

Abstract
There is an urgent need to identify novel therapies for childhood cancers. Neuroblastoma is the most common pediatric solid tumor, and accounts for ~15% of childhood cancer‐related mortality. Neuroblastomas exhibit genetic, morphological and clinical heterogeneity, which limits the efficacy of existing treatment modalities. Gaining detailed knowledge of the molecular signatures and genetic variations involved in the pathogenesis of neuroblastoma is necessary to develop safer and more effective treatments for this devastating disease. Recent studies with advanced high‐throughput “omics” techniques have revealed numerous genetic/genomic alterations and dysfunctional pathways that drive the onset, growth, progression, and resistance of neuroblastoma to therapy. A variety of molecular signatures are being evaluated to better understand the disease, with many of them being used as targets to develop new treatments for neuroblastoma patients. In this review, we have summarized the contemporary understanding of the molecular pathways and genetic aberrations, such as those in MYCN, BIRC5, PHOX2B, and LIN28B, involved in the pathogenesis of neuroblastoma, and provide a comprehensive overview of the molecular targeted therapies under preclinical and clinical investigations, particularly those targeting ALK signaling, MDM2, PI3K/Akt/mTOR and RAS‐MAPK pathways, as well as epigenetic regulators. We also give insights on the use of combination therapies involving novel agents that target various pathways. Further, we discuss the future directions that would help identify novel targets and therapeutics and improve the currently available therapies, enhancing the treatment outcomes and survival of patients with neuroblastoma.
Keywords: clinical, neuroblastoma, preclinical, signaling pathway, targeted therapy
Abbreviations
- 4PB
4‐phenylbutyate
- ADCC
antibody‐dependent cell‐mediated cytotoxicity
- AKT
protein kinase B
- ALCL
anaplastic large‐cell lymphoma
- ALK
anaplastic lymphoma kinase
- ASCL1
Achaete‐scute family bhlh transcription factor 1
- ASCT
autologous stem cell transplantation
- AURKA
Aurora kinase A
- Bcl‐2
B‐cell lymphoma 2
- BCL‐xL
B‐cell lymphoma‐extra large
- BDNF
brain‐derived neurotrophic factor
- BET
bromodomain and extra‐terminal domain
- BIRC5
baculoviral IAP repeat containing 5
- BM
basement membrane
- BMP4
bone morphogenetic protein 4
- BMPs
bone morphogenetic proteins
- BRAF
v‐raf murine sarcoma viral oncogene homolog B1
- BRD
bromodomain
- CAM
chorioallantoic membrane
- CAR
chimeric antigen receptor
- CDK
cyclin‐dependent kinases
- CIC
Capicua transcriptional repressor
- CIN
chromosome instability
- COG
Children's Oncology Group
- CPC
chromosomal passenger complex
- CQ
chloroquine
- DFMO
difluoromethylornithine
- DNMT
DNA methyltransferase
- ECM
extracellular matrix
- EFS
event‐free survival
- EGFR
epidermal growth factor receptor
- ERBB2
Erb‐b2 receptor tyrosine kinases 2
- FDA
Food and Drug Administration
- FGFR1
fibroblast growth factor receptor 1
- FUS
focused ultrasound
- FZD2
Frizzled class receptor 2
- GD2
Disialoganglioside
- GSK3β
glycogen synthase kinase 3β
- HAT
histone acetyltransferase
- HBP1
HMG‐box transcription factor 1
- HDACi
histone deacetylase inhibitor
- HDACs
histone deacetylases
- HDC
high‐dose chemotherapy
- HIF
hypoxia‐inducible factor
- HIFU
high‐intensity focused ultrasound
- IC
induction chemotherapy
- ID2
Iinhibitor of DNA binding 2
- INSM2
insulinoma‐associated 2
- JAK2
Janus kinase 2
- LTLD
lyso‐thermosensitive liposomal doxorubicin
- MAPK
microtubule associated protein kinase
- Mash1
mammalian achaete‐scute homolog 1
- Max
MYC‐associated factor X
- MCL‐1
myeloid cell leukemia 1
- MDM2
mouse double minute 2 homolog
- MDR
multidrug resistance
- MEK
mitogen‐activated protein
- MIBG
metaiodobenzylguanidine
- MR
magnetic resonance
- MRI
magnetic resonance imaging
- MTD
maximum tolerated dose
- mTOR
mammalian target of rapamycin
- NANT
New Advances in Neuroblastoma Therapy
- NB
neuroblastoma
- NCCs
neural crest cells
- NEPENTHE
Next Generation Personalized Therapy
- NF1
neurofibromatosis type I
- NGF
neural growth factor
- NHLH2
Nescient helix‐loop‐helix protein 2
- NMC
NUT Midline Carcinoma
- Notch 1
Notch receptor 1
- NTRK
neurotrophic tropomyosin receptor kinase
- ODC1
ornithine decarboxylase 1
- OS
overall survival
- PDGFR
platelet‐derived growth factor receptor alpha
- PDX
patient‐derived xenograft
- PHOX2
paired‐like homeobox 2b
- PI3K
phosphatidylinositol‐3‐kinase
- PIK3CA
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha
- PKMT
lysine methyltransferases
- PPTP
pediatric preclinical testing program
- PTEN
phosphatase and tensin homolog
- PTPN11
tyrosine‐protein phosphatase non‐receptor type 11
- RAS
rat sarcoma
- RIPK1/3
receptor‐interacting serine/threonine‐protein kinase 1/3
- RP2D
recommended phase 2 dose
- RSPO2
roof plate‐specific spondin 2
- RTK
receptor tyrosine kinase
- SNP
single nucleotide polymorphism
- Sox10
SRY‐related HMG‐box gene 10
- TCF3
transcription factor 3
- TLRs
Toll‐like receptors
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- TRAIL
tumor necrosis factor (TNF)‐related apoptosis‐inducing ligand
- TrK
tropomyosin receptor kinase
- TWIST1
twist‐related protein 1
- UTR
untranslated region
- VEGF
vascular endothelial growth factor
- WNT1
WNT family member 1
- ZA
zoledronic acid
1. INTRODUCTION
Neuroblastoma (NB) is a heterogeneous solid tumor that arises in the sympathetic nervous system. NB tumors most commonly develop in the abdomen and are most frequently localized in the adrenal gland. 1 , 2 NB tumors account for 7%–8% of childhood malignancies, and approximately 650 NB patients are diagnosed each year in the United States. However, NB accounts for approximately 15% of all pediatric cancer deaths. While the survival for patients with low‐ and intermediate‐risk disease approaches 100%, the 5‐year survival rate for high‐risk NB patients is less than 50%. 2 , 3 , 4 , 5 , 6 There are some ethnic differences in NB, with the disease being more prevalent in those with European ancestry, and African‐American children tending to exhibit higher‐risk disease. 7 NB tumors have also been classified as an embryonic tumor, because evidence suggests that such tumors originate from neural crest cells (NCCs) during fetal development. 8
NB develops from the cells of the sympathetic nervous system, particularly sympathoadrenal progenitor cells, which differentiate into adrenal chromaffin cells and sympathetic ganglion cells. 5 The transformation of sympathoadrenal precursors into sympathetic ganglia and adrenal chromaffin cells requires several factors, including overexpression of neural growth factor (NGF) and MYCN, SRY‐related HMG‐box gene 10 (Sox10) and mammalian achaete‐scute homolog 1 (MASH1) induced by bone morphogenetic proteins (BMPs). 9 The transformation of persistent resting progenitor cells into NB cells requires anaplastic lymphoma kinase (ALK) mutations and MYCN amplification, and involves transcription factors including Sox11, nescient helix‐loop‐helix protein 2 (NHIH2), Twist‐related protein 1 (TWIST1), achaete‐scute family bhlh transcription factor 1 (ASCL1), insulinoma‐associated 2 (INSM2), and transcription factor 3 (TCF3). 9 Several transcriptional regulators are involved in deciding the fate of cells with sympathetic lineages, such as MASH1, inhibitor of DNA binding 2 (ID2), dHAND, hypoxia‐inducible factor (HIF), and paired‐like homeobox 2b (PHOX2), all of which likely play roles in the pathogenesis of NB. 10 , 11 , 12 , 13 , 14 , 15 , 16 Elevated levels of N‐Myc protein produced due to MYCN amplification play an important role in the pathogenesis of NB. The MYCN locus encodes MYCNOS (antisense transcript), and this encodes N‐CYM. 17 Inhibition of GSK3β (glycogen synthase kinase 3β)‐driven N‐Myc degradation leads to N‐CYM, stabilizing N‐Myc. 5 ALK also plays a significant role in the transformation of sympathoadrenal cells into NB cells. The expression of ALK correlates with an inferior prognosis. 18 , 19 Overall, it is thought that activated ALK collaborates with MYCN to markedly accelerate NB growth. 20
NB has been divided into four major stages; localized stages L1 and L2, disseminated stage M, and disseminated stage MS, which occurs in patients younger than 18 months of age. Various prognostic parameters have been used to classify NB tumors, including the degree of differentiation, presence or absence of stroma, mitosis‐karyorrhexis index, patient age, NB stage, histological category, MYCN oncogene status, DNA ploidy, and chromosome 11q status. 21 , 22 Further, these parameters also help to classify patients into four groups based on their risk of death: (i) very low, (ii) low, (iii) intermediate, and (iv) high‐risk. 23
Patients with low‐risk NB have a favorable prognosis and a 5‐year survival rate of more than 90%. 9 However, 60% of patients have high‐risk NB, and the prognosis of treatment in such patients remains poor. 9 Patients categorized as having low‐risk NB are typically provided minimal therapy, and some children are curatively treated by surgery alone, or may experience spontaneous tumor regression. 5 , 24 , 25 Milder chemotherapy is administered to patients in the intermediate‐risk group, and they may also be treated by removing the remaining tumor mass. 26 The current standard treatment for high‐risk NB includes three treatment blocks—(i) induction, (ii) consolidation, and (iii) maintenance. 27 Induction chemotherapy (IC) aims to reduce the tumor by shrinking it and also reducing the risk of metastasis via chemotherapy and surgery. 28 , 29 The consolidation block involves the administration of HDC (high dose chemotherapy) accompanied by ASCT (autologous stem cell transplantation) and radiotherapy. 28 , 29 Maintenance involves immunotherapy using anti‐disialoganglioside (GD2) monoclonal antibody (mAb) with cytokines and differentiation therapy using 11‐cis retinol. 28 , 29 The IC generally includes platinum compounds (carboplatin, cisplatin), etoposide, cyclophosphamide, and vincristine (COJEC) 1 , 5 , 25 , 30 , 31 , 32 , 33 , 34 and in North America also includes topoisomerase inhibitors (topotecan) and anthracyclines. 35 , 36 However, the response to IC is not sufficient in 1/3 of children with high‐risk NB. 34 , 37 During induction therapy, surgery is performed to resect primary tumor tissue. 38 , 39 The consolidation phase can provide improved event‐free survival (EFS), especially in NB patients who have undergone tandem ASCT. 6
Approximately half of high‐risk patients do not respond to the first‐line therapy protocol or relapse in the first 2 years after treatment. 34 , 37 , 40 , 41 The outcome for high‐risk NB patients is very poor, with a 5‐year survival rate of less than 50%. 25 In addition, the response to current standard treatments is highly heterogeneous, varying from total regression to the development of multi‐drug resistance and severe toxicities. 1 , 42 NB is a complex disease that exhibits biological, clinical, morphological, and genetic heterogeneity, making it arduous to develop a successful universal therapy. 27 , 43 , 44 The high tumor heterogeneity, drug resistance, and severe toxicities associated with standard treatment in children all lead to relatively poor outcomes for NB treatment. It is also important to note that oncology drugs have the lowest LOA (likelihood of approval) from phase I (6.7%) compared with drugs used for other diseases (allergy, dermatology, urology, autoimmune disease, and ophthalmology). 45 Complicating matters, the current treatments approved for NB have limited targeted specificity. 26 Thus, efforts should be strengthened to understand the tumor biology and develop new, more effective therapies for patients with NB.
Significant progress has been made to comprehend the molecular mechanisms involved in the etiology and pathogenesis of NB, and these investigations have identified new therapeutic targets. In particular, genome‐wide association studies, transcriptomics, genome sequencing, and high‐throughput genome analysis have revealed genetic alterations and disrupted pathways that are responsible for NB growth and development. Many of these are being tested as druggable targets for patients with NB. The use of molecular targeted therapy focused on genomic aberrations and disrupted pathways represents a new approach for the treatment of NB that may result in improved efficacy and reduced toxicity. This review provides an overview of the current state‐of‐the‐art molecular understanding of the development and progression of NB, with a particular emphasis on genetic aberrations and disrupted molecular pathways. As noted, several excellent reviews have been published, covering recent advances made in pathogenesis, diagnosis, and clinical management of NB and interested readers are referred to those excellent publications. 4 , 9 , 22 , 29 , 42 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 In this review, we will emphasize how these advances in knowledge about molecular pathogenesis can be translated to developing molecular targeted therapies for NB management, especially personalized therapies. We also attempt to provide insights into the promise of combination therapy using inhibitors that target many different pathways and their investigation in clinical trials. Further, we point out the future directions that should be taken to improve or develop effective targeted therapy to improve the survival rates of patients with NB while reducing treatment‐associated toxicity.
2. MAJOR MOLECULAR PATHWAYS INVOLVED IN NB TUMORIGENESIS
Recent research has been focused on identifying the molecular mechanisms involved in the NB pathogenesis. Ongoing investigations have identified several signaling pathways required for the growth and progression of NB (Figure 1), or that contribute to the resistance of the disease to conventional treatments. In this section, we provide a comprehensive summary of the role of signaling pathways involved in the pathogenesis of NB.
Figure 1.
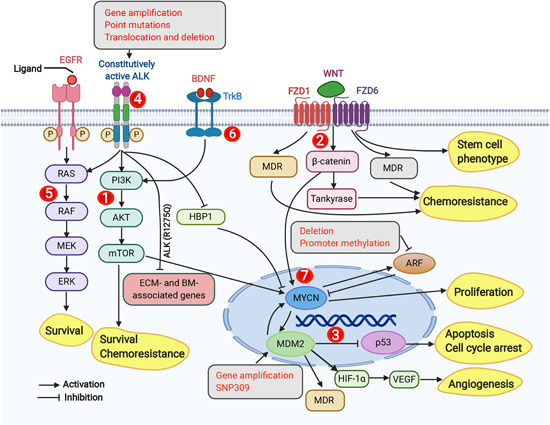
Overview of the molecular signaling pathways implicated in neuroblastoma. The signaling pathways described to play a role in neuroblastoma cells are (1) PI3K/AKT/mTOR pathway—promotes NB cell survival and chemoresistance; (2) Wnt signaling, which is involved in drug resistance, stemness, and increases MYCN levels; (3) p53‐MDM2 pathway, where MDM2 inhibits p53 activity, promotes angiogenesis, increases MYCN translation, and promotes drug resistance. Single nucleotide polymorphisms (i.e., SNP309) and gene amplification increase MDM2 expression. In the p53‐MDM2 pathway, activated p53 is involved in apoptosis and growth arrest; (4) ALK signaling activates PI3K/AKT/mTOR, RAS‐MAPK, and MYCN expression, and an ALK(R1275Q) mutant inhibits the expression of BM‐ and ECM‐associated genes; (5) RAS‐MAPK signaling promotes the survival of neuroblastoma cells and is activated by EGFR signaling; (6) TrkB signaling activates the PIK/AKT/mTOR signaling; (7) MYCN signaling promotes NB cell proliferation and activates MDM2 expression. Gray boxes in the figure represent genetic aberrations (i.e., gene amplification, point mutations, translocations, and deletions) and promoter methylation, and yellow boxes represent downstream biological phenotypes (i.e., survival, chemoresistance, angiogenesis, apoptosis, cell cycle arrest, proliferation, and stemness) in neuroblastoma cells. AKT, protein kinase B; ALK, anaplastic lymphoma kinase; BDNF, brain‐derived neurotrophic factor; BM, basement membrane; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; ERK, extracellular signal‐regulated kinase; FZD1, frizzled‐1; FZD6, Frizzled‐6; HBP1, HMG‐Box transcription factor 1; HIF, hypoxia‐inducible factor; MDR, multidrug resistance; MDM2, mouse double minute 2 homolog; MEK, mitogen‐activated protein; mTOR, mammalian target of rapamycin; NB, neuroblastoma; PI3K, phosphatidylinositol‐3‐kinase; RAS, rat sarcoma; TrKB, tropomyosin receptor kinase B; VEGF, vascular endothelial growth factor [Color figure can be viewed at wileyonlinelibrary.com]
2.1. phosphatidylinositol‐3‐kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway
PI3K/Akt/mTOR pathway is an important pro‐survival signaling pathway which is activated in most NBs. 73 Immunohistochemistry using a human tissue microarray consisting of 116 primary NB specimens has been performed to analyze the phosphorylation of Akt. The study has revealed that Akt phosphorylation at serine 473 (S473) and/or threonine 308 (T308) is a common event in NB tissue. 74 Moreover, Akt is highly phosphorylated, with S473 phosphorylation being present in 61.2% of primary NBs, with T308 being present in 62.9% of primary NBs. 74 Further, that study has also revealed that 66 out of the 116 samples (56.9%) are positive for both antibodies (p‐Akt [S473] and p‐Akt [T308]), while 5 of the 116 (4.3%) exhibit S473 phosphorylation but not T308 phosphorylation, and 7 of 116 (6%) are found to have T308 phosphorylation but not S473 phosphorylation. 74 A small percentage (32.8%; 38 out of 116 NB tumors) have low phosphorylation at both the S473 and T308 sites. 74 In the same study, strong phosphorylation (55.2%) of the S6 ribosomal protein (a target of mTOR) has been observed in NB samples. 74 The study has also confirmed that there is a correlation between Akt phosphorylation and MYCN amplification, which is significant for Akt phosphorylation at T308 or at both sites (S473 and T308), but not S473 phosphorylation alone. 74 A study by Johnsen et al. 75 has shown that Akt expression could be detected in all NB primary samples, but nonmalignant adrenal medullas lack this Akt expression. In addition, it has been found that catalytic p110α and the regulatory p85α isoforms of PI3K are more highly expressed in NB cell lines and primary NB samples compared with normal adrenal gland tissue. 76
The PI3K/Akt pathway is upregulated in many malignancies through several mechanisms, including deletions or mutations of components of the signaling cascade (PIK3CA—phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha or PTEN—phosphatase and tensin homolog). 74 However, deletions of the PTEN tumor suppressor gene (an antagonist of PI3K signaling) affect a small fraction of NB tissues. For instance, a screening study of 45 NB patients has revealed that homozygous deletions of PTEN are only found in 5% of primary tumors (2 of 41). 77 Further, mutation analyses of the PIK3CA gene have also shown that mutations are only present in 2 out of 69 NB samples (27 NB‐derived cell lines and 42 primary tumors). 78 These results suggest that the frequency of genetic aberrations (PTEN and PIK3CA) is relatively low in NBs. Nevertheless, activating mutations in ALK are observed in hereditary NB. 79 , 80 , 81 , 82 A study by Osajima‐Hakomori et al. 83 has shown that suppression of ALK by RNA interference (RNAi) significantly reduces Akt phosphorylation and decreases the survival of NB cells.
NBs have been characterized to exhibit abnormal receptor tyrosine kinase (RTK) activity, which occurs mainly due to mutation or overexpression of growth factor receptors or their ligands. This has been involved in increased activation of the PI3K/Akt/mTOR pathway. 84 Further, various growth factors have been found to affect the PI3K/Akt/mTOR pathways in NB. In particular, BDNF (brain‐derived neurotrophic factor) is a growth factor that transmits its signal via tyrosine kinase receptor tropomyosin‐related kinase B (TrkB) and promotes survival, and also confers chemoresistance by engaging the PI3/Akt/mTOR pathway. 85 , 86 An exogenous supply of BDNF or ectopic expression of TrkB causes increased Akt phosphorylation, which is associated with decreased sensitivity towards DNA damaging agents. 85 , 86 In NB, the PI3/Akt/mTOR pathway has been found to affect several pathways/proteins to enhance the NB phenotype. For example, the PI3/Akt/mTOR pathway contributes to MYCN stabilization, and MYCN, in turn, contributes to several processes associated with malignancy, such as proliferation, angiogenesis, and altering metabolic programming. 87 , 88 A study by Chesler et al. 89 has shown that the PI3/Akt/mTOR pathway inhibition leads to a decrease in the levels of N‐Myc protein in NB.
2.2. WNT signaling pathway
WNT/β‐catenin signaling has been found to be responsible for NB progression and development. Enhanced WNT/β‐catenin signaling augments the MYCN levels in non‐MYCN‐amplified NB cells. 90 A study by Zhang et al. 90 , 91 has shown that silencing WNT family member 1 (WNT1) expression by RNAi reduced the viability of SH‐SY5Y NB cells. Another study by Zins et al. 92 has shown that knockdown of frizzled class receptor 2 (FZD2) inhibits the proliferation of the NB SK‐N‐AS and SK‐N‐DZ cell lines, and reduces the WNT3A‐facilitated SK‐N‐DZ cell migration and WNT5A‐facilitated SK‐N‐AS cell migration. Wnt/β‐catenin signaling also plays a key role in the chemoresistance of NB cells. For instance, higher expression of FZD1 and multidrug resistance (MDR) is present in doxorubicin‐resistant NB cells compared with non‐resistant NB cells. 93 Knockdown of FZD1 in LAN1 NB cells also reduces the expression of MDR1 and restores sensitivity to doxorubicin. 93 WNT3A/roof plate‐specific spondin 2 (RSPO2) signaling plays a critical role in regulating cyclin D1, BMP4, and the phosphorylation of RB protein, suggesting that WNT3a signaling has a role in the differentiation or progression of NB. 94 Additionally, FZD6 (a Wnt receptor) positive NB cells are resistant to doxorubicin, form neurospheres, and express elevated levels of Twist1 and Notch receptor 1 (Notch 1) (mesenchymal markers), 95 implying that FZD6 can be used as a marker of NB with stem cell properties. These observations indicate that Wnt signaling represents a promising target for therapeutic interventions in NB.
