

Abstract
Mitragynine is the most abundant psychoactive alkaloid derived from the leaves of Mitragyna speciosa (kratom), a tropical plant indigenous to regions of Southeast Asia. Mitragynine displays a moderate affinity to opioid receptors, and kratom is often self-prescribed to treat pain and/or opioid addiction. The purpose of this study was to investigate the safety and pharmacokinetic properties of mitragynine in the dog. Single dose oral (5 mg/kg) and intravenous (0.1 mg/kg) pharmacokinetic studies of mitragynine were performed in female beagle dogs. The plasma concentrations of mitragynine were measured using ultra-performance liquid chromatography coupled with a tandem mass spectrometer, and the pharmacokinetic properties were analyzed using non-compartmental analysis. Following intravenous administration, mitragynine showed a large volume of distribution (Vd, 6.3 ± 0.6 L/kg) and high clearance (Cl, 1.8 ± 0.4 L/h/kg). Following oral mitragynine dosing, first peak plasma (Cmax, 278.0 ± 47.4 ng/mL) concentrations were observed within 0.5 h. A potent mu-opioid receptor agonist and active metabolite of mitragynine, 7-hydroxymitragynine, was also observed with a Cmax of 31.5 ± 3.3 ng/mL and a Tmax of 1.7 ± 0.6 h in orally dosed dogs while its plasma concentrations were below the lower limit of quantification (1 ng/mL) for the intravenous study. The absolute oral bioavailability of mitragynine was 69.6%. Administration of mitragynine was well tolerated, although mild sedation and anxiolytic effects were observed. These results provide the first detailed pharmacokinetic information for mitragynine in a non-rodent species (the dog) and therefore also provide significant information for allometric scaling and dose predictions when designing clinical studies.
Keywords: mitragynine, 7-hydroxymitragynine, absolute bioavailability, pharmacokinetics, kratom, Mitragyna speciosa, Rubiaceae
Introduction
Mitragyna speciosa (Korth.) Havil. (Rubiaceae), also known as kratom, is a tropical plant native to Thailand, Malaysia, and Indonesia [1,2]. In the United States, kratom is not a recognized therapy, but surveys show that it is being widely used to treat pain, mood enhancement, and to alleviate opioid withdrawal symptoms [3]. Mitragynine is the most abundant active alkaloid present in kratom and is considered to be largely responsible for its therapeutic activity [2,4]. Several studies have suggested that mitragynine and products containing mitragynine have analgesic, anti-inflammatory, antipyretic, antidiarrheal, euphoric, antidepressant, and anxiolytic effects [5–9]. Mitragynine binds to opioid (mu and kappa), alpha-adrenergic (α1A, α1B, α1D, α2A, α2B, and α2C), adenosine (A2a), dopamine (D2), and serotonin (5-HT2C, and 5-HT7) receptors [10,11]. Mitragynine is a mu-opioid receptor agonist with a moderate affinity (Ki, 161 ± 10 nM), but it is also a kappa-opioid antagonist (Ki, 198 ± 30 nM) [11]. It is a partial and biased agonist at the mu-opioid receptor with an Emax of approximately 40% that of DAMGO [(D-Ala2, N-MePhe4, Gly-ol5)-enkephalin] in assays of G-protein activation, and has almost no effect in β-arrestin-2 assays [12]. Drugs with similar mu-opioid biased effects have been suggested to produce less constipation and respiratory depression [13] than other opioids, and early work suggests this may hold true for mitragynine [9].
The United States Food and Drug Administration (FDA) warns consumers against using kratom due to possible risks of addiction, abuse, and dependence [14], although the two rodent studies that have examined mitragynineʼs abuse liability do not support the alleged high abuse potential [15,16]. Furthermore, mitragynine administration resulted in a reduction of morphine and heroin intake in rodents and therefore may be of therapeutic value for opiate addiction and withdrawal. Dependency on kratom has been described after heavy use in humans [17,18], even so, it has potential to treat opioid and methamphetamine dependency [19]. Only limited studies of mitragynine pharmacokinetics and metabolism have been conducted to date, with even less information concerning penetrance and distribution of the parent drug and metabolites into tissues [20]. There is one human pharmacokinetic study [21] that used a kratom tea rather than mitragynine, but there is no dose-ranging clinical study to examine the safety of mitragynine itself. Such studies are an essential precursor to examination of its efficacy as a treatment for opiate withdrawal symptoms, as an agonist therapy, or as a treatment for chronic pain and anxiety.
Estimation of a first-in-human dose is an essential element in the clinical development of a drug for approval by the FDA [22]. Research and development of a drug must be demonstrated in more than one animal species in order to predict human pharmacokinetics [23,24]. The objective of this study was to investigate the pharmacokinetics and bioavailability of mitragynine in healthy female beagle dogs after single oral and intravenous administration. In addition, this report outlines the pharmacokinetics of the metabolite 7-hydroxymitragynine following mitragynine ingestion. Depending on the metabolite formation and its further secondary metabolism, 7-hydroxymitragynine could represent the lionʼs share of opioid activity following mitragynine administration. However, while this has been examined in mice [25], it has not been examined in other species typically used as toxicological models during preclinical development of a clinical drug product. The dog is particularly commonly used as a “second, non-rodent” species for toxicological and pharmacokinetic studies during medication development. As such, this study provides key pharmacokinetic and safety data for mitragynine, essential information in the development of future first-in-human mitragynine dose-ranging studies.
