

Abstract
X-linked agammaglobulinemia (XLA) is an immune disorder caused by mutations in Bruton’s tyrosine kinase (BTK). BTK is expressed in B and myeloid cells, and its deficiency results in a lack of mature B cells and protective antibodies. We previously reported a lentivirus (LV) BTK replacement therapy that restored B cell development and function in Btk and Tec double knockout mice (a phenocopy of human XLA). In this study, with the goal of optimizing both the level and lineage specificity of BTK expression, we generated LV incorporating the proximal human BTK promoter. Hematopoietic stem cells from Btk−/−Tec−/− mice transduced with this vector rescued lineage-specific expression and restored B cell function in Btk−/−Tec−/− recipients. Next, we tested addition of candidate enhancers and/or ubiquitous chromatin opening elements (UCOEs), as well as codon optimization to improve BTK expression. An Eμ enhancer improved B cell rescue, but increased immunoglobulin G (IgG) autoantibodies. Addition of the UCOE avoided autoantibody generation while improving B cell development and function and reducing vector silencing. An optimized vector containing a truncated UCOE upstream of the BTK promoter and codon-optimized BTK cDNA resulted in stable, lineage-regulated BTK expression that mirrored endogenous BTK, making it a strong candidate for XLA therapy.
Keywords: gene therapy, XLA, UCOE, lentivirus, DNase hypersensitive sites, codon optimization, primary immunodeficiency, hematopoietic stem cells, Bruton's tyrosine kinase, BTK, X-linked Agammaglobulinemia
Graphical Abstract
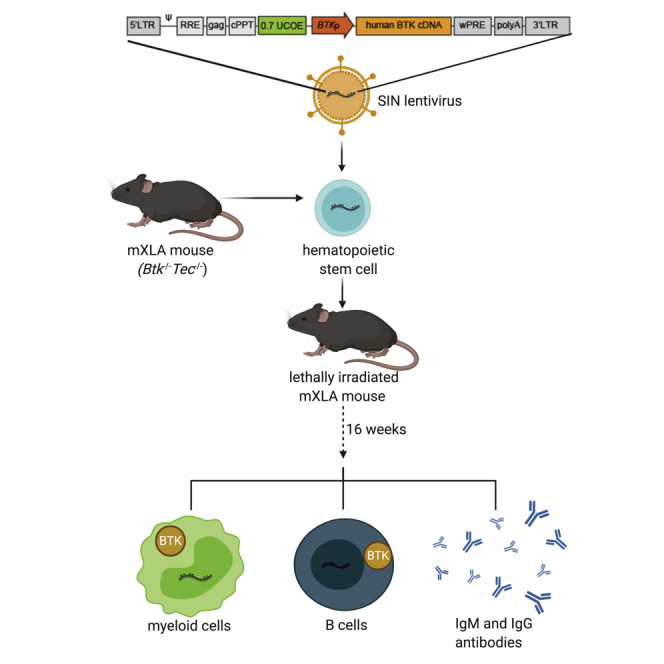
Mutations in BTK lead to XLA characterized by absent B cells and antibody responses. LVs containing the BTK promoter and other candidate elements were tested in a murine XLA model. Use of a 0.7-kb UCOE and codon optimization restored B cell development, antibody responses, and lineage-appropriate expression without evidence of toxicity.
Introduction
X-linked agammaglobulinemia (XLA) is a rare primary immunodeficiency disorder affecting approximately 1 in 100,000 males. The disease is characterized by a block in early B cell development that results in a scarcity of mature B cells and serum antibodies. Patients experience sepsis and/or chronic bacterial infections early in life and usually die during childhood if they do not receive treatment. Currently, XLA is managed by life-long administration of pooled human immunoglobulin (Ig), which has dramatically improved the prognosis of this disorder. Even with treatment, however, patients have a reduced lifespan and suffer frequent and serious health complications, including chronic sinopulmonary disease, inflammatory bowel disease, skin infections, malignancy, and encephalomyelitis.1, 2, 3, 4, 5, 6, 7, 8, 9
XLA results from mutations in the gene Bruton’s tyrosine kinase (BTK). The BTK protein is a non-receptor tyrosine kinase that is a critical component of the precursor (pre-)B and mature B cell receptor (BCR) signaling cascades.10 B cells first express the pre-BCR, with the surrogate light chain, at the pre-B cell stage of development in the bone marrow (BM). The survival and proliferation of B cells that express pre-BCRs or mature BCRs is tightly dependent on tonic BCR signals. BTK deficiency in XLA therefore results in an almost complete block in BM B cell development at the pre-B cell stage, and XLA patients have few peripheral immature and mature B cells. B cells that are in the periphery are largely non-functional, as BTK is also required for the activation of mature B cells in response to BCR engagement.11 BTK is also expressed in all myeloid cell subsets, where it has been shown to function in diverse pathways, including mast cell activation,12,13 osteoclast differentiation,14,15 myeloid cell development,16,17 macrophage survival,18 neutrophil migration,19 production of nitric oxide and reactive oxygen intermediates,17,20,21 and cytokine secretion.22, 23, 24, 25
Gene replacement therapy using a self-inactivating lentivirus (SIN-LV) to deliver the BTK cDNA into autologous hematopoietic stem cells (HSCs) represents a potentially curative treatment option for XLA. Related therapies have demonstrated improved immune function in pediatric and adult subjects with other primary immunodeficiency diseases, including X-linked severe combined immunodeficiency (X-SCID; interleukin [IL]-2 receptor gamma deficiency),26 chronic granulomatous disease (CGD; gp91phox deficiency),27 adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID),28 and Wiskott-Aldrich syndrome (WAS; WAS protein [WASp] deficiency).29,30 XLA is an appealing candidate disease for a gene replacement therapy because gene-corrected BTK+ B cells are predicted to exhibit a selective advantage during early B cell development. This concept is supported by previous studies of non-random X-inactivation in female carrier mice and humans,11,31 and by using limiting HSC transfer experiments in XID mice.32,33 These data suggest that as few as 1%–5% of modified HSCs might be sufficient for restoration of the B cell compartment. Furthermore, because the selective advantage in the BTK+ B cell compartment is not dependent on concentrations of circulating Ig, conventional antibody replacement therapy can be maintained during gene therapy treatment.
We have previously demonstrated restoration of B cell development and function using both gammaretrovirus (γRV) and SIN-LV to replace BTK in ex vivo gene therapy of Btk−/−Tec−/− mice.34,35 The Btk−/−Tec−/− strain mimics XLA phenotypes, including the block in B cell development at the pre-B cell stage;36 for brevity, this strain is denoted herein as murine XLA (mXLA). Note that TEC, a closely related protein to BTK, partially compensates for the loss of BTK in mice, but not in humans. Therefore, knockout of both Tec and Btk expression is required to model human XLA, without discernable additional defects to immune or other tissues. Our previously described SIN-LV used the Ig heavy chain mu gene (IGHM) transcriptional enhancer Eμ37,38 with the CD79B minimal promoter (B29)39 upstream of a human BTK cDNA. Although the Eμ.B29 LV rescued B cell development and function of mXLA mice after ex vivo gene therapy, expression of BTK was confined to the B cell lineage and was absent in myeloid cells. Additionally, we detected evidence of epigenetic silencing following secondary transplantation.35
While the use of SIN-LV has greatly reduced the risk of insertional mutagenesis compared to γRV-based gene transfer vectors, LV long terminal repeats (LTRs; such as γRV LTRs) and their internal promoters are subject to methylation and silencing.40 In addition, following viral integration, transgene expression varies depending on the LV insertion site, a feature referred to as position effect variegation (PEV).41,42 Silencing and PEV are both likely to result in reduced efficacy or potential long-term failure of gene replacement therapy, as illustrated in the extreme in a γRV gene therapy trial for X-liked CGD (X-CGD).43,44 Recently, ubiquitous chromatin opening elements (UCOEs) have been used to prevent methylation and epigenetic silencing, improving transgene expression and stability (as reviewed45). UCOEs are enhancer-less, methylation-resistant CpG islands that have been identified in some bi-directional gene pairs.46 The most well-characterized UCOE (A2UCOE) comprises the closely spaced promoter regions of the differentially expressed housekeeping genes HNRNPA2B1 and CBX3. When inserted upstream of a transgenic expression cassette, the A2UCOE limits methylation of the transgene in cell lines and mouse HSCs, conferring both stable protein expression and minimizing PEV.47,48 Fragments of this UCOE have been shown to be effective in preclinical “LV gene therapy” (LV-GT) studies,47,49,50 as well as in protein producer cell lines.51 Importantly, the A2UCOE does not appear to interfere with the tissue-specific expression profile of associated promoter elements.50 Taken together, these features make the A2UCOE a potentially useful addition to gene therapy vectors, where restriction of transgene expression to selected lineages is critical.
In the current study, we sought to identify genetic elements that could improve the expression profile of BTK delivered by SIN-LV for XLA therapy, including reconstitution of BTK expression to the relevant hematopoietic cell lineages (including both B and myeloid cells) and achieving stable, lasting BTK expression in these lineages. We compared a range of vector elements including candidate enhancers (Eμ versus BTK), alternative UCOE elements, as well as codon optimization of BTK cDNA in combination with the 788-bp BTK minimal promoter (BTKp)52 as the LV internal promoter.
Results
LVs containing the human BTK promoter restore the lineage specificity of BTK expression
We previously reported rescue of BTK expression in B cells, but not myeloid cells, using an LV construct containing a B cell-specific internal enhancer-promoter (Eμ.B29).35 Later, we found that switching the B29 promoter for the BTKp improved expression of a GFP reporter cDNA in myeloid cells originating from vector-treated human and murine HSCs; the BTKp on its own drove low amounts of GFP expression in both B and myeloid cells.53 Our first step here was to evaluate candidate LVs in the mXLA model, first comparing LV with the Eμ enhancer and either the B29 or BTK promoters driving human BTK cDNA expression (Eμ.B29.BTK or Eμ.BTKp.BTK; Figure S1A). We transduced lineage-depleted mXLA BM cells with candidate LVs followed by transplantation into myeloablated mXLA recipient mice (we refer to this procedure hereafter as LV-GT). Mice were immunized with nitrophenol-conjugated chicken γ-globulin (NP-CGG) at 12 and 16 weeks post-transplant. At 25–30 weeks post-transplant, we analyzed cells from the BM, spleen (SP), and peritoneal fluid (PF) by flow cytometry for surface markers and intracellular BTK expression. Mice transplanted with Eμ.BTKp.BTK LV-GT had BTK expression patterns closely resembling those of wild-type (WT) mice, with BTK expression in B and myeloid cells, but not in T cells (Figures S1B and S1C). As previously observed, BTK was expressed only in the B cell compartment in mice receiving Eμ.B29.BTK LV-GT.
UCOE inclusion improves BTK expression, B cell counts, and serum Ig production
We next directly compared the LV Eμ.BTKp.BTK and BTKp.BTK with a series of LVs that were designed to increase transcription from the BTKp (Figure 1A; the vector backbone is identical to that shown in Figure S1A). The first candidate LV added a 1.5-kb A2UCOE47 upstream of the BTKp (1.5UCOE.BTKp.BTK); various configurations of this UCOE have been shown to reduce methylation of integrated proviral sequences.45,48,49,54 We also included a version of the 1.5UCOE.BTKp.BTK construct that used a human BTK cDNA codon optimized for both human and mouse expression (1.5UCOE.BTKp.coBTK). Initial testing using GFP-co-expressing constructs (Figure S2A) showed expression and BCR signaling using codon optimization in BTK−/− chicken DT40 B cells (Figures S2B and S2C), suggesting the production of functional BTK protein.
Figure 1.