2.3. p53‐mouse double minute 2 homolog (MDM2) pathway
The MDM2 oncogene is an E3 ubiquitin ligase that inhibits p53 activity. 96 Under unstressed conditions, MDM2 binds to the transactivation domain of p53 and targets p53 for ubiquitination and subsequent degradation by the proteasome. 97 , 98 , 99 Studies have shown that amplification of MDM2 is found in a variety of malignancies, including NB. 100 In addition, elevated MDM2 levels exist in NB without MDM2 gene amplification, and this is due to the existence of a single nucleotide polymorphism (SNP) in the MDM2 promoter. 101 In general, MDM2 overexpression in human cancers has been found to be correlated to a poor prognosis, and associates with metastasis and advanced stages of the disease. 102 The enhanced MDM2 activity leads to attenuation of p53 activity, and thus results in increased tumor formation. In addition, MDM2 has oncogenic functions independent of p53 that have been reported to be involved in NB growth and progression. In particular, MDM2 binds directly to the 3′‐untranslated region (3′‐UTR) of vascular endothelial growth factor (VEGF) and increases VEGF messenger RNA (mRNA) stabilization and translation, which promotes NB growth. 103 In addition, MDM2 binds MYCN mRNA in the 3′‐UTR and thereby increases the stability and translation of MYCN mRNA in NB cells. 104 Elevated MDM2 has also been found to cause multidrug resistance in NB cells. 105 Thus, the oncogenic potential of MDM2 makes it a potential target for anticancer therapy in NB cells.
2.4. ALK signaling pathway
NB tumor tissues express full‐length ALK and exhibit single‐base missense mutations in the kinase domain of ALK, which promote ligand‐independent signaling. 106 , 107 , 108 Single‐base missense mutations have been found in sporadic as well as familial NB 79 , 82 , 109 ; and the mutations at F1174L, R1275Q, and F1245C comprise around 85% of all ALK mutations present in NB. 109 The most common mutations are R1275Q and F1174L, both of which are present within the kinase domain of ALK, and these mutations lead to ligand‐independent autophosphorylation of ALK and increased kinase activity. 81 , 110 , 111 The activation of ALK in NB leads to enhanced survival, migration, and cell proliferation. 110 Further, ALK mutations have been found to hyperactivate rat sarcoma (RAS)‐microtubule associated protein kinase (MAPK) signaling in NB, thus promoting the development of cancer. 61 Both the wildtype and mutant forms of ALK induce MYCN transcription in NB cells. 112 In fact, a study conducted by Berry et al. 113 has demonstrated that ALK mutations potentiate the oncogenic activity of MYCN in NB cells.
Apart from ALK mutations, around 2%–3% of cases involve gene amplification, leading to increased expression of ALK protein and constitutive kinase activity. 83 , 114 , 115 It is also interesting to note that ALK is coamplified with MYCN, as the two genes are in proximity at 2p23 and 2p24, respectively. 80 , 109 , 116 A study by Chang et al. 117 has demonstrated that high MYCN expression is present in 24 (39.3%) and ALK protein in 25 (41%) of the 61 NB tumors analyzed. A mechanistic study has indicated that ALK plays a role in the positive regulation of MYCN activity through suppression of HMG‐box transcription factor 1 (HBP1) expression in NB cells. 118 Further, large deletions and translocations lead to truncation of the extracellular region of ALK, providing another mechanism of ligand‐independent ALK signaling in NB. 119 , 120 , 121 At relapse, NB tumors have been found to exhibit an increased frequency of ALK mutations. 122 , 123 , 124 In fact, deep sequencing has revealed that F1174 and R1275 ALK mutations are present during diagnosis in 10% of cases, and these mutations are undetected by Sanger sequencing. 125 ALK (R1275Q) is an activating mutation found in sporadic as well as familial NB patients. 126 It downregulates the expression of extracellular matrix (ECM) and basement membrane (BM)‐associated genes in NB tumors. 126 In addition, tumors with ALK (R1275Q)/MYCN have been found to exhibit reduced ECM/BM‐related protein expression compared with tumors with MYCN overexpression alone. 126 Likely due to these changes in ECM/BM proteins, enhanced metastasis and invasion have been found in ALK (R1275Q)/MYCN mice. 126 In NB cells, several miRNAs have been found to regulate ALK protein expression. Both miR‐424‐5p and miR‐503‐5p downregulate the ALK expression levels and decrease cell viability in ALK‐positive NB cells. 127 A phosphoproteomics analysis has shown that ALK also promotes NB growth via the JNK signaling pathway. 128
2.5. RAS/MAPK signaling pathway
The RAS/MAPK pathway is involved in the growth and survival of various pediatric malignancies, including NB. It has been estimated that 3%–5% of patients have mutations at the genetic level in the RAS‐MAPK pathway, while relapsed tumors (~80%) contain genetic mutations associated with this pathway. 53 , 63 In relapsed NB, the activating mutations of the RAS‐MAPK pathway detected include mutations in neurofibromatosis type I (NF1), v‐raf murine sarcoma viral oncogene homolog B1 (BRAF), tyrosine‐protein phosphatase nonreceptor type 11 (PTPN11), fibroblast growth factor receptor 1 (FGFR1), KRAS, NRAS, HRAS, and ALK. 123 , 129 Other nonmutational mechanisms of MAPK pathway activation include signaling via the tyrosine kinase receptors epidermal growth factor receptor (EGFR), ALK, and Erb‐b2 receptor tyrosine kinases 2 (ERBB2). 63 Further, it has been found that mutations in the capicua transcriptional repressor (CIC) gene activate the RAS‐MAPK pathway, and such activation is responsible for increasing the tumorigenicity of NB cells. 130 The effect of mutations of ALK, RAS‐MAPK, RAS, NF1, or BRAF, and their relationship to the sensitivity to MEK inhibitors have also been discussed in the past. In particular, it has been found that a nanomolar concentration of mitogen‐activated protein (MEK) inhibitor is sufficient to cause cell cycle arrest in NB cell lines with mutated RAS or BRAF genes. 130 On the contrary, cell cycle inhibition in NF1‐ and ALK‐mutated NB cells is less effective. 130
3. PRECLINICAL STUDIES OF TARGETED NB THERAPY
3.1. Targeting genetic and protein aberrations for NB therapy
As described above, ongoing investigations have identified numerous genetic and protein aberrations as potential therapeutic targets for NB (Figure 2), many of which have been evaluated in preclinical models of NB. Preclinical studies have identified several inhibitors (Figure 2) that can be pursued in clinical trials for NB patients.
Figure 2.
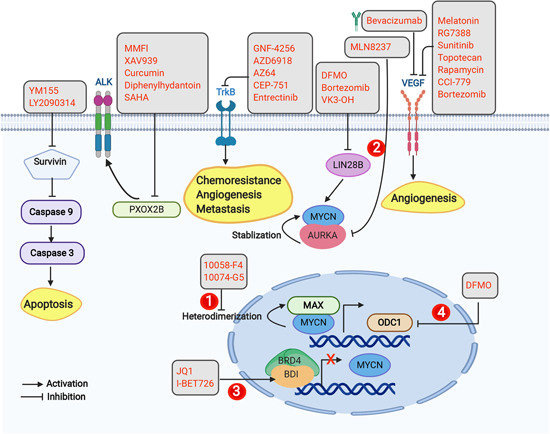
Targeted therapy involving genetic/protein aberrations in neuroblastoma cells. Some of the approaches employed under targeted therapy involve small molecule inhibitors of TrkB, VEGF, LIN28B, survivin, and Phox2b. Another approach is inhibition of MYCN using one of several strategies: (1) inhibition of MYCN/MAX heterodimerization; (2) inhibition of Aurora A kinase; (3) inhibition of bromodomain and extra‐terminal domain (BET) protein; and (4) inhibition of ODC1. Gray boxes represent inhibitors of survivin, PHOX2B, TrkB, LIN28B, VEGF, AURKA, MYCN/MAX heterodimerization, ODC1, and BET; yellow boxes represent downstream effects, including VEGF, TrkB, and caspase 3 activation. AURKA, Aurora A kinase; BDNF, brain‐derived growth factor; DFMO, difluoromethylornithine; Max, MYC‐associated factor X; MMF, mycophenolate mofetil; ODC, ornithine decarboxylase; Phox2b, paired‐like homeobox 2b; SAHA, suberoylanilide hydroxamic acid; TrKB, tropomyosin receptor kinase B; VEGF, vascular endothelial growth factor [Color figure can be viewed at wileyonlinelibrary.com]
3.1.1. TrK inhibitors
TrK belongs to the neurotrophin receptor family and plays a critical role in NB biology. Elevated expression of TrkB is correlated with high‐risk NB and poor survival, while increased TrkA expression is correlated with lower‐risk NB and tumors that are prone to spontaneous regression. 131 , 132 , 133 , 134 In fact, patients with an advanced stage of NB and MYCN amplification have decreased TrkA expression. 132 , 134 , 135 TrkB has been found to promote resistance to etoposide, doxorubicin, and cisplatin in NB. 86 TrkB has also been reported to increase angiogenesis and metastasis. 136 , 137 , 138 Thus, TrkB is a target for NB treatment. In preclinical models, Trk inhibitors GNF‐4256 and AZD6918 slow the growth of xenograft tumors, and combining a Trk inhibitor with chemotherapeutic drugs leads to significantly better effects compared with treatment with either agent alone. 139 , 140 AZ64, an inhibitor of neurotrophic tropomyosin receptor kinase (NTRK), has been found to inhibit TrkB, and enhance the efficacy of both local radiation and chemotherapy in a NB xenograft model. 141 This data provides an indication that Trk inhibition may be a useful adjunct to existing chemotherapy. Further, CEP‐751 (KT‐6587) has been found to exhibit effective antitumor activity against NB cells and xenografts expressing elevated levels of TrkB. 142 , 143 Entrectinib (RXDX‐101) is another inhibitor of TrkA/B/C, and has been found to inhibit NB tumor growth, while entrectinib also augments the tumor growth inhibition of temozolomide when used in combination therapy in a xenograft mouse model. 144
3.1.2. MYCN inhibitors
The MYCN oncogene encodes a transcription factor that controls several cellular processes. MYCN gene amplification has been detected in 20%–30% of NB cases, and this amplification strongly correlates with the stage and aggressiveness of the disease. 145 , 146 A study conducted on 110 infants with stage 4s NB has indicated that patients with MYCN amplification have a worse survival compared with patients without MYCN amplification. 147 Another study involving 2660 stage 1 or 2 NB patients has shown that patients diagnosed with tumors with MYCN amplification have a significantly worse EFS and inferior overall survival (OS) compared with patients without MYCN amplification. 146 In a recent analysis of nearly 6000 patient samples, the presence of both homogenous and heterogenous MYCN amplification confers a worse EFS and OS compared with the prognosis of patients with wild‐type MYCN. 148 These findings have supported that MYCN promotes angiogenesis, survival, and metastasis in NB, and inhibits immune surveillance.
Various strategies have been proposed to downregulate MYCN to decrease NB development, growth, and proliferation. One of the first strategies employed is targeting MYCN/MYC‐associated factor X (Max) interactions. After amplification, MYCN forms heterodimers with MAX to act as a transcription factor and promote NB growth. 149 , 150 Two compounds, 10058‐F4 and 10074‐G5, have been found to block heterodimerization, and treatment of MYCN‐amplified models of NB with these compounds induced differentiation and apoptosis in vitro conditions, and suppressed the growth of xenograft tumors. 151 , 152 , 153
MYCN is stabilized by Aurora A kinase (AURKA) via protein‐protein interactions, which renders MYCN less prone to degradation by the proteasome. 53 Thus, AURKA inhibition is a secondary approach to inhibit MYCN in NB cells. Treatment of IMR32 NB cells with MLN8237 (alisertib) (a specific aurora kinase inhibitor) induced cell senescence, cell growth inhibition, G2/M arrest, and MYC degradation, and induced inhibition of tumor growth in a xenograft mouse model. 154 Another approach to inhibit MYCN includes the use of inhibitors that can inhibit bromodomain and extra‐terminal domain (BET) family of proteins. 53 The BET proteins act as transcriptional regulators of many genes, including MYCN. 53 BET inhibitors such as JQ1 cause bromodomain inhibition, which downregulates MYCN in NB cells, resulting in apoptosis and cell cycle. 155 Further, GSK1324726A (I‐BET726) is involved in BET inhibition and decreases cell growth, induces cytotoxicity, and directly inhibits MYCN expression in NB cells. 156
Yet another approach for targeting MYCN involves inhibition of ornithine decarboxylase 1 (ODC1). 53 The ODC1 gene encodes an enzyme that catalyzes the rate‐limiting step of polyamine synthesis. 53 Polyamines act as cationic chaperones to support the MCYN activity in NB cells via covalent and ionic mechanisms, and hence are responsible for maintaining the phenotype of NB. 53 Difluoromethylornithine (DFMO), an ODC1 inhibitor, is an FDA‐approved treatment for trypanosomiasis. A study by Hogarty et al. 157 has shown that disabling ODC1 by DFMO inhibited the proliferation of NB cell lines. The same study has also shown that the treatment of NB mouse model with DFMO is effective in delaying tumor initiation and enhancing the therapeutic effects of chemotherapy to increase the survival of mice with established tumors. 157 Further, combined treatment using DFMO with celecoxib (a nonsteroidal anti‐inflammatory drug) have synergistic antitumor effects in NB models exhibiting ALK mutation, MYCN amplification, and TP53 mutation with multidrug resistance. 158
3.1.3. Baculoviral IAP repeat containing 5 (BIRC5) and survivin inhibitors
The BIRC5 gene encodes human survivin, which is located on the long arm of chromosome 17 (q25). 159 Advanced‐stage NB often exhibits a gain of the chromosomal 17q25 region, 160 and the BIRC5 gene (present in this 17q25 region) is gained in 49% of NB tumors. 161 Increased survivin expression is correlated with a poor prognosis in NB patients. 160 In fact, the levels of survivin mRNA are higher in individuals older than 12 months, in advanced stages of disease (stages 3 and 4), and have a strong correlation with low levels of TrkA expression. 160 The elevated levels of survivin expression in NB are also correlated with MYCN amplification. 160 Survivin also increases glycolysis and resistance to treatment in NB. 162 , 163 In addition, survivin exerts antiapoptotic effects by inhibiting caspase 9 and enhancing resistance to apoptosis induced by staurosporine in NB cells. 164 Survivin has also been found to provide resistance to immune‐ or drug‐mediated cell death. 165 For example, a study of several NB cell lines has found that NB10, NB cell line that exhibits the least survivin expression, was the most sensitive to both TRAIL (tumor necrosis factor [TNF]‐related apoptosis‐inducing ligand) and etoposide induced cell death. 165 On the contrary, the NB7 and NB16 cell lines, which have an abundance of survivin, were more resistant to TRAIL‐ and etoposide‐induced cell death. 165 Survivin has also been found to cause the stabilization of the microtubules in the chromosomal passenger complex (CPC). 166
Various inhibitors have been found to target survivin in preclinical studies of NB. For example, YM155 decreases the survivin expression, inhibits the proliferation of and induces apoptosis in NB SH‐SY5Y cells. 167 The same study has also shown that reduced expression of survivin after treatment with YM155 is effective to sensitize SH‐SY5Y cells to cisplatin (chemotherapeutic agent), and induces tumor regression and apoptosis in SH‐SY5Y xenograft model. 167 Research conducted by Kunnimalaiyaan et al. 168 has demonstrated that LY2090314 (a GSK‐3 inhibitor) is capable for causing growth inhibition and inducing apoptosis in NB cells, and also reducing the survivin level. Withanolides (WA, WGA, WGB‐DA, WGA‐TA) have also been found to be cytotoxic to NB cells, potentially because they downregulate survivin in NB cells. 169 Noscapine, a nontoxic natural compound, induces apoptosis via downregulation of survivin in both p53 wild type and null NB cells. 170 Interestingly, the antidiabetic drug troglitazone also holds the capacity to sensitize NB cells to TRAIL‐induced apoptosis via downregulation of survivin. 171
3.1.4. VEGF inhibitors
VEGF is a 45 kDa dimeric glycoprotein that plays an important role in the formation of blood vessels (angiogenesis). 172 Apart from the functions of VEGF in angiogenesis and vascular permeability, the autocrine signaling of VEGF plays a role in cancer stem cells, and the resistance of tumor cells to treatments. 173 , 174 The human VEGF‐A gene is positioned on chromosome 6 and contains eight exons. 175 Alternate splicing of the VEGF gene generates several isoforms, including VEGF121, VEGF189, and VEGF165, which are expressed in different human tumors. 176 , 177 Among the different isoforms of VEGF, VEGF165 mRNA is the predominant isoform expressed in human NB cells. 178 Increased expression of VEGF is found more frequently in advanced‐stage (stages 3 and 4) NB tumors compared with low‐stage (stages 1, 2, and 4S) tumors. 179 Increased VEGF‐A levels have been observed in the serum and plasma of NB patients. 180
The activity of several VEGF inhibitors has been investigated in preclinical models. For instance, melatonin has been found to inhibit angiogenesis in human SH‐SY5Y NB cells by downregulating VEGF. 181 RG7388, an MDM2 inhibitor, causes tumor growth inhibition in p53 wildtype NB cells, and inhibits HIF‐1α/VEGF signaling, and alters angiogenesis. 182 Sunitinib, a receptor tyrosine kinase inhibitor, has been found to impair NB growth and enhance the cytotoxic activity of chemotherapeutic drugs, and decreased MYCN and VEGF expression in NB cells. 183 Topotecan (topoisomerase inhibitor), is capable of inhibiting HIF‐2α and HIF‐1α accumulation and also transcriptional activity, and thus inhibits VEGF expression in NB cells. 184 mTOR inhibitors, namely rapamycin and CCI‐779, have been found to reduce VEGF‐A secretion, inhibit mTOR,and induce apoptosis and cell cycle arrest in NB cells. 75 Bortezomib, a proteasome inhibitor, decreases cellular proliferation and induces cell cycle arrest, and also bortezomib treatment leading to a reduction of 76.3% of VEGF levels in treated tumors as compared with controls. 185 Imatinib mesylate has also been found to inhibit the cellular growth of NB both in vitro and in vivo, and the inhibition of cellular growth is correlated with the decrease in expression of platelet‐derived growth factor receptor alpha (PDGFR), c‐kit, and VEGFR. 186
3.1.5. PHOX2B inhibitors
PHOX2B is the first genetic predisposition identified in NB. An estimated 6.4% of patients with hereditary NB have germline mutations of PHOX2B. 16 , 187 , 188 , 189 However, mutations in PHOX2B are rarely seen in the germline and tumor cells of sporadic NBs. 189 , 190 Loss‐of‐function mutations in the PHOX2B gene impede NB differentiation by disrupting calcium regulation. 191 A study by Bachetti et al. 192 has demonstrated that increased expression of PHOX2B in NB cells leads to an increase in ALK protein. In particular, PHOX2B contributes to the pathogenesis of NB by driving ALK gene expression by directly binding the ALK gene promoter. 192 Elevated levels of Phox2b protein have been found in MYCN‐amplified NB cell lines, while no detectable Phox2b expression is found in NB cell lines with low MYCN expression. 193 A real‐time qPCR study has shown higher PHOX2B expression in NB cell lines compared with normal tissues. 192 A study by van Limpt et al. 16 have described several different frameshift mutations of PHOX2B, such as 284‐291del8nt, 633‐670del38nt, 702‐714dup13nt, 721‐755del35nt, 721‐737dup17nt, and 721‐740del20nt in sporadic NBs. A study by Raabe et al. 189 has described point mutations such as c.667G>C in a human NB‐derived cell line and c.299G>T in sporadic NB with multifocal primaries, as well as frameshift mutations (c.676delG and c.691_698dup8) in hereditary NB. Mutation of PHOX2B is correlated to RAS‐MAPK pathway activation in NB cell lines. 130 It has also been found that one of the downstream targets of PHOX2B is the MSX1 homeobox transcriptional factor, and this transcriptional factor activates the Delta‐Notch pathway in NB. 194
At the preclinical level, various molecules have been found to inhibit Phox2b in NB cells. For instance, mycophenolate mofetil decreases PHOX2B mRNA and protein expression in IMR32 NB cells, and induces Caspase 3/7 cleavage and apoptosis in NB cells. 195 In another study by Di Zanni et al., 196 has demonstrated that curcumin, diphenylhydantoin, and suberoylanilide hydroxamic acid aree found to inhibit Phox2b in NB cells. XAV939 treatment has also been found to downregulate PHOX2A and PHOX2B in NB SH‐SY5Y cells, and the same study has also demonstrated that combination treatment with XAV939 significantly increases the sensitivity of IMR‐32 and SH‐SY5Y cells to doxorubicin treatment in two‐dimensional (2D), as well as three‐dimensional (3D), cultures. 197
As mentioned above, PHOX2B activates the Delta‐Notch pathway in NB cells, 194 and various studies have shown that Notch pathway activation induces NB cell growth and proliferation, suggesting that Notch is involved in the pathogenesis of NB. 198 , 199 A study by Funahashi et al. 200 has shown that treatment with a Notch signaling antagonist inhibits angiogenesis and impairs tumor viability in a mouse model of NB, suggesting that Notch blockade or inhibition represents a potential therapy for NB. Several γ‐secretase inhibitors (GSIs) have been found to block Notch receptor cleavage, and thus GSIs have been developed and tested in Alzheimer's disease as Notch pathway inhibitors. For instance, a dipeptide analog called DAPT (a noncompetitive inhibitor of γ‐secretase) has been found to inhibit Aβ generation in the brain and plasma in an APP transgenic mouse model of Alzheimer's disease. 201 , 202 Likewise, indomethacin (a nonsteroidal anti‐inflammatory agent) is capable of lowering Aβ42 in in vitro and in vivo model systems by targeting the γ‐secretase complex. 203 Based on these observations, GSIs are now being repurposed to test their efficacy against various human malignancies, including NB. For example, DAPT has been found to inhibit cellular growth, promote neuronal differentiation, and induce apoptosis in NB cells. 198 Indomethacin has also been found to inhibit the growth of NB cells, and to enhance the chemosensitivity of NB. 204
3.1.6. LIN28B inhibitors
A SNP has been found in Lin28B and is strongly involved in the development of high‐risk NB. 205 Research has shown that NB cells exhibit overexpression and amplification of LIN28B. 205 , 206 In NB, Lin28B increases the expression of N‐Myc via let‐7 miRNAs repression. 206 Lin28 also promotes the stabilization of downstream AURKA and increases the oncogenic activity of RAN GTPase, hence promoting tumorigenesis. 207 These features suggest that targeting LIN28 may be beneficial in treating NB. A study by Lozier et al. 208 has shown that DFMO treatment reduces LIN28B protein levels in SMS‐KCNR, BE(2)‐C, and CHLA90 NB cells. The same study has also shown that the sensitivity to treatment with DFMO correlates with overexpression of LIN28B (BE(2)‐C>SMS‐KCNR>CHLA90). 208 Bortezomib has also been found to inhibit LIN28B expression, and the combination of bortezomib and DFMO leads to more significant inhibition of LIN28B expression in NB cells than treatment with either agent alone. 209 A derivative of vitamin K3 (VK3‐OH) has also been found to suppress LIN28B at the protein as well as mRNA levels in MYCN‐driven NB cells. 210 Further, JQ1 and panobinostat have been found to synergistically downregulate the gene expression of LIN28B and protein expression of N‐Myc in NB cells. 211 Thus, several different compounds exist that show potent effects against LIN28B.
