

Abstract
Oncolytic adenoviruses have become ideal agents in the path toward treating cancer. Such viruses have been engineered to conditionally replicate in malignant cells in which certain signaling pathways have been disrupted. Other than such oncolytic properties, the viruses need to activate the immune system in order to sustain a long-term response. Therefore, oncolytic adenoviruses have been genetically modified to express various immune-stimulatory agents to achieve this. However, genetically modifying adenoviruses is very time consuming and labor intensive with the current available methods. In this paper, we describe a novel method we have called GAMER-Ad to genetically modify adenovirus genomes within 2 days. Our method entails the replacement of the gp19k gene in the E3 region with any given gene of interest (GOI) using Gibson Assembly avoiding the homologous recombination between the shuttle and the parental plasmid. In this manuscript as proof of concept we constructed and characterized three oncolytic adenoviruses expressing CXCL9, CXCL10, and interleukin-15 (IL-15). We demonstrate that our novel method is fast, reliable, and simple compared to other methods. We anticipate that our method will be used in the future to genetically engineer oncolytic but also other adenoviruses used for gene therapy as well.
Keywords: adenoviruses, Gibson Assembly, gene therapy, oncolytic viruses, molecular cloning
Graphical Abstract
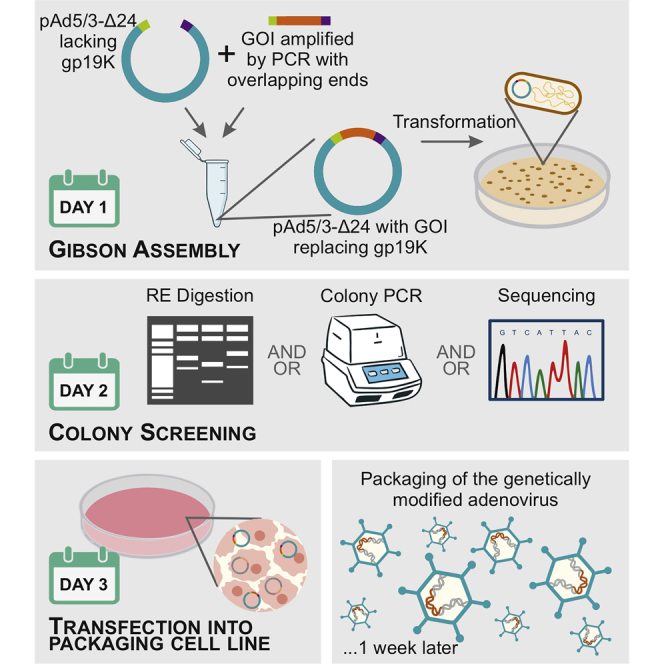
GAMER-Ad is a novel method describing how to engineer adenovirus platforms to express any gene of interest in just 2 days for gene therapy or oncolytic purposes. It consists in using the well-described cloning method Gibson Assembly to replace the gp19k region with a gene of interest.
Introduction
In the clinic, anti-cancer agents target rapidly dividing cells or single genetic mutations. Advancements in genome sequencing have led us to understand that cancer is not monogenic but rather a complex and heterogeneous disease.1 This explains why the use of single genetic mutation agents over the past decades have not yielded significant full response rates or cures as once expected. Therefore, scientists have stopped hunting for individual tumor suppressor genes or oncogenes and started investigating methods in disrupting whole tumorigenic biological pathways.2 Oncolytic viruses are the ideal agents in achieving this, because such viruses are able to thrive in tumor cells, where such malignant pathways have been activated or disrupted, and exploit metabolic pathways that characterize tumorigenesis.2 Also, oncolytic viruses have extensively been shown to stimulate systemic host immune responses. The tumor microenvironment is immunosuppressive and boosting the immune system has been observed to have significant anti-tumor effects.2 Hence, the dual mechanism oncolytic viruses possess makes them interesting therapy agents.
To date, only one oncolytic virus has been granted US Food and Drug Administration (FDA) approval for treatment despite years of extensive investigation.3 One of the reasons is that oncolytic viruses have been generally seen as direct tools for killing cancer due to their tumor-specific tropism. A growing body of evidence has shown that the ability of the virus to activate the immune system is a key attribute with regard to long-term antitumor effects.4 Therefore, to make more significant advances with such therapies there has been a shift in focus from viewing oncolytic viruses not solely as direct oncolytic tools but also as immunotherapies. Scientists have equipped oncolytic viruses with multiple immune-stimulatory molecules that have enhanced anti-tumor effects. For example, oncolytic viruses have been engineered to express molecules like interleukins (IL-2, IL-12, IL-15, and IL-18), chemokines (CCL5, CCL19, CCL20, and CCL21), immune-activating ligands (CD40L), bi-specific T cell engager molecules, and much more (all reviewed in Ylösmäki et al.2).