Comparison of the rescue of BTK expression and B cell development using alternative LV constructs in mXLA gene therapy
(A) SIN-LVs were used to express human BTK cDNA driven by the BTK promoter (BTKp) (top). Variations of the vector contain a 1.5-kb ubiquitous chromatin opening element (1.5UCOE), Eμ enhancer, and/or codon-optimized human BTK cDNA (coBTK). (B and C) Bone marrow (BM), spleen (SP), and peritoneal fluid (PF) from LV gene therapy (LV-GT)-treated mice were analyzed 25–30 weeks post-transplant, and BTK expression in B cells (B) and myeloid cells (C) was determined by flow cytometry. Data represent mean ± SEM from 10 independent experiments; n = 14 (WT mock), 12 (mXLA mock), 5 (BTKp.BTK), 24 (1.5UCOE.BTKp.BTK), 18 (1.5UCOE.BTKp.coBTK), and 21 (Eμ.BTKp.BTK). (D–F) B cells (B220+) in BM, SP, and PF were stained for surface markers characterizing B cell subsets shown as number per mouse (SP and BM) or percentage of live lymphocytes (PF): (D) early B cell development (BM), (E) late B cell development (SP), and (F) peritoneal cavity B cells. Data represent mean ± SEM from 10 independent experiments with n (BM, SP, PF) = 11, 13, 14 (WT mock); 10, 11, 10 (mXLA mock); 5, 5, 5 (BTKp.BTK); 20, 21, 22 (1.5UCOE.BTKp.BTK); 14, 14, 17 (1.5UCOE.BTKp.coBTK); and 18, 19, 20 (Eμ.BTKp.BTK). (G and H) Total serum IgG (G) and IgM (H) in mice receiving LV-GT. Mice were immunized with NP-CGG 12 and 16 weeks post-transplant, and serum was collected 20 weeks post-transplant. Antibodies were measured by ELISA and quantified relative to a standard curve. Data represent mean ± SEM from four independent experiments: n = 9 (WT mock), 10 (mXLA mock), 2 (BTKp.BTK), 19 (1.5UCOE.BTKp.BTK), 14 (1.5UCOE.BTKp. coBTK), and 16 (Eμ.BTKp.BTK). p values were determined using the one-way ANOVA with Sidak’s correction for multiple comparisons. Statistically significant differences between experimental and mXLA mock cohorts are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
To evaluate the utility of these LV vectors in the mXLA model, we analyzed BTK expression in lymphoid tissues in 10 independent LV-GT experiments. Of note, intracellular BTK staining of cells from mXLA LV-GT recipients displayed population shifts in mean fluorescence intensity (MFI) rather than distinct BTK-positive and BTK-negative staining populations (except for Eμ.BTKp.BTK LV). This made comparison difficult because measures of vector marking (% of cells that are BTK+) did not accurately reflect the proportion of cells that were positive for the vector (Figure S3A). Of the four LV cohorts, Eμ.BTKp.BTK had the highest proportion of B cells expressing BTK in the BM, SP, and peritoneum (34%, 64%, and 65% BTK+, respectively; Figure 1B). The remaining vectors resulted in fewer BTK-expressing B cells (6.2%–8.6% in BM, 9.7%–17% in SP, and 23%–25% in PF). In contrast, the proportions of BTK+ myeloid cells were more consistent between the LV groups (Figure 1C). These observations correlated with the MFI of BTK in total B or myeloid cell populations (Figures S3B and S3C). We noted that several Eμ.BTKp.BTK LV-GT mice had substantially higher expression (based on MFI) of BTK in B and myeloid cells than even the WT control.
Because the BCR/BTK signaling axis is important for progression between multiple stages of B cell development, we used immunophenotyping to analyze the major B cell developmental subsets in the BM, SP, and PF. Numerical reconstitution of BM and SP B cells at all stages were improved over mXLA mock-treated animals (transplanted with lineage-negative [Lin]− cells without LV transduction) in all gene therapy-treated groups (Figures 1D and 1E); the proportion of PF B cells improved as well (Figure 1F). Many of these increases were significantly higher than the corresponding mXLA mock cells (p < 0.05 by one-way ANOVA), particularly within the 1.5UCOE.BTKp.coBTK and Eμ.BTKp.BTK cohorts. While all LV groups had improved B cell counts versus mXLA mock, BTKp.BTK LV was consistently the least effective.
XLA patients have a complete or near absence of Ig production due to the absence or dysfunction of B cells. Gene therapy with BTKp.BTK LV failed to increase the serum concentrations of IgM or IgG in mXLA recipients, while gene therapy using the other three LVs led to increases in both isotypes (Figures 1G and 1H). Increases relative to mXLA mock recipient mice were significant (p < 0.05) based on one-way ANOVA for Eμ.BTKp.BTK (IgG and IgM) and 1.5UCOE.BTKp.coBTK (IgG) recipients.
Eμ.BTKp LV-treated mice develop high-titer autoantibodies
BTK overexpression can promote loss of self-tolerance and autoimmunity,55,56 and BTK inhibitors have previously been shown to provide benefit in murine models of systemic lupus erythematosus.57 Notably, a subset of mice receiving Eμ.BTKp.BTK LV-GT expressed BTK at higher amounts than did WT mock in mature B cells and developed total IgG levels that were above those of the WT mock-treated mice. Therefore, we measured anti-double-stranded DNA (dsDNA) antibodies in the sera of all LV-GT mice and compared autoantibody titers to sera from an autoimmune-prone mouse model, i.e., Was−/− B cell chimeric mice,58 as a positive control. Strikingly, most Eμ.BTKp.BTK recipients had elevated anti-dsDNA IgG titers similar to the positive control sera (Figure 2A). We also evaluated the Eμ.BTKp.BTK cohort for IgG2c and IgG3 subclass autoantibodies. Potentially pathogenic IgG2c autoantibodies59,60 were present in nearly all Eμ.BTKp.BTK recipients (Figure 2B). To provide a detailed assessment of IgG subclass distribution, we analyzed nine mice with high anti-dsDNA IgG from the Eμ.BTKp.BTK cohort and demonstrated class switching to all four IgG subclasses in the C57BL/6 strain (Figure S4A), with a bias toward IgG2c in several animals (including three animals with the highest overall anti-dsDNA concentrations).
Figure 2.

Evidence of autoantibody formation and promoter silencing after Eμ.BTKp.BTK LV-GT
(A) Anti-dsDNA IgG in serum from LV-GT-treated mXLA mice measured by ELISA prior to immunization (relative amount as OD450 absorbance). Data represent mean ± SEM from 10 independent experiments: n = 18 (WT mock), 16 (mXLA mock), 6 (BTKp.BTK), 30 (1.5UCOE.BTKp.BTK), 18 (1.5UCOE.BTKp.coBTK), 29 (Eμ.BTKp.BTK), and 2 (WASp chimera control). p values were determined using the one-way ANOVA corrected for multiple comparisons using Sidak’s method. Statistically significant differences between indicated cohorts are shown. ∗p < 0.05, ∗∗∗∗p < 0.0001. (B) Anti-dsDNA IgG2c and IgG3 subclasses in Eμ.BTKp,BTK mice (n = 23). (C) Average viral copy number (VCN) per cell in BM and SP as measured by qPCR. Data represent mean ± SEM from eight independent gene therapy experiments: n = 5 (BTKp.BTK), 18 (1.5UCOE.BTKp.BTK), 10 (1.5UCOE.BTKp.coBTK), and 18 (Eμ.BTKp.BTK) for SP, and n = 5 (BTKp.BTK.), 21 (1.5UCOE.BTKp.BTK), 14 (1.5UCOE.BTKp.coBTK), and 19 (Eμ.BTKp.BTK) for BM. (D) Methylation of BTKp within total splenocytes as quantified by bisulfite sequencing. Data are displayed as a percentage of methylated CpGs within the BTKp. p values were determined using the one-way ANOVA corrected for multiple comparisons using Tukey’s method. Statistically significant differences between experimental and mXLA Mock cohorts are shown. ∗∗∗∗p < 0.0001.
Sera from eight Eμ.BTKp.BTK LV-GT mice with high IgG anti-dsDNA antibodies were also analyzed using an autoantigen microarray (with 88 self-antigens) and compared to four experimentally matched, WT mock or LV 1.5UCOE.BTKp.BTK recipient mice (Figure S4B). Analysis of IgG autoantibodies in the Eμ.BTKp.BTK cohort revealed reactivity with most of the arrayed antigens, although there was much variability between mice for specific antigens. Histopathology and proteinuria analyses did not reveal evidence of autoantibody-related pathology in the Eμ.BTKp.BTK cohort (data not shown), although radiation-related tissue injury was present in all LV-GT mice, making it difficult to distinguish less severe renal injury. Overall, these results led us to discontinue testing the Eμ enhancer for LV BTK expression.
1. 5UCOE protects proviral BTKp from methylation in vivo
Clinical BTK LV-GT should provide sustained rescue of transgene expression, with a minimal number of viral integrations per cell in order to lower the risk of insertional mutagenesis. We found no evidence of selective cell outgrowth in recipient mice based on the normal distribution of hematopoietic cells in the tissues analyzed by flow cytometry after transplant. We used qPCR to measure integrated viral copy numbers (VCNs) in genomic DNA (gDNA) isolated from BM and SP cells. Mice treated with BTKp.BTK and Eμ.BTKp.BTK LV-GT had a relatively high average VCN in both BM and SP (average copies per cell >5), whereas mice treated with 1.5UCOE.BTKp and 1.5UCOE.BTKp.coBTK had a lower average VCN of 2.2 (Figure 2C).
To assess the potential for epigenetic silencing of candidate LVs, we performed bisulfite sequencing of integrated proviral BTKp from splenocyte gDNA samples. BTKp sequences derived from BTKp.BTK and Eμ.BTKp.BTK LV-GT recipients were substantially methylated (48% and 33%, respectively), while minimal methylation (3%) was observed for the 1.5UCOE.BTKp.BTK provirus (Figure 2D).
Due to elevated autoantibody titers after Eμ.BTKp.BTK LV-GT, and CpG methylation of the internal promoters in BTKp.BTK and Eμ.BTKp.BTK LV-GT, we did not further pursue these candidate vectors. In contrast, results from the 1.5UCOE.BTKp.coBTK LV-GT suggested that the UCOE element in conjunction with BTKp improved lineage-specific BTK expression and preserved the BTKp from methylation. Therefore, subsequent efforts were focused toward further optimizing this LV platform.
Incorporation of a truncated UCOE and endogenous regulatory elements to facilitate BTK expression
We wanted to further improve reconstitution of B cells and antibodies in our mXLA model, while also enhancing the efficiency of viral production. For the latter purpose, we decreased the LV size, using a smaller fragment of A2UCOE previously demonstrated to prevent silencing of integrated LV.51 A similar region of the A2UCOE prevented methylation and silencing of internal promoters in LVs, as assessed in pluripotent stem cells differentiated in vitro or in vivo.61 While other 1.5-kb versions of the A2UCOE contain the bidirectional promoter regions of both CBX3 and HNRNPA2B1,62 the UCOE fragment tested here exclusively spanned from the CBX3 promoter to the 3′ of alternative exon 1. Truncation to 0.7 kb eliminated most of the region downstream of alternative exon 1 (Figure 3A). However, 1.5UCOE.BTKp.coBTK LV was designed to place CBX3 transcription in the reverse orientation relative to the BTKp. Cloning 0.7-kb UCOE (0.7UCOE) into the BTKp.coBTK vector in either orientation (forward or reverse) had no impact on viral titer or BTK expression in vitro (data not shown). We therefore performed all subsequent studies with LV containing 0.7UCOE in the reverse orientation relative to BTKp. We observed a 2- to 10-fold increase in titer using LV 0.7UCOE.BTKp.coBTK (2 × 109 infectious units [IU]/mL after 100 × concentration of viral supernatant) compared to LV 1.5UCOE.BTKp.coBTK (∼4 × 108 IU/mL after 100 × concentration).
Figure 3.

Testing novel LV elements to improve BTK expression profile
(A) Depiction of the divergently transcribed housekeeping genes CBX3 and HNRNPA2B1 (A2UCOE) and the location of 1.5-kb UCOE within this region. (B) DNase I hypersensitive sites (DHS) within intronic regions of the BTK gene are shown (DHS1, DHS2, DHS3, DHS4, and DHS5; blue boxes). The ENCODE genome segmentation tool-predicted enhancer element that includes DHS4 is drawn as a yellow bar; exons are shown as black boxes. Various combinations of DHS sequences were cloned into the 0.7UCOE.BTKp.coBTK construct and tested in vitro (data not shown) and in vivo (Figure S5B). (C) Alternative codon optimizations of the BTK cDNA (coBTK and co2BTK) cloned into identical LV vectors were compared after in vitro transduction of mXLA lineage-negative BM cells (HSCs). Representative flow plots show intracellular BTK expression 7 days post-transduction. (D) Depiction of four candidate LV vectors, chosen as top candidates based on BTK expression (%BTK+ and MFI) after in vitro transduction of mXLA HSCs. Constructs contain a 0.7-kb UCOE upstream of BTKp, driving either coBTK or co2BTK expression with or without the BTK DHS4 fragment.
