

Abstract
Membrane Protein Palmitoylated 5 (MPP5) is a highly conserved apical complex protein essential for cell polarity, fate and survival. Defects in cell polarity are associated with neurologic disorders including autism and microcephaly. MPP5 is essential for neurogenesis in animal models, but human variants leading to neurologic impairment have not been described. We identified three patients with heterozygous MPP5 de novo variants (DNV) and global developmental delay (GDD) and compared their phenotypes and magnetic resonance imaging (MRI) to ascertain how MPP5 DNV leads to GDD. All three patients with MPP5 DNV experienced GDD with language delay/regression and behavioral changes. MRI ranged from normal to decreased gyral folding and microcephaly. The effects of MPP5 depletion on the developing brain were assessed by creating a heterozygous conditional knock out (het CKO) murine model with central nervous system (CNS)-specific Nestin-Cre drivers. In the het CKO model, Mpp5 depletion led to microcephaly, decreased cerebellar volume and cortical thickness. Het CKO mice had decreased ependymal cells and Mpp5 at the apical surface of cortical ventricular zone compared with wild type. Het CKO mice also failed to maintain progenitor pools essential for neurogenesis. The proportion of cortical cells undergoing apoptotic cell death increased, suggesting that cell death reduces progenitor population and neuron number. Het CKO mice also showed behavioral changes, similar to our patients. To our knowledge, this is the first report to show that variants in MPP5 are associated with GDD, behavioral abnormalities and language regression/delay. Murine modeling shows that neurogenesis is likely altered in these individuals, with cell death and skewed cellular composition playing significant roles.
Introduction
Membrane Protein Palmitoylated 5 (MPP5), also known as Protein Associated with Lin7 (PALS1), is a highly conserved apical complex protein characterized by its asymmetric localization to the apical side of polarized cells (OMIM 606958). It is expressed in the brain and peripheral nervous system, in addition to the placenta, kidney, heart and skeletal muscle (1). MPP5 is a member of the membrane-associated guanylate kinase family (MAGUK) proteins, which are important for determining cell polarity at tight junctions. It is a scaffolding protein with multiple distinct protein–protein interaction domains, such as PDZ, SH3, GuKc and two L27 domains. The L27 domains are known to bind PatJ and Lin7, proteins important in tight junction formation. PDZ (postsynaptic density 95/discs/large/zona occludens-1 domain) mediates MPP5’s connection to Crumbs proteins to form a complete apical polarity complex (1–3). GuKc, a guanylate kinase homologue domain, lacks recognized kinase activity but has a known function in protein binding and helps to define it as a MAGUK protein. SH3 domains allow protein binding to ligands rich in proline. MPP5 has been shown to act as an adaptor protein connecting the Crumbs–MPP5 (Pals1)–PatJ1 and Par3–Par6–aPKC apical complexes (4). Both of these apical polarity complexes have been extensively studied and their critical functions in the developing brain are well established. (5–7).
During cerebral cortex development, MPP5 is strongly expressed in ventricular zone progenitor cells and concentrated at the cell junctions in the ventricular surface apical to adherens junctions (8). When MPP5 is depleted, ventricular zone progenitor cells fail to self-renew and exit the cell cycle, leading to precocious production of post-mitotic neurons. Most of these cells die prematurely, reducing the size of the cerebral cortex. Complete knockout of MPP5 produces near absence of cerebral cortical neurons. Heterozygous knockout mice have a significant reduction in the size of their cerebral cortex with a notable reduction in the size of their hippocampus. These results suggest a dose-dependent effect (8). MPP5 is also present in all proliferating cell populations of the developing cerebellum and essential for establishing normal cerebellar architecture. Depletion of MPP5 in cerebellar progenitors leads to abnormal cellular layering and folia and overall reduction of cerebellum size. Importantly, MPP5 is required for normal proliferative capacity of progenitors in all cerebellar germinal zones regardless of apico-basal polarity (9).
In addition to its roles in brain development, MPP5 has also been shown to be essential for retina morphogenesis and retinal cell survival. Conditional knockout of Mpp5 in the developing eye mimics the early onset severe form of retinal blindness, Leber congenital amaurosis (LCA), with disrupted lamina structure and profound cell death (10). Mice with conditional knockdown of Mpp5 also display disorganized retinal architecture with abnormal Crumbs apical complex localization and extensive gliosis (11). Furthermore, previous studies of the peripheral nervous system in Mpp5 knockdown mice demonstrated that Mpp5 is important for Schwann cell myelination. It is required for myelin sheath formation because it directs correct trafficking of myelin proteins to the plasma membrane (12). Conditional knockout of Mpp5 in Schwann cells also revealed its function in radial sorting of axons and timely myelination (13).
These studies highlight the broad importance of MPP5 in neural cell polarity, fate, survival, neurogenesis, tissue morphogenesis and myelination. Although defects in cell polarity have been associated with neurologic disorders, including microcephaly, lissencephaly, autism, epilepsy and schizophrenia (14), MPP5 has not previously been implicated in human development. In this study, we describe three patients with de novo variants in MPP5 and global developmental delay (GDD) with language delay or regression and behavioral changes. Using a heterozygous Mpp5 knockout mouse model with CNS-specific Nestin-Cre driver, we assessed how Mpp5 depletion can alter neurodevelopment. Overall, this study highlights the importance of MPP5 in human brain development and cognitive function.
Results
Trio exome sequencing (ES) was performed on a patient presenting with GDD/intellectual disability (ID), speech regression and dysmorphic facial features (Individual 1, Table 1), and a de novo somatic nonsense variant in the MPP5 gene was identified. International data sharing through the MatchMaker Exchange allowed us to identify two additional patients carrying pathogenic variants in this gene with similar clinical spectrum (15–16). In order to better understand the pathogenicity of MPP5 alteration, we first compared the clinical features and magnetic resonance imaging (MRI) of each of the patients. All of the patients were diagnosed with significant GDD and ID, hypertonia, regression of speech or severe language delay and behavioral concerns (Table 1). MRI of the patients varied with findings ranging from normal to a reduction in brain size and decreased gyral folding.
Table 1.
