

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly malignant disease, where even surgical resection and aggressive chemotherapy produce dismal outcomes. Immunotherapy is a promising alternative to conventional treatments, possessing the ability to elicit T cell-mediated killing of tumor cells and prevent disease recurrence. Immunotherapeutic approaches thus far have seen limited success in PDAC due to a poorly immunogenic and exceedingly immunosuppressive tumor microenvironment, which is enriched with dysfunctional and immunosuppressed antigen-presenting cells (APCs). We developed a highly potent immunostimulatory nanoparticle (immuno-NP) to activate and expand APCs in the tumor and induce local secretion of interferon β (IFNβ), which is a pro-inflammatory cytokine that plays a major role in APC recruitment. The effectiveness of the immuno-NP stems from its dual cargo of two synergistic immune modulators consisting of an agonist of the stimulator of interferon genes (STING) pathway and an agonist of the Toll-like receptor 4 (TLR4) pathway. We show the functional synergy of the dual-agonist cargo can be tweaked by adjusting the ratio of the two agonists loaded in the immuno-NP, leading to an increase in IFNβ production (11-fold) compared to any single agonist immuno-NP variant. Using the orthotopic murine Panc02 model of PDAC, we show that systemic administration allowed immuno-NPs to deposit into the perivascular regions of the tumor, which coincided with the APC-rich tumor areas leading to predominant uptake of immuno-NPs by APCs. The immuno-NPs were effectively taken up by a significant portion of dendritic cells in the tumor (>56%). This led to a significant expansion of APCs, resulting in an 11.5-fold increase of dendritic cells and infiltration of lymphocytes throughout the pancreatic tumor compared to untreated animals.
Keywords: cancer immunotherapy, immunostimulatory nanoparticles, pancreatic cancer, systemic delivery, STING and TLR4 agonists, interferon β
Graphical Abstract

INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers characterized by late diagnosis, metastatic spread, chemoresistance and limited treatment options [1, 2]. First-line chemotherapy treatments offer little survival benefit, and even patients who undergo surgery have a high rate of disease recurrence [3]. Cancer immunotherapy has an inherent premise to treat the drug-resistant and metastatic forms of PDAC by eliciting T cell-mediated killing of tumor cells. Immune checkpoint inhibitors, such as anti-PD-1 and anti-CTLA-4 monoclonal antibodies, have received significant attention in recent years, following positive results and FDA approval in other solid tumors, such as melanoma [4–7] and lung cancer [8]. However, immune checkpoint blockade strategies have shown limited success in clinical trials of pancreatic cancer as either monotherapies or in combination with standard chemo- or radiotherapies [9–12]. This stems from the fact that the tumor microenvironment (TME) of PDAC is highly immunosuppressive [13]. Studies have shown that leukocytes can comprise nearly 50% of the cellular component in pancreatic tumors, many of which promote tumor growth and metastasis [14–17]. By producing various cytokines and growth factors, PDAC cells, along with supporting pancreatic stellate cells and cancer-associated fibroblasts, are responsible for re-educating innate and adaptive immune cells into an immunosuppressive phenotype [15, 18, 19]. As a result, the TME of PDAC is enriched with dysfunctional and immunosuppressive antigen-presenting cells (APCs), including dendritic cells (DC) and tumor-associated macrophages (TAMs), and other immunosuppressive cells, namely myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) [18, 20–24]. Due to this highly immunosuppressive TME and the frequent exclusion of cytotoxic T cells, PDAC is characterized as a “cold” tumor, which allows cancer cells to avoid detection by the immune system. An effective approach that targets the immunosuppressive pancreatic TME can facilitate an antitumor response leading to tumor rejection and prevention of disease relapse.
While most immunotherapies focus on the adaptive immune system, we designed a highly potent immuno-stimulatory nanoparticle (immuno-NP) with a focus on modulating the local tumor innate immunity of PDAC. The immuno-NP aims at triggering robust activation and expansion of APCs in the TME, as APCs initiate tumor recognition and present tumor-associated antigens to naïve T cells, which can then launch a tumor-specific cytotoxic response. While traditional approaches for APC activation utilize a single immune-potentiating molecule, nanoparticles can deliver one or more immune-stimulating agonists to the same cell with greater efficacy than free molecules [25, 26]. Specifically, immuno-NPs simultaneously activate two functionally synergistic innate immune pathways by co-delivering two immune agonists (Fig. 1A), both of which are strong inducers of Type I interferons (IFNs) [25]. Type I IFNs, such as IFNβ, are highly potent cytokines that promote recruitment of innate immune cells and activation of T-cell priming [27, 28]. The dual cargo of the immuno-NP includes cyclic diguanylate monophosphate (cdGMP), an agonist of the stimulator of interferon genes (STING) pathway [29], and monophosphoryl lipid A (MPLA), a Toll-like receptor 4 (TLR4) pathway agonist [30]. The 60-nm immuno-NP can be conveniently and stably loaded with the two agonists by encapsulating the hydrophilic cdGMP within the particle core and embedding the hydrophobic MPLA within a lipid bilayer (Fig. 1B). By co-loading the two immune agonists in the same nanoparticle, we ensure uptake of both agonists by the same APC, eliciting remarkable synergy [25].
Fig. 1.
