

Abstract
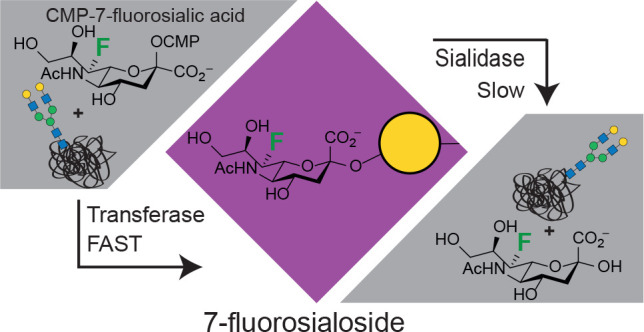
The maintenance of therapeutic glycoproteins within the circulatory system is associated, in large part, with the integrity of sialic acids as terminal sugars on the glycans. Glycoprotein desialylation, either by spontaneous cleavage or through host sialidases, leads to protein clearance, mainly through the liver. Thus, the installation of minimally modified sialic acids that are hydrolysis-resistant yet biologically equivalent should lead to increased circulatory half-lives and improved pharmacokinetic profiles. Here we describe the chemoenzymatic synthesis of CMP–sialic acid sugar donors bearing fluorine atoms at the 7-position, starting from the corresponding 4-deoxy-4-fluoro-N-acetylhexosamine precursors. For the derivative with natural stereochemistry we observe efficient glycosyl transfer by sialyltransferases, along with improved stability of the resultant 7-fluorosialosides toward spontaneous hydrolysis (3- to 5-fold) and toward cleavage by GH33 sialidases (40- to 250-fold). Taking advantage of the rapid transfer of 7-fluorosialic acid by sialyltransferases, we engineered the O-glycan of Interferon α-2b and the N-glycans of the therapeutic glycoprotein α1-antitrypsin. Studies of the uptake of the glyco-engineered α1-antitrypsin by HepG2 liver cells demonstrated the bioequivalence of 7-fluorosialic acid to sialic acid in suppressing interaction with liver cell lectins. In vivo pharmacokinetic studies reveal enhanced half-life of the protein decorated with 7-fluorosialic acid relative to unmodified sialic acid in the murine circulatory system. 7-Fluorosialylation therefore offers considerable promise as a means of prolonging circulatory half-lives of glycoproteins and may pave the way toward biobetters for therapeutic use.
Short abstract
7-Fluorosialylation offers considerable promise as a means of prolonging circulatory half-lives of glycoproteins and may pave the way toward biobetters for therapeutic use.
Therapeutic proteins are a fixture in the treatment of many serious medical conditions, such as cancer,1 diabetes,2 anemia,3 and Hepatitis C.4 Their clinical success has typically been attributed to the highly efficient, specific, and well-tolerated nature of these drugs.5 As such, this pharmaceutical industry sector has grown rapidly, both in terms of the market value and the number of approved drugs, and further increases are projected.5,6 Despite remarkable developments in formulation technology, protein therapeutics have certain drawbacks including intravenous administration, the inherent risk of immunogenicity,7 and generally more complex drug development and production processes.8
The vast majority of clinically relevant proteins are glycosylated.9,10 The biological function, life cycle, and biophysical properties of glycoproteins are highly dependent on their glycosylation patterns.10,11 Sialic acids cap the ends of glycan entities on glycoproteins, and their removal often leads to dramatically reduced plasma half-life.12 Removal of sialic acids occurs either spontaneously or by the action of sialidases (sialic acid hydrolyzing enzymes)13 and leads to exposure of the underlying galactose residues. This in turn triggers interaction with lectin receptors leading to clearance from the circulatory system. One such lectin is the hepatocyte asialoglycoprotein receptor (ASGPR), also called the Ashwell–Morell receptor, which is thought to recognize glycoproteins that present terminal galactose residues and enable their endocytosis. However, its physiological role in glycoprotein turnover is still under debate.14−16 Glycoproteins decorated with hydrolysis-resistant sialosides may evade lectin-mediated clearance and may hold promise for increased elimination half-lives. This may help solve drawbacks as longer circulatory retention could lead to less frequent intravenous injections and lower doses, thereby also reducing costs.