3.2. Targeting signaling pathways for NB therapy
Extensive preclinical studies have been conducted to investigate the potential of targeting signaling pathways for the treatment of NB. Several potential inhibitors have been identified (Figure 3) employing in vitro and in vivo NB models.
Figure 3.
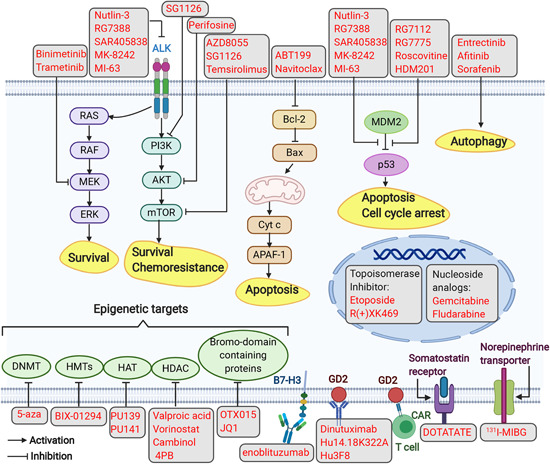
Targeted therapy in neuroblastoma. Several approaches to targeted therapy involve the following modalities: (1) small molecule inhibitors targeting signaling pathways (i.e., PI3K/AKT/mTOR, RAS‐MAPK, p53‐MDM2, Bcl‐2, and ALK); (2) chemical inhibitors inducing autophagy; (3) immunotherapy employing monoclonal antibodies targeting GD2 and B7‐H3, and using CAR T cells targeting GD2; (4) targeting epigenetic regulators; (5) radiopharmaceuticals targeting NET (131I‐MIBG) and the somatostatin receptor (DOTATATE); (6) targeted therapy based on topoisomerase inhibitors or nucleoside analogs. Gray boxes represent inhibitors of MEK, ALK, PI3K, AKT, mTOR, Bcl‐2, p53‐MDM2, epigenetic targets, and topoisomerases; compounds that act as autophagy inducers; nucleoside analogs; monoclonal Abs that target B7‐H3 or GD2; and radiopharmaceuticals targeting the somatostatin receptor and NET; yellow boxes represent downstream biological phenotypes (i.e., survival, chemoresistance, apoptosis, cell cycle arrest, and autophagy). AKT, protein kinase B; ALK, anaplastic lymphoma kinase; APAF‐1, apoptotic peptidase activating factor 1; Bax, BCL2‐associated X; Cyt c, cytochrome c; DNMT, DNA methyltransferases; ERK, extracellular signal‐regulated kinase; HAT, histone acetyltransferases; HDAC, histone deacetylases; HMT, histone methyltransferase; MDM2, mouse double minute 2 homolog; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol‐3‐kinase; RAS, rat sarcoma [Color figure can be viewed at wileyonlinelibrary.com]
3.2.1. Targeting the p53‐MDM2 pathway in NB
In contrast to many other malignancies, NBs generally have intact and wild type p53. 212 , 213 Thus, reactivating the functional activity of wild type p53 by targeting the p53‐MDM2 pathway via MDM2 inhibitors may represent a favorable approach for the NB treatment. Various MDM2 inhibitors have been explored using preclinical NB models. For instance, nutlin‐3 (a p53/MDM2 antagonist) has been found to target the p53/MDM2 interaction and activate the p53 pathway in both chemosensitive and chemoresistant NB cells with wild‐type p53. 214 The same study has also found that nutlin‐3 is effective against chemoresistant p53 wild type NB xenograft tumors in mice. 214 Nutlin‐3a has also been found to increase the antitumor effects of chemotherapeutic drugs via rapid p53 stabilization in NB cells. 215 A second generation nutlin, RG7388, has been developed, which exhibits increased efficacy and lower toxicity than nutlin‐3a. Treatment of NB cells with wild‐type p53 using RG7388 induces p53 activation and apoptosis. 182 Other MDM2 antagonists which have been demonstrated anticancer activity in NB models include SAR405838 (MI‐77301), 216 MK‐8242, 217 MI‐63, 218 RG7112, 219 and RG7775. 220 In addition, a study by Giustiniano et al. 221 has shown that “compound 12” acts as a dual inhibitor of MDM2/p53 and MDM4/p53 complexes and enhances the p53 protein levels in human SHSY‐5Y NB cells. Roscovitine (seliciclib or CYC202) has also been found to inhibit cell viability, activate p53 and p53‐dependent genes (Bax, p21), and inhibit MDM2, N‐Myc, and Akt1 expression. 222
3.2.2. Targeting ALK signaling in NB
Aggressive efforts are underway to develop ALK inhibitors as targeted therapy for NB. Point mutations and amplification of ALK are oncogenic in nature in in vitro as well as in vivo conditions, leading to the constitutive phosphorylation of ALK and other downstream signaling molecules, which is important for the survival and proliferation of NB. 79 , 80 , 81 , 82 Interestingly, in contrast to aberrations like an amplification of MYCN, aberrant ALK is susceptible to inhibition by small molecules. 79 At the preclinical level, it has been found that the R1275Q mutation and ALK amplification in NB cell lines are both sensitive to crizotinib, and regression of xenograft tumors has been observed following treatment. 223 Another compound, CH5424802, inhibits the NB cellular growth, which expresses amplified ALK. 224 Lorlatinib (PF‐06463922), a third‐generation ROS1 and ALK inhibitor, has been found to be effective against NB cells and against ALK‐mutated xenograft mouse models (both F1174L and F124C), as well as crizotinib‐resistant xenografts. 225 In fact, lorlatinib has been found to demonstrate activity surpassing that of crizotinib. 225 Moreover, lorlatinib has been demonstrated strong antitumor in xenograft models containing F1174L, F1245C, and R1275Q mutations. 225
3.2.3. Targeting the PI3K/AKT/mTOR and RAS‐MAPK pathways in NB
AZD8055 is a dual inhibitor of mTORC1‐mTORC2, and has been evaluated in preclinical NB models. It has been found to suppress growth and induce apoptosis in NB cell lines, and also decreases tumor growth in a xenograft model. 226 With regard to targeting the RAS‐MAPK pathway, research has revealed that binimetinib, a MEK1/2 inhibitor, inhibits tumor growth and improves the survival of mouse models of NB. 123 It has also been found that binimetinib exhibits synergistic effects with a ribociclib (cyclin‐dependent kinase [CDK]4/6 inhibitor), and suppresses the growth of tumors in a xenograft mice model. 227
3.2.4. Targeting antiapoptotic proteins and autophagy in NB
Among the numerous antiapoptotic proteins, research has been focused on the Bcl‐2 (B‐cell lymphoma 2) protein in NB cells. Testing new Bcl‐2 inhibitors at the preclinical level is a challenge, as most NB cell lines have low levels of Bcl‐2. 228 , 229 However, two Bcl‐2 inhibitors, venetoclax (ABT199) and navitoclax, have been examined in in vitro and in vivo models of NB. 53 While venetoclax alone had moderate activity, when it was used in combination with alisertib (Aurora A kinase inhibitor), the activity was increased, with NB tumors exhibiting complete regression. 230 Of note, it has been found that Bcl‐2‐dependent NB cells are sensitive to ABT‐199, while myeloid cell leukemia 1 (MCL‐1)‐dependent cells are completely resistant. 231 B‐cell lymphoma‐extra large (BCL‐xL), a transmembrane protein present in the mitochondria, functions as antiapoptotic protein by preventing the release of mitochondrial contents, inducing caspase activation and ultimately apoptosis. 232 Research has been carried out to investigate the combination of a BCL‐xL inhibitor with an Aurora kinase inhibitor in pediatric malignancies. Levesley et al. 232 have demonstrated that when ABT‐263 (a BCL‐xL inhibitor) is used in combination with MLN8237 (an Aurora kinase inhibitor), it increases the sensitivity of MLN8237 cells, apparently via a BCL‐xL inhibition‐based mechanism. ABT‐263 promotes caspase‐dependent apoptosis and impedes cell division in MLN8237 human glioma and pediatric cell lines. 232 The same study has also highlighted that BCL‐xL inhibition causes an increase in apoptosis when used in combination with chemotherapeutic agents. 232 A study by Bate‐Eya et al. 233 has demonstrated that combination treatment with an MCL‐1 inhibitor (1210477) and ABT199 leads to significant synergistic effects against NB cells. The above findings indicate that the use of combinations of targeted therapy may be necessary to overcome the resistance, including amyloid cell leukemia sequence MCL‐1‐dependent resistance, to Bcl‐2 and other targeted inhibitors.
Autophagy is associated with the development of resistance to molecular targeted therapies. The induction of autophagy is an important factor that promotes cancer cells to survive the stress induced by anticancer agents. For instance, entrectinib (an ALK inhibitor) treatment has been shown to induce autophagy in NB cells. 234 The presence of the ALKF1174L mutation in SH‐SY5Y NB cells has been found to make them less sensitive to entrectinib compared with cells without this mutation. 234 Treatment of a cell line harboring mutated ALKF1174L with a combination of entrectinib and chloroquine (CQ) (an autophagy inhibitor) has been found to increase cell death compared with entrectinib treatment alone. 234 Afitinib and Sorafenib (receptor tyrosine kinase inhibitors), also induce autophagy in NB cells. 53 As with entrecitinib, combination treatment using tyrosine kinase inhibitors with an autophagy‐blocking agent (either CQ or Spautin‐1) leads to significant increase in cell death in NB cells. 234 , 235
3.3. Targeting epigenetics in NB
In addition to genetic mutations and SNPs, epigenetic factors may also contribute to disease. Several different agents have been evaluated to target the epigenetic landscape in cancer cells. These include DNA methyltransferases (DNMTs), enzymes responsible for histone modifications (acetylation, methylation, and deacetylation) and chromatin readers.
3.3.1. DNMTs
The expression of DNMTs is altered in NB. 236 Among the different DNMTs, DNMT3A/B expression is higher in high‐risk NBs, and these enzymes are overexpressed in cisplatin‐resistant NB cells. 237 A DNMT inhibitor, 5‐aza‐deoxycytidine (5‐aza or decitabine), has been tested in NB cells and found to reduce their proliferation and colony formation. 238 , 239 It has also been found that 5‐aza can potentiate the efficacy of currently available chemotherapeutic drugs (cisplatin, doxorubicin, and etoposide) in NB cells. 240
3.3.2. Histone modifications
Lysine methyltransferases (PKMT), a specific type of histone methyltransferase (which induce histone modifications), have been targeted in NB. 236 In particular, BIX‐01294, an inhibitor of PKMT, decreases invasion and proliferation and induces apoptosis in NB cells. 241 The addition of acetyl groups to histone lysine residues is another histone modification type, and is catalyzed by histone acetyltransferases (HATs). 242 Various HAT inhibitors, such as PU139 (a HAT pan‐inhibitor) and PU141 (CBP and p300 selective inhibitors), have been investigated and found to reduce the growth of NB cells under both in vitro and in vivo conditions. 243 Histone deacetylases (HDACs) appear to be related to the prognosis of NB. For example, HDAC8 and HDAC10 are overexpressed in NB, and their inhibition significantly reduces the proliferation of NB in vitro 244 , 245 and in vivo. 246 Further, treatment with valproic acid (an HDACi) inhibits cellular proliferation and induces apoptosis and differentiation in NB cells. 247 , 248 Vorinostat (suberoylanilide hydroxamic acid or SAHA), is another HDACi, which results in G2/M phase arrest, followed by activation of the intrinsic apoptosis pathway. 249 In addition, synergistic anticancer effects were observed when SAHA was combined with the proteasome inhibitor MG132 in NB SH‐SY5Y cells. 250
As discussed previously, MYCN amplification is one of the important genetic aberrations associated with NB. HDACs have been found to take part in crosstalk with MYCN in NB. For instance, MYCN directly induces the transcription of SIRT1 (class III HDACs), and pharmacological inhibition via cambinol (a SIRT1 inhibitor) effectively reduces the growth of tumors in an MYCN‐driven transgenic mouse model of NB. 251
3.3.3. Chromatin readers
Chromatin readers identify histone modifications and recruit other proteins to the modification site to initiate or inhibit transcription. These readers include bromodomain‐, tudordomain‐, and chromodomain‐containing proteins. Among these readers, bromodomain (BRD)‐containing proteins can perform several functions, such as chromatin remodeling, histone activation, and transcriptional activation. 252 The effects of BRD inhibition in NB have been evaluated using the BET inhibitor JQ1. The treatment effectively reduces the MYCN levels and cellular growth, and induces apoptosis in vitro as well as in vivo NB models. 155 , 253 In addition, when JQ1 is used in combination with panobinostat (HDACi), there is synergistic growth inhibition and apoptosis in NB cells, which is accompanied by reduced expression of the N‐Myc protein and LIN28B gene. 211 This combination also reduces the expression of N‐Myc in tumor tissues and blocks tumor progression in vivo. 211 Further, a study by Henssen et al. 254 has shown that OTX015, a BRD inhibitor, reduces the viability in MYCN‐amplified NB cells, and shows potency against MYCN‐amplified NB xenografts. The same study has also shown that OTX015 has the potential to disrupt the BRD4‐chromatin interaction and suppress the expression of MYCN in NB cell lines. 254
3.4. Necroptosis induction for NB therapy
Necroptosis is a form of cell death triggered by necrosis. 255 It is driven by the interplay between receptors of necrotic death, their ligands, Toll‐like receptors (TLRs), interferons, and the necrosome complex. 22 In normal cells, necroptosis is impeded via caspase‐8‐mediated cleavage of RIPK1/3 (receptor‐interacting serine/threonine‐protein kinase 1/3). 256 Aggressive NBs frequently lack caspase expression, making them resistant to apoptosis. However, this can be exploited by inducing necroptotic cell death. 22 Necroptosis can be induced in NB cells by increasing cytoplasmic Ca2+ to activate calcium‐calmodulin kinase II, which activates RIPK1. 257 It is important to note that even NB tumors with decreased expression of caspase‐8 may be resistant to cell death instigated by drugs that induce necroptosis, possibly via epigenetic mechanisms. 258 Demethylating agents or HDAC inhibitors may be used to induce cell death in chemoresistant NB tumors with defective caspase 8.
3.5. Targeting HIF in NB
In general, cancer cells are characterized by a hypoxic microenvironment, and such cells adapt to the hypoxic microenvironment via the upregulation of HIF, which mediates the transcription of several target genes that increase cancer progression. 259 Various researchers have described a role for hypoxia in NB tumor initiation, cell survival, and metastasis. 60 A study by Jögi et al. 15 has shown that hypoxia stabilizes both HIF‐1α and HIF‐2α in NB cells. The same study has also demonstrated that pretreatment of SK‐N‐BE(2) NB cells in vitro at 1% O2 cause the tumor cells to have a reduced tumor latency and increases the growth of subcutaneous xenografts compared with SK‐N‐BE(2) cells pretreated under normoxic culture conditions. 15 In addition, a study by Chen et al. 260 has shown that HIF‐1α upregulation in NB promotes the proliferation, invasiveness, and migration of malignant cells via SHH signaling. HIF‐1α activation also provides resistance to antiangiogenic therapies in a xenograft mouse model of NB. 261 Thus, inhibitors targeting HIF‐1α should be developed for NB therapy. Among the various inhibitors under development, PT2385 is a selective antagonist of HIF‐2 that has been found to decrease the expression HIF‐2 target genes and to inhibit tumor growth in a patient‐derived xenograft (PDX) model of clear cell renal cell carcinoma. 262 Persson et al. 263 have investigated the effects of inhibiting ARNT (aryl hydrocarbon receptor nuclear translocator)‐dependent HIF‐2 induced transcription by PT2385, and found that PT2385 treatment inhibits the dimerization between ARNT and HIF‐2α, and also reduces the nuclear HIF‐2α protein levels under hypoxia in the NB PDX. However, the same study has shown that there are no effects on HIF‐2 target gene expression, and no major change is observed in the cell survival in vitro or on the in vivo tumor growth after PT2385 treatment. 263 In the same study, it has also been found that combination treatment using PT2385 with cisplatin or doxorubicin does not enhance the anticancer effects of the chemotherapeutic drugs compared with treatment with the chemotherapeutic drugs alone. 263 Overall, more extensive studies should be conducted to identify new HIF inhibitors that might be useful as anticancer agents for NB treatment.
3.6. Targeting cancer exosomes in NB
Exosomes are small extracellular vesicles with a significant role in intercellular communication associated with cancer. 264 , 265 , 266 There are many lines of evidence indicating that exosomes released from NB cells contribute to the progression of cancer. For instance, a study by Challagundla et al. 267 has demonstrated a role for exosomes containing miRNAs in the resistance of NB to chemotherapy, wherein exosomal miRNAs such as miR‐21 and miR‐155 have critical roles in chemotherapy resistance through the TLR8‐NFкB and TERF1 signaling pathways. Another study by Haug et al. 268 has shown that MYCN‐amplified NB cells release a variety of exosome‐like vesicular particles carrying various miRNAs (e.g., miR‐16, miR‐125b, miR‐21, miR‐23a, miR‐24, miR‐25, miR‐27b, miR‐218, miR‐320a, miR‐320b, and miR‐92a). The exosomal miRNAs released from MYCN‐amplified NB cells do not stimulate TLR8 signaling in recipient cells. 268 However, a functional enrichment analysis reveals that NB exosomal miRNAs affect pathways relates to cell growth and cell death. 268 These studies may indicate that targeting exosomes containing various miRNAs involved in the pathogenesis of NB may represent an effective approach to reduce NB tumorigenesis.
On the contrary, miR‐186 is responsible for the repression of oncogenic proteins (MYCN and AURKA) in NB cells, and is downregulated in NB and transforming growth factor‐β‐treated natural killer (NK) cells. 269 , 270 Thus, an approach to restore the miR‐186 levels in NB through NK cell‐derived exosomes could serve as another approach to reduce the tumor burden, promote survival, and restore the cell‐killing abilities of NK cells. 270
4. CLINICAL STUDIES OF TARGETED NB THERAPY
During the last decade, several clinical trials of new monotherapies or combination protocols exist for high‐risk NB. The molecular targets of the drugs evaluated in clinical trials are depicted in Figures 2 and 3. The progress made in these trials, some of which are still ongoing, has streamlined the development of personalized medicine for children with high‐risk NB. Various combinations of small molecule inhibitors with standard chemotherapy or other agents have been tested for high‐risk NB patients, the details of which are summarized in Table 1.
Table 1.