Results and Discussion
A simple and sensitive bioanalytical method for the simultaneous quantification of mitragynine (Fig. 1) and its active metabolite, 7-hydroxymitragynine, in dog plasma was successfully developed and validated following the FDA guidelines [26]. Ultra-performance liquid chromatography coupled with a tandem mass spectrometer (UPLC-MS/MS) was used for the analysis. Initially, a method reported by Avery et al. was implemented [27] until it was found that several hydroxylated mitragynine metabolites in dog plasma eluted at the same retention time as 7-hydroxymitragynine; consequently, chromatographic conditions were modified. The developed method was linear over a concentration range of 1–200 ng/mL for both mitragynine and 7-hydroxymitragynine, and observed correlation coefficients for both analytes were always ≥ 0.99. Retention times for 7-hydroxymitragynine, mitragynine, and verapamil (internal standard, IS) were 2.8, 4.1, and 4.8 min, respectively. The lower limit of quantification (signal-to-noise ratio > 10, LLOQ) for both mitragynine and 7-hydroxymitragynine was 1 ng/mL. A gradient flow of acetonitrile and 0.1% formic acid in water on an Acquity UPLC BEH C18 column provided the optimum resolution with adequate peak shapes of the analytes and IS. The total analysis time per sample was 5.5 min. A simple, inexpensive protein precipitation method suitable for high-throughput analysis was utilized for sample cleanup and extraction of mitragynine and 7-hydroxymitragynine in biological samples. Due to the basic nature of mitragynine and 7-hydroxymitragynine [28], various combinations of methanol and acetonitrile with and without acid modifiers (formic acid, acetic acid, and trifluoroacetic acid) were attempted to quench the plasma. The best recovery along with good peak shapes for both analytes was observed when spiked dog plasma was quenched with methanol containing 0.05% formic acid. The most specific and sensitive multiple reaction monitoring (MRM) transitions of m/z 399.25 > 174.16, 415.19 > 175.14, and 455.27 > 150.10 were utilized as quantifiers for mitragynine, 7-hydroxymitragynine and the IS, respectively. Additionally, MRM transitions of m/z 399.25 > 226.18 and 415.19 > 190.10 were included in the analytical method as qualifiers for mitragynine and 7-hydroxymitragynine, respectively. Intraday and inter-day precision (%RSD) and accuracy (%bias) of the method were calculated at four different concentrations [LLOQ (1 ng/mL), low quality control (3 ng/mL, LQC), medium quality control (90 ng/mL, MQC) and high quality control (180 ng/mL, HQC), N = 6 each] on 3 different days, and both accuracy and precision of the method for mitragynine and 7-hydroxymitragynine were within the acceptable limits (within 20% for LLOQ and 15% for LQC, MQC, and HQC) (Table 1). Recovery of mitragynine and 7-hydroxymitragynine from pooled dog plasma was determined at low, medium, and high concentrations (N = 5, each), and the mean recoveries for mitragynine and 7-hydroxymitragynine were 82.5 ± 3.4 and 85.3 ± 6.7%, respectively. Mitragynine and 7-hydroxymitragynine were stable in stock solutions after 2 weeks at − 20°C. Mitragynine and 7-hydroxymitragynine were also found to be stable (± 15%) at low and high concentrations (LQC and HQC) after two freeze-thaw cycles, long-term (25 days at − 80°C), benchtop (1 h at room temperature), and autosampler stability (18 h at 10°C) experiments. Dilution integrity experiments were also performed to analyze test samples above the linearity range (5- and 10-X), and nominal values of analytes in dilution integrity quality control (DIQC) standards were within the acceptable limits (109.1 ± 4.3%). Pharmacokinetic studies of mitragynine were conducted in female beagle dogs after a single oral (5 mg/kg) and intravenous (0.1 mg/kg) administration of mitragynine. After administration, animals were observed for behavioral changes and adverse effects. No major adverse events were noted in either study, although all subjects experienced mild transient sedation immediately after dosing. Sedation lasted approximately 2 to 4 h after the oral 5 mg/kg dose and for up to 1 h following the 0.1 mg/kg intravenous dosing. Furthermore, stress-related frenetic signs such as pacing, barking, jumping, and spinning were reduced. Among the orally dosed dogs, two dogs displayed lip licking and excessive drooling, likely due to poor palatability of the oral solution. Although nausea cannot be completely ruled out, excessive drooling resolved within 30 min of dosing, and all dogs ate readily when offered food 2 h after dosing. Panting occurred transiently in one dog. No clinically significant changes in vital signs, physical examinations, or clinical laboratory tests were observed for both oral and intravenous pharmacokinetic studies. The mean complete blood counts (CBC) for oral and intravenous administrations of mitragynine that were obtained following the study remained within normal reference ranges and were similar to baseline values (Table 2). The mean biochemical parameters for oral and intravenous administrations of mitragynine obtained after completion of the study were also largely within reference ranges and similar to baseline values (Table 3). Mild hypoglycemia noted across many of the samples could likely be an artifact from delayed sample processing [29]. In an unpublished pilot study to determine optimal dosing for intravenous mitragynine, generalized hives and hyperthermia were noted in one dog at a dose of 1 mg/kg, likely a secondary effect to endogenous histamine release from mast cells known to occur with administration of various opioids [30–32]. These signs resolved after the administration of the antihistamine, diphenhydramine at 2 mg/kg as an intramuscular injection. As a result, this dog was excluded from the pharmacokinetic analysis.
Fig. 1.
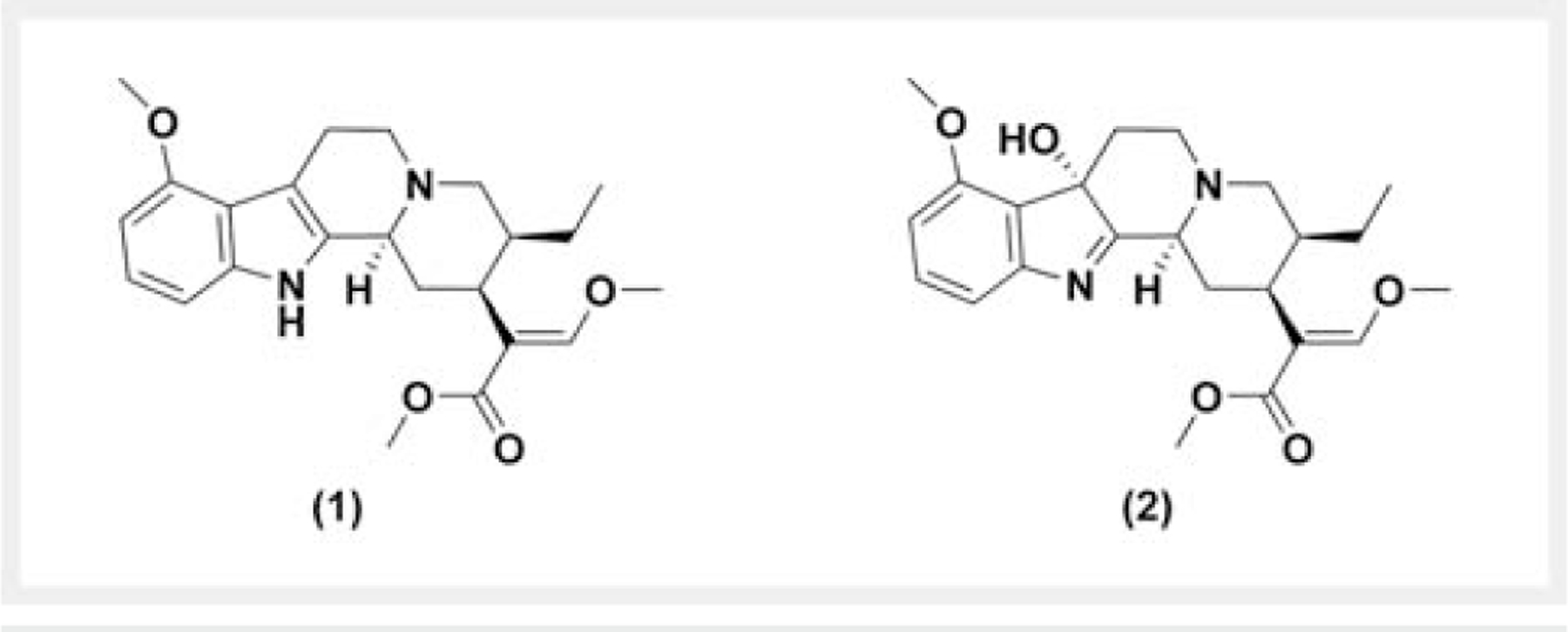
Chemical structure of (1) mitragynine and (2) 7-hydroxymitragynine.