Clinical and neuroradiologic features of individuals with MPP5 pathogenic variants
| Individual | Individual 1 | Individual 2 | Individual 3 |
|---|---|---|---|
| MPP5 variant (GeneBank: NM_022474.4) | c.(1555C>T) p.(Arg519Ter), present in 5/25 reads, confirmed by Sanger sequencing | c.(1289A>G) p.(Glu430Gly) | c.(974A>C) p.(His325Pro) |
| Inheritance | De novo, somatic mosaicism | De novo, heterozygous | De novo, heterozygous |
| gnomAD frequency | 3.98e−6 (1/251 338) | 0 | 0 |
| CADD score | 36 | 25.3 | 24.5 |
| Sequencing method | Clinical ES | Research-based trio ES | Clinical ES |
| Sex | Female | Male | Male |
| Ethnicity | German, Austrian and Ashkenazi Jewish ancestry | Scottish, Irish, English | Unknown |
| Current age, years | 7 | 21 | 13 |
| Gestational age at birth (in weeks) | 38+6 | 37 | 41+4 |
| Birth weight | Unknown | 3306 g | 2975 g |
| Birth length | Unknown | 46 cm | Unknown |
| Head circumference at birth | Unknown | 34 cm | Unknown |
| Prenatal concerns | Intrauterine growth restriction | Preeclampsia | None |
| Failure to thrive | Present | None | None |
| Feeding issues | +, Gastroesophageal reflux with history of recurrent aspiration, g-tube dependence | None | None |
| Global developmental delay | Present | Present | Present |
| Speech | Delayed | Absent as teenager | Delayed |
| Gross motor skills | Delayed | Delayed, walked at 2.5 years old | Normal |
| Fine motor skills | Delayed | Delayed, walked at 2.5 years old | Normal |
| Regression | Regression of speech | Possible regression versus severe language delay; at age 6 was said to have <10 words, absent as an adult | Regression of speech (he lost his first words (mama, papa) and started to talk at 4–5 years of age |
| Intellectual disability | Present | Present, severe | Present, IQ 55 |
| Microcephaly (HC and percentile) | Present, HC 42.5 cm, 1st percentile | Not present, HC 10th percentile | Not present, HC 35th percentile |
| Seizures | None | Present, first generalized tonic–clonic seizure in 20 s | None |
| Ataxia | Not present | Present | Not present |
| Autism spectrum disorder | Not present | Not present | Positive for ASD |
| Hypotonia | Central hypotonia | Present | None reported |
| Hypertonia | +, Bilateral lower extremities | +, Peripheral hypertonia with spasticity | None reported |
| Behavior abnormalities | +, Periods of high energy with a lot of laugher followed by profound hypotonia | +, Anxiety, panic attacks | +, Aggression; anger tantrums; tics; severe behavior problems |
| Structural brain abnormalities | Microcephaly with slightly decreased gyral folding present | MRI in 2010 at 12 years of age with a small midline arachnoid cyst in the posterior fossa | MRI at 4 years of age; reported to be normal |
| Vision/eye abnormalities | Present, cortical visual impairment, retinal dystrophy, nystagmus | None present | None present |
| Hearing loss | Present, bilateral sensorineural hearing loss | None present | None present |
| Dysmorphic features | Bitemporal hollowing, prominent forehead, down-slanting eyes, sparse eyebrows, corners of mouth turn downwards, posteriorly rotated ears, inverted nipples | Widely spaced teeth, fine facial features | Large teeth/fetal finger pads |
Clinical and genotypic findings in patients carrying MPP5 variants
Individual 1 had notable intrauterine growth restriction with no additional prenatal concerns present. She has a history of GDD and ID with speech regression. At 15 months of age, she was noted to have central hypotonia with increased tone in her lower extremities and hyperreflexia. When re-evaluated at 8 years of age, she was noted to be hypotonic overall with absent deep tendon reflexes. Individual 1 is also visually impaired and has been diagnosed with both cortical visual impairment and retinal dystrophy. Retinal dystrophy was diagnosed after electroretinography (ERG) testing was obtained at 15 months of age. Scotopic function testing showed a significant attenuation of B wave amplitudes, suggesting a decrease in Muller and bipolar cell function. Photopic function testing was also performed and demonstrated below average amplitudes with mildly prolonged implicit times, suggesting borderline cone cell dysfunction. Based on these results, she was diagnosed with mild dysfunction of retina bilaterally. Individual 1 also has bilateral sensorineural hearing loss requiring hearing aids. Her MRI is notable for microcephaly with slightly decreased gyral folding (Fig. 1). Her dysmorphic features include bitemporal hollowing with prominent forehead, down-slanting eyes, sparse eyebrows, downturned corners of her mouth, posteriorly rotated ears and inverted nipples (Fig. 1; Table 1). On ES, she carried a de novo somatic mosaic variant in MPP5, c.(1555C>T), resulting in a nonsense variant, p.(Arg519*), in 20% (5/25) of the reads, confirmed by Sanger sequencing. This variant is within the GuKc domain of the protein with a Combined Annotation Dependent Depletion (CADD) score of 36, suggesting loss of function with high confidence. This variant has been seen once in gnomAD with a frequency of 3.98e−6 (1/251 338).
Figure 1.
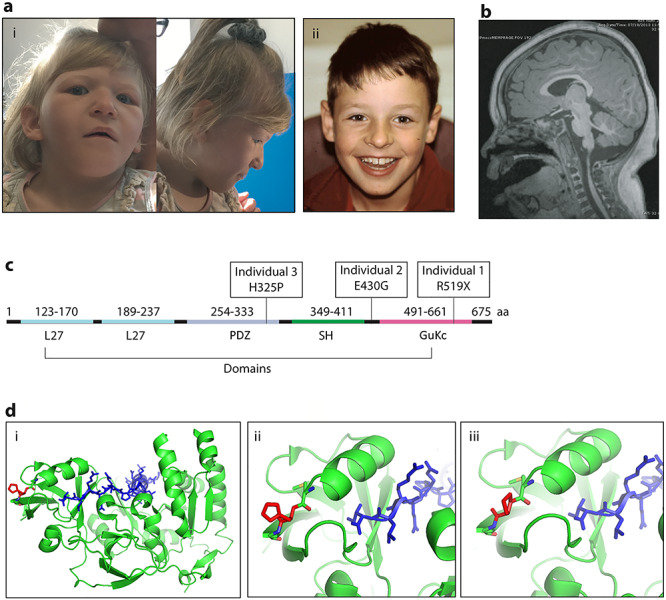
Patient clinical phenotypes and variant localization. (A) Images of dysmorphic features seen in Individual 1 (i) and Individual 2 (ii). (B) Brain MRI image of Individual 1, highlighting the mild decrease in gyral folding. (C) MPP5 gene with domains and patient variants noted. (D) Molecular modeling of the missense variant in individual 3 with (i) showing the overall protein structure of MPP5, (ii) depicting His325 and (iii) showing Pro325.
Individual 2 was born with a normal weight and head circumference, and there were no significant prenatal findings other than maternal preeclampsia. He has a history of GDD and ID with speech delay and regression. He was noted to have less than 10 words at 6 years of age and now has absence of speech as an adult. He is reported to have normal hearing and vision. He has peripheral hypertonia with spasticity and ataxia and has been diagnosed with a mixed cerebral palsy. He has anxiety and panic attacks. His MRI at 12 years of age was notable for a small midline arachnoid cyst in the posterior fossa. His dysmorphic features include widely spaced teeth and fine facial features (Table 1). He has a de novo heterozygous variant on research-based trio ES, c.(1289A>G), resulting in the missense variant, p.(Glu430Gly), which is within the SH3 domain of the protein. This variant has a gnomAD frequency of 0 with a CADD score of 25.3.
Birth history was overall unremarkable with no prenatal concerns for Individual 3. He has since been diagnosed with severe ID with an IQ of 55 and a notable speech delay. He lost his first words of mama and papa and then started to speak at 4–5 years of age. He is reported to have normal hearing and vision. He has both autism spectrum disorder and aggression with a normal brain MRI at 4 years of age. Dysmorphic features include large teeth and prominent finger pads (Table 1). He has a heterozygous de novo variant in MPP5, c.(974A>C), resulting in the missense variant, p.(His325Pro), which is within the PDZ domain of the protein. This variant was found on clinical ES and has a gnomAD frequency of 0, with a CADD score of 24.5.
Molecular modeling was performed for each of the missense variants observed in the cohort. Individual 2 has a missense variant, p.(Glu430Gly), in the long loop of the SH3 domain, linking the SH3 and GuKc domains. This region is highly flexible, preventing further localization of Glu430 and understanding of its role in the structure. Individual 3 has a variant, p.(His325Pro), in the PDZ domain of MPP5. The side chain of His325 points away from the protein–protein binding interface, where PDZ interacts with the C-terminal PDZ binding domain of the Crumbs. His325 creates a loop rather than a secondary structure, suggesting that the missense variant, Pro325, will not affect the local secondary structure of the PDZ binding domain directly but will likely affect the overall stability of the PDZ domain (Fig. 1) (17).