Overview of combination therapy being used in clinical trials for neuroblastoma
| Inhibitor | Molecular target | Agent(s) used in combination | Phase | Status | Identifier | Results |
|---|---|---|---|---|---|---|
| Crizotinib | ALK | Topotecan hydrochloride | I | Completed | NCT01606878 |
|
| Cyclophosphamide | ||||||
| Doxorubicin hydrochloride | ||||||
| Vincristine sulfate | ||||||
| Dexrazoxane hydrochloride | ||||||
| Crizotinib | ALK | Standard therapy (busulfan, carboplatin, cisplatin, cyclophosphamide, dexrazoxane, doxorubicin, etoposide, isotretinoin, melphalan, thiotepa, topotecan, vincristine) | III | Recruiting | NCT03126916 |
|
| Ceritinib | ALK | Ribociclib | I | Recruiting | NCT02780128 |
|
| Lorlatinib | ALK | Chemotherapy (cyclophosphamide, topotecan) | I | Recruiting | NCT03107988 |
|
| RG7388 (Idasanutlin) | MDM2 | Chemotherapy (cyclophosphamide/topotecan/fludarabine/cytarabine/) or venetoclax | I/II | Recruiting | NCT04029688 |
|
| Trametinib | MEK1/2 | Dabrafenib (BRAF kinase inhibitor) | I/II | Recruiting | NCT02124772 |
|
| Vorinostat | Histone deacetylase (HDAC) | Isotretinoin | I | Completed | NCT00217412 |
|
| Vorinostat | HDAC | Bortezomib | I | Completed | NCT01132911 |
|
| Vorinostat | HDAC | 131I‐MIBG | II | Active, but not recruiting | NCT02035137 |
|
| Vorinostat | HDAC | Dinutuximab/GM‐CSF/IL‐2 and isotretinoin, and +/‐DFMO | II | Recruiting | NCT02559778 |
|
| Vorinostat | HDAC | 131I‐MIBG | I | Completed | NCT01019850 |
|
| Vorinostat | HDAC | Isotretinoin | I | Completed | NCT01208454 |
|
| Decitabine | DNA methyltransferase (DNMT) | Doxorubicin and cyclophosphamide | I | Completed | NCT00075634 |
|
| Gemcitabine(pyrimidine nucleoside analog) | DNA | Ribociclib | I | Recruiting | NCT03434262 |
|
| Gemcitabine(pyrimidine nucleoside analog) | DNA | Nab‐paclitaxel | I | Recruiting | NCT03507491 |
|
| Etoposide | Topoisomerase | Monoclonal antibody 3F8 | II | Completed | NCT00004110 |
|
| 131I‐MIBG | Norepinephrine receptor | Carboplatin, etoposide, melphalan, and peripheral blood stem cell infusion, and radiotherapy | II | Completed | NCT00253435 |
|
| 131I‐MIBG | Norepinephrine receptor | Dinutuximab | I | Recruiting | NCT03332667 |
|
| 131I‐MIBG | Norepinephrine receptor | Bevacizumab | I | Completed | NCT00450827 |
|
| Dinutuximab (ch14.18) | GD2 | 131I‐MIBG and nivolumab | I | Recruiting | NCT02914405 |
|
| Dinutuximab beta | GD2 | Temozolomide and topotecan | II | Recruiting | NCT02308527 |
|
| Dinutuximab and sargramostim (GM‐SF) | GD2 | Chemotherapy (carboplatin, cisplatin, cyclophosphamide, dexrazoxane, doxorubicin, etoposide, isotretinoin, melphalan, thiotepa, topotecan, vincristine) | II | Recruiting | NCT03786783 |
|
| Dinutuximab | GD2 | Immunotherapy (isotretinoin + sargramostim + IL‐2) | II | Active, not recruiting | NCT02169609 |
|
| Isotretinoin | Unknown | Dinutuximab, aldesleukin, and sargramostim | III | Active, not recruiting | NCT00026312 |
|
| Hu14.18K32A | GD2 | Cyclophosphamide and topotecan | II | Active, not recruiting | NCT01857934 |
|
| Hu3F8 | GD2 | GM‐CSF | I/II | Recruiting | NCT01757626 |
|
| Hu3F8 | GD2 | Sargramostim | II | Active, not recruiting | NCT00072358 |
|
| Hu3F8 | GD2 | Sargramostim | I | Completed | NCT00450307 |
|
| Bevacizumab | VEGF‐A | Irinotecan and temozolomide | II | Completed | NCT01114555 |
|
| Bevacizumab | VEGF‐A | Cyclophosphamide and topotecan | II | Completed | NCT01492673 |
|
| C7R‐GD2. CART cells | GD2 | Cyclophosphamide and fludarabine | I | Recruiting | NCT03635632 |
|
| Zoledronic acid | Farnesyl pyrophosphate synthase | Cyclophosphamide | I | Completed | NCT00206388 |
|
| Zoledronic acid | Farnesyl pyrophosphate synthase | IL‐2 | I | Terminated | NCT01404702 |
|
| MLN8237 | Aurora A kinase | Irinotecan and temozolomide | I/II | Completed | NCT01601535 |
|
| Difluoromethylornithine (DFMO) | Ornithine decarboxylase | Etoposide | I | Completed | NCT01059071 |
|
| Difluoromethylornithine | Ornithine decarboxylase | Bortezomib | I/II | Active, not recruiting | NCT02139397 |
|
| Temsirolimus | mTOR | Temozolomide | II | Completed | NCT01767194 |
|
| Temsirolimus | mTOR | Perifosine | I | Completed | NCT01049841 |
|
| Sorafenib | Multikinase inhibitor | Cyclophosphamide and topotecan | I | Active, not recruiting | NCT02298348 |
|
| Nifurtimox | DNA | Cyclophosphamide and topotecan | II | Active, not recruiting | NCT00601003 |
|
| Bortezomib | 26S proteasome | Irinotecan | I | Completed | NCT00644696 |
|
4.1. Clinical trials of small molecule inhibitors
4.1.1. ALK inhibitors
The ADVL0912 phase I/II trial involving crizotinib has been completed by the Children's Oncology Group (COG) in pediatric patients with solid tumors (NCT00939770). 281 The early results of that trial have shown that out of 11 NB patients with known ALK mutations, one had a CR (complete response) and two patients had stable disease. 281 Another phase I trial (NCT01606878) has been conducted in 2013 by the COG for anaplastic large‐cell lymphoma (ALCL) or high‐risk NB patients, to study the effects of crizotinib in combination with chemotherapy (dexrazoxane hydrochloride, topotecan hydrochloride, cyclophosphamide, doxorubicin, vincristine sulfate). A phase III trial (NCT03126916) is currently recruiting high‐risk patients to evaluate the effects of combining standard therapy with crizotinib. Another ALK inhibitor, ceritinib, has been examined in a phase I trial that assessed its efficacy as monotherapy against ALK‐activated pediatric malignancies, including NB (NCT01742286). In 2016, the clinical study Next Generation Personalized Neuroblastoma Therapy (NEPENTHE) was initiated and has been currently recruiting patients with NB (NCT02780128). The NEPENTHE study is placing patients in treatment groups on the basis of genetic aberrations identified using deep sequencing. Participants with ALK mutations are being treated with combination therapy using ribociclib and ceritinib. Another ALK inhibitor, ensartinib, has entered clinical trials of patients with non‐Hodgkin's lymphoma, relapsed or refractory NB, or histiocytic disorders with ROS1 or ALK genomic alterations (NCT03213652). Furthermore, a phase I trial of lorlatinib has been initiated by NANT (New Approaches to Neuroblastoma Therapy) Consortium, and in this trial, lorlatinib is either used as a single agent or combined with chemotherapy for relapsed or refractory NB patients (NCT03107988). NCI (National Cancer Institute) started a phase II trial that aims to classify patients into molecularly targeted treatments on the basis of genetic profiling (NCT03155620). In this study, ensartinib (ALK inhibitor) is being used for patients with relapsed or refractory NB (NCT03155620).
4.1.2. MDM2 inhibitors
The p53‐MDM2 inhibitors have not been extensively explored at the clinical stage. The NIH U.S. National Library of Medicine website (https://clinicaltrials.gov/) provides data for RG7388 and HDM201, which have been used as MDM2 inhibitors for NB. The currently recruiting clinical trials for MDM2 inhibitors are focused on RG7388 (NCT04029688) and HDM201 (NCT02780128). The clinical trial of HDM201 (NCT02780128) is part of the NEPENTHE study described above. It is anticipated that some of the newer MDM2 inhibitors, such as RG7112, 219 RITA, 282 and SF1126, 283 that are currently being investigated in preclinical studies for NB may enter clinical testing soon. Initial studies should focus on the pharmacokinetic properties in healthy individuals, and then test the potency in individuals with relapsed or refractory NB.
4.1.3. RAS‐MAPK and MEK inhibitors
Several MEK inhibitors are currently being evaluated in pediatric patients. For instance, a phase I/IIa clinical trial (NCT02124772) is currently ongoing and recruiting patients to investigate the pharmacokinetic, safety, and clinical activity of trametinib monotherapy, and a combination of dabrafenib with trametinib, in cancer patients harboring V600 mutations. This study includes a patient population of n = 10 for individuals with refractory or relapsed NB to assess their response to mono‐ and combination therapy.
4.1.4. PI3K/Akt/mTOR inhibitors
Different PI3K/Akt/mTOR inhibitors have been assessed in NB patients. For instance, the NANT Consortium has conducted a phase I clinical trial with SF1126 (inhibitor of PI3K and mTOR) for relapsed or refractory NB patients (NCT02337309). This trial had two pediatric phases. In the first phase, a dose‐escalation design of 3 + 3 was followed. In the second phase, once a recommended dose was identified, a population of 10 patients with MYCN amplified or Myc‐N expressing tumors was treated. In terms of AKT inhibitors, a clinical study has been conducted involving perifosine, an AKT inhibitor, for children with solid tumors (NCT00776867). This clinical study recruited 27 high‐risk NB patients, and only one patient had MYCN‐amplified high‐risk NB, while none of the tumors had an ALK mutation in 21 tested patients. 284 A total of nine patients remained progression‐free for a median of 54 months from the study entry. 284
4.1.5. Targeting epigenetic regulators
BET (bromodomain and extraterminal domain) proteins are epigenetic readers, and bromodomains of BET proteins are capable to recognize histones and bind on them at acetylated lysine residues and thereby regulating the chromatin structure. Various BET inhibitors are in clinical trials. For instance, a phase I study of GSK525762 (I‐BET726) has been completed in March 2020. This study has evaluated the pharmacokinetics, safety, pharmacodynamics, and clinical activity of GSK525762 in patients with NMC (NUT Midline Carcinoma) and other cancers, including NB and MYCN‐driven solid tumors (NCT01587703). This study had age eligibility of 16 years and older, and all sexes were eligible for the clinical trial. At the epigenetic level, HDAC can be inhibited to increase acetylation and thus induce a less malignant transcriptional profile. 285 Regarding HDAC inhibitors, a phase I clinical and pharmacokinetic trial study of vorinostat has been conducted in individuals with solid tumors including NB and was found to exhibit a maximum tolerated dose of 230 mg/m2/day. 286 Vorinostat (class I and II HDACi) has been investigated in several trials in NB patients (NCT00217412, NCT01132911, NCT02035137, NCT02559778, NCT01019850, and NCT01208454). A varied phase II trial of combination therapy is also ongoing, that is, comparing treatment with 131I‐MIBG alone and vorinostat with 131I‐MIBG for resistant or relapsed NB (NCT02035137). Decitabine, a DNMT pan‐inhibitor, has been studied in a phase I trial in relapsed or refractory solid tumors or NB patients (NCT00075634). Another pan‐HDAC inhibitor, 4‐phenylbutyate (4PB), has also been evaluated in phase I clinical trial in patients with brain tumors or NB (NCT00001565).
4.1.6. Clinical trials of nucleoside analogs and agents targeting DNA synthesis
One of the synthetic pyrimidine nucleoside analogs is gemcitabine that functions as a deoxycytidine triphosphate and incorporates into DNA strands synthesized during cell division. Various clinical trials are ongoing in the use of gemcitabine in NB patients. For instance, St. Jude Children's Research Hospital is recruiting patients for a phase I clinical trial to evaluate molecularly driven doublet therapies for individuals with various cancer conditions, including NB, and involves the use of drugs such as gemcitabine, ribociclib, sonidegib, and trametinib in doublets for patients (NCT03434262). Another phase I study is the one which is based on the combination treatment of gemcitabine with nab‐paclitaxel for relapsed and refractory pediatric solid tumors (NCT03507491). Another purine analog, fludarabine, has been evaluated in different clinical trials in NB patients.
4.1.7. Clinical trials of selected immunotherapy regimens
Baylor College of Medicine, as a sponsor, has a completed phase I trial of a third‐generation GD‐2 chimeric antigen receptor (CAR) and iCaspase suicide safety study in NB patients (GRAIN) (NCT01822652). In this study, genes such as CD28 and OX40 are appended to GD2 T cells to make the cells live longer. One of the goals of the trial is to find out the highest safety dose of iC9‐GD2‐CD28‐OX40 (iC9‐GD2) T cells that can be provided to relapsed/refractory NB patients. The same study is also examining whether it is beneficial to give chemotherapy before the T‐cell infusion, which is referred to as lymphodepletion, and the chemotherapy used in this trial is a combination of cyclophosphamide and fludarabine. mAb therapy is also being combined with chemotherapy to treat NB, and a phase II trial has been conducted to study the effectiveness of the combination of a monoclonal anybody (3F8) against ganglioside GD2 and etoposide in patients with NB (NCT00004110).
4.1.8. Clinical trials using 131I‐MIBG in NB
A norepinephrine analog is metaiodobenzylguanidine (MIBG), which is actively taken up via norepinephrine transporters and accumulates in NB cells. Radioiodine‐labeled MIBG can be used for the treatment and diagnosis of NB. 287 This is because norepinephrine receptors are being targeted by MIBG, and such receptors are expressed in 90% of NB tumors. 288 In addition,131I‐MIBG also holds the ability to target tumors irrespective of their MYCN status or specific histology. 289 There have been several clinical trials of 131I‐MIBG for relapsed or refractory NB patients. 290 The molecule has also been used in combination protocol with stem cell transplantation and chemotherapy, either as a single‐dose treatment or multiple‐dose treatment, for salvage treatment and as induction therapy for refractory or relapsed NB. 57 U.S. FDA has approved 131I‐MIBG as a diagnostic agent in 1994 and for the imaging of pediatric NB in 2008. 291 A dose‐escalation study has been performed by Matthay et al. 292 in phase I trial of 131I‐MIBG to define its dose‐limiting toxicity with and without autologous bone marrow support in refractory NB patients. That study has found that a dose of up to 12 mCi/kg did not require autologous stem cell rescue, and 37% was the response rate at this particular dose. 292 It has also found that doses up to 18 mCi/kg can be used safely, providing there is a possibility of autologous peripheral blood stem cell rescue. 292 A large phase II trial has been performed to evaluate the effects of age, prior therapy, and disease site on the 131I‐MIBG therapy response to refractory NB patients. 293 In that study, patients with (n = 148) and without (n = 16) cryopreserved HSCs (hematopoietic stem cells) have been treated with 18 and 12 mCi/kg of 131I‐MIBG, respectively. The ORR (overall response rate) was 36%, and also the response rate in patients older than 12 years was significantly higher. 293
Apart from monotherapy, various combinations have been studied, such as the use of 131I‐MIBG with chemotherapeutic agents like cisplatin, 294 , 295 topotecan, 296 cyclophosphamide, 297 and melphalan, 298 which yielded response rates of 27%–80%. 294 , 295 , 299 Phase II study has investigated the effects of 131I‐MIBG in combination with chemotherapy (carboplatin, etoposide, melphalan, referred to as CEM) and radiation therapy in patients acquiring bone marrow transplants or autologous peripheral stem cell for relapsed or refractory NB (NCT00253435). 289 In that study, a 10% response rate was observed in primary refractory or progressive disease patients, and the addition of 131I‐MIBG to CEM was found to be tolerable for high‐risk NB patients. Another study by Mastrangelo et al. 297 has shown that patients who received chemotherapy accompanied by treatment with 131I‐MIBG (200 mCi) for relapsed and refractory NB has impressive response rates, where five patients having treatment‐naive had one complete response, two very good partial responses, and two partial responses, thus suggesting that 131I‐MIBG has particular promise for the treatment of previously untreated individuals. 131I‐MIBG is currently being evaluated in the upfront treatment of patients with HR‐NB in a COG‐NCI‐sponsored Phase III clinical trial (NCT03126916). 300
4.1.9. Immunotherapy‐based clinical trials
4.1.9.1. Monoclonal antibodies
Dinutuximab is a chimeric mAb and composed of human constant regions of IgG1 and murine variable regions of IgG3, targets glycolipid GD2 in NB cells. Dinutuximab was FDA approved in 2015 to treat high‐risk NB patients. 9 Various clinical trials are active involving dinutuximab for patients with NB (NCT02743429, NCT04253015, NCT03332667, NCT02169609, NCT02914405, NCT04221035, NCT02308527, NCT00026312, NCT01041638, NCT03786783, NCT02914405, NCT01711554, NCT02169609, NCT02169609, and NCT04253015). Dinutuximab therapy has various side effects, such as fever, neuropathic pain, hypotension, and allergic reactions. 301 In terms of their mechanism of action, anti‐GD2 mAbs employ both antibody‐dependent cell‐mediated cytotoxicity (ADCC) and complement‐dependent cytotoxicity to exert its effects in cancer patients. 302 Hu14.18K322A is a humanized form of dinutuximab, and currently in a phase II trial sponsored by St. Jude Children's Research Hospital for advanced stage NB. It is being given with IC, and early results suggest that it can significantly improve the early response, reduce the volume of tumor, and improve the 2‐year EFS (NCT01857934). Another mAb, enoblituzumab, targets B7‐H3 (a type I transmembrane glycoprotein). It has been used in a phase I, open‐label trial to characterize antitumor activity of enoblituzumab in young adults and children expressing B7‐H3 in relapsed or refractory malignant solid tumors (including NB) (NCT02982941). Naxitamab (hu3F8), a humanized mAb targeting ganglioside GD2, has also been tested in NB patients (NCT01419834, NCT01757626, and NCT03033303). These trials of hu3F8 have been demonstrated that it has favorable pharmacokinetics, low immunogenicity, and improved toxicity profile. 273 , 303 , 304 Bevacizumab (Avastin) is a humanized mAb that targets VEGF, and inhibits tumor growth. 305 This mAb has been tested in phase I and II trials for relapsed or refractory NB (NCT01114555, NCT00885326, NCT00450827, NCT02308527, and NCT01492673). Among the following trials, NCT00885326, NCT00450827, and NCT02308527 have no published results. However, a phase II trial of the combination of bevacizumab, irinotecan, and temozolomide (BIT) in high‐risk NB patients (NCT01114555) has shown 3 patients with complete response (CR), 12 with progressive disease, and 18 with no response. The OS and median progression‐free were 31.5 ± 5.6 and 7.7 ± 1.7 months, respectively. Grade 4 toxicities in patients were thrombocytopenia (24%) and neutropenia (30%), grade 3 toxicities include proteinuria (9%), diarrhea (3%), and hepatic transaminitis (15%). 277 Another trial using the combination of cyclophosphamide, topotecan, and bevacizumab in NB patients (NCT01492673) showed the all‐cause mortality was 66.67%, and serious adverse events include febrile neutropenia (66.67%), and other adverse effects include anemia (11.11%), diarrhea (22.22%), metabolism and nutrition disorders, cystitis noninfective, hematuria and alopecia.
4.1.9.2. Conjugated antibodies
Antibodies can be conjugated with therapeutic agents (radionucleotides, drugs, and cytokines) to enhance their functions against cancer cells. An example of the antibody‐drug conjugate is lorvotuzumab mertansine (IMGN901), which is composed of a humanized anti‐CD56 antibody linked to tubulin‐binding maytansinoid DM1, and is evaluated in an active (but not recruiting) phase II trial for patients with relapsed or refractory solid tumors, including NB (NCT02452554). Hu14.18‐IL2 (EMD273063) is a genetic fusion of interleukin‐2 (IL‐2) attached to the carboxy terminus of each IgG heavy chain of Hu14.18. Hu14.18‐IL2 has been part of various clinical trials, some still ongoing, in NB patients (NCT03209869, NCT00082758, NCT01334515, and NCT00003750).
4.1.10. Targeting the tumor microenvironment (TME) in NB
For therapeutic intervention, clinical trials are following three strategies to target the TME, and include the following:
4.1.10.1. Targeting TME cells
Zoledronic acid (ZA) targets osteoclasts in the TME by inhibiting farnesyl pyrophosphate synthase. New Advances in Neuroblastoma Therapy consortium has conducted phase I trial involving ZA and demonstrated that zoledronic is safe to use in individuals with bone metastasis. In addition, ZA has been evaluated in combination with cyclophosphamide in a phase I study (NCT00206388), and in combination with IL‐2 in another clinical trial (NCT01404702). Endothelial cells are also key targets in clinical trials, and a phase I trial has been conducted using bevacizumab (molecular target is VEGF), ZA, and cyclophosphamide in patients with recurrent or refractory high‐risk NB (NCT00885326).
4.1.10.2. Targeting signaling pathways activated by the TME
Small molecule inhibitors are being used to target signaling pathways that are activated by TME. For instance, sorafenib, a multikinase inhibitor, is used in combination with cyclophosphamide and topotecan in NB patients (NCT02298348). Lestaurtinib (CEP‐701) (inhibitor of TrkA/B/C and Janus kinase 2 (JAK2)) has been tested in individuals with recurrent or refractory high‐risk NB (NCT00084422). Further, ZD6474 (a VEGFR inhibitor) has been tested alone and in combination with the retinoic acid in a phase I trial in pediatric NB patients (NCT00533169). An inhibitor of MEK/MAPK signaling kinase and topoisomerase II‐β, (R+) XK469), has been evaluated in a phase I trial in patients with advanced NB (NCT00028522). Temsirolimus is an inhibitor of Akt pathway that targets mTOR protein and undergoes a phase II trial in individuals with relapsed or refractory NB (NCT01767194).
