kretzschmar, H.A., Cruts, M., Van Broeckhoven, C. et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science, 339, 1335–1338. [DOI] [PubMed] [Google Scholar]
- 7. Stewart, H., Rutherford, N.J., Briemberg, H., Krieger, C., Cashman, N., Fabros, M., Baker, M., Fok, A., DeJesus-Hernandez, M., Eisen, A. et al. (2012) Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol., 123, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Langenhove, T., van der Zee, J., Gijselinck, I., Engelborghs, S., Vandenberghe, R., Vandenbulcke, M., De Bleecker, J., Sieben, A., Versijpt, J., Ivanoiu, A. et al. (2013) Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol., 70, 365–373. [DOI] [PubMed] [Google Scholar]
- 9. Van Mossevelde, S., van der Zee, J., Gijselinck, I., Sleegers, K., De Bleecker, J., Sieben, A., Vandenberghe, R., Van Langenhove, T., Baets, J., Deryck, O. et al. (2017) Clinical evidence of disease anticipation in families segregating a C9orf72 repeat expansion. JAMA Neurol., 74, 445–452. [DOI] [PubMed] [Google Scholar]
- 10. van der Zee, J., Gijselinck, I., Dillen, L., Van Langenhove, T., Theuns, J., Engelborghs, S., Philtjens, S., Vandenbulcke, M., Sleegers, K., Sieben, A. et al. (2013) A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat., 34, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zu, T., Liu, Y., Banez-Coronel, M., Reid, T., Pletnikova, O., Lewis, J., Miller, T.M., Harms, M.B., Falchook, A.E., Subramony, S.H. et al. (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U. S. A., 110, E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen, L., Cleary, J.D. and Ranum, L.P.W. (2019) Repeat-associated non-ATG translation: molecular mechanisms and contribution to neurological disease. Annu. Rev. Neurosci., 42, 227–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hao, Z.B., Liu, L., Tao, Z.T., Wang, R., Ren, H.G., Sun, H.Y., Lin, Z.X., Zhang, Z.X., Mu, C.C., Zhou, J.W. et al. (2019) Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun., 10, 2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang, Y.J., Guo, L., Gonzales, P.K., Gendron, T.F., Wu, Y.W., Jansen-West, K., O'Raw, A.D., Pickles, S.R., Prudencio, M., Carlomagno, Y. et al. (2019) Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science, 363, 707 -+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Atanasio, A., Decman, V., White, D., Ramos, M., Ikiz, B., Lee, H.C., Siao, C.J., Brydges, S., LaRosa, E., Bai, Y. et al. (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep., 6, 23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burberry, A., Suzuki, N., Wang, J.Y., Moccia, R., Mordes, D.A., Stewart, M.H., Suzuki-Uematsu, S., Ghosh, S., Singh, A., Merkle, F.T. et al. (2016) Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med., 8, 347ra393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chew, J., Gendron, T.F., Prudencio, M., Sasaguri, H., Zhang, Y.J., Castanedes-Casey, M., Lee, C.W., Jansen-West, K., Kurti, A., Murray, M.E. et al. (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science, 348, 1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang, J., Zhu, Q., Gendron, T.F., Saberi, S., McAlonis-Downes, M., Seelman, A., Stauffer, J.E., Jafar-Nejad, P., Drenner, K., Schulte, D. et al. (2016) Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron, 90, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D.R., Yachnis, A.T. and Ranum, L.P. (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron, 90, 521–534. [DOI] [PubMed] [Google Scholar]
- 20. O'Rourke, J.G., Bogdanik, L., Muhammad, A.K., Gendron, T.F., Kim, K.J., Austin, A., Cady, J., Liu, E.Y., Zarrow, J., Grant, S. et al. (2015) C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron, 88, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peters, O.M., Cabrera, G.T., Tran, H., Gendron, T.F., McKeon, J.E., Metterville, J., Weiss, A., Wightman, N., Salameh, J., Kim, J. et al. (2015) Human C9ORF72 Hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not Neurodegeneration in BAC transgenic mice. Neuron, 88, 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schludi, M.H., Becker, L., Garrett, L., Gendron, T.F., Zhou, Q., Schreiber, F., Popper, B., Dimou, L., Strom, T.M., Winkelmann, J. et al. (2017) Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol., 134, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang, Y.J., Gendron, T.F., Ebbert, M.T.W., O'Raw, A.D., Yue, M., Jansen-West, K., Zhang, X., Prudencio, M., Chew, J., Cook, C.N. et al. (2018) Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med., 24, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waite, A.J., Baumer, D., East, S., Neal, J., Morris, H.R., Ansorge, O. and Blake, D.J. (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging, 35, 1779 e1775–1779 e1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mizielinska, S., Gronke, S., Niccoli, T., Ridler, C.E., Clayton, E.L., Devoy, A., Moens, T., Norona, F.E., Woollacott, I.O.C., Pietrzyk, J. et al. (2014) C9orf72 