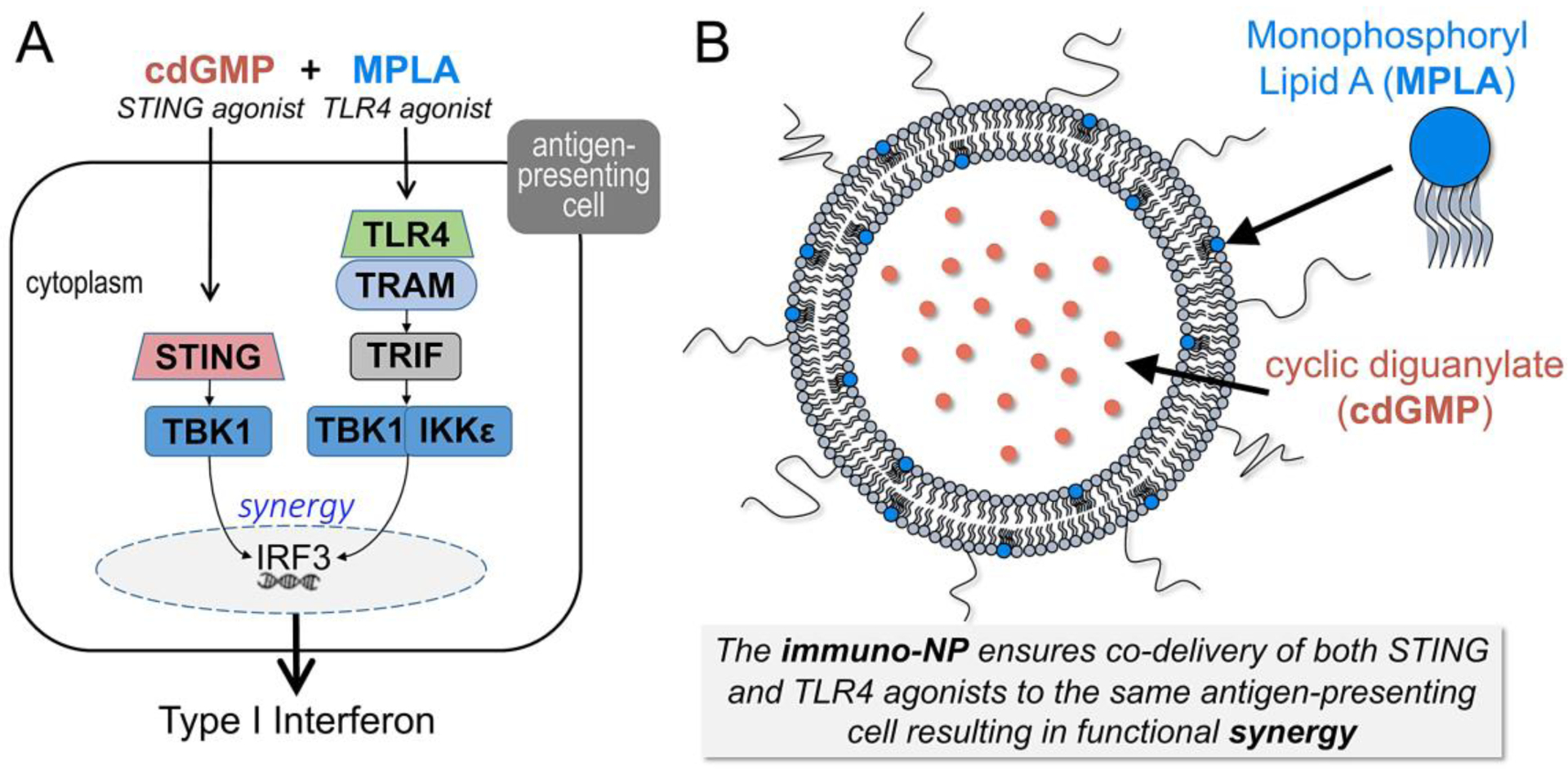
Illustration shows (A) the functional synergy of the STING agonist (cdGMP) and TLR4 agonist (MPLA) to activate the signaling pathways leading to the induction of potent cytokines including Type I interferons, and (B) the immunostimulatory nanoparticle loaded with the dual-agonist cargo of cdGMP and MPLA.
Further, compared to local intratumoral injection, systemic administration allows immuno-NPs to utilize the entire tumor microvasculature and drain into the perivascular regions, which coincide with the APC-rich tumor areas leading to predominant uptake of immuno-NPs by these target subset of immune cells. Systemically-administered nanoparticles loaded with cancer drugs are typically unable to penetrate the dense desmoplastic stroma of pancreatic tumors, resulting in deposition along the tumor vasculature, uptake by resident phagocytes and failure to deliver their drug cargo to cancer cells [31, 32]. While near-perivascular deposition is often viewed as a limitation to nanoparticles in the context of chemotherapy, we show that the APC-rich perivascular region of PDAC is an ideal target for immunotherapy as this location is seamlessly accessible by circulating immuno-NPs. Intratumoral accumulation of immuno-NPs results in activation and expansion of APCs that can continuously process antigens shed from tumor cells. Additionally, our immuno-NP design is highly tunable, offering the likelihood of significant improvement of efficacy, dose reduction and a high safety profile. We show the functional synergy of the dual-agonist cargo and enhanced production of IFNβ can be significantly increased by adjusting the ratio of cdGMP/MPLA loaded in immuno-NPs.
MATERIALS AND METHODS
Materials
The lipids DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) and cholesterol were purchased from Avanti. DSPE-PEG2000 (methoxy-poly(ethylene glycol)-2000 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N) was purchased from Laysan Bio. MPLA was obtained from Sigma Aldrich. Cyclic di-GMP (cdGMP) was purchased from InvivoGen. DiR fluorophore was purchased from Thermo Fisher Scientific. D-luciferin potassium salt was purchased from GoldBio. PDAC cells (Panc02) derived from C57BL/6 mice were graciously donated by the laboratory of Dr. Mark Jackson (Case Comprehensive Cancer Center, Cleveland, OH). A LumiKine Xpress Bioluminescent cytokine ELISA kit was pruchasde from InvivoGen. Dye-conjugated flow cytometry antibodies were purchased from BD Biosciences. Antibodies for immunohistochemistry were obtained from Biolegend. General solvents and chemicals were obtained from Thermo Fisher Scientific.
Preparation of immuno-nanoparticle
The immuno-NPs were synthesized using a previously published method [25]. DPPC, cholesterol and mPEG2000-DSPE were combined at a molar ratio of 77:20:3 and suspended in chloroform. MPLA previously suspended in a mixture of chloroform/methanol/DI water (74:23:3 vol/vol) was added to the solution at either 100 or 300 μg per 42 μmol lipid for the MPLAlow/cdGMP-NP or MPLAhigh/cdGMP-NP formulations, respectively. Lipid films were formed by evaporating the solvents in a chemical hood overnight at room temperature. Cyclic di-GMP (200 μg, 1 mg/mL) in phosphate-buffered saline (PBS) was added to the lipid film, which was heated at 60°C and resuspended with mild vortexing every 5 minutes for 30 minutes. The suspension was then probe sonicated on ice for 5 minutes using a 30/30s cycle between 13 W and 11 W power output. Formulations were diluted to working concentrations and dialyzed against PBS for 2–4 hours. Loading of cdGMP was determined using high performance liquid chromatography (HPLC, Shimadzu). Particle size was measured using dynamic light scattering (DLS) (Brookhaven Instruments). For fluorescent NP experiments, 0.5 mol% of DiR was added to the lipid film during synthesis.
Cell Lines and Animal Models
RAW 264.7 macrophages were obtained from American Type Culture Collection (ATCC). PDAC cells (Panc02) derived from C57BL/6 mice and transfected with the firefly luciferase enzyme were graciously donated by the laboratory of Dr. Mark Jackson (Case Comprehensive Cancer Center). Cells were cultured in high glucose Dulbecco’s modified Eagle medium (DMEM, HyClone) containing 10% fetal bovine serum (FBS, HyClone) and 1% penicillin-streptomycin (Pen-Strep, Gibco). All cells were grown at 37°C with 5% carbon dioxide.