One approach to generation of metabolically stable sialosides is through thioglycosides, which are generally cleaved more slowly by both chemical and enzymatic hydrolysis compared to their natural O-linked counterparts. Accordingly, sulfur-linked sialosides have been used as probes to study binding affinity and specificity of sialic acid binding proteins,17,18 as well as in antigen generation toward vaccine production.19 Another approach to stabilization of glycosides is through incorporation of fluorine close to the anomeric center. Indeed 3-fluorosialic acid (3FSA) derivatives have been used as inhibitors to study both sialidases and sialyltransferases.20−26 Here, the close proximity of the fluorine atom to the anomeric carbon inductively destabilizes oxocarbenium ion-like transition states thereby greatly increasing the activation energy for bond cleavage. This approach works well in that hydrolysis of the 3-fluorosialosides is enormously slowed. However, the very low activity of the sialyltransferases in the transfer of 3FSA renders them essentially useless for enzymatic sialylation of complex glycan structures. Consequently complex chemoenzymatic approaches are needed to assemble the 3-fluorosialylated glycoprotein.27,28
Here, we explore the value of fluorine substitution at the 7-position of sialic acid. We have chosen the 7-position because, like the 3-position,28 it is close to the ring oxygen where there is a relative buildup of positive charge in the transition state. Thus, attachment of the highly electronegative fluorine at this position should have a destabilizing effect on the oxocarbenium ion-like transition state. Survey studies with several sialidases supported this assumption and confirmed that some several sialyltransferases could indeed transfer the modified sugar.29 A fluorine at C-6 would have an even bigger effect, but such substrates are “doubly anomeric” and inherently less stable and thus are not suitable. The non-natural donor CMP-7-deoxy-7-fluorosialic acid (CMP-7FSA) was prepared conveniently via a chemoenzymatic approach starting from 4-F-N-acetylglucosamine (4F-GlcNAc) and kinetic parameters for sialyl transfer determined for an array of sialyltransferases. In parallel, kinetic parameters for hydrolysis of a 7-fluorosialoside and its nonfluorinated parent by a set of sialidases were determined, along with rate constants for spontaneous hydrolysis. Finally, we enzymatically engineered the glycan structures of two proteins, interferon α-2b and α1-antitrypsin (A1AT). We used the glycoengineered A1AT to study uptake by liver cells in vitro and measured blood concentrations over time to derive pharmacokinetic profiles in mice.
Results and Discussion
Synthesis of the 7-fluorosialic acid derivatives was achieved by a variant of a published chemoenzymatic route.29 Our approach used a GlcNAc/ManNAc epimerase to isomerize the more readily synthesized fluoro-GlcNAc derivative and proceeded to the CMP-sialic acid stage. 4-Deoxy-4-fluoro-GlcNAc (1), 4-deoxy-4-fluoro-GalNAc (2), and 4-deoxy-4,4-difluoro-GlcNAc (3) were synthesized on a milligram to gram scale using a previously described concise, divergent route30 and tested as substrates for the two-enzyme (porcine N-acyl-d-glucosamine 2-epimerase (AGE) and sialic acid aldolase) one-pot conversion to the 7-modified sialic acids 4, 5, and 6 in the presence of five equivalents of pyruvate (Figure 1a). Reactions were readily monitored by 19F-NMR over a 48 h period. 95% conversion of 4-deoxy-4-fluoro-GlcNAc (1) to 4 was achieved in these tests and in 91% isolated yield on scale-up. By contrast GalNAc derivative 2 produced 5 in only 18% yield, and the product was not isolated. 4-Deoxy-4,4-difluoro-GlcNAc behaved more like the gluco analogue, exhibiting substantial conversion in the test runs (66%) such that the difluorosialic acid derivative 6 could be isolated in 51% yield. The quite different yields obtained indicate a requirement for an electronegative atom (oxygen or fluorine) or hydrogen-bond acceptor in the equatorial position at the 4-position since, in its absence, the yield of the sialic acid product drops dramatically. This specificity most likely resides in the aldolase since peaks corresponding to the TalNAc derivative were observed in the 19F NMR spectrum of the reaction mixture of 2, indicating that the epimerase was converting the GalNAc starting material but that the aldolase did not convert it efficiently to 5.
Figure 1.