repeat expansions cause neurodegeneration in drosophila through arginine-rich proteins. Science, 345, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Donnelly, C.J., Zhang, P.W., Pham, J.T., Heusler, A.R., Mistry, N.A., Vidensky, S., Daley, E.L., Poth, E.M., Hoover, B., Fines, D.M. et al. (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron, 80, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prudencio, M., Belzil, V.V., Batra, R., Ross, C.A., Gendron, T.F., Pregent, L.J., Murray, M.E., Overstreet, K.K., Piazza-Johnston, A.E., Desaro, P. et al. (2015) Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci., 18, 1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ebbert, M.T.W., Ross, C.A., Pregent, L.J., Lank, R.J., Zhang, C., Katzman, R.B., Jansen-West, K., Song, Y.P., da Rocha, E.L., Palmucci, C. et al. (2017) Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72-associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol., 134, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cooper-Knock, J., Walsh, M.J., Higginbottom, A., Robin Highley, J., Dickman, M.J., Edbauer, D., Ince, P.G., Wharton, S.B., Wilson, S.A., Kirby, J. et al. (2014) Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain, 137, 2040–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Freibaum, B.D., Lu, Y., Lopez-Gonzalez, R., Kim, N.C., Almeida, S., Lee, K.H., Badders, N., Valentine, M., Miller, B.L., Wong, P.C. et al. (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature, 525, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haeusler, A.R., Donnelly, C.J., Periz, G., Simko, E.A., Shaw, P.G., Kim, M.S., Maragakis, N.J., Troncoso, J.C., Pandey, A., Sattler, R. et al. (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature, 507, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jovicic, A., Mertens, J., Boeynaems, S., Bogaert, E., Chai, N., Yamada, S.B., Paul, J.W., 3rd, Sun, S., Herdy, J.R., Bieri, G. et al. (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci., 18, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee, Y.B., Chen, H.J., Peres, J.N., Gomez-Deza, J., Attig, J., Stalekar, M., Troakes, C., Nishimura, A.L., Scotter, E.L., Vance, C. et al. (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep., 5, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sareen, D., O'Rourke, J.G., Meera, P., Muhammad, A.K., Grant, S., Simpkinson, M., Bell, S., Carmona, S., Ornelas, L., Sahabian, A. et al. (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med., 5, 208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu, Z., Poidevin, M., Li, X., Li, Y., Shu, L., Nelson, D.L., Li, H., Hales, C.M., Gearing, M., Wingo, T.S. et al. (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U. S. A., 110, 7778–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang, K., Donnelly, C.J., Haeusler, A.R., Grima, J.C., Machamer, J.B., Steinwald, P., Daley, E.L., Miller, S.J., Cunningham, K.M., Vidensky, S. et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature, 525, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conlon, E.G., Lu, L., Sharma, A., Yamazaki, T., Tang, T., Shneider, N.A. and Manley, J.L. (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife, 5, e17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taylor, J.P., Brown, R.H., Jr. and Cleveland, D.W. (2016) Decoding ALS: from genes to mechanism. Nature, 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Majounie, E., Renton, A.E., Mok, K., Dopper, E.G., Waite, A., Rollinson, S., Chio, A., Restagno, G., Nicolaou, N., Simon-Sanchez, J. et al. (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol., 11, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murphy, N.A., Arthur, K.C., Tienari, P.J., Houlden, H., Chio, A. and Traynor, B.J. (2017) Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep., 7, 2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones, L., Houlden, H. and Tabrizi, S.J. (2017) DNA repair in the trinucleotide repeat disorders. Lancet Neurol., 16, 88–96. [DOI] [PubMed] [Google Scholar]
- 42. Lee, J.M., Wheeler, V.C., Chao, M.J., Vonsattel, J.P.G., Pinto, R.M., Lucente, D., Abu-Elneel, K., Ramos, E.M., Mysore, J.S., Gillis, T. et al. (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell, 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wright, G.E.B., Collins, J.A., Kay, C., McDonald, C., Dolzhenko, E., Xia, Q.W., Becanovic, K., Drogemoller, B.I., Semaka, A., Nguyen, C.M. et al. (2019) Length of uninterrupted CAG, independent of Polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am. J. Hum. Genet., 104, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Mossevelde, S., van der Zee, J., Cruts, M. and Van Broeckhoven, C. (2017) Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev., 44, 117–124. [DOI] [PubMed] [Google Scholar]