All animal experiments were performed using IACUC-approved protocols from Case Western Reserve University. Immune-competent albino female C57BL/6 (B6(Cg)-Tyrc−2J/J, 10–12 weeks) were purchased from Jackson Laboratory. Flank tumors were established by injecting 1×106 Panc02 cells suspended in PBS subcutaneously into the left flank. Animals received three consecutive days of treatment intravenously (i.v.), beginning when the tumor became palpable and measurable with calipers (~30 mm3, approximately 14–18 days after inoculation). To examine treatment efficacy, tumors were measured with calipers at least twice a week and tumor volume was determined using the formula (length × width2)/2. Tumor growth was also monitored using bioluminescent imaging (BLI) the same day as caliper measurements. Each animal was injected with 200 μL of D-luciferin potassium salt in PBS at a concentration of 12.5 mg/mL. Animals were anesthetized and imaged 8 minutes after injection using the IVIS Spectrum In Vivo Imaging System (PerkinElmer, 124262).
For orthotopic models, albino female C57BL/6 (Jackson Laboratories) were anesthetized with 2.5% isoflurane and given subcutaneous carprofen (5 mg/kg) and local bupivacaine (2 mg/kg) prior to surgery. An incision was made in the left flank and 5×105 Panc02 cells were injected into the extracorporealized tail of the pancreas in a 1:1 ratio of cells in PBS and Matrigel. Carprofen (5 mg/kg) was administered subcutaneously for two days post-surgery. Tumor growth was measured using bioluminescent imaging. Animals were treated via intravenous injection for two consecutive days starting day 22 after inoculation.
ELISA Assay of IFNβ
Six million RAW 264.7 macrophages per well were plated in a 24-well plate and each of the following formulations was tested in triplicate: unloaded NP, cdGMP-NP, MPLAlow-NP, MPLAhigh-NP, MPLAlow/cdGMP-NP, MPLAhigh/cdGMP-NP, and untreated controls. Wells treated with agonist-loaded NPs contained 20μg/mL cdGMP and either 17.7 μg/mL or 51.6 μg/mL MPLA for MPLAlow and MPLAhigh formulations, respectively. After a 24-hour incubation period, supernatants were collected via centrifugation and analyzed for IFNβ production using a LumiKine Xpress Bioluminescent cytokine ELISA kit (InvivoGen). Luminescence was measured with a Tecan Infinite 200. Each condition was tested in three biological samples, which were each assayed in triplicate as technical replicates.
Flow Cytometry
Anti-mouse CD3e (145–2C11), CD11b (M1/70), CD11c (HL3), CD19 (1D3), CD45 (30-F11), CD49b (DX5) and F4/80 (T45–2342) dye-conjugated flow cytometry antibodies were purchased from BD Biosciences. Anti-mouse CD4 (GK1.5) and CD8a (53–6.7) antibodies were purchased from Biolegend. Tissues were collected 24 hours after injection for distribution studies of fluorescent nanoparticles and 48 hours after the start of treatment for efficacy studies. After blood collection through retro-orbital bleeding, animals were euthanized and tissues were harvested immediately. The pancreas was digested for 1 hour in 1 mg/mL collagenase (Sigma Aldrich) at 37°C. Organs were gently homogenized and consecutively strained through 70-μm filters to obtain single-cell suspensions. Red blood cells were removed using multiple incubations and washes with ACK lysis buffer (Gibco). Cells were blocked with anti-mouse CD16/CD32 (2.4G2; BD Biosciences) and stained with fluorescent antibodies (Supplementary Table S1) to identify immune cell populations, followed by viable cell staining using DAPI (BD Biosciences). Cells were fixed with 1% PFA, resuspended in buffer and stored at 4°C until analysis. Samples were run on a BD LSR II flow cytometer and analyzed using FlowJo software.
Immunohistochemistry
For examination of particle distribution, fluorescent NPs were injected via tail vein and organs were collected 12 hours after injection. Animals were euthanized via cardiac perfusion of PBS and 4% paraformaldehyde (PFA, Alfa Aesar). Organs were fixed in PFA, transferred to 30% sucrose overnight, followed by optimal cutting temperature (OCT) gel (Thermo Fisher Scientific) and frozen at −80°C. Cryopreserved tissues were sliced into 10-μm sections using a Leica Cryostat and mounted on Fisher SuperFrost glass slides (ThermoFisher Scientific). Tissues were stained with the following primary antibodies: rat anti-mouse IgG CD31 (endothelial cells; BD Biosciences, 1:10), AF647-conjugated hamster anti-mouse CD11c (DCs; Biolegend, 1:100), rat anti-mouse IgM CD49b (NK cells; Biolegend, 1:100), rat anti-mouse IgG F4/80 (macrophages; Biolegend, 1:4). Secondary antibodies include goat anti-rat IgM AF488 and goat anti-rat IgG AF647 (Invitrogen, 1:150). Slides were washed 3x with PBS and blocked with protein blocking solution (4% goat serum, 0.1% Triton-X 100 in PBS) for 20 min. Following overnight incubation of primary antibodies at 4°C, slides were washed 3x with PBS and corresponding secondary antibodies were incubated at room temperature for 1 hour. Slides were washed 3x with PBS, and DAPI mounting media (Vector Laboratories) was added with a 1.5 glass cover slip. Images were collected with a Leica TCS SP8 gated STED Confocal Microscope (Leica Microsystems).
Statistical Analysis
Statistical analysis was performed using Prism 8 (GraphPad) and are detailed in the figure legends. Data was analyzed using one- or two-way ANOVA with Tukey’s post-test, where P-values less than 0.05 were statistically significant. All experiments were performed with at least three independent biological replicates, and values are reported as mean ± SE or SD as noted in figure captions. For animal experiments, at least 4 mice were used per group.