We next hypothesized that inclusion of predicted regulatory elements within the BTK locus could improve the specificity and amplitude of BTK expression while avoiding overexpression observed after Eμ.BTKp.BTK LV-GT. We identified five conserved DNase I hypersensitive sites in B and myeloid cells (Figure S5) within introns 1–5 of BTK (DHS1–DHS5; Figure 3B, blue boxes). These segments varied in length from 149 bp (DHS5) to 453 bp (DHS4). DHS4 was previously identified as a highly conserved region by Oeltjen et al.52 The ENCODE genome segmentation tool identified a predicted enhancer element within DHS4 (Figure 3B, yellow box). We then cloned the DHS sites in various combinations into the 0.7UCOE.BTKp.coBTK vector (Figure S6A) and compared them to 0.7UCOE.BTKp.coBTK LV-GT in mXLA mice (Figure S6B). Results from these preliminary tests suggested that 0.7UCOE.DHS4.BTKp.coBTK yielded the greatest improvement in BTK expression while maintaining a similar titer to 0.7UCOE.BTKp.coBTK. We therefore selected 0.7UCOE.DHS4.BTKp.coBTK for further in vivo analysis and testing.
Finally, it has been shown that codon optimization can greatly alter transgene expression and may also impact protein conformation and function through changing the rate of protein translation, altering protein folding kinetics.63,64 We tested a second codon-optimized BTK with 16% nucleotide divergence from the coBTK used in Figures 1 and 2 (Table S1). We cloned this alternate version (referred to hereafter as co2BTK) into the 0.7UCOE.BTKp construct. In vitro testing of each of these vectors in HSCs from mXLA mice showed a substantial increase in MFI when using co2BTK compared to coBTK (Figure 3C). The co2BTK vector yielded a BTK+ population that was distinct from non-transduced cells whereas the coBTK vector resulted in a more modest population shift.
Restoration of BTK expression and B cell development using the 0.7UCOE.BTKp LV platform
We directly compared the effects of codon optimization and the DHS4 enhancer by using the following vectors in the mXLA GT model (Figure 3D): 0.7UCOE.BTKp.coBTK, 0.7UCOE.BTKp.co2BTK, 0.7UCOE.DHS4.BTKp.coBTK, and 0.7UCOE.DHS4.BTKp.co2BTK. At 19–23 weeks post-transplant, we analyzed BM, SP, and PF lymphocytes for B cell reconstitution and BTK expression as described in Figure 1. Figure 4A shows representative flow plots of BTK expression in splenic B cells. Specifically, it shows the difference in BTK+ MFI shift of 0.7UCOE.DHS4.BTKp.coBTK and 0.7UCOE.DHS4.BTKp.co2BTK. While co2BTK mediated higher BTK expression, the difference was not as striking as in vitro (Figure 3C), likely because most of the splenic B cells were BTK+, largely eliminating the BTK− population for comparison.
Figure 4.

Alternative UCOE.BTKp LV vectors rescue BTK expression and B cell development
Lymphocytes from BM, SP, and PF of mice receiving LV-GT analyzed 19–23 weeks post-transplant. Lymphoid cell subsets were identified by cell surface markers (Figure S7). (A) Representative flow plots showing intracellular BTK staining in splenic B cells at endpoint analysis. (B–D) Percent of BTK+ cells in BM (B), SP (C), and PF (D) lymphocyte subsets from LV-GT groups, determined by flow cytometry. (E–G) Numbers of cells in each B cell subset were calculated as a proportion of total BM (E) and SP (F) counts and are depicted as the group average in stacked bars for each subset. PF B cell subsets are depicted as % of total lymphocytes (G). p values were determined using the one-way ANOVA with Sidak’s correction for multiple comparisons. For a given B cell subset in (E)–(G), statistically significant differences between experimental and mXLA mock cohorts are shown. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (H) BTK expression in B cell subsets, measured by flow cytometry. (I) MFI (geometric mean) of BTK+ cells in each B cell subset, normalized to WT mock. Data represent mean ± SD from four experiments (in each experiment we compared two to three vectors head-to-head, and tested all vectors in at least two independent experiments): n = 13 (WT mock), 13 (mXLA mock), 11 (0.7UCOE.BTKp.co BTK), 11 (0.7UCOE.BTKp.co2BTK), 16 (0.7UCOE.DHS4.BTKp.co BTK), and 11 (0.7UCOE.DHS4.BTKp.co2 BTK). Results of statistical tests between all experimental groups are listed in Table S2.
We measured the reconstitution of specific lymphoid subsets in BM, SP, and PF using immunophenotyping (see Figure S7 for gating strategy). In the BM (Figure 4B), BTK marking in the B cell compartment ranged from 53% to 67% BTK+ in mice treated with each of the four gene therapy vectors, which is substantially higher than the 34% BTK+ B cell compartment observed in mice that received Eμ.BTKp.BTK LV-GT (Figure 1B). Monocytes and neutrophils ranged from 35% to 57% BTK+ in the BM. Each of the vectors rescued BM BTK expression compared to mXLA mock, with a p value <0.0001; however, the fraction of BTK+ cells for all vectors was also lower than WT mock. Detailed statistical analysis is shown in Table S2.
In SP (Figure 4C), the B cell compartment was >95% BTK+ with both co2BTK-expressing vectors (0.7UCOE.BTKp.co2BTK and 0.7UCOE.DHS4.BTKp.co2BTK) and was not statistically significant from WT mock. The proportion of BTK-expressing cells was slightly lower with the coBTK-expressing vectors (0.7UCOE.BTKp.coBTK and 0.7UCOE.DHS4.BTKp.coBTK), averaging 79% and 86% BTK+, respectively. Inclusion of DHS4 in the vector yielded a higher proportion of BTK expression in the B cell compartment compared to 0.7UCOE alone. In the myeloid compartment (both monocytes and neutrophils), this pattern was reversed, with the 0.7UCOE.BTKp vectors mediating a higher proportion of BTK-expressing cells than the 0.7UCOE.DHS4.BTKp vectors. This latter observation suggested that DHS4 might contain an enhancer element that promotes BTK expression in B cells, but not in myeloid cells. All vectors rescued BTK expression compared to mXLA mock, with a p value ≤0.0001. In PF cells (Figure 4D), BTK expression followed a similar pattern as splenic B cells. The co2BTK vectors achieved on average 90% BTK+ expression, whereas the coBTK-expressing vectors (0.7UCOE.BTKp.coBTK and 0.7UCOE.DHS4.BTKp.coBTK) achieved 76% and 79% BTK+ expression, respectively.
Cells in each tissue were stained for markers of distinct B cell subsets (Figure S7). In BM, the most striking rescue of B cell counts was in the mature B cell compartment; this was observed with all four LV vectors (Figure 4E). In SP, follicular mature B cell numbers were also rescued; this subset was statistically significant between mXLA mock and each of the LV vectors (Figure 4F). Finally, in PF, both the B1 and B2 subsets were rescued compared to mXLA mock (Figure 4G). There were no significant differences in cell numbers between experimental LV groups, although mice in the 0.7UCOE.BTKp.coBTK LV-GT groups generally had the highest cell numbers.
B cell subsets were also analyzed for BTK expression (as %BTK+; Figure 4H). The proportion of B cells that are BTK+ increases throughout B cell development (Figure 4H, which shows developmental subsets from left to right), illustrating the importance of BTK in B cell development. BTK is not required in pro- and pre-B cells, and thus pro/pre-B cell compartments show only moderate BTK expression. In general, the BTK expression patterns observed for total B cells were consistent within each B cell subset: mice treated with the co2BTK-expressing LV-GT had an increase in BTK+ cells compared to the coBTK-expressing LV-GT, and LV-GT with the DHS4 element modestly increased BTK+ cells compared to 0.7UCOE alone. These differences were less pronounced in the SP, as BTK expression approached WT (100%).
Similar trends were observed with respect to BTK+ MFI (Figure 4I, plotted as % of WT mock MFI, to normalize experimental differences in MFI). Notably, B cell subsets in some co2BTK LV-GT mice had BTK MFIs exceeding WT B cells. This difference was most evident in the transitional 1 (T1) B cells in the SP and B1 B cells in the PF. Both transitional and B1 B cells can play a role in the development of autoimmunity.65, 66, 67, 68 Thus, overexpression of BTK in this setting may be a safety concern.
0. 7UCOE.BTKp.coBTK platform vectors rescue B cell function and Ig production in primary recipients
XLA subjects experience recurrent infection due to their inability to generate antibodies to foreign antigens. We evaluated mXLA LV-GT-treated mice for the capacity to mount specific antibody responses. At 12–20 weeks post-transplant, LV-GT recipients were immunized with the T cell-dependent antigen NP-CGG in alum, and serum was collected prior to and 10 days after primary immunization. One month following primary challenge, mice were re-challenged with NP-CGG in phosphate-buffered saline (PBS) and serum was collected 10 days after challenge. NP-specific IgG and IgM, as quantified by enzyme-linked immunosorbent assay (ELISA), revealed increased production of low-affinity (Figure S8A) and high-affinity NP-IgG, especially following re-challenge (Figure 5A). Mice receiving each of the alternative LV-GT vectors showed moderate responses to primary challenge, and high-affinity NP-specific IgG responses to secondary challenge were equal to or greater than those of WT mock.
Figure 5.

Alternative UCOE.BTKp LV vectors restore in vivo and in vitro B cell responses
(A) LV-GT-treated mXLA mice were immunized with NP-CGG in alum at 12 weeks post-transplant. Levels of NP-specific IgG in serum was measured by ELISA and quantified relative to an IgG standard. High-affinity NP-IgG (conjugation value of 4) was measured from serum prior to (–) and 10 days after primary immunization (1°). One month following primary challenge, mice were re-challenged with NP-CGG in PBS and serum was collected 10 days later (2°). One-way ANOVA with Tukey’s correction for multiple comparisons was used to determine differences between experimental and mXLA mock cohorts for both the primary and secondary antibody responses. Statistically significant p values are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (B and C) Total serum IgG (B) and IgM (C) in serum from mice receiving LV-GT was measured by ELISA at endpoint analysis (21–23 weeks post-transplant). p values were determined using the one-way ANOVA with Sidak’s correction for multiple comparisons. Statistically significant differences between experimental and mXLA mock cohorts are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Results from statistical tests between experimental groups are listed in Table S2. (D–F) At endpoint analysis, B cells were isolated from splenocytes by CD43 depletion, labeled with CTV, and stimulated in vitro with IgM, LPS, or media (negative control). (D) The percentage of BTK+ B cells that underwent one or more cell divisions 72 h after incubation with anti-mouse IgM antibodies, LPS, or media only (readout by flow cytometry). (E) BTK+ MFI of cells after each division (D0–D4), shown as % of WT mock. (F) Representative flow plots showing BTK staining and CTV dilution in B cells 72 h after IgM stimulation, gated on live B220+ BTK+ cells. Data represent mean ± SD from four experiments: n = 13 (WT mock), 13 (mXLA mock), 11 (0.7UCOE.BTKp.coBTK), 11 (0.7UCOE.BTKp.co2BTK), 16 (0.7UCOE.DHS4.BTKp.coBTK), and 11 (0.7UCOE.DHS4.BTKp.co2BTK).
At endpoint analysis, we again collected serum and then quantified total serum Ig (IgG and IgM) by ELISA. All experimental groups showed an increase in total IgG over mXLA mock (Figure 5B), and the average amount of total IgG in the 0.7UCOE.BTKp.co2BTK, 0.7UCOE.DHS4.BTKp.coBTK, and 0.7UCOE.DHS4.BTKp.co2BTK groups was equal to or greater than the average for WT mock. A similar pattern followed for total IgM (Figure 5C).
