

Abstract
Acute brain ischemia accounts for most of stroke cases and constitutes a leading cause of deaths among adults and permanent disabilities in survivors. Currently, the intravenous thrombolysis is the only available medication for ischemic stroke; mechanical thrombectomy is an emerging alternative treatment for occlusion of large arteries and has shown some promise in selected subsets of patients. However, the overall narrow treatment window and potential risks largely limit the patient eligibility. New druggable targets are needed to innovate the treatment of brain ischemia. As the rate-limiting enzyme in the biosyntheses of prostanoids, cyclooxygenase (COX), particularly the inducible isoform COX-2, has long been implicated in mechanisms of acute stroke-induced brain injury and inflammation. However, the notion of therapeutically targeting COX has been diminished over the past two decades due to significant complications of the cardiovascular and cerebrovascular systems caused by long-term use of COX-2 inhibitor drugs. New treatment strategies targeting the downstream prostanoid signaling receptors regulating the deleterious effects of COX cascade have been proposed. As such, a large number of selective small molecules that negatively or positively modulate these important inflammatory regulators have been evaluated for neuroprotection and other beneficial effects in various animal models of brain ischemia. These timely preclinical studies, though not yet led to clinical innovation, provided new insights into the regulation of inflammatory reactions in the ischemic brain and could guide drug discovery efforts aiming for novel adjunctive strategies, along with current reperfusion therapy, to treat acute brain ischemia with higher specificity and longer therapeutic window.
Keywords: agonist, antagonist, excitotoxicity, infarction, inhibitor, neuroinflammation, neuroprotection
1. Brain ischemia
Stroke is a group of medical conditions in which the blood supply to some part of the brain is reduced or interrupted, thereby preventing the involved brain regions from receiving oxygen, glucose, and other nutrients, and eventually leading to brain cell death. Despite the continual marked advances in stroke unit care, acute stroke remains in leading causes of deaths in adults and neurological disabilities among survivors, as its prevalence continues to rise, particularly in the developing world 1,2. There are two main types of stroke: ischemic stroke associated with the insufficient blood flow and hemorrhagic stroke mainly caused by intracerebral or subarachnoid bleeding. Of all stroke cases, approximately 87% are caused by brain ischemia, 10% are intracerebral hemorrhage stroke, and only 3% are subarachnoid hemorrhage stroke 3,4. The conventional thrombolytic treatment approved by the U.S. Food and Drug Administration (FDA) for ischemic stroke utilizes the tissue plasminogen activator (tPA) to convert plasminogen into active plasmin, which then can breakdown blood clots (thrombi) formed in the ischemic brain tissues. However, tPA is only recommended for administration in 4.5 hours after the onset of a stroke 5,6, or else its benefits are likely outweighed by the potential risks in excitotoxic injury, peripheral bleeding, and even hemorrhagic transformation 7,8. For patients who are over 80 or had a previous stroke, the treatment window is only about 3 hours. For these reasons, up to 65% of ischemic stroke patients are not eligible for tPA-mediated thrombolytic therapy.
As an emerging option for patients with acute brain ischemia, the endovascular therapy by intra-arterial mechanical thrombectomy provides an interventional procedure of removing blood clots from brain blood vessels with a slightly longer therapeutic time window, i.e., 6 hours after the symptom onset 9. Though it can further improve functional outcomes in patients who also receive tPA treatment 10, thrombectomy is currently restricted to occlusions in large arteries and postsurgical complications, such as intracranial bleeding and damage to the blood vessels at the operative sites, raise serious concerns 11,12. Therefore, there remains an unmet high demand for new therapies with longer therapeutic window, higher efficacy, and lower risk in severe side effects for patients who suffer from the long-lasting devastating consequences of acute brain ischemia. Disappointingly, multiple once-promising drug candidates focusing on the early pathogenic targets failed to provide sufficient benefits in recent clinical studies for acute brain ischemia 13,14. An important conclusion one can draw from these monumental efforts is that the early pathogenic targets might be transitory after the stroke onset; thus, for new strategies seeking longer therapeutic windows, one must look into the secondary signaling events that are triggered by the initial excitotoxic cascade and, in turn, aggravate the progression of neuronal injury following the acute brain ischemia 15. A such prominent delayed pathophysiological process that emerged during the past decades is brain inflammation or neuroinflammation 16.
2. Postischemic neuroinflammation
The conventional concept that stroke is simply a disorder of blood vessels has profoundly been expanded during the past two decades largely owing to the recognition of inflammation in the brain. Acute brain ischemia initially triggers neuronal excitotoxicity due to the lack of energy source and oxygen, and later is characterized by a cascade of inflammatory reactions that involve the injured brain tissues and progress for several days following the onset of ischemic symptoms. The inflammatory processes after ischemic stroke are initially highlighted by the induced expression of many inflammatory mediators, such as adhesion molecules, inflammatory cytokines, mitochondria-derived reactive nitrogen species (RNS) and reactive oxygen species (ROS), followed by the breakdown of blood-brain barrier 17–20. The blood-brain barrier disruption can further intensify neuroinflammation by enabling infiltration of the peripheral inflammatory and immune cells into the ischemic brain regions. For instance, the infiltrated neutrophils are a main source of matrix metalloproteinases (MMPs), resulting in further damage to the endothelial tight junction proteins and extracellular matrix of the neurovascular units. Consequently, the activation of microglia and astrocytes along with peripheral myeloid cell influx into the ischemic sites characterize the extended phase, triggering brain edema and potential hemorrhagic transformation. The affected brain areas are therefore subject to the secondary insults, provoking another wave of inflammatory reactions, oxidative stress, neuronal death, and so forth 16,21–25.
Given that inflammatory processes very likely contribute to the delayed ischemic injury and lead to the aggravation of neurological outcomes, harnessing certain key inflammatory pathways may prevent the secondary neuronal injury. As such, interventions aiming to control these inflammatory reactions in the ischemic brain might provide attractive therapeutic strategies with potentially extended treatment window and afford beneficial effects that could complement the thrombolytic therapy in ameliorating postischemic brain damage 15,26. The anti-inflammatory strategies for neuroprotection after acute brain ischemia have been focused on intervening with the innate immune system by decreasing microglial activation 27,28, blocking neutrophil adhesion and infiltration 29,30, diminishing the ROS and RNS levels 31,32, and inhibiting interleukin-1 (IL-1) signaling 33. However, given the uncertainty of translational validity of therapeutically targeting these signaling pathways, additional knowledge of the postischemic neuroinflammation is necessary to the identification of druggable anti-inflammatory targets for delayed interventions of brain ischemia.
3. COX-1/2 as conventional targets
Cyclooxygenase (COX) is a key rate-limiting enzyme in the biosyntheses of prostanoids, which mediate another large complex inflammatory cascade that has been widely studied in acute brain ischemia and many other inflammation-associated neurological conditions. Up to date, two COX isozymes have been discovered: COX-1 and COX-2. The COX-1 is constitutively expressed across the mammalian body to maintain homeostatic prostanoids, which are essential for numerous physiological functions under normal conditions. COX-2, on the other hand, is generally undetectable in most normal tissues and cells but with some exception in vascular endothelial cells of normal hamsters, where a significant amount of COX-2 is found 34. However, COX-2 is induced by fever, infection, inflammation, hypertension 35, and other stimuli from growth factors and excessive neuronal activities 36, and is widely considered as a chief pro-inflammatory mediator (Figure 1). COX-2 is quickly and strongly induced in the brain by acute neurological insults including strokes and seizures 37–41. In the rat model of transient middle cerebral artery occlusion (MCAO), the COX-2 mRNA expression is considerably upregulated in the ischemic but not the contralateral hemisphere as early as 6 hours following ischemia and maximizes with a five-fold induction at 12 hours after MCAO 38. In rats with 20-minute global ischemia, the COX-2 protein level in the hippocampus begins to increase 4 hours after onset of ischemia and can reach a maximum at 24 hours after ischemia 39. The rapid upregulation of COX-2 is also found in infiltrating neutrophils, vascular endothelial cells, and neurons in infarcts of human ischemic brains, raising the possibility that the enzymatic products of COX-2 may contribute to the mechanisms of ischemic injury in the humans 42.
Figure 1.

Cyclooxygenase/prostanoid signaling cascade after brain ischemia. In response to brain ischemia, arachidonic acid, a membrane-bound 20-carbon fatty acid, is freed from phospholipids by phospholipase A2, and then is immediately converted to prostaglandin H2 (PGH2) by cyclooxygenase (COX), which has two isoforms – COX-1 and COX-2. The COX-1 isozyme is constitutively expressed in various mammalian cells and tissues to maintain the normal homeostasis, whereas COX-2 is usually undetectable under physiological conditions but rapidly and robustly induced by pathological stimuli, such as brain ischemia, at the injury sites. Traditional nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin, ibuprofen, and naproxen, act as nonselective COX inhibitors, while the COXIBs selectively inhibit the COX-2 isozyme. Short-lived PGH2 is further rapidly converted to five types of prostanoids, i.e., prostaglandin D2 (PGD2), PGE2, and PGF2α, thromboxane TXA2, and prostacyclin PGI2 by tissue-specific prostanoid synthases. PGD2 is directly synthesized from PGH2 by lipocalin prostaglandin D synthase (L-PGDS) and hematopoietic PGDS (H-PGDS); PGE2 is directly synthesized by microsomal prostaglandin E synthase-1 (mPGES-1), mPGES-2, and cytosolic PGES (cPGES); PGF2α is synthesized by prostaglandin F synthase (PGFS); PGI2 is synthesized by prostaglandin I synthase (PGIS); TXA2 is synthesized by thromboxane A synthase (TBXAS1). Among the three PGES isoforms, mPGES-1 is inducible, membrane-bound, and functionally coupled to COX-2. There are nine currently known G protein-coupled receptors (GPCRs) that are activated by prostanoids as indicated. DP1, EP2, EP4, and IP are coupled to Gs for cAMP signaling pathways mediated by protein kinase A (PKA) and exchange protein activated by cAMP (EPAC); EP1, FP, and TP receptors link to Gq for inositol trisphosphate (IP3)-mediated Ca2+ mobilization and diacylglycerol (DAG)-activated protein kinase C (PKC) signaling; DP2 and EP3 are mainly associated with Gi to downregulate the cAMP signaling. In addition, EP2 and EP4 may form a complex with β-arrestin to mediate G protein-independent signaling; EP3 and TP can couple to G12/13 to activate the Rho factor and Rho-associated protein kinase (ROCK). These prostanoid receptors together regulate neuronal inflammation and injury following brain ischemia. Note that only major pathways are displayed.
The increase in COX-2 expression leads to the biosyntheses of five types of prostanoids (Figure 1): prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin (PGF2α), prostacyclin or prostaglandin I2 (PGI2), and thromboxane A2 (TXA2). These COX products are rapidly (within seconds) and vigorously (~30 fold) increased upon decapitation-induced brain global ischemia 43–45, suggesting an immediate and dramatic increase in COX enzyme activity itself before the long-lasting elevation in COX expression and a key role for prostanoid cascade in response to stroke onset. Though short-lived, these small bioactive lipids in turn dynamically bind and act on a suite of G protein-coupled receptors (GPCRs): two receptors (DP1 and DP2) activated by PGD2; four (EP1, EP2, EP3, and EP4) activated by PGE2; each of the other three prostanoids acts on a single receptor (FP, IP, and TP) (Figure 1). Because prostanoids play essential roles in both initiating and sustaining the inflammatory reactions, the inhibition of COX-2 by a variety of selective and nonselective inhibitors is considered as a general strategy to treat inflammation-associated disease conditions including acute brain ischemia.
3.1. Selective COX-2 inhibitors
The COX-2 enzyme was first identified and sequenced in 1988 and confirmed in 1991 46, leading to the rise for development of selective inhibitors for COX-2. In less than eight years, the first COX-2 inhibitors (COXIBs) were developed and commercialized, with Celebrex (celecoxib) and Vioxx (rofecoxib) launched in 1999 (Figure 2). The COX-2 selective inhibitors are effective for relief from pain and inflammation without causing significant gastrointestinal complications, common side effects of the traditional nonsteroidal anti-inflammatory drug (NSAIDs), which are nonselective COX inhibitors 47,48. The early success in the development of selective COX-2 inhibitor drugs led to the broad interests of targeting COX-2 for inflammation-associated conditions including ischemic stroke (Table 1). The first COX-2 selective inhibitor that was investigated for therapeutic benefits in experimental brain ischemia was NS398 (Figure 2), which is about 42-fold selective on COX-2 over COX-1. Treatment with COX-2 selective inhibitor NS398 twice daily for 3 consecutive days was able to decrease the ischemia-induced PGE2 and reduce the infarct volume without affecting arterial pressure, blood glucose, or body temperature in a rat model of transient MCAO 38. Likewise, treatment with another COX-2 selective inhibitor SC58125 (Figure 2) for 3 days decreased the ischemia-induced hippocampal PGE2 and CA1 neuronal death after global ischemia in rats 39. Furthermore, systemic treatment with NS398 reduced the neurological deficit and infarction in mice following permanent MCAO 49. In line, COX-2-deficient mice showed lower vulnerability to ischemic brain injury and N-methyl-D-aspartate (NMDA)-mediated neurotoxicity when compared to their wild-type cohorts 50. In contrast, the overexpression of COX-2 in postnatal neurons led to a nearly 10-fold increase in PGE2 level in the brain and enlarged the infarct volume in mice after transient MCAO 51, In the same study, pretreatment with selective COX-2 inhibitor SC58236 (Figure 2) shrank the infarct volume in wild-type mice but not in COX-2 transgenic mice, indicating a dose effect of the COX-2 expression on ischemic injury. These early preclinical findings utilizing genetic ablation and pharmacological inhibition reveal that COX-2 induced by ischemic insults contributes to neuronal injury in the ischemic brain, suggesting that pharmacological inhibition of COX-2 might represent a promising anti-inflammatory strategy to treat ischemic stroke.
