

Abstract
Introduction:
Prenatal stress is known to influence fetal hypothalamic-pituitary-adrenal axis (HPA axis) development. Placental 11β-hydroxysteroid dehydrogenase type 2 (HSD11B2) is a central gene in this pathway, but little is known about what influences its functioning. We assess how maternal distress influences HSD11B2 functioning, and how HSD11B2 in turn, is associated with infant HPA axis development.
Methods:
Data come from 24 mother-infant dyads on the Galápagos Islands. Using adjusted linear regression models, we assess the effects of maternal psychosocial (stress and depressive symptoms, measured by the Perceived Stress Scale and the Patient Health Questionnaire-8, respectively) and physiological (HPA axis dysregulation) distress during pregnancy on HSD11B2 methylation and expression and then test how these HSD11B2 measures influence infant HPA axis development.
Results:
Maternal HPA axis dysregulation during pregnancy is associated with lower placental HSD11B2 expression, which is associated with an exaggerated cortisol reactivity in infants. Sex-specific analyses revealed that maternal depressive symptoms may influence the functioning of placental HSD11B2 differently in girls (n = 11, 46%) than in boys (n = 13, 54%), though the sample size was small.
Discussion:
These results support a disrupted adaptive framework, in which the ability to upregulate HSD11B2 expression in response to acute stress diminishes as maternal stress becomes chronic. In this model, chronic stress may exhaust the protective mechanism of HSD11B2, leaving the infant vulnerable to high levels of maternal cortisol, which could injure the fetal HPA axis and disrupt long-term neurobehavioral and metabolic development. While larger studies will be needed to confirm these findings, this study offers exploratory results on the effects of maternal distress on both HSD11B2 methylation and expression and the effect of HSD11B2 on offspring HPA axis development.
Keywords: expression, methylation, 11β-hydroxysteroid dehydrogenase type 2, HSD11B2, stress, HPA axis
Introduction
The developmental origins hypothesis suggests that early life environments can shape long-term disease risk [1]. A growing body of literature supportive of this hypothesis has found that various measures of maternal distress during pregnancy, including stress, anxiety, depression, and other factors, have been associated with long-term effects on metabolic functioning and neurobehavioral disorders in offspring [2]. Specifically, prenatal stressors have been associated with increased risk for schizophrenia [3], autism, ADHD [4], and impaired cognitive development [5]. Despite strong epidemiological evidence for these associations, the underlying biological mechanisms remain unclear.
The activation of the hypothalamic-pituitary-adrenal axis (HPA axis), which regulates glucocorticoid feedback interactions among the mother, placenta, and fetus during pregnancy, has been proposed to be the primary mechanism through which prenatal maternal stress shapes fetal development and subsequent long-term disease risk [6,7]. Though maternal sensitivity to cortisol decreases as cortisol levels rise at approximately the 25th week of pregnancy, women maintain a diurnal rhythm of cortisol throughout pregnancy with a peak at approximately 30 minutes after waking, allowing circadian regulation in mothers to be assessed throughout pregnancy [8,9]. While glucocorticoids play an essential role in fetal development [10], maternal dysregulated cortisol may alter fetal behavioral, immunological, and neurological development, including areas of the brain that regulate the fetal HPA axis [11]. Theorists propose that frequent or prolonged maternal stressors activate and dysregulate the maternal HPA axis, increasing the production of cortisol, which could be transferred to the infant through the placenta and/or trigger an increased production of placental corticotropin-releasing hormone, thereby stimulating the fetal HPA axis to produce more fetal cortisol [11]. Increased fetal cortisol, in turn, has permanent effects on fetal HPA axis development, which could underlie subsequent long-term disease risk [6,7,12]. However, results from studies on prenatal maternal stress and offspring development have demonstrated mixed results, suggesting that other mechanisms may play a role in the relationship between prenatal stress and infant HPA axis development.
One such mechanism may be that of the placental enzyme, 11β-hydroxysteroid dehydrogenase type 2 (HSD11B2), which buffers the level of cortisol that reaches the fetus by catalyzing a reaction that converts active cortisol to inert cortisone in the placenta [13]. In this way, HSD11B2 is hypothesized to be protective for the fetus by minimizing glucocorticoid exposure [14]. Recently, researchers have begun to utilize both animal and human models to investigate the relationship between maternal stress and various measures of HSD11B2, hypothesizing that maternal stress itself may up-regulate the enzyme as an adaptive, protective measure for the fetus or down-regulate the enzyme due to energetic or exhaustive limitations. Nonetheless, studies have reported mixed results, and often sex-specific results, but little remains known about this enzyme and what influences its functioning and expression.
In the present study, we assess how prenatal distress influences the methylation and expression of placental HSD11B2 and how these differences shape infant HPA axis development. Our conceptual model is shown in Figure 1. Specifically, we investigate: Path A) how prenatal maternal psychosocial distress, measured through stress and depressive symptoms, influences placental HSD11B2; Path B) how prenatal maternal physiological distress, measured through maternal cortisol regulation, influences HSD11B2; and Path C) how differences in HSD11B2 are associated with infant cortisol regulation. Analyses were run independently for placental DNA methylation of the HSD11B2 gene and mRNA expression of HSD11B2 on each path.
Figure 1.
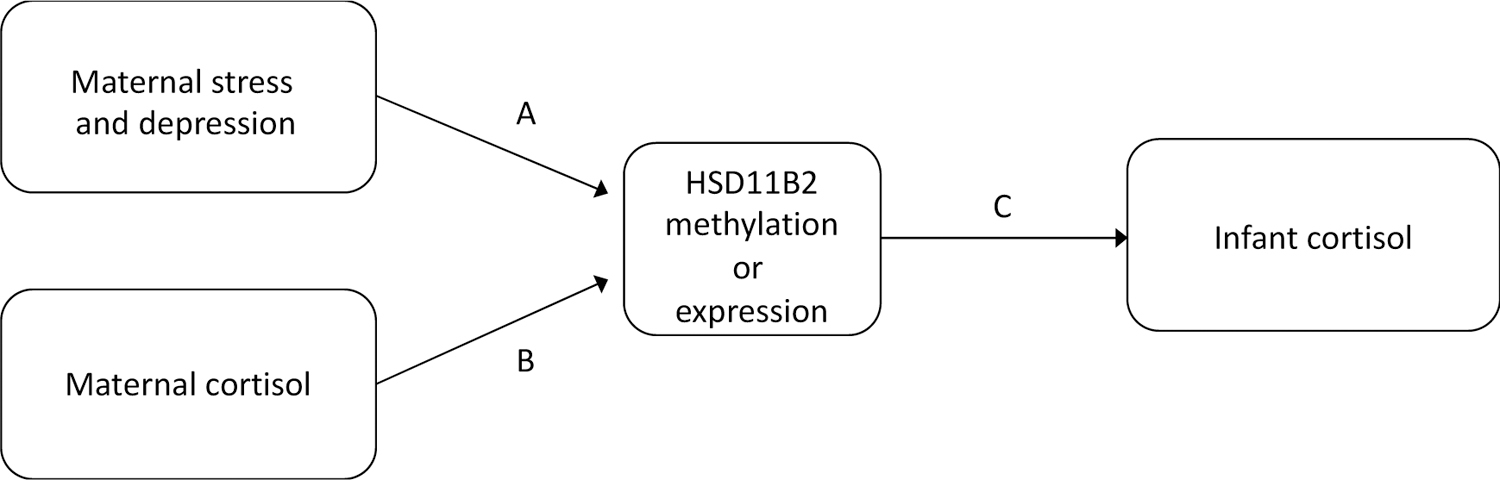
Conceptual model
Methods
Participants
The data were collected over 12 months in 2018 on the Galápagos Islands, where limited infrastructure and geographic isolation contribute to daily distress for residents. Participants were recruited from Hospital Oskar Jandl (HOJ), a public hospital on the Galápagos’ provincial capital island San Cristóbal, using purposive sampling. HOJ is free for all residents and is the only hospital on the island, allowing the research team to screen all pregnant women on the island within the recruitment period. Inclusion criteria included women between the ages of 18 and 50 years who planned to give birth on the island. Of those contacted who were eligible, only four decided to not take part in the study. Of those contacted who were ineligible, 12 planned to give birth on the mainland, four had already moved off the island, and three had serious pregnancy complications, and thus were excluded. Based on annual birth rates, we estimate that we enrolled more than 50% of the births on the island in 2018. Here we present a complete case analysis for those with placental data, which limits our original sample size of 38 participants to 24 participants.