(a) Chemoenzymatic synthesis of 7-deoxy-7-fluorosialic acid derivatives. 4-Modified GlcNAc derivatives (100 mM), sodium pyruvate (500 mM), epimerase (0.1 U/μmol), aldolase (1 U/μmol), 35 °C, 48 h. (b) Synthesis of CMP-donors. cytidine triphosphate (CTP, 1.05 equiv), CMP sialic acid synthetase (1 U/μmol), inorganic pyrophosphatase (1 U/mmol), 86% for 7 and 62% for 8. (c) Enzymatic synthesis of (7F-)sialyl galactosides 10–13. CMP = cytidine monophosphate.
Conversion of the isolated compounds 4 and 6 into their 7-modified CMP–sialic acid derivatives was readily achieved using a CMP–sialic acid synthetase and CTP, in the presence of an inorganic pyrophosphatase to drive the reaction by PPi hydrolysis (Figure 1b).29,31−35 CMP-7FSA (7) and CMP-7-deoxy-7,7-difluorosialic acid (CMP-7,7diFSA) (8) were obtained in yields of 86% and 62% after purification by ion-exchange and size exclusion chromatography (Figure 1b). We further condensed this protocol for the synthesis of CMP-7FSA to a stepwise one-pot-four-enzyme reaction to obtain the donor directly from 4-deoxy-4-fluoro-GlcNAc (1) in an isolated yield of 41%. Here we started off with the epimerase and aldolase at pH 6 to yield 7FSA then added the CMP sialic acid synthetase and inorganic pyrophosphatase in a second step, with tight control of pH between 7 and 8, to generate CMP-7FSA. This one-pot-multienzyme reaction was not further optimized and crude CMP-7FSA instead of isolated material was used as substrate for production of various 7FSA-containing sialosides with no complications. This one-pot conversion allows easy access to large amounts of the activated donor for protein modification applications without the need for complex purification procedures.
Access to the donor analogs enabled us to study the effects of fluorination on kinetic parameters. In general, the substitution of a hydroxyl by a fluorine at the 7-position should affect the transition state for hydrolysis or sialyltransfer in two different ways. The first, affecting both enzyme-catalyzed reactions and spontaneous hydrolysis, is the inductive destabilization of the positive charge that accumulates at the transition state by the more electronegative fluorine. The second is a potential deleterious effect on enzyme–substrate interactions by loss of a potential hydrogen-bond donor upon substituting the 7-hydroxyl for a fluorine atom.
We selected two sialyltransferases from different families for an initial, qualitative assessment of the ability of sialyltransferases to use the modified CMP-sialic acids as substrates, namely CjST123 (α-2,3-sialyltransferase from GT42 family) from Campylobacter jejuni and PmST1 (GT80; mainly an α-2,3-sialyltransferase) from Pasteurella multocida.36 The enzymes were incubated with a fluorescent BODIPY–lactose acceptor and two equivalents of the different donors, and the reaction was analyzed by thin layer chromatography (Figure S2). CjST1 effected complete transfer of the respective sialic acid from natural donor 9 (referred to as CMP-7OHSA) and CMP-7FSA 7 within 10 min while transfer from CMP-7,7diFSA (8) proceeded to ∼20% completion. This enzyme will therefore transfer all the sialic acids to a lactose acceptor, albeit at different rates. A similar picture was obtained for PmST1 at pH 8.5, the reported optimum for α-2,3-sialyltransferase activity: after 5 min, transfers from natural donor 9 and monofluoro donor 7 were mostly complete, while only around 2% conversion with the difluoro donor 8 had occurred. The latter progressed to 25% conversion after 18 h (data not shown).