Figure 2.

Nonselective and selective COX-1/2 inhibitors tested in experimental brain ischemia. The IC50 values on COX-1 and COX-2 of each compound are indicated.
Table 1.
COX inhibition in recent preclinical studies on brain ischemia.
| Animal models | Major therapeutic outcomes | References |
|---|---|---|
| Transient MCAO (2 hr) induced in adult Sprague-Dawley rats | Treatment with COX-2 selective inhibitor NS398 (20 mg/kg, i.p., twice per day) beginning 6 hr after ischemia onset decreased the ischemia-induced PGE2 in the injured brain 24 hr after MCAO and reduced the infarct volume 3 days after MCAO without affecting mean arterial pressure, blood glucose, or rectal temperature. | Nogawa et al., 1997 |
| Global ischemia (20 min) induced in adult male Sprague-Dawley rats | Treatment with COX-2 selective inhibitor SC58125 (30 mg/kg, p.o., once per day) or nonselective inhibitor piroxicam (30 mg/kg) decreased the ischemia-induced PGE2 in the hippocampus 24 hr after global ischemia and reduced CA1 neuronal death 3 days after global ischemia. | Nakayama et al., 1998 |
| MCAO induced in adult C57BL/6 mice | Genetic ablation of COX-2 decreased the infarct volume 4 days after focal ischemia without affecting the cerebral blood flow. | Iadecola et al., 2001 |
| Transient MCAO (1 hr) induced in adult male C57BL/6 mice | COX-2 overexpression in postnatal neurons increased the PGE2 level in the brain and enlarged the infarct volume 4 days after ischemia; pretreatment with COX-2 inhibitor SC58236 (30 mg/kg, p.o., once at 4 hr before MCAO) reduced the infarct volume in wild-type mice but not in COX-2 transgenic mice 4 days after MCAO. | Dore et al., 2003 |
| Permanent MCAO induced in adult male C57BL/6 mice | Treatment with COX-2 selective inhibitor NS398 (10 mg/kg, i.p., twice per day for 3 days) reduced the neurological deficit and the infarct volume 4 days after MCAO; NS398 (10 mg/kg, i.p.), when administered 1 hr before and after MCAO and again 6 hr after MCAO, reduced the infarct volume 24 hr after MCAO. | Sugimoto and Iadecola, 2003 |
| Transient MCAO (2 hr) induced in adult male Sprague-Dawley rats | Oral treatment with COX nonselective inhibitor indomethacin (2.5 mg/kg per day) beginning 3 days before MCAO enhanced the neurogenesis for up to 28 days after ischemia without changing the infarct volume. | Hoehn et al., 2005 |
| Transient forebrain ischemia (10 min) induced in adult male Wistar rats | Treatment with ibuprofen with high dose (100 mg/kg immediately after reperfusion, and 50 mg/kg daily for the next 6 days) led to a long-lasting protection of CA1 hippocampal neurons 4 weeks after ischemia. | Park et al., 2005 |
| Permanent cerebral hypoperfusion induced in adult male Wistar rats | Treatment with the nonselective COX inhibitor indomethacin (3 mg/kg, i.p., for 3 days) did not improve the survival rate or learning disability but aggravated the neuronal injury in the CA1 and CA3 regions; treatment with selective COX-2 inhibitor NS398 (15 mg/kg, i.p., for 3 days) improved the survival rate and abolished the learning disability but exacerbated the neuronal injury and did not prevent the glial activation. | Institoris et al., 2007 |
| Motor cortex stroke ischemia induced in adult male Long-Evans rats | Long-term oral treatment with NS398 (2 mg/kg) only provided limited functional improvement but caused an increase in lesion size. | Silasi and Kolb, 2007 |
3.2. Nonselective COX inhibitors
As selective COX-2 inhibitors capture most attention owing to their promising neuroprotection against ischemic injury, several nonselective COX inhibitors were also tested in animal models of cerebral ischemia and showed some therapeutic potential (Table 1). Oral administration of piroxicam (Figure 2), an NSAID that nonselectively inhibit both COX-1 and COX-2, decreased the ischemic injury-induced PGE2 in the hippocampus and reduced CA1 neuronal death after global ischemia in rats 39. Ibuprofen (Figure 2), another traditional NSAID, when administered with high doses (50-100 mg/kg), caused a long-lasting protection of hippocampal CA1 neurons in a rat model of transient forebrain ischemia presumably via elevating the interleukin-1 receptor antagonist 52. However, it should be noted that the COX inhibition did not always lead to beneficial effects following brain ischemia. Systemic treatment with nonselective COX inhibitor indomethacin (Figure 2) did not decrease the infarct volume in a rat model of transient MCAO, although the NSAID was able to reduce microglial activation and enhance the neurogenesis following brain ischemia 53. Moreover, administration of indomethacin in a rat model of permanent cerebral hypoperfusion did not improve either the animal survival rate or learning disability as anticipated. On the contrary, COX inhibition by indomethacin exacerbated the hypoperfusion-induced neuronal injury in the hippocampal CA1 and CA3 regions 54. In the same study, NS398 increased the proportion of rats displaying neuronal damage and did not prevent the glial activation, although it improved the survival rate and reduced the learning impairment 54. Similarly, long-term treatment with NS398 caused an increase in lesion size in rats with motor cortex stroke, though a limited functional improvement was observed 55.
The contradiction in outcomes of these pharmacological studies is not uncommon, as similar conflicting results were also observed in a large number of animal studies on seizure models 56–58. The differences of ischemic models used in these preclinical studies (Table 1), such as methods for the ischemic induction (focal vs. global), duration of ischemia (transient vs. permanent), animal species (mouse vs. rat), sex (male vs. female), and age (young vs. old), might potentially be contributary factors. In addition, the dose, dosing frequency, dosing duration, as well as the pharmacodynamic and pharmacokinetic properties of these COX inhibitors (Figure 2), including selectivity, off-target activities, in vivo half-lives, and brain penetration, could also affect the therapeutic outcomes. It is worth noting that the selective COX-2 inhibitors overall showed more consistent and profound neuroprotection following brain ischemia than the nonselective COX inhibitors (Table 1), suggestive of a dominant role of the induced COX-2 in ischemic stroke-related neuronal damage. The COX-2 induction leads to the syntheses of five different prostanoids that act on a total of nine GPCRs, which mediate a myriad of detrimental and beneficial effects (Figure 1) 57,59. The past two decades also witnessed an increasing recognition of the notorious adverse effects of COX-2 selective inhibitor drugs that are associated with serious complications such as heart attack and stroke, suggesting that some COX downstream prostanoid signaling pathway might be protective in the microvascular systems 60. The Hyde and Jekyll nature of COX signaling cascade inspired us with a notion that targeting certain downstream prostanoid receptor might represent an alternative anti-inflammatory and neuroprotective strategy for the delayed treatment of brain ischemia with more specificity than the general blockade of the entire COX cascade (Figure 1).
4. Gs-coupled prostanoid receptors
In rodent brains, acute ischemic insult is often followed by rapid and marked increases in prostaglandin products – PGD2, PGE2, and PGF2α, which can be blocked by COX inhibitors, such as indomethacin and ibuprofen 61,62. The elevated prostaglandins by ischemic stroke are also found in the cerebrospinal fluid of human patients and experimental animals 63,64. Likewise, the levels of prostacyclin PGI2 and thromboxane TXA2 also increase along with COX-2 induction in the ischemic brain 65,66. Among the nine GPCRs activated by these five bioactive lipids (Figure 1), DP1, EP2, EP4, and IP receptors are coupled to Gαs that activates adenylyl cyclase to generate cAMP, relax smooth muscles, and thus are also called “relaxant” receptors. As a conventional intracellular second messenger, cAMP binds and activates protein kinase A (PKA) to regulate pleiotropic effects. As such, the cAMP-activated PKA subsequently translocates to the cell nucleus, where it acts on transcription factors including the cAMP-responsive element-binding protein (CREB) 67. The activated CREB in turn binds to CRE sites to regulate the expression of genes that are involved in the neuronal plasticity and regeneration 68. On the other hand, cAMP can enhance the cAMP-regulated guanine nucleotide exchange factors (cAMP-GEFs), also called the exchange factors directly activated by cAMP (EPACs) (Figure 1). The activated EPAC then can stimulate the downstream effectors Rap GTP-binding proteins 1/2 (RAP1/2) to regulate a variety of pathogenic events such as neurotoxicity and neuroinflammation 69–71. However, whether EPAC is directly involved in ischemic injury remains to be determined.
4.1. DP1 receptor
Following a 20-minute episode of global cerebral ischemia in rats, the PGD2 level in the hippocampus can be quickly induced by up to 80-fold 72. Similarly, in neonatal mouse brain after hypoxic ischemia, the PGD2 level rises by 90-fold, but only 6-fold and 45-fold for PGE2 and PGF2α, respectively 73, indicative of PGD2 as the most ample lipid metabolite that is derived from the cell membrane-released arachidonic acid in the ischemic brain. PGD2 is synthesized directly from intermediate PGH2 by PGD2 synthase (PGDS), which has two isozymes: lipocalin PGDS (L-PGDS, or β-trace protein) and hematopoietic PGDS (H-PGDS) 74–76. PGD2 binds to two GPCRs: Gαs-coupled DP1 and Gαi-coupled DP2, which, upon activation by PGD2, respectively upregulates or downregulates the adenylate cyclase that synthesizes cAMP (Figure 1). The PGD2 signaling-induced cAMP in turn activates PKA or other downstream effectors to regulate multiple physiological and pathological functions, such as inhibition of platelet aggregation, relaxation and contraction of smooth muscles, vasodilation, and vasoconstriction 77,78. In the human body (https://www.ncbi.nlm.nih.gov/gene/5729), the DP1 receptor is mainly distributed in colon, endometrium, heart, lung, immune system, etc., whereas its expression in the brain is relatively low under normal conditions. However, both DP1 and H-PGDS in the mouse brain can be induced by hypoxic ischemia 73, presumably contributing to the elevated PGD2 signaling in the ischemic brain (Figure 1).
Interestingly, PGD2 or DP1 selective agonist BW245C (Figure 3) can prevent neuronal death induced by the glutamate toxicity in cultured rat hippocampal organotypic slices and neurons presumably through acting on DP1 receptor in a cAMP/PKA-dependent manner, whereas DP2 activation by DK-PGD2 promoted neuronal loss 79. The neuroprotection of DP1 receptor and detrimental effect of DP2 were also recently confirmed in rat hippocampal cultures that were treated by aluminum for neuronal injury 80. Genetic ablation of the DP1 receptor aggravated ischemic brain injury and enhanced neurological dysfunction following transient ischemia in mice without affecting physiological parameters, such as arterial blood pressure, core body temperature, or cerebral blood flow. In addition, DP1 receptor activation by BW245C showed neuroprotection against glutamate-induced neuronal excitotoxicity in a concentration-dependent manner 81. These findings were confirmed by Taniguchi et al., who also demonstrated that the ischemic lesion in neonatal mouse brain after hypoxic ischemia was increased in H-PGDS/L-PGDS double knock-out mice, but not in L-PGDS or DP2 knock-out mice (Table 2). It was also found that the endothelial cells were severely damaged in H-PGDS/L-PGDS and DP1 knock-out mice; therefore, it is possible that PGD2 signaling via DP1 in the neonatal brain exerts neuroprotective effects via preventing endothelial cell degeneration 73. Interetestingly, the infarct size was in inverse relation to the amount of PGD2 production in L-PGDS, H-PGDS, and H-PGDS/L-PGDS knock-out mice, as the PGD2 levels in L-PGDS, H-PGDS, and H-PGDS/L-PGDS knock-out mice were 46%, 7%, and 1%, respectively, of that in the wild-type cohorts 73. These two preclincal studies provide evidence that the DP1 receptor activaiton might represent a therapeutic strategy for ischemic stroke. However, future study is needed to examine the therapeutic effects of selective DP1 agonists in animal models of transient or permanent MCAO in order to validate the receptor as a potential phamracological target for ischemic injury.
Figure 3.

Small-molecule compounds targeting Gs-coupled prostanoid receptors. The potency of each compound is indicated by KB, Ki, or IC50 value. As a selective agonist for IP receptor, beraprost is about 0.2-0.5 as potent as PGI2.
Table 2.