Visits with participants were conducted once during pregnancy at 34 – 36 weeks gestation and once in the postpartum, when the infant was two months old. For each mother-infant dyad, the research team collected surveys on stress and depressive symptoms, placental samples, and maternal and infant saliva samples. The visits during and after pregnancy took place in the participants’ homes, and all placental collection took place in the hospital at the time of delivery. All participants provided written informed consent prior to participation under appropriate protocols approved by the Institutional Review Boards for the University of North Carolina at Chapel Hill (UNC-Chapel Hill) and Universidad San Francisco de Quito (2017–163E). This project was also approved by Ecuador’s Ministry of Public Health.
Maternal stress and depressive symptoms
Stress.
The Perceived Stress Scale (PSS) was used to assess maternal chronic stress by measuring the degree to which situations are perceived as unpredictable, uncontrollable, and burdensome [15]. The PSS is particularly relevant for pregnant women, since it does not operationalize symptoms that occur frequently in both pregnancy and during times of stress (such as sleep disturbances) to assess chronic stress [16]. We used a Spanish version of this scale that has been tested for reliability, validity, and sensitivity in Spanish-speaking contexts [17]. The PSS is scored from 0 to 40, with scores of 0 – 13 indicating low stress, scores of 14 – 26 indicating moderate stress, and scores 27 – 40 indicating high stress.
Depressive symptoms.
Depressive symptoms were measured by the Patient Health Questionnaire-8 (PHQ-8) [18]. We used a Spanish version of the PHQ-8 that has been validated [19] and tested for reliability [20] in the Spanish language. The PHQ-8 is scored from 0 to 24, with a higher score indicating more depressive symptoms. When analyzed categorically, “high depressive symptoms” was defined using the CDC’s diagnostic cut-point for depression on the PHQ-8, as scores greater than or equal to 10 [21].
Maternal salivary cortisol
Salimetrics guides were used for maternal saliva collection and storage protocols [22]. Maternal saliva samples were collected at 34 – 36 weeks of pregnancy. Over the course of one day, women provided three samples: one immediately upon awakening, one 30 minutes after awakening, and one prior to sleep. Participants were instructed not to eat, drink, or brush their teeth in the 30 minutes prior to collecting samples and to record times of saliva collection. Participants stored samples in their own freezers until a study team member retrieved them the next day and transported them to the GSC where they were frozen at −20° C until analysis.
Cortisol dysregulation in mothers was measured in four ways: elevated morning cortisol, a blunted cortisol awakening response (CAR) at 30 minutes after waking, elevated evening cortisol, and a poor daily cortisol decline. A blunted CAR was defined as a small difference in cortisol levels between waking and 30 minutes post-waking, and poor daily cortisol decline was defined as a large difference between evening cortisol and waking cortisol levels.
Infant salivary cortisol
Infant cortisol was measured when the infant was two months old. Because the HPA axis is not fully developed at two months of age [23], many studies of HPA axis function in infancy have measured cortisol dysregulation through basal cortisol or stress reactivity, that is, in response to a stressor [23,24]. At infant saliva collection, a saliva sample was collected before and 20 – 25 minutes after a stressor per a previously published infant stress reactivity protocol [25]. In our analyses, baseline cortisol is the first of these measures, and cortisol reactivity is the difference between these two measures. In order to apply a stressor to the infant to induce stress reactivity, the research team placed the diapered but otherwise unclothed infant on a metal tray for 60 seconds to mimic an infant’s typical discontented response to being weighed in the hospital. The method for this stressor was developed by the research team and the nurses at HOJ in order to be effective, non-invasive, and culturally appropriate. All infant saliva samples were collected using Salimetrics Infant Swabs and placed into Salimetrics Swab Storage tubes and frozen at −20° C until analysis.
Cortisol dysregulation in infants was measured as elevated baseline cortisol and as a blunted or exaggerated cortisol reactivity at two months old. High baseline cortisol [26] and blunted [25] and exaggerated [27] cortisol reactivity have all been cited as evidence of infant cortisol dysregulation.
Demographic and obstetric characteristics
Maternal demographics and health history were collected at the initial visit, and obstetric and infant characteristics including gestational age, infant sex, infant birth weight, mode of delivery, APGAR score, and placental weight were recorded by hospital staff at HOJ during the birth.
Placental collection
Healthy, intact placentas were collected immediately and dissected within one hour of birth. Umbilical cords were removed, and placentas were weighed and measured. Maternal decidua was removed, and tissue samples were taken from four sampling sites on the fetal side of the placenta. The four sampling sites were selected in each of the four quadrants of the placenta that were at least 2 cm from the umbilical cord insertion site and at least 3 cm from the placental edge, following protocol by Burton and colleagues [28]. Samples were cut from each sampling site and pooled into one storage tube to control for intra-placental variation. The samples were stored in RNAlater (Life Technologies, Grand Island, NY) and stored at −20°C at the hospital. Samples were then sent on ice to UNC-Chapel Hill and Duke University for analysis.
DNA methylation analysis
The analysis of HSD11B2 methylation was conducted at the Murphy Lab at Duke University. Placental genomic DNA was extracted using the Lysing Matrix A from MP Biomedical with the FastPre24 for homogenization, followed by the Solid Tissues Protocol from Purgene. DNA samples were sodium bisulfite modified using the EZ DNA Methylation™ Kit (Zymo Research, Irvine, CA), and pyrosequencing was performed on PCR product amplified from bisulfite-modified DNA based on the region sequenced and displaying differential methylation in human placenta from Alikhani-Koopaei and colleagues [29]. The extent of methylation at the HSD11B2 promoter region was examined with pyrosequencing using the Pyromark Pyrosequencing System (Qiagen Inc.) using the following forward and biotinylated reverse primers for amplification, Sequence (5’-3’) (IDT Inc., Coralville, IA): HSD11B2-F2-AAGTTTTGGAAGGAAAGGGAAGA, HSD11B2-R2-[btn] ACAAAACCTACCTAAAACAAAAACTA, HSD11B2-S-GGGGTAGAGATTTTAA GAA.
The region analyzed contains four CpGs sites of interest [29] with reactions performed in duplicate. Sodium bisulfite–modified, fully methylated referent positive control and fully unmethylated (whole genome amplified) negative control DNA (Qiagen) were examined with each batch. The percent methylation at each CpG site was quantified using the PyroMark CpG software, version 1.0.11. (Qiagen). Methylation across each of the four HSD11B2 CpG sites was averaged to obtain an overall measure of methylation.
mRNA gene expression analysis
The analysis of HSD11B2 expression was conducted at the Microbiome Core Facility at UNC-Chapel Hill. Total RNA was isolated using Qiagen RNeasy Extraction Kit, with the addition of DNaseA digest per the manufacturer’s guidelines. Total RNA was quantified and normalized to 50ng/ul prior to the synthesis of cDNA. 500 ng total RNA was subject to cDNA synthesis via qScript cDNA synthesis kit. Expression of HSD11B2 was analyzed using the following primers [30]: HSD11B2 forward: CTACTCATGGACACATTCAGCT, reverse: TCACTGACTCTGTCTTGAAGC.