4.1.10.3. Chimeric antigen receptor (CAR) T cells
Apart from monoclonal Abs, CAR T cells are developed to target GD2, and the efficacy of such cells has been evaluated in clinical trials. Several clinical trials (NCT02919046, NCT03373097, NCT03635632, NCT02761915, NCT02765243, NCT02311621, NCT02107963, and NCT01822652) have examined the application of CAR T cells in NB patients. Ganglioside GD‐2 is being targeted as a part of immunotherapy‐based approaches for NB. GD‐2 has been targeted by various monoclonal antibodies, such as 3F8, in the combination protocol with sargramostim (NCT00072358), or granulocyte‐macrophage colony‐stimulating factor (NCT00450307). In addition, activated T cells are being armed with GD2‐bispecific antibody, and are being evaluated in phase I/II trials in young adults and children with NB (NCT02173093).
4.1.11. Radionucleotide therapy
DOTATATE is an amino acid peptide with a covalently‐bonded DOTA bifunctional chelator that can also be bound to gallium‐68 and lutetium‐177 to form 68Ga‐DOTATATE and 177Lu‐DOTATATE. This radiolabelled DOTATATE has been used in NB, which expresses somatostatin receptor‐positive, but does not express NET (norepinephrine transporter) or exhibits MIBG resistance. 56 In January 2018, 177Lu‐DOTATATE was approved by FDA for neuroendocrine tumors, and currently in clinical trials for therapy for NB tumors. 56 , 306 A phase III trial of 177Lu‐DOTATATE (NCT01578239) has been demonstrated marked improvements in the EFS of neuroendocrine tumor‐bearing patients in comparison to those treated with long‐acting repeatable octreotide. 307 177Lu‐DOTATATE has also been found to be effective on its use with radiosensitizing agents such as topotecan and nutlin‐3. 308 In addition, an ongoing phase II trial is being performed to assess the efficacy of 68Ga‐DOTATATE as a diagnostic test in patients with somatostatin receptor‐positive NB tumors (NCT03273712).
4.1.12. Other agents
Studies have shown that inhibition of Aurora can destabilize the MYCN. MLN8237 is an Aurora A kinase inhibitor, and was used in combination with temozolomide and irinotecan in a phase I/II trial for NB (NCT01601535). Another Aurora kinase inhibitor, alisertib, has been used as monotherapy in phase I and II trials for NB patients (NCT02444884, NCT01154816).
DFMO (eflornithine) is an inhibitor of ornithine decarboxylase, and has been examined in a phase I trial in both alone conditions and also in combination with etoposide (NCT01059071). 279 The same trial has also studied the dose‐limiting toxicity of DFMO, and found that 500–1500 mg/m2/day doses of DFMO are safe and well‐tolerated in relapsed NB patients. 279 Further, NCT02139397 is a currently active but not recruiting phase I/II trial and this trial will evaluate the combination of bortezomib with DFMO for relapsed or refractory NB.
Entrectinib, a Trk inhibitor, has been evaluated in a phase I trial (STARTRK‐1) to assess its tolerability and safety, and to determine the recommended dose in patients with various malignancies, including NB (NCT02097810). In addition, phase I and II trials of entrectinib are currently ongoing for individuals with solid tumors, including neuroendocrine tumors and NB (NCT02650401 and NCT02568267). The effectiveness of mTOR inhibitors alone and in combination with other treatments have also been investigated. For instance, temsirolimus (mTOR inhibitor) has been tested in combination with temozolomide in clinical trials for NB patients (NCT01767194). Combining an inhibitor of AKT signaling with mTOR inhibition has been found to be more effective than either treatment alone; for instance, a phase I clinical trial has studied the safety and effectiveness of perifosine (AKT inhibitor), and temsirolimus (mTOR inhibitor) in recurrent pediatric solid tumors (NCT01049841).
Sorafenib is a multikinase inhibitor with activity against many protein kinases, such as VEGFR, PDGFR, and RAF. A phase I trial of sorafenib together with topotecan and cyclophosphamide is currently active for relapsed and refractory NB patients (NCT02298348). An antiparasitic agent, nifurtimox, has been used in the treatment of Chagas disease. This drug is tested in a phase II trial, and in this trial, nifurtimox has been used in combination with topotecan and cyclophosphamide for the treatment of relapsed or refractory NB (NCT00601003). The molecular target of Nifurtimox has not been fully elucidated in NB. A study by Sholler et al. 309 has shown that nifurtimox treatment induces the formation of reactive oxygen species, causes DNA fragmentation, and suppresses Akt phosphorylation in NB cells, leading to apoptosis. Another study by Stanchi et al. 310 has shown that nifurtimox can reduce N‐myc expression in NB cells. A study by Kong et al. 311 has indicated that nifurtimox causes deactivation of Akt‐GSK‐3β signaling in NB cells. Different research groups have proposed different mechanisms of anticancer action for nifurtimox; the exact mechanism(s) underlying the cytotoxicity of nifurtimox warrants further investigations. Gefitinib, an EGFR inhibitor, underwent a phase I trial in patients having refractory solid tumors, including NB (NCT00132158). A proteasome inhibitor, bortezomib, has also been investigated in individuals with recurrent or refractory NB (NCT00644696).
As described above, several new drugs/strategies are currently being investigated in patients with NB. However, the antitumor activity observed for these targeted therapies has not been as promising as expected, so it is important to focus on obtaining a comprehensive understanding of the molecular mechanisms involved in the relapse of NB and the lack of the response to existing therapies. Further, most of the existing therapies have been tested in unselected populations, so patient selection methods should be improved to develop more specific, accurate, and efficient therapies for NB.
5. GENERAL DISCUSSION AND FUTURE RESEARCH DIRECTIONS
NB is a heterogeneous disease with varied outcomes, ranging from spontaneous regression to refractory growth. Thus, the most pressing need is to develop novel and effective strategies for the treatment of NB. To improve the clinical outcome for children, it is critical to understand the etiology of NB. Genome analyses suggest that chromosome instability (CIN) is a significant event in the pathogenesis of NB. 4 However, further investigations are needed to confirm whether CIN is the root cause of the gene mutations observed in NB. It is also crucial to understand the cause of CIN in NB and investigate the importance of aberrations in proteins involved in chromosome and centrosome segregation, spindle apparatus machinery, and DNA repair. 4 One of the recurrent genetic alteration observed in the high‐risk NB is the hemizygous deletion of the chromosome 11q 22‐23. 312 ATM is located on the arm of chromosome 11, and has a role in DNA repair, 312 , 313 , 314 and its loss may be related to the induction or progression of NB. It may be useful to inhibit PARP (poly ADP ribose polymerase) to target malignancies that exhibit homologous recombination DNA repair pathway deficiencies.
The currently available targeted therapy for NB includes: (1) targeting genetic aberrations; (2) targeting disrupted signaling molecules; (3) immunology‐based approaches; (4) radiopharmaceutical targeting of norepinephrine and somatostatin receptors; (5) targeting epigenetic regulators; and (6) targeting Bcl‐2 family proteins. Among these strategies, cancer immunotherapy has emerged as an effective strategy. It is also associated with less acute or chronic toxicity than genotoxic therapies. The humanization and modification (by point mutations) of the GD2 antibody are expected to reduce the immunogenicity and side effects of the treatment. 9 For instance, the O‐acetylated form of GD2 reduces off‐tumor target effects on mesenchymal stromal cells, sensory neurons, and the posterior lobe of the pituitary gland. 9 New antibody formats should be developed to deliver high‐dose radiation to cancer cells, reducing toxicities. 61 CAR T cells have also been shown to induce clinical response in NB patients. However, they have only limited success due to difficulties with target antigen selection, a lack of T‐cell persistence, and a suppressive TME. 58 Thus, CAR T cells should be engineered to address the barriers present in cancer patients. Single‐cell RNA sequencing of NBs should also be implemented to identify new leads for immunotherapeutic strategies. 315 Very few compounds targeting epigenetic factors (i.e., HDACi, DNMTi, iBET) have entered clinical trials. As a future direction, the crystal structures of epigenetic regulators should be resolved to develop better inhibitors with fewer side effects. In addition, combination therapy should also be developed by targeting different sites of a pathway, or with the intention of disrupting multiple pathways together.
As a future direction, new therapeutic approaches should be developed to reduce the risk of NB disease progression. Among the new approaches being developed is HIFU (high‐intensity focused ultrasound). 316 Similarly, MR (magnetic resonance)‐HIFU has been developed for the treatment of NB, in which HIFU functions to ablate tumor lesions under the real‐time anatomic guidance and thermal monitoring of MRI (magnetic resonance imaging). 317 MR‐HIFU has been implemented in trials for individuals with relapsed/refractory solid tumors. For instance, AeRang Kim (sponsored by the Children's National Research Institute, Washington, DC) is currently recruiting patients for a phase I study to evaluate the feasibility and safety of MR‐HIFU therapy for individuals with relapsed or refractory solid tumors, including NB (NCT02076906). Further, the same group is recruiting patients for a phase I study to determine the MTD (maximum tolerated dose) and RP2D (recommended phase 2 dose) of LTLD (lyso‐thermosensitive liposomal doxorubicin) administered in combination with MR‐HIFU for patients with relapsed/refractory solid tumors, including NB (NCT02536183). Research has also been conducted to evaluate the anatomic locations of NB tumors to determine the feasibility of MR‐HIFU therapy. 318 In one study, it has been found that only a minority of NB tumors are treatable at the time of diagnosis or relapse, and at diagnosis, all treatable NBs are intra‐abdominal and not‐targetable without the use of respiratory motion compensation. 318 It has also been found that at relapse, many NB patients have intrathoracic, intracranial, or intraabdominal tumors that are not targetable due to adjacent anatomical structures. 318 Thus, the use of MR‐HIFU for drug delivery in NB is primarily limited to a fraction of relapsed tumors, as only a minority of tumors are targetable at diagnosis. 318 To improve the technology of MR‐HIFU, 3D modeling using the MR‐HIFU system should be implemented to improve its accuracy in predicting targetable lesions. 318
At the preclinical level, a study by Eranki et al. 319 has shown that a combination of mechanical HIFU fractionation and checkpoint inhibitors (αCTLA‐4 + αPD‐L1) significantly enhances the systemic antitumor response compared with a previously unresponsive murine NB model, providing evidence that HIFU may have synergistic potential with immunotherapy for the treatment of NB. In addition, combining MR‐HIFU with existing chemotherapy should be implemented to augment the chemoresponse of NB, and increase local control and decrease systemic toxicity. 318
Hyperthermic temperatures also have exhibited therapeutic potential in various applications, including drug/gene delivery and radio/chemosensitization, and such temperatures can be generated in biological tissues by applying pulsed focused ultrasound. 320 Experiments based on theoretical simulations, in vitro validations, and an in vivo subcutaneous murine xenograft model have been conducted in the past to optimize and select pulsed‐focused ultrasound (FUS) exposure parameters for hyperthermia‐based applications. Such experiments can help reduce animal experimentation and are easy to into clinical trials since they are noninvasive or minimally‐invasive. 320 In addition, hyperthermia has also been found to be effective in enhancing the anticancer activity of chemotherapeutic drugs. 321 This is supported by a study carried out by Debes et al. 322 which showed that hyperthermia synergistically enhances the cytotoxicity of an alkylating agent, cisplatin, in NB cells. Targeted image‐guided drug delivery (IGDD) is a technique that has been developed to improve drug deposition in a variety of diseases, including cancer. 323 IGDD employs focused ultrasound along with “microbubble” ultrasound contrast agents (UCAs) to enhance drug delivery into target tissues. 323 This approach is known as sonopermeation, which helps to increase vascular permeability and thus increased penetration of drugs into the target tissue. 323 A report by Bellary et al. 324 has described a novel 2D and 3D quantitative contrast‐enhanced ultrasound imaging (qCEUS) system that could be used to monitor the therapeutic efficacy of sonopermeation in NB tumors. That study has also found that combing ultrasound therapy with UCA significantly enhances the doxorubicin payload to NB in an orthotopic xenograft model, and qCEUS imaging indicates that there is significant doxorubicin uptake induced by increasing the tumor vascular permeability (reduced pericyte coverage) due to microbubble sonopermeation, with no damage to vasculature. 324 Overall, ultrasound‐based approaches already have a significant impact on the diagnosis and management of NB, and may also be useful for the treatment of NB.
Currently, a significant gap exists between in vitro and in vivo testing to identify new drugs for NB. As is typical for drug development, only about 1 in 10 drugs tested in clinical trials are finally approved by the FDA. 45 The models being used for NB research and drug efficacy studies include zebrafish, mice, and chick chorioallantoic membrane (CAM). 325 Using a more physiologically‐relevant and technically‐reproducible model system would facilitate the development of treatments for NB and other disease states. 325 One such system is the 3D tissue‐engineered system, which can model the TME to provide a better understanding of the NB pathogenesis, and also test a drug in more relevant conditions. Further, a patient‐derived xenograft model system can be used to screen and identify effective drugs for NB treatment. The PDX model has high human relevance in terms of exhibiting features of human NB such as the presence of human stromal cells to mimic the TME, genetic complexity, histopathology, and mutational and proteomic profiles of actual patient tumors. 325 With regard to using PDX models in cancer research, a pediatric preclinical testing program (PPTP) has been entrenched to identify effective treatments for pediatric cancers. The aim of this program is to acquire preclinical in vivo data with the help of genomically‐characterized PDX cell lines to test and identify agents that can easily advance in pediatric clinical trials (NCI PPTC, www.ncipptc.org). 326 As a future direction, patient‐derived NB cells should be incorporated in 3D culture systems to test and identify new effective drugs and combinations that can then be tested in PDX models of NB, or in the case of drugs already approved for other indications, in human clinical trials.
Regarding the treatment of high‐risk NB, the patient selection for clinical trials of molecular targeted therapy should be improved by considering tumor heterogeneity. 25 In addition, novel statistical methods should be considered in early phase trials to minimize the requested number of patients, making it easier to recruit a sufficient number of NB patients to analyze. 25 Issues associated with the tumor heterogeneity in NB may be overcome by studying liquid biopsies for cell‐free DNA. 25 This approach would help to detect genetic aberrations that may not be identified by studying a single site of disease, and would ameliorate the need for a tissue biopsy. 25
Overall, it is important to understand the genetic complexity of NB and to develop personalized medicine based on the genetic predisposition of individual patients. Continued attempts should be made to identify new signaling pathways critical for the pathogenesis of NB and resistance to treatment, which can be further exploited as targets for therapy. There is also a need to identify prognostic tumor markers that could be used to assess the prognosis of patients and their response to treatment. The ultimate goal is to apply all of this knowledge to cure children with NB with minimal toxicity.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Study concept and design: Wei Wang, Jia Zhou, Frank McKeon, Jennifer Foster, and Ruiwen Zhang. Drafting of the manuscript: Atif Zafar, Wei Wang, Xinjie Wang, Gang Liu, Jennifer Foster, and Ruiwen Zhang. Revising of the manuscript: Wei Wang, Gang Liu, Xinjie Wang, Frank McKeon, Jennifer Foster, Jia Zhou, and Ruiwen Zhang. Administrative, technical, or material support: Wei Wang and Ruiwen Zhang. Study supervision: Wei Wang and Ruiwen Zhang. All the authors have read and agreed to the published version of the manuscript.
ACKNOWLEDGMENTS
Wei Wang and Ruiwen Zhang were partially supported by the National Institutes of Health (NIH)/National Cancer Institute grants (R01CA186662 and R01CA214019). Wei Wang and Ruiwen Zhang were also supported by the American Cancer Society (ACS) grant RSG‐15‐009‐01‐CDD. Ruiwen Zhang was partially supported by funds for Robert L. Boblitt Endowed Professor in Drug Discovery and research funds from College of Pharmacy and University of Houston. Jia Zhou was supported in part by the John D. Stobo, M.D. Distinguished Chair Endowment Fund at University of Texas Medical Branch. Wa Xian and Frank McKeon were supported by grants from theCancer Prevention Research Institute of Texas(CPRIT; RR150104 to WX and RR1550088 to FM), the National Institutes of Health (1R01DK115445‐01A1 to WX, 1R01CA241600‐01, and U24CA228550 to FM). Wa Xian and Frank McKeon are CPRIT Scholars in Cancer Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funding agencies. We thank Dr. Elizabeth Rayburn for excellent assistance in the preparation of this manuscript.
Biographies
Atif Zafar is a Postdoctoral Research Fellow in the Department of Pharmacological and Pharmaceutical Sciences, Colldege of Pharmacy, University of Houston (UH), Houston, TX. He obtained his Ph.D. in Biochemistry from Aligarh Muslim University, Aligarh, India under the supervision of Prof. Imrana Naseem. Prior to coming to UH, he worked as a Research Associate at Aligarh Muslim University, Aligarh, India, investigating combination therapy for breast cancer. In the same department, he worked as an Assistant Professor and was teaching courses at BS and MS levels in 2018‐19. His current research focus is to identify small molecules as pharamacological agents for human malignancies, and explore the combination therapies in cancer.
Wei Wang is a Research Associate Professor in the Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston (UH), Houston, TX. She obtained her Ph.D. in Molecular Pharmacology from Fudan University, Shanghai, China. Prior to joining the faculty at UH, she completed her post‐doctoral training and worked as a Research Associate at the University of Alabama, School of Medicine, Birmingham, AL, and an assistant professor at Texas Tech University Health Sciences Center (TTUHSC), Amarillo, TX. Dr. Wang's research has been continuously focused on targeted molecular cancer therapy, with particular interests, including signaling pathways, experimental therapeutics, and drug development.
Gang Liu is a Postdoctoral Research Fellow in the Department of Pharmacology and Toxicology at the University of Texas Medical Branch. He graduated from the College of Pharmaceutical Sciences and Chinese Medicine, Southwest University, China in 2010. Then he joined Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences (CAS), Shanghai, China, and worked as an assistant experimentalist for two years. From 2012 to 2017, he obtained his Ph.D. degree from SIMM, CAS, China, under the supervision of Professor Ao Zhang. Dr. Liu is currently pursuing his postdoctoral training at the Chemical Biology Program, Department of Pharmacology and Toxicology at the University of Texas Medical Branch under the supervision of Professor Jia Zhou. His research interests currently focus on the rational design and chemical synthesis of small molecules as novel pharmacological probes and therapeutics for human cancers and other diseases.
Xinjie Wang is a Ph.D. student in the Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, Texas, USA. She received her B.S. degree in pharmaceutical sciences from China Pharmaceutical University, Nanjing, China. Her current research focuses on the discovery of small molecule anticancer therapeutics, combination therapies in cancer, and pharmaceutical development of novel anticancer agents.
Wa Xian is a Research Associate Professor at Stem Cell Center in the Department of Biology and Biochemistry at the University of Houston, Houston, Texas, USA. Dr. Wa Xian received her Bachelor's degree in Biochemistry from Nankai University in China and Ph.D. degree in Molecular Genetics at the University of Texas Graduate School of Biomedical Sciences in Houston. The support of a DOD breast cancer fellowship enabled Dr. Xian's postdoctoral studies with Prof. Jeffrey Rosen at the Baylor College of Medicine. In 2008, Dr. Xian moved to Harvard Medical School under the direction of Prof. Christopher Crum, Director of Women's and Perinatal Pathology at the Brigham and Women's Hospital to study the origin and evolution of ovarian cancer. In 2009, Dr. Xian was appointed Principal Investigator at the Institute of Medical Biology of the Agency for Science, Technology, and Research in Singapore. In 2013, Dr. Xian returned to the US and initiated multiple projects related to inflammatory diseases and cancer at the University of Connecticut Health Center. In 2015, Dr. Xian received the Rising Stars Award from the Cancer Prevention & Research Institute of Texas (CPRIT) and was recruited to Houston, where she is devoted to the cloning of stem cells of regenerative tissues and using advanced technologies to understand the genetics of their self‐renewal, commitment, and differentiation.
Frank McKeon is Professor and Director of Stem Cell Center in the Department of Biology and Biochemsitry at the University of Texas. Dr. McKeon obtained his B.S. in Biology at Pomona College and his Ph.D. in Biochemistry and Biophysics from the University of California, San Francisco. He was Professor of Cell Biology at Harvard University from 1986 to 2012, and Senior Group Leader at the Genome Institute of Singapore from 2008 to 2015. Dr. McKeon is an internationally recognized authority and pioneer in the fields of cell biology, mouse genetics, and adult stem cells. The p63 gene and its function in stem cells of stratified epithelia were discovered in his laboratory, as was the p63 monoclonal antibody that has received FDA approval for prostate cancer diagnosis. In 2015, Dr. McKeon received the Established Investigator Award from the Cancer Prevention & Research Institute of Texas (CPRIT) and was recruited to Houston. His laboratory is presently reconstructing the evolution of esophageal adenocarcinoma from a genomics and epigenetics analyses of stem cells of Barrett's, low‐ and high‐grade dysplasia, and of esophageal adenocarcinoma from individual cases, with the goal of developing both preemptive and ex facto targeting of this and related diseases.
Jennifer Foster is an Assistant Professor in Pediatric Hematology/Oncology at Baylor College of Medicine, Texas Children's Hopsital. She is triple boarded in Pediatrics, Pediatric Hematology/Oncology, and Clinical Pharmacology. As the Clinical Director of Texas Children's Hopsital's Neuroblastoma Program, she develops, implements, and oversees clinical trials for patients with neuroblastoma. She is also an active member of the Developmental Therapeutics Team at Texas Children's Hospital, with expertize in developing and conducting Phase 1 trials for pediatric patients. Her research focuses on pre‐clinical drug development, clinical trial design, clinical pharmacology, and neuroblastoma.