Targeting Gs-coupled prostanoid receptors for experimental brain ischemia.
| Receptor | Animal model | Major therapeutic outcomes | Reference |
|---|---|---|---|
| DP1 | Transient MCAO (90 min) induced in adult male C57BL/6 mice | Genetic ablation of the DP1 receptor enhanced ischemic brain injury and neurological dysfunction after transient ischemia 4 days after MCAO without affecting the mean arterial blood pressure, cerebral blood flow, or core body temperature. | Saleem et al., 2007 |
| Hypoxic ischemia (30 min) induced in C57BL/6 neonatal mice (P7) | The ischemic lesion was increased in H-PGDS/L-PGDS double knock-out mice or DP1 knock-out mice, but not in L-PGDS knock-out mice 24 hr and 7 days after hypoxic ischemia; the infarct size was in inverse relation to the amount of PGD2 production in L-PGDS, H-PGDS, and H-PGDS/L-PGDS knock-out mice. | Taniguchi et al., 2007 | |
| EP2 | Transient MCAO (90 min) induced in adult male C57BL/6 mice | Global deletion of the EP2 receptor exacerbated the infarct volume and neurological deficit 24 hr after MCAO without affecting the cerebral arterial vasculature or cerebral blood flow. | McCullough et al., 2004 |
| Permanent MCAO induced in adult male C57BL/6 mice | Global EP2 deficiency increased the infarct volume 24 hr after MCAO without altering the cerebral blood flow. | Liu et al., 2005 | |
| Transient MCAO (90 min) induced in adult male C57BL/6 mice | Global ablation of the EP2 receptor or pre-treatment with agonist ONO-AE1-259-01 (1 or 2 nmol, i.c.v.) 45-50 min before MCAO exacerbated the infarct volume and neurological deficit 4 days after MCAO without affecting the gross vascular anatomy of the brain, cerebral blood flow, body temperature, or mean arterial blood pressure. | Ahmad et al., 2010 | |
| Permanent MCAO induced in adult male C57BL/6 mice | Global EP2 deficiency increased the infarct volume and neurological deficit 7 days after MCAO. | Ahmad et al., 2010 | |
| Transient MCAO (45 min) induced in adult male C57BL/6 mice | Tamoxifen-induced postnatal deletion of the EP2 receptor reduced the infarct volume, neurological deficit, and neuroinflammation 2 days after MCAO without altering the peripheral immune mobilization in the spleen; treatment with EP2 antagonist benzoxazepine 52 (10 mg/kg, p.o., 2 dose at 4.5 and 24 hr after MCAO onset) reduced the infarct volume, neurological deficit, and post-stroke weight loss 3 days after MCAO. | Liu et al., 2019 | |
| Transient MCAO (45 min) induced in adult male and female C57BL/6 mice | EP2 deletion in neurons but not in myeloid or endothelial cells reduced the infarct volume in male and female mice in a gene dose dependent manner 2 days after MCAO. | Liu et al., 2019 | |
| Transient MCAO (45 min) induced in adult male C57BL/6 mice | Treatment with EP2 antagonist TG6-10-1 (5 or 10 mg/kg, p.o., 3 doses at 4.5, 12 and 24 hr after MCAO onset) reduced the infarct volume, neurological deficit, and cytokine induction 3 days after MCAO. | Li et al., 2020 | |
| EP4 | Transient MCAO (60 min) induced in adult male C57BL/6 mice | Treatment with EP4 selective agonist ONO-AE1-329 (0.03 or 0.3 mg/kg, s.c., once at 3 hr after MCAO) reduced the infarct volume in a dose-dependent manner 24 hr after MCAO; treatment with ONO-AE1-329 (0.3 mg/kg, s.c., twice at 2 and 8 hr after MCAO) reduced the infarct volume and neurological deficit 24 hr after MCAO. | Liang et al., 2011 |
| Transient MCAO (60 min) induced in adult male F1 hybrid B6D2F1/J mice | Treatment with ONO-AE1-329 (0.03 mg/kg, s.c., once at 3 hr after MCAO) improved the rotarod performance at both 2 day and 7 days after MCAO. | Liang et al., 2011 | |
| Transient MCAO (45 or 60 min) induced in adult male and female C57BL/6 mice | Neuronal EP4 ablation increased the infarct volume 24 hr after MCAO, which was reversed by treatment with ONO-AE1-329 (0.3 mg/kg, s.c., once at 3 hr after MCAO). | Liang et al., 2011 | |
| Transient MCAO (30, 45, or 60 min) induced in adult male C57BL/6 mice | Tamoxifen-induced EP4 deletion in endothelial cells increased the infarct volume 24 hr after MCAO, which was reversed by treatment with ONO-AE1-329 (0.3 mg/kg, s.c., once at 3 hr after MCAO); endothelial EP4 deletion did not affect the cerebral blood flow or mean arterial pressure. | Liang et al., 2011 | |
| Transient MCAO (60 min) induced in adult male and female C57BL/6 mice | Treatment with EP4 selective agonist L-902688 (0.75 μg/kg, i.p., 2 doses at 1 and 24 hr after MCAO) decreased the infarct volume and neurological deficit 2 days after MCAO without affecting the post-stroke weight loss, survival, or the cerebral blood flow | Akram et al., 2013 | |
| Transient MCAO (90 min) induced in adult male Sprague-Dawley rats | Treatment with L-902688 (0.3 or 1.0 mg/kg, i.v., once at 1.5 hr after MCAO) reduced the infarct volume, blood-brain barrier breakdown, the induction of IL-1β, IL-6, MMP-3/9, and preserved the tight junction proteins 24 hr after MCAO; treatment with L-902688 (1.0 mg/kg, i.v., once at 1.5 hr after MCAO) reduced the long-term neurological deficit for up to 3 weeks after MCAO. | DeMars et al., 2018 | |
| IP | Transient MCAO (60 min) induced in adult male Long-Evans rats | Overexpression of PGIS by adenovirus-mediated gene delivery into the lateral ventricle 3 days before but not after ischemia reduced infarct volume 24 hr after MCAO. | Fang et al., 2006 |
| Transient MCAO (90 min) and permanent MCAO induced in adult male C57BL/6 mice | Genetic deletion of IP receptor exacerbated the infarct volume and neurological deficit 4 days after transient MCAO and 7 days after permanent MCAO without altering cerebral blood flow, body temperature, or arterial blood pressure; pre- or post-treatment with IP selective agonist beraprost (50 or 100 μg/ml, p.o.) decreased infarct volume and neurological deficit in wild-type mice but not in IP knock-out mice 4 days after transient ischemia; treatment with beraprost (100 μg/ml, p.o.) 4.5 hr after MCAO onset decreased the neurological deficit and infarct volume in wild-type mice 7 days after permanent MCAO began. | Saleem et al., 2010 | |
| Transient global brain ischemia (12 min) induced in adult young and old male C57BL/6 mice | Genetic ablation of IP receptor increased the cognitive deficit, hippocampal CA1 pyramidal neuronal death, microglial activation, and myeloperoxidase activity, and decreased the phosphorylation of CREB in both young and old adult mice. | Shakil and Saleem, 2013 | |
| Transient global brain ischemia (12 min) induced in adult young and old male C57BL/6 mice | Post-treatment with IP agonist beraprost (25, 50, or 100 μg/ml, p.o.) attenuated neuronal death, astrogliosis, microbial invasion, and myeloperoxidase activity, and increased the phosphorylation of CREB in both young and old adult mice in a dose-dependent manner. | Shakil and Saleem, 2014 |
4.2. EP2 receptor
PGE2, another major prostaglandin product of COX within the brain, is directly synthesized from COX-derived intermediate PGH2 by the microsomal prostaglandin E synthase-1 (mPGES-1), mPGES-2, or cytosolic PGES (cPGES). Among these three PGES isozymes, mPGES-1 is highly inducible, membrane-bound, and functionally linked to COX-2 in preference to COX-1 for PGE2 synthesis under pathological conditions (Figure 1) 82. PGE2 is an essential executor of the detrimental effects mediated by induced COX-2 and mPGES-1 in various neurological diseases, such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and epilepsy 57,59,83. The detrimental roles of elevated COX-2 in ischemic stroke is also thought to be largely attributed to the PGE2 signaling. The levels of PGE2 are commonly elevated in ischemic brains of human patients as well as experimental animals 63,64,84,85, with a positive correlation between PGE2 levels and the severity in clinical outcome of the stroke 63. PGE2 exerts a wide variety of physiological and pathological functions via acting on a group of GPCRs, namely, EP1, EP2, EP3, and EP4 71. These membrane-bound receptors are differentially expressed in neurons, endothelial cells, microglia, and astrocytes throughout the brain and engage with divergent second messenger molecules (Figure 1) 71.
Given that targeting some PGE2 downstream signaling pathway might provide more therapeutic specificity than blocking the entire COX cascade by COX inhibitors, these four GPCRs have been investigated as potential therapeutic targets for brain ischemia in preclinical models, with the EP2 receptor as the one studied first and the most by far (Table 2). The EP2 receptor is widely expressed in the human body (https://www.ncbi.nlm.nih.gov/gene/5732), but is mainly found in bone marrow, immune system, reproductive system, and gastrointestinal tract 78,86, where its activation by homeostatic PGE2 leads to multiple important physiological functions, particularly in the immunoregulation 59. Expression of the EP2 receptor in the CNS areas, such as cerebral cortex and hippocampus, is relatively low under normal conditions, but can be quickly induced in response to stimuli from brain ischemia and epileptic seizures 19,87, suggestive of its potential involvement in the mediation of pathophysiology of these acute excitotoxic conditions. Interestingly, activation of the EP2 receptor by excessive PGE2 can strongly promote the brain microglia to generate cytokines and later may contribute to microglial death for the resolution of neuroinflammation 69,88.
The activation of EP2 receptor by its natural ligand PGE2 or selective agonist butaprost (Figure 3) was neuroprotective against glutamate receptor activation-mediated excitotoxicity in neuron-enriched cortical and hippocampal cultures, likely via activating the cAMP-PKA pathway 89. The neuroprotection from EP2 receptor activation has also been demonstrated by us using several positive allosteric modulators of the receptor in primary cultured neurons that were stimulated with NMDA and glycine for excitotoxic induction 90,91. Global congenital deletion of the EP2 receptor in mice led to considerable increases in the cerebral cortical infarction and neurological dysfunction 24 hours after a 90-minute episode of MCAO without affecting the overall brain vasculature or cerebral blood flow 89. Likewise, the genetic ablation of EP2 receptor also enlarged the infarct volume 24 hours after permanent MCAO in mice 92. These short-term exacerbating effects from the EP2 deletion can be extended to 4 days after transient MCAO and 7 days after permanent MCAO in mice 93, accentuating the long-lasting detrimental effects of the EP2 receptor ablation following brain ischemia. In line, intracerebroventricular administration of EP2 selective agonist ONO-AE1-259-01 (Figure 3) before the MCAO onset decreased the infarct volume and relieved the neurological deficit in mice in a dose-dependent manner 93. These earlier studies using genetic global deletion and pharmacological inhibition suggest that the EP2 receptor activation by PGE2 might play some neuroprotective role after brain ischemia (Table 2).
However, the activation of EP2 receptor did not always result in beneficial effects following excitotoxic injury. Treatment with PGE2 or butaprost aggravated the NMDA-induced neuronal excitotoxicity in glia-containing cortical cultures through a cAMP- but not PKA-dependent manner 94. In a recent animal study (Table 2), the tamoxifen-induced deletion of EP2 or the conditional ablation of the neuronal form of the receptor – but not the endothelial or myeloid form – decreased the cortical infarction and neurological deficit after a 45-minute episode of MCAO in mice 19. Markedly, these beneficial effects from postnatal or neuron-specific ablation of the EP2 receptor were able to be recapitulated in wild-type mice that were treated by an EP2-selective antagonist benzoxazepine 52 (Figure 3) in the same animal model of transient focal ischemia 19. We have recently developed selective EP2 antagonists that are highly potent and have adequate pharmacodynamic and pharmacokinetic profiles, and several of these novel compounds showed remarkable neuroprotective and anti-inflammatory effects after prolonged seizures and other acute neurological insults 95–100. Interestingly, post-stroke EP2 inhibition by a bioavailable and highly brain-permeable antagonist TG6-10-1 (Figure 3) was found to decrease the neurological deficit, infarct volume, and expression of three prototypic inflammatory cytokines, i.e., IL-1β, IL-6, and tumor necrosis factor α (TNF-α), in the brain after a 45-minute episode of transient ischemia in mice 101.
These apparently contradicting outcomes from the early and recent studies might result from the perplexing complications caused by the congenital global ablation of the receptor (Table 2). Widely expressed in the periphery and the brain, the Gs-coupled EP2 exerts many fundamental functions in normal physiological conditions. The global EP2 knock-out mice are widely known for developmental and other homeostatic adjustments that lead to complications including impaired fertility and hypertension 102–104. Mouse transcript database reveals that the EP2 expression in the brain is dynamically regulated (https://www.ncbi.nlm.nih.gov/gene/19217). During the early stages of development, the EP2 mRNA level in the CNS continuously rises from E11.5 to E18 by nearly eight-fold. However, its mRNA expression in adult cortex dramatically decreases to only about 25% of that at E18, indicating that the EP2 expression is high in the developing brain but remains very low in the adult brain. As such, the EP2 receptor might play some essential but unidentified roles during the early brain development when its expression is high. Indeed, the global congenital ablation of EP2 impairs some fundamental neural functions, such as sensorimotor gating, synaptic plasticity, and hippocampal long-term depression (LTD) and long-term potentiation (LTP), leading to considerable neurological dysfunctions 105,106. On the contrary, postnatally induced deletion or conditional ablation of EP2 preserves the normal functions of the receptor during early development of the brain, and thus do not phenocopy the behavioral and cognitive impairments found in the global EP2 knock-out mice 19. Nonetheless, the overall neuroprotective and anti-inflammatory effects from two well-characterized small-molecule EP2-selective antagonists – benzoxazepine 52 and TG6-10-1 – in mouse MCAO models provide promising evidence that pharmacological inhibition of the EP2 receptor by small-molecule compounds is a feasible strategy to mitigate the neuronal inflammation and damage after acute brain ischemia (Table 2).