Heterozygous murine model of nervous system-specific Mpp5 deficiency shows neuroanatomical changes
In order to understand the effects of heterozygous MPP5 variants on our patients’ developing brains, we utilized the Mpp5 (Pals1) heterozygous conditional allele with a nervous system-specific Nestin-Cre driver, expressed in both the central and peripheral nervous systems beginning at embryonic day (E) 12. Nervous system-specific haploinsufficiency of Mpp5 (hereafter designated het CKO) leads to mice that were small in stature with their body length 17% shorter than Cre-negative wild-type (WT) littermate controls at postnatal day (P) 21 (P = 0.012) (Fig. 2A and B). Notably, their brain size was markedly reduced (Fig. 2C). To measure this, we compared the cortical thickness of het CKO with that of WT littermate controls and found it reduced by 47% (P < 0.001) (Fig. 2D, E and H). Although the brains of all of the het CKO mice were undersized, about half of them showed both microcephaly and ventriculomegaly, while the others displayed only microcephaly (Fig. 2C–E). Since functional and structural defects, including absence of the ependymal cell layer, are implicated in the pathogenesis of hydrocephalus, we examined ependymal cells with cilia markers, Arl13B and AC3, and the ependymal cell-specific nuclear marker, FoxJ1 (Supplementary Material, Fig. S1). Het CKO animals had decreased ependymal cells along the ventricular lining of the lateral ventricle at P21; the reduction was particularly notable along the dorsal side (Supplementary Material, Fig. S1B and D). In contrast, FoxJ1+ ependymal cells with multiple motile cilia (AC3+ and Arl13B+) were consistently present on the ventricular lining of WT mice (Supplementary Material, Fig. S1A and C). A reduction in the number of ependymal cells may contribute to the het CKO mouse development of ventriculomegaly. The size of the cerebellum and cerebellar lobes was also 42% smaller in the het CKO mice than in WT, and the cerebellum was not fully developed (P ≤ 0.0001); lobes II and III were not separated and the molecular layer was absent in the ventral half of lobe X (Fig. 2G–J). These results show that heterozygous mutation of Mpp5 causes brain malformations in mice, including microcephaly with or without ventriculomegaly (Fig. 2E and F).
Figure 2.
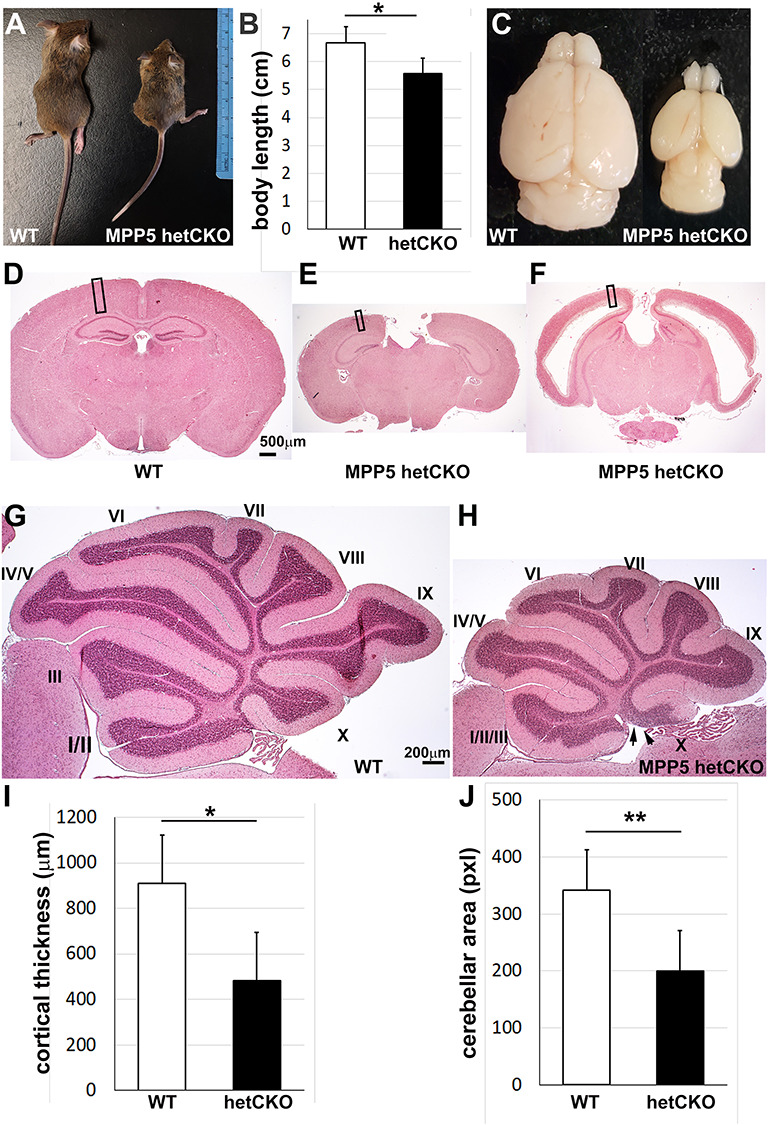
Mpp5 heterozygous CKO mice are smaller and have a reduced number of neurons. (A–C) At P21, body length and brain size were measured in Mpp5 het CKO mice and WT littermate controls (B, −17%, P = 0.012). (D–F) Histology with hematoxylin and eosin shows two microcephaly phenotypes—with and without ventriculomegaly. (G, H) Histology of cerebellum of WT and Mpp5 het CKO comparing size and lobe formation at P21. Arrows indicate the absence of molecular layer in the lobe X in Mpp5 het CKO. (I) Cortical thickness of Mpp5 het CKO compared with that of WT (−47%, P = 0.000002). (J) Comparison of cerebellar area between Mpp5 het CKO and WT (−42%, P = 0.0001). Data are presented as mean ± SEM. WT, n = 5; Mpp5 het CKO, n = 5.
Behavioral changes and visual deficits present in Mpp5 het CKO murine model
Since all three human patients with MPP5 de novo variants show behavioral changes, we investigated Mpp5 het CKO mice for behavioral abnormalities. The het CKO mice were hyperactive and moved around much more frequently than WT mice at P21. The degree of the hyperactivity was measured by comparing the number of times that the mice crossed the grids in an open field chamber over 5 min (Fig. 3A and B). Het CKO mice crossed the grids 2.89 times more than WT (P < 0.05) (Fig. 3C). Interestingly, they also spent 2.42 times the duration in the center of the open field (P < 0.05) (Fig. 3D). Typically, mice avoid the center of the field. This absence of the normal avoidance suggests that the vision of the het CKO mice may be impaired, as it was in Individual 1 in our cohort. She carries a nonsense variant and has visual impairment secondary to both cortical blindness and retinal dystrophy. Although a visual impairment in the mice could be fully explained by cortical impairment, we reasoned that retinal dysfunction may also contribute to their decreased vision. This hypothesis is supported by prior studies that demonstrated that MPP5 loss causes early onset retinal cell degeneration (10) and that mutations of the gene encoding the MPP5 interacting partner Crb1 are responsible for LCA, the most severe form of early onset retinal blindness (18). We assessed retinal function in the het CKO and WT mice with ERG at P21. The scotopic ERG of the het CKO mice demonstrated a downward trend in the amplitude of the a-wave, which measures rod cell function, but the difference was statistically not significant (P = 0.068) (Fig. 4A and C). Remarkably, the ERG of the b-wave of the het CKO was decreased by 73% (P < 0.001), indicating functional impairment of Muller and bipolar cells (Fig. 4A and D). Photopic ERG of these mice revealed that the b-wave signal was also close to baseline (P = 0.012), indicating cone cell dysfunction (Fig. 4B and E). Together, these ERG studies provide evidence that het CKO mice have retinal blindness, which likely contributes to their inability to distinguish between the center and the edges of the open field chamber.
Figure 3.