In addition, we assessed whether B cells in mXLA LV-GT mice regained responsiveness to BCR crosslinking. Clonal proliferation is a critical response of B cells encountering cognate antigen and is dependent on BTK signaling. We isolated splenic B cells via CD43 magnetic depletion and cultured them with anti-mouse IgM (to mimic BCR crosslinking), lipopolysaccharide (LPS), or media only (control) for 72 h. We then measured B cell proliferation by dilution of Cell Trace Violet (CTV), with intracellular staining for BTK expression performed in parallel. Figure 5D shows the percentage of BTK+ B220+ cells that had undergone less than one cell division. While no cells proliferated in media-only control, cells from all LV experimental groups underwent proliferation during the 72-h culture in response to IgM stimulation. All groups proliferated equivalently to LPS stimulation.
Proliferation in response to IgM engagement was highest with 0.7UCOE.BTKp.co2BTK and 0.7UCOE.BTKp.DHS4.co2BTK, and we observed a positive correlation between BTK+ MFI and proliferation; that is, B cells with the highest BTK+ MFI underwent the most rounds of proliferation. In contrast, WT mock cells had consistent BTK MFI throughout each round of cell division. While it is intuitive that cells with the highest BTK expression may proliferate the most, this raised the concern that a subset of cells with supra-endogenous BTK expression may be hyper-proliferative. Figure 5E shows the MFI of BTK+ proliferating cells after each round of cell division, normalized to WT Mock. After division 1 (D1), 0.7UCOE.BTKp.co2BTK and 0.7UCOE.DHS4.BTKp.co2BTK surpassed the BTK+ MFI of WT mock. Following D2, 0.7UCOE.DHS4.BTKp.coBTK also surpassed WT mock. Strikingly, the co2BTK-expressing constructs demonstrated a higher MFI at each subsequent round of cell division, reaching MFIs that were as high as 311% of WT mock (0.7UCOE.DHS4.co2BTK, D4). Representative flow plots (Figure 5F) of cells stained at 72 h from each of the experimental groups illustrate the gating strategy for each round of division. BTK on the y axis shows the increase in MFI. Notably, although 0.7UCOE.BTKp.coBTK had the lowest BCR-triggered cell proliferation, the BTK+ MFI at each round of cell division in 0.7UCOE.BTKp.coBTK remained closest to WT mock.
Additional safety considerations for 0.7UCOE.BTKp LV vectors
We next determined whether the various 0.7UCOE.BTKp-containing vectors correlated with dsDNA autoantibody production (Figures 6A and 6B). Representative serum samples from Eμ.BTKp.BTK (Figure 2) were analyzed in parallel for comparison. Mice from a subset of experimental groups had elevated serum amounts of anti-dsDNA IgG as follows: 0.7UCOE.BTKp.co2BTK (3 of 11), 0.7UCOE.DHS4.BTKp.coBTK (4 of 16), and 0.7UCOE.DHS4.BTKp.co2BTK (5 of 11) (Figure 6A). In the 0.7UCOE.DHS4.BTKp.coBTK and 0.7UCOE.DHS4.BTKp.co2BTK groups, most of these mice had dsDNA-binding antibody concentrations (reported as optical density [OD]) similar to Eμ.BTKp.BTK mice and control samples from the autoimmune Was−/− chimeric mouse model. Elevated amounts of anti-dsDNA IgG2c were also present in many of these animals such as that seen in Eμ.BTKp.BTK mice (Figure 6B). In contrast, mice receiving 0.7UCOE.BTKp.coBTK LV-GT did not exhibit anti-dsDNA antibodies (0 of 11).
Figure 6.

0.7UCOE.BTKp.co LV exhibits selective advantage and functional rescue without evidence for toxicity
(A and B) Relative serum concentrations of anti-dsDNA IgG (A) and anti-dsDNA IgG2c (B) in serum from the indicated mXLA LV-GT cohorts, as measured by ELISA (as OD450). Sera from autoimmune WAS chimeric mice and mice receiving Eμ.BTKp.BTK LV-GT were included as positive controls. p values were determined using the one-way ANOVA with Sidak’s method for multiple comparisons. Significant differences between the indicated groups are shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ns, not significant. (C and D) Genomic DNA (gDNA) was isolated from total BM (C) and SP (D) at endpoint analysis (21–23 weeks post-transplant), and VCN per cell was quantified by qPCR. Data represent mean ± SD from four independent experiments: n = 13 (WT mock), 13 (mXLA mock), 11 (0.7UCOE.BTKp.co), 11 (0.7UCOE.BTKp.co2), 16 (0.7UCOE.DHS4.BKTp.co), and 11 (0.7UCOE.DHS4.BKTp.co2).
In contrast to initial experiments, in which Eμ.BTKp.BTK mice averaged a VCN of 5 in the BM and 6.5 in the SP (Figure 2C), the alternative 0.7UCOE.BTKp.-containing constructs achieved functional rescue at a much lower VCN in BM (0.9–1.4; Figure 6C) and SP (1–1.5; Figure 6D). Thus, using the 0.7UCOE derivative LVs, we have achieved higher BTK expression, while simultaneously decreasing the VCN to a clinically relevant VCN of ∼1 per cell.69
We performed retroviral integration site (RIS) analysis of SP gDNA from two mice that underwent 0.7UCOE.BTKp.coBTK LV-GT. As shown in Figure S9A, splenocytes in both mice were polyclonal. None of the RIS events was within 140 kb of transcription start sites (TSSs) corresponding to known genes involved in insertional mutagenesis. The 0.7UCOE-BTKp-coBTK-treated mice displayed slightly higher frequencies of insertions in or near cancer genes compared to the reference dataset by QuickMap analysis70 (Table S3). There also appeared to be a preference for insertion on certain chromosomes in these mice compared to a reference dataset of random LV integration sites (Figure S9B).
0. 7UCOE.BTKp.coBTK LV mediates sustained BTK expression and function in primary and secondary recipient mice
Based on the promising functional activity and safety profile observed for the 0.7UCOE.BTKp.coBTK LV, we conducted seven additional experiments comparing 0.7UCOE.BTKp.coBTK LV-GT to WT mock and mXLA mock (Figures S10A–S10C). These expanded studies demonstrated consistent rescue of B cell development and total B cell numbers in the BM, SP, and PF, findings consistent with data shown in Figures 4, 5, and 6. Importantly, among 26 additional 0.7UCOE.BTKp.coBTK LV-GT-treated mice, only 2 had modest elevations in anti-dsDNA IgG and IgG2c (Figures S10D and S10E). Thus, across the entire 0.7UCOE.BTKp.coBTK LV-GT cohort (37 recipients), consistent functional rescue was observed with little evidence for dysregulated B cell function.
To determine whether the truncated 0.7-kb UCOE element supports stable and long-term transgene expression, we harvested BM from 0.7UCOE.BTKp.coBTK LV-GT primary recipient mice and transplanted 10 × 106 total BM cells into secondary recipient mXLA mice in two independent experiments. Secondary recipients were sacrificed at 16 weeks post-transplant, and cell populations were assessed for BTK expression in BM and SP. The percentage of BTK+ cells in the BM and SP was variable, even among the WT mock cohort, indicating a low dose of HSCs in the transferred marrow. In the 0.7UCOE.BTKp.coBTK LV cohort, the average BTK marking of neutrophils, monocytes, and B cells was similar, i.e., ∼25%–31% in the BM and ∼26%–29% in the SP, but not statistically different from mXLA mock due to the large standard deviation (SD). IgM levels were also variable, but they clearly improved in some mice (Figure S11B), without the appearance of anti-DNA IgG antibodies (Figure S11C). We did not observe any skewing in lymphoid populations in the secondary recipients (data not shown), and survival curves were not statistically different between the cohorts (Figure S11D). Note that the 4 deaths in the 29 secondary recipients all occurred within the first 6 weeks post-transplant, timing that suggests irradiation as the causal factor.
Maintenance of BTK expression in many of the secondary recipients suggested that the integrated 0.7UCOE.BTKp.coBTK LV was resistant to epigenetic vector silencing. To directly assess this question, bisulfite sequencing was performed to quantify CpG methylation within the BTK promoter. We found minimal CpG methylation in DNA from both primary and secondary recipients of 0.7UCOE.BTKp.coBTK LV-GT (Figure S11E). These data were consistent with the initial observations in 1.5UCOE.BTKp.BTK-treated mice, suggesting that the truncated 0.7UCOE can function equivalently to the larger UCOE element to preserve open chromatin. Taken together, these data demonstrate sustained functional activity of 0.7UCOE.BTKp.coBTK LV vector in engraftable, long-term repopulating stem cells in the mouse XLA model.
0. 7UCOE.BTKp.coBTK efficiently transduces CD34+ cells from control and XLA donors
We next determined whether HSCs from XLA patients could be effectively transduced by the 0.7UCOE.BTKp.coBTK LV. We obtained CD34+-enriched apheresis products from two granulocyte colony-stimulating factor (G-CSF)-mobilized healthy donors and XLA patients. CD34+ cells were transduced with LV 0.7UCOE.BTKp.coBTK at a range of viral doses (multiplicity of infection [MOI] of 0–20). Of note, both XLA patients had missense mutations in BTK that resulted in the expression of non-functional BTK; thus, expression of the BTK transgene was not distinguishable based on antibody staining. At 16 h post-transduction, 10,000 cells from each sample were placed in MethoCult for colony-forming unit (CFU) assays and cultured for 1 week. Cells were collected from the CFU colonies and their VCNs were determined by digital droplet PCR (ddPCR) of gDNA (Figure 7). An MOI of 5 yielded an average VCN of ∼1.0 for both healthy donor and XLA cells; the VCN increased incrementally with MOI, achieving an average of ∼1.5 for each at an MOI of 20. These data suggest that a relatively low MOI (5–20) can achieve a clinically relevant VCN in human CD34+ cells using LV 0.7UCOE.BTKp.coBTK.
Figure 7.

LV 0.7UCOE.BTKp.coBTK transduces human XLA CD34+ cells at a clinically relevant VCN
CD34+ PBSCs from mobilized healthy donors or XLA patients were thawed and, after a 48-h pre-stimulation in culture, transduced with LV at the indicated MOI. 16 h later, 10,000 cells from each condition were transferred to CFU media and cultured. After 1 week, cells from CFU colonies were collected and VCN was determined on gDNA by ddPCR. Black shapes indicate discrete healthy donors (used in two independent experiments); orange shapes indicate XLA patients (each patient repeated in two independent experiments). Bars and lines indicate mean ± SD.
Discussion
B cells have multiple mechanisms for tightly regulating BTK expression and function,71, 72, 73, 74 an investment in resources implying consequences for both overexpression and underexpression. The clinical consequences of BTK overexpression are pleiotropic, as shown by studies linking dysregulated expression to autoimmunity (reviewed by Crofford et al.55) and the expanding use of BTK inhibitors in B lymphoid malignancies and autoimmunity.75, 76, 77, 78 The results presented herein argue that the amount of BTK protein generated by a gene therapy must strike a balance: too low amounts compromise the success of reconstitution (e.g., LV BTKp.BTK), and too high amounts increase the risk of developing self-reactive B cells (e.g., LVs Eμ.BTKp.BTK and 0.7UCOEDHS4.BTKp.co2BTK). These concerns could possibly be addressed using a homology-directed repair (HDR)-based gene editing approach. Inserting a BTK cDNA downstream of the endogenous BTK promoter in theory would allow endogenous transcriptional regulation of BTK (although differences in BTK protein expression related to mRNA processing, stability, and translation could remain). However, to date, HDR-based gene editing of HSCs has been hampered by their low engraftment in vivo.79,80 While our laboratory and others are working toward overcoming this issue, the safety of LV-GT demonstrated in clinical trials for other immunodeficiency diseases supports the development of an LV therapy for XLA.
We approached the challenge of designing an LV-GT for XLA through iterative testing of multiple candidate designs in a murine model of XLA. Note that the mXLA background strain was ∼50% 129, an autoimmune-prone strain,81 increasing our ability to detect shifts in self-tolerance. By assessing vector components such as chromatin opening elements, candidate enhancers, and codon-optimized cDNA in the mXLA LV-GT model, we were able to measure their impacts on transduction efficiency, BTK expression, BCR responses, vector silencing, and auto-antibody production. We identified LV 0.7UCOE.BTKp.coBTK as one that balanced sustained BTK expression and functional rescue, while avoiding vector silencing and increases in self-reactive antibodies.