4.3. EP4 receptor
As the other currently known Gs-coupled receptor of PGE2, the EP4 subtype resembles the EP2 subtype in both expression and signaling transduction (Figure 1). However, the functional coupling to cAMP appears less efficient for the EP4 than that for EP2, and the EP4 receptor also mediates several other pathways, such as extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB or Akt)/mammalian target of rapamycin (mTOR), and p38 mitogen-activated protein kinase (MAPK) pathways 78,107. Similar to EP2, the EP4 receptor is widely distributed in the human body, especially in the CNS, dorsal root ganglion, bone marrow, thyroid, heart, kidney, lung, pancreas, spleen, intestine, colon, uterus, and urinary bladder (https://www.ncbi.nlm.nih.gov/gene/5734), and has been implicated in both physiological and pathological conditions. In the CNS, the EP4 receptor is constitutively expressed in neurons of hippocampus, cortex, and striatum, whereas its expression in endothelial cells is elevated during the early reperfusion after brain ischemia 108.
The differential expression of the EP4 receptor in various types of brain cells inspired investigation to reveal the cell type-specific functions of the receptor in ischemic injury (Table 2). Conditional EP4 ablation in neurons aggravated the injurious outcomes of transient MCAO in mice, and similar results were demonstrated in mice with postnatal ablation of the receptor in endothelial cells109, suggesting that the EP4 receptor activation in these two cell types aggravates the ischemic injury. Subcutaneous administration of an EP4-selective agonist, ONO-AE1-329 (Figure 3), following brain ischemia lessened the infarction and neurological deficit, causing a long-term substantial improvement in rotarod performance of the animals 109. Similarly, L-902688 (Figure 3), another potent EP4-selective agonist, when interperitoneally administered immediately after transient brain ischemia, reduced the infarct volume and improved neurological functions but did not reverse the weight loss or improve the survival after a one-hour MCAO in mice. Further, the post-stroke EP4 receptor activation by L-902688 recapitulated the benefits of COX-2 selective inhibitor NS-398 in the same study 110. Similarly in rats, the post-stroke treatment with L-902688 dose-dependently decreased the cerebral infarction, disruption of the blood-brain barrier, and induction of MMPs and cytokines after transient MCAO, causing a chronic reduction in neurological dysfunction for up to 3 weeks after ischemia 111. Taken together, these interesting findings suggest that the EP4 receptor activation by selective small-molecule agonists is neuroprotective following acute ischemic insults presumably via lessening the neuroinflammatory reactions and blood-brain barrier damage.
4.4. IP receptor
Prostacyclin PGI2 is synthesized from PGH2 by tissue-specific prostaglandin I synthase (PGIS) (Figure 1). PGI2 – via Gs-coupled IP receptor – mainly inhibits platelet activation to prevent formation of the platelet plug that is involved in primary hemostasis. Additionally, the endothelial IP receptor activation leads to smooth muscle relaxation and vasodilation 112. Given that PGI2 counteracts the thromboxane TXA2’s effects of vasoconstriction and platelet aggregation, the balance between TXA2 and PGI2 has long been known essential to the homeostasis of cerebral blood flow 113. Following a transient global cerebral ischemia, the level of PGI2 in the rat hippocampus began to increase along with the COX-2 expression as early as 30 min after the onset of brain ischemia 66. The hippocampal PGI2 level then maximized at two days after the ischemia and remained well above that of the sham conditions for more than 2 weeks, indicating its quick induction and long-lasting effect in the ischemic brain following acute brain ischemia.
As the direct enzyme for PGI2 biosynthesis, PGIS showed a delayed increase in both mRNA and protein expression levels in the ischemic cortex at 2-3 days after transient ischemia in rats. PGIS protein is mainly found in endothelial cells with slightly less expression in neurons, microglia, and astrocytes 65. Interestingly, the overexpression of PGIS through adenovirus-mediated gene delivery into the rat lateral ventricle 3 days before the introduction of focal cerebral ischemia-reperfusion reduced infarct volume (Table 2), indicating a neuroprotective role of PGIS via enhancing PGI2 synthesis that leads to a favorable PGI2/TXA2 ratio. These results together with early findings that some PGI2 analogs showed considerable protective effects against ischemic damages suggest that the PGI2 signaling via its IP receptor may represent a physiological mechanism of self-redemption of the brain against acute ischemic injury 114,115.
The IP receptor is overall highly expressed in human brain and is also readily detected in most other organs, such as heart, lymph node, endometrium, placenta, prostate, intestine, thyroid, and urinary bladder (https://www.ncbi.nlm.nih.gov/gene/5739). Genetic deletion of the PGI2 receptor IP exacerbated the infarct volume and neurological deficit after both permanent and transient ischemia but did not change the cerebral blood flow, body core temperature, or arterial blood pressure during the reperfusion 116. In contrast, treatment with IP selective agonist beraprost (Figure 3) decreased the infarct volume and neurological deficit in wild-type mice but not in IP knock-out mice after transient or permanent ischemia 116, validating the IP as the therapeutic target of beraprost. The genetic deficiency of IP receptor also led to cognitive deficit, hippocampal CA1 pyramidal neuronal death, microglial activation, and myeloperoxidase activity, and a decrease in the phosphorylation of CREB in both young and old mice after transient global brain ischemia 117. However, treatment with beraprost attenuated the ischemia-associated neuronal death, astrogliosis, microbial invasion, and myeloperoxidase activity, and increased the phosphorylated CREB in the same mouse model of global ischemia 118. These findings using combined genetic and pharmacological strategies together reveal a neuroprotective role for the PGIS-PGI2-IP signaling axis in the ischemic brain.
5. Gq-coupled prostanoid receptors
The EP1, FP, and TP receptors are coupled to Gq and their activation by their corresponding cognate prostanoids stimulates phospholipase C (PLC) that hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (Figure 1). IP3 then promotes the release of Ca2+ from the endoplasmic reticulum into cytoplasm via acting on specific IP3 receptors on the endoplasmic reticulum membrane that function as Ca2+ channels. On the other hand, DAG directly activates certain Ca2+-dependent isoforms of protein kinase C (PKC) that lead to the Ca2+ influx current via the voltage dependent Ca2+ channels 119. The EP1, FP, and TP receptors are also classified as “contractile” type of prostanoid receptors in that the increase in cytosolic Ca2+ often leads to the contraction of smooth muscle cells.
5.1. EP1 receptor
The human EP1 receptor is highly expressed in colon, kidney, lung, spleen, and stomach (https://www.ncbi.nlm.nih.gov/gene/5731). In the brain, EP1 receptor is primarily found in neurons and endothelial cells, and its expression is dynamically regulated by ischemic injury in a time-dependent manner. The mRNA level of EP1 receptor is upregulated by nearly 50% in ischemic brain tissues at 4 hours after the occlusion begins, and then resides to basal levels 10-20 hr later. Intriguingly, by 48 hr after the ischemia onset, EP1 mRNA in the affected brain areas increases again by more than 50% above the basal levels 85,120. Though EP1 is also expressed in microglia of the brain, the microglial form of EP1 is much less abundant than its neuronal and endothelial counterparts and may not necessarily be relevant to the post-stroke inflammation-associated brain injury 121,122. Therefore, it is unlikely that the well-established detrimental role of the EP1 receptor is directly mediated by neuroinflammatory mechanisms after ischemic stroke. Instead, the EP1 receptor mediates neurotoxicity after brain ischemia via dysregulating the intracellular Ca2+ homeostasis, as the Ca2+ influx into neurons via the L-type calcium channels is a common molecular mechanism underlying excitotoxic injury, and modulating these calcium channels can lead to neuroprotection 123,124. As such, EP1 receptor activation likely impairs the Na+-Ca2+ exchanger that constitutes an essential self-redemption mechanism by which neurons dispose of the excessive cytosol Ca2+ during excitotoxic insults 122.
The genetic deletion of EP1 receptor consistently alleviated various neuronal injuries that are caused by excitotoxicity, oxygen-glucose deprivation, and the transient cerebral ischemia (Table 3) 122. The reduction in brain infarction and neurological deficit found in EP1 knock-out mice after ischemic injury has been validated by several research groups using MCAO models that differ in the duration of occlusion (Table 3) 120,125,126. These studies also suggest that the EP1 deficiency does not noticeably change the cerebral blood flow 122,127, post-stroke weight loss 125, or the brain major vasculature 126. Pharmacological inhibition of the EP1 receptor by selective antagonists ONO-8713 or SC51089 (Figure 4) considerably decreased the infarction and neurological dysfunction in rodent models of both permanent MCAO 128 and transient MCAO 120,122,127,128. It should be noted that treatment with EP1 antagonist SC51089 seems to have a long therapeutic time window (up to 12 hours after reperfusion begins), and its benefits sustain over time (up to 2 weeks) and do not show any sex difference 128. Moreover, the EP1 receptor may also provide a mechanistic link between the neuroinflammatory responses and the MMPs-mediated blood-brain barrier breakdown after brain ischemia 120. These important preclinical studies together provide evidence that targeting the EP1 receptor might represent an emerging strategy to relieve the destructive outcomes of acute brain ischemia and the subsequent blood-brain barrier damage (Table 3).
Table 3.
Targeting Gq-coupled prostanoid receptors for experimental brain ischemia.
| Receptor | Animal model | Major therapeutic outcomes | Reference |
|---|---|---|---|
| EP1 | Transient MCAO (20 min) induced in adult male C57BL/6 mice | Genetic ablation of the EP1 receptor or treatment with EP1 selective antagonist SC51089 (10 μg/kg, i.p., twice per day) beginning at 5 min or 6 hr after ischemia attenuated the infarct volume, neurological deficit, and brain swelling 3 days after MCAO without affecting cerebral blood flow. | Kawano et al., 2006 |
| Transient MCAO (90 min) induced in adult male C57BL/6 mice | EP1 deficiency reduced infarct volume 4 days after MCAO without affecting the post-stroke weight loss. | Ahmad et al., 2006 | |
| Transient MCAO (90 min) induced in adult male C57BL/6 mice | EP1 deficiency decreased the arterial partial pressure of oxygen level during the ischemic stage and improved the ipsilateral cerebral blood flow during the reperfusion without affecting the brain vasculature network. | Saleem et al., 2007 | |
| Transient MCAO (90 min) induced in adult male C57BL/6 mice | Treatment with EP1 antagonist ONO-8713 (0.1, 1, or 10 nmol, i.c.v.) immediately before MCAO reduced the infarct volume 4 days after ischemia without affecting cerebral blood flow. | Ahmad et al., 2008 | |
| Transient MCAO (25 min) induced in adult male and female C57BL/6 mice | Female mice had smaller infarcts than male mice 3 days after transient ischemia; treatment with EP1 antagonist SC51089 (5, 10, or 20 μg/kg, i.p., twice per day) beginning at 5 min, 6 hr, or 12 hr, but not 24 hr after reperfusion decreased the infarct volume in male mice 3 days after ischemia; treatment with SC51089 (10 μg/kg, i.p., twice per day) beginning at 5 min after reperfusion reduced the infarct volume by 41% in female mice 3 days after ischemia. | Abe et al., 2009 | |
| Permanent MCAO induced in adult male C57BL/6 mice | SC51089 (10 μg/kg, i.p., twice per day) beginning at 6 hr after MCAO onset reduced the infarct volume by 39% 24 hr after ischemia. | Abe et al., 2009 | |
| Transient MCAO (25 min) induced in adult male C57BL/6 mice | Treatment with SC51089 (10 μg/kg, i.p., twice per day for 3 days) beginning at 6 hr after reperfusion led to long-term reduction (2 weeks after ischemia) in the infarct volume, the atrophy of the ipsilateral hemisphere, and functional deficit. | Abe et al., 2009 | |
| Transient MCAO (60 min) induced in adult male C57BL/6 mice | Genetic deletion of EP1 decreased the infarct volume, blood-brain barrier breakdown, MMP-9/-3 activity, and ICAM-1 expression in the cortex 24 hr after ischemia. | Frankowski et al., 2015 | |
| Transient MCAO (90 min) induced in adult male Wistar rats | Treatment with EP1 antagonist SC51089 (3 mg/kg, i.v.) at 3, 18, and 28 hr after reperfusion reduced infarct volume, decreased the blood-brain barrier permeability, preserved the tight-junction proteins, prevented the hemorrhagic transformation, attenuated the MMP-9/-3 activity, and reduced the MPO and ICAM-1 levels in the cortex 48 hr after ischemia. | Frankowski et al., 2015 | |
| FP | Transient MCAO (90 min) induced in adult male C57BL/6 mice | Genetic ablation of the FP receptor reduced infarct volume and improved neurological dysfunction 4 days after transient ischemia without affecting cerebral blood flow, body temperature, or arterial blood pressure; treatment with FP selective agonist latanoprost (100 μg/kg, p.o., once) 30 min after reperfusion began aggravated neurological dysfunction and brain infarction in wild-type mice 4 days after MCAO but not in FP knock-out mice. | Saleem et al., 2009 |
| Permanent MCAO induced in adult male C57BL/6 mice | Genetic ablation of the FP receptor or treatment with antagonist AL-8810 (1 or 10 mg/kg, i.v., once) immediately after permanent MCAO began reduced neurological dysfunction and infarct volume in a dose-dependent manner 2 days after MCAO began. | Kim et al., 2012 | |
| TP | Spontaneously hypertensive stroke prone rats (SHRSP) | Oral treatment with TP antagonist terutroban (30 mg/kg per day for 6 weeks) improved survival, delayed the appearance of brain damage and proteinuria, and reduced the expression of IL-1β, TGF-β, and MCP-1 without affecting the thromboxane in serum and urine. | Gelosa et al., 2010 |
| Transient MCAO (90 min) induced in adult male ICR mice | The expression of TP receptor was upregulated by ischemia/reperfusion injury for up to 7 days after MCAO; post-treatment with TP selective antagonist SQ29548 (10 μl, 2.6 μmol/ml, i.c.v. twice) immediately after reperfusion and again 24 hr later attenuated the activation of microglia/macrophages, induction of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) and iNOS, blood-brain barrier destruction, infarct volume, and neurological deficit 3 days and 7 days after MCAO without affecting the cerebral blood flow. | Yan et al., 2016 |
Figure 4.