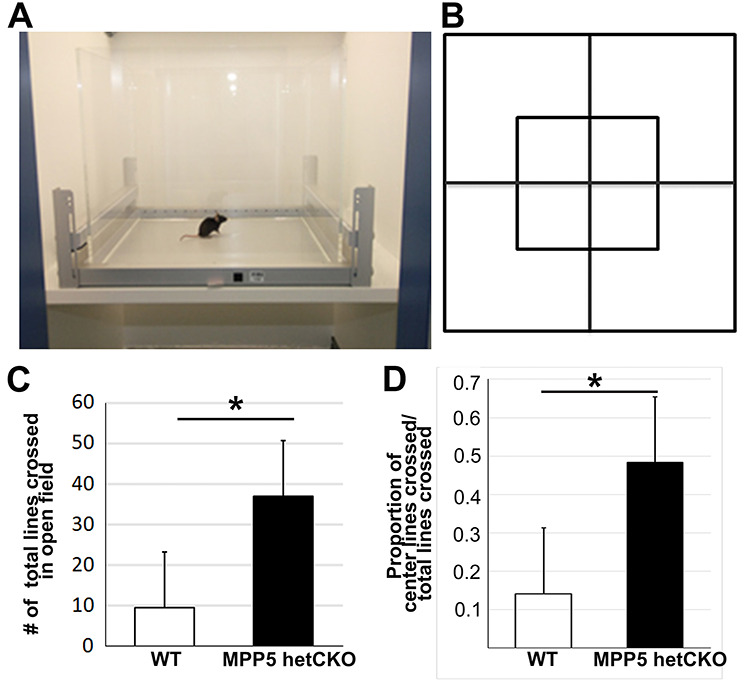
Behavioral testing shows hyperactivity in Mpp5 het CKO mice. (A, B) Diagrams depicting the open field test chamber and grid used in the study. (C, D) Comparison of total grid lines crossed (C, 289% more in het CKO, P = 0.031) and of the proportion of grid lines crossed in the center of the open field chamber among total crossings (D, 242% more in het CKO, P = 0.012). Data are presented as mean ± SEM. WT, n = 4; Mpp5 het CKO, n = 4.
Figure 4.
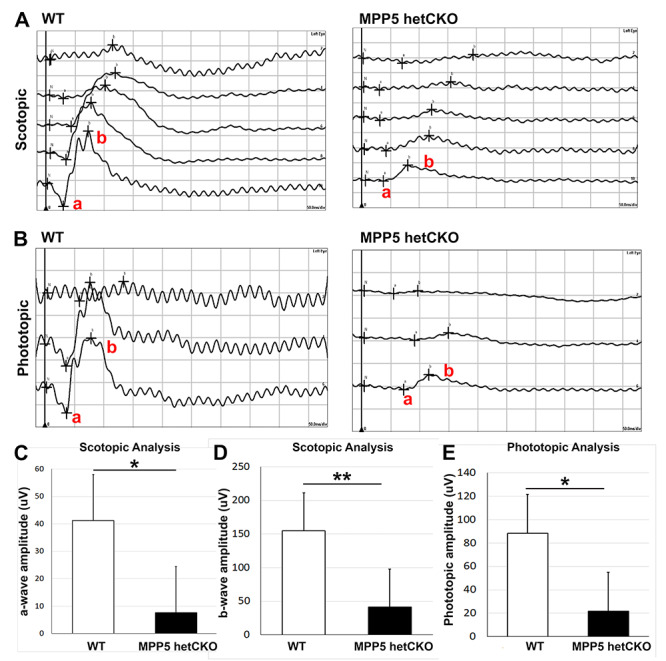
Mpp5 heterozygous CKO mice display retinal blindness. (A) Representative scotopic ERG traces of Mpp5 het CKO and WT, which assess function of rod photoreceptors (a-wave) and Muller and bipolar cells (b-wave). (B) Representative photopic ERG traces of Mpp5 het CKO and WT, which assess retinal cone function. (C, D) Quantification of the a-wave and b-wave amplitude (C, −81%, P = 0.063; D, −73%, P = 0.0005). (E) Quantification of the b-wave amplitude for photopic ERGs (E, −75%, P = 0.012). Data are presented as mean ± SEM. E14: WT, n = 4; Mpp5 het CKO, n = 4.
Mpp5 reduction leads to decrease in cortical neuron number, while neuronal migration is preserved
To better understand the brain pathology, neuronal composition of the het CKO was examined. The cerebral cortex was found to be smaller than expected at P21 (Fig. 2C). Cortical layer-specific markers, including Cux1+ (layer II–IV), Ctip2+ (layer V) and FoxP2+ (layer VI), revealed that neuronal numbers were reduced in all layers of the het CKO compared with WT (Cux1 by 61%, Ctip2 by 42%, Foxp2 by 49%, P < 0.001) (Fig. 5A–D). This global reduction accounts for the significant decrease in cortical thickness observed in het CKO mice (reduced by 47%, P = 0.012) (Fig. 2I). Although neuronal numbers were notably reduced, cortical lamination did not appear to be disrupted. Similarly, the cerebellum was hypoplastic and its lobes deformed (Fig. 1), but cell type-specific marker analyses did not reveal obvious perturbation of cerebellar lamina structure (Supplementary Material, Fig. S2). Neuronal reduction was therefore the major consequence of Mpp5 reduction; neuronal migration did not appear to be critically affected. Importantly, reduction of Cux1+ late-born neurons was the most prominent type of cell decrease in the het CKO cerebral cortex (Fig. 5A and D, suggesting a gradual decrease of neural progenitor populations.
Figure 5.
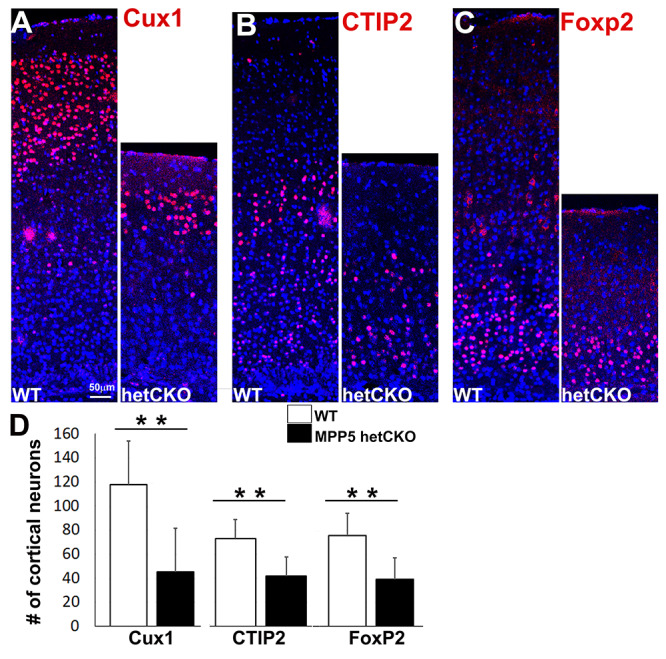
Mpp5 heterozygous CKO mice have a reduced number of neurons. (A–C) Immunostaining of cortical layer-specific neurons (Cux1+, layers 2–4, A; CTIP2+, layer 5, B; FoxP2+, layer 6, C) is shown in Mpp5 het CKO and WT control at P21 (D). The number of layer-specific neurons is compared between Mpp5 het CKO and WT (Cux1+ neurons, −61%, P = 0.0001; CTIP2 + neurons, −42%, P = 0.0001; FoxP2+ neurons, −49%, P = 0.0002). Data are presented as mean ± SEM. WT, n = 5; Mpp5 het CKO, n = 5.
Mpp5 reduction leads to decreased apical protein complex localization at the apical junction
To investigate progenitor loss and defects in neurogenesis during cortical development, E14.5, the peak of cortical neurogenesis, was examined. At this time, cortical progenitors are undergoing numerous cell divisions both to self-renew and to produce the differentiating neurons of the developing cortex. Western blot analysis showed a 35% reduction in Mpp5 protein expression in cortical lysates of Mpp5 het CKO animals (P < 0.005) (Fig. 6A–E) and a similar Mpp5 reduction in cultured fibroblasts derived from Individual 1 (with nonsense variant) was noted (Fig. 6C). Immunostaining of Mpp5 also consistently showed profound decrease of Mpp5 expression at the apical surface (Fig. 6A and B′). Since Mpp5 plays an important role in assembling scaffolding proteins for apical protein complexes (4), we assessed whether Mpp5 reduction would affect the localization of apical polarity complex proteins. Pan Crb antibody, which detects all three Crb proteins, was utilized to examine localization of Crb proteins, the essential interacting proteins in most epithelia, including neuroepithelium (8). Unlike WT, in which they were mainly located at the ventricular surface, Crb proteins in the het CKO appeared to be reduced at the ventricular surface and apical junction with some punctate staining shifted to the basal side (Fig. 6F and G′). The level of Par3, another apical polarity complex protein, was also reduced at the ventricular surface of het CKO with a few puncta displaced to the basal side (Fig. 6H and I′). These results show that Mpp5 is reduced at the apical junction of the het CKO; this reduction leads to decreased localization of Mpp5 interacting proteins at the apical junction at the peak of neurogenesis.
