

Abstract
Epileptic activity in genetic generalized epilepsy (GGE) patients preferentially appears during sleep and its mechanism remains unknown. Here, we found that sleep-like slow-wave oscillations (0.5 Hz SWOs) potentiated excitatory and inhibitory synaptic currents in layer V cortical pyramidal neurons from wild-type (wt) mouse brain slices. In contrast, SWOs potentiated excitatory, but not inhibitory, currents in cortical neurons from a heterozygous (het) knock-in (KI) Gabrg2+Q/390X model of Dravet epilepsy syndrome. This created an imbalance between evoked excitatory and inhibitory currents to effectively prompt neuronal action potential firings. Similarly, physiologically similar up-/down-state induction (present during slow-wave sleep) in cortical neurons also potentiated excitatory synaptic currents within brain slices from wt and het KI mice. Moreover, this state-dependent potentiation of excitatory synaptic currents entailed some signaling pathways of homeostatic synaptic plasticity. Consequently, in het KI mice, in vivo SWO induction (using optogenetic methods) triggered generalized epileptic spike-wave discharges (SWDs), being accompanied by sudden immobility, facial myoclonus, and vibrissa twitching. In contrast, in wt littermates, SWO induction did not cause epileptic SWDs and motor behaviors. To our knowledge, this is the first mechanism to explain why epileptic SWDs preferentially happen during non rapid eye-movement sleep and quiet-wakefulness in human GGE patients.
Keywords: genetic generalized epilepsy, homeostatic synaptic plasticity, inhibitory synaptic currents, seizure onset, slow-wave oscillation
Introduction
Epilepsy affects more than 3 million people in the United States of America and causes cognitive deficits and other comorbidities. In two thirds of patients, the causes are unknown (possible genetic origins, termed genetic generalized epilepsy [GGE]). The Epi4K consortium has identified many novel genetic mutations in epilepsy patients (Allen et al. 2013; Striano and Zara 2017), making it imperative for researchers to clarify seizure onset mechanisms in patients with different genetic mutations and varied epileptic symptoms. While it is widely acknowledged that seizures occur unexpectedly, epileptic spike-wave discharges (SWDs) preferentially appear during slow-wave sleep (SWS) or quiet-wakeful states (motor immobility) (Halasz et al. 2002; Shouse et al. 2004; Ng and Pavlova 2013; Ahmed and Vijayan 2014; Bagshaw et al. 2014; Arain et al. 2015). During SWS or quiet-wakeful states, brain activity in EEG recordings exhibits large amplitude alteration at low delta frequency (around 0.5 Hz, slow-wave oscillations [SWOs]) (Petersen et al. 2003; Demanuele et al. 2010; Lagarde et al. 2019) and cortical neurons reveal decreased firing states, engaging homeostatic plasticity of ion channels, and synaptic currents (Kurotani et al. 2008; Turrigiano 2008; Liu et al. 2010). Based on hemostatic synaptic plasticity, decreasing firing activity in neurons for a short period can increase synaptic strength (both excitatory and inhibitory) (Ibata et al. 2008; Chen et al. 2014; Li et al. 2019), while elevating firing activity can depress synaptic strength (Turrigiano 2008; Sun et al. 2013). Thus during SWS, homeostatic synaptic plasticity can be engaged to alter excitatory and inhibitory synaptic currents in neurons, promoting state-dependent interaction between these currents (Shu et al. 2003; Haider et al. 2006; Dehghani et al. 2016), which might play a role in seizure onset in GGE patients.
We have studied the GABAA receptor mutations Q390X in γ2 (Gabrg2Q390X) associated with Dravet epilepsy syndrome (Kang et al. 2015; Warner et al. 2019). Dravet syndrome patients carrying SCN1A (Heron et al. 2010; Steel et al. 2017) or GABAA receptor mutations have medically refractory seizures and exhibit epileptic SWDs during nonrapid eye-movement sleep (NREM sleep) (Halasz et al. 2013; Bagshaw et al. 2014; Verbeek et al. 2015), cognitive deficits (Bender et al. 2012). These mutations expressed in heterozygous (het) knock-in (KI) mice (Gabrg2+/Q390X heterozygosity simulates human patient conditions) can cause mice to generate febrile seizures, generalized epilepsy (Kang and Macdonald 2016), absence epilepsy (Arain et al. 2012), and juvenile myoclonic seizures (Arain et al. 2015; Warner et al. 2019). Moreover, epileptic SWDs in these het KI mice are present while no ostensible motor behaviors are observed (Arain et al. 2012; Kang and Macdonald 2016), very similar to quiet-wakeful states. Moreover, this Gabrg2Q390X mutation causes dysfunctional GABAA receptor subunit aggregation inside cells (Kang et al. 2010; Zhou et al. 2013), which can influence both homeostatic synaptic plasticity and long-term synaptic plasticity.
In this study, we examined the hypothesis that SWOs during NREM sleep and quiet-wakeful state could generate state-dependent dysfunctional homeostatic potentiation of synaptic currents, particularly inhibitory currents, and eventually trigger seizure onset in a mouse model of Dravet epilepsy syndrome with Gabrg2Q390X mutation. Our results suggest that sleep-like SWOs (0.5 Hz) or up-/down-states in brain slices from het KI mice can scale up/potentiate only excitatory, but not inhibitory synaptic currents in cortical neurons. Consequently, in vivo induction of SWOs or up-/down-states can trigger epileptic SWDs in het KI mice.
Materials and Methods
Mouse Brain Slice Preparation
All procedures were in accordance with guidelines set by the Institutional Animal Care and Use Committee of Vanderbilt University Medical Center. The brain slice preparation method used in this study had been described in previous studies (Zhou et al. 2012, 2013). Wt littermate and het Gabrg2+/Q390X KI mice aged P60–120 (both male and female) were used in this study. Transgenic animals expressing halorhodopsin were also used when necessary by crossing our het Gabrg2+/Q390X KI mice with Thy1-eNpHR2.0-EYFP transgenic mice (#012334, The Jackson Laboratory) (see EYFP expression in Fig. 7 inset). Mice were deeply anesthetized, cardiacally perfused with a sucrose-based ice-cold solution (components below) and decapitated. Coronal brain slices (300–320 μm thickness) containing somatosensory cortex were prepared with a vibratome (Leica VT 1200S, Leica Biosystems Inc.) in a sucrose-based ice-cold solution (containing [in mM]): 214 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 10 MgSO4, 24 NaHCO3, and 11 d-glucose, pH 7.4) and slices were later incubated in a chamber at 35–36 °C for 40 min with continuously oxygenated ACSF (see below for components). Finally slices remained at room temperature for at least 1 h before electrophysiological recordings at 32 °C.
Figure 7.
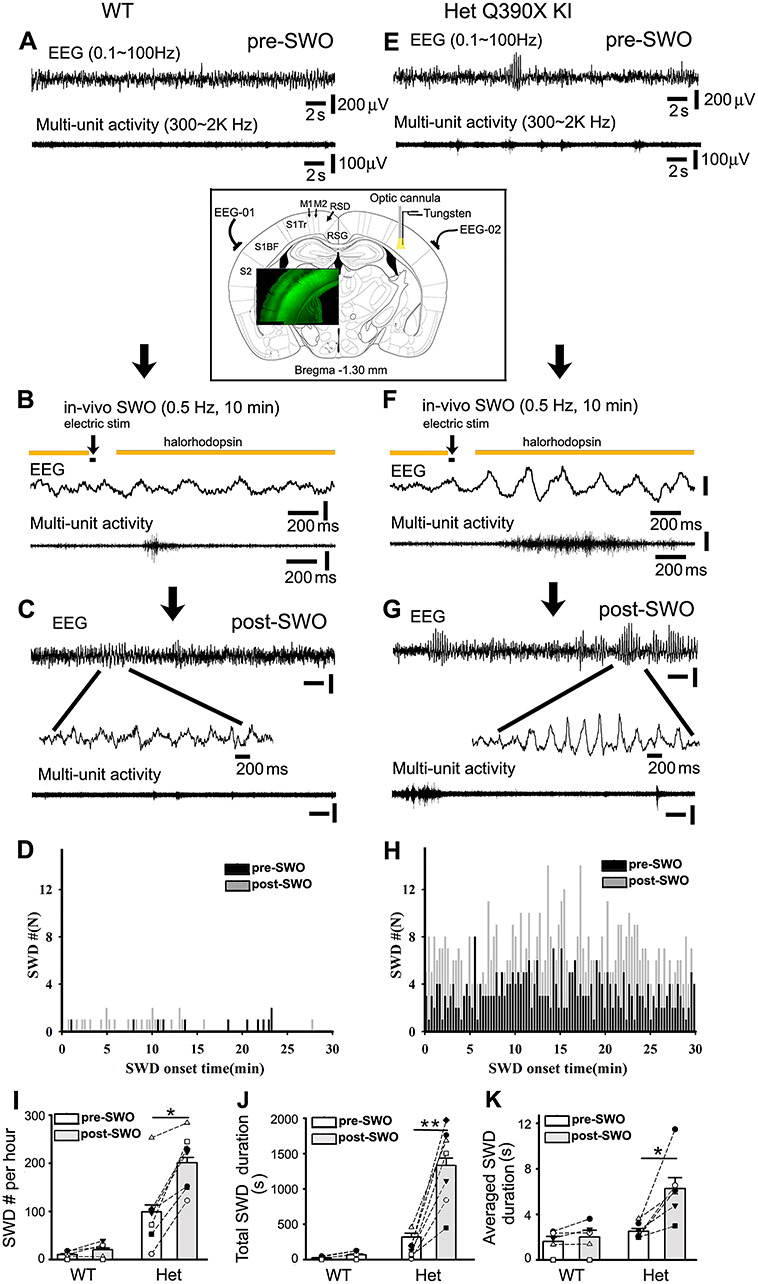
In vivo induction of SWOs or up-/down-states triggers epileptic SWDs in het Gabrg2+/Q390X KI mice. Panels A (wt) and E (het) are representative traces for simultaneous pre-SWO EEG (top) and multiunit (below) recordings (30 s long). Panels B (wt) and F (het) show the time course of in vivo SWO or up-/down-state induction with yellow color indicating laser delivery, and black bar for intracortical stimulation (20 ms). Below are representative traces (2 s long) for simultaneous EEG/multiunit activity during in vivo SWO induction. Panels C (wt) and G (het) show representative traces for simultaneous post-SWO EEG (top) and multiunit (below) recordings. Below are expanded short episode EEG recordings (wt) or epileptic SWDs (het). Scale bars (panels A, C, E, and G) are similarly indicated as labeled except time-scale bars in panels B and F. The downward arrows indicate experiment sequential steps. Panels D (wt) and H (het) show graphs of pre- (black) and post-SWO (gray) epileptic SWD onset time distribution (cumulative data from all wt n = 6 mice or het mice n = 7 mice). SWD events in these two panels were obtained during 30 min right before and after in vivo SWO induction. Panels I–K show summary data for SWD # per h, total SWD duration and averaged single SWD duration for pre-SWO (blank bars) and post-SWO (gray bars) (het KI mice n = 7 each, SWD #/h, paired t-test P = 0.001; total SWD duration/h, paired t-test P = 0.0008; averaged single SWD duration, paired t-test P = 0.005) (wt mice n = 6 each, SWD #/h, paired t-test P = 0.104; total SWD duration/h, paired t-test P = 0.088; averaged single SWD duration, paired t-test P = 0.165). The insert panel shows the implantation of two epidural EEG-electrodes above S1 cortex and one optic cannula/one bipolar tungsten electrode in the S1 cortex (Bregma −1.30 mm, between Figures 41 and 42, The Mouse Brain in Stereotaxic Coordinates, compact 3rd edition by Franklin and Paxinos (2008). The grounding EEG electrode was implanted above cerebellum surface (not shown here). Green fluorescence imaging (overlay insert) shows Thy1-halorhodopsin expression in the cortex (layer 2/3 and 5/6). Labels S1, M1, and RSD/RSG indicate primary somatosensory cortex, motor cortex, and retrosplenial cortex, respectively.