Quantitative PCR was performed on QuantStudio Q6, using BioRad PowerSyber qPCR kit. The thermal cycling conditions were as follows: one cycle at 50°C for 20 sec, 95°C for 10 minutes, followed by 40 cycles of 15 seconds at 95°C, 1 minute at 60°C. Melting curve analysis was carried out using the continuous method from the Q6 Software (Applied Biosystems) conducted at 60°C, with increments of 1°C for 15 seconds. Data analysis was carried out with Q6 Software (Applied Biosystems). The auto threshold and baseline options were used for the calculations of cycle threshold (CT) values per well. Gene expression within placental tissue was calculated using ΔCT to correct for internal variation between the placentae by adjusting for a well-regulated and stable housekeeping gene, RPL19. In order to ease interpretation of models, ΔCT scores have been inverted [x(-1)], so that a higher ΔCT value indicates higher HSD11B2 gene expression.
Statistical analysis
Stress and depressive symptoms scores were normally distributed. Neither placental HSD11B2 methylation nor expression were normally distributed, so non-parametric tests, including Spearman’s correlation and Mann-Whitney U tests, were used for preliminary analyses of these data. Statistical analyses were conducted using Stata version Stata/MP 16.0 (StataCorp, College Station, TX). In linear regression models, stress and depressive symptoms scores, salivary cortisol measures, HSD11B2 methylation and expression were analyzed continuously.
First, we assessed the relationship between placental HSD11B2 methylation and expression using linear regression with robust standard errors to account for concerns about normality in expression. We then tested associations on each pathway using adjusted linear regression models with robust standard errors. Each path was first assessed for HSD11B2 methylation. A second round of analyses was then done for HSD11B2 expression. Analyses were adjusted for each pathway individually. Covariates were selected as they were significantly associated with either the predictor or the outcome variables at p <0.10 on each pathway. We assessed gestational age, maternal age, birth weight, placental weight, infant sex as potential covariates for all paths. Ultimately, in Paths A and B, models for DNA methylation were adjusted for infant sex and models for mRNA expression were unadjusted. On Path C, models for both DNA methylation and mRNA expression were adjusted for gestational age and infant sex. Sex-specific associations were assessed via stratification of adjusted linear regression models as well as an assessment of interaction effects.
Results
Sample characteristics
Maternal, infant, and placental characteristics are detailed in Table 1. Participants ranged in age from 18 to 39 years, and the vast majority (92%) identified as ethnically Mestizo. The majority of women were married (87.5%), and 20.8% had completed college. On psychosocial distress measures, 29.2% of the women in the study scored as depressed on the PHQ-8, and 58.3% scored as moderately or highly stressed on the PSS. The average gestational age was 38.4 weeks (95% CI: 37.85, 38.85), and 58.3% of infants were born by Caesarean. None of the Caesareans were elective, and none of the participants in this sample labored before Caesarean. Just over half of the infants (54%) were male, and the average birth weight was 3380 grams.
Table 1.
Characteristics of the study sample
| Maternal Characteristics | Mean (SD)/range or no. (%) |
|---|---|
| Age (years) | 29.2 (5.9) / 18 – 39 |
| Ethnicity | |
| Mestizo | 22 (91.7%) |
| Indigenous | 1 (4.2%) |
| Afro-Ecuadorian | 1 (4.2%) |
| Parity1 | 0.92 (0.78)/ 0 – 2 |
| Married | 21 (87.5%) |
| Education | |
| Less than high school | 3 (12.5%) |
| Completed high school | 16 (66.7%) |
| Completed college | 5 (20.8%) |
| Born and raised on Galápagos | 9 (37.5%) |
| Food security | |
| Secure | 13 (54.2%) |
| Mild food insecurity | 8 (33.3%) |
| Moderate – High food insecurity | 3 (12.5% |
| Depressed (PHQ-8) | 7 (29.2%) |
| Moderate – High stress (PSS) | 15 (62.5%) |
| Obstetric and Infant Characteristics | |
| Gestational age at delivery (weeks) | 38.4 (1.2) / 36 – 41 |
| Caesarean delivery | 14 (58.3%) |
| APGAR 5 min | 9 (0) / 9 – 9 |
| Male offspring | 13 (54.2%) |
| Infant birth weight (g) | 3380 (383.8) / 2465 – 3900 |
| Placental Characteristics | |
| Placental weight (g) | 530.8 (101.5) / 398 – 896 |
| % methylation, average all HSD11B2 CpG sites | 11.7 (4.9) |
| CpG1, % methylated | 7.9 (3.6) |
| CpG2, % methylated | 17.6 (7.0) |
| CpG3, % methylated | 7.8 (3.7) |
| CpG4, % methylated | 13.4 (5.4) |
Does not include current pregnancy
Placental HSD11B2 characteristics
Quantitative bisulfite sequencing was used to determine methylation of a CpG island region in the promoter of the HSD11B2 gene. The average methylation across the four CpG loci was 11.7% (95% CI: 9.61, 13.72). In order to assess the functional signficance of variation of methylation, we also quantified HSD11B2 gene expression using PCR. High DNA % methylation was significantly associated with lower mRNA gene expression (β = −0.16, p ≤ 0.01) (Figure 2).
Figure 2.
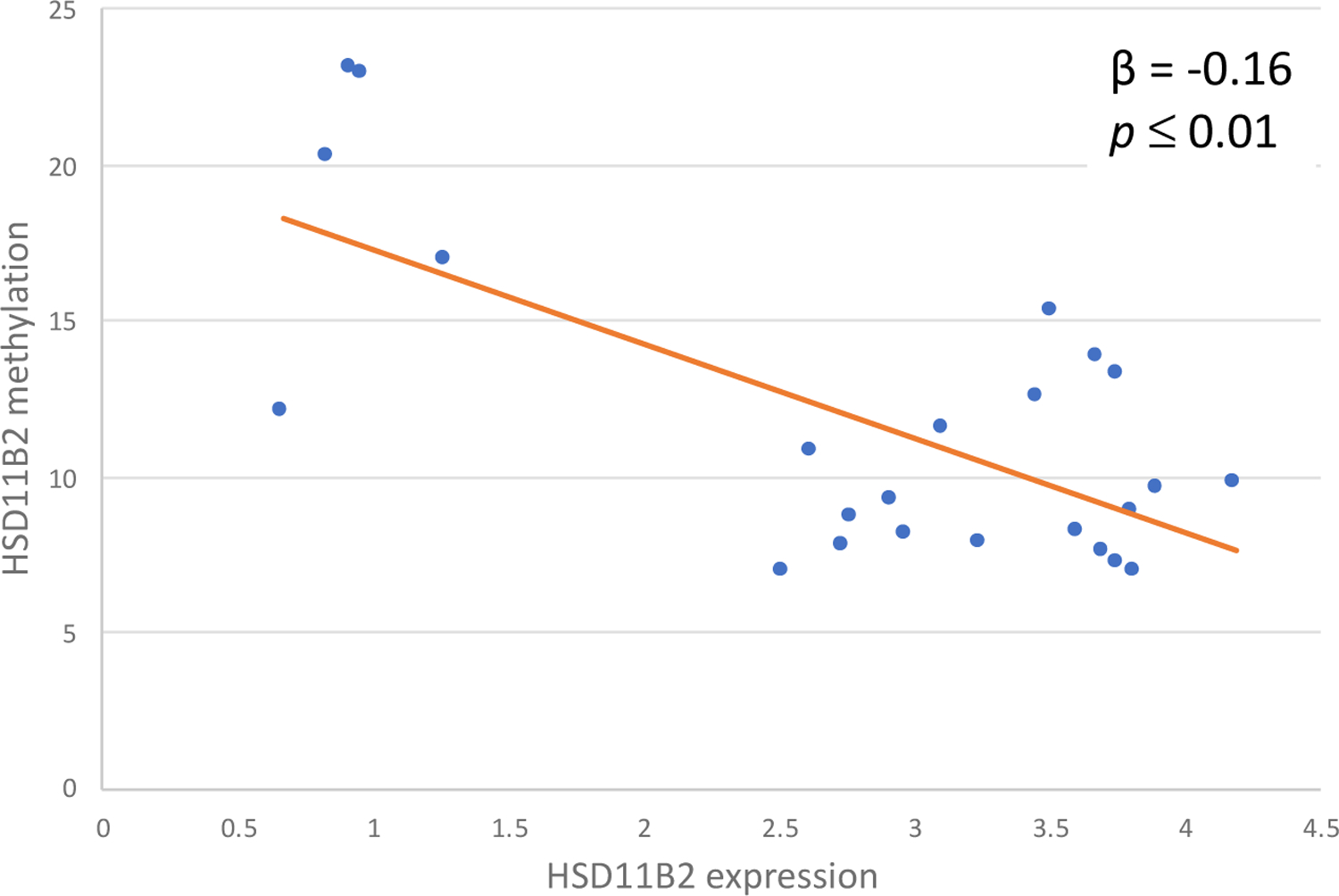
Placental HSD11B2 methylation predicts HSD11B2 expression
Maternal distress and HSD11B2 outcomes
The results of all adjusted linear regression models for our conceptual model are shown in Table 2. In our analyses, neither maternal stress nor depressive symptoms were significantly associated with differences in HSD11B2 methylation or gene expression.