Such oncolytic viruses range from adenoviruses, herpes simplex viruses, vaccinia, Newcastle disease virus, and much more. However, adenoviruses have drawn much of the attention due to their numerous advantages. First, the adenoviral genome is highly stable, which leads to easier large-scale production.5 Also, they are able to infect most human cells due to the high expression of the viral coxsackievirus and adenovirus receptor (CAR) throughout the human body. This high infectivity subsequently leads to higher expression levels and sustainability of transgenes. Moreover, anti-adeno immunity can be circumvented by attenuating adenovirus vectors. In addition, because of the extensive research done on adenoviruses, the safety dosing and routes of administrations have been established.6 Finally, the tropism of the virus can be easily manipulated by modifying its viral capsid.7 For example, Zsolt and colleagues8, 11 were able to redirect adenoviruses to expand solely in tumor cells by replacing the adenoviral fiber knob (responsible for binding to CAR on target cells) with a T cell receptor specific to a unique class of tumor antigens.
Despite all the advantages, starting from a cDNA of interest to modifying an adenovirus able to express such genes involves many steps and significant time to be invested. The most common method used is homologous recombination using a shuttle plasmid and the full adenovirus genome. However, this methodology is very time consuming, inefficient, and labor intensive. Here we present GAMER-Ad (Gibson Assembly Mediated Recombination), a novel method for engineering recombinant viruses that is simple, highly efficient, and takes around 4 days to obtain an adeno full genome with the gene of interest (GOI) inserted (Figure 1). Our method is based on directly substituting the GOI into the E3-gp19k region via the well-known molecular cloning method Gibson Assembly (GA).9,10 Gibson Assembly is a molecular cloning method allowing the assembly of multiple DNA fragments in one isothermal reaction containing three enzymes: exonuclease, DNA polymerase, and DNA ligase. The method requires a base overlap of 20–40 nucleotides among the fragments to be assembled. This overlap will allow the exonucleases to chew the 5′ ends, allowing the seamless joining of adjacent fragments. In this study, we added overlapping regions of the adenovector backbone into the 3′ and 5′ ends of the GOI, and, using the already commercially available GA Master Mix, we were able to replace the gp19k gene in the E3 region of the adenovector backbone with our GOI. In this paper, we describe the use of this method for the creation of three genetically modified adenoviruses to express CXCL9, CXCL10, and IL-15.
Figure 1.

GAMER-Ad versus homologous recombination
A schematic representation comparing the time required to genetically engineer adenoviruses using GAMER-Ad or the commonly used homologous recombination. The GAMER-Ad method starts by first excising E3 using BarI and SrfI following the amplification and insertion of 40 overlapping nucleotides to the fragment to be inserted (GOI) into the adenovector. Both components are then assembled using the Gibson Assembly (GA) reaction and transformed in competent E. coli. To select positive colonies, screening of recombinant plasmids can be done using a colony PCR. This technique considerably simplifies and speeds up the process and shows very high efficiency. As for homologous recombination, the optimized procedure requires the cloning of the shuttle plasmid with homologous arms by PCR, linearization of the shuttle plasmid, transformation of E. coli BJ5183 (a recombination proficient E. coli strain) with the shuttle plasmid and backbone plasmid by electroporation, screening for positive bacteria colonies, and finally transformation of the successfully recombined plasmid into a recombination-incompetent strain of E. coli to obtain high yields of plasmid DNA.
Results
Excising the E3 region and constructing the GOI
The genetic modification of the described viruses was possible by using a vector containing the Ad5/3-Δ24-E3+ genomic DNA. Our novel methodology consists of replacing the gp19k region of the E3 gene with our GOI using the GA method. First, the E3 region from the adenovirus was removed by taking advantage of the convenient and inherent restriction enzymes flanking the gene, BarI and SrfI (Figure 3A). After digestion, products were loaded on an agarose gel, and a clear band between 3 kb and 4 kb can be observed, indicating that the E3 gene (3,398 bp) was excised successfully (Figure 3B).
Figure 3.

Releasing the E3 region and constructing the GOI
(A) A schematic representation of the linearization of the Ad5/3 genome using BarI and SrfI. (B) Ad5/3 was linearized using the mentioned restriction enzymes and loaded onto an agarose gel in lane 2. Lane 1 represents a 1 kb gene ruler from Thermo Fisher. (C) The CMV, poly(A), and chemokine fragments were amplified and loaded onto an agarose gel in lanes 2, 3, and 5, respectively. Lanes 1 and 4 were loaded with 1 kb gene ruler from Thermo Fisher. (D) Schematic representation of the assembly of the GOI. (E) After assembling all three fragments of the GOI using the GA, the final fragment containing CMV-cytokine-poly(A) was amplified using PCR and loaded on a gel. Lane 2 represents the amplified GOI, while lane 1 represents a 1 kb gene ruler from Thermo Fisher.