Brain Slice Electrophysiology
Whole-cell patch-clamp recordings (voltage- or current-clamp) were made from somatosensory cortex layer V pyramidal neurons [controlling cortical epileptic activity propagation (Polack et al. 2007; Wester and Contreras 2012)] by using a Nikon infrared/DIC microscope (Eclipse FN1, Nikon Corp. Inc.), and slices were continuously superfused (flow speed 1–1.5 mL/min) with an ACSF solution (containing [in mM]: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 10 d-glucose, pH 7.4) bubbled with 95% O2/5% CO2. Filled electrodes had resistances of 2–5 MΩ with one internal solution (consisted of [in mM]: 120 K-gluconate, 11 KCl, 1 MgCl2, 1 CaCl2, 0.6 EGTA, 10 HEPES, 2 Na-ATP, 0.6 Na-GTP, 10 K-creatine-phosphate, pH 7.3 (Huguenard and Prince 1994; Schofield et al. 2009)) for spontaneous (s) excitatory synaptic currents (sEPSCs) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated sEPSCs were recorded at a holding potential −55.8 mV (Cl− reversal potential). The internal solution for spontaneous inhibitory synaptic currents (sIPSCs) contained [(in mM): 65 K-gluconate, 65 KCl, 10 NaCl, 5 MgSO4, 0.6 EGTA, 10 HEPES, 2 Na-ATP, 0.6 Na-GTP, 10 K-creatine-phosphate, pH 7.3]. sIPSCs were recorded at a holding potential −60 mV with AMPAR/kainate receptors being blocked (20 μM NBQX in ACSF) (Kurotani et al. 2008).
To record action potentials (APs), the same internal solution for sEPSC recordings was used. A concentric bipolar tungsten electrode for stimulation was placed into the neighboring area around neurons being recorded. The stimulus pulses were 0.1 ms duration and stimulus intensity was adjusted to have some AP evoked failures, and the same stimulus intensity was repeated 30–40 times with a 20–30 s interval.
SWOs (0.5 Hz) were induced by injecting sinusoidal currents (200–300 pA at 0.5 Hz) into cells (current-clamp mode) to cause neuron membrane potentials oscillating between resting membrane potentials (between −70 and −75 mV) and −50 mV (with 4–5 AP firings) for 10 min. For in vivo SWO induction in mouse brains using optogenetic method, in vitro experiments were carried out to determine the feasibility of this optogenetic method and the laser light intensity for in vivo application. Thus, SWOs (0.5 Hz) were induced by injecting currents (150–300 pA, 200–300 ms duration) into cells (current-clamp mode) together with 589 nm laser delivery to activate halorhodopsin for 10 min and these data with laser delivery in ex vivo slices were combined with experiments using SWO induction. Also as one surrogate of in vivo neuron up/down states, up-/down-states of neurons within ex vivo brain slices were induced by using a modified ACSF (containing [in mM] 3.5 or 5 KCl, 1 Ca2+, 1 Mg2+ and 3.5 μM carbachol, other components were same as regular ACSF components) and fast flow perfusion (submerged slices, 6–7 mL/min, at 32–33 °C) (Reid et al. 1988; Sanchez-Vives and McCormick 2000; Hajos and Mody 2009; Neske et al. 2015). And the K+ and carbachol concentration were chosen to be much less than the concentration for seizure induction in brain slices (Frohlich et al. 2008; Cataldi et al. 2011; Hashimoto et al. 2017). Access resistance (Ra) was continuously monitored during recordings and recordings with Ra larger than 25 MΩ or 20% change were discarded. Input–output curves of AP firings in neurons were measured by injecting step-currents into neurons with all synaptic currents blocked (20 μM NBQX, 100 μM D-AP5, and 60 μM picrotoxin in ACSF).
To examine the signaling pathway of homeostatic synaptic potentiation of sEPSCs by SWOs, 4-(diethylamino)-benzaldehyde (DEAB, blocking retinoid acid synthesis), KN-93 (βCaMKII antagonist), BAPTA-AM (calcium buffer), and nifedipine (L-type calcium channel blocker) were applied in experiments. For DEAB experiments, all incubation solutions and superfusing ACSF contained 40 μM DEAB, and brain slices were maintained in these solutions for at least 1 h before recordings. KN-93 (2 μM) (Hello Bio Inc.) was dissolved in DMSO and added into pipette internal solutions. BAPTA-AM (15 μM) was administrated in brain slice incubation solution for at least 40 min before recordings. Nifedipine (20 μM) was dissolved in the ACSF during experiments. All chemicals were purchased from Sigma-Aldrich Inc. except where specifically stated.
Evoked EPSCs (eEPSCs) and IPSCs (eIPSCs) in neurons were simultaneously recorded with one Cs-based internal solution (containing [in mM]: 145 Cs-gluconate, 2 MgCl2, 0.5 EGTA, 10 HEPES, 2 Tris-ATP, 0.2 Na-GTP, 5 QX-314, pH 7.3) and Cl− reversal potential was −89.1 mV (Nanou et al. 2018) to measure charge transfers of eEPSCs/eIPSCs with D-AP5 (100 μM) in ACSF. All evoked synaptic currents were in linear ranges of stimulating input–output curves (same intensity for paired eEPSCs/eIPSCs) and showed smooth rising phases without overlapped multisynaptic events. Recordings with the same stimulus intensity (0.1 ms duration) were repeated 10 times (interval 20–30 s) to be averaged to remove synaptic fluctuation. Instead of using the SWO induction protocol which needs potassium-based internal solutions, Wt and het up/down protocols were used with a modified ACSF (containing [in mM] 3.5 or 5 KCl, 1 Ca2+, 1 Mg2+ and 3.5 μM carbachol) and fast flow perfusion (6–7 mL/min, at 32–33 °C). After up-/down-state induction in neurons (current-clamp mode) within brain slices was successfully confirmed by intracellular recordings using the K-gluconate-based internal solution for sEPSC plasticity (see paragraph before), subsequently eEPSCs/eIPSCs in neurons (using Cs-based solutions) were then recorded at −40 mV (for eEPSC/eIPSC charge transfer) or at −89.1 (for eEPSC peak) and 0 mV (for eIPSC peak). The ratios of inhibition and excitation were calculated as the ratios of eIPSC and eEPSC area under curves or as the ratios of eIPSC to eEPSC peaks.
Data Collection and Analysis
Data were collected using one multiClamp 700B amplifier and Clampex 10 software (Molecular Devices Inc.) and filtered at 2 kHz, and digitized at 20 kHz using a Digidata 1440A (Molecular Devices Inc.). Both sEPSCs and sIPSCs were analyzed with a threshold detection method (5–6 pA, 2.5× baseline RMS) using Clampfit 10.0 software program (Schofield et al. 2009; Rakhade et al. 2012; Zhou et al. 2012). Pre- and post-SWO sEPSC or sIPSC consecutive sweeps (each sweep 5 s long) from the same neurons were concatenated as one file to be analyzed by using the same threshold setting for all synaptic events. All detected sEPSC and sIPSC events were confirmed visually to ensure that their waveforms had normal rising and decaying phases. sEPSC and sIPSC histogram and cumulative distribution graphs were constructed with the Clampfit 10.0 software and Kolmogorov–Smirnov (K–S) nonparametric test was performed with same number of random synaptic events from pre- and post-SWO groups. All figures were prepared with Microsoft Excel, SigmaPlot/Stat, and Adobe Photoshop softwares. Data were expressed as mean ± SEM (standard error of mean). Two-way ANOVA and Holm-Sidak test were used to compare wt and het neurons input–output of AP data.
Mouse Surgery and In vivo EEG/Multiunit Recordings
Wt and het KI mice were mated with mice expressing halorhodopsin under the Thy1-promoter [mice #012334 from The Jackson Laboratory (Gradinaru et al. 2008; Zhao et al. 2008)]. Epidural screw electrodes were surgically implanted (anesthesia 1–3% isoflurane [vol/vol]) (Fig. 7 insert, each EEG electrode for one hemisphere [anterior–posterior between −1.82 and −0.46 mm reference to bregma, midline-lateral −4.0 or +4.0 mm] and one grounding EEG electrode over cerebellum surface, #8201 Pinnacle Technology, Lawrence KS). Also one concentric bipolar tungsten electrode was implanted in somatosensory cortex for intracortical stimulation (S1 cortex, depth 0.8–1 mm in laminar V, using fiber optic cannula coordinates) with one fiber optic cannula (0.2–0.4 mm diameter, Thorlabs Inc.) for in vivo laser light delivery (within S1 cortex, anterior–posterior −1.30 mm reference to bregma, midline-lateral between +2.6 and +3.0 mm, dorsal-ventral 0.7–1.1 mm reference to pia surface). The tungsten electrode was parallelly bundled with one fiberoptic cannula and both were implanted in the same somatosensory cortex location. The tungsten electrode tip in S1 cortex (depth 0.7–1.1 mm, layer V) was inserted a slightly deeper (~100 μm, using stereotaxic ultra-precise manipulator) than the tip of the fiberoptic cannula to ensure that all cortical neurons being recorded were activated by laser delivery (589–680 nm). We also tried one pair of unipolar tungsten electrodes with one (bundled together with one fiber optic cannula) within S1 cortex and another in posterior brain (anterior–posterior −2.5 to 2.6 mm reference to bregma and midline-lateral 0–0.3 mm, no optic cannula at this location) and similar effects were obtained as that of one concentric bipolar tungsten electrodes implanted in somatosensory cortex. Implantation of optic cannula in the S1 cortex did not influence the epileptic SWD incidence and interspike activity in het KI mice (n = 3). EMG leads were inserted into neck trapezius muscles to record muscle activity. After surgery, mice were continuously monitored for recovery from anesthesia and remained in the animal care facility for at least 1 week with a normal sleep/wake cycle. Simultaneous in vivo EEG/EMG/multiunit activity recordings were conducted during day-light period (10:00 am to 4:00 pm, mouse sleep period) after mice acclimated to the apparatus and exhibited quiet-wakeful behavior for at least 1 h in one animal behavior chamber. After in vivo EEG recordings, the mice were euthanized and placement of tungsten electrode/optic cannula within S1 cortex was confirmed visually. Simultaneous EEG (two channels, band filtered at 0.1–100 Hz) and multiunit recordings (one-channel, band filtered 300–2000 Hz) (all in current-clamp mode) data, along with one channel EMG recordings (400 Hz), were collected by using two multiClamp 700B amplifiers (total 4 channels, Molecular devices Inc.) and Clampex 10 software (Molecular Devices Inc.), and digitized at 20 kHz using a Digidata 1440A.
The laser light was delivered through a fiberoptic cable connected to the optic cannula, controlled by a DPSS laser (MGL-III-589-50 [50 mW, Ultralazers Co., Inc.]) and the timing of laser delivery was controlled by Clampex 10 software (Molecular Devices Inc.). Intracortical stimulations (300–400 pA, 20 ms) were applied through implanted tungsten electrodes by Clampex 10 software (current-clamp mode). In vivo SWOs or up-/down-states (0.5 Hz, for 10 min) were induced by alternating laser delivery (for hyperpolarizing down-state, 1800 ms) and no laser delivery (for up-state 200 ms, at the beginning of this up-state, electrical stimulations were applied [simulating sleep spindle duration (Kandel and Buzsaki 1997; Levenstein et al. 2017)]). The injected current amplitudes (300–400 pA) and duration were chosen to avoid kindling effect, which needs larger stimulating currents of ~1 μA amplitudes (Racine 1972a, 1972b; Lothman et al. 1990). Mouse behavior were simultaneously video-recorded, and synchronized with EEG recordings, and classified according to the Racine-scale (Racine 1972a, 1972b) (eyes not closed during epileptic SWDs, based on video-recordings). Bilateral synchronous SWDs (typical absence epilepsy SWD, 6–12 Hz) and slow SWD [atypical absence epilepsy SSWD, 3–6 Hz (Cortez et al. 2001; Velazquez et al. 2007)] were defined as trains (>1 s) of rhythmic biphasic spikes, with a voltage amplitude at least 2-fold higher than baseline amplitudes (Velazquez et al. 2007; Arain et al. 2012). All atypical and typical absence seizures and general tonic–clonic seizures (GTCS) started with SWDs or SSWDs, accompanied by characteristic motor behaviors consisting of sudden immobility, facial myoclonus and vibrissa twitching. These behavior characteristics were also used to distinguish atypical absence epilepsy SSWDs from sleep theta-wave EEG activity. The SWDs/SSWDs and animal behaviors were also confirmed by investigators blinded to animal genotypes. The onset times of SWD or SSWDs were determined by their leading edge points crossing (either upward or downward) the precedent EEG baseline. High-frequency activity was obtained by postexperiment band-filtering (400–800 Hz) tungsten activity originally recorded at 300–3 KHz in vivo. A threshold method of at least 2× baseline amplitude was used to detect high-frequency activity events using Clampfit 10 software (Molecular Devices Inc., Molecular Devices Inc.) and their durations were analyzed. Any high-frequency activity associated with motor behaviors (video monitored) was removed from this analysis.