Figure 6.
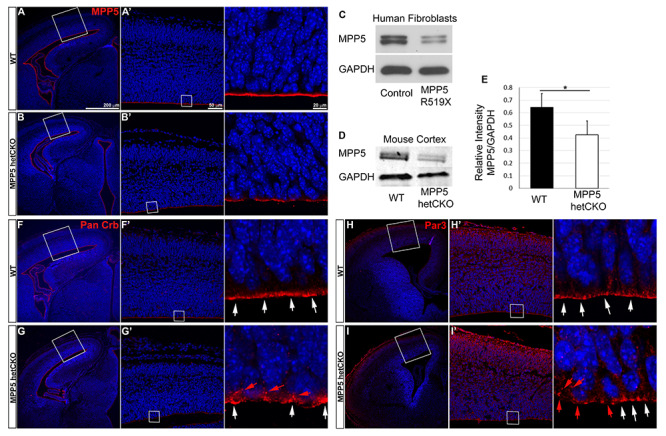
Mpp5 reduction decreases apical localization of apical polarity complex proteins in Mpp5 het CKO at E14.5. (A–B′) Representative image of Mpp5 immunostaining in WT and Mpp5 het CKO cerebral cortex at E14.5. (C) Western blot showing Mpp5 protein levels in fibroblasts derived from patient #1 and normal control. (D, E) Western blot and quantification of Mpp5 protein in Mpp5 het CKO and WT cortical lysates at E14.5 (E, −35%, P = 0.005). (F–I′) Representative images of apical polarity complex protein staining in developing cortex (Pan-Crb, F–G′; Par3, H–I′). White arrows indicate these proteins at the apical surface; red arrows indicate basally shifted ones. Data are presented as mean ± SEM. WT, n = 4; Mpp5 het cKO, n = 4.
Mpp5 reduction leads to decreased progenitor populations secondary to apoptosis
Having confirmed a substantial decrease in Mpp5 expression at E14.5, the composition of cortical cells, including progenitors and neurons, was examined in detail. The size of the cortex was decreased in the het CKO, and the numbers of radial glia progenitors marked by Pax6+ and of intermediate progenitors labeled by Tbr2+ were significantly reduced compared with WT (reduced 26%, P < 0.001 and reduced 30%, P < 0.05, respectively) (Supplementary Material, Fig. S3). Although there were significantly fewer CTIP2+ neurons in the het CKO than in WT at P21, the number of CTIP2+ neurons was unaffected at E14.5 (Supplementary Material, Fig. S3). This suggests that CTIP2+ cells decreased over time through either degeneration or reduced production of CTIP2+ cells after E14.5. These results highlight that the major phenotype caused by Mpp5 reduction is the failure to maintain progenitor pools.
To determine why progenitor populations are decreased, progenitor cells were labeled by injecting BrdU at E13.5. BrdU is incorporated into newly synthesized DNA and marks cells in S-phase at the time of injection. We examined the fate of the labeled cells 24 h later by double immunostaining for BrdU and cell type-specific markers: Sox9+ (radial glia progenitors), Tbr2+ (intermediate progenitors) or p27+ (post-mitotic cells). At this time, the cells that are presumably progenitors would have undergone one cell cycle. There were significantly fewer BrdU-marked cells expressing each of these markers in het CKO than in WT (P < 0.05) (Fig. 7). The proportion of Sox9+ and Tbr2+ radial glia progenitors among total BrdU+ cells was markedly reduced compared with WT (reduced 50%, P = 0.00002 and reduced 29%, P = 0.0001, respectively) (Fig. 7A–C). The proportion of post-mitotic cells (p27+) was also reduced (−11%), but the extent of reduction was much less than that of progenitor populations (P < 0.05) (Fig. 7G and H). This result suggests that progenitor cells at E13.5 are no longer cycling and become post-mitotic cells or undergo cell death. We hypothesized that the cells underwent cell death, given prior studies showing that Mpp5 loss causes neuroepithelial progenitors to exit the cell cycle prematurely and leads to massive cell death (8). As expected, there was a significant increase in the number of cells undergoing apoptotic cell death (442% increase, P < 0.001) in the het CKO mice, marked by immunostaining for cleaved caspase (CC3), among total BrdU+ cells (Fig. 7J–L). To investigate the cell death further, we counted CC3+ dying cells and cells with double-strand DNA damage marked by gH2AX+ in cortical sections. Cell death and DNA damage were found to be significantly increased by 313% in the het CKO cortex compared with WT (P = 0.001) (Fig. 8). To determine whether progenitors as well as post-mitotic neurons undergo apoptosis, a marker for post-mitotic cells (p27+) and a marker for proliferating cells (PCNA+) in conjunction with CC3 were utilized. Since many dying cells are likely to stop expressing cell type-specific markers, the detection of dying cells with cell-specific markers is probably incomplete. However, subsets of CC3+ cells clearly overlapped with P27 or PCNA (Fig. 8I and J′), which provides evidence of their apoptotic cell death. In summary, these results suggest that reduction in progenitor cells and neurons in the Mpp5 het CKO is primarily caused by massive apoptotic cell death, emphasizing that the pathogenesis of Mpp5 haploinsufficiency is from compromised cell survival.
Figure 7.
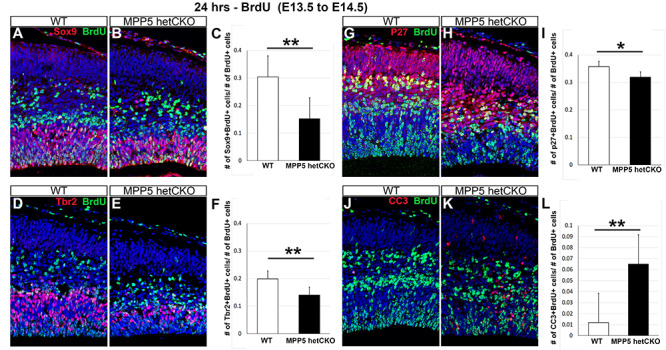
Cell death reduces Mpp5 het CKO progenitor pools. (A–L) Mice injected with BrdU at E13.5 analyzed at E14.5 with double immunostaining for BrdU and additional markers. The proportion of radial glia progenitors (Sox9+, A–C, −50%, P = 0.00002), intermediate progenitors (Tbr2+, D–F, −29%, P = 0.0001) and post-mitotic cortical cells (p27+, G–H, −11%, P = 0.02) among total BrdU+ cells is compared in WT and Mpp5 het CKO. The proportion of cells undergoing apoptotic cell death marked by CC3 among total BrdU+ cells is also assessed (J–L, 442%, P = 0.00016). Data are presented as mean ± SEM. E14: WT, n = 4; Mpp5 het cKO, n = 4.
Figure 8.
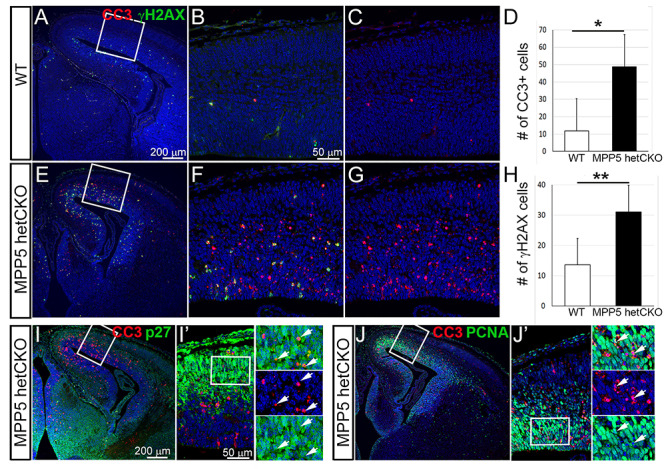
Developing cerebral cortex of Mpp5 het CKO mice displays massive cell death. (A–H) Analysis of the numbers of cells undergoing apoptotic cell death labeled by CC3 (D, 313%, P = 0.12) or double-strand DNA damage marked by gH2AX (H, 128%, P = 0.001) in cortical sections of Mpp5 het CKO (E–G) and WT (A–C) at E14.5 (I–J′). Overlapping expression of CC3 and post-mitotic cell marker, p27 (I, I′) or proliferating cell marker, PCNA (J, J′) is present in Mpp5 het CKO (arrows). Data are presented as mean ± SEM. WT, n = 4; Mpp5 het CKO, n = 4.
