

Abstract
Although the type 4 secretion system of the integrating and conjugative elements (tfs ICE) is common in Helicobacter pylori, its clinical association with the cag pathogenicity island (cagPAI) have not yet been well-investigated. In this study, Vietnamese patient H. pylori samples (46 duodenal ulcer (DU), 51 non-cardia gastric cancer (NCGC), 39 chronic gastritis (CG)) were fully sequenced using next-generation sequencing and assembled into contigs. tfs3, tfs4, and cagPAI genes were compared with the public database. Most (94%) H. pylori strains possessed a complete cagPAI, which was the greatest risk factor for clinical outcomes, while the prevalences of tfs3 and tfs4 were 45% and 77%, respectively. Complete tfs3 and tfs4 were found in 18.3% and 17.6% of strains, respectively. The prevalence of H. pylori strains with complete tfs3 ICE in DU patients was significantly higher than that in NCGC patients (30.4% vs 11.7%, P < 0.05). In addition, the prevalence of strains with complete tfs3 ICE and cagPAI was significantly higher in DU patients than that in NCGC (28.4% vs 9.8%, P = 0.038) and CG patients (28.2% vs 7.7%, P = 0.024). cagPAI and complete tfs3 increased the risk of DU compared to NCGC (OR = 3.56, 95%CI: 1.1–14.1, P = 0.038) and CG (OR = 4.64, 95%CI: 1.1–27.6, P = 0.024). A complete cluster of tfs3 ICE was associated with gastroduodenal diseases in Vietnam. However, there was a low prevalence of the dupA/complete dupA cluster (15.4%) in the Vietnam strains. The prevalence of cagPAI in Vietnam strains was significantly higher than in US (P = 0.01) and Indonesia (P < 0.0001); the prevalence of the dupA cluster was also higher in the Vietnam strains than in the Indonesian strains (P < 0.05). In addition, the prevalence of ctkA, an accessory gene of tfs3, was significantly different between Vietnam and US strains (28% vs 2%, P = 0.0002). In summary, the acquisition of tfs3/4 ICE was common in H. pylori strains in patients with gastroduodenal disease in Vietnam, and the complete cluster of tfs3 ICE was a reliable marker for the severity of disease in the H. pylori infected population.
Subject terms: Microbiology, Medical research
Introduction
Helicobacter pylori is a gram-negative bacterium that consistently infects the human stomach. The prevalence of H. pylori infection in the human population is approximately 50% and is responsible for a subset of 15–20% of clinical cases, with less than 5% of patients developing gastric cancer1,2. Together with the environment and host factors, the virulence of H. pylori is believed to be an important factor increasing the risk of gastroduodenal diseases3.
The type 4 secretion system (T4SS) is a protein complex found in prokaryotes used to transport DNA, proteins, or effector molecules from the cytoplasm to the extracellular space beyond the cell. The H. pylori genome is known to encode up to four T4SSs, and each plays an independent role during host infection4. The first discovered T4SS was located within the cag pathogenicity island (cagPAI), which plays a crucial role in H. pylori infection by forming a T4SS assembly, interacting with integrin receptor (α5β1), and supporting the injection of CagA, an oncogenic protein of H. pylori, into the host cell5,6. After translocating into gastric epithelial cells, CagA localized to the inner surface of the plasma membrane, in which it undergoes tyrosine phosphorylation at the Glu-Pro-Ile-Tyr-Ala (EPIYA) motif7. The length of the cagPAI is about 40 kb and is comprised of 26 to 30 genes, depending on the strains that encode a complete set of T4SS (virB1-virB11), a coupling-protein virD4, and a subset of the cag genes, which contribute to a functional cagPAI8. The presence of the cagPAI had been found to be associated with severe inflammation and the development of gastroduodenal diseases6. The second T4SS was named as comB, which comprises all T4SS core components (comB2 to comB4, and comB6 to comB10) and plays an important role during the natural transformation of H. pylori9. The DNA transformation and homologous recombination substantially maintain the genome variability, which is crucial for promoting chronic infection of H. pylori10. In a different manner than the other T4SSs, comB-T4SS was unique in DNA uptake, especially in the efficiency of DNA transfer, which enabled the survival of H. pylori during long-term infection10. The third T4SS was previously called TnPZ (transposon element of plasticity region), and had been identified within the plasticity regions in which the GC content was lower (34–35%) than that in the rest of the genome (39%)11,12. The full length of TnPZ was determined to be 37 kb to 46 kb and is composed of a cluster of T4SS genes, a tyrosine recombinase family gene (xerT), a long open reading frame (> 2800 codon) encoded to helicase and DNA methylase domain, and a subset of accessory genes with unknown function12. Since there was evidence for horizontal gene transfer of TnPZ in a conjugation manner, they were termed as integrating and conjugating elements (ICE) and referred to as ICEHptfs or tfs ICE4,12,13. Similar to cagPAI, tfs ICE possessed all core genes of T4SS (virB2-virB4, virB6-virB11, and virD4 coupling-protein) in addition to a virD2 relaxase12. Because of the nucleotide diversity between vir genes of tfs ICEs in H. pylori strains, they were classified into 2 types: tfs3 and tfs413. Sequence analysis showed that the nucleotide diversity of tfs3 ICE genes from virB2 to virB11 was highly similar among H. pylori strains13. In contrast, tfs4 ICEs are divided into 3 subtypes: 4a, 4b, and 4c based on the sequence diversity of virB2, virB3, virB4, virB6, virB7, and virB8 discriminating 4a to 4b, while the diversity of virB11, virD2, and virD4 discriminates 4a/4b to 4c13. However, each H. pylori strain differently presented the combination of tfs4 modules, which included the left (L1/L2), center (C1/C2), and right (R1/R2) modules14. In addition, a full set of T4SS genes was located within each module: L1/L2 (xerT, virB6), C1/C2 (virD2, virD4, virB11, virB10, virB9), and R1/R2 (virB2, virB3, virB4), and the tfs4 subtypes were thus classified based on the following module combinations: tfs4a (L2C1R2), tfs4b (L1C1R1), tfs4a/4b (L2C1R1) and tfs4c (L2C2R2)14,15.