Table 1.
Accuracy and precision for mitragynine and 7-hydroxymitragynine of the assay method in dog plasma.
| Concentration (ng/mL) | Mitragynine | 7-Hydroxymitragynine | ||||||
|---|---|---|---|---|---|---|---|---|
| Accuracy (%Bias) | Precision (%RSD) | Accuracy (%Bias) | Precision (%RSD) | |||||
| Intraday | Inter-day | Intraday | Inter-day | Intraday | Inter-day | Intraday | Inter-day | |
| 1 | 14.4 | 4.6 | 6.9 | 12.5 | 1.6 | − 1.4 | 6.8 | 11.3 |
| 3 | 5.1 | − 1.6 | 11.2 | 11.8 | 4.4 | 0.9 | 12.8 | 11.5 |
| 90 | 5.1 | 6 | 5.3 | 5.6 | 6.4 | 6.4 | 4.5 | 4.9 |
| 180 | 13.5 | 10.9 | 4.1 | 4.4 | 14.6 | 10.3 | 6.4 | 7.7 |
Table 2.
Mean complete blood count parameters before and after oral (5 mg/kg) and intravenous (0.1 mg/kg) administration of mitragynine.
| Complete blood count (Reference range) | Orala | Intravenousb | ||
|---|---|---|---|---|
| Pre-dose | Post-dose | Pre-dose | Post-dose | |
| WBC (5.0–13.0 K/uL) | 8.0 ± 1.5 | 8.1 ± 1.5 | 8.0 ± 1.5 | 8.1 ± 1.5 |
| RBC (5.7–8.3 M/uL) | 7.1 ± 0.5 | 7.1 ± 0.5 | 7.1 ± 0.5 | 7.1 ± 0.5 |
| Hct (40–56%) | 47.7 ± 2.2 | 49.2 ± 2.5 | 47.7 ± 2 | 49.2 ± 2.3 |
| MCV (64–74 fL) | 67.4 ± 1.7 | 69.0 ± 2.1 | 67.6 ± 2 | 69.0 ± 1.8 |
| Platelets 134–396 K/uL) | 356.6 ± 60.9 | 263.8 ± 26.6 | 356.6 ± 60.9 | 263.8 ± 25.8 |
| Neutrophils (2.7–8.9 K/uL) | 4.9 ± 1.9 | 5.1 ± 1.4 | 4.9 ± 1.9 | 5.1 ± 1.3 |
| Lymphocytes (0.9–3.4 K/uL) | 2.4 ± 0.4 | 2.1 ± 0.4 | 2.4 ± 0.4 | 2.1 ± 0.4 |
| Monocytes (0.1–0.8 K/uL) | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 |
| Eosinophils (0.1–1.3 K/uL) | 0.2 ± 0.2 | 0.3 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.1 |
| Basophils (0–0.1 K/mL) | 10.0 ± 20.0 | 30.0 ± 8.0 | 10.0 ± 20.0 | 30.0 ± 10.0 |
N = 5, values are the mean ± SEM;
N=4, values are themean ± SEM
Table 3.
Mean biochemistry parameters before and after oral (5mg/kg) and intravenous (0.1mg/kg) administration of mitragynine.