Several other investigators have described development of candidate LV vectors for XLA. Ng et al.82 described LV-GT in Btk knockout (Btk−/−) mice using a LV with a codon-optimized BTK cDNA driven by either a B cell-specific CD19 promoter or non-specific promoters including the EF1α short or SP focus-forming virus (SFFV) promoters. LV using the SFFV promoter drove a polyclonal erythroid myeloproliferation. Nonetheless, LV with the CD19 and EF1α short promoters resulted in partial reconstitution of B cell development and T cell-independent antibody responses in primary and secondary recipients, although the B cell block in Btk−/−mice used is less restrictive than the mXLA model. Notably, functional restoration with the ubiquitous EF1α LV vector required very high VCNs (VCN of 11, in secondary recipients). For safety reasons and other rationale noted below, we opted to not pursue a non-tissue-specific promoter such as EF1α short. Additionally, the CD19 promoter, also tested for LV-GT in human CD34+ cells by Moreau et al.,83 was not active in myeloid cells, a finding also described in our previous work evaluating candidate B lineage-restricted LV vectors.53
Although XLA myeloid cells do not exhibit a selective disadvantage in vivo, as evidenced by mostly random X-inactivation in female carriers,84 signaling pathways important in myeloid cell function (e.g., Toll-like receptors and Fc receptors) are impacted in XLA.85, 86, 87, 88 Thus, rescue of BTK expression in both B and myeloid cells is desirable for optimal clinical benefit. In a previous study we showed that the use of the BTKp as an internal promoter could direct transgene expression in both B and myeloid cells in vitro. In the present study, however, we determined that the BTKp when used alone to drive expression of canonical human BTK cDNA did not rescue B cell defects in the mXLA model and was extensively methylated in peripheral splenic cells. Addition of the 1.5-kb A2UCOE upstream of the BTKp incrementally improved BTK expression and antibody responses to a T cell-dependent immunogen, which was further improved by codon optimization of the cDNA. Notably, the strong transcriptional enhancer Eμ, and later a BTK gene regulatory element, DHS4, while improving BTKp-driven expression and the development of mature B cells, also showed increased autoantibody production, illustrating that a single regulatory element may skew B cell tolerance.
Addition of the UCOE element correlated with both protection from CpG methylation and improved the proportion of BTKp-expressing cells, consistent with preventing epigenetic silencing of transgene expression.61 Because the viral titers and transduction efficiencies were poor when using the 1.5-kb UCOE, we generated a truncated 0.7-kb element that improved viral titers and transduction efficiency (% BTK+ cells). Importantly, the 0.7-kb element maintained the ability to protect the BTKp from methylation, permitting sustained BTK expression following serial transplantation.
Codon optimization also highly impacted BTK expression. Codon usage is an important way of regulating protein expression; within a species, highly available codons are used more frequently in genes that are highly expressed.89 As optimized using different algorithms, LV driving co2BTK expression (0.7UCOE.BTKp.co2BTK and 0.7UCOE.DHS4.BTKp.co2BTK) displayed increased protein expression relative to identical LV constructs utilizing the alternative codon-optimized BTK cDNA construct, coBTK. There are various mechanisms by which codon usage impacts protein translation, including altering the rates of translation elongation and co-translational protein folding,64 and codon usage also impacts mRNA stability.90 For tightly regulated proteins, codon optimization may alter the safety and efficacy of protein expression;63 selection of a codon-optimized cDNA should therefore be closely evaluated when designing a gene therapy vector. Another codon-optimized BTK cDNA sequence was described in a Btk−/− mouse LV-GT model.82 In that study, codon optimization of BTK cDNA alone resulted in 14- to 48-fold increased transgene RNA per viral integration. However, BTK protein expression using the WT cDNA was not assessed in parallel, so it is unclear what effect the codon optimization had on BTK protein expression.
Despite having lower BTK expression compared to the co2BTK-expressing vector, 0.7UCOE.BTKp.coBTK gene therapy restored BTK expression in both B and myeloid cells, rescued BM, splenic, and peritoneal B cell development, and improved Ig responses in the mXLA model with an average VCN of 1–1.5. There was no evidence of clonal expansion in mice treated with this vector, as evidenced by lack of skewed lymphocyte populations or clonal expansion by RIS. Although RISs within oncogenes were slightly increased relative to random insertion dataset, these biases may be specific to the LV transduction in the mXLA disease background rather than the specific LV tested. However, the size of the dataset is too small to draw conclusions. Our findings suggest that 0.7UCOE.BTKp.coBTK may also rescue the human XLA phenotype. Consistent with this concept, 0.7UCOE.BTKp.coBTK efficiently transduced control and XLA peripheral blood CD34+ HSCs at a low MOI (10–20 transduction units/cell).
Importantly, our combined data support the concept that achieving the highest possible expression of BTK is not a safe strategy for treating XLA. Lineage regulated, stable, and sub- or near-endogenous BTK expression is likely the safest and most effective approach for a future curative therapy. Taken together, these combined data demonstrate that 0.7UCOE.BTKp.coBTK LV mediates sustained BTK expression and recues B cell development at a clinically relevant VCN without evidence for autoimmunity or genotoxicity. In order to rigorously qualify this vector for early phase clinical trials, we are planning an extensive safety and efficacy dataset including the following: in vitro murine BM immortalization assays; an endogenous promoter transactivation assay in human cell lines; nonclinical toxicology and efficacy studies in the mXLA model and in NSG mice using murine and control human HSCs, respectively; and assessment of B cell development in NSG mice transplanted using LV-transduced, XLA CD34+ cells.
Materials and methods
LV vector construction and viral production
All SIN-LVs were based on the pRRL backbone derived from pRRLSIN.cPPT.PGK-GFP.WPRE, a gift from Didier Trono (plasmid #12252, Addgene, Watertown, MA, USA). pRRL Eμ.B29.BTK and pRRL Eμ.B29.BTK.2A.eGFP, containing the reference human BTK cDNA coding sequence, were described previously.35 Other genetic elements were substituted or inserted into these plasmids, as well as plasmids pRRL BTKp.eGFP and pRRL Eμ.BTKp.eGFP,53 using standard molecular biology techniques, including restriction enzyme and T4 DNA ligation (New England Biolabs, Ipswich, MA, USA) and InFusion HD (Takara Bio USA, Mountain View, CA, USA), with oligonucleotides synthesized by Eurofins (Louisville, KY, USA) or Integrated DNA Technologies (Coralville, IA, USA). Other genetic elements (DNA sequences of these elements are provided in Table S1) used in plasmids shown here include: (1) a human BTK cDNA codon optimized for expression in human and mouse cells, coBTK, that was designed and synthesized by GenScript (Piscataway, NJ, USA); (2) a cDNA using an alternative codon optimization of human BTK, co2BTK (the parental plasmid was a gift from C.I. Edvard Smith); (3) a 1.5-kb segment of the A2UCOE, made by cleaving at the BspEI site upstream of the HNRNPA2B1 promoter in the 2.0-kb UCOE described previously;51 (4) the 0.7-kb UCOE described previously;51 and (5) BTK DHS elements 1–5 synthesized as gBlocks from GenScript. The DNA sequences of all plasmids were verified using a BigDye Terminator v3.1 cycle sequencing kit and a 3730xl DNA analyzer (both Applied Biosystems, Waltham, MA, USA).
The LVs were produced by transient transfection of the above pRRL plasmids into 293T cells along with second-generation helper plasmids pMD2.G and psPAX2 (both from Didier Trono; Addgene #12259 and #12260, respectively) using polyethylenimine. Viral supernatants collected 48 h post-transfection were concentrated by overnight centrifugation at 4°C. Viral titers were determined by transduction of NALM-6 cells with serial dilutions of viral stocks and detection of proviral sequences by qPCR (the qPCR method is described below for VCN determination). Viral titers were calculated using a standard curve generated from gDNA of a NALM6 clone with a single integrated provirus.35
Cell culture
All mammalian cell cultures were performed in a 37°C incubator with 5% CO2. Lymphocyte media were composed of RPMI 1640 (HyClone, Logan, UT, USA) with 10% fetal calf serum (FCS; Omega Scientific, Tarzana, CA, USA), 10 mM HEPES (Gibco/Thermo Fisher Scientific, Waltham, MA, USA), 55 μM 2-mercaptoethanol (Gibco), and 100 IU/mL penicillin-streptomycin (HyClone). Murine HSC media were either StemSpan serum-free expansion medium (SFEM) (STEMCELL Technologies, Vancouver, BC, Canada; Figures 1 and 2) or stem cell growth medium (SCGM) (CellGenix, Portsmouth, NH, USA; Figures 3, 4, 5, and 6; Figure S10), with both having the following additives: 2% FCS, 1× GlutaMAX (Gibco), 100 IU/mL penicillin-streptomycin, and recombinant murine cytokines stem cell factor (SCF) (50 ng/mL) and thyroperoxidase (TPO) (20 ng/mL; both PeproTech, Cranbury, NJ, USA). Human HSC media were SCGM with 100 ng/mL each recombinant human TPO, SCF, and Flt3 (PeproTech).
Immunophenoyping and flow cytometry
To characterize lymphocytes and BTK expression, single-cell suspensions in PBS with and without 0.5% BSA were prepared from blood, BM, SP, and peritoneal wash, and incubated with biotin- or fluorochrome-conjugated antibodies against cell surface proteins for 30 min at 4°C. Antibodies used for immunophenotyping are listed in Table S4. Cells were then fixed and permeabilized using a Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA, USA) following the manufacturer’s instructions (Figures 1 and 2). Alternatively, following surface stains, cells were fixed for 15 min at 37°C using a Cytofix/Cytoperm kit. Cells were then washed and resuspended in a 1:1 mixture of Phosflow Perm Buffer II and 1× Perm/Wash buffer (both BD Biosciences). After incubating on ice for 30 min, cells were washed twice in PBS and then stained with anti-human BTK (clone 53/BTK)-Alexa Fluor 647 (BD Biosciences; 1:250 in 1× Perm Buffer I) for 60 min, followed by two washes in 1× Perm Buffer I. All samples were acquired on a five-laser LSR II flow cytometer using FACSDiva software (BD Biosciences) and analyzed using FlowJo software versions 9 and 10 (Tree Star, Ashland, OR, USA). Only cells within the lymphocyte gate (based on forward and side scatter) were used in analysis (after excluding doublets). The numbers of a cell of interest were determined for BM, SP, and peritoneal cavity by multiplying subset percentages (expressed as percentage of live cells) by the total number of cells recovered from the tissue.
Murine stem cell isolation and LV transduction
C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) and Btk−/−Tec−/− (mXLA) mice36 in a mixed background strain (C57BL/6:.129, ∼50:50) were housed in a specific pathogen-free facility at the Seattle Children’s Research Institute (SCRI). All handling of animals and experimental procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and experiments were approved by the SCRI Institutional Animal Care and Use Committee. After euthanization, BM cells were isolated from the femurs, tibias, hips, and vertebrae of C57BL/6 WT and mXLA mice (6–16 weeks of age) as previously described.91 HSCs were then enriched by depleting lineage-committed cells using the EasySep mouse hematopoietic progenitor cell enrichment kit (STEMCELL Technologies). Lineage-negative cells were plated at 2 × 106 cells/mL (Figures 1 and 2) or 4 × 106 cells/mL (Figures 3, 4, 5, and 6) in HSC media. Cells were transduced overnight (10–14 h) with LV along with 4 μg/mL Polybrene (MilliporeSigma, St. Louis, MO, USA), while mock-treated WT and mXLA cells were cultured overnight without the addition of LV. In Figures 1 and 2, the dose of LV added was an MOI (determined using qPCR titer) of 5 or 10 infectious units/cell; in Figures 3, 4, 5, and 6, we added 10 μL of each concentrated LV supernatant per million cells (MOI of ∼10–20). After viral transduction, cells were rinsed and then resuspended in PBS at 1–5 × 106 cells/200 μL for injection into recipient mice.
mXLA transplantation, tissue collection, and processing
mXLA recipient mice (9–17 weeks of age; average 12.4 weeks) were conditioned with total-body irradiation given as a single dose (9.5 Gy; Figures 3, 4, 5, and 6; Figure S10) or split dose (2 × 4.5 Gy, 16–24 h apart; Figures 1 and 2). Recipient mice were then given 1–5 × 106 (Figures 1 and 2) or 1–2 × 106 (Figures 3, 4, 5, and 6; Figure S10) WT mock, mXLA mock, or LV-treated mXLA cells via the retro-orbital plexus. All mice were provided with antibiotic-treated drinking water ad libitum (0.5 mg/mL Baytril, Bayer Healthcare, Pittsburgh, PA, USA) for 2 weeks post-irradiation.