Table 2.
Adjusted regression analyses for paths in conceptual model
| Exposure | Outcome | DNA Methylation | mRNA Expression | |||||
|---|---|---|---|---|---|---|---|---|
| β | 95% CI | p-value | β | 95% CI | p-value | |||
| Path A | Stress | HSD11B2 | −0.21 | −0.64, 0.22 | 0.31 | 0.01 | −0.08, 0.11 | 0.77 |
| Depressive symptoms, continuous | HSD11B2 | 0.15 | −0.23, 0.53 | 0.41 | −0.07 | −0.17, 0.02 | 0.13 | |
| High depressive symptoms, categorical | HSD11B2 | 2.40 | −2.53, 7.33 | 0.32 | −0.92 | −1.99, 0.16 | 0.09 | |
| Path B | Maternal morning cortisol | HSD11B2 | 1.38 | −1.75, 4.51 | 0.37 | −0.98 | −1.73, 0.23 | 0.01* |
| Maternal CAR | HSD11B2 | 0.15 | −4.02, 4.31 | 0.94 | 0.82 | 0.01, 1.63 | 0.05* | |
| Maternal evening cortisol | HSD11B2 | −0.67 | −5.36, 4.03 | 0.77 | −0.13 | −1.13, 0.86 | 0.78 | |
| Maternal daily cortisol decline | HSD11B2 | −1.07 | −3.11, 0.97 | 0.29 | 0.44 | −0.22, 1.10 | 0.18 | |
| Path C | HSD11B2 | Infant baseline cortisol | 0.00 | −0.09, 0.09 | 0.97 | 0.18 | −0.10, 0.47 | 0.18 |
| HSD11B2 | Infant cortisol reactivity | 0.03 | −0.07, 0.12 | 0.54 | −0.32 | −0.63, −0.01 | 0.04* | |
For DNA methylation, Path A and Path B were adjusted for infant sex. Path C was adjusted for gestational age and infant sex.
For mRNA expression, Path A and Path B were unadjusted. Path C was adjusted for gestational age and infant sex.
Indicates a p-value of ≤0.05
While maternal cortisol dysregulation during pregnancy was not associated with differences in HSD11B2 methylation, two measures of maternal cortisol dysregulation were associated with HSD11B2 expression. High maternal morning cortisol (β = −0.98, p = 0.01, 95% CI: −1.73, 0.23) and a blunted maternal CAR (β = 0.82, p = 0.05, 95% CI: 0.01, 1.63) were associated with lower mRNA expression of HSD11B2. These associations are shown in Figure 3. Neither evening cortisol nor daily cortisol decline were associated with differences in HSD11B2 methylation or expression.
Figure 3.
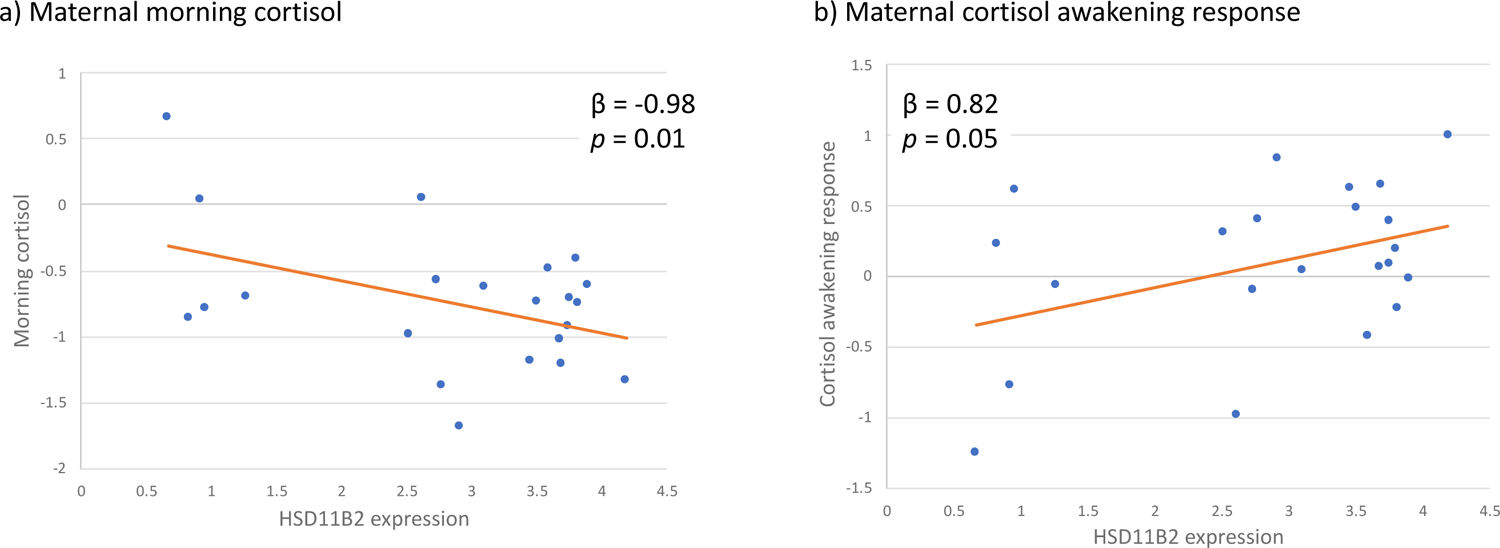
Maternal cortisol regulation is associated with differences in placental HSD11B2 expression
HSD11B2 exposures and infant cortisol
DNA methylation was not associated with either measure of infant cortisol regulation, but lower mRNA expression of HSD11B2 was associated with a higher cortisol reactivity (β = −0.32 p = 0.04, 95% CI: −0.63, − 0.01) (Figure 4), but not with higher baseline cortisol when infants were two months old.
Figure 4.