| Biochemistry (ref. range) | Orala | Intravenousb | ||
|---|---|---|---|---|
| Pre-dose | Post-dose | Pre-dose | Post-dose | |
| ALP (7–117U/L) | 33.6 ± 9.8 | 38.0 ± 9.9 | 33.6 ± 9.8 | 38.0 ± 9.9 |
| ALB (2.62–3.91 g/dL) | 3.0 ± 0.1 | 3.3 ± 0.2 | 3.1 ± 0.2 | 3.3 ± 0.2 |
| ALT (23–93U/L) | 24.6 ± 2.4 | 27.2 ± 2.4 | 24.6 ± 2.4 | 27.2 ± 2.4 |
| AST (16–53U/L) | 32.6 ± 6.8 | 29.4 ± 4.5 | 32.6 ± 6.8 | 29.4 ± 4.4 |
| Chol (102–340mg/dL) | 179.2 ± 27.7 | 190.8 ± 25.2 | 179.2 ± 27.7 | 190.8 ± 25.2 |
| Glob (1.8–4.0 g/dL) | 2.4 ± 0.1 | 2.54 ± 0.1 | 2.4 ± 0.1 | 2.5 ± 0.1 |
| Glucose (78–124mg/dL) | 71.3 ± 6.4 | 77.3 ± 10.7 | 71.3 ± 6.4 | 82.0 ± 9.9 |
| Magnesium(1.7–2.4mg/dL) | 2.2 ± 0.2 | 2.0 ± 0.1 | 2.2 ± 0.2 | 2.02 ± 0.1 |
| Phosphorus (2.2–4.8mg/dL) | 4.4 ± 0.2 | 4.1 ± 0.6 | 4.4 ± 0.2 | 4.1 ± 0.6 |
| Total Protein (5.0–7.4 g/dL) | 5.5 ± 0.1 | 5.8 ± 0.2 | 5.5 ± 0.1 | 5.8 ± 0.2 |
| Calcium (8.7–10.4mg/dL) | 9.0 ± 0.3 | 8.7 ± 0.3 | 9.0 ± 0.3 | 8.5 ± 0.3 |
| Sodium(141.9–150.6mEq/L) | 146.1 ± 2.1 | 147.7 ± 0.6 | 146.1 ± 2.1 | 147.7 ± 0.6 |
| Potassium(3.8–5.0mEq/L) | 4.9 ± 0.4 | 5.0 ± 0.1 | 4.9 ± 0.4 | 4.9 ± 0.1 |
| T Bilirubin (0.1–0.4mg/dL) | 0.3 ± 0.4 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 |
| Chloride (107.8–117.1mEq/L) | 110.5 ± 4.1 | 109.5 ± 1.8 | 110.5 ± 4.1 | 109.4 ± 1.9 |
| Creatinine (0.6–1.5mg/dL) | 0.9 ± 0.2 | 0.8 ± 0.1 | 0.9 ± 0.2 | 0.8 ± 0.1 |
| BUN (7–27mg/dL) | 25.0 ± 4.5 | 19.2 ± 5.2 | 25.0 ± 4.5 | 19.2 ± 5.2 |
N = 5, values are the mean ± SEM;
N=4, values are themean ± SEM
Following the oral dose of mitragynine, very fast absorption of mitragynine was observed, and the first peak plasma concentrations (Cmax) in all the animals occurred within 0.5 h post-dose (Fig. 2). During the absorption phase, a multiple peak phenomenon (Cmax1, 278.0 ± 47.4 ng/mL and Cmax2, 236.9 ± 50.2 ng/mL) was also observed (Tmax1, 0.3 ± 0.1 h and Tmax2, 1.9 ± 0.7 h) (Fig. 14S, Supporting Information). Mitragynine showed similar absorption across species as the Cmax occurred within 2 h post-dose in rats, dogs, and humans [21,27,33]. Opioid receptor-mediated delayed gastric emptying and/or absorption through multiple sites in the gastrointestinal tract could be potential causes for the multiple peaks [11,34]. Also, the insoluble compound may be solubilized by bile salts and phospholipid release in the bile, and it may be responsible for the Cmax2. The possibility of enterohepatic recirculation can be refuted due to the absence of multiple peaks in the concentration-time profile after the intravenous administration of mitragynine and the basicity of the compound [34,35]. The similarity of the gastrointestinal tract in rodents, humans, and dogs as well as comparable opioid pharmacology of mitragynine and/or 7-hydroxymitragynine might have resulted in a multiple peak phenomenon of mitragynine across these species [21,27]. Following intravenous administration, mitragynine showed a large volume of distribution (Vd, 6.3 ± 0.6 L/kg) and high clearance (Cl, 1.8 ± 0.4 L/h/kg) in dogs (Table 4). The Vd of mitragynine was greater than the total blood volume (0.09 L/kg) in dogs, indicating extravascular distribution of mitragynine [27]. Absolute oral bioavailability of mitragynine in dogs was 69.6%. The oral bioavailability of mitragynine in dogs was found to be 2.5- and 4.2-fold higher than that of the oral bioavailability of mitragynine in rats [27,33]. A CYP3A-mediated oxidative metabolite of mitragynine, 7-hydroxymitragynine, has shown potent mu-opioid (Ki, 7.2 ± 0.9 nM) activity [11,36]. Mitragynine appears to be a promising treatment for opioid self-administration without any abuse or addiction potential [15,16]. However, 7-hydroxymitragynine exhibits a 22.5-fold increase in binding affinity at the mu-opioid receptor compared to mitragynine (Ki, 161.0 ± 10.0 nM) [11], which raises concerns of opioid-mediated abuse or addiction potential. Therefore, plasma levels of 7-hydroxymitragynine were also measured in the test samples. Interestingly, the resulting concentrations of 7-hydroxymitragynine in dogs receiving intravenously administered mitragynine were always below the LLOQ (1 ng/mL). In orally dosed dogs, the percentage of the metabolite area under the curve (%AUC) ratio of 7-hydroxymitragynine to mitragynine was 12.6 ± 1.6%. Significant conversion of mitragynine to 7-hydroxymitragynine in oral study samples could be attributed to the CYP3A-mediated conversion of mitragynine in the enterocytes. Following the oral dose of mitragynine, the Cmax (31.5 ± 3.3 ng/mL) of 7-hydroxymitragynine occurred at 1.7 ± 0.6 h (Tmax) post-dose (Table 5). As previously reported, multiple oxidative metabolites of mitragynine were found in human liver microsomes, and circulating levels of five other mono-oxidative metabolites of mitragynine sharing mass transitions of 7-hydroxymitragynine were also detected in the oral study samples [36] (Fig. 15S, Supporting Information). Pharmacokinetic studies were performed in female dogs, so the estrus cycle may have an effect on the pharmacokinetics [37], but gender-related differences in the pharmacokinetics of mitragynine are not expected due to the similar expression of CYP3A/CYP3A12 in male and female dogs [38,39].
Fig. 2.