Component genes of tfs3 and tfs4 are associated with the pro-inflammatory activity in the gastric mucosa and increased risk of gastroduodenal diseases16,17. We previously showed that dupA, a virB4 homolog located within the right module (R1) of tfs4b ICE, was considered to be a specific marker for duodenal ulcer (DU)18. Furthermore, the presence of dupA in combination with its neighbor T4SS genes forming an intact dupA cluster (C1R1) might be a more reliable marker for disease risk than incomplete dupA cluster or dupA alone19. The in vitro study showed that the expression of some tfs4b T4SS genes (virB2, virB4, virB8, and virB10) was more up-regulated in response to low pH and contact with the human gastric cell line, which supported the role of tfs4b in host colonization20. These studies suggested that tfs3 and tfs4b might form an alternative T4SS for DNA or effector protein in a similar manner to cagPAI and were considered to be virulence factors of H. pylori. tfs3 and tfs4b ICEs could distribute differently, suggesting that the risk of these clusters in gastroduodenal diseases should be considered within in each country and ethnicity12–15. Although the association of tfs4 ICE with gastroduodenal diseases has been determined in some countries, the clinical association of tfs3 ICE with gastroduodenal diseases has not yet been adequately investigated. Moreover, a comprehensive study about the prevalence and status of tfs3 and tfs4 in clinical outcomes has not yet been conducted. Hence, we conducted a study to examine the distribution and status of tfs3 and tfs4 in H. pylori strains isolated from Vietnamese patients with gastroduodenal diseases including DU, non-cardia gastric cancer (NCGC), and chronic gastritis (CG).
The prevalence of H. pylori infection in Vietnam was reported to be 65.6%, and the incidence of gastric cancer in Vietnam was classified as an intermediate risk in Asia, but the highest in Southeast Asia (age- standardized rate (ASR) of gastric cancer, 16.3/100,00 in both sex)21–23. Although several previous studies have investigated the virulence factors in Vietnam, these studied only examined some well-known virulence factors (cagA, vacA, and several cagPAI genes)23–25. It is still unknown why some infected subjects develop severe diseases like DU and gastric cancer. Therefore, we aimed to investigate the association of tfs ICE and cagPAI with clinical outcomes in Vietnam via whole-genome analyses using next-generation sequencing.
Results
Sequence comparison of vir T4SS genes of tfs3, tfs4, cagPAI, and comB
The genomes of 136 H. pylori strains were newly assembled into 23–129 contigs (Supplementary-Table S1), and then tfs3 and tfs4 were identified by the common 12 genes: xerT, virB2, virB3, virB4, virB6, virB7, virB8, virB9, virB10, virB11, virD2, and virD4 using the ABRICATE pipeline. Similarly, the cagPAI and comB were identified by 10 (virB2-B11, virD4) and 6 (virB2-virB4, virB7-virB10) T4SS genes by the same approach, respectively.
Since all four types of tfs gene clusters contained xerT and 11 conserved vir genes, first, the nucleotide identities of each of these 12 genes of the tfs3, tfs4a, tfs4b, and tfs4c ICE were compared using pairwise sequence alignment. Their sequence identity is shown in Table S4 and visualized in Fig. 1. There was a low identity of T4SS nucleotide diversity between tfs3 ICE and either of tfs4a/b/c ICE (less than 60%). For tfs3 ICE, there were only virB6 and virB7, which was diverse in nucleotide sequence between strain Gambia94/24 and strain India7 (less than 80%), and the other vir T4SS genes had high identity (from 84–100%). We denoted virB6 and virB7 of strain Gambia94/24 as variant 1, while strain India7 possessed variant 2 of those. In contrast to tfs3 ICE, there were only 2 T4SS genes, which were encoded by all three tfs4 subtypes (a/b/c) with high identity: virB9 with high identity (> 90%) and virB10 with moderate identity (> 80%). The T4SS genes of tfs4a (strain P12) and tfs4b (strain Shi470) had high identity with more than 90% in virB11, virD2, and virD4, while tfs4a (strain P12) and tfs4c (strain R036d) shared identity between 83.5% and 98.0% in virB2, virB3, virB4, virB6, virB8, and virB10. Therefore, the nucleotide diversity in 3 genes (virB11, virD2, and virD4) could distinguish tfs4a and tfs4b from tfs4c, while that in 6 genes (virB2, virB3, virB4, virB6, virB7, virB8) could distinguish tfs4a/4c from tfs4b. From this analysis, we used tfs3 (strains Gambia94/24 and India7), tfs4a (strain P12), tfs4b (strain Shi470), and tfs4c (strain R036d) to construct a reference database. These strains possessed the prototypical tfs3/4a/b/c in addition to cagPAI and comB clusters with no frameshift or premature stop codon in any gene. In addition, the module combinations of tfs4 in these strains were L2C1R2, L1C1R1, L2C1R1, and L2C2R2 for tfs4a, tfs4b, tfs4a/4b and tfs4c, respectively14,15. The annotation number of these tfs genes was retrieved from reference strains from Genbank database and is shown in Table S2 (https://www.ncbi.nlm.nih.gov/nuccore).
Figure 1.

Gene arrangement of cagPAI, tfs3, tfs4a, tfs4b, and tfs4c ICE, which were retrieved from H. pylori strain 26,695, Gambia94/24, P12, Shi470, and R036d, respectively. The red color indicates vir homolog T4SS genes in which nucleotide identity is more than 80% to corresponding genes of tfs4b ICE, while horizontal dashed red and dashed gray colors indicate that nucleotide identity was less than 80%, respectively, in a sequence comparison between tfs4b and others (Table 1 and Table S4). The black color indicates the other cag genes of cagPAI (ζ, ε, δ, Z, U, S, Q, P, M, N, I, H, G F, D, B, A). Gray color indicates accessory genes of tfs3 and tfs4 ICE, which included DNA processing genes (xerT and topA) and unknown function genes.
Also, the 11 T4SS genes in tfs3 ICE (strain Gambia94/24), tfs4a ICE (strain P12), tfs4b ICE (strain Shi470), and tfs4c (strain R036d) were applied for pairwise sequence comparison to double-check identity against comB and cagPAI, the other two T4SS clusters, in the same strain background. The low identity from the pairwise alignment of T4SS nucleotide sequences of tfs3/tfs4a/tfs4b/tfs4c against comB (less than 61%) and cagPAI (less than 50%), indicated the distinct evolution from tfs3 and tfs4 ICE (Table S3 – Supplementary).