Results
To address the roles of SWOs on epileptic SWD generation, we used wt and het Gabrg2+/Q390X KI mice, and transgenic wt or het Gabrg2+/Q390X KI mice expressing halorhodopsin protein in cortical neurons for ex vivo and in vivo optogenetic SWO induction. The expression of halorhodopsin in layer V cortex was confirmed by enhanced EYFP fluorescent protein expression in cortical neurons and ex vivo physiology experiments. SWOs (0.5 Hz) were produced in layer V cortical pyramidal neurons within somatosensory cortex by injecting sinusoidal currents (current-clamp) to cause neuronal membrane potential oscillating between resting membrane potential (between −70 and −75 mV) and −50 mV, very similar to neuronal up-/down-state alteration during NREM sleep (Steriade et al. 2001; Petersen et al. 2003). We did not find significant difference between wt and het KI mice in neuronal physiological properties (wt: n = 33, n = 29 mice, capacitance 108.95 ± 6.67 pF; input resistance 87.87 ± 7.75 MΩ, resting membrane potential −72.07 ± 0.57 mV, AP amplitude 79.64 ± 1.54 mV [measured from precedent resting membrane potential]) (het: n = 37, n = 34 mice, capacitance 106.89 ± 4.72 pF, t-test P = 0.798; input resistance 77.47 ± 6.63 MΩ, t-test P = 0.309; resting membrane potential −71.52 ± 0.54 mV, t-test P = 0.491; AP amplitudes 82.84 ± 1.35 mV, t-test P = 0.119).
SWO Induction State-dependently Increases sEPSCs in Layer V Cortical Neurons from wt and het KI Mice
AMPAR-mediated sEPSCs were recorded by clamping cortical neurons at the chloride reversal potential (−55.8 mV). Compared with pre-SWO baseline sEPSCs, SWO induction (0.5 Hz) for 10 min significantly increased sEPSC amplitudes in neurons from wt littermates with stable access resistances (Fig. 1A–C, from 16.69 ± 1.85 to 27.90 ± 4.13 pA, n = 8 cells [total n = 5 mice], paired t-test P = 0.004). Potentiated/scaled-up sEPSCs reached their peak ~10 min after SWO induction and then decayed to a plateau at ~30 min, remaining larger than baseline sEPSC amplitudes (Fig. 1B). Both summary data of pre- and post-SWO sEPSC amplitudes and cumulative sEPSC amplitude distribution indicated that SWO induction did significantly increase AMPAR-mediated sEPSCs in neurons from wt littermates (Fig. 1C, K–S test P = 0.00001). Similarly, post-SWO sEPSCs in cortical neurons from het KI mice also exhibited significant potentiation following SWO induction (Fig. 1D–F, from 18.89 ± 1.52 to 28.12 ± 3.47 pA, n = 9 cells [total n = 6 mice], paired t-test P = 0.009; K–S test P = 0.00001), indicating that SWO induction could also potentiate sEPSCs in cortical neurons from het KI mice. Moreover, SWO induction in neurons from both wt and het KI mice led to a similar percentage of potentiation of sEPSCs (wt 119.40 ± 4.00% [n = 8 cells] vs. het 143.38 ± 10.68% [n = 9 cells], at 28.5 min, t-test P = 0.064, Supplementary Fig. 1). We did not observe any significant changes between pre- and post-SWO sEPSC frequency and sEPSC decay in neurons from wt or het KI mice (wt [n = 8 cells, n = 5 mice], frequency from 2.43 ± 0.69 to 1.71 ± 0.36 Hz, paired t-test P = 0.36; decay τ from 18.57 ± 5.35 to 25.13 ± 3.89 ms, paired t-test P = 0.304) (het [n = 9 cells, n = 6 mice], frequency from 1.67 ± 0.25 to 3.06 ± 0.84 Hz, paired t-test P = 0.10; decay τ from 12.80 ± 1.62 to 34.15 ± 10.89 ms, paired t-test P = 0.078).
Figure 1.
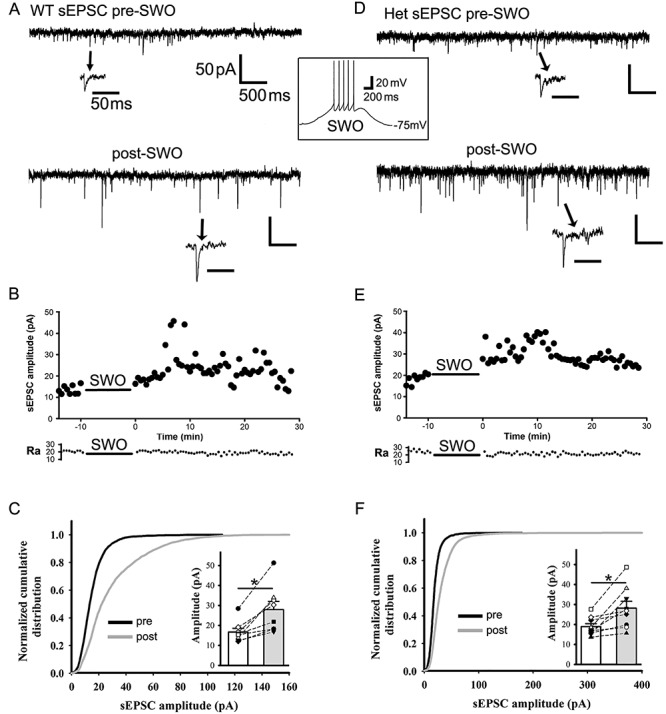
SWO induction potentiates sEPSCs in layer V cortical pyramidal neurons from wt and het Gabrg2+/Q390X KI mice. Panels A and D are representative traces pre-SWO (top) and post-SWO sEPSCs (lower) for wt littermates (left column) and het KI mice (right column). The arrows point to individual sEPSC events in expanded time scales. The middle inset is one representative SWO induction trace from rest membrane potentials (−75 mV). Scale bars are indicated as labeled. Panels B and E show time courses of pre-SWO and post-SWO sEPSC amplitudes for corresponding recordings from panels A (wt) and D (het). Each data point in the panels B/E was obtained by averaging all sEPSC events during continuous 30 s recordings. The lower panels of access resistance (Ra) show their accompanied values during the whole 42.5 min recordings. Panels C and F show normalized cumulative histogram of sEPSC events for wt and het KI mice. Insets are summary data of pre-SWO and post-SWO sEPSC values (wt n = 8 neurons, n = 5 mice, paired t-test P = 0.004; het n = 9 neurons, n = 6 het KI mice, paired t-test P = 0.009) (wt cumulative curves used 4905 synaptic events for pre-SWO[during 4.2 min baseline recordings for each neuron, total n = 8 neurons] and 4506 synaptic events for post-SWO [during final 4.2 min recordings for each neurons, total n = 8 neurons], while het cumulative curves used 3886 synaptic events for pre-SWO [during 4.2 min baseline recordings for each neuron, total n = 9 neurons] and 9519 synaptic events for post-SWO [during final 4.2 min recordings for each neuron, total n = 9 neurons]). Each data point for inset graphs was obtained by averaging sEPSC events during baseline or post-SWO last 4.2 min recordings.
Next, we tested synaptic scaling-down (depression) by high-frequency stimulating oscillations (50 Hz). After 10 min of injected 50 Hz oscillating currents, cortical neurons from both wt and het KI mice exhibited attenuation in sEPSC amplitudes for 30 min compared with baseline sEPSCs (Supplementary Fig. 2, wt and het data combined [n = 6 cells, n = 4 mice], from 14.22 ± 0.77 to 10.00 ± 0.76 pA, paired t-test P = 0.004), without changes in sEPSC frequency and decay (frequency from 2.34 ± 0.49 Hz to 1.73 ± 0.14 Hz, paired t-test P = 0.218; decay τ from 15.40 ± 4.48 to 21.88 ± 4.62 ms, paired t-test P = 0.285). This suggested that the classic homeostatic synaptic plasticity mechanism involved in scaling-up/potentiation or scaling-down/depression of sEPSCs (Turrigiano and Nelson 2004; Turrigiano 2008). To determine the involvement of similar signaling pathways for homeostatic plasticity mechanism involving low-level intracellular Ca2+, retinoid acid and βCaMKII activation, we examined the effects of DEAB (40 μM, blocking retinoid acid synthesis) (Turrigiano and Nelson 2004; Aoto et al. 2008; Chen et al. 2014; Li et al. 2019), low-level BAPTA-AM (15 μM, maintaining low-level Ca2+) (Ibata et al. 2008; Wang et al. 2011), KN93 (βCaMKII antagonist) (Thiagarajan et al. 2002) and nifedipine (L-type calcium channel blocker) (Kurotani et al. 2008) (Fig. 2). These chemical agents can either block (DEAB and KN93) or enhance (low-level Ca2+ and nifedipine) homeostatic synaptic plasticity in neurons. With 40 μM DEAB in ACSF, SWOs did not enhance any sEPSCs in cortical neurons from wt and het KI mice (Fig. 2A,B [n = 10 cells, n = 4 mice for each comparison], amplitudes from 15.05 ± 1.48 to 15.52 ± 2.30 pA, paired t-test P = 0.632; frequency from 4.45 ± 0.96 to 4.17 ± 1.00 Hz, paired t-test P = 0.733; decay τ from 15.50 ± 1.96 to 27.80 ± 8.15 ms, paired t-test P = 0.112), confirming that retinoid acid-mediated homeostatic synaptic plasticity did contribute to SWO-induced sEPSC potentiation/scaling-up in cortical neurons from wt and het KI mice. Similar to DEAB suppressing action, administration of KN-93 (2 μM) inside cells also suppressed the sEPSC potentiation in cortical neurons from wt and het mice (Fig. 2B [n = 7 cells, n = 3 mice], sEPSC amplitudes from 12.78 ± 1.17 to 11.04 ± 1.04 pA, paired t-test P = 0.013; no change in sEPSC frequency from 2.91 ± 0.92 to 1.41 ± 0.36 Hz, paired t-test P = 0.117, but produced sEPSCs with a longer decay τ 9.75 ± 1.45 to 25.02 ± 3.88 ms, paired t-test P = 0.005). Forty minute BAPTA-AM (15 μM) treatment or nifedipine (20 μM) in ACSF could enhance the potentiation of sEPSCs in cortical neurons from wt and het KI mice (BAPTA-AM [n = 5 cells, n = 3 mice], sEPSC amplitudes from 9.55 ± 0.56 to 12.99 ± 0.88 pA, paired t-test P = 0.006; sEPSC frequency from 0.86 ± 0.20 to 1.88 ± 0.41 Hz, paired t-test P = 0.021, while longer decay τ 10.98 ± 2.28 to 26.80 ± 2.26 ms, paired t-test P = 0.009) (nifedipine [n = 7 cells, n = 4 mice], sEPSC amplitudes from 12.14 ± 1.21 to 21.49 ± 3.38 pA, paired t-test P = 0.044; sEPSC frequency from 4.40 ± 0.93 to 3.03 ± 0.55 Hz, paired t-test P = 0.063 and decay τ 15.99 ± 3.02 to 24.89 ± 3.61 ms, paired t-test P = 0.105).
Figure 2.
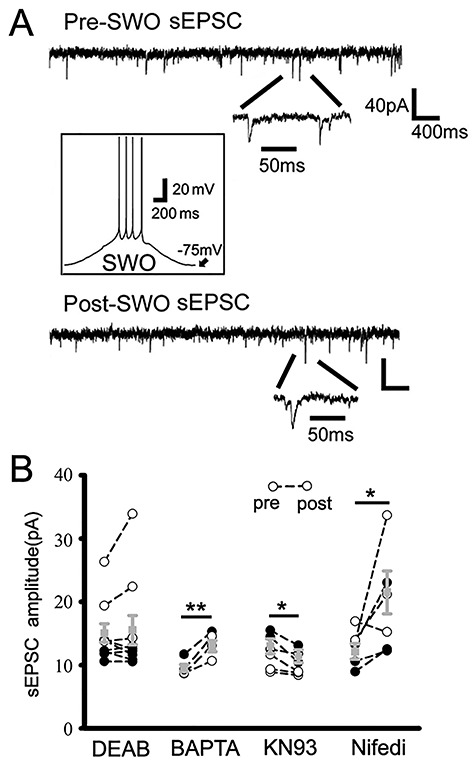
Pharmacology of SWO-induced potentiation of sEPSCs in cortical neurons. Panel A shows representative traces for pre-(top) and post-SWO sEPSCs (bottom) in cortical neurons for DEAB treatment group. Individual sEPSC events are expanded to show their rising and decaying phases. The inset shows SWO induction trace from resting membrane potential −75 mV. Scale bars are indicated as labeled. Panel B shows summary data for pre-SWO and post-SWO sEPSCs with DEAB treatment (40 μM, n = 10 cells, n = 4 mice, paired t-test P = 0.632), BAPAT-AM (15 μM, n = 5 cells, n = 3 mice, paired t-test P = 0.006), KN93 (2 μM, n = 7 cells, n = 3 mice, paired t-test P = 0.013) and nifedipine (20 μM, n = 7 cells, n = 4 mice, paired t-test P = 0.044).