Discussion
Rare de novo heterozygous variants in MPP5 were identified in three patients with GDD/ID, language delay or regression and behavioral changes in the present study. MPP5 is an apical complex protein essential for the development of cell polarity, survival and neurogenesis. While variants in other proteins associated with cell polarity, such as LIS1, ARFGEF2, FLNA and DCX, have been associated with neurodevelopment, this is the first instance of an apical complex protein altering human brain development (14,19). To further understand the pathogenicity of MPP5 depletion, a heterozygous murine model with a CNS-specific Nestin-Cre driver was utilized. This study demonstrates that there is significant phenotypic overlap between the mouse model and human patients.
A unifying phenotype in the cohort was GDD with each patient ultimately diagnosed with ID, once they reached an age that testing was validated it. Microcephaly is a physical exam finding that can be associated with GDD/ID and suggests a possible underlying neuroanatomic abnormality, defined as an occipital frontal circumference of two or more standard deviations below the mean, or less than the second percentile (20). Individual 1 had significant microcephaly with her head circumference at the first percentile. Individual 2's head circumference was at the 10th percentile and individual 3's was the 35th percentile. Neuroanatomical changes are also associated with global development delay, with abnormal findings seen in approximately 30% of children with GDD/ID (21). MRI is recommended by the American Academy of Pediatrics as part of the diagnostic approach to understanding the etiology of GDD/ID in children (21). Each of the individuals in the cohort had an MRI completed, and their neuroimaging, like their head circumferences, demonstrated a spectrum of severity. Individual 1’s MRI showed a slight decrease in gyral folding in addition to microcephaly. Individual 2’s MRI showed a small midline arachnoid cyst in the posterior fossa, and Individual 3’s MRI was reported as normal. Individual 1 has a mosaic nonsense variant, leading to loss of MPP5 function, which has caused the most severe phenotype. In contrast, Individuals 2 and 3 both have missense variants, which may result in hypomorphic alleles, preserving some MPP5 function necessary for cell survival, thereby preventing neuronal apoptosis and resultant alterations in neurogenesis. The murine model showed even more significant neuroanatomic findings, with microcephaly, reduced cortical thickness, ventriculomegaly and hypoplastic and dysmorphic cerebellum present. The differences in neuroanatomy between the affected humans and the murine model highlights the dose dependent effect of MPP5 expression. Further investigations using molecular and genetic modeling of the patients’ missense variants will likely help to explain the cellular deficiencies caused by these variants, which ultimately lead to their GDD, ID and behavior changes.
Seizure occurrence was another difference in clinical features appreciated in the cohort. Individual 2 has a history of epilepsy, starting when he was in his early 20s. Neither of the other two individuals has had a seizure; however, they were notably younger in age at the time of the study (7 and 13 years old). In the mouse model, some of the het CKO mice had seizures, with some seizures fatal. Variability in the occurrence and severity of the observed phenotypes suggests a spectrum of deficits due to MPP5 haploinsufficiency.
Individual 1 had notable hypotonia that progressed over time; she now has absent reflexes, suggesting a peripheral neuropathy. This is likely secondary to altered myelination of her peripheral nerves, as has been seen in a prior mouse model (12). This may be due to increased progenitor cell apoptosis over time. However, our murine model was specific to the central nervous system and therefore can only provide a limited understanding of her peripheral findings.
One of the three individuals in the cohort with a somatic nonsense variant (Individual 1) had a visual impairment and was diagnosed with both cortical blindness and bilateral retinal dystrophy. ERG studies from her showed a significant reduction in scotopic B wave amplitude with a borderline photopic response, suggesting that she has mild retinal dysfunction. A significant number of Mpp5 het CKO mice studied were also found to have visual deficits. ERG data in the het CKO mice showed a reduction in both scotopic and photopic b-wave amplitudes, indicating Muller, bipolar and cone cell dysfunction, which suggests that the mice have retinal blindness as well. Taken together with the patient data, visual impairment in the mouse model may be due to defects in both cortical and retinal development, contributing to the inability of the het CKO mice to distinguish between the center and the edges of the open field chamber. Future patients with MPP5 variants would likely benefit from early visual testing.
Another unifying clinical feature in the cohort is abnormal behavior, ranging in severity from anxiety with panic attacks to aggression. Behavioral changes were also evident in the mouse model, with het CKO mice crossing the grids significantly more times than WT, suggesting that the mice were more hyperactive.
Consistent with prior studies, the murine model demonstrates that cell death is a major contributor to reduction in cortical cell number and ultimate brain size and neuroanatomy. Although it is not yet clear whether precocious differentiation precedes cell death, it is notable that increased cell death is a major common feature of complete Mpp5 conditional knockout (8) and heterozygous loss, suggesting a critical alteration in neurogenesis that is dose dependent. Importantly, the current pan-nervous system heterozygote Mpp5 model offers new insights into the function of MPP5 in human pathology. Prior models deleted Mpp5 from a limited brain area rather than from the entire nervous system, with their primary focus on analysis of null mutants rather than on heterozygotes. These models do not address the MPP5 haploinsufficiency postulated to be the disease mechanism in human cases (8,9). Earlier models and analyses also overlooked global Mpp5 functional deficits, such as reduced body size, seizures, ventriculomegaly and hyperactivity. It is plausible that the reduction of MPP5 in brain regions, such as the basal ganglia or in GABAergic interneurons derived from ventral pallidum, contributes to changes in neurological outcomes. Examples include seizures and behavioral alterations, which are seen in both our mouse model and human cases. Through this study, we also gained a new understanding of the dose-dependent Mpp5 requirements at the cellular level. For instance, lamination is not disturbed in either the cerebellum or cortex of the Mpp5 het CKO model; however, cerebellar lamination is severely disrupted in homozygote mutants and is associated with glial scaffold defects (9, Mpp5 CKO;hGFAP-Cre). The intact lamination in het CKO suggests that the development of Bergmann glia is not Mpp5 dose sensitive. In contrast, the ability of cerebellar progenitors to produce a sufficient number of neurons and establish proper foliation does demonstrate a dose-dependent requirement for Mpp5 (9).
The mouse model therefore demonstrates that haploinsufficiency is the likely mechanism of action and helps to explain the difference in the clinical features and neuroanatomy seen in the human individuals and murine model. Fibroblasts from the individuals with missense variants were unfortunately unavailable and may have provided further evidence for this dose-dependent mechanism. Another possible, though unlikely, mechanism of the missense variants is that they can exert dominant negative effects in certain cellular contexts. For instance, the missense variants could encode a mutant protein that abnormally interacts with another protein that has yet to be identified. This interaction could lead to ectopic localization or association of MPP5 with another protein complex, preventing its typical function. However, this mechanism is unlikely, given the milder clinical findings seen in our two patients with missense variants. Future studies could evaluate this further, by introducing mutated MPP5 proteins into WT cells and determining whether a dominant negative effect is seen. Alternatively, MPP5 mutant proteins could be introduced into Mpp5 het CKOs to determine if any functional restoration is produced by the mutant protein-expressing cells. These future experiments could provide further evidence that the missense variants act through haploinsufficiency rather than a dominant negative mechanism.
In summary, this functional analysis in a murine model provides a foundation for understanding how rare de novo variants of MPP5 may lead to GDD, language delay or regression and behavioral changes in patients.