The GOI to replace the gp19k region consisted of a Citomegalovirus (CMV), coding sequence of IL-15, and a poly(A). The CMV and poly(A) entailed 40 overlapping nucleotides to the 5′ and 3′ ends of the excised Ad-5/3-Δ24-E3+, respectively, to ensure a correct assembly. Additionally, the poly(A) fragment also contained all the coding sequences of the E3 region except for the gp19k gene. As shown in Figure 2, all three components were amplified via PCR. Moreover, using our primers, 40 overlapping nucleotides were added to the 5′ and 3′ ends of the IL-15 fragment allowing assembly with poly(A) and CMV fragments (Figure 3C). Using the GA Master Mix, all three components were assembled together. A PCR using a forward primer flanking the 5′ end of the CMV and reverse primer flanking the 3′ of poly(A) demonstrated that assembly was successful and in the correct orientation (Figure 3D).
Figure 2.

Cloning strategy of GAMER-Ad
The GOI was constructed made up of three components: a CMV starting sequencing, followed by the coding sequencing of the chemokines, and finally ending in a poly(A) tail. The chemokine coding sequences contained 40 nucleotides in their 5′ and 3′, which are homologous to the ends of the CMV and poly(A) fragment, respectively. The CMV and poly(A) tail also contain all the genes required for E3 except the gp19k gene. All three fragments were then assembled together using the GA method. The Ad-5/3 genome was excised with BarI and SrfI to liberate the E3 region. The GOI was then inserted into the excised genome, again using the GA.
Assembling the GOI into the adenovirus genome
Following adenovector digestion and GOI construction, the two components were assembled together using again the GA Master Mix. The GA products were transformed in E. coli cells, and eight colonies were taken for colony PCR. Using primers flanking the CMV and poly(A) tail (only found in the GOI), we saw that from eight colonies, six of them had the GOI (Figure 4A). Three positive colonies (based on colony PCR) were selected, grown in Lysogeni Broth (LB) medium, and the DNA was isolated. To further check whether replacement of E3 with the GOI in the adenovector was successful, constructs underwent restriction enzyme analysis and Sanger sequencing. Isolated DNA from the three colonies and unmodified adenovector were digested using EcoRI. In case of success in replacing gp19k with the GOI, a band should be observed at 2,075 bp, compared to a 2,718 bp band when gp19k remains (Figure 4B). Digestion with EcoRI demonstrated that gp19k was replaced in all three selected colonies (Figure 4C). Sanger sequencing was then performed and confirmed that all the three selected clones had the gp19k gene replaced by the GOI (Figure 4D). To further illustrate the rapidness and simplicity of our method, we engineered two new Ad5/3 viruses expressing CXCL9 and CXCL10. The same process was used, and each virus took an average of 2 days to be cloned.
Figure 4.

Screening for positive colonies for the IL-5-expressing virus
(A) After transformation of GA products, eight colonies were chosen (lanes 2–9) and a colony PCR was conducted. Primers flanking the CMV and IL-15 coding region were used. As a positive control, the GA of CMV-IL-5-poly(A) (lane 10) was used. Lane 1 represents a 1 kb gene ruler from Thermo Fisher. (B) Simulation of expected lanes where unmodified adenovector is cut with EcoRI (lane 3) compared to positive adenovirus clones successfully having the gp19k replaced with the GOI (lane 2). Lane1 represents a 1 kb gene ruler from Thermo Fisher. (B) Actual representation of three colonies transformed with the assembled adenovirus containing the GOI (lanes 2, 3, and 4). Lane 5 represents wild-type adenovirus cut with EcoRI, while lane 1 represents a 1 kb gene ruler from Thermo Fisher. (C) Three samples of Sanger sequencing from one of the positive sequences. Original sequence represents the reference sequence of IL-15.
Viral production and Ad5/3-Δ24 contamination check
After successfully constructing adenovectors expressing CXCL9, CXCL19, and IL-15, we linearized the genomes with PacI to release the vector sequence and expose the viral ITRs required for the initiation of the viral DNA replication in the host cell. The digestion product was transfected into A549. Viral plaques appeared on the cell layer 7 days post transfection, suggesting successful rescue of recombinant viruses (Figure 5A). Packaging cells were lysed to release the virus, and the viral crude was utilized for further amplification of the virus. Viruses were successfully purified using CsCl, and the viral particle titer and infectious unit titer were determined.
Figure 5.

Viral production with lack of Ad5/3-Δ24 contamination
(A) Representative images of A549 cells transfected with IL-15-expressing adenovirus and wild-type virus. Images were taken 9 days post-transfection at two different magnification, 10× and 40×. (B) Ad5/3-Δ24 contamination was checked by amplifying the gp19k region (Ad5/3-Δ24) or the CMV and poly(A) region (GOI). A simulation of what should be expected is presented and type of samples are annotated. Lane MW represents a 1 kb gene ruler from Thermo Fisher. Lanes 4 and 9 represent Ad5/3-Δ24 virus. Lanes 1 and 6 represent CXCL9-expressing adenovirus. Lanes 2 and 7 represent CXCL10-expressing adenovirus. Lanes 3 and 8 represent IL-15-expressing adenovirus. (C) The actual representation of the wild-type contamination PCR assay. Lane 1 represents a 1 kb gene ruler from Thermo Fisher. Lanes 5 and 9 represent the unmodified Ad5/3-Δ24 virus. Lanes 2 and 6 represent CXCL9-expressing adenovirus. Lanes 3 and 7 represent CXCL10-expressing adenovirus. Lanes 4 and 8 represent IL-15-expressing adenovirus.