These pharmacological studies indicated that SWO-induced sEPSC potentiation/scaling-up shared similar signaling pathways as the classic homeostatic synaptic plasticity. In addition, treating brain slices with BAPTA-AM (15 μM) slightly decreased baseline sEPSC amplitudes of cortical neurons, which might be due to BAPTA buffering intracellular Ca2+ on transmitter release (Rozov et al. 2001).
SWO Induction State-Dependently Increases sIPSCs in Cortical Neurons from wt, but not het KI Mice
In cortical neurons, synaptic excitation and inhibition are proportionally balanced to avoid cortical hyperexcitability (Shu et al. 2003; Haider et al. 2006; Dehghani et al. 2016). Thus, we determined whether SWO induction could change GABAA receptor-mediated sIPSCs in cortical neurons. sIPSCs were isolated by using AMPAR/kainate receptor antagonist NBQX (20 μM) in ACSF while neurons were voltage-clamped at −60 mV (Cl− reversal potential −15 mV). SWO induction in cortical neurons from wt mice significantly increased sIPSC amplitudes compared with pre-SWO baseline (Fig. 3A-C, 27.14 ± 3.79 to 39.06 ± 6.06 pA, n = 10 cells [total n = 8 mice], paired t-test P = 0.001; K–S test P = 0.00001), similar to the potentiation of miniature IPSCs in neurons from Kurotani et al. (2008). Moreover, the time course of post-SWO sIPSCs exhibited a peak ~10 min after SWO induction (Fig. 3B), very similar to the time course of post-SWO sEPSCs in neurons from wt littermates (Fig. 1B). However, in neurons from het KI mice, SWO induction did not change any sIPSCs compared with pre-SWO baseline (with stable access resistances) (Fig. 3D–F). Both summary data of pre- and post-SWO sIPSCs (Fig. 3F inset, from 18.60 ± 2.08 to 16.86 ± 1.85 pA, n = 11 cells [total n = 7 mice], paired t-test P = 0.1076) and cumulative distribution analysis (Fig. 3F, K–S test P = 0.061) confirmed no sIPSC potentiation/scaling-up in neurons from het KI mice, indicating that SWO-induced potentiation/scaling-up of sIPSCs in het KI mice was impaired. Additionally, we observed a significant increase in post-SWO sIPSC frequency in neurons from wt littermates (from 3.41 ± 0.87 to 7.90 ± 1.70 Hz, n = 10 cells [total n = 8 mice], paired t-test P = 0.021), but not from het KI mice (from 4.28 ± 1.20 to 2.45 ± 0.47 Hz, n = 11 cells [total n = 7 mice], paired t-test P = 0.148). Meanwhile, sIPSC decay in neurons from both wt and het KI mice became longer after SWO induction (decay τ, wt from 10.18 ± 2.24 to 29.62 ± 7.86 ms, n = 10 cells [total n = 8 mice], paired t-test P = 0.0229; het from 10.46 ± 2.58 to 31.37 ± 8.09 ms, n = 11 cells [total n = 7 mice], paired t-test P = 0.0278), suggesting that GABAA receptors might change their subunit composition after SWO induction.
Figure 3.
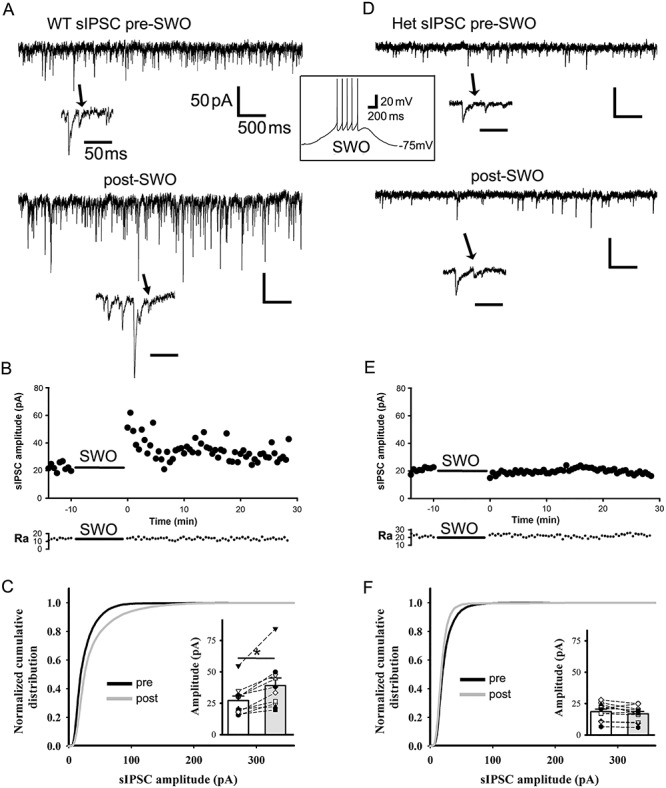
SWO induction potentiates sIPSCs in layer V cortical pyramidal neurons from wt, but not from het Gabrg2+/Q390X KI mice. Panels A and D are representative traces pre-SWO (top) and post-SWO sIPSCs (lower) for wt littermates (left column) and het KI mice (right column). The arrows indicate individual sIPSC events in expanded time scales. The middle inset is one representative SWO induction trace from resting membrane potentials (−75 mV). Scale bars are indicated as labeled. Panels B and E show time courses of pre-SWO and post-SWO sIPSC amplitudes for corresponding recordings from panel A (wt) and D (het). Each data point in the panels B/E was obtained by averaging all sIPSC events during continuous 30 s recordings. The low panels of access resistance (Ra) show their values during the whole 42.5 min recordings. Panels C and F show normalized cumulative histogram of sIPSC events for wt and het KI mice. Insets are summary data of pre-SWO and post-SWO sIPSC values (wt n = 10 neurons, n = 8 mice, paired t-test P = 0.001; het n = 11 neurons, n = 7 het KI mice, paired t-test P = 0.1076) (wt cumulative curves used 6293 synaptic events for pre-SWO [during 4.2 min baseline recordings for each neurons, total n = 10 neurons] and 14 299 events for post-SWO [during final 4.2 min recordings for each neuron, total n = 10 neurons], while het cumulative curves used 7124 synaptic events for pre-SWO [during 4.2 min baseline recordings for each neurons, total n = 11 neurons] and 6290 synaptic events for post-SWO[during final 4.2 min recordings for each neurons, total n = 11 neurons]). Each data point for inset graphs was obtained by averaging sIPSC events during baseline or post-SWO last 4.2 min recordings.
We next examined the depression/scaling-down effect of elevated neuron firing activity on sIPSCs from wt littermates, as we did for sEPSC depression/scaling-down by elevated frequency stimulating oscillations. After inducing 5 Hz membrane potential oscillation for 10 min, post-5 Hz sIPSCs were decreased in amplitudes compared with pre-5 Hz baseline (Supplementary Fig. 3 [n = 5 cells, n = 4 mice], amplitude from 20.37 ± 1.58 to 13.73 ± 1.11 pA, paired t-test P = 0.004; frequency from 4.27 ± 1.14 to 1.60 ± 0.41 Hz, paired t-test P = 0.048; sEPSC decay τ from 9.68 ± 1.17 to 23.36 ± 3.92 ms, paired t-test P = 0.041), suggesting that the classic homeostatic synaptic plasticity mechanism contributed to SWO-induced sIPSC potentiation/scaling-up in neurons from wt littermates.
Physiologically Similar Up-/Down-State Induction Increases sEPSCs in Cortical Neurons
Cortical neurons exhibit up-/down-state alteration in vivo during NREM sleep (Steriade et al. 2001; Petersen et al. 2003), similar to SWO induction in ex vivo brain slices. As a surrogate for in vivo physiological up-/down-state, we examined whether up-/down-state induction within ex vivo brain slices increased synaptic sEPSCs. Using a modified ACSF (containing 3.5 or 5 KCl, 1 Ca2+, 1 Mg2+ and 3.5 μM carbachol) and fast flow perfusion (6–7 mL/min) (Sanchez-Vives and McCormick 2000; Hajos and Mody 2009; Neske et al. 2015) (K+ and carbachol concentration were chosen to be much less than the concentration for seizure induction in brain slices [Frohlich et al. 2008; Cataldi et al. 2011; Hashimoto et al. 2017]), we could induce long episodes (10s) of membrane depolarization (up-state, around −50 mV, with simultaneous AP firing) from resting membrane potentials (down-state, around −70 mV) (Fig. 4A middle, n = 7 cells, n = 3 for wt and n = 4 het KI mice). Compared with preup-/down-state baseline, up-/down-state induction (10 min) significantly increased sEPSCs in amplitudes (Fig. 4A–C [n = 7 cells, n = 7 mice], from 18.10 ± 2.19 to 27.69 ± 2.98 pA, paired t-test P = 0.003), frequency (from 1.21 ± 0.22 to 2.52 ± 0.58 Hz, paired t-test P = 0.014), and decay τ (from 16.12 ± 1.68 to 51.01 ± 14.61 ms, paired t-test P = 0.069), which was very similar to SWO-induced potentiation/scaling-up of sEPSCs in neurons. We also noted that this modified ACSF within slices induced many cortical neurons’ up-/down-states at the same time and could change the whole network activity to increase synaptic sEPSC frequency. Furthermore, up-/down-induced sEPSC potentiation within ex vivo slices could be blocked by DEAB treatment (40 μM), without any changes in sEPSC frequency and decay τ ([n = 6 cells, n = 6 mice], amplitude from 18.06 ± 1.96 to 16.14 ± 1.83 pA, paired t-test P = 0.003; frequency from 2.49 ± 0.78 to 2.18 ± 0.50 Hz, paired t-test P = 0.691; decay τ from 21.73 ± 10.69 to 52.41 ± 11.63 ms, paired t-test P = 0.191). Together, these results suggested that in vivo up-/down-state induction could engage a similar homeostatic plasticity mechanism as ex vivo SWOs to increase sEPSCs in neurons (both wt and het KI mice) or sIPSCs in neurons (wt only).
Figure 4.
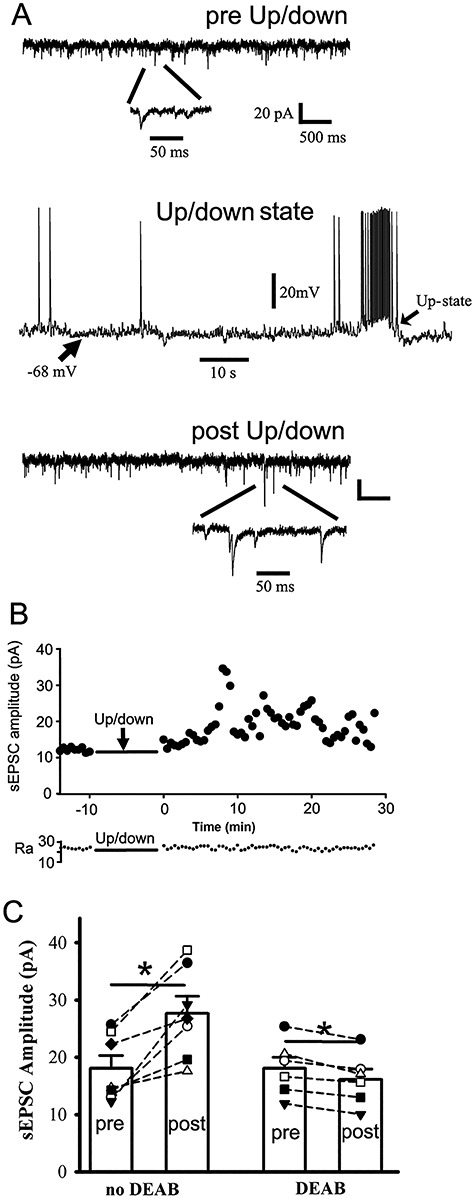
Physiologically similar up-/down-state induction potentiates sEPSCs in cortical neurons from wt and het Gabrg2+/Q390X KI mice. Panel A (top and bottom) shows representative traces for pre/post-up/down sEPSCs in cortical neurons. Below are expanded sEPSC events. Panel A (middle) shows one representative up-/down-state induced by using fast flow perfusion (6–7 mL/min) with a modified ACSF ([mM] 3.5 or 5 KCl, 1 Ca2+, 1 Mg2+ and 3.5 μM carbachol). Scale bars are indicated as labeled. Panel B shows time courses of pre/post-up-/down-state sEPSC amplitudes for corresponding recordings from panel A. Each data point in these panels B was obtained by averaging all sIPSC events during continuous 30 s recordings. The panel B (lower part) shows access resistance (Ra) values during the whole 42.5 min recordings. Panel C shows summary data of pre-/postup-/down-state sEPSCs (n = 7 neurons, n = 7 mice, paired t-test P = 0.003) and DEAB blockade effect (n = 6 neurons, n = 6 mice, paired t-test P = 0.003) (each data point for panel C was obtained by averaging sEPSC events during baseline or postup-/down-state last 4.2 min recordings).