Prevalence of tfs3, tfs4, cagPAI, and comB in Vietnam isolates
Next, all 136 strains were applied to identify the T4SS genes of tfs3, tfs4a/b/c, cagPAI, and comB using the ABRICATE pipeline and the reference sequences from the above step. The sequence was identified and denoted as tfs3, tfs4, cagPAI, or comB based on whether the coverage was more than 60% and percentage identity more than 80% against the query. Based on the criteria from the above analysis, the subtypes of tfs4 (a/b/c) and tfs4 modules (L1/L2/C1/C2/R1/R2) were also determined. As shown in Table S5, the number of strains possessing tfs3 and tfs4 was 62 (45.5%) and 105 (77%), respectively. These results indicated the high prevalence of tfs3/4 in H. pylori strains isolated from gastroduodenal patients in Vietnam. The distribution of the tfs4 module in Vietnam strains skewed towards L1 (30.1%) and L2 (47.0%) compared to R1 (19.8%) and R2 (27.9%); the C1 module (29.4%) was more prevalent than the C2 module (2.9%). The number and percentage identity of each vir T4SS gene of tfs3, tfs4 are also shown in Table S5.
In this study, we selected all 11 vir genes to access the status of cagPAI. Based on the criteria, 128 (94.1%) strains possessed the complete cagPAI and 2 strains had incomplete cagPAI (1.4%), while only 6 strains were cagPAI–negative (4.4%). Also, all strains possessed the comB cluster in their genome. The coverage and identity percentage of each tfs3, tfs4, cagPAI, and comB T4SS gene in each strain are shown in the additional data (Table S6).
Distribution of the cagA-EPIYA motif in terms of disease severity and geographical population
The cagA gene mostly harbored the ABD-type Glu-Pro-Ile-Tyr-Ala (EPIYA) motif: 43 (93%) in strains from DU patients, 48 (94%) in strains from NCGC patients, and 31 (79%) in strains from CG patients (Fig. 2). In contrast, a minority of patients harbored BD- (1 in NCGC), ABC- (2 DU, 2 NCGC, and 1 CG), and ABCC- (1 in CG) type EPIYA motifs. Seven strains were cagA-negative: 1 (2%) from DU patients and 6 (15.3%) from CG patients. The presence of cagA, regardless of the EPIYA motif, was associated with the severity of clinical outcomes in Vietnam: 97% in DU vs 84% in CG (P = 0.04) and 100% in NCGC vs 84% in CG (P = 0.005).
Figure 2.
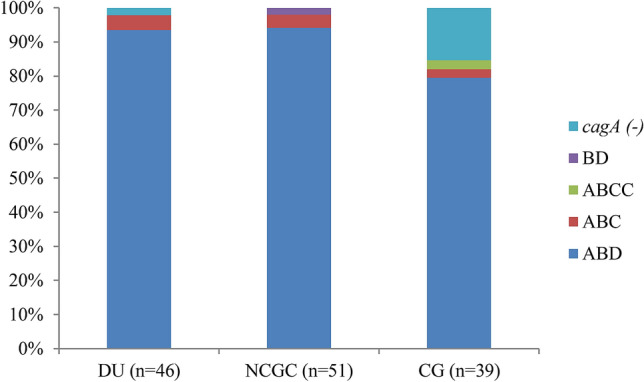
Distribution of cagA-EPIYA motifs in three clinical outcomes: duodenal ulcer (DU), non-cardia gastric cancer (NCGC), and chronic gastritis (CG). The EPIYA motifs of cagA included ABD, BD, ABC, and ABCC.
The distribution of the cagA-EPIYA motifs was mostly in agreement with the population-specificity of Vietnam strains: 124 (95.3%) of the hspEAsia strains harbored the East Asian-type cagA, and three (50%) of the hpEurope strains were cagPAI (-)/cagA(-) (Fig. 3). However, there was an exchange in the population-specific cagA genotype: 3 of 6 hpEurope strains possessed East Asian-type cagA, while 6 of 130 hspEAsia strains possessed Western-type cagA (Fig. 3).
Figure 3.

A phylogenetic tree was constructed through the concatenation of seven housekeeping genes from 136 Vietnam strains (olive circle) and 379 reference strains from PubMLST (http://pubmlst.org/helicobacter/). The circle indicates our studied strains: blue (cagA-negative), red (Western-type cagA), and olive (East Asian-type cagA).
Distribution and status of tfs3 and tfs4a/b/c in H. pylori isolated from patients with gastroduodenal diseases in Vietnam
As for the cagPAI, the strains that possessed all 11 vir T4SS genes (virB2, virB3, virB4, virB6, virB7, virB8, virB9, virB10, virB11, virD2, and virD4) were defined as a complete form (Table S5), while the other cases were an incomplete form. In contrast, the strain was defined as tfs-negative if tfs sequences were absent.
Based on this criterion, there was a high prevalence of tfs-positive strains in Vietnam (90.4%): DU (87.0%), NCGC (98.0%), and CG (84.6%) (Table 1). There was a dominant prevalent of single tfs compared to double tfs (58.1% vs 32.3%, P < 0.001), which were divided into: tfs3 (13.2%), tfs4 (44.8%), and tfs3-4 (32.3%). Together with the high prevalence of tfs3 and tfs4, the co-existence of tfs3-4 would be a result of horizontal gene transfer during H. pylori mixed-infection in the host. The prevalence of tfs3 was similar between DU (50%), CG (41.1%), and NCGC (46.1%) patients, while the prevalence of tfs4 was higher in NCGC (88.2%) and CG (74.3%) patients than in DU (67.4%) patients. Considering the tfs4 module, there were no differences in the prevalences of the L2/C2/R2/L1/C1 module between DU, NCGC, and CG patients. In contrast, the prevalence of R1 module was significantly different between the NCGC and CG patients and the DU patients (25.5% vs 8.7% and 25.6% vs 8.7%, respectively, all P < 0.05).
Table 1.
Distribution of tfs3 and tfs4 ICE in DU, NCGC, and CG patients.