After, we sought to determine whether there was an Ad5/3-Δ24 contamination in the purified preparations of the cloned chemokine expressing Ad-5/3. We designed two different PCR protocols, with the first protocol having primers flanking the gp19k gene and the second having primers binding to CMV and poly(A) (only found in the cloned Ad5/3-Δ24) (Figure 5B). Purified virus samples from the cytokine-expressing viruses and unmodified Ad5/3-Δ24 were boiled to release the DNA from the viral capsid and were used as template for the PCRs. When primers flanking the gp19k gene were used, a band was only seen with the Ad5/3-Δ24 virus. As expected, when primers binding to the CMV and poly(A) were added, corresponding bands were seen in all cloned Ad-5/3 viruses, but no band was observed with the Ad-5/3-Δ24 virus (Figure 5C). Hence, this demonstrates that our method does lead to Ad5/3-Δ24 virus contamination.
Viral replication and fitness of cloned viruses
Following the cloning and purification of Ad-5/3 viruses expressing CXCL9, CXL10, or IL-15, we wanted to check that method and modification used did not affect the oncolytic fitness or replication of the viruses. All viruses have a well-known 24-bp deletion in the E1A region conditioning such viruses to replicate only in Rb-deficient cells. As a result, two tumor models, lung carcinoma cell (A549) and triple-negative breast cancer cells (MDA-MB-436), were infected with our cloned viruses and unmodified Ad5/3-Δ24 virus. Oncolysis was observed at day 3 post infection in a dose-dependent fashion in both cell lines (Figures 6A and 6B). Oncolysis levels of the cloned viruses resembled those of the unmodified virus, indicating that our cloning method did not affect oncolytic potency or virus replication. To further corroborate this, two murine cell lines, B16F10 and 4T1, were also studied. Human adenoviruses serotype 5 can infect murine cell lines but are unable to replicate. As expected, no cell death was observed with either murine cell line when infected with the unmodified and cytokine-expressing Ad-5/3 viruses (Figures 6C and 6D). These data illustrate that our novel cloning methodology does not affect oncolytic fitness or replication.
Figure 6.

Oncolytic fitness
(A–D) Cell viability assay of (A) A549, (B) MDA-MB-436, (C) B16F10, and (D) B16F1 cell lines. Cell lines were infected with CXCL9 (blue), CXCL10 (red), and IL-15 (green)-expressing adenoviruses along with wild-type virus (purple). Cell viability was checked after 3 days using an (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay. The data are presented as mean ± SD (n = 3).
Chemokine expression and migration induction from cloned viruses
The next step of the cloned viruses’ in vitro characterization was to investigate the levels of expression of the cytokine upon virus infection. MDA-MB-436 cells were infected with the unarmed and the cytokine viruses, separately. The cell’s supernatant was collected at day 2 post infection. All cloned viruses were able to induce expression and secretion of the designated chemokine or cytokine at levels ranging from 0.9–37 ng (Figure 7A).
Figure 7.

Functional activity of chemokine-expressing viruses
(A) Chemokines from CXCL9, CXCL10, and IL-15-expressing viruses were measured using an ELISA. A549 cells were infected with 100 MOI of the viruses indicated, and at day 2 post-infection the supernatant was collected and tested. (B) Migration was assessed using a Transwell system. Chemokine-expressing viruses were added to MDA-MB-436 cells at MOI 100 at the lower chamber. To the top chamber, calcein green-labeled PBMCs were added, and at day 2 the number of cells at the bottom (migrated cells) were counted manually. (C) Visual representation of migrated green labeled PBMCs. Scale bar, 400 nm.
CXCL9, CXCL10, and IL-15 have a common role in T cell migration. To test whether the secreted chemokines/cytokines from our viruses were functional, we infected MDA-MB-436 cells with the cytokine-encoding viruses and tested peripheral blood mononuclear cell (PBMC) migration using a Transwell system. Calcein green-labeled PBMCs were placed on top of Transwells and migrated green PBMCs were then counted on the lower chamber. When MDA-MB-436 cells were infected with our cloned viruses, an increase in migratory PBMCs was observed compared to uninfected cells or cells infected with the unarmed virus (Figures 7B and 7C). The highest increase in PBMC migration was observed with Ad5/3-CXCL9, followed by Ad5/3-CXCL10. In conclusion, our cloned viruses are able to express functional chemokines/cytokines that induce lymphocyte migration.
Discussion
Oncolytic adenoviruses have become ideal agents to treat cancer due to their specific tumor tropism. To achieve a long-term response, such viruses need to induce an anti-tumor response.4 In doing so, scientist have armed oncolytic adenoviruses with various immune-stimulating agents. Yet, the current methods to genetically modify the adenoviruses are rather complex, time consuming, and expensive. In spite of this, we adapted a novel method to genetically modify oncolytic adenoviruses using GA, which is faster, cheaper, and more convenient than other methods.