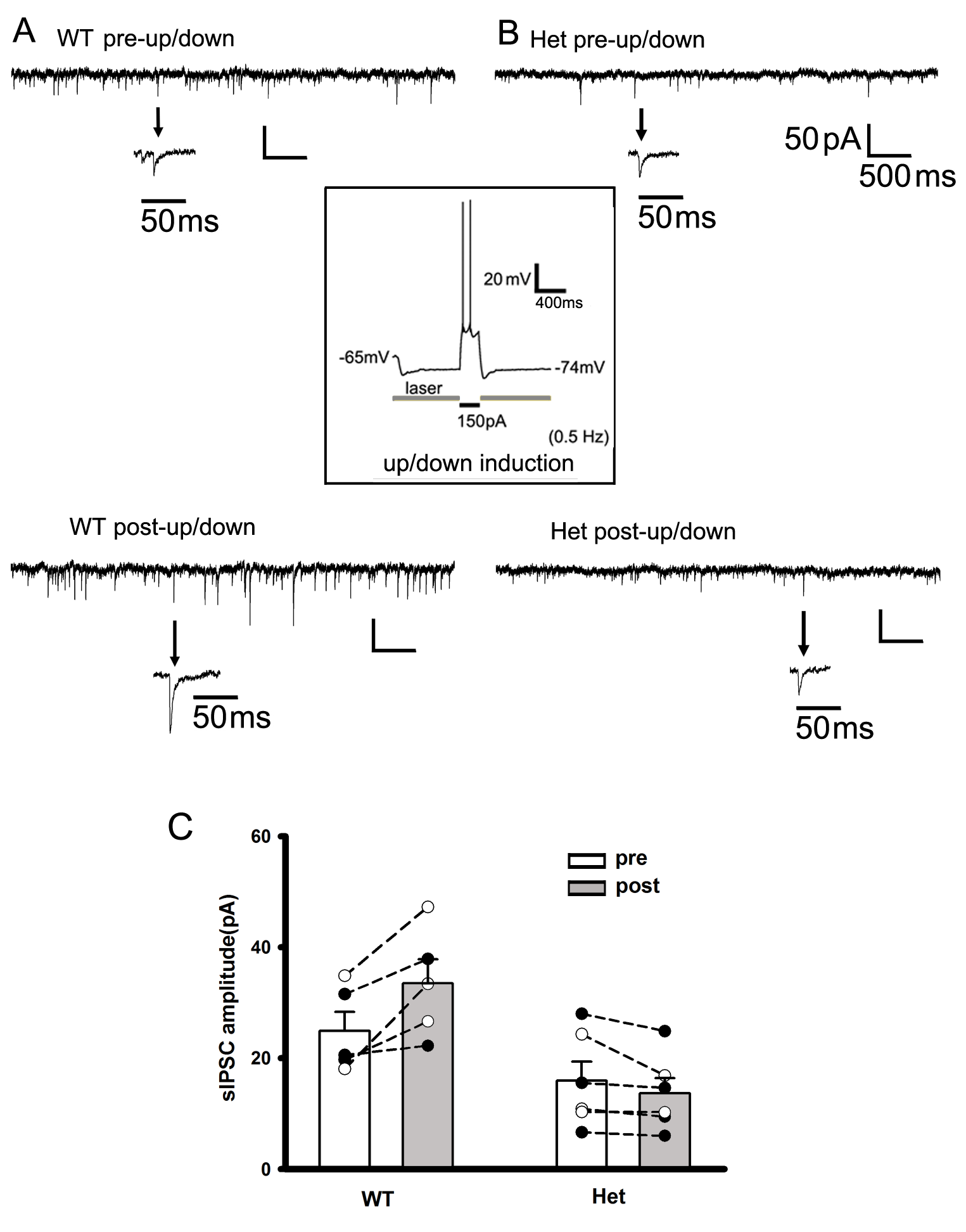
Physiologically Similar Up-/Down-State Induction Creates an Imbalance Between eEPSCs and eIPSCs in Cortical Neurons from het KI Mice
Given that SWOs increase sEPSC amplitudes, but do not change sIPSCs, in cortical neurons from het KI mice, we determined if there was an imbalance between IPSCs and EPSCs. To assess the evoked(e) IPSC/EPSC balance in neurons from wt and het KI mice, neurons were at clamped at −40 mV to simultaneously compare their charge transfers as Nanou et al.’s study [(Nanou et al. 2018), Fig. 7 in their paper]. Within brain slices from wt mice, up-/down-state induction significantly increased the charge transfer ratios of eIPSC/eEPSC (Fig. 5A,B, wt 1.38 ± 0.24, n = 11 [total n = 3 mice]; wt up/down 3.64 ± 0.88, n = 7 cells [total n = 3 mice], t-test P = 0.0113). In contrast, het cortical neurons (het control group) exhibited large charge transfer ratios of eIPSC/eEPSC (Fig 5B, 4.11 ± 0.99, n = 7 cells [total n = 3 mice], wt control group vs. het control group t-test P = 0.006). And up-/down-state induction significantly decreased these ratios (Fig. 5B, 0.619 ± 0.259, n = 10 [total n = 3 mice], t-test P = 0.0119), indicating that, following up-/down-state induction, IPSC charge transfers greatly reduced compared with potentiated/scaled-up EPSC charge transfers, creating an imbalance between eEPSCs and eIPSCs in cortical neurons from het KI mice. Moreover, the ratios of eIPSC peak (holding at 0 mV) to eEPSC peak (holding at chloride reversal potential −89.1 mV) showed similar results (Fig. 5C and D) (wt control [n = 7 cells, n = 2 mice] vs. wt up/down [n = 8 cells, n = 2 mice] from 1.02 ± 0.20 to 2.65 ± 0.49, t-test P = 0.012; het control [n = 8 cells, n = 2 mice] vs. het up/down [n = 6 cells, n = 2 mice] from 4.38 ± 0.73 to 0.64 ± 0.227, t-test P = 0.0012), indicating that following up/down induction, eIPSCs in neurons from het KI mice were not able to suppress potentiated eEPSCs. In addition, similar to pre-up/down charge transfer ratios, there was significant difference in pre-up/down IPSC/EPSC peak ratios (t-test P = 0.001) between wt and het KI mice, suggesting that neural network activity in het KI mice may recruit more IPSCs to compensate elevated EPSCs in neurons while SWOs were not present or induced.
Figure 5.
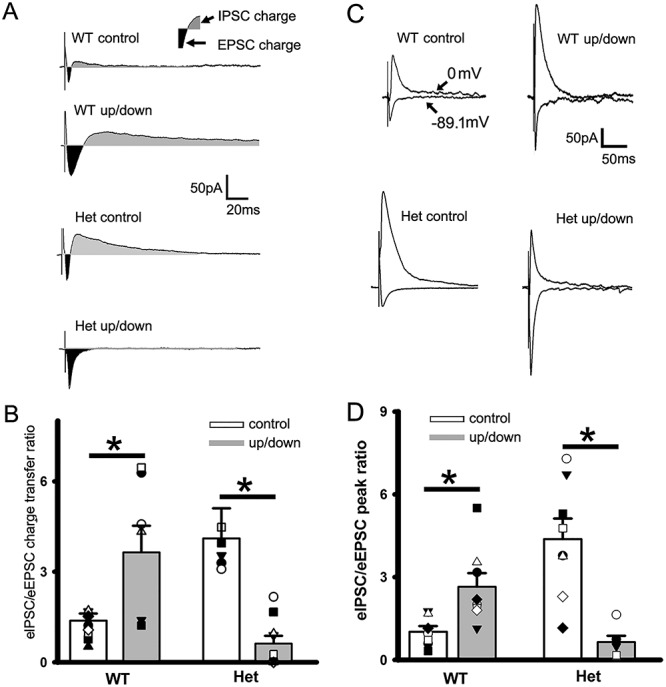
Balanced eIPSCs/eEPSCs following up-/down-state induction in cortical neurons from wt mice, while imbalanced eIPSCs/eEPSCs from het Gabrg2+/Q390X KI mice. (A) Representative traces of paired eEPSCs and eIPSCs in neurons (clamped at −40 mV) from wt control and wt up/down, het control and het up/down groups. The dark areas between inward current traces and baselines represent the excitatory current charges and the gray areas between outward current traces and baselines represent the inhibitory current charges. Stimulus artifacts are shown right before eEPSCs. Scale-bars are indicated as labeled. (B) Summarized data for these groups regarding eIPSC/eEPSC ratios (wt control n = 11, n = 3 mice and wt up/down n = 7 cells, n = 3 mice, t-test P = 0.0113; het control n = 7 cells, n = 3 mice and het up/down n = 10, n = 3 mice, t-test P = 0.0119). Vertical box and bars are averaged summary data (* mean significant difference). (C) Representative traces of paired eEPSCs (holding at −89.1 mV) and eIPSCs (holding at 0 mV) in neurons from wt control and wt up/down, het control and het up/down groups. Stimulus artifacts are shown right before eEPSCs and eIPSCs. Scale-bars are indicated as labeled. (D) Summarized data for these groups regarding eIPSC/eEPSC peak ratios for wt control (n = 7 cells) versus wt up/down (n = 8 cells) (n = 2 mice each, t-test P = 0.012) and for het control (n = 8 cells) versus het up/down (n = 6 cells) (n = 2 mice each, t-test P = 0.0012).
Impaired sIPSC Potentiation Increases APs in Cortical Neurons from het KI Mice
Neuronal APs were evoked by local stimulation with stimulus intensity adjusted to have some AP failures. In wt cortical neurons, SWO-induction (0.5 Hz) decreased the success rates of evoked APs compared with pre-SWO baseline (Fig. 6A and C, from 55.10 ± 0.097% to 20.05 ± 0.09%, n = 8 cells [total n = 5 mice], paired t-test P = 0.005), compatible with post-up/down potentiated eIPSC charge transfers (also larger peak ratios eIPSC/eEPSC) in wt neurons. However, in het cortical neurons, SWO induction significantly increased the success rate of evoked APs compared with pre-SWO baseline (Fig. 6B,C, from 30.89 ± 10.16% to 65.10 ± 10.53%, n = 8 cells [total n = 4 mice], paired t-test P = 0.001), indicating that more neurons generated AP following SWOs and contributed to synchronous neuron discharges in het KI mice, as in vivo SWOs reflect the whole-brain macroscopic activity (Massimini et al. 2004; Volgushev et al. 2006). This is compatible with post-up/-down potentiated eEPSC charge transfers (not balanced by unpotentiated eIPSCs) (smaller peak ratios eIPSC/eEPSC) in neurons from het KI mice. However, the number of evoked APs per stimulus (during successful trials) was not significant difference between wt and het KI mice. In addition, there was no significant difference in neuronal AP input–output between wt and het KI mice (Supplementary Fig. 4A–C, wt n = 5 cells [total n = 2 mice]; het n = 6 [total n = 3 mice], two-way ANOVA, P = 0.989).
Figure 6.
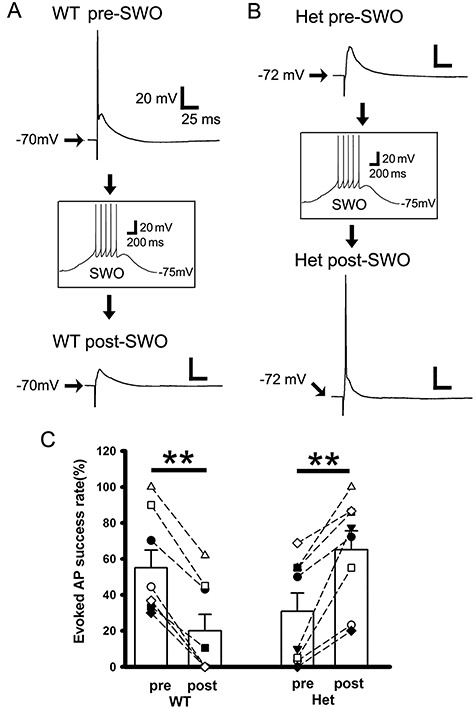
Impaired sIPSC potentiation following SWOs prompts neurons to more successfully generate APs from het Gabrg2+/Q390X KI mice. Panels A and B are representative traces of pre-SWO (top) and post-SWO (lower) evoked APs or EPSPs in neurons from wt littermate mice (panel A) and het KI mice (panel B). Top (A or B) and lower panels (A or B) are two individual traces from the same neurons being recorded with successfully evoked APs or postsynaptic potentials. The middle insets (A or B) show one SWO induction from rest membrane potentials (−75 mV). Scale bars are indicated as labeled. Panel C shows summary data for the success rate of evoked APs in wt littermate (n = 8 cells, n = 5 mice, paired t-test P = 0.005) and het KI mice (n = 8 cells, n = 4 mice, paired t-test P = 0.001). Each pair of pre- and post-SWO dots being connected with a line corresponds to the same neuronal pre- and post-SWO AP success rates. Vertical box and bars are averaged summary data (mean ± SEM).