| Type of T4SS | DU (%), n = 46 | NCGC (%), n = 51 | CG (%), n = 39 | Total (%), n = 136 | ||||
|---|---|---|---|---|---|---|---|---|
| tfs negative | 6 (13.0) | 1 (0.2) | 6 (15.3) | 13 (9.6) | ||||
| tfs positive | 40 (87.0) | 50 (98.0) | 33 (84.6) | 123 (90.4) | ||||
| Incomplete tfs | Complete tfs* | 21 (45.6) | 19 (41.3) | 35 (68.6) | 15 (29.4) | 19 (48.7) | 14 (35.9) | 75 (55.1) | 48 (35.3) |
| Single tfs | ||||||||
| tfs3 | 2 (4.3) | 7 (15.2) | 2 (3.9) | 3 (5.8) | 4 (10.2) | 0 | 8 (5.8) | 10 (7.3) |
| tfs4 | 13 (28.2) | 4 (8.7) | 20 (39.2) | 9 (17.6) | 8 (20.5) | 7 (17.9) | 41 (30.1) | 20 (14.7) |
| Double tfs** | ||||||||
| Incomplete tfs3 and tfs4 | Complete tfs3/tfs4 | 6 (13.0) | 8 (17.4) | 13 (25.5) | 3 (5.8) | 7 (17.9) | 7 (17.9) | 26 (19.1) | 18 (13.2) |
| tfs4 module | ||||||||
| L1 | 10 (21.0) | 18 (35.3) | 13 (33.3) | 41 (30.1) | ||||
| L2 | 21 (45.6) | 27 (52.9) | 16 (41.0) | 64 (47.0) | ||||
| C1 | 9 (19.5) | 16 (31.3) | 15 (38.4) | 40 (29.4) | ||||
| C2 | 2 (4.3) | 0 | 2 (5.1) | 4 (2.9) | ||||
| R1 | 4 (8.7) | 13 (25.5)a | 10 (25.6)b | 27 (19.8) | ||||
| R2 | 14 (30.4) | 14 (27.4) | 10 (25.6) | 38 (27.9) | ||||
| Other T4SS | ||||||||
| cagPAI | 45 (97.8) | 51 (100) | 34 (87.1) | 130 (96.1) | ||||
| comB*** | 46 (100) | 51 (100) | 39 (100) | 136 (100) | ||||
80% < Identity of gene only counted.
Complete tfs harbored 11 vir genes (virB2, virB3, virB4, virB6, virB7, virB8, virB9, virB10, virB11, virD4, and virD2)
** Double tfs were divided into: both of complete tfs, one of complete tfs, and both of tfs incomplete form cagPAI harbored all ten vir genes (virB2, virB3, virB4, virB6, virB7, virB8, virB9, virB10, virB11, and virD4).
***comB harbored all seven vir genes (virB2, virB3, virB4, virB7, virB8, virB9, and virB10).
aIndicated the significant different between NCGC to DU at P < 0.05.
bIndicated the significant different between CG to DU at P < 0.05.
Furthermore, we assessed the status of tfs among different types. Among 136 strains, the complete forms of tfs3 and tfs4 were observed in 18% and 19.1% of strains. The prevalences of the complete tfs4 forms were as follows: 15.4% for L1C1R1, 0.7% for L2C1R1, and 2.9% for L2C2R2.
The prevalence of vir T4SS genes of the tfs3 cluster in gastroduodenal diseases
Although more than 45% of Vietnam strains possessed tfs3 ICE, they dominantly harbored the incomplete form or only a fragment in the genome (Table 2). Among these incomplete tfs3 clusters, there was a tendency to have the xerT, virD2, and virB6, while the virB2-virD4 gene cluster was lost. Moreover, it is possible that the complete form of tfs3 ICE formed an alternative functional assembly compared to the incomplete form or fragment. The prevalence of complete tfs3 was significantly higher in DU patients compared to NCGC patients (30.4% vs 11.7%, P = 0.026) and CG patients (30.4% vs 12.8%, P = 0.068). This result showed the association of tfs3 with gastroduodenal diseases in Vietnam. In addition, we assessed the status of both tfs3 ICE and cagPAI. Interestingly, the prevalences of strains that possessed both cagPAI and complete tfs3 was significant higher in DU patients compared to that in CG (28.2% vs 7.7%, P = 0.024) and NCGC (28.2% vs 9.8%, P = 0.038) patients.
Table 2.
Prevalence of tfs3 ICE T4SS genes in DU, NCGC, and CG.
| DU (%), n = 46 | NCGC (%), n = 51 | CG (%), n = 39 | Total (%), n = 136 | |
|---|---|---|---|---|
| virB2 | 15 (32.6) | 11 (21.5) | 10 (25.6) | 36 (26.4) |
| virB3 | 15 (32.6) | 11 (21.5) | 10 (25.6) | 36 (26.4) |
| virB4 | 15 (32.6) | 11 (21.5) | 10 (25.6) | 36 (26.4) |
| virB6 | 18 (39.1) | 14 (27.4) | 13 (33.3) | 45 (33.0) |
| virB7 | 16 (34.7) | 9 (17.6) | 7 (17.9) | 32 (23.5) |
| virB8 | 15 (32.6) | 9 (17.6) | 7 (17.9) | 31 (22.8) |
| virB9 | 17 (36.9) | 10 (19.6) | 7 (17.9) | 34 (25.0) |
| virB10 | 17 (36.9) | 10 (19.6) | 7 (17.9) | 34 (25.0) |
| virB11 | 16 (34.8) | 11 (21.5) | 7 (17.9) | 34 (25.0) |
| virD4 | 16 (34.8) | 11 (21.5) | 7 (17.9) | 34 (25.0) |
| virD2 | 19 (41.3) | 10 (19.6) | 15 (38.4) | 44 (32.3) |
| xerT | 21 (45.6) | 20 (39.2) | 18 (46.1) | 59 (43.3) |
| ctkA | 14 (30.4) | 15 (29.4) | 10 (25.6) | 39 (28.6) |
| Incomplete tfs3 | 9 (19.5) | 15 (30.0) | 13 (33.3) | 37 (27.2) |
| Complete tfs3 | 14b (30.4) | 6 (11.7) | 5 (12.8) | 25 (18.4) |
| Complete tfs3 and ctkA | 7 (15.2) | 5 (9.8) | 1 (2.5) | 12 (8.8) |
| Incomplete tfs3 and cagPAI | 9 (19.5) | 13 (25.5) | 11 (28.2) | 33 (24.3) |
| Complete tfs3 and cagPAI | 13a,b (28.2) | 5 (9.8) | 3 (7.7) | 21 (15.4) |
aIndicated significant difference in prevalence between DU and CG at P < 0.05.
bIndicated significant difference in prevalence between DU and NCGC at P < 0.05.
In addition, the prevalence of ctkA (cell-translocating kinase A), an accessory gene of tfs3 ICE, was 28.6% in Vietnam strains; this prevalence was divided into 23.9% for DU patients, 29.4% for NCGC patients, and 25.6% for CG patients. According to Delahay et al., the pro-inflammatory activity of ctkA was supported by the T4SS genes of tfs3 ICE14. In our study, the prevalence of ctkA( +)/complete tfs3 ICE tended to be higher in DU patients than in CG patients (15.2% vs 2.5%, P = 0.064) but not higher in NCGC patients than in CG patients (9.8% vs 2.5%, P = 0.228).