Compounds modulating Gq-coupled prostanoid receptors. The potency of each compound is indicated by EC50, IC50, or Ki value.
5.2. FP receptor
PGF2α in the brain also can be vigorously induced by brain ischemia in both human patients and experimental animals. PGF2α is the most induced and abundant prostaglandin in the cerebrospinal fluid of rabbits with focal ischemia, though PGD2 is the dominant one under normal condition 64. In the cat brain, the resting level of PGF2α is higher than that of PGE2; following a 15-minute episode of global ischemia, the brain PGF2α level remains higher than PGE2, though its induction fold is only about half of that for PGE2 84. However, in neonatal mouse brain after hypoxic ischemia, the PGF2α level in the cerebral cortex can increase up to 45-fold, whereas PGE2 only rises only about 6-fold 73. PGF2α is synthesized from intermediate PGH2 by prostaglandin F synthase (PGFS) 129, and activates the sole currently known receptor FP, which is coupled to Gαq 130. FP receptor is widely distributed in the human body (https://www.ncbi.nlm.nih.gov/gene/5737), especially in colon, CNS, endometrium, placenta, cardiovascular system, myometrium, ovarian, etc. 78 When bound by PGF2α or other selective agonists, the FP receptor becomes active, followed by the release of Gαq from the G protein heterotrimeric complex to activate PLC. The activated PLC then is able to hydrolyze PIP2 to DAG and IP3, which in turn activates certain PKC-dependent pathways and initiates Ca2+-sensitive signaling, respectively (Figure 1) 78,131.
Genetic ablation of the FP receptor in mice was shown to reduce the infarct volume and to improve neurological dysfunction after transient ischemia without affecting cerebral blood flow, core temperature, or arterial blood pressure 132. Contrariwise, oral treatment with FP receptor selective agonist latanoprost (Figure 4) after the ischemia-reperfusion exacerbated the neurological dysfunction and brain infarction in wild-type mice while had no effect in FP knock-out mice, indicative of the FP as the target of latanoprost’s therapeutic action. In addition, the FP deficiency partially diminished the NMDA-induced excitotoxic brain damage (Table 3) 132. Interestingly, the genetic ablation of the FP receptor in mice also alleviated neurological dysfunction and reduced brain infarction following permanent MCAO, and these benefits in FP knock-out mice were recapitulated in their wild-type peers that were systemically treated by FP selective antagonist AL-8810 (Figure 4) in a dose-dependent manner 133. Furthermore, blockade of the FP receptor led to neuroprotection from glucose-oxygen deprivation-induced cell death and ROS formation in cultured hippocampal slices neurons presumably through downregulating the FP receptor-related Ca2+ signaling 133. These preclinical findings suggest that PGF2α signaling via FP receptor contributes to the brain damage and functional deficit caused by acute ischemic injury (Table 3), thereby providing proof-of-concept evidence to support the FP receptor as a promising molecular target for brain ischemia.
5.3. TP receptor
Thromboxane TXA2 is synthesized from PGH2 by tissue-specific thromboxane A synthase 1 (TBXAS1), and activates Gq/G12/13-coupled TP receptor to stimulate new platelets and to increase platelet aggregation (Figure 1) 78. TXA2 is also a well-known vasoconstrictor via regulating Ca2+-sensitive signaling pathways in smooth muscle cells 134,135. Therefore, PGI2 and TXA2 in general act as physiological antagonists to each other 60. However, there might be some crosstalk between these two main prostanoid signaling pathways, as PGI2 can activate the TP receptor in blood vessels of hypertensive rats although PGI2 usually relaxes arteries in normal rodents 136. Likewise, PGF2α also binds to the TP receptor to trigger vascular contraction in healthy rodents 34. In the CNS, the balance between TXA2 and PGI2 plays an essential role in the regulation of cerebral blood flow in response to ischemic injury 113,137. Following a transient global cerebral ischemia, the levels of PGI2 and TXA2 in the rat hippocampus increase, and the induction of PGI2 is faster and higher than that of TXA2 66, resulting in an long-lasting increase in the PGI2/TXA2 ratio and imbalance between these two important blood flow regulators in the ischemic brain. Similarly, following transient focal ischemia, TXA2 is also elevated in the brain but less than PGI2 65,66, suggesting a pathogenic role of TXA2 signaling via TP receptor in the ischemic brain. In addition, a genetic associate study identified some specific single nucleotide polymorphisms (SNPs) and haplotypes of human TP gene that are highly correlated to cerebral infarction 138, suggesting the TP gene may be associated with the increased susceptibity to ischemic stroke in humans. The TP receptor is widely distributed in the human body (https://www.ncbi.nlm.nih.gov/gene/6915), particularly in colon, endometrium, kidney, spleen, placenta, heart, liver, etc., and its expression in the brain is moderate under normal conditions. Interestingly, the expression of TP receptor has been found to be upregulated for up to 7 days mainly in microglia by ischemia-reperfusion injury in mice 139,140, suggesting its involvement in postischemic brain inflammation and injury.
TP receptor activation by selective agonist U46619 (Figure 4) increased the expression of several pro-inflammatory molecules, such as IL-1β, IL-6, and the inducible nitric oxide synthase (iNOS), as well as the phosphorylation of ERK, which can be attenuated by TP selective antagonist SQ-29548 (Figure 4) or MEK inhibitor U0126 139. This interesting study revealed a role of TP receptor in microglial activation via the ERK signaling pathway after cerebral ischemia. Intracerebroventricular treatment with SQ-29548 after transient ischemia in mice reduced the infarct volume and neurological deficit without affecting the cerebral blood flow, accompanied by decreases in microglial activation, induction of pro-inflammatory cytokines and iNOS, and blood-brain barrier disruption (Table 3) 140. Oral treatment with another TP antagonist terutroban (Figure 4) in stroke-prone rats for 6 weeks led to promising benefits including improved survival, delayed appearance of brain damage and proteinuria, and reduced expression of IL-1β, transforming growth factor β (TGF-β), and monocyte chemoattractant protein 1 (MCP1) or chemokine (C-C motif) ligand 2 (CCL2), without affecting the thromboxane levels in serum and urine 141. Future research utilizing TP knock-out mice is required to further validate the role of the TXA2 signaling via TP receptor in ischemic injury. Nonetheless, these promising findings from pharmacological studies suggest that the TP receptor antagonism could play an important role in the clinical prevention or treatment for cerebral ischemia (Table 3). Indeed, terutroban showed a better antithrombotic activity than that of aspirin and caused a significant reduction in endothelial/platelet activation in patients with previous cerebral ischemia and/or carotid stenosis in a small-scale double-blind, parallel-group study 142. However, a large-scale randomized, double-blind, parallel-group trial failed to show a superiority of terutroban over aspirin in secondary prevention of cardiovascular and cerebrovascular events among stroke patients with terutroban showing a slight increase in minor bleeding 143.
6. Gi-coupled prostanoid receptors
The DP2 and EP3 receptors are currently known prostanoid receptors that are primarily coupled to Gαi, and their activation leads to downregulation of the cAMP signaling (Figure 1); therefore, they are classified as “inhibitory” type of prostanoid receptors. However, they may also mediate other signaling pathways through the alternative splicing forms or some other unidentified mechanisms.
6.1. DP2 receptor
As the other currently known receptor activate by PGD2, the DP2 was originally identified as chemoattractant receptor-homologous molecule expressed on the T helper type 2 (Th2) cells (CRTH2) as a biomarker for Th2 cells 144. In the meantime, this proved to be the same as the orphan G protein-coupled receptor GPR44 expressed in both the CNS and periphery with a high similarity to typical chemoattractant receptors 145. PGD2 was subsequently identified as the natural ligand for the GPR44/CRTH2 receptor 146, and its current official name DP2 was thus introduced. The phylogenetic relationship analysis also suggest that the DP2 receptor belongs to the superfamily of leukocyte chemoattractant receptors and may not evolve from the same phylogenetic tree as other types of prostanoid receptors 147. The DP2 receptor primarily couples to the Gi in a manner similar to other chemoattractant receptors, as its activation by PGD2 leads to pertussis toxin-sensitive decreases in cAMP levels and Ca2+ mobilization (Figure 1). In addition, the DP2 receptor has been implicated in initiating the PI3K signaling 78. In the human body, the DP2 receptor is widely expressed in brain, colon, duodenum, heart, lung, placenta, intestine, stomach, and testis (https://www.ncbi.nlm.nih.gov/gene/11251). There were several studies demonstrating some detrimental and inflammatory effects mediated by PGD2 in the periphery mainly through the DP2 receptor 148–150. The selective DP2 agonist 13,14-dihydro-15-keto PGD2 (DK-PGD2) (Figure 5) increased, whereas the DP2 antagonist CAY10471 (Figure 5) decreased, the neuronal damage and Ca2+ mobilization in cultured rat hippocampal neurons treated with aluminum maltolate 80, suggesting a deleterious role of DP2 in neuronal injury. However, the genetic deletion of DP2 in mice has no considerable effect on the infarct volume in either cerebral cortex or basal ganglia after hypoxic ischemia (Table 4) 73. Future studies are needed to expand current limited understanding of the PGD2/DP2-mediated signaling transduction pathways, particularly in the pathogenesis of ischemic stroke.
Figure 5.
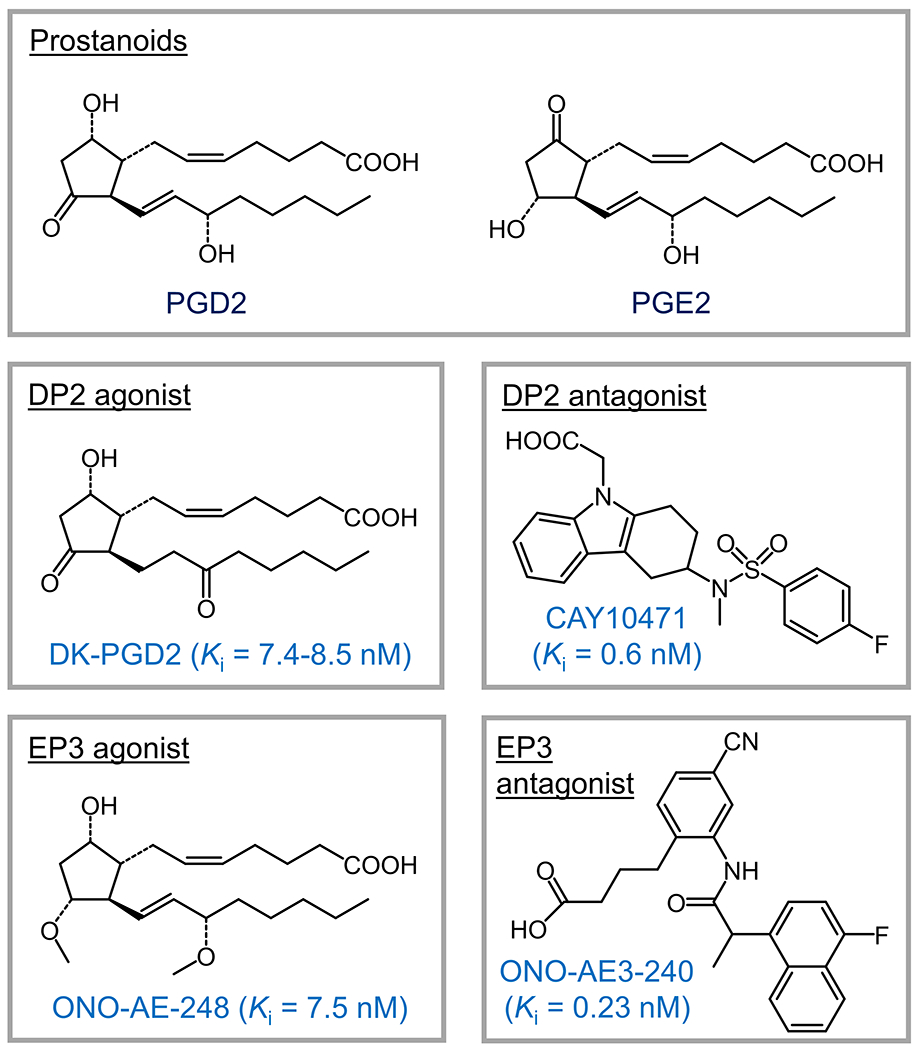
Small molecules targeting Gi-coupled prostanoid receptors. The Ki value of each compound is indicated.
Table 4.