- 45. Hsiung, G.Y., DeJesus-Hernandez, M., Feldman, H.H., Sengdy, P., Bouchard-Kerr, P., Dwosh, E., Butler, R., Leung, B., Fok, A., Rutherford, N.J. et al. (2012) Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain, 135, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nguyen, L., Montrasio, F., Pattamatta, A., Tusi, S.K., Bardhi, O., Meyer, K.D., Hayes, L., Nakamura, K., Banez-Coronel, M., Coyne, A. et al. (2020) Antibody therapy targeting RAN proteins rescues C9 ALS/FTD phenotypes in C9orf72 mouse model. Neuron, 105, 645–662 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zu, T., Guo, S., Bardhi, O., Ryskamp, D.A., Li, J., Khoramian Tusi, S., Engelbrecht, A., Klippel, K., Chakrabarty, P., Nguyen, L. et al. (2020) Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. U. S. A., 117, 18591–18599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiao, S., MacNair, L., McGoldrick, P., McKeever, P.M., McLean, J.R., Zhang, M., Keith, J., Zinman, L., Rogaeva, E. and Robertson, J. (2015) Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol., 78, 568–583. [DOI] [PubMed] [Google Scholar]
- 49. Laflamme, C., McKeever, P.M., Kumar, R., Schwartz, J., Kolahdouzan, M., Chen, C.X., You, Z., Benaliouad, F., Gileadi, O., McBride, H.M. et al. (2019) Implementation of an antibody characterization procedure and application to the major ALS/FTD disease gene C9ORF72. Elife, 8, e48363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Butti, Z. and Patten, S.A. (2018) RNA Dysregulation in amyotrophic lateral sclerosis. Front. Genet., 9, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prudencio, M., Gonzales, P.K., Cook, C.N., Gendron, T.F., Daughrity, L.M., Song, Y., Ebbert, M.T.W., van Blitterswijk, M., Zhang, Y.J., Jansen-West, K. et al. (2017) Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet., 26, 3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bray, N.L., Pimentel, H., Melsted, P. and Pachter, L. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 54. Katz, Y., Wang, E.T., Airoldi, E.M. and Burge, C.B. (2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods, 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang, Y., Chen, K., Sloan, S.A., Bennett, M.L., Scholze, A.R., O'Keeffe, S., Phatnani, H.P., Guarnieri, P., Caneda, C., Ruderisch, N. et al. (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci., 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ince-Dunn, G., Okano, H.J., Jensen, K.B., Park, W.Y., Zhong, R., Ule, J., Mele, A., Fak, J.J., Yang, C., Zhang, C. et al. (2012) Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron, 75, 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scotti, M.M. and Swanson, M.S. (2016) RNA mis-splicing in disease. Nat. Rev. Genet., 17, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Paulson, H. (2018) Repeat expansion diseases. Handb. Clin. Neurol., 147, 105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gusella, J.F. and MacDonald, M.E. (2000) Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci., 1, 109–115. [DOI] [PubMed] [Google Scholar]
- 60. Mancuso, R., Olivan, S., Osta, R. and Navarro, X. (2011) Evolution of gait abnormalities in SOD1(G93A) transgenic mice. Brain Res., 1406, 65–73. [DOI] [PubMed] [Google Scholar]
- 61. Wooley, C.M., Sher, R.B., Kale, A., Frankel, W.N., Cox, G.A. and Seburn, K.L. (2005) Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve, 32, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jiang, J. and Cleveland, D.W. (2016) Bidirectional transcriptional inhibition as therapy for ALS/FTD caused by repeat expansion in C9orf72. Neuron, 92, 1160–1163. [DOI] [PubMed] [Google Scholar]
- 63. Gusella, J.F. and MacDonald, M.E. (2009) Huntington's disease: the case for genetic modifiers. Genome Med., 1, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Blitterswijk, M., Baker, M.C., DeJesus-Hernandez, M., Ghidoni, R., Benussi, L., Finger, E., Hsiung, G.Y., Kelley, B.J., Murray, M.E., Rutherford, N.J. et al. (2013) C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology, 81, 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nordin, A., Akimoto, C., Wuolikainen, A., Alstermark, H., Jonsson, P., Birve, A., Marklund, S.L., Graffmo, K.S., Forsberg, K., Brannstrom, T. et al. (2015) Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet., 24, 3133–3142. [DOI] [PubMed] [Google Scholar]
- 66. Dandelot, E. and Gourdon, G. (2018) The flash-small-pool PCR: how to transform blotting and numerous hybridization steps into a simple denatured PCR. Biotechniques, 64, 262–265. [DOI] [PubMed] [Google Scholar]
- 67. Gomes-Pereira, M., Bidichandani, S.I. and Monckton, D.G. (2004) Analysis of unstable triplet repeats using small-pool polymerase chain reaction. Methods Mol. Biol., 277, 61–76. [DOI] [PubMed] [Google Scholar]
- 68. Lagier-Tourenne, C., Polymenidou, M. and Cleveland, D.W. (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet., 19, R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tada, M., Doi, H., Koyano, S., Kubota, S., Fukai, R., Hashiguchi, S., Hayashi, N., Kawamoto, Y., Kunii, M., Tanaka, K. et al. (2018) Matrin 3 is a component of neuronal cytoplasmic inclusions of motor neurons in sporadic amyotrophic lateral sclerosis. Am. J. Pathol., 188, 507–514. [DOI] [PubMed] [Google Scholar]