In vivo SWO or Up-/Down-State Induction Causes Generalized Epileptic SWDs in het KI Mice
Using wt and het KI mice expressing halorhodopsin (Fig. 7 overlay green fluorescent inset) and intracortical stimulation through implanted tungsten electrodes within primary somatosensory cortex (layer V, Fig. 7 insert), we induced SWOs or up-/down-state (0.5 Hz) in vivo. Electrical currents of 300–400 pA were chosen to avoid kindling effect (kindling currents are within approximate 1 μA range, Supplementary Fig. 5, n = 3 mice each) (Racine 1972a, 1972b; Lothman et al. 1990) and one 20 ms stimulating duration was used to simulate sleep spindle duration during neuron up-state (Kandel and Buzsaki 1997). Neuronal down-states were controlled by laser delivery (590 nm) to activate halorhodopsin. With enough light intensity, neurons hyperpolarized by 5–10 mV from resting membrane potentials (Supplementary Fig. 6 inset). After SWO or up-/down-state induction in neurons within ex vivo brain slices (both wt and het KI mice), sEPSCs in cortical neurons exhibited increased amplitudes, without changes in sEPSC frequency or decay, compared with pre-SWO baseline ([wt and het sEPSC data together] wt n = 3 cells and het KI n = 4 cells, total 5 mice, amplitudes from 15.75 ± 1.95 to 22.30 ± 3.24 pA, paired t-test P = 0.011; frequency from 1.54 ± 0.71 to 1.30 ± 0.34 Hz, paired t-test P = 0.725; decay τ from 10.13 ± 1.92 to 44.75 ± 15.30 ms, paired t-test P = 0.078), indicating that the laser protocol could be used for in vivo SWO or up-/down-state induction. Moreover, sIPSCs in wt cortical neurons, but not het, were potentiated by laser-generated SWO or up/down induction (Supplementary Fig. 6, wt sIPSC amplitudes from 24.92 ± 3.43 to 33.47 ± 4.37 pA, n = 5 cells [total n = 4 mice], paired t-test P = 0.023; het sIPSCs amplitudes from 15.92 ± 3.46 to 13.66 ± 2.74 pA, n = 6 cells [total n = 4 mice], paired t-test P = 0.100). And there was no significant changes in sIPSC frequency or decay τ between baseline and postup-/down-states (wt [n = 5 cells, n = 4 mice]: frequency from 3.99 ± 1.05 to 8.86 ± 2.76 Hz, paired t-test P = 0.214; decay τ from 15.17 ± 3.87 to 39.07 ± 11.04 ms, paired t-test P = 0.085) (het [n = 6 cells, n = 4 mice]: frequency from 5.39 ± 2.00 to 2.64 ± 0.88 Hz, paired t-test P = 0.210; decay τ from 13.60 ± 7.84 to 15.92 ± 3.76 ms, paired t-test P = 0.715).
Using laser delivery (intensity determined by those used in ex vivo brain slices, Fig. 7 insert) and intracortical stimulation to induce in vivo SWOs or up-/down-states in wt mice (0.5 Hz for 10 min) (Fig. 7B), only short EEG oscillation activity was observed (band filtered between 0.1 and 100 Hz), accompanied by a few multiunit activity events (band filtered between 300 and 3 kHz). Compared with pre-SWO baseline EEG/multiunit activity, post-SWO EEG/multiunit activity exhibited no significant changes in wt mice (Fig. 7A, C and D). In contrast, during pre-SWO baseline in het KI mice (Fig. 7E), EEG/multiunit activity exhibited bilateral epileptic SWDs accompanied by animal immobility and slightly prolonged multiunit burst activity events (Fig. 7E). During the induction of in vivo laser-generated SWOs or up-/down-states (Fig. 7F), epileptic SWDs were observed and multiunit activity became longer in het KI mice (by comparing with non-SWD EEG and shorter multiunit activity in wt mice [Fig. 7B]). Furthermore, compared with pre-SWO baseline EEG/multiunit activity (Fig. 7E), more epileptic SWDs (accompanied by animal immobility, facial myoclonus and vibrissa twitching) and longer multiunit activity events appeared following laser-generated SWO induction in het KI mice (Fig. 7G,H). In summary, our results indicated that in het KI mice, the number (#) of post-SWO epileptic SWDs, total post-SWO SWD duration and averaged post-SWO individual SWD duration all were significantly increased, compared with pre-SWO baseline (Fig. 7I–K) (het KI mice [n = 7 each], SWD incidence from 98.95 ± 14.29 to 201.01 ± 11.26/h, paired t-test P = 0.001; total SWD duration from 317.71 ± 55.95 to 1331.56 ± 103.98 s/h, paired t-test P = 0.0008; averaged individual SWD duration from 2.52 ± 0.24 to 6.26 ± 0.97 s, paired t-test P = 0.005). In addition, in wt mice, we did not observe significant difference in post-SWO SWD incidence, total SWD duration and averaged individual SWD duration compared with the pre-SWO baseline (wt mice [n = 6 each], SWD incidence from 8.5 ± 1.62 to 16.83 ± 3.45/h, paired t-test P = 0.104; total SWD duration from 19.74 ± 4.19 to 54.66 ± 12.3 s/h, paired t-test P = 0.088; averaged individual SWD duration from 2.70 ± 0.46 to 3.34 ± 0.60 s, paired t-test P = 0.165).
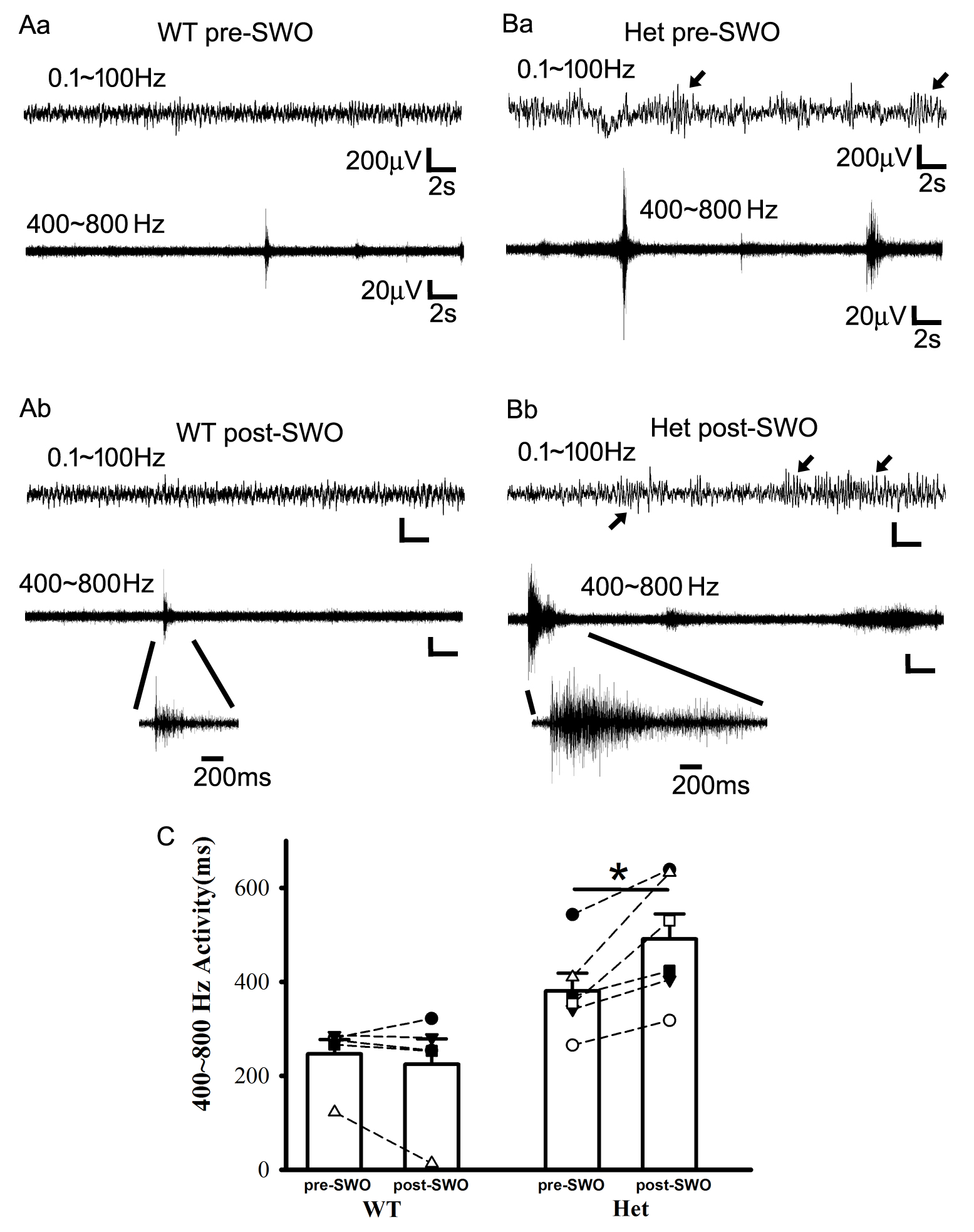
Additionally, high-frequency synaptic activity was recorded with tungsten electrodes (band filtered 400–800 Hz) (Lasztoczi et al. 2004) and always preceded epileptic SWDs in het KI mice (Supplementary Fig. 7) (wt pre-SWO 246.25 ± 31.12 ms [n = 5 mice]; het KI pre-SWO 380.38 ± 37.88 ms [n = 6 mice], t-test P = 0.026), implying that high-frequency synaptic activity could cause these epileptic SWDs. Following in vivo SWOs or up-/down-state induction, post-SWO high-frequency synaptic activity became longer in het KI mice, while there were not significant changes in wt mice (Supplementary Fig. 7C) (averaged episode duration, het from 380.38 ± 37.88 to 491.16 ± 53.43 ms, paired t-test P = 0.013, n = 6 mice) (wt from 246.25 ± 31.12 to 224.42 ± 54.24 ms, paired t-test P = 0.422, n = 5 mice). This implies the involvement of SWO-induced potentiation/ scaling-up of excitatory synaptic currents in cortical neurons from het KI mice.
Discussion
In this study (for summary, Fig. 8), we found that sleep-like SWOs (0.5 Hz) potentiated sEPSCs and sIPSCs in cortical pyramidal neurons (layer V) from wt mice. In contrast, only sEPSCs, but not sIPSCs, were enhanced in cortical neurons from het Gabrg2+/Q390X KI mice. Similar to signaling pathways of homeostatic synaptic potentiation, SWO-induced state-dependent potentiation/scaling-up of sEPSCs in cortical neurons depended on low-level intracellular calcium, βCaMKII activation and retinoid acid synthesis, but not activation of L-type calcium channels. Moreover, similar to SWOs, up-/down-like states induced state-dependent potentiation of sEPSCs in cortical neurons. Following up-/down-state induction, cortical neurons from het KI mice exhibited smaller eIPSC/eEPSC charge transfer ratios and smaller eIPSC/eEPSC peak ratios, prompting more neurons to generate APs, while cortical neurons from wt mice exhibited larger failure of APs. Consequently, in vivo SWO or up-/down-state induction by optogenetic manipulation triggered epileptic SWDs with higher incidence and longer duration in het KI, but not in wt mice. These epileptic SWDs were accompanied by sudden immobility, facial myoclonus, and vibrissa twitching, suggesting that in vivo SWOs or up-/down-states can trigger epileptic SWD onset in GGE patients while they are in NREM sleep or quiet-wakefulness state. In addition, due to very low incidence of GTCSs and the short duration of our in vivo experiments, we could not firmly determine the effect of SWO induction on GTCS generation. Also, in vivo SWO induction did not cause any changes in juvenile myoclonic seizures (O'Muircheartaigh et al. 2012; Arain et al. 2015; Ding et al. 2019).
Figure 8.