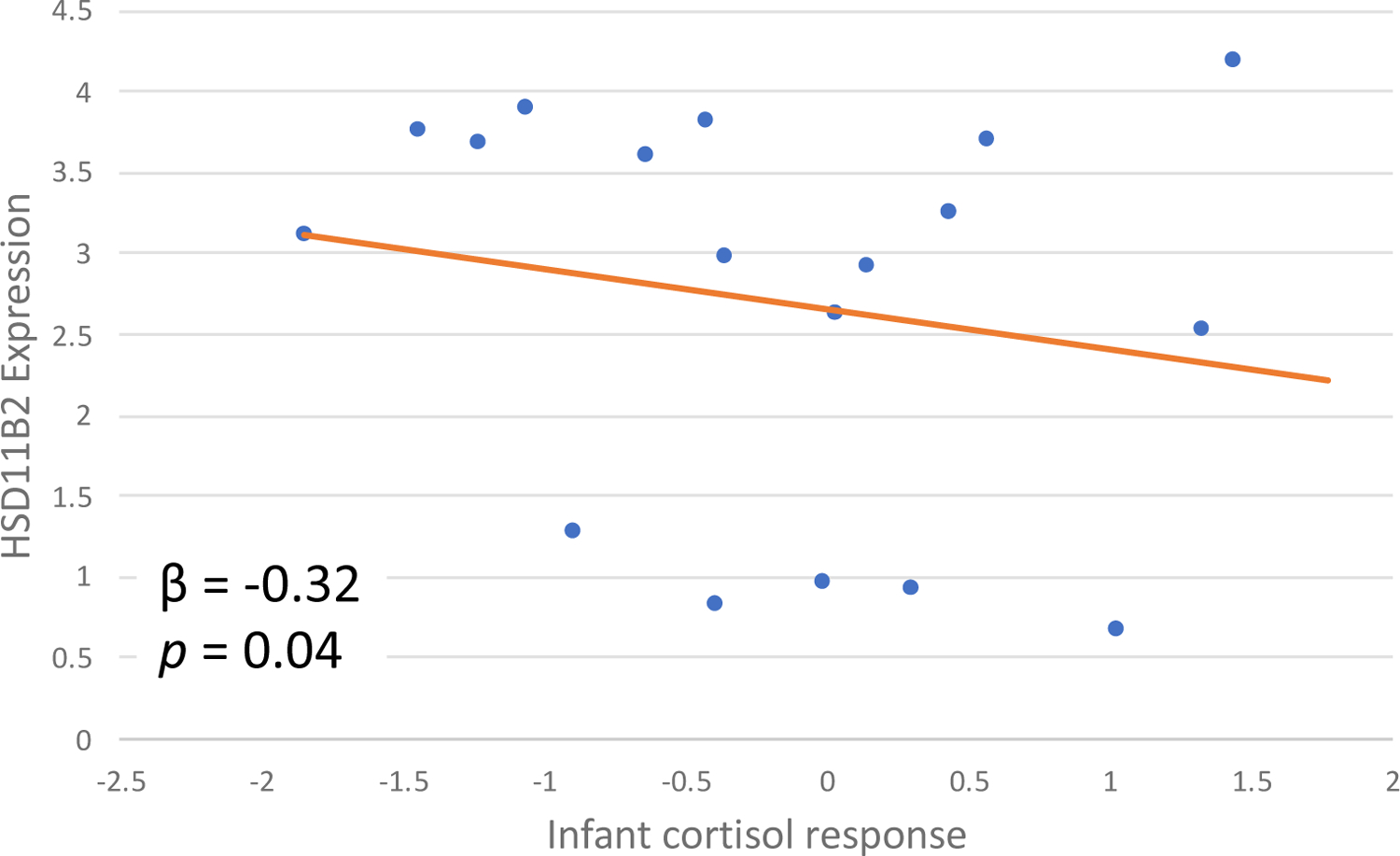
Placental HSD11B2 expression is associated with infant cortisol regulation
Sex-specific associations
Infant sex was associated with differences in placental HSD11B2 methylation (p = 0.04), but not expression. In girls, the average percent methylation of the HSD11B2 gene in the placenta was 14.2% (95% CI: 10.30, 18.13) and in boys it was 9.5% (95% CI: 8.0, 11.0). Due to these results, and many others’ findings of sexually dimorphic responses to glucocorticoids in the fetus and the placenta [31–33], we stratified our models by infant sex and tested interaction effects to examine these pathways. While high maternal depressive symptoms were not associated with differences in methylation or expression of HSD11B2 with the whole sample, we did observe associations in sex-specific models. When the sample was limited to just girls, high maternal depressive symptoms were marginally associated with higher methylation of the HSD11B2 gene (β = 6.74, p = 0.06, 95% CI: −0.32, 13.80) and significantly associated with lower HSD11B2 expression (β = −1.74, p = 0.01, 95% CI: −3.03, −0.45). These differences were not observed in the boys’ sample, where depressive symptoms were neither associated with methylation (β = −2.4, p = 0.15, 95% CI: −5.80, 1.03) nor expression (β = 0.19, p = 0.76, 95% CI: −1.17, 1.56). To further test these results, we assessed interaction effects and found that there was evidence of a significant interaction between high maternal depressive symptoms and infant sex in predicting HSD11B2 methylation (β = −9.13, p = 0.02, 95% CI: −16.28, −1.98) and expression (β = 1.94, p = 0.03, 95% CI: 0.17, 3.70) (Figure 5). No other paths demonstrated significant differences by infant sex in stratified models.
Figure 5.
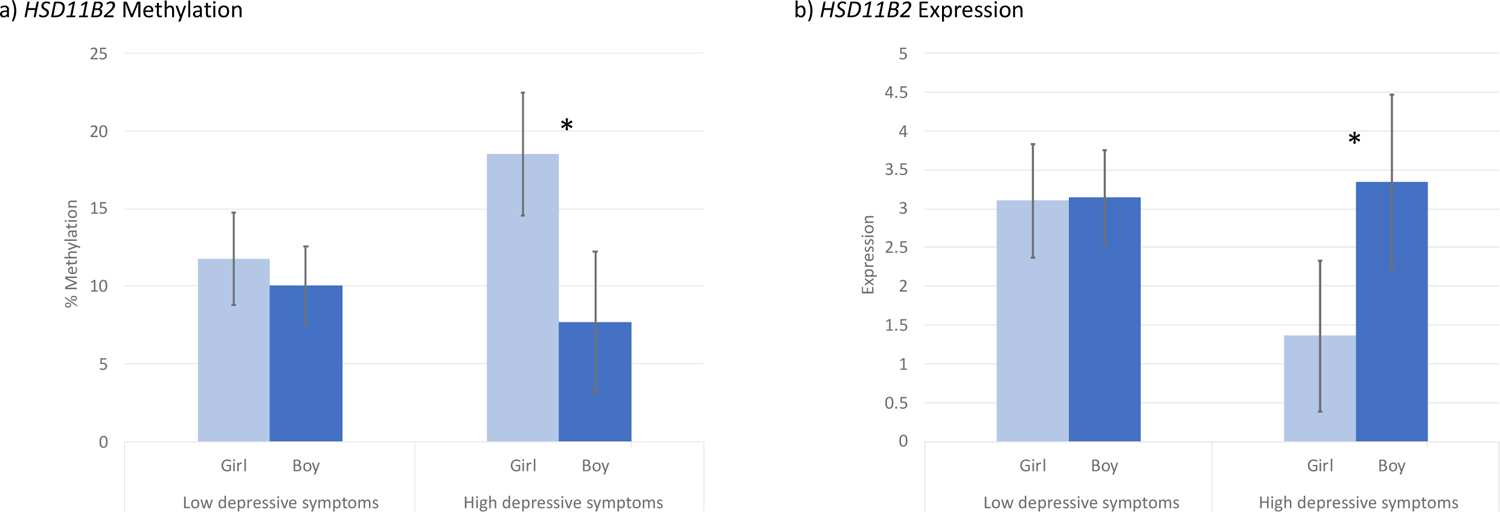
Interaction between maternal depressive symptoms and infant sex in predicting HSD11B2 methylation and expression
* Indicates a significant interaction at p<0.05
Discussion
This study aimed to assess how maternal psychosocial and physiological distress (measured through cortisol) influence epigenetic regulation of the HSD11B2 gene in the placenta, and how these differences then can shape HPA axis regulation in infants, which has long-term consequences for health [6,7,12]. This study incorporated the investigation of both HSD11B2 gene expression and methylation.
Maternal distress and HSD11B2 measures
Our finding that higher placental HSD11B2 methylation was associated with lower HSD11B2 expression is consistent with the results from other studies on this relationship [29,34]. In the full sample, we did not observe differences in placental HSD11B2 methylation or HSD11B2 expression based on psychosocial indicators of distress (stress and depressive symptoms). Nonetheless, we did observe associations between maternal physiological distress and HSD11B2 expression. Two measurements of HPA axis dysregulation, high morning cortisol and a blunted cortisol awakening response, were each associated with lower HSD11B2 expression. Further, in sex-specific analyses, prenatal maternal depressive symptoms were marginally associated with higher HSD11B2 methylation and significantly lower HSD11B2 expression for girls, but not for boys.