Prevalence of dupA and its neighboring vir homologous genes in the tfs4b cluster in gastroduodenal diseases
The dupA cluster, which included the C1 and R1 modules, was combined with the L1 module forming tfs4b (L1C1R1) in 23 strains (16.9%) or the L2 module forming L2C1R1 in 4 strains (2.9%) in our study (Table 3). In contrast to tfs3, the complete dupA cluster (15.4%) was more prevalent than the incomplete form (4.4%) (Table 3). The incomplete dupA cluster tended to have the L1/L2 module (xerT and virB6) and, to a lesser extent, the C1 module (virD2, virD4, virB11, virB10, and virB9), while the R1 module (virB2, virB3, and virB4) was almost absent. It has been confirmed that dupA is the virB4 of tfs4b ICE, and the presence of dupA and its neighboring vir homologous genes (from virB2 to virD2), which form a complete dupA cluster, is a more reliable disease marker than the presence of dupA alone19. Considering each disease group, a complete dupA cluster was found in 15.4% of the total strains (8.7% in DU, 17.6% in NCGC, and 20.5% in CG). In addition, the prevalence of strains with complete dupA cluster and cagPAI was 14.7% in Vietnamese patients, 8.7% in DU patients, 17.6% in NCGC patients, and 17.9% in CG patients.
Table 3.
Prevalence of dupA cluster in DU, NCGC, and CG.
| Module | T4SS genes | DU (%), n = 46 | NCGC (%), n = 51 | CG (%), n = 39 | Total (%), n = 136 |
|---|---|---|---|---|---|
| L1 (n = 23) | xerT | 4 (8.7) | 9 (17.6) | 10 (25.6) | 23 (16.9) |
| virB6 | 4 (8.7) | 9 (17.6) | 10 (25.6) | 23 (16.9) | |
| L2 (n = 4) | xerT | 0 | 3 (5.8) | 1 (2.5) | 4 (2.9) |
| virB6 | 0 | 3 (5.8) | 1 (2.5) | 4 (2.9) | |
| C1 (n = 27) | virD2 | 4 (8.7) | 13 (25.5) | 10 (25.6) | 27 (19.8) |
| virD4 | 4 (8.7) | 10 (25.6) | 9 (23.0) | 23 (16.9) | |
| virB11 | 4 (8.7) | 10 (25.6) | 9 (23.0) | 23 (16.9) | |
| virB10 | 4 (8.7) | 11 (21.5) | 10 (25.6) | 25 (18.3) | |
| virB9 | 4 (8.7) | 12 (23.5) | 10 (25.6) | 26 (19.1) | |
| R1 (n = 27) | virB8 | 4 (8.7) | 12 (23.5) | 10 (25.6) | 26 (19.1) |
| virB7 | 4 (8.7) | 13 (25.5) | 10 (25.6) | 27 (19.8) | |
| virB4 (dupA) | 4 (8.7) | 9 (17.6) | 8 (20.5) | 21 (15.4) | |
| virB3 | 4 (8.7) | 9 (17.6) | 8 (20.5) | 21 (15.4) | |
| virB2 | 4 (8.7) | 9 (17.6) | 8 (20.5) | 21 (15.4) | |
| Incomplete dupA cluster | 0 | 4 (7.8) | 2 (5.1) | 6 (4.4) | |
| Complete dupA cluster | 4 (8.7) | 9 (17.6) | 8 (20.5) | 21 (15.4) | |
| Incomplete dupA cluster and cagPAI | 0 | 3 (5.8) | 1 (2.5) | 4 (2.9) | |
| Complete dupA cluster and cagPAI | 4 (8.7) | 9 (17.6) | 7 (17.9) | 20 (14.7) | |
Previous studies showed that the long-intact form of dupA (2499 bp) with no mutation, which caused a frameshift or premature stop codon in the sequence, was determined to be associated with the severity of clinical outcomes in comparison to the short-form dupA26. Among 21 dupA-positive strains (15.4%), there were 10 strains (7.3%) possessing the long intact-dupA, while 11 strains (8.1%) possessed the mutation, which might be the non-functional dupA. The type of dupA mutation and gastroduodenal diseases are shown in Table 4. Non-intact dupA was more frequently observed in NCGC (4/51; 7.8%) and CG patients (6/39; 15.3%) but not in DU patients (1/46; 2.2%). In contrast, the prevalence of long-intact dupA was divided into: DU (3/46; 6.5%), NCGC (5/51; 9.8%), and CG (2/39; 5.1%).
Table 4.
The mutation of dupA in gastroduodenal patients in Vietnam.
| Strains | Mutation | Type of mutation | Diseases |
|---|---|---|---|
| VN0355 | E750* | Premature stop codon | NCGC |
| VN0434 | E65* | Premature stop codon | NCGC |
| VN0448 | S291fs | Frameshift | NCGC |
| VN0472 | D601fs | Frameshift | NCGC |
| VN0754 | 2030_2256del | Deletion | DU |
| VN1158 | F397fs | Frameshift | CG |
| VN1165 | G458fs | Frameshift | CG |
| VN1192 | G458fs | Frameshift | CG |
| VN1196 | G434fs | Frameshift | CG |
| VN1212 | K113fs | Frameshift | CG |
| VN1251 | 494_528del | Deletion | CG |
Comparison of tfs3, dupA cluster, and cagPAI prevalence between Vietnam and the other geographical regions
The prevalence of cagPAI, tfs3, and dupA cluster in Vietnam strains was compared to those in strains from other geographical regions: Indonesia15, Cambodia (KH)27, and US (MPH)28 strains (Table S6). There were no cases of gastric cancer in previous studies conducted in Cambodia, Indonesia, and the US. In addition to our previous study on the distribution of tfs/cagPAI in Indonesia, we conducted the same analysis of whole-genome contigs of Cambodia and US strains (Table S6). The prevalence of cagPAI in Vietnam strains (94%) was highest among strains from 4 countries and significant compared to the prevalence of cagPAI in US (81%, P = 0.01) and Indonesian (55%, P < 0.0001) strains but not significant compared to the prevalence of Cambodian strains (91%) (Fig. 4). In contrast, the prevalence of tfs3 in Vietnam strains (46%) was not statistically different from that in the US (58%), Indonesia (41%), and Cambodia (45%) strains. The prevalence of ctkA in Vietnam strains was statistically higher than that in the US strains (29% vs 2%, P = 0.0002).
Figure 4.

Distribution of tfs3, dupA cluster, and cagPAI across four geographical regions: Vietnam, Indonesia, Cambodia, and the US (Bronx, NY). The prevalences of ctkA and dupA were included for the comparison.