Jia Zhou is a professor in the Department of Pharmacology and Toxicology at University of Texas Medical Branch. He received his Ph.D. in organic chemistry from Nankai University, China in 1997. Then he joined the chemistry faculty in the same university and was promoted to Associate Professor there. He started his postdoctoral research in organic chemistry with Dr. Sidney M. Hecht at the University of Virginia in 1999. After further postdoctoral training in medicinal chemistry with Dr. Alan P. Kozikowski at Georgetown University Medical Center, he worked in the pharmaceutical industry at Acenta Discovery, and PsychoGenics, Inc. as a Senior Principal Scientist for 7 years. Dr. Zhou is currently a tenured Professor leading a drug discovery team with research programs for human cancers, central nervous system disorders, and infectious diseases. He is also a faculty member of the Center for Addiction Research, Center for Biodefense and Emerging Infectious Diseases, Sealy Center for Structural Biology and Biophysics, and Sealy Center for Molecular Medicine at UTMB. He is an author of more than 170 peer‐reviewed papers, 7 book chapters, and an inventor of 23 patents.
Ruiwen Zhang has been Robert L. Boblitt Endowed Professor in Drug Discovery, Professor of pharmacology and toxicology, Director of Drug Discovery Institute, University of Houston, Houston, TX, since 2017. He is certified by the American Board of Toxicology (D.A.B.T.) and was on its Board of Directors from 2009 until 2013. Dr. Zhang was elected as a Fellow of the American Association for the Advancement of Science (AAAS) in 2009 and a Fellow of the National Academy of Inventors (NAI) in 2018. He obtained his M.D. and Ph.D. in Toxicology from Shanghai Medical University (now Fudan University Shanghai Medical College), Shanghai, China and completed his Postdoctoral/Clinical Pharmacology Fellowship at the University of Alabama, School of Medicine, Birmingham, AL, where he joined the faculty in the Division of Clinical Pharmacology and the Department of Pharmacology and Toxicology and then became Professor and Director of the Cancer Pharmacology Laboratory. In 2010, he moved to Department of Pharmaceutical Sciences, Texas Tech University Health Sciences Center (TTUHSC), as Professor and Department Chair. His major research interests include translational medicine, cancer biology, gene silencing, drug discovery and experimental therapy, clinical pharmacology and toxicology, focusing on targeting oncogenes and tumor suppressor genes.
Zafar A, Wang W, Liu G, et al. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med Res Rev. 2021;41:961–1021. 10.1002/med.21750
Atif Zafar and Wei Wang are co‐first authors.
REFERENCES
- 1. Twist CJ, Schmidt ML, Naranjo A, et al. Maintaining outstanding outcomes using response‐ and biology‐based therapy for intermediate‐risk neuroblastoma: a report from the Children's Oncology Group Study ANBL0531. J Clin Oncol. 2019;37(34):3243‐3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mlakar V, Jurkovic Mlakar S, Lopez G, Maris JM, Ansari M, Gumy‐Pause F. 11q deletion in neuroblastoma: a review of biological and clinical implications. Mol Cancer. 2017;16(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatta G, Botta L, Rossi S, et al. Childhood cancer survival in Europe 1999‐2007: results of EUROCARE‐5—a population‐based study. Lancet Oncol. 2014;15(1):35‐47. [DOI] [PubMed] [Google Scholar]
- 4. Tonini GP, Capasso M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. 2020;39(1):275‐285. [DOI] [PubMed] [Google Scholar]
- 5. Matthay KK, Maris JM, Schleiermacher G, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;2:16078. [DOI] [PubMed] [Google Scholar]
- 6. Park JR, Kreissman SG, London WB, et al. Effect of tandem autologous stem cell transplant vs single transplant on event‐free survival in patients with high‐risk neuroblastoma: a randomized clinical trial. JAMA. 2019;322(8):746‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henderson TO, Bhatia S, Pinto N, et al. Racial and ethnic disparities in risk and survival in children with neuroblastoma: a Children's Oncology Group study. J Clin Oncol. 2011;29(1):76‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delloye‐Bourgeois C, Castellani V. Hijacking of embryonic programs by neural crest‐derived neuroblastoma: from physiological migration to metastatic dissemination. Front Mol Neurosci. 2019;12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kholodenko IV, Kalinovsky DV, Doronin II, Deyev SM, Kholodenko RV. Neuroblastoma origin and therapeutic targets for immunotherapy. J Immunol Res. 2018;2018:7394268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isogai E, Ohira M, Ozaki T, Oba S, Nakamura Y, Nakagawara A. Oncogenic LMO3 collaborates with HEN2 to enhance neuroblastoma cell growth through transactivation of Mash1. PLOS One. 2011;6(5):e19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ichimiya S, Nimura Y, Seki N, Ozaki T, Nagase T, Nakagawara A. Downregulation of hASH1 is associated with the retinoic acid‐induced differentiation of human neuroblastoma cell lines. Med Pediatr Oncol. 2001;36(1):132‐134. [DOI] [PubMed] [Google Scholar]
- 12. Lasorella A, Boldrini R, Dominici C, et al. Id2 is critical for cellular proliferation and is the oncogenic effector of N‐myc in human neuroblastoma. Cancer Res. 2002;62(1):301‐306. [PubMed] [Google Scholar]
- 13. Gestblom C, Grynfeld A, Ora I, et al. The basic helix‐loop‐helix transcription factor dHAND, a marker gene for the developing human sympathetic nervous system, is expressed in both high‐ and low‐stage neuroblastomas. Lab Invest. 1999;79(1):67‐79. [PubMed] [Google Scholar]
- 14. Pietras A, Hansford LM, Johnsson AS, et al. HIF‐2alpha maintains an undifferentiated state in neural crest‐like human neuroblastoma tumor‐initiating cells. Proc Natl Acad Sci USA. 2009;106(39):16805‐16810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jogi A, Ora I, Nilsson H, et al. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest‐like phenotype. Proc Natl Acad Sci USA. 2002;99(10):7021‐7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Limpt V, Schramm A, Lakeman A, et al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene. 2004;23(57):9280‐9288. [DOI] [PubMed] [Google Scholar]
- 17. Armstrong BC, Krystal GW. Isolation and characterization of complementary DNA for N‐cym, a gene encoded by the DNA strand opposite to N‐myc. Cell Growth Differ. 1992;3(6):385‐390. [PubMed] [Google Scholar]
- 18. Duijkers FAM, Gaal J, Meijerink JPP, et al. High anaplastic lymphoma kinase immunohistochemical staining in neuroblastoma and ganglioneuroblastoma is an independent predictor of poor outcome. Am J Pathol. 2012;180(3):1223‐1231. [DOI] [PubMed] [Google Scholar]
- 19. Wang M, Zhou C, Sun Q, et al. ALK amplification and protein expression predict inferior prognosis in neuroblastomas. Exp Mol Pathol. 2013;95(2):124‐130. [DOI] [PubMed] [Google Scholar]
- 20. Zhu S, Lee JS, Guo F, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 2012;21(3):362‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shimada H, Chatten J, Newton WA, et al. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age‐linked classification of neuroblastomas. J Natl Cancer Inst. 1984;73(2):405‐416. [DOI] [PubMed] [Google Scholar]
- 22. Valter K, Zhivotovsky B, Gogvadze V. Cell death‐based treatment of neuroblastoma. Cell Death Dis. 2018;9(2):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohn SL, Pearson ADJ, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009;27(2):289‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shohet J, Foster J. Neuroblastoma. BMJ. 2017;357:j1863. [DOI] [PubMed] [Google Scholar]
- 25. Berlanga P, Cañete A, Castel V. Advances in emerging drugs for the treatment of neuroblastoma. Expert Opin Emerging Drugs. 2017;22(1):63‐75. [DOI] [PubMed] [Google Scholar]
- 26. Johnsen JI, Dyberg C, Wickström M. Neuroblastoma—a neural crest derived embryonal malignancy. Front Mol Neurosci. 2019;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pinto NR, Applebaum MA, Volchenboum SL, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. 2015;33(27):3008‐3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coughlan D, Gianferante M, Lynch CF, Stevens JL, Harlan LC. Treatment and survival of childhood neuroblastoma: Evidence from a population‐based study in the United States. Pediatr Hematol Oncol. 2017;34(5):320‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith V, Foster J. High‐risk neuroblastoma treatment review. Children (Basel, Switzerland). 2018;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24(1):65‐86. [DOI] [PubMed] [Google Scholar]
- 31. Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2202‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu AL, Gilman AL, Ozkaynak MF, et al. Anti‐GD2 antibody with GM‐CSF, interleukin‐2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363(14):1324‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheung NV, Heller G. Chemotherapy dose intensity correlates strongly with response, median survival, and median progression‐free survival in metastatic neuroblastoma. J Clin Oncol. 1991;9(6):1050‐1058. [DOI] [PubMed] [Google Scholar]
- 34. Castel V, Cañete A, Navarro S, et al. Outcome of high‐risk neuroblastoma using a dose intensity approach: improvement in initial but not in long‐term results. Med Pediatr Oncol. 2001;37(6):537‐542. [DOI] [PubMed] [Google Scholar]
- 35. Park JR, Scott JR, Stewart CF, et al. Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high‐risk neuroblastoma: a Children's Oncology Group study. J Clin Oncol. 2011;29(33):4351‐4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simon T, Längler A, Harnischmacher U, et al. Topotecan, cyclophosphamide, and etoposide (TCE) in the treatment of high‐risk neuroblastoma. Results of a phase‐II trial. J Cancer Res Clin Oncol. 2007;133(9):653‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. London WB, Castel V, Monclair T, et al. Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol. 2011;29(24):3286‐3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holmes K, Pötschger U, Pearson ADJ, et al. Influence of surgical excision on the survival of patients with stage 4 high‐risk neuroblastoma: a report from the HR‐NBL1/SIOPEN study. J Clin Oncol. 2020;38(25):2902‐2915. [DOI] [PubMed] [Google Scholar]
- 39. Berthold F, Boos J, Burdach S, et al. Myeloablative megatherapy with autologous stem‐cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high‐risk neuroblastoma: a randomised controlled trial. Lancet Oncol. 2005;6(9):649‐658. [DOI] [PubMed] [Google Scholar]
- 40. Pearson AD, Pinkerton CR, Lewis IJ, Imeson J, Ellershaw C, Machin D. High‐dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. Lancet Oncol. 2008;9(3):247‐256. [DOI] [PubMed] [Google Scholar]
- 41. Matthay KK, Reynolds CP, Seeger RC, et al. Long‐term results for children with high‐risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13‐cis‐retinoic acid: a Children's Oncology Group study. J Clin Oncol. 2009;27(7):1007‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tolbert VP, Matthay KK. Neuroblastoma: clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018;372(2):195‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luksch R, Castellani MR, Collini P, et al. Neuroblastoma (peripheral neuroblastic tumours). Crit Rev Oncol Hematol. 2016;107:163‐181. [DOI] [PubMed] [Google Scholar]
- 44. Bosse KR, Maris JM. Advances in the translational genomics of neuroblastoma: from improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer. 2016;122(1):20‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32(1):40‐51. [DOI] [PubMed] [Google Scholar]
- 46. Zhao Q, Liu Y, Zhang Y, et al. Role and toxicity of radiation therapy in neuroblastoma patients: a literature review. Crit Rev Oncol Hematol. 2020;149:102924. [DOI] [PubMed] [Google Scholar]
- 47. George SL, Parmar V, Lorenzi F, et al. Novel therapeutic strategies targeting telomere maintenance mechanisms in high‐risk neuroblastoma. J Exp Clin Cancer Res. 2020;39(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trigg RM, Turner SD. ALK in neuroblastoma: biological and therapeutic implications. Cancers. 2018;10(4):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Becker J, Wilting J. WNT Signaling in neuroblastoma. Cancers. 2019;11:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Titov A, Valiullina A, Zmievskaya E, et al. Advancing CAR T‐cell therapy for solid tumors: lessons learned from lymphoma treatment. Cancers. 2020;12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szanto CL, Cornel AM, Vijver SV, Nierkens S. Monitoring immune responses in neuroblastoma patients during therapy. Cancers. 2020;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barr EK, Applebaum MA. Genetic predisposition to neuroblastoma. Children (Basel, Switzerland). 2018;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Greengard EG. Molecularly targeted therapy for neuroblastoma. Children (Basel, Switzerland). 2018;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Meany HJ. Non‐high‐risk neuroblastoma: classification and achievements in therapy. Children (Basel, Switzerland). 2019;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Arendonk KJ, Chung DH. Neuroblastoma: tumor biology and its implications for staging and treatment. Children (Basel, Switzerland). 2019;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pastor ER, Mousa SA. Current management of neuroblastoma and future direction. Crit Rev Oncol Hematol. 2019;138:38‐43. [DOI] [PubMed] [Google Scholar]
- 57. Olecki E, Grant CN. MIBG in neuroblastoma diagnosis and treatment. Semin Pediatr Surg. 2019;28(6):150859. [DOI] [PubMed] [Google Scholar]
- 58. Richards RM, Sotillo E, Majzner RG. CAR T cell therapy for neuroblastoma. Front Immunol. 2018;9:2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Higashi M, Sakai K, Fumino S, Aoi S, Furukawa T, Tajiri T. The roles played by the MYCN, Trk, and ALK genes in neuroblastoma and neural development. Surg Today. 2019;49(9):721‐727. [DOI] [PubMed] [Google Scholar]
- 60. Huertas‐Castaño C, Gómez‐Muñoz MA, Pardal R, Vega FM. Hypoxia in the initiation and progression of neuroblastoma tumours. Int J Mol Sci. 2019;21:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Park JA, Cheung NV. Targets and antibody formats for immunotherapy of neuroblastoma. J Clin Oncol. 2020;38(16):1836‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marengo B, Pulliero A, Izzotti A, Domenicotti C. miRNA regulation of glutathione homeostasis in cancer initiation, progression and therapy resistance. MicroRNA (Shariqah, United Arab Emirates). 2020;9(3):187‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Johnsen JI, Dyberg C, Fransson S, Wickström M. Molecular mechanisms and therapeutic targets in neuroblastoma. Pharmacol Res. 2018;131:164‐176. [DOI] [PubMed] [Google Scholar]
- 64. Cavalli E, Ciurleo R, Petralia MC, et al. Emerging role of the macrophage migration inhibitory factor family of cytokines in neuroblastoma. Pathogenic effectors and novel therapeutic targets? Molecules (Basel, Switzerland). 2020;25:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Newman EA, Abdessalam S, Aldrink JH, et al. Update on neuroblastoma. J Pediatr Surg. 2019;54(3):383‐389. [DOI] [PubMed] [Google Scholar]
- 66. Tomolonis JA, Agarwal S, Shohet JM. Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res. 2018;372(2):245‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ritenour LE, Randall MP, Bosse KR, Diskin SJ. Genetic susceptibility to neuroblastoma: current knowledge and future directions. Cell Tissue Res. 2018;372(2):287‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Voeller J, Sondel PM. Advances in Anti‐GD2 immunotherapy for treatment of high‐risk neuroblastoma. J Pediatr Hematol Oncol. 2019;41(3):163‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Swift CC, Eklund MJ, Kraveka JM, Alazraki AL. Updates in diagnosis, management, and treatment of neuroblastoma. Radiographics. 2018;38(2):566‐580. [DOI] [PubMed] [Google Scholar]
- 70. Ryan AL, Akinkuotu A, Pierro A, Morgenstern DA, Irwin MS. The role of surgery in high‐risk neuroblastoma. J Pediatr Hematol Oncol. 2020;42(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 71. Jones DTW, Banito A, Grünewald TGP, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat Rev Cancer. 2019;19(8):420‐438. [DOI] [PubMed] [Google Scholar]
- 72. Tsubota S, Kadomatsu K. Origin and initiation mechanisms of neuroblastoma. Cell Tissue Res. 2018;372(2):211‐221. [DOI] [PubMed] [Google Scholar]
- 73. Fulda S. The PI3K/Akt/mTOR pathway as therapeutic target in neuroblastoma. Curr Cancer Drug Targets. 2009;9(6):729‐737. [DOI] [PubMed] [Google Scholar]
- 74. Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67(2):735‐745. [DOI] [PubMed] [Google Scholar]
- 75. Johnsen JI, Segerström L, Orrego A, et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008;27(20):2910‐2922. [DOI] [PubMed] [Google Scholar]
- 76. Boller D, Schramm A, Doepfner KT, et al. Targeting the phosphoinositide 3‐kinase isoform p110delta impairs growth and survival in neuroblastoma cells. Clin Cancer Res. 2008;14(4):1172‐1181. [DOI] [PubMed] [Google Scholar]
- 77. Muñoz J, Lázcoz P, del Mar Inda M, et al. Homozygous deletion and expression of PTEN and DMBT1 in human primary neuroblastoma and cell lines. Int J Cancer. 2004;109(5):673‐679. [DOI] [PubMed] [Google Scholar]
- 78. Dam V, Morgan BT, Mazanek P, Hogarty MD. Mutations in PIK3CA are infrequent in neuroblastoma. BMC Cancer. 2006;6:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971‐974. [DOI] [PubMed] [Google Scholar]
- 82. Janoueix‐Lerosey I, Lequin D, Brugières L, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455(7215):967‐970. [DOI] [PubMed] [Google Scholar]
- 83. Osajima‐Hakomori Y, Miyake I, Ohira M, Nakagawara A, Nakagawa A, Sakai R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol. 2005;167(1):213‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Blume‐Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355‐365. [DOI] [PubMed] [Google Scholar]
- 85. Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain‐derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy‐induced apoptosis via phosphatidylinositol 3'‐kinase pathway. Cancer Res. 2002;62(22):6756‐6763. [PubMed] [Google Scholar]
- 86. Ho R, Eggert A, Hishiki T, et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 2002;62(22):6462‐6466. [PubMed] [Google Scholar]
- 87. Hogarty MD, Maris JM. PI3King on MYCN to improve neuroblastoma therapeutics. Cancer Cell. 2012;21(2):145‐147. [DOI] [PubMed] [Google Scholar]
- 88. King D, Yeomanson D, Bryant HE. PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol. 2015;37(4):245‐251. [DOI] [PubMed] [Google Scholar]
- 89. Chesler L, Schlieve C, Goldenberg DD, et al. Inhibition of phosphatidylinositol 3‐kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006;66(16):8139‐8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu X, Mazanek P, Dam V, et al. Deregulated Wnt/beta‐catenin program in high‐risk neuroblastomas without MYCN amplification. Oncogene. 2008;27(10):1478‐1488. [DOI] [PubMed] [Google Scholar]
- 91. Zhang L, Li K, Lv Z, Xiao X, Zheng J. The effect on cell growth by Wnt1 RNAi in human neuroblastoma SH‐SY5Y cell line. Pediatr Surg Int. 2009;25(12):1065‐1071. [DOI] [PubMed] [Google Scholar]
- 92. Zins K, Schäfer R, Paulus P, et al. Frizzled2 signaling regulates growth of high‐risk neuroblastomas by interfering with β‐catenin‐dependent and β‐catenin‐independent signaling pathways. Oncotarget. 2016;7(29):46187‐46202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Flahaut M, Meier R, Coulon A, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta‐catenin pathway. Oncogene. 2009;28(23):2245‐2256. [DOI] [PubMed] [Google Scholar]
- 94. Szemes M, Greenhough A, Melegh Z, et al. Wnt signalling drives context‐dependent differentiation or proliferation in neuroblastoma. Neoplasia. 2018;20(4):335‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cantilena S, Pastorino F, Pezzolo A, et al. Frizzled receptor 6 marks rare, highly tumourigenic stem‐like cells in mouse and human neuroblastomas. Oncotarget. 2011;2(12):976‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Moll UM, Petrenko O. The MDM2‐p53 interaction. Mol Cancer Res. 2003;1(14):1001‐1008. [PubMed] [Google Scholar]
- 97. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296‐299. [DOI] [PubMed] [Google Scholar]
- 98. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299‐303. [DOI] [PubMed] [Google Scholar]
- 99. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420(1):25‐27. [DOI] [PubMed] [Google Scholar]
- 100. Corvi R, Savelyeva L, Breit S, et al. Non‐syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene. 1995;10(6):1081‐1086. [PubMed] [Google Scholar]
- 101. Cattelani S, Defferrari R, Marsilio S, et al. Impact of a single nucleotide polymorphism in the MDM2 gene on neuroblastoma development and aggressiveness: results of a pilot study on 239 patients. Clin Cancer Res. 2008;14(11):3248‐3253. [DOI] [PubMed] [Google Scholar]
- 102. Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5(1):27‐41. [DOI] [PubMed] [Google Scholar]
- 103. Zhou S, Gu L, He J, Zhang H, Zhou M. MDM2 regulates vascular endothelial growth factor mRNA stabilization in hypoxia. Mol Cell Biol. 2011;31(24):4928‐4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gu L, Zhang H, He J, Li J, Huang M, Zhou M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene. 2012;31(11):1342‐1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Keshelava N, Zuo JJ, Chen P, et al. Loss of p53 function confers high‐level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001;61(16):6185‐6193. [PubMed] [Google Scholar]
- 106. Lee CC, Jia Y, Li N, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010;430(3):425‐437. [DOI] [PubMed] [Google Scholar]
- 107. Lamant L, Pulford K, Bischof D, et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000;156(5):1711‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dirks WG, Fähnrich S, Lis Y, Becker E, MacLeod RA, Drexler HG. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100(1):49‐56. [DOI] [PubMed] [Google Scholar]