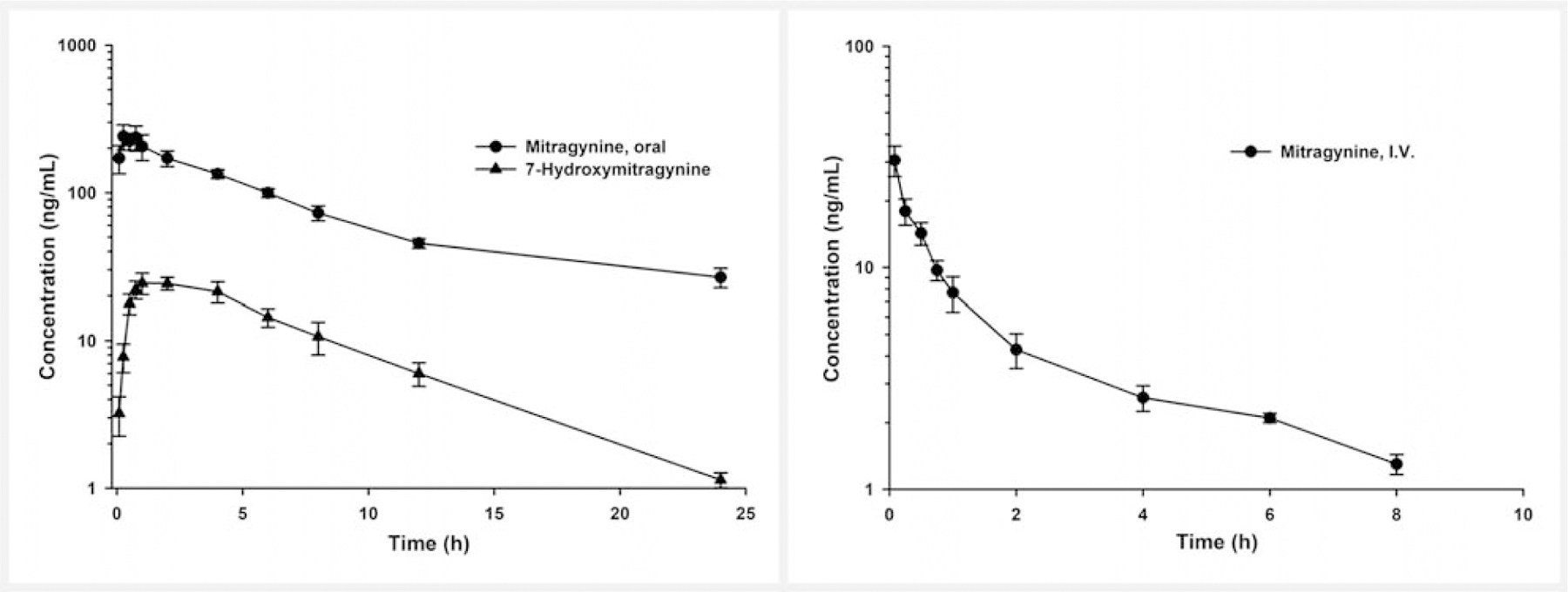
Mean plasma concentration-time profile of (right panel) mitragynine after intravenous (I.V.) administration (0.1 mg/kg, N = 4), and (left panel) mitragynine and 7-hydroxymitragynine after a single oral dose (5 mg/kg, N = 5) of mitragynine in female beagle dogs. Bars represent SEM.
Table 4.
Pharmacokinetic parameters of mitragynine after oral (5 mg/kg) and intravenous (0.1 mg/kg) administration in female beagle dogs.
| Parameter | Orala | Intravenousb | |
|---|---|---|---|
| Cmax (ng/mL) | 1 | 278.0 ± 47.4 | – |
| 2 | 236.9 ± 50.2 | – | |
| Tmax (h) | 1 | 0.3 ± 0.1 | – |
| 2 | 1.9 ± 0.7 | – | |
| AUC/Dose (h*kg*ng/mL/mg) | 443.3 ± 33.4 | 637.1 ± 135.4 | |
| Cl (L/h/kg) | – | 1.8 ± 0.4 | |
| Vd (L/kg) | – | 6.3 ± 0.6 | |
| T1/2 (h) | 8.7 ± 0.2 | 2.7 ± 0.6 | |
| Bioavailability (%) | 69.6 | – | |
N = 5, values are the mean ± SEM;
N = 4, values are the mean ± SEM. Abbreviations: AUC = area under the plasma concentration-time curve, Cl = clearance, Cmax = plasma peak concentration, T1/2 = elimination half-life, Vd = volume of distribution, Tmax = time to reach Cmax
Table 5.
Pharmacokinetic parameters of 7-hydroxymitragynine after oral (5mg/kg) dose of mitragynine in female beagle dogsa.
| Parameter | 7-Hydroxymitragynine |
|---|---|
| Cmax (ng/mL) | 31.5 ± 3.3 |
| Tmax (h) | 1.7 ± 0.6 |
| %Metabolite ratio (AUC7-hydroxymitragynine/AUCmitragynine)*100 | 12.6 ± 1.6 |
N = 5, values are themean ± SEM. Abbreviations: AUC = area under the plasma concentration-time curve, Cmax = plasma peak concentration, Tmax = time to reach Cmax
Mitragynine is metabolized in human liver microsomes predominantly by cytochrome P450 (CYP) 3A4 with minor contributions from CYP2D6 and CYP2C9 [36]. Dogs are the closest species to humans in terms of total liver CYP content and CYP3A4-mediated hydroxylase activity [40–42]. The dog CYP3A12 isoform expressed in both the liver and intestine also showed substrate specificity to human CYP3A4 [42]. Therefore, pharmacokinetic parameters derived from the dog study could be allometrically scaled for its pharmaceutical development in humans for the treatment of pain and opioid withdrawal symptoms.
Materials and Methods
Reagents and chemicals
Mitragynine (as mitragynine sulfate salt; purity ≥ 98%) was isolated and purified from a kratom alkaloid-enriched extract (Choice Organics) as described in our earlier published papers [27,43]. 7-Hydroxymitragynine (purity ≥ 98%) was obtained through mitragynine semi-synthesis as previously reported [43]. Purity and structural characterization of mitragynine, mitragynine sulfate, and 7-hydroxymitragynine were established by proton nuclear magnetic resonance spectroscopy (1H NMR), carbon (13C) NMR, 2D NMR, ultrahigh-performance liquid chromatography-photodiode array detection (UHPLC-PDA), and liquid chromatography high-resolution quadrupole time of flight mass spectrometry (LC-Q-TOF) [43]. LC-MS grade acetonitrile, methanol, formic acid, and water were procured from Fisher Scientific. Verapamil (purity ≥ 98%, IS) was purchased from Sigma-Aldrich. Canine plasma was obtained from Innovative Research, Inc.