Although the prevalence of the dupA cluster in the Vietnam and Cambodia strains tended to be lower than that in the US strains (15% vs 23%, P = 0.22), the prevalence of the dupA cluster in strains in these three countries was higher than that in Indonesia strains (all P < 0.05). The prevalence of dupA( +) in the US strains (60%) was higher compared to those of Vietnam (15%), Indonesian (5%), and Cambodian (19%) strains (all P < 0.0001). However, 37% of the US strains possessed the short-form dupA (78% in coverage compared to long-form dupA) (Table S6); dupA was in the long form in Vietnam and Cambodia strains (Table 4).
Discussion
In this study, the prevalence of strains possessing the tfs ICE was high among Vietnam strains (90.4%), which were divided into tfs3 ICE (45%) and tfs4 ICE (77%) (Table S6). The distribution of tfs ICE was dependent on the ethnic and geographical phylogeny of strains14,15. This could explain the difference in tfs ICE from that observed in our previous study in Indonesia (54.3%), in which the strains were isolated from several ethnicities15; all strains in this study were isolated from people of the Kinh ethnicity in Vietnam and predominantly belonged to the hspEAsia population (95.5%) (Fig. 3). The pooled prevalence of tfs ICE in H. pylori samples selected from seven distinct phylogeographic populations was 92.8% for tfs4 ICE and 62.2% for tfs3 ICE14. In addition, the distributions of tfs3 and tfs4 in hpEurope/hpAfrica1 strains (139 strains) were 69% and 96%, respectively, and were different from those of the hspEAsia strains (28 strains): 53% and 71%, respectively14. Additionally, although there were a few whole-genome hpAfrica2 strains available, all possessed tfs4c (L2C2R2) (100%)14. As shown in Tables S6 and S7, the prevalences of tfs3 and tfs4 were 43% and 76%, respectively, in our hspEAsia strains, which is consistent with the results of previous studies. Furthermore, the prevalence of tfs ICE in our previous study in Indonesia, which was composed of hpNEAfrica/hpEurope/hpAsia2 populations, was significantly lower than that in Vietnam (54.3% vs 90.4%, P < 0.001), also supporting the different prevalence of tfs ICE between populations15.
dupA, a virB4 homolog of tfs4b, was considered to be a marker of DU, but was not universally associated with disease development29,30. Our previous study indicated that the presence of dupA and its neighboring vir genes, which form the complete tfs4b cluster (or dupA cluster), is considered to be a more reliable disease-marker compared with single dupA19,20. Vietnam strains possessed a low prevalence of dupA (15.4%) compared to those from the US (69.3%), Japan (37%), Korea (37%), and Columbia (55%)18,19. In this study, the prevalence of the dupA cluster in Vietnam strains was significantly lower than that in the US strains (15.4% vs 69.3%, P < 0.0001)19 but was slightly higher than that in the Indonesia strains15 (15.4% vs 5%, P < 0.01); Indonesia has a low risk of gastric cancer. Moreover, there was a remarkably low incidence of H. pylori-related disease in the Nigerian population (hspWAfrica), which possesses a high prevalence of the truncated R1 module of tfs4b (L1/L2C1R1f.), in which is a short-form dupA with a lack of virB2 and virB3; although this population contained the high prevalences of cagA and vacA s1m1 genotype31. In our analysis, most of the US strains (hspWAfrica) similarly possessed L1/L2C1R1f., which supported the role of this tfs4 subtype in hspWAfrica1 strains. In addition, our previous study in Okinawa (Japan) showed that long-form and intact long-form dupA increased the risk of gastroduodenal diseases (gastric ulcer and gastric cancer) but not cagA26. These studies suggested that the deficient of dupA cluster would be reversibly associated with the severity of diseases and have a protective role in some settings. The function of dupA cluster has not yet been determined, but it might support the translocation of effector protein(s) similarly to CagA injection by the cagPAI. In our study, only 21/136 strains (15.4%) had the dupA, and 11 of them (8.1%) were non-functional dupA caused by mutation (frameshift, premature stop codon, and deletion). In addition, non-intact dupA was more frequently observed in CG (15.3%) patients than in NCGC patients (7.8%), while there was a negative correlation between dupA and DU patients. Our data suggested that there was an effect of dupA on pathogenesis, despite its low prevalence in Vietnam. The dupA gene encodes homologs of VirB4 ATPase. The Pfam search shows that dupA contains the CagE_TrbE_VirB domain and FtsK/SpoIIIE family. The FtsK/SpoIIIE domain contains a putative ATP- binding P-loop motif, is involved in cell division and peptidoglycan synthesis or modification and is implicated in intracellular chromosomal DNA transfer. Members of the TraG/TraD family are potential NTP hydrolases that are essential for DNA processing and the mating pair formation system. The in vitro study showed that there was a more positive correlation between IL-8 expression and dupA-positive strains than dupA-negative strains18.
The prevalence of ctkA (jhp0940 in strain J99), a cell-translocating kinase, was 41.2% in gastric cancer patients but zero among the strains isolated from patients with gastritis (P < 0.0006) in Costa Rica and was the first disease-marker of tfs3 ICE32. In addition, ctkA is located within the tfs3 ICE and is variably distributed in diverse H. pylori strains33. There was evidence that the tfs3 T4SS supported and increased the pro-inflammatory activity of effector protein (CtkA) in gastric epithelial cells, and it is suggested that tfs3 may form a novel T4SS assembly for protein secretion similar to cagPAI33. However, there was no association between ctkA alone or in combination with complete tfs3 with the severity of clinical outcomes in Vietnam. Compared to the US strains, the high prevalence of ctkA in Vietnam strains might indicate population-dependent variation between geographical regions. Our previous study in Indonesia revealed that intact cagPAI/tfs ICE-positive strains had significantly higher antral activity than non-intact cagPAI/tfs-negative and non-intact cagPAI/tfs-positive strains; no difference was observed between intact cagPAI/tfs-negative and non-intact cagPAI/tfs-negative15. In this study, the prevalences of complete cagPAI/incomplete tfs3 as well as complete cagPAI/complete tfs3 were 24.3% and 15.4%, respectively, which implied that functional cagPAI, regardless of tfs3 status, correlated with disease progression. These results indicate that cagPAI is the factor most affecting the development of gastroduodenal diseases, and its combination with other virulence factors, such as tfs3/tfs4, might increase the severity of clinical outcomes. The role of tfs ICE in pathogenesis could be associated with inflammation induction in gastric epithelial cells15, because T4SS encoded by this element promotes IL-8 expression independent of the presence of cagPAI12 and was reinforced when tfs ICE was present20. Although the association of the intact dupA cluster with clinical outcomes had been determined, the association of tfs3 ICE with gastroduodenal diseases is still not yet determined. In our study, there was a dominant complete tfs3 ICE in DU patients, which was the first report of tfs3 ICE in diseases. In the future, more studies about the prevalence and association of tfs3/4 ICE and cagPAI with clinical outcomes need to be conducted to clarify the role of tfs3 ICE in gastroduodenal diseases.