GA has been previously used, to a certain extent, to construct adenovirus genomes. However, the novelty of GAMER-Ad is that it can be done with routinely used Ad-5, Ad-5/3, and Ad-3 viruses. Moreover, the method entails replacing the gp19k gene from E3 region with a GOI, which has not been done before by using GA. Usually, genetic manipulations are done in limited regions that are not essential for viral production, such as E1, E2A, E3, and E4.5,11,12 Yet, much attention has been drawn to the E3 region for genetic manipulation. The E3 region is a complex region, with multiple-gene transcription units expressing around seven different E3 proteins.13 This complexity causes such proteins to be expressed at varying times and levels during the course of infection. If these genes were to be substituted by a therapeutic GOI, this would provide a flexible system in which multiple transgenes could be expressed in varying and predictable levels.14 Additionally, the E3 proteins have well-known roles in immunomodulation; hence, removing such genes creates an opportunity to direct the immune responses to synergize with the therapeutic GOI.15
Currently, many labs replace the adenoviral genes with GOI via homologous recombination using a shuttle plasmid and full-length adenoviral backbones.16 However, this process has a low efficiency, has wild-type (WT) contamination, and is time and labor intensive. Moreover, recombination takes place in a specialized E. coli strain, BJ5183, containing a Rec-A deficiency.17,18 Even though higher yields of homologous recombination are achieved, there is also a higher chance of secondary recombination events giving rise to unwanted repeat regions or secondary structures to the adenoviral genome. Hence, scientists have directed their efforts in developing easier and more efficient methods for generating recombinant adenoviruses. These methods include directly ligating cDNA into E1-deleted adenoviral genomes using Cre-loxP shuttles19 and E. coli-recombinant systems.20 These methods have simplified this process and fixed wild-type viral DNA input problems. Nevertheless, these methods are still time consuming and not very efficient and require several steps in planning.
We have shown that our method is robust, with minimal false positives during the screening stage. An average of 75% of the colonies from the transformed GA products of Ad5/3-Il-15 tested had IL-15 assembled to the excised adenovirus genome. Usually, the official GA method entails that the overlapping ends have around 15–20 bp.21 In our method, 40 overlapping nucleotides seem to be very efficient in assembling products onto the adenovirus genome. Whether this could be further optimized by increasing or decreasing the overlapping region remains to be tested. Moreover, we have shown that no Ad5/3-Δ24 contamination is found while expanding the viruses. Finally, with three different viruses we demonstrated that our method is rapid and time efficient.
The full GOI fragments to be inserted into the adenovirus genome had a size of more than 4,000 bp. Designing a GOI every time can be inconvenient with respect to time and economic efficiency. To help overcome this, we amplified the CMV and poly(A) fragments separately and assembled them with the three different cytokine/chemokine coding sequences. This was done to demonstrate the flexibility of this method and the adaptability of it where only the coding sequence of the therapeutic gene is required for future manipulations.
Beyond being one of the most widely studied viruses in clinical development for cancer therapy (source: ClinicalTrials.gov, URL: https://clinicaltrials.gov/; search criteria: condition or disease: cancer; other terms: oncolytic virus; search date: November 26, 2019), adenoviruses have taken the main stage as the most effective vector for gene delivery due to their numerous advantages. Due to the appealing advantages adenoviruses provide, they have become the most-used gene-delivery vectors in clinical trials, accounting for more than 20% of all gene therapy trials.22 Due to the presence of the inherent restriction enzymes SrfI and BarI on multiple adenovirus serotypes (such as Ad5, Ad3, Ad6, and Ad2), our method can be applied in most adenoviruses used for gene therapy.
In conclusion, we have demonstrated a novel method to replace the gp19k gene with a therapeutic GOI. The present work compiles and describes the main steps necessary, from cloning to the production of three novel armed oncolytic adenoviruses, followed by an extensive characterization of the constructed viruses. Considering the discussed improvements, this methodology can be adopted for the generation of a limitless range of viruses to be used for different applications. We believe that our method will help the scientific community progress in the design and construction of novel armed oncolytic adenoviruses.
Material and methods
Cell lines and viral genomes
Human lung cancer cell line A549, human breast cancer cell line MDA-MB-436, and murine skin cancer cell lines B16F1 and B1610 were purchased from the American Type Culture Collection (ATCC). All cell lines were cultured under appropriate conditions and regularly checked for mycoplasma contamination. In brief, A549 and MDA-MB-436 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), while B16F1 and B16F10 were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium. All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
In this study, a plasmid containing the adenovirus genome pAd5/3-Δ24-E3+ was used as starting material. The plasmid contains all machinery necessary for plasmid amplification, including kanamycin resistance gene, and the genome of adenovirus serotype 5. This plasmid in particular contains a 24 bp deletion in the viral early gene E1A (Δ24) that enables the virus to replicate in Rb-deficient cells, and the knob of the fiber in the late gene L5 is replaced by the knob of a serotype 3 adenovirus.