RESULTS
Characterization of the immuno-nanoparticles
We designed a 60-nm nanoparticle loaded with different ratios of cdGMP and MPLA [25]. We synthesized the lipid-based immuno-NP with the lipid matrix being DPPC, cholesterol and mPEG2000-DSPE containing either 100 or 300 μg MPLA per 42 μmol lipids, termed MPLAlow/cdGMP-NP or MPLAhigh/cdGMP-NP, respectively (Fig. 2A). Characterization of the nanoparticles showed that the two immuno-NP variants were similar in loading and sizing. The MPLAhigh/cdGMP-NP contained a ~3-fold higher MPLA content than the MPLAlow/cdGMP-NP (Fig. 2B), while both formulations contained similar cdGMP content after dialysis (Supplementary Fig. S1A). Dynamic light scattering (DLS) measurements showed that the size of MPLAlow/cdGMP-NPs (mean ± SD = 53.2 ± 11.8 nm) was close to that of MPLAhigh/cdGMP-NPs (mean ± SD = 60.9 ± 7.9 nm) (Fig. 2C) and that these sizes remained constant over time (Supplementary Fig. S1B). To assess functional synergy, we treated the RAW macrophage cell line in vitro with equivalent amounts of immuno-NPs containing cdGMP only, MPLA only, or both agonists. Specifically, we incubated RAW 264.7 macrophages for 24 hours with MPLAlow/cdGMP-NP, MPLAhigh/cdGMP-NP, cdGMP-NP, MPLAlow-NP, MPLAhigh-NP or empty NPs. Supernatants were tested for IFNβ secretion using an ELISA. Quantification of IFNβ showed that single-agonist NPs containing either MPLA or cdGMP resulted in a significant increase in IFNβ production (4–14-fold) compared to untreated controls (Fig. 2D). On the other hand, the dual-agonist immuno-NP variants significantly outperformed any single-agonist NP, showcasing the synergy of efficient and simultaneous delivery of MPLA and cdGMP to APCs. For example, MPLAhigh/cdGMP-NPs resulted in an 11 and 22-fold higher IFNβ levels than the cdGMP-NP and MPLAhigh-NP, respectively. Notably, the ratio of the two agonists in the immuno-NP had a significant impact with MPLAhigh/cdGMP-NPs, displaying a 1.6-fold higher IFNβ secretion than MPLAlow/cdGMP-NPs.
Fig. 2.

Characterization of immuno-NPs. A) Schematic depicting the nanoparticle formulations. B) Loading of cdGMP and MPLA (μg per mmol lipid bilayer components) following 3 hours dialysis against PBS. C) Representative particle size measured by dynamic light scattering (DLS). D) ELISA of IFNβ production from murine RAW 264.7 macrophages following 24-hour exposure to immuno-NP formulations. Data is plotted as mean ± SD. Statistics for the ELISA were calculated using one-way ANOVA with Tukey’s post-test. **P < 0.01; ***P < 0.001; ****P < 0.0001
Microdistribution of immuno-NPs in the tumor microenvironment of an orthotopic pancreatic tumor model
We evaluated the uptake of fluorescent empty-NPs (no immune agonists) by various immune cell subsets in tumors using flow cytometry and immunofluorescence. In the orthotopic Panc02 mouse model, the fluorescent NPs were intravenously administered on day 22 after tumor inoculation, and organs were harvested 24 hours later. Strikingly, flow cytometry revealed ~56% of dendritic cells (DCs), 79% of macrophages and 52% of NK cells contained immuno-NPs at this time point (Fig. 4A, left panel). The fluorescent signal per NP+ cell in the tumor was very high for all innate immune cell subpopulations (Fig. 4A, right panel). We also used flow cytometry to assess the uptake of NPs in organs of the reticuloendothelial system (RES). NPs were found in the liver and spleen in comparable levels to the tumor number (Fig. 4B,C). Confocal microscopy confirms immuno-NPs were mainly deposited in the APC-rich perivascular regions of the tumor in the pancreas within 10 hours after injection, where they were co-localized with dendritic cells in perivascular regions (Fig. 4D, left panel). Confocal imaging showed minimal NP accumulation in healthy pancreatic tissue (Fig. 4D, right panel). Overall, these data show the ability of NPs to preferentially accumulate in the perivascular space of pancreatic tumors and their high efficiency in targeting APCs cells in the perivascular TME niche.
Fig. 4.

Flow cytometry analysis of immune cells in the tumor-bearing pancreas. (A) Analysis of IFNβ-secreting immune cells in the pancreas was performed 48 hours after the start of two days of treatment with MPLAhigh/cdGMP-NPs, MPLAlow/cdGMP-NPs or empty NPs. Comparison of MPLAhigh/cdGMP-NPs to free agonists was performed in terms of cell content in the tumor and other major organs (7 μg cdGMP, 16.5 μg MPLA, i.v.). Analysis of innate immune cells is shown for (B) pancreas, (D) blood, (E) spleen, and (F) and liver. (C) Confocal images of a Panc02 pancreatic tumor 48 hours after treatment with MPLAhigh/cdGMP-NPs show intratumoral infiltration of DCs (10x magnification). Analysis of adaptive immune cells is shown for (G) pancreas, (H) blood, (I) spleen, and (J) and liver. Immune subsets represented as box and whisker plots (5–95 percentile; + mean) with statistics by one-way ANOVA with Tukey post-test (N=5 mice per group). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In a different study, we compared the microdistribution of the two immuno-NP variants to empty NPs. Mice bearing orthotopic Panc02 tumors were injected with two doses of NPs on two consecutive days and flow cytometry analysis was performed 24 h after the second dose. In terms of percent of NP-containing dendritic cells and macrophages in the tumor, the cell uptake of the two immuno-NP variants was comparable to the empty NPs (Fig. 4E, left panel). However, the uptake of the high MPLA immuno-NP by immune cells in the tumor was greater and at higher quantities than the other NP formulations (Fig. 4E, right panel). The overall cell uptake of the NP formulations exhibited a similar trend in major organs with all three formulations having comparable microdistributions (Fig. 4F).
Dual-agonist NPs increase immune cell populations in orthotopic PDAC tumors
We performed mechanistic studies to evaluate the in vivo efficacy of immuno-NP and its dual-agonist cargo in activating the immune system in the orthotopic Panc02 tumors and compared to empty NPs and free immune agonists. First, we compared the MPLAhigh/cdGMP to the MPLAlow/cdGMP-NP formulations in terms of activation of APCs in the tumor microenvironment. We administered i.v. the two immuno-NP variants for two consecutive days, then harvested blood and organs 24 hours after the last treatment for analysis by flow cytometry. Compared to empty NPs, both immuno-NP variants activated APCs in the tumor with the MPLAhigh/cdGMP-NP having higher potency as indicated by increased number of IFNβ-secreting dendritic cells and macrophages (Fig. 5A). We then compared the MPLAhigh/cdGMP NP to free agonists. The immuno-NP treatment triggered a significant innate immune response in the pancreas showing a rapid accumulation of APCs in the TME compartment (Fig. 5B). Compared to untreated and free agonist-treated controls, we observed an increase in CD45+ immune cells (~4.2-fold), DCs (~11.5-fold) and macrophages (~8-fold) within the pancreas following immuno-NP treatment. Confocal microscopy shows the microdistribution of dendritic cells showing DC infiltration throughout the tumor volume 48 hours after treatment with immuno-NPs (Fig. 5C). While the total number of CD45+ cells in the blood was unchanged between the untreated and treatment groups, it is worth noting the significant increase in blood DCs in the case of the free agonists (Fig. 5D). The number of splenic DCs significantly increased in the immuno-NP group, and macrophages increased with both treatments (Fig. 5E). Importantly, immuno-NPs did not significantly elevate the number of immune cells in the liver, whereas treatment with the free agonists at the same dose drastically increased DCs (~28-fold), macrophages (~23-fold) and NK cells (~9-fold) compared to untreated controls (Fig. 5F). These levels were also at least 3 times greater than those observed in the immuno-NP group. These results indicate that off-target toxicity of cdGMP and MPLA can be abrogated by delivering these agents with immuno-NPs. Additionally, within 48 hours of immuno-NP treatment, T and B cells increased in the pancreas (Fig. 4G), suggesting infiltration of lymphocytes into the tumor. The expansion of T and B cells is also shown in blood, spleen and liver (Fig. 4H–J). Not surprisingly, the immuno-NPs used as a monotherapy was not able to significantly increase survival in the orthotopic Panc02 model (Supplementary Fig. S2), indicating the need to follow up activation of innate immunity with other immunotherapies. Overall, these data suggest the immuno-NP treatment effectively activated the local innate immune compartment within the TME, showing its potential to work synergistically with other immunotherapies and dramatically improve antitumor immune responses.