- 70. Mitchell, J.C., McGoldrick, P., Vance, C., Hortobagyi, T., Sreedharan, J., Rogelj, B., Tudor, E.L., Smith, B.N., Klasen, C., Miller, C.C. et al. (2013) Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol., 125, 273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wils, H., Kleinberger, G., Janssens, J., Pereson, S., Joris, G., Cuijt, I., Smits, V., Ceuterick-de Groote, C., Van Broeckhoven, C. and Kumar-Singh, S. (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U. S. A., 107, 3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Malik, A.M., Miguez, R.A., Li, X., Ho, Y.S., Feldman, E.L. and Barmada, S.J. (2018) Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife, 7, e35977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Levine, T.P., Daniels, R.D., Gatta, A.T., Wong, L.H. and Hayes, M.J. (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics, 29, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nassif, M., Woehlbier, U. and Manque, P.A. (2017) The enigmatic role of C9ORF72 in autophagy. Front. Neurosci., 11, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lall, D. and Baloh, R.H. (2017) Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest., 127, 3250–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shao, Q., Liang, C., Chang, Q., Zhang, W., Yang, M. and Chen, J.F. (2019) C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun., 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hayes, L.R. and Rothstein, J.D. (2016) C9ORF72-ALS/FTD: transgenic mice make a come-BAC. Neuron, 90, 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reddy, K., Zamiri, B., Stanley, S.Y., Macgregor, R.B., Jr. and Pearson, C.E. (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem., 288, 9860–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lin, Y., Mori, E., Kato, M., Xiang, S., Wu, L., Kwon, I. and McKnight, S.L. (2016) Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell, 167, 789–802.e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee, K.H., Zhang, P., Kim, H.J., Mitrea, D.M., Sarkar, M., Freibaum, B.D., Cika, J., Coughlin, M., Messing, J., Molliex, A. et al. (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell, 167, 774–788.e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jain, A. and Vale, R.D. (2017) RNA phase transitions in repeat expansion disorders. Nature, 546, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Couthouis, J., Hart, M.P., Shorter, J., DeJesus-Hernandez, M., Erion, R., Oristano, R., Liu, A.X., Ramos, D., Jethava, N., Hosangadi, D. et al. (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. U. S. A., 108, 20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yin, S.Y., Lopez-Gonzalez, R., Kunz, R.C., Gangopadhyay, J., Borufka, C., Gygi, S.P., Gao, F.B. and Reed, R. (2017) Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause Mis-splicing in ALS/FTD patients. Cell Rep., 19, 2244–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kwon, I., Xiang, S., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., Kim, J., Yun, J., Xie, Y. and McKnight, S.L. (2014) Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science, 345, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shi, K.Y., Mori, E., Nizami, Z.F., Lin, Y., Kato, M., Xiang, S., Wu, L.C., Ding, M., Yu, Y., Gall, J.G. et al. (2017) Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. U. S. A., 114, E1111–E1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Protter, D.S.W. and Parker, R. (2016) Principles and properties of stress granules. Trends Cell Biol., 26, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jacobsen, J.C., Erdin, S., Chiang, C., Hanscom, C., Handley, R.R., Barker, D.D., Stortchevoi, A., Blumenthal, I., Reid, S.J., Snell, R.G. et al. (2017) Potential molecular consequences of transgene integration: the R6/2 mouse example. Sci. Rep., 7, 41120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nguyen, L., Laboissonniere, L.A., Guo, S., Pilotto, F., Scheidegger, O., Oestmann, A., Hammond, J.W., Li, H., Hyysalo, A., Peltola, R. et al. (2020) Survival and motor phenotypes in FVB C9-500 ALS/FTD BAC transgenic mice reproduced by multiple labs. Neuron, 108, 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Martin, M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, 17, 10–12. [Google Scholar]
- 90. Marco-Sola, S., Sammeth, M., Guigo, R. and Ribeca, P. (2012) The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods, 9, 1185–1188. [DOI] [PubMed] [Google Scholar]
- 91. Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v1 [q-bio.GN], in press.
- 92. Picard . A set of command line tools (in java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. http://picard.sourceforge.net. in press.
- 93. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M. and DePristo, M.A. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.