Plasmid construction
A schematic of the plasmid construction can be found in Figure 2. First, poly(A) and CMV sequences were synthesized by Thermo Fisher. The poly(A) and CMV sequences contained 40 overlapping nucleotides to the adenovirus genome in the 3′ and 5′ ends, respectively. Also, the poly(A) fragment contained in all the genes of the E3 region except for the gp19k gene. Sequences of the poly(A) and CMV fragments can be found in Figure S1. The poly(A) and CMV fragments were amplified with CMV-Fwd and poly(A)-reverse (Rv) primers Phusion High-Fidelity DNA Polymerase (Thermo Fisher). Coding sequences of CXCL9, CXCL10, and IL-5 were synthesized and cloned in pcDNA 3.3 topo vector by Thermo Fisher. CXCL9, CXCL10, and IL-5 were then amplified, and 40 overlapping nucleotides to the CMV and poly(A) fragments were added to the 5′ and 3′ ends, respectively, using the primers mentioned in Table 1 using Phusion High-Fidelity DNA Polymerase (Thermo Fisher). Each chemokine fragment was then assembled with the poly(A) and CMV fragments as one fragment using the GA Master Mix (New England Biolabs) and according to manufacturer’s instructions. Products were added in a 3:1:1 (poly[A]:CMV:chemokine) molar ratio, where 50 ng of poly(A) was added. The assembled products were further amplified with the CMV-Fwd and poly(A)-Rv using Phusion High-Fidelity DNA Polymerase (Thermo Fisher) to produce enough DNA quantities to proceed to the next step.
Table 1.
Primers used for cloning the chemokine-expressing viruses
| Primer name | Primer sequence, 5′–3′ | Product (bp) | Aim |
|---|---|---|---|
| Fw-CXCL9 | GATAGGCAGCCTGCACCTGAGGAGTGCGGCCGCTTTATCAGGTGGTCTTCTTCTGCCT | 461 | amplify CXCL9 sequence and add overlaps |
| Rv-CXCL9 | ggtaggcgtgtacggtgggaggtctatataagcagagctgGCCACCATGAAGAAGAGCGGCGTGCT | ||
| Fw-CXCL10 | GATAGGCAGCCTGCACCTGAGGAGTGCGGCCGCTTTATCAGGGGCTCCTCTTGCTCCT | 374 | amplify CXCL10 sequence and add overlaps |
| Rv-CXCL10 | cgtgtacggtgggaggtctatataagcagagctgGCCACCATGAACCAGACCGCCATC | ||
| Fw-IL15 | GATAGGCAGCCTGCACCTGAGGAGTGCGGCCGCTTTATCAGCTGGTGTTGATGAACAT | 568 | amplify IL-15 sequence and add overlaps |
| Rv-IL15 | cgtgtacggtgggaggtctatataagcagagctgGCCACCATGAGGATCAGCAAGCCCCA | ||
| Rv-CMV | ATAGTGGGTGCGGATGGACAG | 692 | amplify CMV sequence |
| Fw-CMV | cagctctgcttatatagacctcccaccg | ||
| Fw-PolyA | GCCGAAGTTCAGATGACTAACTCAG | 2,543 | amplify poly(A) sequence |
| Rv-polyA | TGATAAAGCGGCCGCACTCCTCAGGTGC |
At the same time, the pAd5/3-Δ24-E3+ was then linearized with SrfI (New England Biolabs) and BarI (SibEnzyme), releasing the E3 region, and DNA was recovered by ethanol precipitation. The assembled CMV-cytokine-poly(A) fragments were inserted into the linearized adenovirus genome using the GA Master Mix (New England Biolabs) according to manufacturer’s instructions. A 3:1 (insert:vector) molecular ratio was used, where 1.3 μg of the linearized adenovirus genome was added. Approximately 2 μL of assembled products were transformed into DH5-alpha E. coli cells, spread on kanamycin-containing LB agar and incubated overnight at 37°C. Positive colonies were picked and grown on kanamycin selection LB medium. Plasmid DNA was isolated from each colony and analyzed using Sanger sequencing or restriction enzyme analysis using EcoRI.
Packaging and amplification of adenoviruses
Adenovirus genomes were first digested with PacI to release the kanamycin resistance gene. The linearized genome was then rescued by ethanol precipitation, and approximately 3 μg was transfected into A549 cells seeded in a T25 cell culture flask using Effectene (QIAGEN). When cytopathic effect (CPE) was observed, cultures were harvested, subjected to four freeze-thaw cycles, and centrifuged to remove the cellular debris, and the supernatant was preserved at 80°C and used as the seed virus. The next round of amplification was performed similarly, but this time using a T175 cell culture flask, and the last round of amplification was performed in a 10-layer cell culture multi-flask.