Targeting Gi-coupled prostanoid receptors for experimental brain ischemia.
| Receptor | Animal model | Major therapeutic outcomes | Reference |
|---|---|---|---|
| DP2 | Hypoxic ischemia (30 min) induced in BALB/cAJcl neonatal mice (P7) | The ischemic lesion in cerebral cortex and basal ganlia was not changed in DP2 knock-out mice 24 hr after hypoxic ischemia. | Taniguchi et al., 2007 |
| EP3 | Transient MCAO (90 min) induced in adult male C57BL/6 mice | Treatment with EP3 agonist ONO-AE-248 (0.5, 2.5, and 5.0 nmol, i.c.v.) 50-60 min before MCAO exacerbated the infarct volume in a dose-dependent manner 4 days after MCAO, but did not affect core body temperature, cerebral blood flow, or mean arterial blood pressure. | Ahmad et al., 2007 |
| Transient MCAO (90 min) induced in adult male C57BL/6 mice | Genetic deletion of the EP3 receptor did not change the infarct volume or cerebral blood flow 24 hr after MCAO. | Li et al., 2008 | |
| Transient MCAO (90 min) induced in adult male C57BL/6 mice | EP3 deficiency decreased the infarct volume and neurological deficit 2 days but not 4 days after MCAO without affecting the mean arterial blood pressure, pH, blood gases (CO2 and O2), cerebral blood flow, or body temperature. | Saleem et al., 2009 | |
| Transient MCAO (120 min) induced in adult male and female C57BL/6 mice | Genetic deletion of mPGES-1 diminished the infarct volume, edema, and neurological deficit 24 hr after MCAO; treatment with EP3 selective antagonist ONO-AE3-240 (3 mg/kg, i.p., 3 doses at 2, 8, and 14 hr after MCAO) reduced infarct volume, edema, and neurological deficit 24 hr after MCAO in wild-type but not in mPGES-1 knockout mice. | Ikeda-Matsuo et al., 2010 | |
| Transient MCAO (120 min) induced in adult male and female C57BL/6 mice | EP3 deficiency or treatment with ONO-AE3-240 (1 or 3 mg/kg, i.p., 3 doses at 2, 8, and 14 hr after MCAO) decreased the infarct volume, edema, neurological deficit, neuronal apoptosis, and blood-brain barrier breakdown, preserved the tight junction proteins, and attenuated the microglial activation 24 hr after MCAO without affecting the cerebral blood flow, mean arterial blood pressure, rectal temperature, or brain vasculature. | Ikeda-Matsuo et al., 2011 |
6.2. EP3 receptor
The EP3 receptor subtype of PGE2 mainly couples to Gi for an inhibitory action on the adenylate cyclase, so its activation downregulates the cAMP-mediated signaling (Figure 1). EP3 has several alternative splicing forms that merely differ in their intracellular C-terminus, enabling the receptor to link with other G proteins in addition to Gi. In humans, the EP3-I and EP3-II forms also couple to Gq; the EP3-II and EP3-IV may be associated with Gs 151. Similarly, there are three EP3 variants in mice – α, β, and γ, among which the EP3γ variant appears to also connect with Gs. All α, β, and γ variants can couple to G12/13 to activate the Rho pathway and Rho-associated protein kinase (ROCK) 152. As such, the G protein complexes associated with EP3 receptor, in response to PGE2 binding, dissociate into Gαi, Gα12/13, Gαs, and Gβγ components that are capable of initiating multiple signaling pathways (Figure 1), depending on the cellular and molecular context. Therefore, a differential variant expression pattern of the EP3 is able to provide another level of mediation of the receptor-regulated signaling in different types of cells and tissues. Widely expressed in the human body (https://www.ncbi.nlm.nih.gov/gene/5733), these EP3 variants are particularly abundant in the CNS, cardiovasculature, reproductive system, intestinal epithelium, kidney, and urinary bladder, where they have been implicated in a wide range of physiological and pathological functions 78. The expression of EP3 receptor appears to be differentially regulated by excitotoxic insults, as it is upregulated by kainate-induced seizures 153, but not by glutamate or ischemic stroke 85. However, in the periphery, the EP3 receptor is upregulated together with COX-2 by uterine artery ischemia-reperfusion 154.
Owing to its well-known contribution to the ischemia-reperfusion injuries in peripheral organs, the EP3 receptor was believed to play a similar pathogenic role in the brain 154,155, inspiring studies on the receptor in experimental brain ischemia (Table 4). ONO-AE-248, an EP3-selective agonist (Figure 5), when administered intracerebroventricularly before a 90-minute ischemia in mice, increased the infarct volume without affecting the animal body temperature, cerebral blood flow, or arterial blood pressure 155. Interestingly, genetic deletion of the EP3 receptor in mice decreased the infarct volume and neurological dysfunction at 2 days after ischemia-reperfusion 108,156. In line, intraperitoneal administration of another EP3-selective antagonist ONO-AE3-240 (Figure 5) attenuated the infarct size, brain swelling, and neurological dysfunction following transient MCAO in mice without showing any sexual dimorphism 85. Given the less ischemic injury in mPGES-1 knock-out mice compared to the wild-type cohorts and the lack of benefit of ONO-AE3-240 in mPGES-1-deficient mice after transient MCAO, the detrimental effects mediated by mPGES-1 were thought to be at least partially attributed to the PGE2 signaling via the EP3 receptor. Interestingly, the genetic deficiency in EP3 or post-stroke inhibition of the receptor by ONO-AE3-240 in mice reduced the neuronal apoptosis, blood-brain barrier disruption, and microglial activation, as well as maintained the tight junction proteins in the neurovascular units after transient focal forebrain ischemia 157. Taken together, these results suggest that the EP3 receptor activation by PGE2 contributes to ischemia-reperfusion injury via enhancing the apoptotic and inflammatory processes. As such, pharmacological inhibition of the EP3 receptor with small-molecule compounds might provide a promising strategy to relieve brain injury triggered by acute brain ischemia (Table 4).
7. Concluding remarks and perspectives
During the past two decades, mounting concerns over the cardiovascular and cerebrovascular incidents caused by long-term use of COX-2 inhibitor drugs have motivated tremendous efforts for preclinical studies to determine the downstream prostanoid receptors that mediate the deleterious effects of COX-2 in various inflammation-associated conditions. In this regard, the acute brain ischemia is a hot topic for preclinical research owing to enormous impact of the disease and the lack of delayed treatment. Recent marked advances in chemical biology and genetic approaches for gene manipulation provided valuable tools to better understand the physiological and pathological functions of prostanoids and their receptors, which are found far more complex in ischemic injury than we originally believed.
7.1. The devil is in the detail
As the currently known prostanoid receptors that are coupled to Gαs, the DP1, EP2, EP4, and IP receptors mediate the excitatory signaling pathways via elevating the cytosol cAMP levels (Figure 1). Early studies consistently suggest that global deletion of these receptors exacerbated brain injury in various models of brain ischemia, whereas their activation by their cognate prostanoids or selective small-molecule agonists overall led to neuroprotective and other beneficial effects against ischemic injury (Table 2). However, using genetic strategies with more specificity recently revealed some discrepancy. Conditional deletion of the EP4 receptor in neurons increased the infarct volume 24 hours after MCAO 109; however, the neuronal EP2 ablation has recently been revealed to reduce the infarct volume 2 days after MCAO 19, and the results were validated by pharmacological inhibition of the receptor via two independent small-molecule antagonists 19,101. This incongruity can at least partially be explained by their downstream signaling pathways that might be involved in the regulation of ischemic injury, considering that the activation of Gαs-coupled receptors can lead to multiple signaling pathways: G protein-dependent PKA and EPAC pathways; G protein-independent β-arrestin signaling (Figure 1) 147. Intriguingly, systemic treatment with the EP4-selective agonist ONO-AE1-329 reversed the ischemic injury in the neuronal EP4 knockout mice 109, indicating a yet-to-be-identified molecular mechanism following the EP4 receptor activation. Future studies are required to determine the downstream effectors of Gs-coupled receptors that specifically mediate neuroprotection or deleterious effects following acute brain ischemia (Figure 1). It will also be important to examine the effects of the condition deletion of DP1 and IP receptors in neurons, microglia, and endothelial cells on ischemic injury, and one would not be surprised if the cell-type specific ablations of these two receptors lead to divergent outcomes as they did in the cases of EP2 and EP4.
7.2. Three is better than one
The genetic deletion of the three Gq-coupled prostanoid receptors – EP1, FP, and TP consistently led to decreases in the infarct volume, neurological deficit, and inflammatory damage after various types of ischemic injuries without affecting cerebral blood flow, core body temperature, or arterial blood pressure (Table 3). These broad beneficial effects suggest that inhibition of these “contractile” receptors by small molecules with well-defined pharmacodynamic and pharmacokinetic profiles might represent a promising strategy to treat acute brain ischemia. However, it should also be noted that genetic ablation or pharmacological inhibition of a single of these Gq-coupled receptors cannot fully eliminate the ischemic injury (Table 3), indicating synergistic effects from these prostanoid receptors on postischemic inflammation and damage. Given that their natural ligands PGE2, PGF2α, and TXA2 are all rapidly and robustly induced following acute brain ischemia, combined therapies utilizing small-molecule antagonists that concomitantly target these three receptors should be evaluated in the future for improved outcomes if not a full reverse of the ischemic injury.
7.3. Prostanoid synthases matter
As the inducible form of PGES, mPGES-1 is often co-expressed with COX-2 in neurons, endothelial cells, and microglia, and can be quickly elevated in the cerebral cortex and hippocampus by ischemic injury, contributing to the post-stroke cortical infarction and neurological dysfunction presumably through producing excessive PGE2 from COX-2-derived PGH2 (Figure 1) 82,158. The COX-2 inhibition increases risks in cardiovascular toxicity mainly because it may also lead to nonspecific suppression of the syntheses for other types of prostanoids that might play essential roles in normal physiological conditions, e.g., PGI2, a well-known important and effective vasodilator 60. As such, pharmacological inhibition of mPGES-1 might represent an alternative strategy to treat acute brain ischemia without affecting these beneficial prostanoids 159,160. The deficiency of the two PGD2 synthases – H-PGDS and L-PGDS exacerbated the postischemic lesion in both short term and long term after hypoxic ischemia, and the infarct size was in inverse relation to the PGD2 level in the brain (Table 2). Therefore, selective direct potentiation of H-PGDS and L-PGDS by small-molecule activators might provide a novel strategy to relieve the ischemic injury through elevating the PGD2 level in the ischemic brain. Because PGD2 mediates neuroprotection through the DP1 receptor, whereas the DP2 receptor has negligible implication in ischmic injury 73, increasing in PGD2 level would specifically lead to the DP1-mediate beneficail effects in the ischemic brain. As such, future studies should also be directed to determine the feasiblity targeting prostaglndin synthases, particulary the inducible mPGES-1 and H-PGDS (Figure 1), for new therapis to treat acute brain ischemia.
Acknowledgements
This work was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) grants R01NS100947 (J.J.) and R21NS109687 (J.J.).