- 109. Bresler SC, Weiser DA, Huwe PJ, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26(5):682‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gonzalez Malagon SG, Liu KJ. ALK and GSK3: shared features of neuroblastoma and neural crest cells. J Exp Neurosci. 2018;12:1179069518792499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Schönherr C, Ruuth K, Kamaraj S, et al. Anaplastic lymphoma kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012;31(50):5193‐5200. [DOI] [PubMed] [Google Scholar]
- 113. Berry T, Luther W, Bhatnagar N, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22(1):117‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Miyake I, Hakomori Y, Shinohara A, et al. Activation of anaplastic lymphoma kinase is responsible for hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene. 2002;21(38):5823‐5834. [DOI] [PubMed] [Google Scholar]
- 115. Carpenter EL, Mossé YP, Targeting ALK. in neuroblastoma—preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9(7):391‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. De Brouwer S, De Preter K, Kumps C, et al. Meta‐analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010;16(17):4353‐4362. [DOI] [PubMed] [Google Scholar]
- 117. Chang HH, Lu MY, Yang YL, et al. The prognostic roles of and correlation between ALK and MYCN protein expression in neuroblastoma. J Clin Pathol. 2020;73(3):154‐161. [DOI] [PubMed] [Google Scholar]
- 118. Claeys S, Denecker G, Durinck K, et al. ALK positively regulates MYCN activity through repression of HBP1 expression. Oncogene. 2019;38(15):2690‐2705. [DOI] [PubMed] [Google Scholar]
- 119. Fransson S, Hansson M, Ruuth K, et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosomes Cancer. 2015;54(2):99‐109. [DOI] [PubMed] [Google Scholar]
- 120. Okubo J, Takita J, Chen Y, et al. Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene. 2012;31(44):4667‐4676. [DOI] [PubMed] [Google Scholar]
- 121. Cazes A, Louis‐Brennetot C, Mazot P, et al. Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res. 2013;73(1):195‐204. [DOI] [PubMed] [Google Scholar]
- 122. Schleiermacher G, Javanmardi N, Bernard V, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. 2014;32(25):2727‐2734. [DOI] [PubMed] [Google Scholar]
- 123. Eleveld TF, Oldridge DA, Bernard V, et al. Relapsed neuroblastomas show frequent RAS‐MAPK pathway mutations. Nat Genet. 2015;47(8):864‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Padovan‐Merhar OM, Raman P, Ostrovnaya I, et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PLOS Genet. 2016;12(12):e1006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bellini A, Bernard V, Leroy Q, et al. Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clin Cancer Res. 2015;21(21):4913‐4921. [DOI] [PubMed] [Google Scholar]
- 126. Ueda T, Nakata Y, Yamasaki N, et al. ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene. 2016;35(34):4447‐4458. [DOI] [PubMed] [Google Scholar]
- 127. De Mariano M, Stigliani S, Moretti S, et al. A genome‐wide microRNA profiling indicates miR‐424‐5p and miR‐503‐5p as regulators of ALK expression in neuroblastoma. Oncotarget. 2017;8(34):56518‐56532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chen K, Lv F, Xu G, Zhang M, Wu Y, Wu Z. Phosphoproteomics reveals ALK promote cell progress via RAS/JNK pathway in neuroblastoma. Oncotarget. 2016;7(46):75968‐75980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high‐risk neuroblastoma. Nat Genet. 2013;45(3):279‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Eleveld TF, Schild L, Koster J, et al. RAS‐MAPK pathway‐driven tumor progression is associated with loss of CIC and other genomic aberrations in neuroblastoma. Cancer Res. 2018;78(21):6297‐6307. [DOI] [PubMed] [Google Scholar]
- 131. Brodeur GM. TRK‐a expression in neuroblastomas: a new prognostic marker with biological and clinical significance. J Natl Cancer Inst. 1993;85(5):344‐345. [DOI] [PubMed] [Google Scholar]
- 132. Nakagawara A, Arima‐Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328(12):847‐854. [DOI] [PubMed] [Google Scholar]
- 133. Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK‐B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14(1):759‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kogner P, Barbany G, Dominici C, Castello MA, Raschellá G, Persson H. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 1993;53(9):2044‐2050. [PubMed] [Google Scholar]
- 135. Nakagawara A, Arima M, Azar CG, Scavarda NJ, Brodeur GM. Inverse relationship between trk expression and N‐myc amplification in human neuroblastomas. Cancer Res. 1992;52(5):1364‐1368. [PubMed] [Google Scholar]
- 136. Brodeur GM, Minturn JE, Ho R, et al. Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res. 2009;15(10):3244‐3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Desmet CJ, Peeper DS. The neurotrophic receptor TrkB: a drug target in anti‐cancer therapy? Cell Mol Life Sci. 2006;63(7‐8):755‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE, Brodeur GM. Expression of the neurotrophin receptor TrkA down‐regulates expression and function of angiogenic stimulators in SH‐SY5Y neuroblastoma cells. Cancer Res. 2002;62(6):1802‐1808. [PubMed] [Google Scholar]
- 139. Croucher JL, Iyer R, Li N, et al. TrkB inhibition by GNF‐4256 slows growth and enhances chemotherapeutic efficacy in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2015;75(1):131‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Li Z, Zhang Y, Tong Y, Tong J, Thiele CJ. Trk inhibitor attenuates the BDNF/TrkB‐induced protection of neuroblastoma cells from etoposide in vitro and in vivo. Cancer Biol Ther. 2015;16(3):477‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Iyer R, Varela CR, Minturn JE, et al. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother Pharmacol. 2012;70(3):477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Evans AE, Kisselbach KD, Liu X, et al. Effect of CEP‐751 (KT‐6587) on neuroblastoma xenografts expressing TrkB. Med Pediatr Oncol. 2001;36(1):181‐184. [DOI] [PubMed] [Google Scholar]
- 143. Evans AE, Kisselbach KD, Yamashiro DJ, et al. Antitumor activity of CEP‐751 (KT‐6587) on human neuroblastoma and medulloblastoma xenografts. Clin Cancer Res. 1999;5(11):3594‐3602. [PubMed] [Google Scholar]
- 144. Iyer R, Wehrmann L, Golden RL, et al. Entrectinib is a potent inhibitor of Trk‐driven neuroblastomas in a xenograft mouse model. Cancer Lett. 2016;372(2):179‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Seeger RC, Brodeur GM, Sather H, et al. Association of multiple copies of the N‐myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313(18):1111‐1116. [DOI] [PubMed] [Google Scholar]
- 146. Bagatell R, Beck‐Popovic M, London WB, et al. Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol. 2009;27(3):365‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Katzenstein HM, Bowman LC, Brodeur GM, et al. Prognostic significance of age, MYCN oncogene amplification, tumor cell ploidy, and histology in 110 infants with stage D(S) neuroblastoma: the pediatric oncology group experience—a pediatric oncology group study. J Clin Oncol. 1998;16(6):2007‐2017. [DOI] [PubMed] [Google Scholar]
- 148. Campbell K, Naranjo A, Hibbitts E, et al. Association of heterogeneous MYCN amplification with clinical features, biological characteristics and outcomes in neuroblastoma: a report from the Children's Oncology Group. Eur J Cancer (Oxford, England: 1990). 2020;133:112‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wenzel A, Schwab M. The mycN/max protein complex in neuroblastoma. Short review. Eur J Cancer (Oxford, England: 1990). 1995;31(4):516‐519. [DOI] [PubMed] [Google Scholar]
- 150. Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res. 2010;107:163‐224. [DOI] [PubMed] [Google Scholar]
- 151. Müller I, Larsson K, Frenzel A, et al. Targeting of the MYCN protein with small molecule c‐MYC inhibitors. PLOS One. 2014;9(5):e97285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Ribeiro D, Klarqvist MDR, Westermark UK, et al. Regulation of nuclear hormone receptors by MYCN‐driven miRNAs impacts neural differentiation and survival in neuroblastoma patients. Cell Rep. 2016;16(4):979‐993. [DOI] [PubMed] [Google Scholar]
- 153. Zirath H, Frenzel A, Oliynyk G, et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci USA. 2013;110(25):10258‐10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Yang Y, Ding L, Zhou Q, et al. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int. 2020;20:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Puissant A, Frumm SM, Alexe G, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discovery. 2013;3(3):308‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wyce A, Ganji G, Smitheman KN, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLOS One. 2013;8(8):e72967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hogarty MD, Norris MD, Davis K, et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008;68(23):9735‐9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Evageliou NF, Haber M, Vu A, et al. Polyamine antagonist therapies inhibit neuroblastoma initiation and progression. Clin Cancer Res. 2016;22(17):4391‐4404. [DOI] [PubMed] [Google Scholar]
- 159. Ambrosini G, Adida C, Altieri DC. A novel anti‐apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917‐921. [DOI] [PubMed] [Google Scholar]
- 160. Islam A, Kageyama H, Takada N, et al. High expression of survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19(5):617‐623. [DOI] [PubMed] [Google Scholar]
- 161. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3(3):203‐216. [DOI] [PubMed] [Google Scholar]
- 162. Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013;32(40):4748‐4757. [DOI] [PubMed] [Google Scholar]
- 163. Hagenbuchner J, Kiechl‐Kohlendorfer U, Obexer P, Ausserlechner MJ. BIRC5/Survivin as a target for glycolysis inhibition in high‐stage neuroblastoma. Oncogene. 2016;35(16):2052‐2061. [DOI] [PubMed] [Google Scholar]
- 164. Chandele A, Prasad V, Jagtap JC, Shukla R, Shastry PR. Upregulation of survivin in G2/M cells and inhibition of caspase 9 activity enhances resistance in staurosporine‐induced apoptosis. Neoplasia. 2004;6(1):29‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Azuhata T, Scott D, Takamizawa S, et al. The inhibitor of apoptosis protein survivin is associated with high‐risk behavior of neuroblastoma. J Pediatr Surg. 2001;36(12):1785‐1791. [DOI] [PubMed] [Google Scholar]
- 166. Lamers F, van der Ploeg I, Schild L, et al. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocr Relat Cancer. 2011;18(6):657‐668. [DOI] [PubMed] [Google Scholar]
- 167. Liang H, Zhang L, Xu R, Ju XL. Silencing of survivin using YM155 induces apoptosis and chemosensitization in neuroblastomas cells. Eur Rev Med Pharmacol Sci. 2013;17(21):2909‐2915. [PubMed] [Google Scholar]
- 168. Kunnimalaiyaan S, Schwartz VK, Jackson IA, Clark Gamblin T, Kunnimalaiyaan M. Antiproliferative and apoptotic effect of LY2090314, a GSK‐3 inhibitor, in neuroblastoma in vitro. BMC Cancer. 2018;18(1):560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Subramanian C, Grogan PT, Opipari VP, Timmermann BN, Cohen MS. Novel natural withanolides induce apoptosis and inhibit migration of neuroblastoma cells through down regulation of N‐myc and suppression of Akt/mTOR/NF‐κB activation. Oncotarget. 2018;9(18):14509‐14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Li S, He J, Li S, et al. Noscapine induced apoptosis via downregulation of survivin in human neuroblastoma cells having wild type or null p53. PLOS One. 2012;7(7):e40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Schultze K, Böck B, Eckert A, et al. Troglitazone sensitizes tumor cells to TRAIL‐induced apoptosis via down‐regulation of FLIP and Survivin. Apoptosis. 2006;11(9):1503‐1512. [DOI] [PubMed] [Google Scholar]
- 172. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4‐10. [DOI] [PubMed] [Google Scholar]
- 173. Stanton MJ, Dutta S, Zhang H, et al. Autophagy control by the VEGF‐C/NRP‐2 axis in cancer and its implication for treatment resistance. Cancer Res. 2013;73(1):160‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13(12):871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mattei MG, Borg JP, Rosnet O, Marmé D, Birnbaum D. Assignment of vascular endothelial growth factor (VEGF) and placenta growth factor (PLGF) genes to human chromosome 6p12‐p21 and 14q24‐q31 regions, respectively. Genomics. 1996;32(1):168‐169. [DOI] [PubMed] [Google Scholar]
- 176. Roskoski R, Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62(3):179‐213. [DOI] [PubMed] [Google Scholar]
- 177. Tischer E, Mitchell R, Hartman T, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266(18):11947‐11954. [PubMed] [Google Scholar]
- 178. Meister B, Grünebach F, Bautz F, et al. Expression of vascular endothelial growth factor (VEGF) and its receptors in human neuroblastoma. Eur J Cancer (Oxford, England: 1990). 1999;35(3):445‐449. [DOI] [PubMed] [Google Scholar]
- 179. Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GM, Himelstein BP. High‐level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6(5):1900‐1908. [PubMed] [Google Scholar]
- 180. Sköldenberg EG, Larsson A, Jakobson A, et al. The angiogenic growth factors HGF and VEGF in serum and plasma from neuroblastoma patients. Anticancer Res. 2009;29(8):3311‐3319. [PubMed] [Google Scholar]
- 181. González A, González‐González A, Alonso‐González C, Menéndez‐Menéndez J, Martínez‐Campa C, Cos S. Melatonin inhibits angiogenesis in SH‐SY5Y human neuroblastoma cells by downregulation of VEGF. Oncol Rep. 2017;37(4):2433‐2440. [DOI] [PubMed] [Google Scholar]
- 182. Lakoma A, Barbieri E, Agarwal S, et al. The MDM2 small‐molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild‐type neuroblastoma. Cell Death Discov. 2015;1:15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Calero R, Morchon E, Johnsen JI, Serrano R. Sunitinib suppress neuroblastoma growth through degradation of MYCN and inhibition of angiogenesis. PLOS One. 2014;9(4):e95628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Puppo M, Battaglia F, Ottaviano C, et al. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia‐inducible factor‐1 alpha and ‐2 alpha. Mol Cancer Ther. 2008;7(7):1974‐1984. [DOI] [PubMed] [Google Scholar]
- 185. Hamner JB, Dickson PV, Sims TL, et al. Bortezomib inhibits angiogenesis and reduces tumor burden in a murine model of neuroblastoma. Surgery. 2007;142(2):185‐191. [DOI] [PubMed] [Google Scholar]
- 186. Beppu K, Jaboine J, Merchant MS, Mackall CL, Thiele CJ. Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J Natl Cancer Inst. 2004;96(1):46‐55. [DOI] [PubMed] [Google Scholar]
- 187. Trochet D, Bourdeaut F, Janoueix‐Lerosey I, et al. Germline mutations of the paired‐like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74(4):761‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Mosse YP, Laudenslager M, Khazi D, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75(4):727‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Raabe EH, Laudenslager M, Winter C, et al. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008;27(4):469‐476. [DOI] [PubMed] [Google Scholar]
- 190. Serra A, Häberle B, König IR, et al. Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol. 2008;30(10):728‐732. [DOI] [PubMed] [Google Scholar]
- 191. Wang W, Zhong Q, Teng L, et al. Mutations that disrupt PHOXB interaction with the neuronal calcium sensor HPCAL1 impede cellular differentiation in neuroblastoma. Oncogene. 2014;33(25):3316‐3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Bachetti T, Di Paolo D, Di Lascio S, et al. PHOX2B‐mediated regulation of ALK expression: in vitro identification of a functional relationship between two genes involved in neuroblastoma. PLOS One. 2010;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ke XX, Zhang D, Zhao H, et al. Phox2B correlates with MYCN and is a prognostic marker for neuroblastoma development. Oncol Lett. 2015;9(6):2507‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Revet I, Huizenga G, Chan A, et al. The MSX1 homeobox transcription factor is a downstream target of PHOX2B and activates the Delta‐Notch pathway in neuroblastoma. Exp Cell Res. 2008;314(4):707‐719. [DOI] [PubMed] [Google Scholar]
- 195. Di Zanni E, Bianchi G, Ravazzolo R, Raffaghello L, Ceccherini I, Bachetti T. Targeting of PHOX2B expression allows the identification of drugs effective in counteracting neuroblastoma cell growth. Oncotarget. 2017;8(42):72133‐72146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Di Zanni E, Fornasari D, Ravazzolo R, Ceccherini I, Bachetti T. Identification of novel pathways and molecules able to down‐regulate PHOX2B gene expression by in vitro drug screening approaches in neuroblastoma cells. Exp Cell Res. 2015;336(1):43‐57. [DOI] [PubMed] [Google Scholar]
- 197. Suebsoonthron J, Jaroonwitchawan T, Yamabhai M, Noisa P. Inhibition of WNT signaling reduces differentiation and induces sensitivity to doxorubicin in human malignant neuroblastoma SH‐SY5Y cells. Anti‐Cancer Drugs. 2017;28(5):469‐479. [DOI] [PubMed] [Google Scholar]
- 198. Ferrari‐Toninelli G, Bonini SA, Uberti D, et al. Targeting Notch pathway induces growth inhibition and differentiation of neuroblastoma cells. Neuro‐Oncology. 2010;12(12):1231‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Påhlman S, Stockhausen MT, Fredlund E, Axelson H. Notch signaling in neuroblastoma. Sem Cancer Biol. 2004;14(5):365‐373. [DOI] [PubMed] [Google Scholar]
- 200. Funahashi Y, Hernandez SL, Das I, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008;68(12):4727‐4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Dovey HF, John V, Anderson JP, et al. Functional gamma‐secretase inhibitors reduce beta‐amyloid peptide levels in brain. J Neurochem. 2001;76(1):173‐181. [DOI] [PubMed] [Google Scholar]
- 202. Lanz TA, Himes CS, Pallante G, et al. The gamma‐secretase inhibitor N‐[N‐(3,5‐difluorophenacetyl)‐L‐alanyl]‐S‐phenylglycine t‐butyl ester reduces A beta levels in vivo in plasma and cerebrospinal fluid in young (plaque‐free) and aged (plaque‐bearing) Tg2576 mice. J Pharmacol Exp Ther. 2003;305(3):864‐871. [DOI] [PubMed] [Google Scholar]
- 203. Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. γ‐Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828(12):2898‐2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Lau L, Hansford LM, Cheng LS, et al. Cyclooxygenase inhibitors modulate the p53/HDM2 pathway and enhance chemotherapy‐induced apoptosis in neuroblastoma. Oncogene. 2007;26(13):1920‐1931. [DOI] [PubMed] [Google Scholar]
- 205. Diskin SJ, Capasso M, Schnepp RW, et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet. 2012;44(10):1126‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Molenaar JJ, Domingo‐Fernández R, Ebus ME, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let‐7 suppression. Nat Genet. 2012;44(11):1199‐1206. [DOI] [PubMed] [Google Scholar]
- 207. Schnepp RW, Khurana P, Attiyeh EF, et al. A LIN28B‐RAN‐AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer Cell. 2015;28(5):599‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Lozier AM, Rich ME, Grawe AP, et al. Targeting ornithine decarboxylase reverses the LIN28/Let‐7 axis and inhibits glycolytic metabolism in neuroblastoma. Oncotarget. 2015;6(1):196‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Rich ME, Zhao P, Nagulapally AB, Bond J, Sholler GS. Inhibition of neuroblastoma cell growth by difluoromethylornithine (DFMO) and bortezomib through suppression of LIN28 and MYCN. J Cancer Ther. 2017;8(6):561‐578. [Google Scholar]
- 210. Yoda H, Nakayama T, Miura M, Toriyama M, Motohashi S, Suzuki T. Vitamin K3 derivative induces apoptotic cell death in neuroblastoma via downregulation of MYCN expression. Biochem Biophys Rep. 2019;20:100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Shahbazi J, Liu PY, Atmadibrata B, et al. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N‐Myc expression and induce anticancer effects. Clin Cancer Res. 2016;22(10):2534‐2544. [DOI] [PubMed] [Google Scholar]
- 212. Goldman SC, Chen CY, Lansing TJ, Gilmer TM, Kastan MB. The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. Am J Pathol. 1996;148(5):1381‐1385. [PMC free article] [PubMed] [Google Scholar]
- 213. Moll UM, LaQuaglia M, Bénard J, Riou G. Wild‐type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci USA. 1995;92(10):4407‐4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Van Maerken T, Ferdinande L, Taildeman J, et al. Antitumor activity of the selective MDM2 antagonist nutlin‐3 against chemoresistant neuroblastoma with wild‐type p53. J Natl Cancer Inst. 2009;101(22):1562‐1574. [DOI] [PubMed] [Google Scholar]
- 215. Barbieri E. MDM2 inhibition sensitizes neuroblastoma to chemotherapy‐induced apoptotic cell death. Mol Cancer Ther. 2006;5(9):2358‐2365. [DOI] [PubMed] [Google Scholar]
- 216. Lu J, Guan S, Zhao Y, et al. Novel MDM2 inhibitor SAR405838 (MI‐773) induces p53‐mediated apoptosis in neuroblastoma. Oncotarget. 2016;7(50):82757‐82769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Kang MH, Reynolds CP, Kolb EA, et al. Initial testing (stage 1) of MK‐8242—a novel MDM2 inhibitor‐by the pediatric preclinical testing program. Pediatr Blood Cancer. 2016;63(10):1744‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Gamble LD, Kees UR, Tweddle DA, Lunec J. MYCN sensitizes neuroblastoma to the MDM2‐p53 antagonists Nutlin‐3 and MI‐63. Oncogene. 2012;31(6):752‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Al‐Ghabkari A, Narendran A. In vitro characterization of a potent p53‐MDM2 inhibitor, RG7112 in neuroblastoma cancer cell lines. Cancer Biother Radiopharm. 2019;34(4):252‐257. [DOI] [PubMed] [Google Scholar]
- 220. Chen L, Pastorino F, Berry P, et al. Preclinical evaluation of the first intravenous small molecule MDM2 antagonist alone and in combination with temozolomide in neuroblastoma. Int J Cancer. 2019;144(12):3146‐3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Giustiniano M, Daniele S, Pelliccia S, et al. Computer‐aided identification and lead optimization of dual murine double minute 2 and 4 binders: structure‐activity relationship studies and pharmacological activity. J Med Chem. 2017;60(19):8115‐8130. [DOI] [PubMed] [Google Scholar]