Purification of adenoviruses
Cells infected with the different adenoviruses were observed under the microscope daily, and once they displayed a clear CPE, cells were further detached from the flask by strongly tapping the cell culture flask. Cells were collected by centrifugation, and the cell pellets were resuspended in 7 mL of cultured medium and subjected to four freeze-thaw cycles. The lysates were further centrifuged to remove cell debris, and supernatant was collected. Viruses were purified using a two-stage CsCl gradient procedure followed by ultracentrifugation. The purified adenoviruses were dialyzed overnight against A195 buffer, formulated by Evans et al.23 Dialyzed viruses were aliquoted and stored at −80°C for further use.
Colony PCR
Single colonies were picked and added to 20 μL of MilliQ (MQ) water. Samples were then boiled at 95°C for 5 min. One microliter was used as a DNA template, and the following primers were used to conduct the PCR:
Forward (FW) primer: 5′-TGATGTTCTTCTCCTCCAGC-3′
RV primer: 5′-ATTATGCCCAGTACATGACC-3′
Viral particle titering
Viral particles were titered by measuring the optical density (OD) at 260 nm. First, virus preparations were mixed with virus lysis buffer (VBL) containing 10 mM Tris-HCL, 1 mM EDTA, and 0.5% SDS in a 1:3 (virus:VBL) ratio. Samples were incubated at 95°C for 15 min and absorbance at 260 nm was measured. VP was calculated using the following formula:
Infectious titer determination
A549 cells were seeded in a 24-well plate and infected with series of 10-fold dilutions of the virus. After 48 h of incubation at 37°C, cells were fixed with ice-cold methanol and washed with PBS containing 1% bovine serum albumin. Cells were incubated with mouse anti-hexon antibody, followed by secondary horseradish peroxidase (HRP)-conjugated antibody against mouse. Cells were incubated with 3,3' Diaminobenzidine (DAB) solution according to manufacturer’s instructions until a clear color formation was detected by naked eye. At this point, the reaction was quenched with PBS and the stained cells were counted under a BF microscope. The calculations of the infectious titer were done using the following formula:
where x is the average of infected cells per field, A(well) is the surface area of the well (in this case is equal to 190 mm2), A(field) is the surface area of the field, d is the dilution factor used, and V is the volume of virus dilution applied per well in milliliters.
Cell viability assay
Cells were plated in a 96-well plate, and on the following day cells were infected at different multiplicities of infection (MOIs). Three-day post-infection cell viability was assessed using a MTS assay and performed according to manufacturer’s protocol (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega) according to manufacturer’s instructions. Absorbance was measured at 490 nm using Varioskan LUX multimode microplate reader (Thermo Fisher).
Wild-type contamination assay
Virus preparations were first incubated at 95°C for 15 min to release the genomic DNA from the viral capsid. A PCR was performed using DreamTaq Polymerase (Thermo Fisher).
To analyze wild-type viruses, the following primers were used: 5′-tttcctcaataaagctgcgtc-3′ (reverse) and 5′-attcaagcaactctacgggct-3′ (forward). To analyze genetically modified viruses, the following primers were used: 5′-ccagtacatgaccttatg-3′ (reverse) and 5′-ttgtcccagccaatcag-3′ (forward).
Transwell migration assays
Migration was tested using a Transwell (Corning) with a 6.5 μM pore membrane. In the bottom (apical side) 100,000 MDA-MB-436 cells were added and infected at MOI 100. On the top (basolateral side), 105 PBMCs stained with calcein green (Thermo Fisher) according to manufacturer’s instructions, were added. After two days, green-labeled PBMCs that translocated to the bottom of the Transwell were counted manually.
Concentrations of cytokines
A549 cells were infected with 100 MOI of virus, and supernatant was collected. From the supernatant, the concentrations of CXCL9, CXCL10, and IL-15 were measured using the respective ELISA Duo Set test (R&D Systems) according to the manufacturer’s instructions.
Acknowledgments
F.H. thanks the Research Foundation of the University of Helsinki for funding his doctoral studies at the Faculty of Pharmacy, Helsinki University. E.Y. acknowledges the Academy of Finland (project no. 1317206). V.C. acknowledges the European Research Council under the Horizon 2020 framework (https://erc.europa.eu); ERC consolidator grant (agreement no. 681219); Jane and Aatos Erkko Foundation (project no. 4705796); HiLIFE Fellow (project no. 797011004); Cancer Finnish Foundation (project no. 4706116); and Magnus Ehrnrooth Foundation (project no. 4706235).
Author contributions
F.H., B.M., E.Y., and V.C. conceived and planned all the experiments. F.H., M.F., and Y.G. carried out most of the experiments. E.Y., M.F., S.F., J.C., B.M., M.F., and M.G. helped in carrying out most of the experiments. E.Y. and V.C. supervised the project. F.H., B.M., E.Y., M.G., and V.C. wrote and corrected the paper. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2021.01.014.