UPLC-MS/MS method for the simultaneous quantification of mitragynine and 7-hydroxymitragynine in dog plasma
A UPLC-MS/MS method reported by Avery et al. for the quantification of mitragynine in rat plasma was modified for the simultaneous quantification of mitragynine and 7-hydroxymitragynine in dog plasma [27]. The developed method was validated following the FDA Guidelines for the Bioanalytical Method Validation [26]. Bioanalysis was performed on a Waters Acquity UPLC I-Class Plus coupled with a Waters Xevo TQ-S Micro triple-quadrupole mass spectrometer. An MRM-based mass spectrometric detection of mitragynine and 7-hydroxymitragynine was achieved using the positive electrospray ionization mode. Collision-induced dissociation of precursor ions (m/z) to product ion-based (m/z) ion transitions m/z 399.25 > 174.16, 415.19 > 175.14, and 455.27 > 150.10, applying collision energy 32, 46, and 42 V, were used to perform the analysis of mitragynine, 7-hydroxymitragynine, and the IS, respectively. The IS was selected on the basis of comparative physicochemical properties, and ionization efficiency similar to that of mitragynine, and 7-hydroxymitragynine. Cone voltages for mitragynine, 7-hydroxymitragynine, and the IS were held at 60, 42, and 28 V, respectively. Capillary voltage, cone gas flow, desolvation gas flow, desolvation temperature, and source temperature were set to 0.5 V, 50 L/h, 900 L/h, 450°C, and 150°C, respectively. Nitrogen was used as the cone and desolvation gas, while argon was utilized as the collision gas.
Chromatographic separations of mitragynine and 7-hydroxymitragynine were achieved on a Waters Acquity UPLC BEH C18 column (1.7 µm, 2.1 × 50 mm) using a gradient method. Column and autosampler temperatures were maintained at 55 and 10°C, respectively. The mobile phase consisted of 0.1% formic acid in water (A solvent) and acetonitrile (B solvent), and the flow rate was set to 0.35 mL/min. The gradient was started with pump A supplying 80% until 0.5 min, and the composition of the A solvent in the mobile phase was linearly decreased to 68 and 62% reaching 2.2 and 3.5 min, respectively. The composition of component A in the mobile phase was further increased to 80% by 3.6 min, and was maintained until 5.5 min. Compositions and volumes of weak and strong needle washes were implemented from the method of Sharma et al. [43]. A simple and fast protein precipitation method was used for the removal of endogenous substances and extraction of mitragynine and 7-hydroxymitragynine from dog plasma. Plasma samples (25 µL) were quenched with 100 µL of methanol containing 0.05% formic acid and the IS (10 ng/mL). Quenched samples were vortex mixed and filtered through a 0.45 micron 96-well filtration plate by centrifugation (2000 rpm, 5 min) at 4°C. The filtrates were transferred to the autosampler and analyzed for mitragynine and 7-hydroxymitragynine content using the UPLC-MS/MS method. An eight-point (1, 2.5, 5, 20, 50, 100, 150, and 200 ng/mL) calibration curve for both mitragynine and 7-hydroxymitragynine along with the four concentrations of quality control standards [1 (LLOQ), 3 (LQC), 90 (MQC) and 180 ng/mL (HQC)] was used during the analysis and the validation. The acquisition of the UPLC-MS/MS system was controlled by MassLynx XS software version 4.2 and the data was processed using TargetLynx XS. The developed method was validated according to the FDA guidelines for selectivity, accuracy, precision, recovery, dilution integrity, matrix effect, linearity, and stability. The validated method was further implemented for the analysis of oral and intravenous pharmacokinetic study samples.
Pharmacokinetic studies of mitragynine in dogs
Single dose oral (5 mg/kg, N = 5) and intravenous (0.1 mg/kg, N = 4) pharmacokinetic studies of mitragynine were performed in female beagle dogs. The study was approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (protocol number: 201910731, dated July 11, 2019) at the University of Florida. Healthy adult female, purpose-bred research beagles, born in January 2017 (age 2.7 years), weighing 8.8–10.3 kg (mean 9.2 kg) were housed in a temperature- and light-controlled environment. All dogs were fasted overnight and were fed 2 h post-dose. Prior to the start of each part of the study, a central line was placed in the right or left jugular vein of each dog. Heparinized saline was used to maintain the patency of the cannula. CBC and serum biochemical panels were performed prior to the study and at the end of the study. Hematologic abnormalities were graded in accordance with the VCOG Common Terminology Criteria for Adverse Events (VCOG-CTCAE) v 1.0.14 [44]. Clinical monitoring of vital signs (mentation, temperature, heart rate, and respiratory rate) began 5 min after the administration of mitragynine. General health was assessed throughout the study period, including food and water consumption, elimination, activity level, and subjective clinical observations of attitude, level of consciousness, and hydration.
A crossover study design was implemented for the pharmacokinetic studies. The oral pharmacokinetic study was performed first, and the intravenous study was conducted after a washout period of 3 weeks. The first dog oral dose of mitragynine was derived from a clinical study performed following the oral doses of kratom tea in Thailand [21]. As recommended by the FDA, a dog equivalent dose was calculated first as: human equivalent dose * (average human weight/average dog weight)0.33 [22]. The human equivalent dose, average human weight, and average dog weight were 0.3 mg/kg, 60 kg, and 10 kg, respectively. A tenfold of the dog equivalent dose of mitragynine (5 mg/kg) was administered to compensate for the pharmacological activity mediated by other kratom alkaloids. The formulations for the oral and intravenous pharmacokinetic studies were prepared by dissolving mitragynine sulfate in water and saline, respectively. The formulation for the intravenous study was filtered through a sterile 0.2 µm syringe filter prior to the administration. Mitragynine content from the sulfate salt in formulations was normalized to the free base equivalent, and both formulations were analyzed for mitragynine content after the study. Mitragynine was dosed orally to five dogs, and blood samples (200 µL) were collected from an indwelling catheter (IVC) at pre-dose, and 0.083, 0.25, 0.50, 0.75, 1, 2, 4, 6, 8, 12, and 24 h post-dose. For the intravenous study, a solution formulation was administered to four dogs, and blood samples (200 µL) were collected from IVC at pre-dose, and 0.083, 0.25, 0.50, 0.75, 1, 2, 4, 6, 8, and 12 h post-dose. Blood samples were collected in labeled heparinized tubes, and plasma was harvested after centrifugation at 1500 rpm for 10 min. The collected plasma samples were stored at − 80°C until analysis using the UPLC-MS/MS method.