Starting at 6 weeks post-transplant, peripheral blood samples were periodically collected via the retro-orbital plexus. For flow cytometry analyses, peripheral blood samples were mixed with heparin and subjected to erythrocyte lysis and antibody staining. For analysis of serum antibodies, peripheral blood was either allowed to clot for 1 h at room temperature (RT), centrifuged for 5 min at 1,600 × g, and clear serum was collected, or blood was collected in Microtainer serum collection tubes (BD Biosciences) and spun at 10,000 × g for 1 min to separate serum.
Comprehensive analyses of immune cell reconstitution and phenotype were performed 16–30 weeks post-transplant after sacrificing animals. Peripheral blood was obtained by cardiac puncture, and sera were collected as described above. Hematopoietic tissues were isolated and placed in ice-cold PBS, with or without the addition of 2% FCS and 100 IU/mL penicillin-streptomycin. Peritoneal lymphocytes were obtained by injecting 8 mL of PBS into the peritoneal cavity and collecting the fluid with a syringe. BM was collected by flushing femurs and tibias with media using a syringe. Splenocytes were isolated by homogenization of SPs between frosted microscope slides. BM and SP cells were subject to erythrocyte lysis and removal of tissue debris by passaging through a 40-μm cell strainer, then used in subsequent assays. For serial transplants, 10 × 106 BM cells (without lineage depletion) from WT mock, mXLA mock, or LV-GT mice were transplanted into conditioned mXLA secondary recipient mice (as above), with 2 secondary recipients/donor.
T cell-dependent immunization and antibody detection
Immunizations and ELISAs were performed as previously described.35 For T cell-dependent (T-D) immunization, mice were given 100 μg of NP(45)-CGG (Biosearch Technologies, Novato, CA, USA) in 100 μL of Imject alum adjuvant (Thermo Fisher Scientific) via intraperitoneal (i.p.) injection 12–20 weeks post-transplant and peripheral blood samples were collected pre-injection (day 0) and 10 days after NP-CGG injection. Re-challenge was performed approximately 4 weeks later, again collecting peripheral blood samples before and 10 days after NP-CGG injection. For ELISA, 96-well MaxiSorp Immuno plates (Nalge Nunc, Rochester, NY, USA) were coated overnight at 4°C with either 100 ng of goat anti-mouse IgM or IgG (SouthernBiotech, Birmingham, AL, USA), 2.5 μg of NP(4)-BSA (to determine high-affinity IgG; Biosearch Technologies), or 2.5 μg of NP(30)-BSA (to determine low-affinity IgG; Biosearch Technologies), all diluted in PBS. Plates were washed three times (wash buffer PBS + 0.05% Tween 20 (Pierce Biotechnology, Rockford, IL, USA) using the AquaMax 2000 (Molecular Devices, San Jose, CA, USA), blocked for 2 h at RT with PBS + 2% BSA, and then washed three times. Sera and standards were diluted in wash buffer with 1% BSA and plated in duplicate wells with dilution series starting at 1:1,250 (sera) or 400 ng/mL (unlabeled murine IgM, IgG, or IgG3) for 2 h at RT. Following six washes, detection antibodies (horseradish peroxidase [HRP]-conjugated goat anti-mouse IgM, IgG, or IgG3, SouthernBiotech) diluted 1:2,000 in PBS with 1% BSA and 0.05% Tween 20 were added and incubated for 1 h at RT. Following six washes, plates were incubated with OptEIA tetramethylbenzidine (TMB) substrate reagent (BD Biosciences), reactions were stopped with 2 N H2SO4, and 450 nm absorbance was measured with 570 nm subtraction on a SpectraMax plate reader (Molecular Devices). Antibody concentrations were calculated from IgM and IgG standard curves using SoftMax Pro software (Molecular Devices) with a four-parameter curve fit.
Anti-DNA antibody ELISA
For anti-DNA antibody ELISA, 96-well MaxiSorp Immuno plates were coated with 0.005% poly-l-lysine (MilliporeSigma) in PBS for 30 min at RT. Wells were aspirated and coated with 5 μg of calf thymus DNA in Tris-EDTA (10 mM Tris, 1 mM EDTA [pH 8.0]) buffer overnight at 4°C. After aspirating the solution, wells were then blocked with PBS + 0.5% BSA for at least 1 h at RT. Plates were washed three times (as above). Sera were diluted 1:200 in PBS + 0.5% BSA + 0.05% Tween 20, plated in duplicate wells, and incubated at RT for 2 h. After washing six times, detection antibodies were added (HRP goat anti-mouse IgM, IgG, IgG2c, or IgG3, SouthernBiotech) and diluted 1:2,000 in PBS + 0.5% BSA + 0.05% Tween 20 + 1% normal goat serum (Jackson ImmunoResearch, West Grove, PA, USA) and incubated for 1 h at RT. After washing six times, plates were incubated with OptEIA TMB substrate reagent, reactions were stopped with 2 N H2SO4, and absorbance was measured at 450 nm with 570 nm subtraction on the SpectraMax plate reader.
Proliferation assay of murine splenic B cells in vitro
For cell proliferation assays, untouched splenic B cells were isolated with CD43 MicroBeads (Miltenyi Biotec, Auburn, CA, USA), and ∼2 × 106 B cells from each mouse were labeled with 1 μM CellTrace Violet (Invitrogen/Thermo Fisher Scientific, Carlsbad, CA, USA) in 1 mL of PBS, according to the manufacturer’s instructions. Labeled B cells were then plated in 96-well flat-bottom plates at 0.5–1.0 × 106 cells/mL in lymphocyte media and treated with one of the following conditions: media alone, 20 μg/mL goat anti-mouse IgM F(ab′)2 fragment (Jackson ImmunoResearch), or 10 μg/mL LPS (Salmonella minnesota Re 595, MilliporeSigma). Following 72 h in culture, cells were stained for B220-phycoerythrin (PE)-Cy7 (BioLegend, San Diego, CA, USA) with or without Live/Dead fixable near-infrared (IR) dead cell stain (Invitrogen). Cells were fixed and permeabilized as described above, and intracellular staining for BTK was performed. Cells were analyzed on an LSR II (BD Biosciences).
VCN analysis by qPCR
Quantification of viral integrations was performed as previously described.53 Briefly, gDNA was isolated from BM, SP, and thymus from LV-GT-treated mXLA mice at endpoint analysis. Primer/probe sets were used to normalize cell DNA content. In mouse cells, a murine β-actin primer/probe set (Invitrogen) was used. In human cells, an RNase P primer/probe set was used (Invitrogen). A Gag-specific primer/probe set was used to amplify viral integrations from murine cells using forward primer (5′-GGAGCTAGAACGATTCGCAGTTA-3′), reverse primer (5′-GGTTGTAGCTGTCCCAGTATTTGTC-3′), and probe (5′-[6FAM]-ACAGCCTTCTGATGTTTCTAACAGGCCAGG-[BHQ1]-3′). A primer-binding site (PBS)/Gag-specific primer/probe set was used for determining VCNs in human cells using forward primer (5′-ACTTGAAAGCGAAAGGGAAAC-3′), reverse primer (5′-CACCCATCTCTCTCCTTCTAGCC-3′), and probe (5′-[6FAM]-AGCTCTCTCGACGCAGGACTCGGC-[BHQ1]-3′). Viral integrations were quantified relative to a standard curve: gDNA from an A20 murine B cell line with six copies of integrated LV, or the human NALM6 B cell line harboring single LV integration.
Human stem cell transduction
Collection and use of human cells were conducted according to protocols approved by the Institutional Review Boards of the National Institutes of Health (XLA patient BTK V535F; protocol 05-I-0213) and the SCRI (XLA E589G; protocol FH7649). CD34+ peripheral blood stem cells (PBSCs) from XLA patient BTK V535F were a gift from Harry Malech. XLA patient BTK E589G was G-CSF-mobilized and leukapheresed at the Seattle Cancer Care Alliance. Immediately following collection, CD34+ cells were enriched by a CliniMACS column (Miltenyi Biotec) and viably frozen by the Co-operative Center for Excellence in Hematology (CCEH) at the Fred Hutchinson Cancer Research Center (Seattle, WA, USA). CD34+ PBSCs from G-CSF-mobilized healthy donors were purchased from the CCEH. CD34+ PBSCs were thawed and cultured in human HSC media for 48 h. Then, cells were resuspended to a cell density of 1 × 106 cells/mL in human HSC media with 4 mg/mL Polybrene (MilliporeSigma), and then cultured for 24 h with various MOIs of 0.7UCOE.BTKp.coBTK (titer, 4 × 109 viral particles [vp]/mL) in duplicate wells of a 96-well plate. After 16 h, media with LV were removed and cells were resuspended in fresh human HSC media at 1 million cells/mL. 20,000 cells were divided and transferred to two 35-mm cell culture plates containing 1.1 mL of human MethoCult semi-solid culture media (STEMCELL Technologies). After incubation for 1 week, CFU colonies were collected with PBS, gDNA extracted using the DNeasy mini kit (QIAGEN, Germantown, MD, USA), and then VCNs were determined using ddPCR.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (GraphPad, San Diego, CA, USA). p values were determined using the one-way ANOVA and the Sidak’s or Tukey’s corrections for multiple comparisons, or the unpaired t test, and are described in the figure legends.
Acknowledgments
We are grateful to our patients and families for their assistance in this work. We thank Harry Malech for the gift of XLA patient CD34+ PBSCs; Tara Bumgarner, Suzanne Skoda-Smith, Christiana Smith, Kathleen Teaderman, and Morgan Withrow for coordinating and/or performing XLA patient leukapheresis; Didier Trono for the lentiviral backbone and packaging plasmids; and Edvard Smith for the co2BTK-containing plasmid. This work was supported by NIH R01 AI084457 and the Program for Cell and Gene Therapy at the Seattle Children’s Research Institute.
Author contributions
B.J.S, S.S., H.M.C., B.D.S., S.K., C.C., M.H., J.P., B.Y.R., I.F.K, and J.E.A. conducted the experiments; B.J.S, S.S., H.M.C., K.S., B.D.S., J.E.A., and D.J.R. designed the experiments; B.J.S., H.M.C., J.E.A., D.J.R., and K.S. wrote the paper.
Declaration of interests
D.J.R., S.S., and K.S. are inventors on a patent filed by Seattle Children’s Research Institute related to the lentiviral vectors described in this study. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2021.01.007.