Fig. 5.
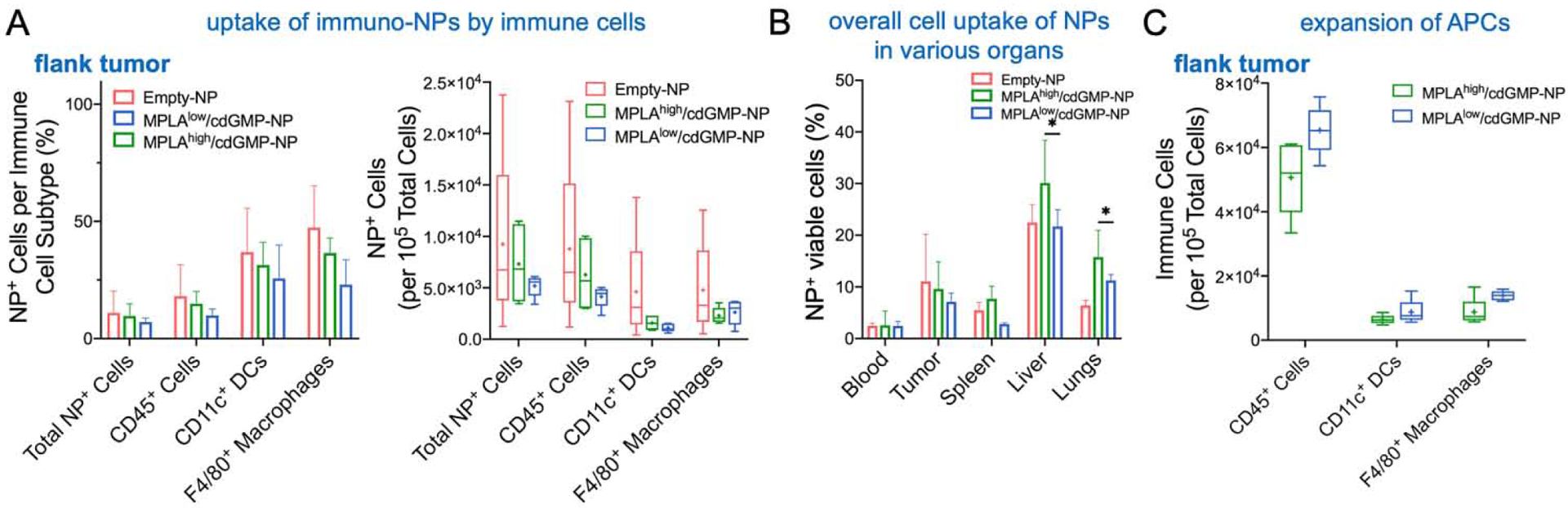
Microdistribution and uptake of immuno-NPs by APCs in a flank pancreatic tumor model. Flow cytometry analysis was performed 24 h after two doses for two consecutive days of fluorescent NP (empty, no immune agonists) or the two immuno-NP variants to compare their uptake by immune cells in the tumor. The accumulation of NPs within the flank tumor in terms of (A) percent of immune cells in the tumor containing NPs (left panel) and number of NP+ cells (right panel). (B) Overall cell uptake of immuno-NPs is shown for major organs. Data plotted as mean ± SE with N=5 animals. Statistics were calculated using one-way ANOVA with Tukey’s post-test. *P < 0.05. (C) Analysis of innate immune cells in the tumor is shown. Immune subsets represented as box and whisker plots (5–95 percentile; + mean) with statistics by one-way ANOVA with Tukey post-test (N=5 mice per group).
Therapeutic efficacy of the immuno-NP variants in a flank pancreatic tumor model
First, we compared the microdistribution of the two immuno-NP variants to empty NPs. Mice bearing orthotopic Panc02 tumors were injected with two doses of NPs on two consecutive days and flow cytometry analysis was performed 24 h after the second dose. In terms of percent of NP-containing dendritic cells and macrophages in the flank tumor, the cell uptake of the two immuno-NP variants was comparable to the empty NPs (Fig. 5A). Similarly, the uptake of the empty or immuno-NPs by immune cells was comparable in organs of the reticuloendothelial system (Fig. 5B). Notably, both immuno-NP formulations triggered a similar innate immune response in the flank tumor in terms of accumulation of APCs in the TME compartment (Fig. 5C).
The therapeutic efficacy of the two dual-agonist immuno-NP variants and free agonists was assessed in the flank model of Panc02 in immune-competent C57BL/6 mice. Animals were treated via intravenous (i.v.) injection for three consecutive days beginning when their tumors reached 30 mm3 (about 14–18 days after tumor inoculation). Using the dose of cdGMP (14μg cdGMP per mouse) constant for all treatments, we administered either MPLAlow/cdGMP-NPs, MPLAhigh/cdGMP-NPs or free agonists (at the high MPLA dose). Fig. 6A shows representative BLI images for each group. By the third day of treatment, quantification of the BLI signal indicated that the tumor signal of the group treated with MPLAhigh/cdGMP-NPs decreased by as much as 94% compared to the untreated control (Fig. 6B). Importantly, the tumor BLI signal of the mice treated with MPLAhigh/cdGMP-NPs remained close to baseline signal for at least 20 days, whereas the tumor growth of mice treated with free agonists or MPLAlow/cdGMP-NPs was rapid and similar to the untreated group. Caliper measurements of the tumor volume were in agreement with the BLI findings (Fig. 6C). In terms of survival, 14% of the MPLAhigh/cdGMP-NP group was cancer free after 100 days, whereas median survival time of the other groups was 22.5–24 days (Fig. 6D). Fig. 7A shows a comparison indicating that the uptake of immuno-NPs by immune cells was significantly greater in the orthotopic tumor than the flank tumor. Most notably, the number of IFNβ-secreting immune cells and overall expansion of immune cells was higher in the flank than the orthotopic tumor (Fig. 7B,C).