Although the biology of tfs ICE in H. pylori is not yet well understood, tfs ICE might have an impact on the fitness benefit, which helps these bacteria adapt to the gastric environment11. There is a long and complex history of acquisition, module exchange, and rearrangement of tfs ICEs within various H. pylori populations13,14. A limitation of our study was the unfinished, whole draft genome, which was fragmented into several contigs. Because of the fragmentary nature of the draft genome, it has remained refractory to the study of long chromosomal segments such as tfs ICE. It is necessary to utilize both the Illumina sequencer and the 3rd generation sequencer (PacBio, Nanopore), which generates a full-genome contig, to study the structure and location of tfs ICE in the H. pylori genome34. Secondly, our study was based on the comparison between the DNA sequences, and the expression of the T4SS gene cluster of tfs ICE at the mRNA/protein level has not yet been determined.
In summary, the acquisition of tfs3/4 ICE was common in H. pylori strains isolated from patients with gastroduodenal disease in Vietnam, and the complete cluster of tfs3 ICE was a reliable marker for the severity of diseases in the infected population.
Methods
Sample collection
A total of 136 H. pylori strains were isolated from patients with gastroduodenal diseases in Ha Noi and Ho Chi Minh, Vietnam from 2009–2017. H. pylori culture was performed as previously described22. The 109 strains were used in our previous epidemiological studies21,22,27 strains were additionally cultured in this study. All strains were isolated from patients with Kinh ethnicity and were divided into 3 disease-groups: NCGC (51 strains), DU (46 strains), and CG (39 strains). These strains had never been evaluated before to assess the status of cagPAI and tfs ICE. Local ethics approval was obtained from the Ethics Committee of Cho Ray Hospital and 108 Military Hospital, and written informed consent was obtained from all patients. The study was also approved by the Ethics Committee of Oita University Faculty of Medicine, Japan and was carried out in accordance with the Declaration of Helsinki (https://doi.org/10.1515/9783110208856.233).
DNA preparation and next-generation sequencing
The total genomic DNA of isolates were extracted using the QIAamp DNA Mini Kit (QIAGEN, UK) and quantified by Quantus Fluorometer (Promega Corporation). The DNA library was prepared using the Nextera XT DNA sample kit (Illumina, San Diego, CA, USA) allowing paired-end sequencing techniques. Short-read sequences of H. pylori were obtained from next-generation sequencing (NGS); Hiseq and Miseq platform (Illumina, Inc., San Diego, CA, USA).
Software tools for H. pylori genome analysis
The quality of raw sequencing reads was checked with FASTQC (Babraham Bioinformatics)35, filtered, and low-quality bases trimmed (< Q30) using Trimmomatic36. Trimmed paired-read sequences were subsequently de novo assembled to generate contigs using Shovill assembly35 (https://github.com/tseemann/shovill). The quality of de novo assembly included: contig numbers; total length; and N50, N75, L50, L75, and GC percentage, and the completeness of whole-genome contigs was checked by QUAST37 and BUSCO37. Pairwise sequence alignment was performed for sequence comparison between tfs3 ICE (strain Gambia94/24 and strain India7), tfs4a ICE (strain P12), tfs4b ICE (strain Shi470), tfs4c ICE (strain R036d), cagPAI (strain 26,695), and comB (strain 26,695) (https://www.ebi.ac.uk/Tools/psa/)13, which was used to make custom databases. To detect the presence of tfs ICE, cagPAI, and comB in each strain, assembled contigs were analyzed by ABRICATE (https://github.com/tseemann/abricate), a pipeline for determining virulence factor with the user’s custom databases.
Genotyping and phylogenetic analysis
Phylogeny and population assignment were constructed based on 7 house-keeping genes (atpA, efp, trpC, ppa, mutY, yphC, and ureI). Each gene sequence was retrieved and concatenated from assembled contig into FASTA format by the MLSTcheck package (https://github.com/sanger-pathogens/mlst_check/). Additionally, a total of 379 H. pylori strains with known origins available at PubMLST (http://pubmlst.org/helicobacter/), originally described by Falush et al.38, were included in the analysis. Sequences were aligned using MAFFT alignment algorithm39. The Newick tree format was generated using the neighbor-joining algorithm (Kimura-2 method) of MEGA software version 740, and a phylogenetic tree was constructed using the same software.
Statistical method
Fisher’s exact test and proportion test were used. A logistic regression model was used to calculate the odds ratio (OR) and 95% confidence interval (CI) between virulence factors and clinical outcomes. For all statistical tests, P values < 0.05 were accepted as statistically significant. Data analysis was performed with RStudio v1.1.4 (RStudio, Inc, USA).
Supplementary Information
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (26640114, 15H02657, and 16H05191) (Y.Y.), the Special Coordination Funds for Promoting Science and Technology from the MEXT of Japan (Y.Y.), and National Institutes of Health Grants DK62813 (Y.Y.). This work was also supported by the Okinawa Prefectural Government. B.H.P and V.P.T are doctoral students supported by the Japanese Government (Monbukagakusho: MEXT) Scholarship Program for 2015 and 2017, respectively.
Author contributions
Study design, B.H.P., V.P.T, J.A., and Y.Y.; Formal analysis, B.H.P, V.P.T., J.A., T.M., and Y.Y.; Funding acquisition, T.M., and Y.Y.; Investigation, H.D.Q.D., T.T.B., T.D.T., P.H.T., N.P.M.T., V.P.T., V.V.K., and T.T.H.T; Resources, H.D.Q.D., V.V.K., T.T.B., T.D.T., P.H.T., V.P.T., and N.P.M.T. Methodology, B.H.P., V.P.T., J.A., and T.M.; Writing the original draft, B.H.P., J.A., and Y.Y. All authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-83862-1.