While these results are seemingly contradictory to an adaptive framework, where maternal distress would decrease HSD11B2 methylation thus increasing HSD11B2 expression to provide a protective effect to the infant in times adversity, our results are consistent with findings from many other studies and may fit into a broader physiological model where adaptive responses become disrupted through overuse. Animal models have found that chronic stress and anxiety may diminish the protection of placental HSD11B2. One study found that prenatal stress increased the activity of HSD11B2 in low-anxiety rats, but not in high-anxiety rats [35], while another study found that while acute maternal stress up-regulated HSD11B2 activity, chronic stress diminished the capacity of placental HSD11B2 to up-regulate in the face of an acute stressor [36]. Other studies on chronic prenatal stress in rats have found that chronic restraint is associated with lower placental HSD11B2 expression [37,38] as well as increased methylation of specific CpG sites of the HSD11B2 gene promoter [37].
Studies on this pathway in humans report mixed findings. Like in animal studies, many studies in humans have found that chronic maternal distress (including anxiety and/or depression) is associated with greater methylation of the HSD11B2 promoter region [39,40] as well as lower expression of HSD11B2 [41–43]. Other distress exposures have also been associated with these differences. In one study, prenatal life events were associated with a downregulation in HSD11B2 expression, though only in Caucasian women [30]. In a few studies, though, the opposite effect is observed. One study found that women of low socioeconomic status (SES) had lower HSD11B2 methylation [44], which would promote HSD11B2 expression. Nonetheless, some studies report no differences in placental HSD11B2 measures based on anxiety [30], depression [30,40,45,46], or prenatal natural disasters [47]. While results are mixed, findings from animal and human models most often suggest that in healthy, low-stress individuals, the placenta increases HSD11B2 expression in the face of distress, protecting the fetus from excess cortisol, but that in chronically-stressed individuals, this protection may diminish, exposing the infant to high levels of cortisol that have long-term consequences for neurological development [36].
HSD11B2 measures and infant cortisol
Our analyses of the effects of HSD11B2 measures on infant cortisol found that lower HSD11B2 expression was associated with an higher cortisol reactivity in infants, which has been cited as evidence of infant HPA axis dysregulation [27]. Nonetheless, we did not observe differences in baseline cortisol when infants were two months old. While this result may indicate that HSD11B2 expression does not have an effect on infant baseline cortisol, it is also possible that postpartum mood or other exposures have attenuated prenatal insults to HPA axis development [48]. However, another study found that HSD11B2 methylation moderated the relationship between prenatal depression and infant baseline cortisol at one month of age, such that a 1% decrease in methylation was associated with a 9% increase in baseline cortisol in infants whose mothers were depressed during pregnancy [26].
Few studies in humans have assessed the relationship between placental HSD11B2 measures and infant cortisol, but many have assessed the relationship between HSD11B2 measures and neurobehavioral outcomes in infants. One study, which used factor analysis, found that high HSD11B2 methylation reduced the risk that infants were into a reactive, poorly regulated neurobehavioral profile [49], while another study found that higher depression scores during pregnancy were significantly associated with higher levels of negative affectivity among infants with low placental HSD11B2 expression, but not among infants with high HSD11B2 expression [45]. Our results fit into these frameworks, where higher levels of placental HSD11B2 methylation and lower levels of HSD11B2 expression may contribute to neurological changes in infants.
Sex-specific placental HSD11B2 measures
Our sex-specific findings revealed that on average, the placentas of girls had significantly more HSD11B2 methylation than those of boys. Further, maternal depressive symptoms were marginally associated with higher HSD11B2 methylation and significantly associated with lower HSD11B2 expression in the placentas of girls, but not of boys. These results are consistent with others’ findings that infant sex is often associated with differences in placental function and physiology [28]. Further, our finding that girls have lower HSD11B2 expression than boys aligns with the general finding that girls are typically more susceptible to insults to the HPA axis in early life [50], since higher HSD11B2 expression could provide a protective effect against excess maternal cortisol. Though not all studies found sex differences in placental measures [51], a study among small for gestational age infants showed that placentas of girls exhibit lower HSD11B2 activity than those of boys [52], while another found that among placentas of women with untreated asthma, those of girls exhibit lower HSD11B2 activity than those of boys [53]. Although the mechanisms behind sex differences in fetal programming remain unknown, others have suggested that these differences in early life may be the consequence of a “viability-vulnerability tradeoff” [54], in which males do not adjust to early life adversity, and thus only the most fit survive, while females modify their growth in response to adversity, improving their viability but increasing their vulnerability to the deleterious effects of these adjustments later in life [54].
Strengths and Limitations
This study has some limitations that should be noted. First, our small sample size decreases our statistical power, so our results should be considered to be exploratory. Second, maternal stress and depressive symptoms were measured through self-assessment on validated surveys, but scores were not confirmed by clinical assessment or diagnosis, as these services were unavailable on the island. Further, the results of Path C should be interpreted with caution, as there are a variety of postnatal exposures, including maternal mood, that could influence the infant’s developing HPA axis. We also did not collect data on infant sleep or last feed before collecting salivary cortisol, so these measures were not incorporated into models. Finally, while we assess the epigenetic profile and expression of HSD11B2, which plays a central role in the conversion of cortisol to cortisone, there are other pathways that might be contributing to these effects. Here, we do not test methylation or expression of NRC31, a glucocorticoid receptor in the placenta that may be an upstream regulator placental HSD11B2 [30]. Further, we did not assess the placental expression of monoamine oxidase A (MAOA), which has also been linked to maternal prenatal stress and infant temperament [55]. Future research should work to integrate these potential biological mechanisms.
Despite these limitations, our study shows clear and significant relationships between maternal distress, HSD11B2 regulation, and infant HPA axis development, contributing to evolutionary theories of early life adaptation and serving as foundational exploratory research on the understudied placental influence on fetal programming. To our knowledge, this is one of the first studies to examine how both HSD11B2 methylation and expression in the placenta respond to maternal distress and shape infant cortisol. Further, after Stroud and colleagues [26], we believe this is only the second study to examine how differences in HSD11B2 measures shape infant HPA axis regulation in humans. Last, while the majority of studies on HSD11B2 methylation and expression have focused on Caucasian populations, our study examines these pathways with a majority Mestizo population from Ecuador. This distinction is especially important since others have found differences in HSD11B2 measures based on ethnicity [30].
Our findings indicate that maternal physiological distress during pregnancy, measured through HPA axis dysregulation, is associated with lower placental HSD11B2 expression, which in turn, is associated with an exaggerated cortisol reactivity in infants. Sex-specific analyses revealed that maternal psychosocial distress during pregnancy, measured through depressive symptoms, is marginally associated with more placental HSD11B2 methylation and significantly associated with less HSD11B2 expression for the mothers of girls, but not boys. Together with others’ findings, our results support a disrupted adaptive framework, in which the ability to upregulate HSD11B2 expression in response to acute stress diminishes as maternal stress becomes chronic. In these cases, chronic stress, and potentially the overuse of this biological mechanism, can cause the hypermethylation of HSD11B2 and thus transcriptional repression of the gene, which downregulates HSD11B2 expression, leaving the infant vulnerable to high maternal cortisol. In turn, overexposure to maternal cortisol injures the fetal brain and HPA axis [56], which could permanently alter the infant’s neurobehavioral and metabolic pathways [2]. As results from some similar studies have been inconsistent, further research on these important pathways are necessary to better understand the protective role of HSD11B2 in infant development.
Placenta Highlights.
Maternal HPA axis dysregulation is associated with lower HSD11B2 expression
Low HSD11B2 expression is associated with higher cortisol reactivity in infants
Maternal depression results suggest sex-specific HSD11B2 functioning
Acknowledgements
Thank you to the leadership and staff at Hospital Oskar Jandl for their collaboration on this project. Thank you to our participants and to the Galapagos Science Center for support.