Supplemental information
References
- 1.Hyman D.M., Taylor B.S., Baselga J. Implementing Genome-Driven Oncology. Cell. 2017;168:584–599. doi: 10.1016/j.cell.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ylösmäki E., Cerullo V. Design and application of oncolytic viruses for cancer immunotherapy. Curr. Opin. Biotechnol. 2020;65:25–36. doi: 10.1016/j.copbio.2019.11.016. [DOI] [PubMed] [Google Scholar]
- 3.Pitt J.M., Marabelle A., Eggermont A., Soria J.C., Kroemer G., Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016;27:1482–1492. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- 4.Andtbacka R.H.I., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 5.Marelli G., Howells A., Lemoine N.R., Wang Y. Oncolytic viral therapy and the immune system: A double-edged sword against cancer. Front. Immunol. 2018;9:866. doi: 10.3389/fimmu.2018.00866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy M.A., Parks R.J. Adenovirus virion stability and the viral genome: size matters. Mol. Ther. 2009;17:1664–1666. doi: 10.1038/mt.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wold W.S., Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013;13:421–433. doi: 10.2174/1566523213666131125095046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon A.R., Hong J., Kim S.W., Yun C.O. Redirecting adenovirus tropism by genetic, chemical, and mechanical modification of the adenovirus surface for cancer gene therapy. Expert Opin. Drug Deliv. 2016;13:843–858. doi: 10.1517/17425247.2016.1158707. [DOI] [PubMed] [Google Scholar]
- 9.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 10.Lee C.S., Bishop E.S., Zhang R., Yu X., Farina E.M., Yan S., Zhao C., Zheng Z., Shu Y., Wu X. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017;4:43–63. doi: 10.1016/j.gendis.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sebestyen Z., de Vrij J., Magnusson M., Debets R., Willemsen R. An oncolytic adenovirus redirected with a tumor-specific T-cell receptor. Cancer Res. 2007;67:11309–11316. doi: 10.1158/0008-5472.CAN-07-0739. [DOI] [PubMed] [Google Scholar]
- 12.Flint S.J., Wewerka-Lutz Y., Levine A.S., Sambrook J., Sharp P.A. Adenovirus transcription. II. RNA sequences complementary to simian virus 40 and adenovirus 2DNA in AD2+ND1- and AD2+ND3-infected cells. J. Virol. 1975;16:662–673. doi: 10.1128/jvi.16.3.662-673.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly T.J., Jr., Lewis A.M., Jr. Use of nondefective adenovirus-simian virus 40 hybrids for mapping the simian virus 40 genome. J. Virol. 1973;12:643–652. doi: 10.1128/jvi.12.3.643-652.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkner K.L., Sharp P.A. Generation of adenovirus by transfection of plasmids. Nucleic Acids Res. 1983;11:6003–6020. doi: 10.1093/nar/11.17.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wold W.S.M., Tollefson A.E., Hermiston T.W. E3 transcription unit of adenovirus. Curr. Top. Microbiol. Immunol. 1995;199:237–274. doi: 10.1007/978-3-642-79496-4_13. [DOI] [PubMed] [Google Scholar]
- 16.Hawkins L.K., Johnson L., Bauzon M., Nye J.A., Castro D., Kitzes G.A., Young M.D., Holt J.K., Trown P., Hermiston T.W. Gene delivery from the E3 region of replicating human adenovirus: evaluation of the 6.7 K/gp19 K region. Gene Ther. 2001;8:1123–1131. doi: 10.1038/sj.gt.3301507. [DOI] [PubMed] [Google Scholar]
- 17.Wold W.S.M., Doronin K., Toth K., Kuppuswamy M., Lichtenstein D.L., Tollefson A.E. Immune responses to adenoviruses: viral evasion mechanisms and their implications for the clinic. Curr. Opin. Immunol. 1999;11:380–386. doi: 10.1016/S0952-7915(99)80064-8. [DOI] [PubMed] [Google Scholar]
- 18.He T.C., Zhou S., Da Costa L.T., Yu J., Kinzler K.W., Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West S.C. The processing of recombination intermediates: mechanistic insights from studies of bacterial proteins. Cell. 1994;76:9–15. doi: 10.1016/0092-8674(94)90168-6. [DOI] [PubMed] [Google Scholar]
- 20.Camerini-Otero R.D., Hsieh P. Homologous recombination proteins in prokaryotes and eukaryotes. Annu. Rev. Genet. 1995;29:509–552. doi: 10.1146/annurev.ge.29.120195.002453. [DOI] [PubMed] [Google Scholar]
- 21.Hardy S., Kitamura M., Harris-Stansil T., Dai Y., Phipps M.L. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan R., Li C., Jiang S., Ma L. A novel and simple method for construction of recombinant adenoviruses. Nucleic Acids Res. 2006;34:e89. doi: 10.1093/nar/gkl449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans R.K., Nawrocki D.K., Isopi L.A., Williams D.M., Casimiro D.R., Chin S., Zhu D.M., Shiver J.W., Volkin D.B. Development of stable liquid formulations for adenovirus-based vaccines. J. Pharm. Sci. 2004;93:2458–2475. doi: 10.1002/jps.20157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.