Fig. 6.

Treatment with immuno-NPs decreases tumor burden and prolongs survival in murine flank model of PDAC. (A) Representative bioluminescent images of Panc02 flank tumor-bearing mice following three consecutive days of treatment. Units of radiance are photons/sec. (B) Quantification of tumor luminescent signal (photons/sec) during treatment monitoring. Black arrows along x-axis indicate treatment days. (C) Caliper measurements of tumor volume (mm3), calculated using the formula length×(width/2)2. (D) Kaplan-Meier plots of survival analysis along with median survival for each group and statistics by the log rank test. Treatment groups included MPLAhigh/cdGMP-NPs (N=7), MPLAlow/cdGMP-NPs (N=4), free agonists (N=6), and untreated controls (N=5). Mean ± SD are plotted with statistics using two-way ANOVA with Tukey post-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Fig. 7.

Comparison of the immune cell response to the immuno-NP treatments between the orthotopic and flank Panc02 tumor model. (A) Uptake of the two immuno-NPs and empty NP by APCs in the orthotopic and flank tumor in terms of percent of immune cells in the tumor containing NPs (left panel) and number of immune cells containing NPs (right panel). Analysis of (B) IFNβ-secreting immune cells and (C) expansion of APCs is compared in the orthotopic and flank tumor. Statistics by one-way ANOVA with Tukey post-test (N=5 mice per group). *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
Immunotherapeutic approaches to induce anti-tumor responses largely fail in immunologically ignorant “cold” tumors, such as pancreatic cancer. The tumor microenvironment of PDAC is dominated by non-cancerous stromal cells and leukocytes with immunosuppressive phenotypes, including MDSCs and regulatory T cells [18, 20, 22–24]. These cells produce suppressive cytokines and express inhibitory surface receptors, which contribute to the dysfunction of tumor-resident APCs, allowing cancer cells to proliferate undetected. Inhibition of immune checkpoint markers with monoclonal antibodies, such as anti-PD-1/PD-L1 and anti-CTLA-4, relies on the presence of cytotoxic T cells and pre-existing anti-tumor immune responses, which are often excluded and absent in advanced tumors [33]. Cellular immunotherapies introduce tumor antigen-responsive immune cells to the patient, but it requires prior knowledge of tumor-specific antigens, which can vary largely between cancer cells [34]. To augment existing immunotherapies and generate robust and consistent therapeutic outcomes, activation of innate immune pathways within a tumor is a promising approach to ultimately induce adaptive cytotoxic responses, tumor clearance and immunologic memory.
Regarding activation of innate immunity, type I IFNs are potent pro-inflammatory cytokines that play a crucial role in recruitment of APCs and natural killer (NK) cells, activation of CD8+ cytotoxic T cells, and subsequent anti-tumor responses [35, 36]. Both MPLA and cdGMP target pattern recognition receptors that detect pathogenic markers of bacteria and viruses and initiate production of type I IFNs through overlapping downstream pathways. MPLA, derived from lipopolysaccharide (LPS) on bacterial cell walls, was the first clinically approved molecular vaccine adjuvant for use in humans [30]. The small molecule cdGMP activates the STING pathway, which naturally detects cytosolic nucleic acids that result from cell damage or bacterial infection [29]. STING agonists, such as cdGMP and other cyclic dinucleotides, have gained increasing interest as vaccine adjuvants in many different cancers [37, 38]. Recent studies have demonstrated remarkable STING-mediated antitumor immunity in preclinical tumor models resistant to PD-1 checkpoint inhibition [37, 39, 40]. In light of these exciting results, clinical trials of STING agonists are underway in multiple cancers (clinicaltrials.gov identifier: NCT03172936, NCT04144140, NCT04109092).
In this paper, we examined immuno-NPs containing cdGMP along with varying amounts of MPLA in the context of pancreatic cancer. Both TNBC and PDAC are poorly immunogenic adenocarcinomas, often containing extensive desmoplasia, tumor-promoting immune cells and a milieu of surface receptors and soluble factors, which contribute to their immunosuppressive tumor microenvironments. We show effective delivery of immuno-NPs to tumor-resident leukocytes that resulted in significant expansion of the APC population in the pancreatic TME. More than 50% of leukocytes in orthotopic pancreatic tumors contained immuno-NPs within 24 hours of injection. Confocal imaging shows immuno-NPs co-localized with tumor-resident DCs and macrophages, which are mainly located on the infiltrating edges of the tumor, as shown in other studies [14, 41]. Delivery of immuno-NPs along vasculature and peri-tumoral regions was sufficient to induce remarkable APC infiltration and immune cell recruitment throughout the tumor in the Panc02 orthotopic and flank models. Our lab previously developed and tested 60-nm dual agonist immuno-stimulatory nanoparticles (immuno-NPs) in a 4T1 murine model of metastatic triple negative breast cancer (TNBC) [25]. Systemic administration of these particles produced a lasting IFNβ-driven anti-tumor immune response, which resulted in initial primary tumor reduction and prevention of lung metastasis. Treatment with immuno-NPs caused only transient weight loss and was well-tolerated by animals. Furthermore, liver enzyme assays of serum AST and ALT demonstrated no long-term hepatotoxicity, indicating the safety of immuno-NPs [25].
Differences in the growth and morphology of pre-clinical orthotopic versus heterotopic pancreatic models have been described [42]; however, variations in immune cell content between the models has not been widely studied. This was evident in our study with the overall survival benefits from the immuno-NP treatment being limited in the orthotopic model. This can be partially attributed to an immunosuppressive TME that is influenced by the presence of stromal cells within the orthotopic landscape that is absent in heterotopic flank tumors. Although more NPs were taken up by immune cells within the pancreas, any pro-inflammatory immune cell activity may not be sufficient to overcome the surrounding immunosuppressive TME. It has been shown that the immunosuppressive TME of pancreatic tumors includes MDSCs [15, 19, 43] and the immunosuppressive cytokine transforming growth factor beta (TGF-β) [44–46]. It has also been shown that IFNβ-driven inflammation causes an increase in populations of regulatory PD-1/PD-L1-expressing leukocytes and tumor cells, and CTLA-4-expressing Tregs, which already are common in PDAC [47–49]. While evaluation of these mechanisms of immune cell inhibition were beyond the scope of this paper, future studies should examine the long-term expression of checkpoint markers and cytokines following initial immune cell recruitment to the tumor. These findings can educate different combinations of immunotherapies, which can facilitate more robust and lasting antitumor immune responses. Our data suggests that activation of the local innate immune compartment using the immuno-NP treatment can be augmented and work together with TGF-β inhibitors [44–46] or immune checkpoint inhibitors, such as anti-PD-1 (against PD-1+ cells) and anti-CTLA-4 (against Tregs), which have shown promising efficacy in PDAC following vaccine therapy [50–53].