Funding:
This work was supported by the National Science Foundation [BCS-1730297], Fulbright-Hays [P022A170061], the Center for Galápagos Studies at the University of North Carolina at Chapel Hill, the University of North Carolina at Chapel Hill and the National Institutes of Health Eunice Kennedy Shriver National Institute of Child Health and Human Development Training Grant [T32-HD007168] through the Carolina Population Center. The funders had no role in study design, data collection, data analysis, data interpretation, writing, or publication submission decision-making.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: None
References
- [1].Gluckman PD, Cutfield W, Hofman P, Hanson MA, The fetal, neonatal, and infant environments-the long-term consequences for disease risk, Early Hum. Dev (2005) 51–59. 10.1016/j.earlhumdev.2004.10.003. [DOI] [PubMed]
- [2].Reynolds RM, Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis - 2012 Curt Richter Award Winner, Psychoneuroendocrinology 38 (2013) 1–11. 10.1016/j.psyneuen.2012.08.012. [DOI] [PubMed] [Google Scholar]
- [3].Khashan AS, Abel KM, Mcnamee R, Pedersen MG, Webb RT, Baker PN, Kenny LC, M.; Preben, Mortensen B, Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events, Arch Gen Psychiatry 65 (2008) 146–152. [DOI] [PubMed] [Google Scholar]
- [4].Ronald A, Happé F, Dworzynski K, Bolton P, Plomin R, Exploring the relation between prenatal and neonatal complications and later autistic-like features in a representative community sample of twins, Child Dev 81 (2010) 166–182. [DOI] [PubMed] [Google Scholar]
- [5].King S, Laplante DP, The effects of prenatal maternal stress on children’s cognitive development: Project Ice Storm, Stress 8 (2005) 35–45. 10.1080/10253890500108391. [DOI] [PubMed] [Google Scholar]
- [6].Pike IL, Maternal stress and fetal responses: Evolutionary perspectives on preterm delivery, Am. J. Hum. Biol 17 (2005) 55–65. 10.1002/ajhb.20093. [DOI] [PubMed] [Google Scholar]
- [7].Seckl JR, Glucocorticoids, developmental “programming” and the risk of affective dysfunction, Prog. Brain Res 167 (2008) 17–34. [DOI] [PubMed] [Google Scholar]
- [8].Kivlighan KT, DiPietro JA, Costigan KA, Laudenslager ML, Diurnal rhythm of cortisol during late pregnancy: Associations with maternal psychological well-being and fetal growth, Psychoneuroendocrinology 33 (2008) 1225–1235. 10.1016/j.psyneuen.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Christian LM, Physiological reactivity to psychological stress in human pregnancy: Current knowledge and future directions, Prog. Neurobiol 99 (2012) 106–116. 10.1016/j.pneurobio.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Drake AJ, McPherson RC, Godfrey KM, Cooper C, Lillycrop KA, Hanson MA, Meehan RR, Seckl JR, Reynolds RM, An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and foetal growth, Clin. Endocrinol. (Oxf) 77 (2012) 808–815. 10.1111/j.1365-2265.2012.04453.x. [DOI] [PubMed] [Google Scholar]
- [11].Beijers R, Buitelaar JK, de Weerth C, Mechanisms underlying the effects of prenatal psychosocial stress on child outcomes: beyond the HPA axis, Eur. Child Adolesc. Psychiatry 23 (2014) 943–956. 10.1007/s00787-014-0566-3. [DOI] [PubMed] [Google Scholar]
- [12].Chrousos GP, Stress and disorders of the stress system, Nat. Publ. Gr 5 (2009) 374–381. 10.1038/nrendo.2009.106. [DOI] [PubMed] [Google Scholar]
- [13].Murphy VE, Smith R, Giles WB, Clifton VL, Endocrine regulation of human fetal growth: The role of the mother, placenta, and fetus, Endocr. Rev 27 (2006) 141–169. 10.1210/er.2005-0011. [DOI] [PubMed] [Google Scholar]
- [14].Edwards CRW, Benediktsson R, Lindsay RS, Seckl JR, Dysfunction of placental glucocorticoid barrier: Link between fetal environment and adult hypertension?, Lancet 341 (1993) 355–357. [DOI] [PubMed] [Google Scholar]
- [15].Cohen S, Kamarck T, Mermelstein R, A global measure of perceived stress, J. Health Soc. Behav 24 (1983) 385–396. http://www.jstor.org/stable/2136404. [PubMed] [Google Scholar]
- [16].Nast I, Bolten M, Meinlschmidt G, Hellhammer DH, How to measure prenatal stress? A systematic review of psychometric instruments to assess psychosocial stress during pregnancy, Paediatr. Perinat. Epidemiol 27 (2013) 313–322. 10.1111/ppe.12051. [DOI] [PubMed] [Google Scholar]
- [17].Remor E, Psychometric properties of a European Spanish version of the Perceived Stress Scale (PSS), Span. J. Psychol 9 (2006) 86–93. 10.1017/S1138741600006004. [DOI] [PubMed] [Google Scholar]
- [18].Kroenke K, Spitzer RL, Williams JBW, The PHQ-9 validity of a brief depression severity measure, J. Gen. Intern. Med 16 (2001) 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Baader TM, Molina JLF, Rojas CC, Farías RS, Fierro-Freixenet C, Backenstrass M, Mundt C, Validación y utilidad de la encuesta PHQ-9 (Patient Health Questionnaire) en el diagnóstico de depresión en pacientes usuarios de atención primaria en Chile, Rev. Chil. Neuropsiquiatr 50 (2012) 10–22. [Google Scholar]
- [20].Cassiani-Miranda CA, Vargas-Hernández C, Pérez-Aníbal E, Isabel M, Vargas-Hernández MC, Herazo-Bustos MI, Hernández-Carrillo M, Confiabilidad y dimensión del cuestionario de salud del paciente (PHQ-9) para la detección de síntomas de depresión en estudiantes de ciencias de la salud en Cartagena, 2014, Biomédica 37 (2017) 112–20. 10.7705/biomedica.v37i0.3221. [DOI] [PubMed] [Google Scholar]
- [21].Kroenke K, Strine TW, Spitzer RL, Williams JBW, Berry JT, Mokdad AH, The PHQ-8 as a measure of current depression in the general population, J. Affect. Disord 114 (2009) 163–173. 10.1016/j.jad.2008.06.026. [DOI] [PubMed] [Google Scholar]
- [22].Salimetrics, SalivaBio, Saliva Collection and Handling Advice, 2015.