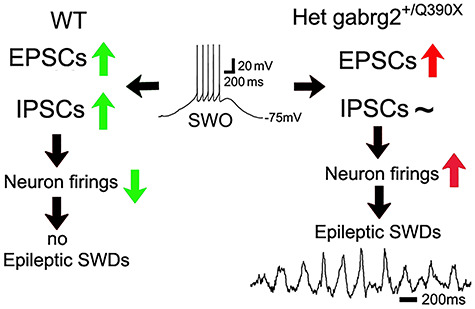
Summary diagram shows that SWO-induced homeostatic scaling-up/potentiation of excitatory synaptic currents in cortical neurons triggers epileptic SWDs in het KI mice. Green up-arrows represent balanced potentiated EPSCs and IPSCs. Green down-arrow represents decreased AP firings. Red up-arrows represent unbalanced potentiated EPSCs or increased neuron firings, respectively. Tilde symbol represents no change of IPSCs. Black arrows represent the sequential directions of causal interaction. Inset is one representative SWO traces with necessary labels. All other labels are self-explanatory.
Through homeostatic synaptic plasticity, neuronal firing activity manipulation has been used to regulate (scale up or down) neuronal synaptic strength to maintain firing stability (Turrigiano and Nelson 2004; Watson et al. 2016; Levenstein et al. 2017). Within in vitro culture neurons, decreasing neuronal firings by using TTX and NBQX potentiates/scales up miniature EPSCs and elevating neuronal firings by using picrotoxin attenuates/scales down miniature EPSCs (Kurotani et al. 2008; Turrigiano 2008; Chen et al. 2014). Moreover, in ex vivo brain slices from rats experiencing hypoxic seizures, elevated neuron firing activity also scales down sEPSCs in neurons (Sun et al. 2013). However, whether physiological cortical neuron activity during in vivo sleep states (NREM or REM sleep) can directly engage homeostatic synaptic plasticity or not remains questionable (Liu et al. 2010; Chauvette et al. 2012; Watson et al. 2016; Levenstein et al. 2017; Gonzalez-Rueda et al. 2018; Levenstein et al. 2019). In our study, 0.5-Hz SWOs resemble in vivo EEG delta wave oscillation (0.5 Hz) during NREM sleep and neuronal membrane potential alterations (Figs 1 and 3), and mirror neuronal up-/down-states (decreased neuronal firings) during NREM sleep or quiet wakefulness (Petersen et al. 2003). Moreover, as a surrogate of in vivo up-/down-state in neurons, physiologically similar up-/down-states in cortical neurons (Fig. 4, 10 s long up-states) can be generated by using a modified ACSF ([mM]3.5 or 5 K+, 1 Mg2+ and 1 Ca2+) as other groups (Sanchez-Vives and McCormick 2000; Rigas and Castro-Alamancos 2007; Neske 2015; Neske et al. 2015). And these up-/down-states can enhance sEPSCs in neurons from both wt and het KI mice. Thus, our results indicate that in vivo SWOs (0.5 Hz) or up-/down-states can engage homeostatic potentiation/scaling-up of sEPSCs/sIPSCs in cortical neurons from wt and of sEPSCs in neurons from het KI mice. Consistent with this argument, our pharmacology results of SWO-induced potentiation of sEPSCs (Fig. 2) indicate the involvement of similar signaling pathways for homeostatic synaptic potentiation that requires low-level calcium and βCaMKII activation, retinoid acid synthesis, and that blocking of L-type calcium channels enhances this synaptic potentiation (Thiagarajan et al. 2002; Turrigiano and Nelson 2004; Turrigiano 2008; Li et al. 2019). Similar to SWO-induced sEPSC potentiation, SWO-induced sIPSC potentiation in cortical neurons also shows Ca2+ dependency and can be enhanced by nifedipine in other study (Kurotani et al. 2008). Moreover, this SWO-induced sIPSC potentiation in cortical neurons uses an intracellular GABAA receptor forward trafficking mechanism as other study (Kurotani et al. 2008), which is disrupted in our het KI mice (Kang et al. 2015). This explains why SWO-induced sIPSC potentiation remains intact in wt, but not het neurons in our study.
Other than the synaptic potentiation/scaling-up by low-frequency stimulating oscillations (0.5 Hz SWOs or up-/down-state), high-frequency (50 Hz) stimulating oscillations in cortical neurons can induce synaptic depression/scaling-down of sEPSCs, which is very similar to neuronal hyperexcitability states during hypoxic epilepsy (Sun et al. 2013). Similarly, slightly elevated activity of 5 Hz in neurons suppresses sIPSCs, indicating that homeostatic plasticity of sEPSCs and sIPSCs in cortical neurons may have different set points of activity-dependent homeostatic synaptic plasticity.
In addition, 10 min SWO induction in most cortical neurons (80–90%) is able to engage homeostatic synaptic potentiation, in agreement with other reports that 5–10 (Frank et al. 2006), 30 min (Hou et al. 2011) or more (up to hours) manipulation of neuron activity (Sutton et al. 2006; Aoto et al. 2008; Ibata et al. 2008) can induce hemostatic synaptic plasticity. We did observe that in some neurons (<10–20%), SWO induction for 5–6 min still can engage homeostatic synaptic potentiation, suggesting that engagement of homeostatic synaptic potentiation can be a fast process in our study. Moreover, the 10-min duration of SWO or up/down activity in this study is within temporal ranges of in vivo neuronal up/down durations and SWO–EEG duration during SWS, making our low-frequency stimulating oscillations for SWOs and up–down states more physiologically relevant in vivo.
Interestingly, SWOs or up-/down-states (0.5 Hz) effectively induce both sEPSC and sIPSC potentiation in neurons (Figs 1 and 3). This indicates that sEPSCs and sIPSCs in neurons may be kept in a balance during SWS sleep or quiet-wakefulness, as some in vivo studies have suggested (Shu et al. 2003; Adesnik and Scanziani 2010; Dehghani et al. 2016; Takahashi et al. 2016; Moore et al. 2018). This ensures that sIPSCs suppress sEPSCs within neuronal networks, which creates one macroscopic balance within neuronal networks without escaped excitation. Our results of eIPSC/eEPSC ratios (charge transfer or peak ratios) in wt littermates are consistent with this macroscopic balance mechanism (Fig. 5), similar to other findings (Haider et al. 2006; Dehghani et al. 2016). During nonepileptic states in het KI mice (Fig. 5 het control), pre-up/-down eIPSCs (large amplitudes) in neurons are able to suppress pre-up/-down eEPSCs (small amplitude) to stop any escaped synaptic excitation. However, during SWS, up-/down-states scale up/potentiate sEPSCs, but not sIPSCs, in cortical neurons from het KI mice (Fig. 3D,E), causing smaller eIPSC/eEPSC ratios (Fig. 5, het up–down). This indicates that the post-up/-down eIPSCs cannot suppress post-up/down eEPSCs. Consequently, these state-dependently potentiated EPSCs eventually trigger epileptic SWDs in GGE models (McCormick and Contreras 2001; Huguenard and McCormick 2007; Bateup et al. 2013). Thus, SWS or quiet-wakeful states in het KI mice become a very critical state with unbalanced potentiated EPSCs in cortical neurons, likely leading to seizure onset. In contrast, other states such as REM sleep and active-wakeful states in het KI mice maintain balanced EPSCs and IPSCs in cortical neurons (Fig. 5, het control, large eEPSC/eIPSC charge transfer or peak ratios), leading to seizure-free status. In accordance with this argument, the peak of post-SWO epileptic SWD incidence in het KI mice (Fig. 7H het, gray trace, post-SWO) almost coincides the peak of potentiated post-SWO sEPSC amplitudes in het cortical neurons (Fig. 1E het), implying that it is the SWO-induced imbalance between potentiated sEPSCs and nonpotentiated sIPSCs that eventually triggers epileptic SWDs and other epileptic motor behaviors (Fig. 7H het post-SWO). Moreover, the post-SWO high-frequency field potential activity (400–800 Hz, due to synaptic activity) becomes significantly longer to drive subsequent epileptic SWDs (Supplementary Fig. 7), consistent with one in vitro epilepsy study that high-frequency synaptic activity causes epileptic activity (Lasztoczi et al. 2004).
GGE patients have genetic causes for their seizure activity generation (Allen et al. 2013; Striano and Zara 2017). However, seizure onset remains unpredictable. Recent review papers in one journal (review articles in Epileptic disorders, volume 21 supplement 1, 2019) have discussed the SWS association with seizure incidence. In addition, other acquired epileptic patients also show SWO activity right before seizure onset (Lagarde et al. 2019). However, to our knowledge, there are no any mechanisms to explain this SWO preference in neuroscience and epilepsy fields. Our findings of the SWO-initiating seizure onset mechanism in het KI mice offer a likely mechanism for preferential incidence of epileptic SWDs during NREM sleep and quiet-wakefulness that generate SWOs (Halasz et al. 2002; Shouse et al. 2004; Ng and Pavlova 2013; Ahmed and Vijayan 2014; Bagshaw et al. 2014; Arain et al. 2015). Since in vivo SWOs are synchronized in the whole-brain (Contreras et al. 1996; Massimini et al. 2004; Volgushev et al. 2006), SWO-induced unbalanced/potentiated EPSCs in cortical neurons of human GGE patients synchronously occur in many epileptic foci within the whole brain and prompt simultaneous neuron firings, eventually trigger epileptic SWD activity (Beenhakker and Huguenard 2009). In contrast, during REM sleep and active-wakeful period, since neuronal firing rates are elevated and synaptic sEPSC scaling-down/depression (or LTD) in neurons will be engaged. This creates one brain state with attenuated sEPSCs and large eIPSC charge transfers in het KI mice (Fig. 5, het control), likely leading to suppression of epileptic SWD activity. Consistent with this mechanism, we have never observed any epileptic SWDs and epileptic behaviors while animals actively perform locomotive, eating and grooming behaviors during day-light period. This may explain why many epileptic patients (including those Dravet epilepsy syndrome and GGE absence epilepsy) are seizure-free during most active wakeful period and why seizure incidence becomes much higher during NREM sleep and quiet-wakefulness (Halasz et al. 2013; Bagshaw et al. 2014; Verbeek et al. 2015). This SWD onset mechanism also applies to other acquired seizure onset preference during NREM sleep (Lagarde et al. 2019) if GABAergic synaptic potentiation/scaling-up is impaired.
In addition, SWS sleep and quiet-wakefulness are the period when theta-oscillations for memory consolidation in cortex and hippocampus occur (Cantero et al. 2003). During the same SWS sleep and quiet-wakefulness, scaling-up of sEPSCs by SWOs or up/down states trigger epileptic SWD onset in het KI mice, which consequently and catastrophically disrupts memory consolidation and chronically generates learning/cognitive deficits in Dravet epilepsy syndrome and other GGE patients (Bender et al. 2012).
In conclusion, sleep-like SWOs induce state-dependent potentiation/scaling-up of sEPSCs and sIPSCs in cortical neurons, requiring some signaling pathways of homeostatic synaptic plasticity. And, the potentiated sEPSCs and sIPSCs remain in a balance in neurons from wt mice. However, sEPSCs, but not sIPSCs, can be potentiated in neurons from het Gabrg2+/Q390X KI mice (one model of Dravet epilepsy syndrome), indicating that the dynamic balance between sEPSCs and sIPSCs is disrupted in het KI and neurons become more successful to generate APs. Consequently, in vivo SWO or up-/down-state induction (by optogenetic method and intracortical stimulation) triggers epileptic SWDs with significantly higher incidence frequency and longer duration in het KI mice, accompanied by sudden immobility, facial myoclonus, and vibrissa twitching. This suggests that in vivo SWOs can trigger epileptic seizure onset in GGE patients during NREM sleep or quiet-wakefulness, potentially leading to new translational therapeutic treatments of GGE patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Chun-Qing Zhang, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Mackenzie A Catron, Department of Neuroscience Graduate Program, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Li Ding, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Caitlyn M Hanna, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Martin J Gallagher, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Department of Neuroscience Graduate Program, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Robert L Macdonald, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Department of Neuroscience Graduate Program, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Chengwen Zhou, Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Department of Neuroscience Graduate Program, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Funding
This work was supported by National Institute of Neurological Disorders and Stroke grants R21 NS096483 M.J.G., R.L.M., C.Z.[contact] and R01 NS107424 (C.Z.). In addition, Chun-Qing Zhang's visiting scholarship (2013-2015) was supported by National Natural Science Foundation of Science of China grant 81771217 (C-Q.Z.).
Notes
C.Z. and C.-Q.Z. designed experiments; C.Z. performed experiments; L.D. and C.M.H. provided animal care and surgery. C.-Q.Z., M.A.C. and C.Z. analyzed data; and C.Z. wrote the manuscript and discussed with M.J.G. and R.L.M. Competing Interests: All authors declare that no competing interests exist for this study.