Pharmacokinetics analysis
Plasma concentration-time data of both mitragynine (oral and intravenous) and 7-hydroxymitragynine (oral only) was subjected to non-compartmental analysis using Phoenix, version 7.1. The Cmax and Tmax for mitragynine and 7-hydroxymitragynine for the oral study were directly observed from the concentration-time profile. The AUC for mitragynine and 7-hydroxymitragynine were calculated using the linear trapezoidal method. The absolute bioavailability (F) of mitragynine in dogs was calculated using the following equation: F = [(AUC/Dose)oral/(AUC/Dose)intravenous]*100. In vivo exposure of 7-hydroxymitragynine formed from the metabolism of mitragynine was calculated as %Metabolite Ratio = (AUC7-hydroxymitragynine/AUCmitragynine)*100.
Supplementary Material
Acknowledgements
This study was supported by UG3 DA048353 and R01 DA047855 grants from the National Institute on Drug Abuse and the University of Florida Clinical and Translational Science Institute, which is supported in part by the NIH National Center for Advancing Translational Sciences under award number UL1TR001427. We would like to thank Dr. Aidan Hampson, who was the NIDA Scientific Officer on the award number UG3DA048353 and was substantially involved in the studies described in this report. He had no substantial involvement in the other cited grants. Additionally, the authors would like to thank Ms. Paige Maxwell for providing technical support.
Footnotes
Supporting information available online at http://www.thieme-connect.de/products
Supporting information
Isolation of mitragynine, synthesis of 7-hydroxymitragynine, 1H NMR, 13C NMR, 2D HSQC, and UHPLC-PDA-Q-TOF spectrometric analysis, representative chromatograms of mitragynine and 7-hydroxymitragynine, and concentration-time profiles for the oral study are available as Supporting Information.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- [1].Adkins JE, Boyer EW, McCurdy CR. Mitragyna speciosa, a psychoactive tree from Southeast Asia with opioid activity. Curr Top Med Chem 2011; 11: 1165–1175 [DOI] [PubMed] [Google Scholar]
- [2].Jansen KL, Prast CJ. Ethnopharmacology of kratom and the Mitragyna alkaloids. J Ethnopharmacol 1988; 23: 115–119 [DOI] [PubMed] [Google Scholar]
- [3].Prozialeck WC, Avery BA, Boyer EW, Grundmann O, Henningfield JE, Kruegel AC, McMahon LR, McCurdy CR, Swogger MT, Veltri CA, Singh D. Kratom policy: The challenge of balancing therapeutic potential with public safety. Int J Drug Policy 2019; 70: 70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Takayama H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem Pharm Bull 2004; 52: 916–928 [DOI] [PubMed] [Google Scholar]
- [5].Hazim AI, Ramanathan S, Parthasarathy S, Muzaimi M, Mansor SM. Anxiolytic-like effects of mitragynine in the open-field and elevated plus-maze tests in rats. J Physiol Sci 2014; 64: 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mossadeq WS, Sulaiman M, Mohamad TT, Chiong H, Zakaria Z, Jabit M, Baharuldin M, Israf D. Anti-inflammatory and antinociceptive effects of Mitragyna speciosa Korth methanolic extract. Med Princ Pract 2009; 18: 378–384 [DOI] [PubMed] [Google Scholar]
- [7].Idayu NF, Hidayat MT, Moklas M, Sharida F, Raudzah AN, Shamima A, Apryani E. Antidepressant-like effect of mitragynine isolated from Mitragyna speciosa Korth in mice model of depression. Phytomedicine 2011; 18: 402–407 [DOI] [PubMed] [Google Scholar]
- [8].McWhirter L, Morris S. A case report of inpatient detoxification after kratom (Mitragyna speciosa) dependence. Eur Addict Res 2010; 16: 229–231 [DOI] [PubMed] [Google Scholar]
- [9].Macko E, Weisbach J, Douglas B. Some observations on the pharmacology of mitragynine. Arch Int Pharmacodyn Ther 1972; 198: 145. [PubMed] [Google Scholar]
- [10].Prozialeck WC, Jivan JK, Andurkar SV. Pharmacology of kratom: an emerging botanical agent with stimulant, analgesic and opioid-like effects. J Am Osteopath Assoc 2012; 112: 792–799 [PubMed] [Google Scholar]
- [11].Obeng S, Kamble SH, Reeves ME, Restrepo LF, Patel A, Behnke M, Chear N, Ramanathan S, Sharma A, Leon F. Investigation of the adrenergic and opioid binding affinities, metabolic stability, plasma protein binding properties and functional effects of selected indole-based kratom alkaloids. J Med Chem 2020; 63: 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Váradi A, Marrone GF, Palmer TC, Narayan A, Szabó MR, Le Rouzic V, Grinnell SG, Subrath JJ, Warner E, Kalra S. Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit β-arrestin-2. J Med Chem 2016; 59: 8381–8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Madariaga-Mazon A, Marmolejo-Valencia AF, Li Y, Toll L, Houghten RA, Martinez-Mayorga K. Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discov Today 2017; 22: 1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Food and Drug Administration. FDA and Kratom. Available at https://www.fda.gov/news-events/public-health-focus/fda-and-kratom. Accessed March 31, 2020
- [15].Yue K, Kopajtic TA, Katz JL. Abuse liability of mitragynine assessed with a self-administration procedure in rats. Psychopharmacol 2018; 235: 2823–2829 [DOI] [PubMed] [Google Scholar]
- [16].Hemby SE, McIntosh S, Leon F, Cutler SJ, McCurdy CR. Abuse liability and therapeutic potential of the Mitragyna speciosa (kratom) alkaloids mitragynine and 7-hydroxymitragynine. Addict Biol 2019; 24: 874–885 [DOI] [PubMed] [Google Scholar]
- [17].Singh D, Muller CP, Vicknasingam BK. Kratom (Mitragyna speciosa) dependence, withdrawal symptoms and craving in regular users. Drug Alcohol Depend 2014; 139: 132–137 [DOI] [PubMed] [Google Scholar]