References
- 1.Uemura N, et al. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001;345(11):784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 2.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010;7(11):629–641. doi: 10.1038/nrgastro.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer W, et al. Strain-specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res. 2010;38(18):6089–6101. doi: 10.1093/nar/gkq378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015;10(6):955–965. doi: 10.2217/fmb.15.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell. Microbiol. 2008;10(8):1573–1581. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- 7.Higashi H, et al. EPIYA motif is a membrane-targeting signal of Helicobacter pylori virulence factor CagA in mammalian cells. J. Biol. Chem. 2005;280(24):23130–23137. doi: 10.1074/jbc.M503583200. [DOI] [PubMed] [Google Scholar]
- 8.Fischer W. Assembly and molecular mode of action of the Helicobacter pylori Cag type IV secretion apparatus. FEBS J. 2011;278(8):1203–1212. doi: 10.1111/j.1742-4658.2011.08036.x. [DOI] [PubMed] [Google Scholar]
- 9.Karnholz A, et al. Functional and topological characterization of novel components of the comB DNA transformation competence system in Helicobacter pylori. J. Bacteriol. 2006;188(3):882–893. doi: 10.1128/JB.188.3.882-893.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorer MS, et al. Natural competence promotes Helicobacter pylori chronic infection. Infect. Immun. 2013;81(1):209–215. doi: 10.1128/IAI.01042-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kersulyte D, et al. Cluster of type IV secretion genes in Helicobacter pylori's plasticity zone. J. Bacteriol. 2003;185(13):3764–3772. doi: 10.1128/JB.185.13.3764-3772.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kersulyte D, et al. Helicobacter Pylori's plasticity zones are novel transposable elements. PLoS ONE. 2009;4(9):e6859. doi: 10.1371/journal.pone.0006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer W, et al. A comprehensive analysis of Helicobacter pylori plasticity zones reveals that they are integrating conjugative elements with intermediate integration specificity. BMC Genomics. 2014;15:310. doi: 10.1186/1471-2164-15-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delahay RM, Croxall NJ, Stephens AD. Phylogeographic diversity and mosaicism of the Helicobacter pylori tfs integrative and conjugative elements. Mob. DNA. 2018;9:5. doi: 10.1186/s13100-018-0109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waskito LA, et al. Distribution and clinical associations of integrating conjugative elements and cag pathogenicity islands of Helicobacter pylori in Indonesia. Sci. Rep. 2018;8(1):6073. doi: 10.1038/s41598-018-24406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Jonge R, et al. The Helicobacter pylori plasticity region locus jhp0947-jhp0949 is associated with duodenal ulcer disease and interleukin-12 production in monocyte cells. FEMS Immunol. Med. Microbiol. 2004;41(2):161–167. doi: 10.1016/j.femsim.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Sugimoto M, et al. Role of Helicobacter pylori plasticity region genes in development of gastroduodenal diseases. J. Clin. Microbiol. 2012;50(2):441–448. doi: 10.1128/JCM.00906-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H, et al. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology. 2005;128(4):833–848. doi: 10.1053/j.gastro.2005.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung SW, et al. The intact dupA cluster is a more reliable Helicobacter pylori virulence marker than dupA alone. Infect. Immun. 2012;80(1):381–387. doi: 10.1128/IAI.05472-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva B, et al. The expression of Helicobacter pylori tfs plasticity zone cluster is regulated by pH and adherence, and its composition is associated with differential gastric IL-8 secretion. Helicobacter. 2017;22(4):e12390. doi: 10.1111/hel.12390. [DOI] [PubMed] [Google Scholar]
- 21.Binh TT, et al. Advanced non-cardia gastric cancer and Helicobacter pylori infection in Vietnam. Gut Pathog. 2017;9:46. doi: 10.1186/s13099-017-0195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen TL, et al. Helicobacter pylori infection and gastroduodenal diseases in Vietnam: a cross-sectional, hospital-based study. BMC Gastroenterol. 2010;10:114. doi: 10.1186/1471-230X-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binh TT, et al. Molecular epidemiology of Helicobacter pylori infection in a minor ethnic group of Vietnam: a multiethnic, population-based study. Int. J. Mol. Sci. 2018;19(3):708. doi: 10.3390/ijms19030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida T, et al. Analysis of virulence factors of Helicobacter pylori isolated from a Vietnamese population. BMC Microbiol. 2009;9:175. doi: 10.1186/1471-2180-9-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen LT, et al. Clinical relevance of cagPAI intactness in Helicobacter pylori isolates from Vietnam. Eur. J. Clin. Microbiol. Infect. Dis. 2010;29(6):651–660. doi: 10.1007/s10096-010-0909-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi A, et al. Intact long-type dupA as a marker for gastroduodenal diseases in Okinawan subpopulation, Japan. Helicobacter. 2013;18(1):66–72. doi: 10.1111/j.1523-5378.2012.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuan VP, et al. A next-generation sequencing-based approach to identify genetic determinants of antibiotic resistance in Cambodian Helicobacter pylori clinical isolates. J. Clin. Med. 2019;8(6):858. doi: 10.3390/jcm8060858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saranathan R, et al. Helicobacter pylori infections in the Bronx, New York: surveying antibiotic susceptibility and strain lineage by whole-genome sequencing. J. Clin. Microbiol. 2020 doi: 10.1128/JCM.01591-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Argent RH, et al. The presence of dupA in Helicobacter pylori is not significantly associated with duodenal ulceration in Belgium, South Africa, China, or North America. Clin. Infect. Dis. 2007;45(9):1204–1206. doi: 10.1086/522177. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt HM, et al. The prevalence of the duodenal ulcer promoting gene (dupA) in Helicobacter pylori isolates varies by ethnic group and is not universally associated with disease development: a case-control study. Gut Pathog. 2009;1(1):5. doi: 10.1186/1757-4749-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison U, et al. Helicobacter pylori strains from a Nigerian cohort show divergent antibiotic resistance rates and a uniform pathogenicity profile. PLoS ONE. 2017;12(5):e0176454. doi: 10.1371/journal.pone.0176454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Occhialini A, et al. Distribution of open reading frames of plasticity region of strain J99 in Helicobacter pylori strains isolated from gastric carcinoma and gastritis patients in Costa Rica. Infect. Immun. 2000;68(11):6240–6249. doi: 10.1128/IAI.68.11.6240-6249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alandiyjany MN, et al. A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the Helicobacter pylori Ser/Thr kinase, CtkA. PLoS ONE. 2017;12(7):e0182144. doi: 10.1371/journal.pone.0182144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wick RR, et al. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017;13(6):e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simao FA, et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 38.Falush D, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299(5612):1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 39.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.