References
- Adesnik H, Scanziani M. 2010. Lateral competition for cortical space by layer-specific horizontal circuits. Nature. 464:1155–U1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed OJ, Vijayan S. 2014. The roles of sleep-wake states and brain rhythms in epileptic seizure onset. J Neurosci. 34:7395–7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Yet al. 2013. De novo mutations in epileptic encephalopathies. Nature. 501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L. 2008. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 60:308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arain F, Zhou C, Ding L, Zaidi S, Gallagher MJ. 2015. The developmental evolution of the seizure phenotype and cortical inhibition in mouse models of juvenile myoclonic epilepsy. Neurobiol Dis. 82:164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arain FM, Boyd KL, Gallagher MJ. 2012. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor alpha1 subunit. Epilepsia. 53:e161–e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw AP, Rollings DT, Khalsa S, Cavanna AE. 2014. Multimodal neuroimaging investigations of alterations to consciousness: the relationship between absence epilepsy and sleep. Epilepsy Behav. 30:33–37. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. 2013. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. 78:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. 2009. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 62:612–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini PP. 2012. SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. 23:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero JL, Atienza M, Stickgold R, Kahana MJ, Madsen JR, Kocsis B. 2003. Sleep-dependent theta oscillations in the human hippocampus and neocortex. J Neurosci. 23:10897–10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldi M, Panuccio G, Cavaccini A, D'Antuono M, Taglialatela M, Avoli M. 2011. Involvement of inward rectifier and M-type currents in carbachol-induced epileptiform synchronization. Neuropharmacology. 60:653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvette S, Seigneur J, Timofeev I. 2012. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 75:1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lau AG, Sarti F. 2014. Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology. 78:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Destexhe A, Sejnowski TJ, Steriade M. 1996. Control of spatiotemporal coherence of a thalamic oscillation by corticothalamic feedback. Science. 274:771–774. [DOI] [PubMed] [Google Scholar]
- Cortez MA, McKerlie C, Snead OC 3rd. 2001. A model of atypical absence seizures: EEG, pharmacology, and developmental characterization. Neurology. 56:341–349. [DOI] [PubMed] [Google Scholar]
- Dehghani N, Peyrache A, Telenczuk B, Le Van Quyen M, Halgren E, Cash SS, Hatsopoulos NG, Destexhe A. 2016. Dynamic balance of excitation and inhibition in human and monkey neocortex. Sci Rep. 6:23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demanuele C, Sonuga-Barke EJ, James CJ. 2010. Slow neuronal oscillations in the resting brain vs task execution: a BSS investigation of EEG recordings. Conf Proc IEEE Eng Med Biol Soc. 2010:1638–1641. [DOI] [PubMed] [Google Scholar]
- Ding L, Satish S, Zhou C, Gallagher MJ. 2019. Cortical activation in generalized seizures. Epilepsia. 60:1932–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. 2006. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 52:663–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. 2008. The mouse brain in stereotaxic coordinates (compact 3rd). Cambridge (MA): Academic Press. [Google Scholar]
- Frohlich F, Bazhenov M, Iragui-Madoz V, Sejnowski TJ. 2008. Potassium dynamics in the epileptic cortex: new insights on an old topic. Neuroscientist. 14:422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rueda A, Pedrosa V, Feord RC, Clopath C, Paulsen O. 2018. Activity-dependent downscaling of subthreshold synaptic inputs during slow-wave-sleep-like activity in vivo. Neuron. 97:1244–1252 e1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Thompson KR, Deisseroth K. 2008. eNpHR: a natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 36:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. 2006. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 26:4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Mody I. 2009. Establishing a physiological environment for visualized in vitro brain slice recordings by increasing oxygen supply and modifying aCSF content. J Neurosci Methods. 183:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasz P, Kelemen A, Szucs A. 2013. The role of NREM sleep micro-arousals in absence epilepsy and in nocturnal frontal lobe epilepsy. Epilepsy Res. 107:9–19. [DOI] [PubMed] [Google Scholar]
- Halasz P, Terzano MG, Parrino L. 2002. Spike-wave discharge and the microstructure of sleep-wake continuum in idiopathic generalised epilepsy. Neurophysiol Clin. 32:38–53. [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Sawada T, Natsume K. 2017. The change of picrotoxin-induced epileptiform discharges to the beta oscillation by carbachol in rat hippocampal slices. Biophys Physicobiol. 14:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron SE, Scheffer IE, Iona X, Zuberi SM, Birch R, McMahon JM, Bruce CM, Berkovic SF, Mulley JC. 2010. De novo SCN1A mutations in Dravet syndrome and related epileptic encephalopathies are largely of paternal origin. J Med Genet. 47:137–141. [DOI] [PubMed] [Google Scholar]
- Hou Q, Gilbert J, Man HY. 2011. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron. 72:806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. 2007. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 30:350–356. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. 1994. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J Neurosci. 14:5485–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. 2008. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 57:819–826. [DOI] [PubMed] [Google Scholar]
- Kandel A, Buzsaki G. 1997. Cellular-synaptic generation of sleep spindles, spike-and-wave discharges, and evoked thalamocortical responses in the neocortex of the rat. J Neurosci. 17:6783–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. 2016. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 73:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. 2010. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci. 30:13895–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL. 2015. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat Neurosci. 18:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotani T, Yamada K, Yoshimura Y, Crair MC, Komatsu Y. 2008. State-dependent bidirectional modification of somatic inhibition in neocortical pyramidal cells. Neuron. 57:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde S, Buzori S, Trebuchon A, Carron R, Scavarda D, Milh M, McGonigal A, Bartolomei F. 2019. The repertoire of seizure onset patterns in human focal epilepsies: determinants and prognostic values. Epilepsia. 60:85–95. [DOI] [PubMed] [Google Scholar]
- Lasztoczi B, Antal K, Nyikos L, Emri Z, Kardos J. 2004. High-frequency synaptic input contributes to seizure initiation in the low-[Mg2+] model of epilepsy. Eur J Neurosci. 19:1361–1372. [DOI] [PubMed] [Google Scholar]
- Levenstein D, Buzsaki G, Rinzel J. 2019. NREM sleep in the rodent neocortex and hippocampus reflects excitable dynamics. Nat Commun. 10:2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenstein D, Watson BO, Rinzel J, Buzsaki G. 2017. Sleep regulation of the distribution of cortical firing rates. Curr Opin Neurobiol. 44:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Park E, Zhong LR, Chen L. 2019. Homeostatic synaptic plasticity as a metaplasticity mechanism—a molecular and cellular perspective. Curr Opin Neurobiol. 54:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Faraguna U, Cirelli C, Tononi G, Gao XB. 2010. Direct evidence for wake-related increases and sleep-related decreases in synaptic strength in rodent cortex. J Neurosci. 30:8671–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Kapur J, Stringer JL. 1990. Recurrent spontaneous hippocampal seizures in the rat as a chronic sequela to limbic status epilepticus. Epilepsy Res. 6:110–118. [DOI] [PubMed] [Google Scholar]
- Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G. 2004. The sleep slow oscillation as a traveling wave. J Neurosci. 24:6862–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Contreras D. 2001. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 63:815–846. [DOI] [PubMed] [Google Scholar]
- Moore AK, Weible AP, Balmer TS, Trussell LO, Wehr M. 2018. Rapid rebalancing of excitation and inhibition by cortical circuitry. Neuron. 97:1341–1355 e1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanou E, Lee A, Catterall WA. 2018. Control of excitation/inhibition balance in a hippocampal circuit by calcium sensor protein regulation of presynaptic calcium channels. J Neurosci. 38:4430–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neske GT. 2015. The slow oscillation in cortical and thalamic networks: mechanisms and functions. Front Neural Circuits. 9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neske GT, Patrick SL, Connors BW. 2015. Contributions of diverse excitatory and inhibitory neurons to recurrent network activity in cerebral cortex. J Neurosci. 35:1089–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Pavlova M. 2013. Why are seizures rare in rapid eye movement sleep? Review of the frequency of seizures in different sleep stages. Epilepsy Res Treat. 2013:932790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Muircheartaigh J, Vollmar C, Barker GJ, Kumari V, Symms MR, Thompson P, Duncan JS, Koepp MJ, Richardson MP. 2012. Abnormal thalamocortical structural and functional connectivity in juvenile myoclonic epilepsy. Brain. 135:3635–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC, Hahn TT, Mehta M, Grinvald A, Sakmann B. 2003. Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc Natl Acad Sci USA. 100:13638–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. 2007. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 27:6590–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. 1972a. Modification of seizure activity by electrical stimulation. I. After-discharge threshold. Electroencephalogr Clin Neurophysiol. 32:269–279. [DOI] [PubMed] [Google Scholar]
- Racine RJ. 1972b. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 32:281–294. [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Fitzgerald EF, Klein PM, Zhou C, Sun H, Huganir RL, Jensen FE. 2012. Glutamate receptor 1 phosphorylation at serine 831 and 845 modulates seizure susceptibility and hippocampal hyperexcitability after early life seizures. J Neurosci. 32:17800–17812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid KH, Edmonds HL Jr, Schurr A, Tseng MT, West CA. 1988. Pitfalls in the use of brain slices. Prog Neurobiol. 31:1–18. [DOI] [PubMed] [Google Scholar]
- Rigas P, Castro-Alamancos MA. 2007. Thalamocortical up states: differential effects of intrinsic and extrinsic cortical inputs on persistent activity. J Neurosci. 27:4261–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. 2001. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 531:807–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. 2000. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 3:1027–1034. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Kleiman-Weiner M, Rudolph U, Huguenard JR. 2009. A gain in GABAA receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures. Proc Natl Acad Sci U S A. 106:7630–7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouse MN, Scordato JC, Farber PR. 2004. Sleep and arousal mechanisms in experimental epilepsy: epileptic components of NREM and antiepileptic components of REM sleep. Ment Retard Dev Disabil Res Rev. 10:117–121. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. 2003. Turning on and off recurrent balanced cortical activity. Nature. 423:288–293. [DOI] [PubMed] [Google Scholar]
- Steel D, Symonds JD, Zuberi SM, Brunklaus A. 2017. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. 58:1807–1816. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. 2001. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 85:1969–1985. [DOI] [PubMed] [Google Scholar]
- Striano P, Zara F. 2017. Epilepsy: common and rare epilepsies share genetic determinants. Nat Rev Neurol. 13:200–201. [DOI] [PubMed] [Google Scholar]
- Sun H, Kosaras B, Klein PM, Jensen FE. 2013. Mammalian target of rapamycin complex 1 activation negatively regulates polo-like kinase 2-mediated homeostatic compensation following neonatal seizures. Proc Natl Acad Sci USA. 110:5199–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. 2006. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 125:785–799. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kobayashi C, Ishikawa T, Ikegaya Y. 2016. Subcellular imbalances in synaptic activity. Cell Rep. 14:1348–1354. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. 2002. Alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 36:1103–1114. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. 2008. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 135:422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. 2004. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 5:97–107. [DOI] [PubMed] [Google Scholar]
- Velazquez JL, Huo JZ, Dominguez LG, Leshchenko Y, Snead OC 3rd. 2007. Typical versus atypical absence seizures: network mechanisms of the spread of paroxysms. Epilepsia. 48:1585–1593. [DOI] [PubMed] [Google Scholar]
- Verbeek NE, Wassenaar M, Campen JS, Sonsma A, Gunning B, Knoers N, Lindhout D, Jansen FE, Leijten F, Brilstra EHet al. 2015. Seizure precipitants in Dravet syndrome: what events and activities are specifically provocative compared with other epilepsies? Epilepsy Behav. 47:39–44. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Chauvette S, Mukovski M, Timofeev I. 2006. Precise long-range synchronization of activity and silence in neocortical neurons during slow-wave sleep. J Neurosci. 26:5665–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Zhang Z, Hintze M, Chen L. 2011. Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. J Neurosci. 31:17764–17771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TA, Smith NK, Kang JQ. 2019. The therapeutic effect of stiripentol in Gabrg2(+/Q390X) mice associated with epileptic encephalopathy. Epilepsy Res. 154:8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson BO, Levenstein D, Greene JP, Gelinas JN, Buzsaki G. 2016. Network homeostasis and state dynamics of neocortical sleep. Neuron. 90:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester JC, Contreras D. 2012. Columnar interactions determine horizontal propagation of recurrent network activity in neocortex. J Neurosci. 32:5454–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Cunha C, Zhang F, Liu Q, Gloss B, Deisseroth K, Augustine GJ, Feng G. 2008. Improved expression of halorhodopsin for light-induced silencing of neuronal activity. Brain Cell Biol. 36:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Z, Ding L, Deel ME, Arain FM, Murray CR, Patel RS, Flanagan CD, Gallagher MJ. 2013. Altered cortical GABAA receptor composition, physiology, and endocytosis in a mouse model of a human genetic absence epilepsy syndrome. J Biol Chem. 288:21458–21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Lippman JJ, Sun H, Jensen FE. 2012. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J Neurosci. 31:18211–18222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.