REFERENCES
- 1.Di Carlo A Human and economic burden of stroke. Age and ageing. 2009;38:4–5. [DOI] [PubMed] [Google Scholar]
- 2.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosamond W, Flegal K, Furie K, et al. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. [DOI] [PubMed] [Google Scholar]
- 4.Writing Group M, Mozaffarian D, Benjamin EJ, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. [DOI] [PubMed] [Google Scholar]
- 5.Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. The New England journal of medicine. 2008;359:1317–1329. [DOI] [PubMed] [Google Scholar]
- 6.Emberson J, Lees KR, Lyden P, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet (London, England). 2014;384:1929–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab. 2004;24:945–963. [DOI] [PubMed] [Google Scholar]
- 8.Del Zoppo GJ, Saver JL, Jauch EC, Adams HP Jr., American Heart Association Stroke C. Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke. 2009;40:2945–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albers GW, Marks MP, Kemp S, et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. The New England journal of medicine. 2018;378:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saver JL, Goyal M, Bonafe A, et al. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. The New England journal of medicine. 2015;372:2285–2295. [DOI] [PubMed] [Google Scholar]
- 11.Balami JS, White PM, McMeekin PJ, Ford GA, Buchan AM. Complications of endovascular treatment for acute ischemic stroke: Prevention and management. International journal of stroke : official journal of the International Stroke Society. 2018;13:348–361. [DOI] [PubMed] [Google Scholar]
- 12.Jadhav AP, Molyneaux BJ, Hill MD, Jovin TG. Care of the Post-Thrombectomy Patient. Stroke. 2018;49:2801–2807. [DOI] [PubMed] [Google Scholar]
- 13.Cheng YD, Al-Khoury L, Zivin JA. Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2004;1:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Wang K. The fate of medications evaluated for ischemic stroke pharmacotherapy over the period 1995–2015. Acta pharmaceutica Sinica B. 2016;6:522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CJ, Denes A, Tyrrell PJ, Di Napoli M. Phase II anti-inflammatory and immune-modulating drugs for acute ischaemic stroke. Expert Opin Investig Drugs. 2015;24:623–643. [DOI] [PubMed] [Google Scholar]
- 16.Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. Journal of neuroinflammation. 2019;16:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turner RJ, Sharp FR. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci. 2016;10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hafez S, Abdelsaid M, Fagan SC, Ergul A. Peroxynitrite-Induced Tyrosine Nitration Contributes to Matrix Metalloprotease-3 Activation: Relevance to Hyperglycemic Ischemic Brain Injury and Tissue Plasminogen Activator. Neurochem Res. 2018;43:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Liang X, Wang Q, et al. PGE2 signaling via the neuronal EP2 receptor increases injury in a model of cerebral ischemia. Proc Natl Acad Sci U S A. 2019;116:10019–10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang C, Hawkins KE, Dore S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2019;316:C135–C153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 22.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jickling GC, Liu D, Stamova B, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci. 2014;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura K, Shichita T. Cellular and molecular mechanisms of sterile inflammation in ischaemic stroke. J Biochem. 2019;165:459–464. [DOI] [PubMed] [Google Scholar]
- 27.Guan YZ, Jin XD, Guan LX, et al. Nicotine inhibits microglial proliferation and is neuroprotective in global ischemia rats. Mol Neurobiol. 2015;51:1480–1488. [DOI] [PubMed] [Google Scholar]
- 28.Eldahshan W, Fagan SC, Ergul A. Inflammation within the neurovascular unit: Focus on microglia for stroke injury and recovery. Pharmacological research. 2019;147:104349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connolly ES Jr, Winfree CJ, Springer TA, et al. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gelderblom M, Weymar A, Bernreuther C, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793–3802. [DOI] [PubMed] [Google Scholar]
- 31.Parmentier S, Bohme GA, Lerouet D, et al. Selective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. Br J Pharmacol. 1999;127:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nash KM, Schiefer IT, Shah ZA. Development of a reactive oxygen species-sensitive nitric oxide synthase inhibitor for the treatment of ischemic stroke. Free Radic Biol Med. 2018;115:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veltkamp R, Gill D. Clinical Trials of Immunomodulation in Ischemic Stroke. Neurotherapeutics. 2016;13:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong SL, Leung FP, Lau CW, et al. Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ Res. 2009;104:228–235. [DOI] [PubMed] [Google Scholar]
- 35.Wong WT, Tian XY, Chen Y, et al. Bone morphogenic protein-4 impairs endothelial function through oxidative stress-dependent cyclooxygenase-2 upregulation: implications on hypertension. Circ Res. 2010;107:984–991. [DOI] [PubMed] [Google Scholar]
- 36.Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a platelet-activating factor antagonist. J Biol Chem. 1996;271:24794–24799. [DOI] [PubMed] [Google Scholar]
- 38.Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakayama M, Uchimura K, Zhu RL, et al. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci U S A. 1998;95:10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du Y, Kemper T, Qiu J, Jiang J. Defining the therapeutic time window for suppressing the inflammatory prostaglandin E2 signaling after status epilepticus. Expert review of neurotherapeutics. 2016;16:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartings JA, York J, Carroll CP, et al. Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain. 2017;140:2673–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iadecola C, Forster C, Nogawa S, Clark HB, Ross ME. Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta neuropathologica. 1999;98:9–14. [DOI] [PubMed] [Google Scholar]
- 43.Brose SA, Thuen BT, Golovko MY. LC/MS/MS method for analysis of E(2) series prostaglandins and isoprostanes. Journal of lipid research. 2011;52:850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brose SA, Baker AG, Golovko MY. A fast one-step extraction and UPLC-MS/MS analysis for E2/D 2 series prostaglandins and isoprostanes. Lipids. 2013;48:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seeger DR, Golovko SA, Golovko MY. Blood-Brain Barrier Is the Major Site for a Rapid and Dramatic Prostanoid Increase upon Brain Global Ischemia. Lipids. 2020;55:79–85. [DOI] [PubMed] [Google Scholar]
- 46.Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A. 1991;88:2692–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flower RJ. The development of COX2 inhibitors. Nature reviews Drug discovery. 2003;2:179–191. [DOI] [PubMed] [Google Scholar]
- 48.Pereira-Leite C, Nunes C, Jamal SK, Cuccovia IM, Reis S. Nonsteroidal Anti-Inflammatory Therapy: A Journey Toward Safety. Medicinal research reviews. 2017;37:802–859. [DOI] [PubMed] [Google Scholar]
- 49.Sugimoto K, Iadecola C. Delayed effect of administration of COX-2 inhibitor in mice with acute cerebral ischemia. Brain research. 2003;960:273–276. [DOI] [PubMed] [Google Scholar]
- 50.Iadecola C, Niwa K, Nogawa S, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci U S A. 2001;98:1294–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dore S, Otsuka T, Mito T, et al. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol. 2003;54:155–162. [DOI] [PubMed] [Google Scholar]
- 52.Park EM, Cho BP, Volpe BT, Cruz MO, Joh TH, Cho S. Ibuprofen protects ischemia-induced neuronal injury via up-regulating interleukin-1 receptor antagonist expression. Neuroscience. 2005;132:625–631. [DOI] [PubMed] [Google Scholar]
- 53.Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–2724. [DOI] [PubMed] [Google Scholar]
- 54.Institoris A, Farkas E, Berczi S, Sule Z, Bari F. Effects of cyclooxygenase (COX) inhibition on memory impairment and hippocampal damage in the early period of cerebral hypoperfusion in rats. Eur J Pharmacol. 2007;574:29–38. [DOI] [PubMed] [Google Scholar]
- 55.Silasi G, Kolb B. Chronic inhibition of cyclooxygenase-2 induces dendritic hypertrophy and limited functional improvement following motor cortex stroke. Neuroscience. 2007;144:1160–1168. [DOI] [PubMed] [Google Scholar]
- 56.Rojas A, Jiang J, Ganesh T, et al. Cyclooxygenase-2 in epilepsy. Epilepsia. 2014;55:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dey A, Kang X, Qiu J, Du Y, Jiang J. Anti-Inflammatory Small Molecules To Treat Seizures and Epilepsy: From Bench to Bedside. Trends in pharmacological sciences. 2016;37:463–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dhir A An update of cyclooxygenase (COX)-inhibitors in epilepsy disorders. Expert Opin Investig Drugs. 2019;28:191–205. [DOI] [PubMed] [Google Scholar]
- 59.Yu Y, Nguyen DT, Jiang J. G protein-coupled receptors in acquired epilepsy: Druggability and translatability. Prog Neurobiol. 2019;183:101682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaudet RJ, Alam I, Levine L. Accumulation of cyclooxygenase products of arachidonic acid metabolism in gerbil brain during reperfusion after bilateral common carotid artery occlusion. Journal of neurochemistry. 1980;35:653–658. [DOI] [PubMed] [Google Scholar]
- 62.Pappius HM, Wolfe LS. Effects of indomethacin and ibuprofen on cerebral metabolism and blood flow in traumatized brain. J Cereb Blood Flow Metab. 1983;3:448–459. [DOI] [PubMed] [Google Scholar]
- 63.Carasso RL, Vardi J, Rabay JM, Zor U, Streifler M. Measurement of prostaglandin E2 in cerebrospinal fluid in patients suffering from stroke. Journal of neurology, neurosurgery, and psychiatry. 1977;40:967–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katz B, Sofonio M, Lyden PD, Mitchell MD. Prostaglandin concentrations in cerebrospinal fluid of rabbits under normal and ischemic conditions. Stroke. 1988;19:349–351. [DOI] [PubMed] [Google Scholar]
- 65.Fang YC, Wu JS, Chen JJ, et al. Induction of prostacyclin/PGI2 synthase expression after cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2006;26:491–501. [DOI] [PubMed] [Google Scholar]
- 66.Yu L, Yang B, Wang J, et al. Time course change of COX2-PGI2/TXA2 following global cerebral ischemia reperfusion injury in rat hippocampus. Behavioral and brain functions : BBF. 2014;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu Y, Jiang J. COX-2/PGE2 axis regulates hippocampal BDNF/TrkB signaling via EP2 receptor after prolonged seizures. Epilepsia Open. 2020;5:418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem. 2013;288:9293–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chang MC, Lin SI, Lin LD, et al. Prostaglandin E2 Stimulates EP2, Adenylate Cyclase, Phospholipase C, and Intracellular Calcium Release to Mediate Cyclic Adenosine Monophosphate Production in Dental Pulp Cells. J Endod. 2016;42:584–588. [DOI] [PubMed] [Google Scholar]
- 71.Jiang J, Qiu J, Li Q, Shi Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends in cancer. 2017;3:75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huttemeier PC, Kamiyama Y, Su M, Watkins WD, Benveniste H. Microdialysis measurements of PGD2, TXB2 and 6-KETO-PGF1 alpha in rat CA1 hippocampus during transient cerebral ischemia. Prostaglandins. 1993;45:177–187. [DOI] [PubMed] [Google Scholar]
- 73.Taniguchi H, Mohri I, Okabe-Arahori H, et al. Prostaglandin D2 protects neonatal mouse brain from hypoxic ischemic injury. J Neurosci. 2007;27:4303–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mohri I, Eguchi N, Suzuki K, Urade Y, Taniike M. Hematopoietic prostaglandin D synthase is expressed in microglia in the developing postnatal mouse brain. Glia. 2003;42:263–274. [DOI] [PubMed] [Google Scholar]
- 75.Post JM, Loch S, Lerner R, et al. Antiepileptogenic Effect of Subchronic Palmitoylethanolamide Treatment in a Mouse Model of Acute Epilepsy. Frontiers in molecular neuroscience. 2018;11:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Urade Y, Eguchi N, Hayaishi O. Lipocalin-type prostaglandin D synthase as an enzymic lipocalin. In: Madame Curie Bioscience Database [Internet]. Landes Bioscience; 2013. [Google Scholar]
- 77.Garza LA, Liu Y, Yang Z, et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Science translational medicine. 2012;4:126ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woodward DF, Jones RL, Narumiya S. International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacological reviews. 2011;63:471–538. [DOI] [PubMed] [Google Scholar]
- 79.Liang X, Wu L, Hand T, Andreasson K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. Journal of neurochemistry. 2005;92:477–486. [DOI] [PubMed] [Google Scholar]
- 80.Ma J, Yang Q, Wei Y, et al. Effect of the PGD2-DP signaling pathway on primary cultured rat hippocampal neuron injury caused by aluminum overload. Sci Rep. 2016;6:24646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saleem S, Zhuang H, de Brum-Fernandes AJ, Maruyama T, Narumiya S, Dore S. PGD(2) DP1 receptor protects brain from ischemia-reperfusion injury. Eur J Neurosci. 2007;26:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ikeda-Matsuo Y, Ota A, Fukada T, Uematsu S, Akira S, Sasaki Y. Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:11790–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cimino PJ, Keene CD, Breyer RM, Montine KS, Montine TJ. Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr Med Chem. 2008;15:1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stevens MK, Yaksh TL. Time course of release in vivo of PGE2, PGF2 alpha, 6-keto-PGF1 alpha, and TxB2 into the brain extracellular space after 15 min of complete global ischemia in the presence and absence of cyclooxygenase inhibition. J Cereb Blood Flow Metab. 1988;8:790–798. [DOI] [PubMed] [Google Scholar]
- 85.Ikeda-Matsuo Y, Tanji H, Ota A, et al. Microsomal prostaglandin E synthase-1 contributes to ischaemic excitotoxicity through prostaglandin E2 EP3 receptors. Br J Pharmacol. 2010;160:847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug discovery today. 2017;22:57–71. [DOI] [PubMed] [Google Scholar]
- 87.Jiang J, Yang MS, Quan Y, Gueorguieva P, Ganesh T, Dingledine R. Therapeutic window for cyclooxygenase-2 related anti-inflammatory therapy after status epilepticus. Neurobiol Dis. 2015;76:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fu Y, Yang MS, Jiang J, Ganesh T, Joe E, Dingledine R. EP2 Receptor Signaling Regulates Microglia Death. Molecular pharmacology. 2015;88:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McCullough L, Wu L, Haughey N, et al. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang J, Van TM, Ganesh T, Dingledine R. Discovery of 2-Piperidinyl Phenyl Benzamides and Trisubstituted Pyrimidines as Positive Allosteric Modulators of the Prostaglandin Receptor EP2. ACS Chem Neurosci. 2018;9:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang J, Ganesh T, Du Y, et al. Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc Natl Acad Sci U S A. 2010;107:2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. [DOI] [PubMed] [Google Scholar]