In conclusion, we designed an immunostimulatory NP that incorporates systemic delivery targeting the APC-rich perivascular regions of PDAC, and delivery of two synergistic immune agonists that reprogram and activate local APCs into T cell-stimulatory cells. The effective delivery of a dual-agonist cargo using the immuno-NP effectively boosted innate immunity in the hard-to-reach and highly immunosuppressive microenvironment of pancreatic tumors.
Supplementary Material
Fig. 3.

Microdistribution and uptake of immuno-NPs by APCs in an orthotopic model of pancreatic tumor. Flow cytometry analysis was performed 24 h after single IV injection of fluorescent NP (empty, no immune agonists) to evaluate the accumulation of NPs within tumor-bearing pancreas in terms of (A) percent of immune cells in the tumor containing NPs (left panel) and relative NP fluorescence for NP+ cell populations measured as median fluorescence intensity (right panel), (B) percent of immune cells in major organs containing NPs and (C) NP fluorescence for NP+ cell populations in the liver and spleen. (D) Confocal images visualizing NP deposition within an orthotopic Panc02 tumor 10 hours after i.v. injection. Fluorescent NPs (yellow) coincide with the location of DCs (CD11c, green) in near-perivascular regions (CD31, red). Nuclei shown in blue, 40x magnification. (E) Flow cytometry analysis was performed 24 h after two subsequent daily doses of fluorescent NP (empty, no immune agonists) or the two immuno-NP variants to compare their uptake by immune cells in the tumor. (F) Overall cell uptake of immuno-NPs is shown for major organs. Flow cytometry data plotted as mean ± SE with N=5 animals. Statistics were calculated using one-way ANOVA with Tukey’s post-test. **P < 0.01.
Highlights.
Nanoparticle is loaded with two potent innate immune agonists.
The dual-agonist cargo provides remarkable functional synergy.
Systemic delivery enables uptake by APCs in perivascular regions of tumors.
Significant activation and expansion of APCs in pancreatic tumor.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Cancer Institute (U01CA198892, R01CA253627), Case Comprehensive Cancer Center GI SPORE 2P50CA150964-07A1, the Case Comprehensive Cancer Center Support Grant (P30CA043703) and the Shiverick Family Fund, the Clinical Translational Science Collaborative of Cleveland (UL1TR002548), and the Alex’s Lemonade Stand Foundation (E.K.). M.L. was supported by a fellowship from the NIH Interdisciplinary Biomedical Imaging Training Program (T32EB007509) administered by the Department of Biomedical Engineering, Case Western Reserve University. P.A.B. was supported by a graduate research fellowship from NSF. We acknowledge the Case Center for Imaging Research, Case Comprehensive Cancer Center Flow Cytometry Core, and the Case School of Medicine Light Microscopy Core. Purchase of the Leica TCS SP8 confocal microscope used in this study was supported by funding from NIH ORIP S10-OD024996.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020, CA Cancer J Clin, 70 (2020) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Christenson ES, Jaffee E, Azad NS, Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: a bright future, Lancet Oncol, 21 (2020) e135–e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH, Recent progress in pancreatic cancer, CA Cancer J Clin, 63 (2013) 318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gong J, Chehrazi-Raffle A, Reddi S, Salgia R, Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations, J Immunother Cancer, 6 (2018) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ, Improved survival with ipilimumab in patients with metastatic melanoma, N Engl J Med, 363 (2010) 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, Mateus C, Postow MA, Gergich K, Elassaiss-Schaap J, Li XN, Iannone R, Ebbinghaus SW, Kang SP, Daud A, Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial, Lancet, 384 (2014) 1109–1117. [DOI] [PubMed] [Google Scholar]
- [7].Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A, Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma, N Engl J Med, 369 (2013) 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G Jr., Garrido M, Lubiniecki GM, Shentu Y, Im E, Dolled-Filhart M, Garon EB, Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial, Lancet, 387 (2016) 1540–1550. [DOI] [PubMed] [Google Scholar]
- [9].Kabacaoglu D, Ciecielski KJ, Ruess DA, Algul H, Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options, Front Immunol, 9 (2018) 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS, Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients, Nature, 515 (2014) 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, Drengler R, Chen C, Smith L, Espino G, Gergich K, Delgado L, Daud A, Lindia JA, Li XN, Pierce RH, Yearley JH, Wu D, Laterza O, Lehnert M, Iannone R, Tolcher AW, Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors, Clin Cancer Res, 21 (2015) 4286–4293. [DOI] [PubMed] [Google Scholar]
- [12].Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM, Safety and activity of anti-PD-L1 antibody in patients with advanced cancer, N Engl J Med, 366 (2012) 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Johnk C, Henne-Bruns D, Kremer B, Kalthoff H, Systemic and local immunosuppression in pancreatic cancer patients, Clin Cancer Res, 7 (2001) 925s–932s. [PubMed] [Google Scholar]
- [14].Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH, Dynamics of the immune reaction to pancreatic cancer from inception to invasion, Cancer Res, 67 (2007) 9518–9527. [DOI] [PubMed] [Google Scholar]
- [15].Fan JQ, Wang MF, Chen HL, Shang D, Das JK, Song J, Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma, Molecular cancer, 19 (2020) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yao W, Maitra A, Ying H, Recent insights into the biology of pancreatic cancer, EBioMedicine, 53 (2020) 102655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ren B, Cui M, Yang G, Wang H, Feng M, You L, Zhao Y, Tumor microenvironment participates in metastasis of pancreatic cancer, Molecular cancer, 17 (2018) 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Anderson KG, Stromnes IM, Greenberg PD, Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies, Cancer Cell, 31 (2017) 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Karamitopoulou E, Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features, Br J Cancer, 121 (2019) 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hiraoka N, Onozato K, Kosuge T, Hirohashi S, Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions, Clin Cancer Res, 12 (2006) 5423–5434. [DOI] [PubMed] [Google Scholar]
- [21].Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan X, Wei L, Abrams Z, Luedke E, Lesinski GB, Mundy-Bosse B, Bekaii-Saab T, Carson WE 3rd, Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease, Cancer immunology, immunotherapy : CII, 64 (2015) 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D, Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer, Cell Rep, 20 (2017) 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Komura T, Sakai Y, Harada K, Kawaguchi K, Takabatake H, Kitagawa H, Wada T, Honda M, Ohta T, Nakanuma Y, Kaneko S, Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4+ T cells with clinical impact, Cancer Sci, 106 (2015) 672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pergamo M, Miller G, Myeloid-derived suppressor cells and their role in pancreatic cancer, Cancer Gene Ther, 24 (2017) 100–105. [DOI] [PubMed] [Google Scholar]