Materials and Methods
Human subjects
Individual 1 was enrolled in a research study at the Manton Center for Orphan Disease Research at Boston Children’s Hospital, approved by the Boston Children’s Hospital Institutional Review Board. ES was performed by GeneDx. The MPP5 variant was added to GeneMatcher, and two additional individuals were identified with de novo MPP5 variants on ES (22). The individuals were each enrolled in research studies by their own hospitals, The University of Calgary and The Radboud University Hospital Nijmegen, which had been approved by their Institutional Review Boards.
Mice
Animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Lewis Katz School of Medicine at Temple University. The Mpp5f/f mice were genotyped as previously described (8). Nestin-Cre mice were obtained from Jackson Lab (stock# 003771) and genotyped accordingly. Since we detected visual impairment in Mpp5 het CKO, we assessed whether their genetic background contains genetic alleles known to cause retinal degeneration when homozygotic. Some mouse embryonic stem cells of C57BL6/N origin used to generate flox or Cre driver and commonly used WT mouse strains such as C57BL6/N substrains carry mutant alleles of RD1 (mutations in the Pde6b, beta subunit of cGMP-PDE) and RD8 (a mutation in Crb1 gene). Therefore, we used PCR genotyping to examine whether these alleles are present in the genomes of mice used in the study (23–25). The Nestin-Cre mice do not include RD1 or RD8 alleles but some Mpp5 floxed animals carry one or two copies of RD1 and/or RD8. Because the Mpp5 heterozygote CKO model is generated by intercross between Nestin-Cre and Mpp5 floxed allele, there is a possibility that one copy of RD1 and/or RD8 is present in the Mpp5 het CKO. As it is known that none of the RD1 or RD8 heterozygotes at P21 show ERG findings, the ERG phenotype is not due to their heterozygosity (26–29). However, we cannot completely eliminate the possibility that the genetic interaction between Mpp5 and/or RD1 or RD8 may contribute to the severity of the vision compromise observed in Mpp5 het CKO animals.
Immunohistochemistry
Tissue samples were embedded in paraffin and sliced into 7 μm sections. Prior to immunohistochemistry, samples were rehydrated in a series of xylene and ethanol washes and rinsed in distilled water. Antigen retrieval was performed by boiling samples in a solution of sodium citrate with Tween-20. Samples were then washed in phosphate-buffered saline (PBS). Samples were incubated in primary antibodies with 5% normal goat serum at 4°C overnight and rinsed again in PBS the next morning. Samples were incubated in secondary antibodies (Alexa Flour 488 anti-mouse, Alexa Flour 488 anti-rat, Cy3 anti-rabbit, Alexa Flour 647 anti-mouse) with 5% normal goat serum and Hoechst 33258 at room temperature for 3 h. Samples were washed in PBS four times before being mounted with coverslips using Fluoromount G. Finally, samples were imaged with a confocal microscope (SP8, Leica) and image analysis was performed using LAS AF (Leica) and Photoshop (Adobe).
Electroretinography
Scotopic and photopic ERGs were performed using the Ganzfeld ERG (Retiport21, Germany) as previously described (10). Two animals per genotype were used for testing (WT, Mpp5 het CKO) resulting in four datasets for each genotype (two eyes from two mice each).
Western blot
Cortices from E14 Mpp5 het CKO or WT and fibroblast cultures from Individual 1 and control were lysed to obtain protein for western blot analysis. The protein concentration of each sample was measured via BCA (Pierce BCA Protein Assay kit). 15 μg of protein from each sample was separated on an SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane. After blocking in Odyssey blocking bugger for 1 h, blots were probed with antibodies for Mpp5 (1:2000) and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:5000) at 4°C overnight. The next day, blots were incubated in the corresponding secondary antibodies—donkey anti-mouse IRDye-800CW and goat anti-rabbit IRdye-680CW (1:2500; LI-COR Biosciences, Nebraska, USA)—for 1 h at room temperature. Western blots were scanned using fluorescence on a LI-COR Odyssey CLx. The signal intensity of the bands was measured using Image Studio software and normalized to GAPDH.
Behavior analyses
An open field testing chamber was used for behavioral analysis. The testing chamber had a grid on the bottom dividing the chamber into four quadrants, as well as two concentric squares denoting the peripheral and center of the open field. Animals at P21 were placed into the southeast quadrant of the open field chamber and observed for 5 min. Every time a line was crossed in the periphery or in the center of the open field chamber, it was recorded.
Quantification and statistical analyses
Quantification, including cell counting, was performed manually on two non-consecutive sections from four or five animals per genotype (WT, Mpp5 het CKO). Student’s t-test was used to determine statistical significance of the data.
Conflict of Interest statement. K.M. and J.J. are employees of GeneDx, Inc. The other authors declare no competing interests.
Funding
National Institute of Neurological Disorders and Stroke (R01NS104038 to S.K.); Shriners Hospitals for Children (86100, 85109 to S.K.); National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR068429-01 to P.B.A.); National Institutes of Health (T32HD098061 to A.R.D.); Alberta Children’s Hospital Foundation, Calgary (to A.M.I.).
Supplementary Material
Contributor Information
Noelle Sterling, Department of Anatomy and Cell Biology, Shriners Hospitals Pediatric Research Center, Lewis Katz School of Medicine. Temple University, Philadelphia, PA, 19140, USA.
Anna R Duncan, Division of Newborn Medicine, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA.
Raehee Park, Department of Anatomy and Cell Biology, Shriners Hospitals Pediatric Research Center, Lewis Katz School of Medicine. Temple University, Philadelphia, PA, 19140, USA.
David A Koolen, Department of Human Genetics, Donders Institute for Brain, Cognition and Behavior, Radboud University Medical Center, 6500 HB Nijmegen, The Netherlands.
Jiahai Shi, Department of Biomedical Sciences, City University of Hong Kong, Hong Kong, Hong Kong SAR.
Seo-Hee Cho, Department of Anatomy and Cell Biology, Shriners Hospitals Pediatric Research Center, Lewis Katz School of Medicine. Temple University, Philadelphia, PA, 19140, USA.
Paul J Benke, Division of Clinical Genetics, Joe DiMaggio Children’s Hospital, Hollywood, FL 33021, USA.
Patricia E Grant, Division of Newborn Medicine, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA; Department of Radiology, Boston Children’s Hospital, Boston, MA 02115, USA.
Casie A Genetti, Division of Genetics & Genomics, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA; Manton Center for Orphan Disease Research, Boston Children’s Hospital, Boston, MA 02115, USA.
Grace E VanNoy, Division of Genetics & Genomics, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA; Manton Center for Orphan Disease Research, Boston Children’s Hospital, Boston, MA 02115, USA; Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
Jane Juusola, Clinical Genomics Program, GeneDx, Gaithersburg, MD 20877, USA.
Kirsty McWalter, Clinical Genomics Program, GeneDx, Gaithersburg, MD 20877, USA.
Jillian S Parboosingh, Department of Medical Genetics and Alberta Children's Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1A4, Canada.
Ryan E Lamont, Department of Medical Genetics and Alberta Children's Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1A4, Canada.
Francois P Bernier, Department of Medical Genetics and Alberta Children's Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1A4, Canada.
Christopher Smith, Department of Medical Genetics and Alberta Children's Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1A4, Canada.
David J Harris, Division of Genetics & Genomics, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA.
Alexander P A Stegmann, Department of Clinical Genetics, Maastricht University Medical Center, 6229 HX Maastricht, The Netherlands; Department of Human Genetics, Radboud University Medical Center, 6525 GA Nijmegen, The Netherlands.
A Micheil Innes, Department of Medical Genetics and Alberta Children's Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1A4, Canada.
Seonhee Kim, Department of Anatomy and Cell Biology, Shriners Hospitals Pediatric Research Center, Lewis Katz School of Medicine. Temple University, Philadelphia, PA, 19140, USA.