- [23].Howland MA, Sandman CA, Glynn LM, Developmental origins of the human hypothalamic-pituitary-adrenal axis, Expert Rev. Endocrinol. Metab 12 (2017) 321–339. 10.1080/17446651.2017.1356222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].O’Connor TG, Bergman K, Sarkar P, Glover V, Prenatal cortisol exposure predicts infant cortisol response to acute stress, Dev. Psychobiol 55 (2012) 145–155. 10.1002/dev.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tollenaar MS, Beijers R, Jansen J, Riksen-Walraven JMA, De Weerth C, Maternal prenatal stress and cortisol reactivity to stressors in human infants, Stress 14 (2011) 53–65. 10.3109/10253890.2010.499485. [DOI] [PubMed] [Google Scholar]
- [26].Stroud LR, Papandonatos GD, Parade SH, Salisbury AL, Phipps MG, Lester BM, Padbury JF, Marsit CJ, Prenatal major depressive disorder, placenta glucocorticoid and serotonergic signaling, and infant cortisol response, Psychosom. Med 78 (2016) 979–990. 10.1097/PSY.0000000000000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Davis EP, Glynn LM, Waffarn F, Sandman CA, Prenatal maternal stress programs infant stress regulation, J. Child Psychol. Psychiatry Allied Discip 52 (2011) 119–129. 10.1111/j.1469-7610.2010.02314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Burton GJ, Sebire NJ, Myatt L, Tannetta D, Wang YL, Sadovsky Y, Staff AC, Redman CW, Optimising sample collection for placental research, Placenta 35 (2014) 9–22. 10.1016/j.placenta.2013.11.005. [DOI] [PubMed] [Google Scholar]
- [29].Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM, Epigentic regulation of 11β-hydroxysteroid dehydrogenase type 2 expression, J. Clin. Invest 114 (2004) 1146–1157. 10.1172/JCI21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Capron LE, Ramchandani PG, Glover V, Maternal prenatal stress and placental gene expression of NR3C1 and HSD11B2: The effects of maternal ethnicity, Psychoneuroendocrinology 87 (2018) 166–172. 10.1016/j.psyneuen.2017.10.019. [DOI] [PubMed] [Google Scholar]
- [31].Clifton VL, Review: Sex and the human placenta: Mediating differential strategies of fetal growth and survival, Placenta 24 (2010) S33–S39. 10.1016/j.placenta.2009.11.010. [DOI] [PubMed] [Google Scholar]
- [32].Gabory A, Roseboom TJ, Moore T, Moore LG, Junien C, Placental contribution to the origins of sexual dimorphism in health and diseases: Sex chromosomes and epigenetics, Biol. Sex Differ 4 (2013). 10.1186/2042-6410-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bale TL, Sex differences in prenatal epigenetic programing of stress pathways, Stress 14 (2011) 348–356. 10.3109/10253890.2011.586447. [DOI] [PubMed] [Google Scholar]
- [34].Marsit CJ, Maccani MA, Padbury JF, Lester BM, Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome, PLoS One 7 (2012) e33794. 10.1371/journal.pone.0033794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lucassen PJ, Bosch OJ, Jousma E, Krömer SA, Andrew R, Seckl JR, Neumann ID, Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: Possible key role of placental 11β-hydroxysteroid dehydrogenase type 2, Eur. J. Neurosci 29 (2009) 97–103. 10.1111/j.1460-9568.2008.06543.x. [DOI] [PubMed] [Google Scholar]
- [36].Welberg LAM, Thrivikraman KV, Plotsky PM, Chronic maternal stress inhibits the capacity to up-regulate placental 11-beta-hydroxysteroid dehydrogenase type 2 activity, J. Endocrinol 186 (2005) R7–R12. 10.1677/joe.1.06374. [DOI] [PubMed] [Google Scholar]
- [37].Peña CJ, Monk C, Champagne FA, Epigenetic effects of Prenatal stress on 11β-Hydroxysteroid Dehydrogenase-2 in the Placenta and fetal brain, PLoS One 7 (2012) e39791. 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mairesse J, Lesage J, Breton C, Bréant B, Hahn T, Darnaudéry M, Dickson SL, Seckl J, Blondeau B, Vieau D, Maccari S, Viltart O, Maternal stress alters endocrine function of the feto-placental unit in rats, Am. J. Physiol. Endocrinol. Metab 292 (2007) E1526–E1533. [DOI] [PubMed] [Google Scholar]
- [39].Monk C, Feng T, Lee S, Krupska I, Champagne FA, Tycko B, Distress during pregnancy: Epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior, Am. J. Psychiatry 173 (2016) 705–713. 10.1176/appi.ajp.2015.15091171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Conradt E, Lester BM, Appleton AA, Armstrong DA, Marsit CJ, The roles of DNA methylation of NR3C1 and 11β-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior, Epigenetics 8 (2013) 1321–1329. 10.4161/epi.26634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Togher KL, O’Keeffe MM, Khashan AS, Gutierrez H, Kenny LC, O’Keeffe GW, Epigenetic regulation of the placental HSD11B2 barrier and its role as a critical regulator of fetal development, Epigenetics 9 (2014) 816–822. 10.4161/epi.28703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Seth S, Lewis AJ, Saffery R, Lappas M, Galbally M, Maternal prenatal mental health and placental 11β-HSD2 gene expression: Initial findings from the mercy pregnancy and emotional wellbeing study, Int. J. Mol. Sci 16 (2015) 27482–27496. 10.3390/ijms161126034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].O’Donnell KJ, Bugge Jensen A, Freeman L, Khalife N, O’Connor TG, Glover V, Maternal prenatal anxiety and downregulation of placental 11β-HSD2, Psychoneuroendocrinology 37 (2012) 818–826. 10.1016/j.psyneuen.2011.09.014. [DOI] [PubMed] [Google Scholar]
- [44].Appleton AA, Armstrong DA, Lesseur C, Lee J, Padbury JF, Lester BM, Marsit CJ, Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity, PLoS One 8 (2013) e74691. 10.1371/journal.pone.0074691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang W, Finik J, Dana K, Glover V, Ham J, Nomura Y, Prenatal depression and infant temperament: The moderating role of placental gene expression, Infancy 23 (2018) 211–231. 10.1111/infa.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Reynolds RM, Pesonen AK, O’Reilly JR, Tuovinen S, Lahti M, Kajantie E, Villa PM, Laivuori H, Hämäläinen E, Seckl JR, Räikkönen K, Maternal depressive symptoms throughout pregnancy are associated with increased placental glucocorticoid sensitivity, Psychol. Med 45 (2015) 2023–2030. 10.1017/S003329171400316X. [DOI] [PubMed] [Google Scholar]
- [47].St-Pierre J, Laplante DP, Elgbeili G, Dawson PA, Kildea S, King S, Vaillancourt C, Natural disaster-related prenatal maternal stress is associated with alterations in placental glucocorticoid system: The QF2011 Queensland Flood Study, Psychoneuroendocrinology 94 (2018) 38–48. 10.1016/j.psyneuen.2018.04.027. [DOI] [PubMed] [Google Scholar]
- [48].Jahnke JR, Madres Sanas, Bebés Sanos: The Intergenerational Effects of Maternal Stress on the Galápagos Islands, University of North Carolina at Chapel Hill, 2020. [Google Scholar]
- [49].Paquette AG, Lester BM, Lesseur C, Armstrong DA, Guerin DJ, Appleton AA, Marsit CJ, Placental epigenetic patterning of glucocorticoid response genes is associated with infant neurodevelopment, Epigenomics 7 (2015) 767–779. 10.2217/epi.15.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Carpenter T, Grecian SM, Reynolds RM, Sex differences in early-life programming of the hypothalamic–pituitary–adrenal axis in humans suggest increased vulnerability in females: a systematic review, J. Dev. Orig. Health Dis 8 (2017) 244–255. 10.1017/S204017441600074X. [DOI] [PubMed] [Google Scholar]
- [51].Demendi C, Börzsönyi B, Pajor A, Rigó J, Nagy ZB, Szentpéteri I, Joó JG, Abnormal fetomaternal glucocorticoid metabolism in the background of premature delivery: Placental expression patterns of the 11β-hydroxysteroid dehydrogenase 2 gene, Eur. J. Obstet. Gynecol. Reprod. Biol 165 (2012) 210–214. 10.1016/j.ejogrb.2012.08.009. [DOI] [PubMed] [Google Scholar]
- [52].Mericq V, Medina P, Kakarieka E, Márquez L, Johnson MC, Iñ Iguez G, Differences in expression and activity of 11b-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender, Eur. J. Endocrinol 161 (2009) 419–425. 10.1530/EJE-09-0308. [DOI] [PubMed] [Google Scholar]
- [53].Murphy VE, Gibson PG, Giles WB, Zakar T, Maternal asthma is associated with reduced female fetal growth, Am. J. Respir. Crit. Care Med 168 (2003) 1317–1323. [DOI] [PubMed] [Google Scholar]
- [54].Sandman CA, Glynn LM, Davis EP, Is there a viability-vulnerability tradeoff? Sex differences in fetal programming, J. Psychosom. Res 75 (2013) 327–335. 10.1016/j.jpsychores.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pehme PM, Zhang W, Finik J, Pritchett A, Buthmann J, Dana K, Hao K, Nomura Y, Placental MAOA expression mediates prenatal stress effects on temperament in 12-month-olds HHS Public Access, Infant Child Dev 27 (2018). 10.1002/icd.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].O’Donnell KJ, Meaney MJ, Fetal origins of mental health: The developmental origins of health and disease hypothesis, Am. J. Psychiatry 174 (2017) 319–328. 10.1176/appi.ajp.2016.16020138. [DOI] [PubMed] [Google Scholar]