- 93.Ahmad M, Saleem S, Shah Z, Maruyama T, Narumiya S, Dore S. The PGE2 EP2 receptor and its selective activation are beneficial against ischemic stroke. Exp Transl Stroke Med. 2010;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takadera T, Ohyashiki T. Prostaglandin E2 deteriorates N-methyl-D-aspartate receptor-mediated cytotoxicity possibly by activating EP2 receptors in cultured cortical neurons. Life Sci. 2006;78:1878–1883. [DOI] [PubMed] [Google Scholar]
- 95.Jiang J, Ganesh T, Du Y, et al. Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proc Natl Acad Sci U S A. 2012;109:3149–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang J, Quan Y, Ganesh T, Pouliot WA, Dudek FE, Dingledine R. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc Natl Acad Sci U S A. 2013;110:3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kang X, Qiu J, Li Q, et al. Cyclooxygenase-2 contributes to oxidopamine-mediated neuronal inflammation and injury via the prostaglandin E2 receptor EP2 subtype. Sci Rep. 2017;7:9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang J, Yu Y, Kinjo ER, Du Y, Nguyen HP, Dingledine R. Suppressing pro-inflammatory prostaglandin signaling attenuates excitotoxicity-associated neuronal inflammation and injury. Neuropharmacology. 2019;149:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qiu J, Li Q, Bell KA, et al. Small-molecule inhibition of prostaglandin E receptor 2 impairs cyclooxygenase-associated malignant glioma growth. Br J Pharmacol. 2019;176:1680–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nagib MM, Yu Y, Jiang J. Targeting prostaglandin receptor EP2 for adjunctive treatment of status epilepticus. Pharmacology & therapeutics. 2020;209:107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li L, Yu Y, Hou R, Hao J, Jiang J. Inhibiting the PGE2 Receptor EP2 Mitigates Excitotoxicity and Ischemic Injury. ACS Pharmacology & Translational Science. 2020;3:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hizaki H, Segi E, Sugimoto Y, et al. Abortive expansion of the cumulus and impaired fertility in mice lacking the prostaglandin E receptor subtype EP(2). Proc Natl Acad Sci U S A. 1999;96:10501–10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kennedy CR, Zhang Y, Brandon S, et al. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5:217–220. [DOI] [PubMed] [Google Scholar]
- 104.Tilley SL, Audoly LP, Hicks EH, et al. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999;103:1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Savonenko A, Munoz P, Melnikova T, et al. Impaired cognition, sensorimotor gating, and hippocampal long-term depression in mice lacking the prostaglandin E2 EP2 receptor. Experimental neurology. 2009;217:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang H, Zhang J, Breyer RM, Chen C. Altered hippocampal long-term synaptic plasticity in mice deficient in the PGE2 EP2 receptor. Journal of neurochemistry. 2009;108:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Majumder M, Xin X, Liu L, et al. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem cells (Dayton, Ohio). 2016;34:2290–2305. [DOI] [PubMed] [Google Scholar]
- 108.Li J, Liang X, Wang Q, Breyer RM, McCullough L, Andreasson K. Misoprostol, an anti-ulcer agent and PGE2 receptor agonist, protects against cerebral ischemia. Neurosci Lett. 2008;438:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liang X, Lin L, Woodling NS, et al. Signaling via the prostaglandin E(2) receptor EP4 exerts neuronal and vascular protection in a mouse model of cerebral ischemia. J Clin Invest. 2011;121:4362–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Akram A, Gibson CL, Grubb BD. Neuroprotection mediated by the EP(4) receptor avoids the detrimental side effects of COX-2 inhibitors following ischaemic injury. Neuropharmacology. 2013;65:165–172. [DOI] [PubMed] [Google Scholar]
- 111.DeMars KM, McCrea AO, Siwarski DM, Sanz BD, Yang C, Candelario-Jalil E. Protective Effects of L-902,688, a Prostanoid EP4 Receptor Agonist, against Acute Blood-Brain Barrier Damage in Experimental Ischemic Stroke. Front Neurosci. 2018;12:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gryglewski RJ, Dembinska-Kiec A, Korbut R. A possible role of thromboxane A2 (TXA2) and prostacyclin (PGI2) in circulation. Acta biologica et medica Germanica. 1978;37:715–723. [PubMed] [Google Scholar]
- 114.Kainoh M, Ueno S, Endoh T, Nakadate T. Inhibitory effect of beraprost sodium on formation of lipid peroxides in ischemia and recirculation-induced cerebral injury. Pharmacological research. 1993;28:249–258. [DOI] [PubMed] [Google Scholar]
- 115.Matsuda S, Wen TC, Karasawa Y, et al. Protective effect of a prostaglandin I2 analog, TEI-7165, on ischemic neuronal damage in gerbils. Brain research. 1997;769:321–328. [DOI] [PubMed] [Google Scholar]
- 116.Saleem S, Shah ZA, Maruyama T, Narumiya S, Dore S. Neuroprotective properties of prostaglandin I2 IP receptor in focal cerebral ischemia. Neuroscience. 2010;170:317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shakil H, Saleem S. Genetic Deletion of Prostacyclin IP Receptor Exacerbates Transient Global Cerebral Ischemia in Aging Mice. Brain sciences. 2013;3:1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shakil H, Saleem S. Prostaglandin I2 IP Receptor Agonist, Beraprost, Prevents Transient Global Cerebral Ischemia Induced Hippocampal CA1 Injury in Aging Mice. Journal of neurological disorders. 2014;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yagami T, Koma H, Yamamoto Y. Pathophysiological Roles of Cyclooxygenases and Prostaglandins in the Central Nervous System. Mol Neurobiol. 2016;53:4754–4771. [DOI] [PubMed] [Google Scholar]
- 120.Frankowski JC, DeMars KM, Ahmad AS, et al. Detrimental role of the EP1 prostanoid receptor in blood-brain barrier damage following experimental ischemic stroke. Sci Rep. 2015;5:17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Caggiano AO, Kraig RP. Prostaglandin E receptor subtypes in cultured rat microglia and their role in reducing lipopolysaccharide-induced interleukin-1beta production. Journal of neurochemistry. 1999;72:565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kawano T, Anrather J, Zhou P, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. [DOI] [PubMed] [Google Scholar]
- 123.Li XM, Yang JM, Hu DH, et al. Contribution of downregulation of L-type calcium currents to delayed neuronal death in rat hippocampus after global cerebral ischemia and reperfusion. J Neurosci. 2007;27:5249–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hu HH, Li SJ, Wang P, et al. An L-type calcium channel agonist, bay K8644, extends the window of intervention against ischemic neuronal injury. Mol Neurobiol. 2013;47:280–289. [DOI] [PubMed] [Google Scholar]
- 125.Ahmad AS, Saleem S, Ahmad M, Dore S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–270. [DOI] [PubMed] [Google Scholar]
- 126.Saleem S, Li RC, Wei G, Dore S. Effects of EP1 receptor on cerebral blood flow in the middle cerebral artery occlusion model of stroke in mice. J Neurosci Res. 2007;85:2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ahmad AS, Yun YT, Ahmad M, Maruyama T, Dore S. Selective blockade of PGE2 EP1 receptor protects brain against experimental ischemia and excitotoxicity, and hippocampal slice cultures against oxygen-glucose deprivation. Neurotox Res. 2008;14:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Abe T, Kunz A, Shimamura M, Zhou P, Anrather J, Iadecola C. The neuroprotective effect of prostaglandin E2 EP1 receptor inhibition has a wide therapeutic window, is sustained in time and is not sexually dimorphic. J Cereb Blood Flow Metab. 2009;29:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Suzuki-Yamamoto T, Nishizawa M, Fukui M, et al. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS letters. 1999;462:335–340. [DOI] [PubMed] [Google Scholar]
- 130.Basu S Novel cyclooxygenase-catalyzed bioactive prostaglandin F2alpha from physiology to new principles in inflammation. Medicinal research reviews. 2007;27:435–468. [DOI] [PubMed] [Google Scholar]
- 131.Abramovitz M, Boie Y, Nguyen T, et al. Cloning and expression of a cDNA for the human prostanoid FP receptor. J Biol Chem. 1994;269:2632–2636. [PubMed] [Google Scholar]
- 132.Saleem S, Ahmad AS, Maruyama T, Narumiya S, Dore S. PGF(2alpha) FP receptor contributes to brain damage following transient focal brain ischemia. Neurotox Res. 2009;15:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim YT, Moon SK, Maruyama T, Narumiya S, Dore S. Prostaglandin FP receptor inhibitor reduces ischemic brain damage and neurotoxicity. Neurobiol Dis. 2012;48:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamamoto K, Ebina S, Nakanishi H, Nakahata N. Thromboxane A2 receptor-mediated signal transduction in rabbit aortic smooth muscle cells. General pharmacology. 1995;26:1489–1498. [DOI] [PubMed] [Google Scholar]
- 135.Ding X, Murray PA. Cellular mechanisms of thromboxane A2-mediated contraction in pulmonary veins. American journal of physiology Lung cellular and molecular physiology. 2005;289:L825–833. [DOI] [PubMed] [Google Scholar]
- 136.Tian XY, Wong WT, Leung FP, et al. Oxidative stress-dependent cyclooxygenase-2-derived prostaglandin f(2alpha) impairs endothelial function in renovascular hypertensive rats. Antioxidants & redox signaling. 2012;16:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nishimaki S, Seki K. An imbalance between prostacyclin and thromboxane in relation to cerebral blood flow in neonates with maternal preeclampsia. Prostaglandins Other Lipid Mediat. 1999;58:43–49. [DOI] [PubMed] [Google Scholar]
- 138.Kaneko Y, Nakayama T, Saito K, et al. Relationship between the thromboxane A2 receptor gene and susceptibility to cerebral infarction. Hypertension research : official journal of the Japanese Society of Hypertension. 2006;29:665–671. [DOI] [PubMed] [Google Scholar]
- 139.Yang W, Yan A, Zhang T, et al. Thromboxane A2 Receptor Stimulation Enhances Microglial Interleukin-1beta and NO Biosynthesis Mediated by the Activation of ERK Pathway. Front Aging Neurosci. 2016;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yan A, Zhang T, Yang X, et al. Thromboxane A2 receptor antagonist SQ29548 reduces ischemic stroke-induced microglia/macrophages activation and enrichment, and ameliorates brain injury. Sci Rep. 2016;6:35885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gelosa P, Ballerio R, Banfi C, et al. Terutroban, a thromboxane/prostaglandin endoperoxide receptor antagonist, increases survival in stroke-prone rats by preventing systemic inflammation and endothelial dysfunction: comparison with aspirin and rosuvastatin. J Pharmacol Exp Ther. 2010;334:199–205. [DOI] [PubMed] [Google Scholar]
- 142.Bal Dit Sollier C, Crassard I, Simoneau G, Bergmann JF, Bousser MG, Drouet L. Effect of the thromboxane prostaglandin receptor antagonist terutroban on arterial thrombogenesis after repeated administration in patients treated for the prevention of ischemic stroke. Cerebrovascular diseases (Basel, Switzerland). 2009;28:505–513. [DOI] [PubMed] [Google Scholar]
- 143.Bousser MG, Amarenco P, Chamorro A, et al. Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet (London, England). 2011;377:2013–2022. [DOI] [PubMed] [Google Scholar]
- 144.Nagata K, Tanaka K, Ogawa K, et al. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999;162:1278–1286. [PubMed] [Google Scholar]
- 145.Marchese A, Sawzdargo M, Nguyen T, et al. Discovery of three novel orphan G-protein-coupled receptors. Genomics. 1999;56:12–21. [DOI] [PubMed] [Google Scholar]
- 146.Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. The Journal of experimental medicine. 2001;193:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hirata T, Narumiya S. Prostanoid receptors. Chem Rev. 2011;111:6209–6230. [DOI] [PubMed] [Google Scholar]
- 148.Nagata K, Hirai H. The second PGD(2) receptor CRTH2: structure, properties, and functions in leukocytes. Prostaglandins, leukotrienes, and essential fatty acids. 2003;69:169–177. [DOI] [PubMed] [Google Scholar]
- 149.Kagawa S, Fukunaga K, Oguma T, et al. Role of prostaglandin D2 receptor CRTH2 in sustained eosinophil accumulation in the airways of mice with chronic asthma. International archives of allergy and immunology. 2011;155 Suppl 1:6–11. [DOI] [PubMed] [Google Scholar]
- 150.Almishri W, Cossette C, Rokach J, Martin JG, Hamid Q, Powell WS. Effects of prostaglandin D2, 15-deoxy-Delta12,14-prostaglandin J2, and selective DP1 and DP2 receptor agonists on pulmonary infiltration of eosinophils in Brown Norway rats. J Pharmacol Exp Ther. 2005;313:64–69. [DOI] [PubMed] [Google Scholar]
- 151.Kotani M, Tanaka I, Ogawa Y, et al. Molecular cloning and expression of multiple isoforms of human prostaglandin E receptor EP3 subtype generated by alternative messenger RNA splicing: multiple second messenger systems and tissue-specific distributions. Molecular pharmacology. 1995;48:869–879. [PubMed] [Google Scholar]
- 152.Macias-Perez IM, Zent R, Carmosino M, Breyer MD, Breyer RM, Pozzi A. Mouse EP3 alpha, beta, and gamma receptor variants reduce tumor cell proliferation and tumorigenesis in vivo. J Biol Chem. 2008;283:12538–12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Takemiya T, Matsumura K, Sugiura H, et al. Endothelial microsomal prostaglandin E synthase-1 exacerbates neuronal loss induced by kainate. J Neurosci Res. 2010;88:381–390. [DOI] [PubMed] [Google Scholar]
- 154.Yamazaki K, Endo T, Kitajima Y, et al. Elevation of both cyclooxygenase-2 and prostaglandin E2 receptor EP3 expressions in rat placenta after uterine artery ischemia-reperfusion. Placenta. 2006;27:395–401. [DOI] [PubMed] [Google Scholar]
- 155.Ahmad M, Ahmad AS, Zhuang H, Maruyama T, Narumiya S, Dore S. Stimulation of prostaglandin E2-EP3 receptors exacerbates stroke and excitotoxic injury. J Neuroimmunol. 2007;184:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Saleem S, Kim YT, Maruyama T, Narumiya S, Dore S. Reduced acute brain injury in PGE2 EP3 receptor-deficient mice after cerebral ischemia. J Neuroimmunol. 2009;208:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ikeda-Matsuo Y, Tanji H, Narumiya S, Sasaki Y. Inhibition of prostaglandin E2 EP3 receptors improves stroke injury via anti-inflammatory and anti-apoptotic mechanisms. J Neuroimmunol. 2011;238:34–43. [DOI] [PubMed] [Google Scholar]
- 158.Ikeda-Matsuo Y, Hirayama Y, Ota A, Uematsu S, Akira S, Sasaki Y. Microsomal prostaglandin E synthase-1 and cyclooxygenase-2 are both required for ischaemic excitotoxicity. Br J Pharmacol. 2010;159:1174–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bahia MS, Katare YK, Silakari O, Vyas B, Silakari P. Inhibitors of microsomal prostaglandin E2 synthase-1 enzyme as emerging anti-inflammatory candidates. Medicinal research reviews. 2014;34:825–855. [DOI] [PubMed] [Google Scholar]
- 160.Ikeda-Matsuo Y The Role of mPGES-1 in Inflammatory Brain Diseases. Biological & pharmaceutical bulletin. 2017;40:557–563. [DOI] [PubMed] [Google Scholar]