- [18].Singh D, Muller CP, Vicknasingam BK, Mansor SM. Social functioning of kratom (Mitragyna speciosa) users in Malaysia. J Psychoactive Drugs 2015; 47: 125–131 [DOI] [PubMed] [Google Scholar]
- [19].Singh D, Yeou Chear NJ, Narayanan S, Leon F, Sharma A, McCurdy CR, Avery BA, Balasingam V. Patterns and reasons for kratom (Mitragyna speciosa) use among current and former opioid poly-drug users. J Ethnopharmacol 2020; 249: 112462. [DOI] [PubMed] [Google Scholar]
- [20].Ya K, Tangamornsuksan W, Scholfield CN, Methaneethorn J, Lohitnavy M. Pharmacokinetics of mitragynine, a major analgesic alkaloid in kratom (Mitragyna speciosa): A systematic review. Asian J Psychiatr 2019; 43: 73–82 [DOI] [PubMed] [Google Scholar]
- [21].Trakulsrichai S, Sathirakul K, Auparakkitanon S, Krongvorakul J, Sueajai J, Noumjad N, Sukasem C, Wananukul W. Pharmacokinetics of mitragynine in man. Drug Des Devel Ther 2015; 9: 2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Food and Drug Administration. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthyvolunteers. Accessed March 31, 2020
- [23].Nair A, Morsy MA, Jacob S. Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Develop Res 2018; 79: 373–382 [DOI] [PubMed] [Google Scholar]
- [24].Food and Drug Administration. Product development under the animal rule: guidance for industry. Available at https://www.fda.gov/media/88625/download. Accessed March 31, 2020
- [25].Kruegel AC, Uprety R, Grinnell SG, Langreck C, Pekarskaya EA, Le Rouzic V, Ansonoff M, Gassaway MM, Pintar JE, Pasternak GW, Javitch JA, Majumdar S, Sames D. 7-Hydroxymitragynine is an active metabolite of mitragynine and a key mediator of its analgesic effects. ACS Cent Sci 2019; 5: 992–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Food and Drug Administration. Bioanalytical method validation guidance for industry. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry. Accessed March 31, 2020
- [27].Avery BA, Boddu SP, Sharma A, Furr EB, Leon F, Cutler SJ, McCurdy CR. Comparative pharmacokinetics of mitragynine after oral administration of Mitragyna speciosa (Kratom) leaf extracts in rats. Planta Med 2019; 85: 340–346 [DOI] [PubMed] [Google Scholar]
- [28].ChemAxon. Marvin 17.14.0. Available at https://www.chemaxon.com. Accessed March 31, 2020
- [29].Gilor S, Gilor C. Common laboratory artifacts caused by inappropriate sample collection and transport: how to get the most out of a sample. Top Companion Anim Med 2011; 26: 109–118 [DOI] [PubMed] [Google Scholar]
- [30].Casale TB, Bowman S, Kaliner M. Induction of human cutaneous mast cell degranulation by opiates and endogenous opioid peptides: evidence for opiate and nonopiate receptor participation. J Allergy Clin Immunol 1984; 73: 775–781 [DOI] [PubMed] [Google Scholar]
- [31].Saljoughian M Opioids: allergy vs. pseudoallergy. US Pharm 2006; 7: HS-5–HS-9 [Google Scholar]
- [32].Ennis M, Schneider C, Nehring E, Lorenz W. Histamine release induced by opioid analgesics: a comparative study using porcine mast cells. Agents Actions 1991; 33: 20–22 [DOI] [PubMed] [Google Scholar]
- [33].Kruegel AC, Grundmann O. The medicinal chemistry and neuropharmacology of kratom: A preliminary discussion of a promising medicinal plant and analysis of its potential for abuse. Neuropharmacol 2018; 134: 108–120 [DOI] [PubMed] [Google Scholar]
- [34].Jaiswal S, Shukla M, Sharma A, Rangaraj N, Vaghasiya K, Malik MY, Lal J. Preclinical pharmacokinetics and ADME characterization of a novel anti-cancer chalcone, cardamonin. Drug Test Anal 2017; 9: 1124–1136 [DOI] [PubMed] [Google Scholar]
- [35].Godfrey KR, Arundel PA, Dong Z, Bryant R. Modelling the double peak phenomenon in pharmacokinetics. Comput Methods Programs Biomed 2011; 104: 62–69 [DOI] [PubMed] [Google Scholar]
- [36].Kamble SH, Sharma A, King TI, León F, McCurdy CR, Avery BA. Metabolite profiling and identification of enzymes responsible for the metabolism of mitragynine, the major alkaloid of Mitragyna speciosa (kratom). Xenobiotica 2019; 49: 1279–1288 [DOI] [PubMed] [Google Scholar]
- [37].Kulkarni KH, Yang Z, Niu T, Hu M. Effects of estrogen and estrus cycle on pharmacokinetics, absorption, and disposition of genistein in female Sprague-Dawley rats. J Agric Food Chem 2012; 60: 7949–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Martinez SE, Shi J, Zhu HJ, Jimenez TEP, Zhu Z. Absolute quantitation of drug-metabolizing cytochrome p450 enzymes and accessory proteins in dog liver microsomes using label-free standard-free analysis reveals interbreed variability. Drug Metab Dispos 2019; 47: 1314–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nishibe Y, Wakabayashi M, Harauchi T, Ohno K. Characterization of cytochrome P450 (CYP3A12) induction by rifampicin in dog liver. Xenobiotica 1998; 28: 549–557 [DOI] [PubMed] [Google Scholar]
- [40].Court MH. Canine cytochrome P450 (CYP) pharmacogenetics. Vet Clin North Am Small Anim Pract 2013; 43: 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2006; 2: 875–894 [DOI] [PubMed] [Google Scholar]
- [42].Pasanen M Species Differences in CYP Enzymes. In: Angosto MC, Gomez-Lechon MJ, eds. Citocromo P450. Madrid: Instituto De Espana, Real Academia Nacional De Farmacia Madrid; 2004: 3–90 [Google Scholar]
- [43].Sharma A, Kamble SH, Leon F, Chear NJ, King TI, Berthold EC, Ramanathan S, McCurdy CR, Avery BA. Simultaneous quantification of ten key Kratom alkaloids in Mitragyna speciosa leaf extracts and commercial products by ultra-performance liquid chromatography-tandem mass spectrometry. Drug Test Anal 2019; 11: 1162–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Veterinary cooperative oncology group – common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1. Vet Comp Oncol 2016; 14: 417–446 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.