Supplemental information
References
- 1.Brosens L.A., Tytgat K.M., Morsink F.H., Sinke R.J., Ten Berge I.J., Giardiello F.M., Offerhaus G.J., Keller J.J. Multiple colorectal neoplasms in X-linked agammaglobulinemia. Clin. Gastroenterol. Hepatol. 2008;6:115–119. doi: 10.1016/j.cgh.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Lee P.P., Chen T.X., Jiang L.P., Chan K.W., Yang W., Lee B.W., Chiang W.C., Chen X.Y., Fok S.F., Lee T.L. Clinical characteristics and genotype-phenotype correlation in 62 patients with X-linked agammaglobulinemia. J. Clin. Immunol. 2010;30:121–131. doi: 10.1007/s10875-009-9341-5. [DOI] [PubMed] [Google Scholar]
- 3.Plebani A., Soresina A., Rondelli R., Amato G.M., Azzari C., Cardinale F., Cazzola G., Consolini R., De Mattia D., Dell’Erba G., Italian Pediatric Group for XLA-AIEOP Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin. Immunol. 2002;104:221–230. doi: 10.1006/clim.2002.5241. [DOI] [PubMed] [Google Scholar]
- 4.Misbah S.A., Spickett G.P., Ryba P.C., Hockaday J.M., Kroll J.S., Sherwood C., Kurtz J.B., Moxon E.R., Chapel H.M. Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J. Clin. Immunol. 1992;12:266–270. doi: 10.1007/BF00918150. [DOI] [PubMed] [Google Scholar]
- 5.Hermaszewski R.A., Webster A.D. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q. J. Med. 1993;86:31–42. [PubMed] [Google Scholar]
- 6.van der Meer J.W., Weening R.S., Schellekens P.T., van Munster I.P., Nagengast F.M. Colorectal cancer in patients with X-linked agammaglobulinaemia. Lancet. 1993;341:1439–1440. doi: 10.1016/0140-6736(93)90883-i. [DOI] [PubMed] [Google Scholar]
- 7.Kainulainen L., Varpula M., Liippo K., Svedström E., Nikoskelainen J., Ruuskanen O. Pulmonary abnormalities in patients with primary hypogammaglobulinemia. J. Allergy Clin. Immunol. 1999;104:1031–1036. doi: 10.1016/s0091-6749(99)70085-0. [DOI] [PubMed] [Google Scholar]
- 8.Rudge P., Webster A.D., Revesz T., Warner T., Espanol T., Cunningham-Rundles C., Hyman N. Encephalomyelitis in primary hypogammaglobulinaemia. Brain. 1996;119:1–15. doi: 10.1093/brain/119.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Lougaris V., Soresina A., Baronio M., Montin D., Martino S., Signa S., Volpi S., Zecca M., Marinoni M., Baselli L.A. Long term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J. Allergy Clin. Immunol. 2020;146:429–437. doi: 10.1016/j.jaci.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Tsukada S., Saffran D.C., Rawlings D.J., Parolini O., Allen R.C., Klisak I., Sparkes R.S., Kubagawa H., Mohandas T., Quan S. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 11.Hendriks R.W., de Bruijn M.F., Maas A., Dingjan G.M., Karis A., Grosveld F. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre-B cell stage. EMBO J. 1996;15:4862–4872. [PMC free article] [PubMed] [Google Scholar]
- 12.Hata D., Kawakami Y., Inagaki N., Lantz C.S., Kitamura T., Khan W.N., Maeda-Yamamoto M., Miura T., Han W., Hartman S.E. Involvement of Bruton’s tyrosine kinase in FεRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 1998;187:1235–1247. doi: 10.1084/jem.187.8.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyer A.S., Morales J.L., Huang W., Ojo F., Ning G., Wills E., Baines J.D., August A. Absence of Tec family kinases interleukin-2 inducible T cell kinase (Itk) and Bruton’s tyrosine kinase (Btk) severely impairs FcεRI-dependent mast cell responses. J. Biol. Chem. 2011;286:9503–9513. doi: 10.1074/jbc.M110.165613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S.H., Kim T., Jeong D., Kim N., Choi Y. The Tec family tyrosine kinase Btk Regulates RANKL-induced osteoclast maturation. J. Biol. Chem. 2008;283:11526–11534. doi: 10.1074/jbc.M708935200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinohara M., Koga T., Okamoto K., Sakaguchi S., Arai K., Yasuda H., Takai T., Kodama T., Morio T., Geha R.S. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806. doi: 10.1016/j.cell.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 16.Fiedler K., Sindrilaru A., Terszowski G., Kokai E., Feyerabend T.B., Bullinger L., Rodewald H.R., Brunner C. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood. 2011;117:1329–1339. doi: 10.1182/blood-2010-04-281170. [DOI] [PubMed] [Google Scholar]
- 17.Mangla A., Khare A., Vineeth V., Panday N.N., Mukhopadhyay A., Ravindran B., Bal V., George A., Rath S. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104:1191–1197. doi: 10.1182/blood-2004-01-0207. [DOI] [PubMed] [Google Scholar]
- 18.Melcher M., Unger B., Schmidt U., Rajantie I.A., Alitalo K., Ellmeier W. Essential roles for the Tec family kinases Tec and Btk in M-CSF receptor signaling pathways that regulate macrophage survival. J. Immunol. 2008;180:8048–8056. doi: 10.4049/jimmunol.180.12.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller H., Stadtmann A., Van Aken H., Hirsch E., Wang D., Ley K., Zarbock A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) γ2 and PI3Kγ pathways. Blood. 2010;115:3118–3127. doi: 10.1182/blood-2009-11-254185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honda F., Kano H., Kanegane H., Nonoyama S., Kim E.S., Lee S.K., Takagi M., Mizutani S., Morio T. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat. Immunol. 2012;13:369–378. doi: 10.1038/ni.2234. [DOI] [PubMed] [Google Scholar]
- 21.Mukhopadhyay S., Mohanty M., Mangla A., George A., Bal V., Rath S., Ravindran B. Macrophage effector functions controlled by Bruton’s tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J. Immunol. 2002;168:2914–2921. doi: 10.4049/jimmunol.168.6.2914. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt N.W., Thieu V.T., Mann B.A., Ahyi A.N., Kaplan M.H. Bruton’s tyrosine kinase is required for TLR-induced IL-10 production. J. Immunol. 2006;177:7203–7210. doi: 10.4049/jimmunol.177.10.7203. [DOI] [PubMed] [Google Scholar]
- 23.Horwood N.J., Page T.H., McDaid J.P., Palmer C.D., Campbell J., Mahon T., Brennan F.M., Webster D., Foxwell B.M. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J. Immunol. 2006;176:3635–3641. doi: 10.4049/jimmunol.176.6.3635. [DOI] [PubMed] [Google Scholar]
- 24.Horwood N.J., Mahon T., McDaid J.P., Campbell J., Mano H., Brennan F.M., Webster D., Foxwell B.M. Bruton’s tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor α production. J. Exp. Med. 2003;197:1603–1611. doi: 10.1084/jem.20021845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.González-Serrano M.E., Estrada-García I., Mogica-Martínez D., González-Garay A., López-Herrera G., Berrón-Ruiz L., Espinosa-Padilla S.E., Yamazaki-Nakashimada M.A., Vargas-Hernández A., Santos-Argumedo L. Increased pro-inflammatory cytokine production after lipopolysaccharide stimulation in patients with X-linked agammaglobulinemia. J. Clin. Immunol. 2012;32:967–974. doi: 10.1007/s10875-012-9706-z. [DOI] [PubMed] [Google Scholar]
- 26.De Ravin S.S., Wu X., Moir S., Anaya-O’Brien S., Kwatemaa N., Littel P., Theobald N., Choi U., Su L., Marquesen M. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016;8:335ra57. doi: 10.1126/scitranslmed.aad8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohn D.B., Booth C., Kang E.M., Pai S.Y., Shaw K.L., Santilli G., Armant M., Buckland K.F., Choi U., De Ravin S.S., Net4CGD consortium Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med. 2020;26:200–206. doi: 10.1038/s41591-019-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrua F., Brigida I., Aiuti A. Update on gene therapy for adenosine deaminase-deficient severe combined immunodeficiency. Curr. Opin. Allergy Clin. Immunol. 2010;10:551–556. doi: 10.1097/ACI.0b013e32833fea85. [DOI] [PubMed] [Google Scholar]
- 29.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hacein-Bey Abina S., Gaspar H.B., Blondeau J., Caccavelli L., Charrier S., Buckland K., Picard C., Six E., Himoudi N., Gilmour K. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313:1550–1563. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conley M.E., Brown P., Pickard A.R., Buckley R.H., Miller D.S., Raskind W.H., Singer J.W., Fialkow P.J. Expression of the gene defect in X-linked agammaglobulinemia. N. Engl. J. Med. 1986;315:564–567. doi: 10.1056/NEJM198608283150907. [DOI] [PubMed] [Google Scholar]
- 32.Quintáns J., McKearn J.P., Kaplan D. B cell heterogeneity. I. A study of B cell subpopulations involved in the reconstitution of an X-linked immune defect of B cell differentiation. J. Immunol. 1979;122:1750–1756. [PubMed] [Google Scholar]
- 33.Rohrer J., Conley M.E. Correction of X-linked immunodeficient mice by competitive reconstitution with limiting numbers of normal bone marrow cells. Blood. 1999;94:3358–3365. [PubMed] [Google Scholar]
- 34.Yu P.W., Tabuchi R.S., Kato R.M., Astrakhan A., Humblet-Baron S., Kipp K., Chae K., Ellmeier W., Witte O.N., Rawlings D.J. Sustained correction of B-cell development and function in a murine model of X-linked agammaglobulinemia (XLA) using retroviral-mediated gene transfer. Blood. 2004;104:1281–1290. doi: 10.1182/blood-2003-09-3044. [DOI] [PubMed] [Google Scholar]
- 35.Kerns H.M., Ryu B.Y., Stirling B.V., Sather B.D., Astrakhan A., Humblet-Baron S., Liggitt D., Rawlings D.J. B cell-specific lentiviral gene therapy leads to sustained B-cell functional recovery in a murine model of X-linked agammaglobulinemia. Blood. 2010;115:2146–2155. doi: 10.1182/blood-2009-09-241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellmeier W., Jung S., Sunshine M.J., Hatam F., Xu Y., Baltimore D., Mano H., Littman D.R. Severe B cell deficiency in mice lacking the Tec kinase family members Tec and Btk. J. Exp. Med. 2000;192:1611–1624. doi: 10.1084/jem.192.11.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerji J., Olson L., Schaffner W. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell. 1983;33:729–740. doi: 10.1016/0092-8674(83)90015-6. [DOI] [PubMed] [Google Scholar]
- 38.Cockerill P.N., Garrard W.T. Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell. 1986;44:273–282. doi: 10.1016/0092-8674(86)90761-0. [DOI] [PubMed] [Google Scholar]
- 39.Thompson A.A., Wood W.J., Jr., Gilly M.J., Damore M.A., Omori S.A., Wall R. The promoter and 5′ flanking sequences controlling human B29 gene expression. Blood. 1996;87:666–673. [PubMed] [Google Scholar]
- 40.Herbst F., Ball C.R., Tuorto F., Nowrouzi A., Wang W., Zavidij O., Dieter S.M., Fessler S., van der Hoeven F., Kloz U. Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol. Ther. 2012;20:1014–1021. doi: 10.1038/mt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lotti F., Menguzzato E., Rossi C., Naldini L., Ailles L., Mavilio F., Ferrari G. Transcriptional targeting of lentiviral vectors by long terminal repeat enhancer replacement. J. Virol. 2002;76:3996–4007. doi: 10.1128/JVI.76.8.3996-4007.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Persons D.A., Hargrove P.W., Allay E.R., Hanawa H., Nienhuis A.W. The degree of phenotypic correction of murine β-thalassemia intermedia following lentiviral-mediated transfer of a human γ-globin gene is influenced by chromosomal position effects and vector copy number. Blood. 2003;101:2175–2183. doi: 10.1182/blood-2002-07-2211. [DOI] [PubMed] [Google Scholar]
- 43.Ott M.G., Schmidt M., Schwarzwaelder K., Stein S., Siler U., Koehl U., Glimm H., Kühlcke K., Schilz A., Kunkel H. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 44.Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 45.Neville J.J., Orlando J., Mann K., McCloskey B., Antoniou M.N. Ubiquitous chromatin-opening elements (UCOEs): applications in biomanufacturing and gene therapy. Biotechnol. Adv. 2017;35:557–564. doi: 10.1016/j.biotechadv.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Antoniou M., Harland L., Mustoe T., Williams S., Holdstock J., Yague E., Mulcahy T., Griffiths M., Edwards S., Ioannou P.A. Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics. 2003;82:269–279. doi: 10.1016/s0888-7543(03)00107-1. [DOI] [PubMed] [Google Scholar]