- [25].Atukorale PU, Raghunathan SP, Raguveerγ V, Zheng C, Moon TJ, Weiss ML, Bielecki PA, Goldberg AL, Covarrubias G, Hoimes CJ, Karathanasis E, Nanoparticle encapsulation of synergistic immune agonists enables systemic co-delivery to tumor sites and interferon β-driven anti-tumor immunity, Cancer Research, 79 (2019) 5394–5406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ, Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants, J Clin Invest, 125 (2015) 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF, Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells, J Exp Med, 208 (2011) 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I, Direct effects of type I interferons on cells of the immune system, Clin Cancer Res, 17 (2011) 2619–2627. [DOI] [PubMed] [Google Scholar]
- [29].Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE, STING is a direct innate immune sensor of cyclic di-GMP, Nature, 478 (2011) 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC, The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4, Science, 316 (2007) 1628–1632. [DOI] [PubMed] [Google Scholar]
- [31].Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nat Nanotechnol, 6 (2011) 815–823. [DOI] [PubMed] [Google Scholar]
- [32].Tanaka HY, Kano MR, Stromal barriers to nanomedicine penetration in the pancreatic tumor microenvironment, Cancer Sci, 109 (2018) 2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pardoll DM, The blockade of immune checkpoints in cancer immunotherapy, Nat Rev Cancer, 12 (2012) 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, Wells DK, Cary CIO, Grbovic-Huezo O, Attiyeh M, Medina B, Zhang J, Loo J, Saglimbeni J, Abu-Akeel M, Zappasodi R, Riaz N, Smoragiewicz M, Kelley ZL, Basturk O, Australian I Pancreatic Cancer Genome, R. Garvan Institute of Medical, H. Prince of Wales, H. Royal North Shore, G. University of, H. St Vincent’s, Q.B.M.R. Institute, C.f.C.R. University of Melbourne, I.f.M.B. University of Queensland, H. Bankstown, H. Liverpool, C.O.B.L. Royal Prince Alfred Hospital, H. Westmead, H. Fremantle, H. St John of God, H. Royal Adelaide, C. Flinders Medical, P. Envoi, H. Princess Alexandria, H. Austin, I. Johns Hopkins Medical, A.R.-N.C.f.A.R.o. Cancer, Gonen M, Levine AJ, Allen PJ, Fearon DT, Merad M, Gnjatic S, Iacobuzio-Donahue CA, Wolchok JD, DeMatteo RP, Chan, Greenbaum BD, Merghoub T, Leach SD, Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer, Nature, 551 (2017) 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Muller L, Aigner P, Stoiber D, Type I Interferons and Natural Killer Cell Regulation in Cancer, Front Immunol, 8 (2017) 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF, Type I interferon response and innate immune sensing of cancer, Trends Immunol, 34 (2013) 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M, STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity, Proc Natl Acad Sci U S A, 112 (2015) 15408–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gray PM, Forrest G, Wisniewski T, Porter G, Freed DC, DeMartino JA, Zaller DM, Guo Z, Leone J, Fu TM, Vora KA, Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities, Cell Immunol, 278 (2012) 113–119. [DOI] [PubMed] [Google Scholar]
- [39].Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW Jr., Kim Y, STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade, Science translational medicine, 7 (2015) 283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW Jr., Gajewski TF, Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity, Cell Rep, 11 (2015) 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S, Significance of M2-polarized tumor-associated macrophage in pancreatic cancer, The Journal of surgical research, 167 (2011) e211–219. [DOI] [PubMed] [Google Scholar]
- [42].Erstad DJ, Sojoodi M, Taylor MS, Ghoshal S, Razavi AA, Graham-O’Regan KA, Bardeesy N, Ferrone CR, Lanuti M, Caravan P, Tanabe KK, Fuchs BC, Orthotopic and heterotopic murine models of pancreatic cancer and their different responses to FOLFIRINOX chemotherapy, Dis Model Mech, 11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, Miller HE, Olson M, Rajasekaran K, Ernstoff MS, Wang D, Malarkannan S, Wang L, Immune-Checkpoint Protein VISTA Regulates Antitumor Immunity by Controlling Myeloid Cell-Mediated Inflammation and Immunosuppression, Cancer Immunol Res, 7 (2019) 1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lee HM, Kim KS, Kim J, A comparative study of the effects of inhibitory cytokines on human natural killer cells and the mechanistic features of transforming growth factor-beta, Cell Immunol, 290 (2014) 52–61. [DOI] [PubMed] [Google Scholar]
- [45].Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, Corrias MV, Moretta A, Bottino C, Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells, J Immunol, 190 (2013) 5321–5328. [DOI] [PubMed] [Google Scholar]
- [46].Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, Trojan J, Kozloff M, Simionato F, Cleverly A, Smith C, Wang S, Man M, Driscoll KE, Estrem ST, Lahn MMF, Benhadji KA, Tabernero J, TGFbeta receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer, Cancer Chemother Pharmacol, 83 (2019) 975–991. [DOI] [PubMed] [Google Scholar]
- [47].Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, Nakajima Y, Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer, Clin Cancer Res, 13 (2007) 2151–2157. [DOI] [PubMed] [Google Scholar]
- [48].Basso D, Fogar P, Falconi M, Fadi E, Sperti C, Frasson C, Greco E, Tamburrino D, Teolato S, Moz S, Bozzato D, Pelloso M, Padoan A, De Franchis G, Gnatta E, Facco M, Zambon CF, Navaglia F, Pasquali C, Basso G, Semenzato G, Pedrazzoli S, Pederzoli P, Plebani M, Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory costimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study, PLoS One, 8 (2013) e54824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade, Science, 359 (2018) 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, Woolman S, Vithayathil T, Glickman LH, Ndubaku CO, McWhirter SM, Dubensky TW Jr., Armstrong TD, Jaffee EM, Zaidi N, Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer, JCI Insight, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, Yao S, Anders RA, Laheru D, Wolfgang CL, Edil BH, Schulick RD, Jaffee EM, Zheng L, PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors, J Immunother, 38 (2015) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA Jr., Donehower RC, Jaffee EM, Laheru DA, Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer, J Immunother, 36 (2013) 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zaidi N, Quezada SA, Kuroiwa JMY, Zhang L, Jaffee EM, Steinman RM, Wang B, Anti-CTLA-4 synergizes with dendritic cell-targeted vaccine to promote IL-3-dependent CD4(+) effector T cell infiltration into murine pancreatic tumors, Annals of the New York Academy of Sciences, 1445 (2019) 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.