- 222. Ramaiah MJ, Pushpavalli SNCVL, Lavanya A, et al. Novel anthranilamide‐pyrazolo[1,5‐a]pyrimidine conjugates modulate the expression of p53‐MYCN associated micro RNAs in neuroblastoma cells and cause cell cycle arrest and apoptosis. Bioorg Med Chem Lett. 2013;23(20):5699‐5706. [DOI] [PubMed] [Google Scholar]
- 223. Bresler SC, Wood AC, Haglund EA, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3(108):108ra114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679‐690. [DOI] [PubMed] [Google Scholar]
- 225. Infarinato NR, Park JH, Krytska K, et al. The ALK/ROS1 inhibitor PF‐06463922 overcomes primary resistance to crizotinib in ALK‐driven neuroblastoma. Cancer Discov. 2016;6(1):96‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Xu DQ, Toyoda H, Yuan XJ, et al. Anti‐tumor effect of AZD8055 against neuroblastoma cells in vitro and in vivo. Exp Cell Res. 2018;365(2):177‐184. [DOI] [PubMed] [Google Scholar]
- 227. Hart LS, Rader J, Raman P, et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin Cancer Res. 2017;23(7):1785‐1796. [DOI] [PubMed] [Google Scholar]
- 228. Van Goethem A, Yigit N, Moreno‐Smith M, et al. Dual targeting of MDM2 and BCL2 as a therapeutic strategy in neuroblastoma. Oncotarget. 2017;8(34):57047‐57057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Lamers F, Schild L, Koster J, Versteeg R, Caron HN, Molenaar JJ. Targeted BIRC5 silencing using YM155 causes cell death in neuroblastoma cells with low ABCB1 expression. Eur J Cancer (Oxford, England: 1990). 2012;48(5):763‐771. [DOI] [PubMed] [Google Scholar]
- 230. Ham J, Costa C, Sano R, et al. Exploitation of the apoptosis‐primed state of MYCN‐amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell. 2016;29(2):159‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Tanos R, Karmali D, Nalluri S, Goldsmith KC. Select Bcl‐2 antagonism restores chemotherapy sensitivity in high‐risk neuroblastoma. BMC Cancer. 2016;16:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Levesley J, Steele L, Brüning‐Richardson A, et al. Selective BCL‐XL inhibition promotes apoptosis in combination with MLN8237 in medulloblastoma and pediatric glioblastoma cells. Neuro‐Oncology. 2018;20(2):203‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Bate‐Eya LT, den Hartog IJM, van der Ploeg I, et al. High efficacy of the BCL‐2 inhibitor ABT199 (venetoclax) in BCL‐2 high‐expressing neuroblastoma cell lines and xenografts and rational for combination with MCL‐1 inhibition. Oncotarget. 2016;7(19):27946‐27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Aveic S, Pantile M, Seydel A, et al. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget. 2016;7(5):5646‐5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Aveic S, Pantile M, Polo P, et al. Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell Int. 2018;18:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Jubierre L, Jiménez C, Rovira E, et al. Targeting of epigenetic regulators in neuroblastoma. Exp Mol Med. 2018;50(4):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Qiu YY, Mirkin BL, Dwivedi RS. Inhibition of DNA methyltransferase reverses cisplatin induced drug resistance in murine neuroblastoma cells. Cancer Detect Prev. 2005;29(5):456‐463. [DOI] [PubMed] [Google Scholar]
- 238. Bartolucci S, Rossi M, Longo A, et al. 5‐Aza‐2'‐deoxycytidine as inducer of differentiation and growth inhibition in mouse neuroblastoma cells. Cell Differ Dev. 1989;27(1):47‐55. [DOI] [PubMed] [Google Scholar]
- 239. Carpinelli P, Granata F, Augusti‐Tocco G, Rossi M, Bartolucci S. Antiproliferative effects and DNA hypomethylation by 5‐aza‐2'‐deoxycytidine in human neuroblastoma cell lines. Anti‐Cancer Drugs. 1993;4(6):629‐635. [DOI] [PubMed] [Google Scholar]
- 240. Charlet J, Schnekenburger M, Brown KW, Diederich M. DNA demethylation increases sensitivity of neuroblastoma cells to chemotherapeutic drugs. Biochem Pharmacol. 2012;83(7):858‐865. [DOI] [PubMed] [Google Scholar]
- 241. Lu Z, Tian Y, Salwen HR, et al. Histone‐lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anti‐Cancer Drugs. 2013;24(5):484‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Sun XJ, Man N, Tan Y, Nimer SD, Wang L. The role of histone acetyltransferases in normal and malignant hematopoiesis. Front Oncol. 2015;5:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Gajer JM, Furdas SD, Gründer A, et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis. 2015;4(2):e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Oehme I, Deubzer HE, Lodrini M, Milde T, Witt O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Invest Drugs. 2009;18(11):1605‐1617. [DOI] [PubMed] [Google Scholar]
- 245. Oehme I, Linke JP, Bock BC, et al. Histone deacetylase 10 promotes autophagy‐mediated cell survival. Proc Natl Acad Sci U S A. 2013;110(28):E2592‐E2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Rettig I, Koeneke E, Trippel F, et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid‐mediated differentiation. Cell Death Dis. 2015;6(2):e1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Rocchi P, Tonelli R, Camerin C, et al. p21Waf1/Cip1 is a common target induced by short‐chain fatty acid HDAC inhibitors (valproic acid, tributyrin and sodium butyrate) in neuroblastoma cells. Oncol Rep. 2005;13(6):1139‐1144. [PubMed] [Google Scholar]
- 248. Stockhausen MT, Sjölund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br J Cancer. 2005;92(4):751‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. De los Santos M, Zambrano A, Aranda A. Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH‐SY5Y cells. Mol Cancer Ther. 2007;6(4):1425‐1432. [DOI] [PubMed] [Google Scholar]
- 250. Wu JC, Jiang HM, Yang XH, Zheng HC. ING5‐mediated antineuroblastoma effects of suberoylanilide hydroxamic acid. Cancer Med. 2018;7(9):4554‐4569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 251. Marshall GM, Liu PY, Gherardi S, et al. SIRT1 promotes N‐Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N‐Myc protein stability. PLOS Genet. 2011;7(6):e1002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Tamkun JW, Deuring R, Scott MP, et al. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68(3):561‐572. [DOI] [PubMed] [Google Scholar]
- 253. Lee S, Rellinger EJ, Kim KW, et al. Bromodomain and extraterminal inhibition blocks tumor progression and promotes differentiation in neuroblastoma. Surgery. 2015;158(3):819‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Henssen A, Althoff K, Odersky A, et al. Targeting MYCN‐driven transcription by BET‐bromodomain inhibition. Clin Cancer Res. 2016;22(10):2470‐2481. [DOI] [PubMed] [Google Scholar]
- 255. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311‐320. [DOI] [PubMed] [Google Scholar]
- 256. Duprez L, Takahashi N, Van Hauwermeiren F, et al. RIP kinase‐dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35(6):908‐918. [DOI] [PubMed] [Google Scholar]
- 257. Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014;74(4):1056‐1066. [DOI] [PubMed] [Google Scholar]
- 258. Nicolai S, Pieraccioli M, Peschiaroli A, Melino G, Raschellà G. Neuroblastoma: oncogenic mechanisms and therapeutic exploitation of necroptosis. Cell Death Dis. 2015;6(12):e2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Semenza GL. Defining the role of hypoxia‐inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Chen S, Zhang M, Xing L, Wang Y, Xiao Y, Wu Y. HIF‐1α contributes to proliferation and invasiveness of neuroblastoma cells via SHH signaling. PLOS One. 2015;10(3):e0121115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Hartwich J, Orr WS, Ng CY, Spence Y, Morton C, Davidoff AM. HIF‐1α activation mediates resistance to anti‐angiogenic therapy in neuroblastoma xenografts. J Pediatr Surg. 2013;48(1):39‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Wallace EM, Rizzi JP, Han G, et al. A small‐molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 2016;76(18):5491‐5500. [DOI] [PubMed] [Google Scholar]
- 263. Persson CU, von Stedingk K, Fredlund E, et al. ARNT‐dependent HIF‐2 transcriptional activity is not sufficient to regulate downstream target genes in neuroblastoma. Exp Cell Res. 2020;388(2):111845. [DOI] [PubMed] [Google Scholar]
- 264. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science (New York, NY). 2020;367:6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer. 2019;18(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Challagundla KB, Wise PM, Neviani P, et al. Exosome‐mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. 2015;107:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Haug BH, Hald ØH, Utnes P, et al. Exosome‐like extracellular vesicles from MYCN‐amplified neuroblastoma cells contain oncogenic miRNAs. Anticancer Res. 2015;35(5):2521‐2530. [PubMed] [Google Scholar]
- 269. Neviani P, Wise PM, Murtadha M, et al. Natural killer‐derived exosomal miR‐186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019;79(6):1151‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Schmittgen TD. Exosomal miRNA cargo as mediator of immune escape mechanisms in neuroblastoma. Cancer Res. 2019;79(7):1293‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Geoerger B, Bourdeaut F, DuBois SG, et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res. 2017;23(10):2433‐2441. [DOI] [PubMed] [Google Scholar]
- 272. Nguyen R, Sahr N, Sykes A, et al. Longitudinal NK cell kinetics and cytotoxicity in children with neuroblastoma enrolled in a clinical phase II trial. J Immunother Cancer. 2020;8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK. Humanized 3F8 anti‐GD2 monoclonal antibody dosing with granulocyte‐macrophage colony‐stimulating factor in patients with resistant neuroblastoma: a phase 1 clinical trial. JAMA Oncol. 2018;4(12):1729‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Kushner BH, Modak S, Basu EM, Roberts SS, Kramer K, Cheung NK. Posterior reversible encephalopathy syndrome in neuroblastoma patients receiving anti‐GD2 3F8 monoclonal antibody. Cancer. 2013;119(15):2789‐2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Cheung IY, Hsu K, Cheung NK. Activation of peripheral‐blood granulocytes is strongly correlated with patient outcome after immunotherapy with anti‐GD2 monoclonal antibody and granulocyte‐macrophage colony‐stimulating factor. J Clin Oncol. 2012;30(4):426‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Kushner BH, Kramer K, Modak S, Cheung NK. Successful multifold dose escalation of anti‐GD2 monoclonal antibody 3F8 in patients with neuroblastoma: a phase I study. J Clin Oncol. 2011;29(9):1168‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Modak S, Kushner BH, Basu E, Roberts SS, Cheung NK. Combination of bevacizumab, irinotecan, and temozolomide for refractory or relapsed neuroblastoma: results of a phase II study. Pediatr Blood Cancer. 2017;64:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. DuBois SG, Mosse YP, Fox E, et al. Phase II trial of alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Cancer Res. 2018;24(24):6142‐6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Saulnier Sholler GL, Gerner EW, Bergendahl G, et al. A phase I trial of DFMO targeting polyamine addiction in patients with relapsed/refractory neuroblastoma. PLOS One. 2015;10(5):e0127246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Mody R, Naranjo A, Van Ryn C, et al. Irinotecan‐temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open‐label, randomised, phase 2 trial. Lancet Oncol. 2017;18(7):946‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Mossé YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large‐cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Burmakin M, Shi Y, Hedström E, Kogner P, Selivanova G. Dual targeting of wild‐type and mutant p53 by small molecule RITA results in the inhibition of N‐Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin Cancer Res. 2013;19(18):5092‐5103. [DOI] [PubMed] [Google Scholar]
- 283. Peirce SK, Findley HW, Prince C, Dasgupta A, Cooper T, Durden DL. The PI‐3 kinase‐Akt‐MDM2‐survivin signaling axis in high‐risk neuroblastoma: a target for PI‐3 kinase inhibitor intervention. Cancer Chemother Pharmacol. 2011;68(2):325‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Kushner BH, Cheung NKV, Modak S, et al. A phase I/Ib trial targeting the Pi3k/Akt pathway using perifosine: long‐term progression‐free survival of patients with resistant neuroblastoma. Int J Cancer. 2017;140(2):480‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Applebaum MA, Desai AV, Glade Bender JL, Cohn SL. Emerging and investigational therapies for neuroblastoma. Expert Opin Orphan Drugs. 2017;5(4):355‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Fouladi M, Park JR, Stewart CF, et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children's Oncology Group phase I consortium report. J Clin Oncol. 2010;28(22):3623‐3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Parisi MT, Eslamy H, Park JR, Shulkin BL, Yanik GA. ¹³¹I‐Metaiodobenzylguanidine theranostics in neuroblastoma: historical perspectives; practical applications. Semin Nucl Med. 2016;46(3):184‐202. [DOI] [PubMed] [Google Scholar]
- 288. Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012;18(10):2740‐2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Yanik GA, Villablanca JG, Maris JM, et al. 131I‐metaiodobenzylguanidine with intensive chemotherapy and autologous stem cell transplantation for high‐risk neuroblastoma. A new approaches to neuroblastoma therapy (NANT) phase II study. Biol Blood Marrow Trans. 2015;21(4):673‐681. [DOI] [PubMed] [Google Scholar]
- 290. DuBois SG, Matthay KK. Radiolabeled metaiodobenzylguanidine for the treatment of neuroblastoma. Nucl Med Biol. 2008;35(Suppl 1):S35‐S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Jacobson AF, Deng H, Lombard J, Lessig HJ, Black RR. 123I‐meta‐iodobenzylguanidine scintigraphy for the detection of neuroblastoma and pheochromocytoma: results of a meta‐analysis. J Clin Endocrinol Metab. 2010;95(6):2596‐2606. [DOI] [PubMed] [Google Scholar]
- 292. Matthay KK, DeSantes K, Hasegawa B, et al. Phase I dose escalation of 131I‐metaiodobenzylguanidine with autologous bone marrow support in refractory neuroblastoma. J Clin Oncol. 1998;16(1):229‐236. [DOI] [PubMed] [Google Scholar]
- 293. Matthay KK, Yanik G, Messina J, et al. Phase II study on the effect of disease sites, age, and prior therapy on response to iodine‐131‐metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol. 2007;25(9):1054‐1060. [DOI] [PubMed] [Google Scholar]
- 294. Mastrangelo R, Tornesello A, Lasorella A, et al. Optimal use of the 131‐I‐metaiodobenzylguanidine and cisplatin combination in advanced neuroblastoma. JNO. 1997;31(1‐2):153‐158. [DOI] [PubMed] [Google Scholar]
- 295. Mastrangelo S, Tornesello A, Diociaiuti L, Riccardi R, Rufini V, Troncone L. Treatment with meta‐[131I]iodobenzylguanidine and cisplatin in stage IV neuroblastoma. Q J Nucl Med. 1995;39(4 Suppl 1):69‐71. [PubMed] [Google Scholar]
- 296. Gaze MN, Chang YC, Flux GD, Mairs RJ, Saran FH, Meller ST. Feasibility of dosimetry‐based high‐dose 131I‐meta‐iodobenzylguanidine with topotecan as a radiosensitizer in children with metastatic neuroblastoma. Cancer Biother Radiopharm. 2005;20(2):195‐199. [DOI] [PubMed] [Google Scholar]
- 297. Mastrangelo S, Tornesello A, Diociaiuti L, et al. Treatment of advanced neuroblastoma: feasibility and therapeutic potential of a novel approach combining 131‐I‐MIBG and multiple drug chemotherapy. Br J Cancer. 2001;84(4):460‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Gaze MN, Wheldon TE, O'Donoghue JA, et al. Multi‐modality megatherapy with [131I]meta‐iodobenzylguanidine, high dose melphalan and total body irradiation with bone marrow rescue: feasibility study of a new strategy for advanced neuroblastoma. Eur J Cancer (Oxford, England: 1990). 1995;31a(2):252‐256. [DOI] [PubMed] [Google Scholar]
- 299. Matthay KK, Tan JC, Villablanca JG, et al. Phase I dose escalation of iodine‐131‐metaiodobenzylguanidine with myeloablative chemotherapy and autologous stem‐cell transplantation in refractory neuroblastoma: a new approaches to Neuroblastoma Therapy Consortium Study. J Clin Oncol. 2006;24(3):500‐506. [DOI] [PubMed] [Google Scholar]
- 300. Kraal KC, Tytgat GA, van Eck‐Smit BL, Kam B, Caron HN, van Noesel M. Upfront treatment of high‐risk neuroblastoma with a combination of 131I‐MIBG and topotecan. Pediatr Blood Cancer. 2015;62(11):1886‐1891. [DOI] [PubMed] [Google Scholar]
- 301. Ozkaynak MF, Gilman AL, London WB, et al. A comprehensive safety trial of chimeric antibody 14.18 with GM‐CSF, IL‐2, and isotretinoin in high‐risk neuroblastoma patients following myeloablative therapy: Children's Oncology Group Study ANBL0931. Front Immunol. 2018;9:1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Sait S, Modak S. Anti‐GD2 immunotherapy for neuroblastoma. Expert Rev Anticancer Ther. 2017;17(10):889‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY. Humanizing murine IgG3 anti‐GD2 antibody m3F8 substantially improves antibody‐dependent cell‐mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology. 2012;1(4):477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Cheung IY, Kushner BH, Modak S, Basu EM, Roberts SS, Cheung NV. Phase I trial of anti‐GD2 monoclonal antibody hu3F8 plus GM‐CSF: impact of body weight, immunogenicity and anti‐GD2 response on pharmacokinetics and survival. Oncoimmunology. 2017;6(11):e1358331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti‐VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333(2):328‐335. [DOI] [PubMed] [Google Scholar]
- 306. Gains JE, Bomanji JB, Fersht NL, et al. 177Lu‐DOTATATE molecular radiotherapy for childhood neuroblastoma. J Nucl Med. 2011;52(7):1041‐1047. [DOI] [PubMed] [Google Scholar]
- 307. Strosberg J, El‐Haddad G, Wolin E, et al. Phase 3 trial of (177)Lu‐Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Tesson M, Vasan R, Hock A, et al. An evaluation in vitro of the efficacy of nutlin‐3 and topotecan in combination with (177)Lu‐DOTATATE for the treatment of neuroblastoma. Oncotarget. 2018;9(49):29082‐29096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Saulnier Sholler GL, Brard L, Straub JA, et al. Nifurtimox induces apoptosis of neuroblastoma cells in vitro and in vivo. J Pediatr Hematol Oncol. 2009;31(3):187‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Cabanillas Stanchi KM, Bruchelt G, Handgretinger R, Holzer U. Nifurtimox reduces N‐Myc expression and aerobic glycolysis in neuroblastoma. Cancer Biol Ther. 2015;16(9):1353‐1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Kong E, Zhu J, Wu W, et al. Nifurtimox inhibits the progression of neuroblastoma in vivo. J Cancer. 2019;10(10):2194‐2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Attiyeh EF, London WB, Mossé YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353(21):2243‐2253. [DOI] [PubMed] [Google Scholar]
- 313. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15(8):1781‐1791. [DOI] [PubMed] [Google Scholar]
- 314. Radhakrishnan SK, Bebb DG, Lees‐Miller SP. Targeting ataxia‐telangiectasia mutated deficient malignancies with poly ADP ribose polymerase inhibitors. Transl Cancer Res. 2013;2(3):155‐162. [Google Scholar]
- 315. Wu T, Wu X, Wang HY, Chen L. Immune contexture defined by single cell technology for prognosis prediction and immunotherapy guidance in cancer. Cancer Commun (London, England). 2019;39(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Yang R, Reilly CR, Rescorla FJ, et al. Effects of high‐intensity focused ultrasound in the treatment of experimental neuroblastoma. J Pediatr Surg. 1992;27(2):246‐250. [DOI] [PubMed] [Google Scholar]
- 317. Tung S, Fahy AS, Lamberti‐Pasculli M, Waspe AC, Pichardo S, Gerstle JT. Magnetic resonance‐guided high‐intensity focused ultrasound (MRgHIFU) virtual treatment planning for abdominal neuroblastoma utilizing retrospective diagnostic 3D CT images. J Pediatr Hematol Oncol. 2019;41(7):e443‐e449. [DOI] [PubMed] [Google Scholar]
- 318. Shim J, Staruch RM, Koral K, Xie XJ, Chopra R, Laetsch TW. Pediatric sarcomas are targetable by MR‐guided high intensity focused ultrasound (MR‐HIFU): anatomical distribution and radiological characteristics. Pediatr Blood Cancer. 2016;63(10):1753‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Eranki A, Srinivasan P, Ries M, et al. High‐intensity focused ultrasound (HIFU) triggers immune sensitization of refractory murine neuroblastoma to checkpoint inhibitor therapy. Clin Cancer Res. 2020;26(5):1152‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Wang S, Frenkel V, Zderic V. Optimization of pulsed focused ultrasound exposures for hyperthermia applications. J Acoust Soc Am. 2011;130(1):599‐609. [DOI] [PubMed] [Google Scholar]
- 321. Matsumoto H. [Revisiting sensitization mechanisms in cancer thermochemotherapy]. Fukuoka igaku zasshi = Hukuoka acta medica. 2009;100(4):95‐103. [PubMed] [Google Scholar]
- 322. Debes A, Willers R, Göbel U, Wessalowski R. Role of heat treatment in childhood cancers: distinct resistance profiles of solid tumor cell lines towards combined thermochemotherapy. Pediatr Blood Cancer. 2005;45(5):663‐669. [DOI] [PubMed] [Google Scholar]
- 323. Tandon P, Farahani K. NCI image‐guided drug delivery summit. Cancer Res. 2011;71(2):314‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Bellary A, Villarreal A, Eslami R, et al. Perfusion‐guided sonopermeation of neuroblastoma: a novel strategy for monitoring and predicting liposomal doxorubicin uptake in vivo. Theranostics. 2020;10(18):8143‐8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Nolan JC, Frawley T, Tighe J, Soh H, Curtin C, Piskareva O. Preclinical models for neuroblastoma: advances and challenges. Cancer Lett. 2020;474:53‐62. [DOI] [PubMed] [Google Scholar]
- 326. Houghton PJ, Morton CL, Tucker C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49(7):928‐940. [DOI] [PubMed] [Google Scholar]