- 47.Zhang F., Thornhill S.I., Howe S.J., Ulaganathan M., Schambach A., Sinclair J., Kinnon C., Gaspar H.B., Antoniou M., Thrasher A.J. Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells. Blood. 2007;110:1448–1457. doi: 10.1182/blood-2006-12-060814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams S., Mustoe T., Mulcahy T., Griffiths M., Simpson D., Antoniou M., Irvine A., Mountain A., Crombie R. CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnol. 2005;5:17. doi: 10.1186/1472-6750-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang F., Frost A.R., Blundell M.P., Bales O., Antoniou M.N., Thrasher A.J. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol. Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brendel C., Müller-Kuller U., Schultze-Strasser S., Stein S., Chen-Wichmann L., Krattenmacher A., Kunkel H., Dillmann A., Antoniou M.N., Grez M. Physiological regulation of transgene expression by a lentiviral vector containing the A2UCOE linked to a myeloid promoter. Gene Ther. 2012;19:1018–1029. doi: 10.1038/gt.2011.167. [DOI] [PubMed] [Google Scholar]
- 51.Bandaranayake A.D., Correnti C., Ryu B.Y., Brault M., Strong R.K., Rawlings D.J. Daedalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res. 2011;39:e143. doi: 10.1093/nar/gkr706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oeltjen J.C., Malley T.M., Muzny D.M., Miller W., Gibbs R.A., Belmont J.W. Large-scale comparative sequence analysis of the human and murine Bruton’s tyrosine kinase loci reveals conserved regulatory domains. Genome Res. 1997;7:315–329. doi: 10.1101/gr.7.4.315. [DOI] [PubMed] [Google Scholar]
- 53.Sather B.D., Ryu B.Y., Stirling B.V., Garibov M., Kerns H.M., Humblet-Baron S., Astrakhan A., Rawlings D.J. Development of B-lineage predominant lentiviral vectors for use in genetic therapies for B cell disorders. Mol. Ther. 2011;19:515–525. doi: 10.1038/mt.2010.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ackermann M., Lachmann N., Hartung S., Eggenschwiler R., Pfaff N., Happle C., Mucci A., Göhring G., Niemann H., Hansen G. Promoter and lineage independent anti-silencing activity of the A2 ubiquitous chromatin opening element for optimized human pluripotent stem cell-based gene therapy. Biomaterials. 2014;35:1531–1542. doi: 10.1016/j.biomaterials.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 55.Crofford L.J., Nyhoff L.E., Sheehan J.H., Kendall P.L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016;12:763–773. doi: 10.1586/1744666X.2016.1152888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kil L.P., de Bruijn M.J., van Nimwegen M., Corneth O.B., van Hamburg J.P., Dingjan G.M., Thaiss F., Rimmelzwaan G.F., Elewaut D., Delsing D. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. 2012;119:3744–3756. doi: 10.1182/blood-2011-12-397919. [DOI] [PubMed] [Google Scholar]
- 57.Katewa A., Wang Y., Hackney J.A., Huang T., Suto E., Ramamoorthi N., Austin C.D., Bremer M., Chen J.Z., Crawford J.J. Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNα-driven lupus nephritis. JCI Insight. 2017;2:e90111. doi: 10.1172/jci.insight.90111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Becker-Herman S., Meyer-Bahlburg A., Schwartz M.A., Jackson S.W., Hudkins K.L., Liu C., Sather B.D., Khim S., Liggitt D., Song W. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J. Exp. Med. 2011;208:2033–2042. doi: 10.1084/jem.20110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson S.W., Scharping N.E., Kolhatkar N.S., Khim S., Schwartz M.A., Li Q.Z., Hudkins K.L., Alpers C.E., Liggitt D., Rawlings D.J. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J. Immunol. 2014;192:4525–4532. doi: 10.4049/jimmunol.1400098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ehlers M., Fukuyama H., McGaha T.L., Aderem A., Ravetch J.V. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J. Exp. Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Müller-Kuller U., Ackermann M., Kolodziej S., Brendel C., Fritsch J., Lachmann N., Kunkel H., Lausen J., Schambach A., Moritz T., Grez M. A minimal ubiquitous chromatin opening element (UCOE) effectively prevents silencing of juxtaposed heterologous promoters by epigenetic remodeling in multipotent and pluripotent stem cells. Nucleic Acids Res. 2015;43:1577–1592. doi: 10.1093/nar/gkv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knight S., Zhang F., Mueller-Kuller U., Bokhoven M., Gupta A., Broughton T., Sha S., Antoniou M.N., Brendel C., Grez M. Safer, silencing-resistant lentiviral vectors: optimization of the ubiquitous chromatin-opening element through elimination of aberrant splicing. J. Virol. 2012;86:9088–9095. doi: 10.1128/JVI.00485-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mauro V.P., Chappell S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014;20:604–613. doi: 10.1016/j.molmed.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou Z., Dang Y., Zhou M., Li L., Yu C.H., Fu J., Chen S., Liu Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA. 2016;113:E6117–E6125. doi: 10.1073/pnas.1606724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Du S.W., Jacobs H.M., Arkatkar T., Rawlings D.J., Jackson S.W. Integrated B cell, Toll-like, and BAFF receptor signals promote autoantibody production by transitional B cells. J. Immunol. 2018;201:3258–3268. doi: 10.4049/jimmunol.1800393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Enghard P., Humrich J.Y., Chu V.T., Grussie E., Hiepe F., Burmester G.R., Radbruch A., Berek C., Riemekasten G. Class switching and consecutive loss of dsDNA-reactive B1a B cells from the peritoneal cavity during murine lupus development. Eur. J. Immunol. 2010;40:1809–1818. doi: 10.1002/eji.200940050. [DOI] [PubMed] [Google Scholar]
- 67.Hardy R.R. B-1 B cell development. J. Immunol. 2006;177:2749–2754. doi: 10.4049/jimmunol.177.5.2749. [DOI] [PubMed] [Google Scholar]
- 68.Jacobs H.M., Thouvenel C.D., Leach S., Arkatkar T., Metzler G., Scharping N.E., Kolhatkar N.S., Rawlings D.J., Jackson S.W. Cutting edge: BAFF promotes autoantibody production via TACI-dependent activation of transitional B cells. J. Immunol. 2016;196:3525–3531. doi: 10.4049/jimmunol.1600017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X., Rivière I. Genetic engineering and manufacturing of hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2017;5:96–105. doi: 10.1016/j.omtm.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Appelt J.U., Giordano F.A., Ecker M., Roeder I., Grund N., Hotz-Wagenblatt A., Opelz G., Zeller W.J., Allgayer H., Fruehauf S., Laufs S. QuickMap: a public tool for large-scale gene therapy vector insertion site mapping and analysis. Gene Ther. 2009;16:885–893. doi: 10.1038/gt.2009.37. [DOI] [PubMed] [Google Scholar]
- 71.Mohamed A.J., Yu L., Bäckesjö C.M., Vargas L., Faryal R., Aints A., Christensson B., Berglöf A., Vihinen M., Nore B.F., Smith C.I. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009;228:58–73. doi: 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 72.Nisitani S., Satterthwaite A.B., Akashi K., Weissman I.L., Witte O.N., Wahl M.I. Posttranscriptional regulation of Bruton’s tyrosine kinase expression in antigen receptor-stimulated splenic B cells. Proc. Natl. Acad. Sci. USA. 2000;97:2737–2742. doi: 10.1073/pnas.050583597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rawlings D.J., Witte O.N. The Btk subfamily of cytoplasmic tyrosine kinases: structure, regulation and function. Semin. Immunol. 1995;7:237–246. doi: 10.1006/smim.1995.0028. [DOI] [PubMed] [Google Scholar]
- 74.Yu L., Mohamed A.J., Simonson O.E., Vargas L., Blomberg K.E., Björkstrand B., Arteaga H.J., Nore B.F., Smith C.I. Proteasome-dependent autoregulation of Bruton tyrosine kinase (Btk) promoter via NF-κB. Blood. 2008;111:4617–4626. doi: 10.1182/blood-2007-10-121137. [DOI] [PubMed] [Google Scholar]
- 75.da Cunha-Bang C., Niemann C.U. Targeting Bruton’s tyrosine kinase across B-cell malignancies. Drugs. 2018;78:1653–1663. doi: 10.1007/s40265-018-1003-6. [DOI] [PubMed] [Google Scholar]
- 76.Lucas F., Woyach J.A. Inhibiting Bruton’s tyrosine kinase in CLL and other B-cell malignancies. Target. Oncol. 2019;14:125–138. doi: 10.1007/s11523-019-00635-7. [DOI] [PubMed] [Google Scholar]
- 77.Wu J., Liu C., Tsui S.T., Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J. Hematol. Oncol. 2016;9:80. doi: 10.1186/s13045-016-0313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hutcheson J., Vanarsa K., Bashmakov A., Grewal S., Sajitharan D., Chang B.Y., Buggy J.J., Zhou X.J., Du Y., Satterthwaite A.B., Mohan C. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res. Ther. 2012;14:R243. doi: 10.1186/ar4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu K.R., Natanson H., Dunbar C.E. Gene editing of human hematopoietic stem and progenitor cells: promise and potential hurdles. Hum. Gene Ther. 2016;27:729–740. doi: 10.1089/hum.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Z.Y., Thrasher A.J., Zhang F. Gene therapy and genome editing for primary immunodeficiency diseases. Genes Dis. 2019;7:38–51. doi: 10.1016/j.gendis.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dai X., James R.G., Habib T., Singh S., Jackson S., Khim S., Moon R.T., Liggitt D., Wolf-Yadlin A., Buckner J.H., Rawlings D.J. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J. Clin. Invest. 2013;123:2024–2036. doi: 10.1172/JCI66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ng Y.Y., Baert M.R., Pike-Overzet K., Rodijk M., Brugman M.H., Schambach A., Baum C., Hendriks R.W., van Dongen J.J., Staal F.J. Correction of B-cell development in Btk-deficient mice using lentiviral vectors with codon-optimized human BTK. Leukemia. 2010;24:1617–1630. doi: 10.1038/leu.2010.140. [DOI] [PubMed] [Google Scholar]
- 83.Moreau T., Bardin F., Barlogis V., Le Deist F., Chabannon C., Tonnelle C. Hematopoietic engraftment of XLA bone marrow CD34+ cells in NOG/SCID mice. Cytotherapy. 2009;11:198–205. doi: 10.1080/14653240802716616. [DOI] [PubMed] [Google Scholar]
- 84.Futatani T., Miyawaki T., Tsukada S., Hashimoto S., Kunikata T., Arai S., Kurimoto M., Niida Y., Matsuoka H., Sakiyama Y. Deficient expression of Bruton’s tyrosine kinase in monocytes from X-linked agammaglobulinemia as evaluated by a flow cytometric analysis and its clinical application to carrier detection. Blood. 1998;91:595–602. [PubMed] [Google Scholar]
- 85.Maglione P.J., Simchoni N., Cunningham-Rundles C. Toll-like receptor signaling in primary immune deficiencies. Ann. N Y Acad. Sci. 2015;1356:1–21. doi: 10.1111/nyas.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marron T.U., Martinez-Gallo M., Yu J.E., Cunningham-Rundles C. Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines. J. Allergy Clin. Immunol. 2012;129:184–190.e1–4. doi: 10.1016/j.jaci.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pal Singh S., Dammeijer F., Hendriks R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer. 2018;17:57. doi: 10.1186/s12943-018-0779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ren L., Campbell A., Fang H., Gautam S., Elavazhagan S., Fatehchand K., Mehta P., Stiff A., Reader B.F., Mo X. Analysis of the effects of the Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib on monocyte Fcγ receptor (FcγR) function. J. Biol. Chem. 2016;291:3043–3052. doi: 10.1074/jbc.M115.687251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Novoa E.M., Jungreis I., Jaillon O., Kellis M. Elucidation of codon usage signatures across the domains of life. Mol. Biol. Evol. 2019;36:2328–2339. doi: 10.1093/molbev/msz124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K.E., Graveley B.R., Coller J. Codon optimality is a major determinant of mRNA stability. Cell. 2015;160:1111–1124. doi: 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Astrakhan A., Sather B.D., Ryu B.Y., Khim S., Singh S., Humblet-Baron S., Ochs H.D., Miao C.H., Rawlings D.J. Ubiquitous high-level gene expression in hematopoietic lineages provides effective lentiviral gene therapy of murine Wiskott-Aldrich syndrome. Blood. 2012;119:4395–4407. doi: 10.1182/blood-2011-03-340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.