Pankaj B Agrawal, Division of Newborn Medicine, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA; Division of Genetics & Genomics, Department of Pediatrics, Boston Children’s Hospital, Boston, MA 02115, USA; Manton Center for Orphan Disease Research, Boston Children’s Hospital, Boston, MA 02115, USA.
References
- 1. Kamberov, E., Makarova, O., Roh, M., Liu, A., Karnak, D., Straight, S. and Margolis, B. (2000) Molecular cloning and characterization of pals, proteins associated with mLin-7. J. Biol. Chem., 275, 11425–11431. [DOI] [PubMed] [Google Scholar]
- 2. Roh, M.H., Makarova, O., Liu, C.J., Shin, K., Lee, S., Laurinec, S., Goyal, M., Wiggins, R. and Margolis, B. (2002) The Maguk protein, Pals1, functions as an adapter, linking mammalian homologues of crumbs and discs lost. J. Cell Biol., 157, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Makarova, O., Roh, M.H., Liu, C.J., Laurinec, S. and Margolis, B. (2003) Mammalian Crumbs3 is a small transmembrane protein linked to protein associated with Lin-7 (Pals1). Gene, 302, 21–29. [DOI] [PubMed] [Google Scholar]
- 4. Hurd, T.W., Gao, L., Roh, M.H., Macara, I.G. and Margolis, B. (2003) Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat. Cell Biol., 5, 137–142. [DOI] [PubMed] [Google Scholar]
- 5. Liu, W.A., Chen, S., Li, Z., Lee, C.H., Mirzaa, G., Dobyns, W.B., Ross, M.E., Zhang, J. and Shi, S.H. (2018) PARD3 dysfunction in conjunction with dynamic HIPPO signaling drives cortical enlargement with massive heterotopia. Genes Dev., 32, 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Costa, M.R., Wen, G., Lepier, A., Schroeder, T. and Gotz, M. (2008) Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development, 135, 11–22. [DOI] [PubMed] [Google Scholar]
- 7. Dudok, J.J., Murtaza, M., Henrique Alves, C., Rashbass, P. and Wijnholds, J. (2016) Crumbs 2 prevents cortical abnormalities in mouse dorsal telencephalon. Neurosci. Res., 108, 12–23. [DOI] [PubMed] [Google Scholar]
- 8. Kim, S., Lehtinen, M.K., Sessa, A., Zappaterra, M.W., Cho, S.H., Gonzalez, D., Boggan, B., Austin, C.A., Wijnholds, J., Gambello, M.J. et al. (2010) The apical complex couples cell fate and cell survival to cerebral cortical development. Neuron, 66, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park, J.Y., Hughes, L.J., Moon, U.Y., Park, R., Kim, S.B., Tran, K., Lee, J.S., Cho, S.H. and Kim, S. (2016) The apical complex protein Pals1 is required to maintain cerebellar progenitor cells in a proliferative state. Development, 143, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cho, S.H., Kim, J.Y., Simons, D.L., Song, J.Y., Le, J.H., Swindell, E.C., Jamrich, M., Wu, S.M. and Kim, S. (2012) Genetic ablation of Pals1 in retinal progenitor cells models the retinal pathology of Leber congenital amaurosis. Hum. Mol. Genet., 21, 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park, B., Alves, C.H., Lundvig, D.M., Tanimoto, N., Beck, S.C., Huber, G., Richard, F., Klooster, J., Andlauer, T.F., Swindell, E.C. et al. (2011) PALS1 is essential for retinal pigment epithelium structure and neural retina stratification. J. Neurosci., 31, 17230–17241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ozcelik, M., Cotter, L., Jacob, C., Pereira, J.A., Relvas, J.B., Suter, U. and Tricaud, N. (2010) Pals1 is a major regulator of the epithelial-like polarization and the extension of the myelin sheath in peripheral nerves. J. Neurosci., 30, 4120–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zollinger, D.R., Chang, K.J., Baalman, K., Kim, S. and Rasband, M.N. (2015) The polarity protein Pals1 regulates radial sorting of axons. J. Neurosci., 35, 10474–10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hakanen, J., Ruiz-Reig, N. and Tissir, F. (2019) Linking cell polarity to cortical development and malformations. Front. Cell. Neurosci., 13, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Philippakis, A.A., Azzariti, D.R., Beltran, S., Brookes, A.J., Brownstein, C.A., Brudno, M., Brunner, H.G., Buske, O.J., Carey, K., Doll, C. et al. (2015) The Matchmaker exchange: a platform for rare disease gene discovery. Hum. Mutat., 36, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li, Y., Wei, Z., Yan, Y., Wan, Q., Du, Q. and Zhang, M. (2014) Structure of crumbs tail in complex with the PALS1 PDZ-SH3-GK tandem reveals a highly specific assembly mechanism for the apical crumbs complex. Proc. Natl. Acad. Sci. U. S. A., 111, 17444–17449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. den Hollander, A.I., Roepman, R., Koenekoop, R.K. and Cremers, F.P. (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res., 27, 391–419. [DOI] [PubMed] [Google Scholar]
- 19. Barkovich, A.J., Guerrini, R., Kuzniecky, R.I., Jackson, G.D. and Dobyns, W.B. (2012) A developmental and genetic classification for malformations of cortical development: update 2012. Brain, 135, 1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shevell, M., Ashwal, S., Donley, D., Flint, J., Gingold, M., Hirtz, D., Majnemer, A., Noetzel, M., Sheth, R.D., Quality Standards Subcommittee of the American Academy of Neurology et al. (2003) Practice parameter: evaluation of the child with global developmental delay: report of the quality standards Subcommittee of the American Academy of neurology and the practice Committee of the Child Neurology Society. Neurology, 60, 367–380. [DOI] [PubMed] [Google Scholar]
- 21. Moeschler, J.B., Shevell, M. and Committee on Genetics (2014) Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics, 134, e903–e918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buchert, R., Tawamie, H., Smith, C., Uebe, S., Innes, A.M., Al Hallak, B., Ekici, A.B., Sticht, H., Schwarze, B., Lamont, R.E. et al. (2014) A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency. Am. J. Hum. Genet., 95, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cho, S.H., Nahar, A., Kim, J.H., Lee, M., Kozmik, Z. and Kim, S. (2019) Targeted deletion of Crb1/Crb2 in the optic vesicle models key features of Leber congenital amaurosis 8. Dev. Biol., 453, 141–154. [DOI] [PubMed] [Google Scholar]
- 24. Mattapallil, M.J., Wawrousek, E.F., Chan, C.C., Zhao, H., Roychoudhury, J., Ferguson, T.A. and Caspi, R.R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci., 53, 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gimenez, E. and Montoliu, L. (2001) A simple polymerase chain reaction assay for genotyping the retinal degeneration mutation (Pdeb(rd1)) in FVB/N-derived transgenic mice. Lab. Anim., 35, 153–156. [DOI] [PubMed] [Google Scholar]
- 26. Mehalow, A.K., Kameya, S., Smith, R.S., Hawes, N.L., Denegre, J.M., Young, J.A., Bechtold, L., Haider, N.B., Tepass, U., Heckenlively, J.R. et al. (2003) CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet., 12, 2179–2189. [DOI] [PubMed] [Google Scholar]
- 27. Low, B.E., Krebs, M.P., Joung, J.K., Tsai, S.Q., Nishina, P.M. and Wiles, M.V. (2014) Correction of the Crb1rd8 allele and retinal phenotype in C57BL/6N mice via TALEN-mediated homology-directed repair. Invest. Ophthalmol. Vis. Sci., 55, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Low, J.C. (1987) The corneal ERG of the heterozygous retinal degeneration mouse. Graefes Arch. Clin. Exp. Ophthalmol., 225, 413–417. [DOI] [PubMed] [Google Scholar]
- 29. Nivison-Smith, L., Zhu, Y., Whatham, A., Bui, B.V., Fletcher, E.L., Acosta, M.L. and Kalloniatis, M. (2014) Sildenafil alters retinal function in mouse carriers of retinitis pigmentosa. Exp. Eye Res., 128, 43–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.