A coupled enzymatic assay37 that monitors CMP release was used to obtain detailed kinetic data on the transfer of the sialic acid derivatives by CjST1, PmST1, the α-2,6-sialyltransferase from Photobacterium sp. JT-ISH-224 (PsST; GT80)38 as well as the sialyltransferase CjST2 (GT42), which has strong α-2,8 and weak α-2,3 activity.24 The results (Km, kcat, and catalytic efficiency kcat/Km; Table 1) confirm the initial assessment that sialyltransferases can transfer at least the monofluoro donor with high efficiency. Only minor effects on Km values were observed when substituting natural CMP-7OHSA for CMP-7FSA. Crystallographic data on three of the sialyltransferases in complex with CMP-3FSA revealed one hydrogen bond to the 7OH group for CjST1 (with Asn66),23 for CjST2 (with Asn51),24 and for PmST1 (with Trp270),25 all in their closed forms after conformational changes resulting from donor binding. According to our data, loss of these interactions does not significantly interfere with the enzymes’ ability to bind the glycan donor. The kcat value was affected to a different extent for each enzyme, but effects remained relatively small in each case. CjST2 was most affected by monofluorination, being slowed to less than a third for both acceptor substrates while the PsST exhibited a slightly higher kcat value for the CMP-7FSA compared to CMP-7OHSA. CjST1 and PmST1 were slowed by a factor of less than 2. Interestingly, difluorination affected the turnover rates of PmST1 much more strongly than those of CjST1 suggesting that the transition state for the GT80 Pasteurella sialyltransferase has more oxocarbenium ion character than that for the GT42 Campylobacter enzyme. Since CjST2 also exhibits α-2,8-sialyltransferase activity, we were able to show that 7FSA also functions as a glycosyl acceptor, with transfer occurring at comparable rates to that with 7OHSA when 7-fluorosialyl lactose was used as acceptor (Table 1).
Table 1. Kinetic Parameters for Sialyltransferase-Catalyzed Reaction of CMP 7-Fluorosialic Acid Derivatives.
| enzyme | donor | acceptor | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) |
|---|---|---|---|---|---|
| PmST1 (GT80) | CMP-7OHSA | lactose | 89 (±1) | 180 (±2) | 2045 |
| CMP-7FSA | lactose | 52 (±1) | 99 (±1) | 1900 | |
| CMP-7,7-diFSA | lactose | 77 (±1) | 10 (±0.3) | 130 | |
| PsST (GT80) | CMP-7OHSA | lactose | 70 (±1) | 44 (±1) | 656 |
| CMP-7FSA | lactose | 50 (±1) | 75 (±1) | 1590 | |
| CjST1 (GT42) | CMP-7OHSA | lactose | 210 (±10) | 3.6 (±0.1) | 17.3 |
| CMP-7FSA | lactose | 610 (±40) | 2.6 (±0.2) | 4.2 | |
| CMP-7,7-diFSA | lactose | 420 (±40) | 1.1 (±0.1) | 2.5 | |
| CjST2 (GT42) | CMP-7OHSA | 3′-sialyllactose | 320 (±20) | 1.5 (±0.2) | 4.8 |
| CMP-7FSA | 3′-sialyllactose | 150 (±10) | 0.40 (±0.03) | 2.6 | |
| CMP-7OHSA | 3′-(7F)sialyllactose | 210 (±20) | 1.3 (±0.01) | 6.4 | |
| CMP-7FSA | 3′-(7F)sialyllactose | 100 (±20) | 0.42 (±0.04) | 4.3 |
These kinetic data highlight the fact that 7FSA can be efficiently transferred by sialyltransferases, allowing facile assembly of glycoproteins or other sialylated species that are decorated with this non-natural analogue for biotechnological or medical applications.
Having assessed the effects of 7-fluorination on sialyltransferase kinetic parameters, we set out to determine the effect of 7-fluorination on kinetic parameters for both enzymatic and nonenzymatic hydrolysis of sialosides, as well as the CMP sialic acid derivatives themselves. Sialoside substrates were prepared by using CjST1 to transfer 7FSA to commercially available aryl β-galactosides with o-nitrophenol (oNP) and 4-methylumbelliferyl alcohol (MU) as the aglycones to access disaccharides 10–13 (Figure 1c). The rates of spontaneous hydrolysis of the different CMP donors were measured at 60 °C, in a high salt pH 7.2 buffer, by observing the changes in intensities of diagnostic protons over time, using 1H NMR spectroscopy in a previously described protocol (see Supporting Information for detailed protocol).39 The decrease in intensity of each of these protons was monitored and fitted to a single exponential decay (Supporting Information, Figure S3). Substitution of a single fluorine atom at the 7-position caused a 3-fold decrease in hydrolysis rate constant (0.313 ± 0.002 h–1; krel = 0.33) relative to the natural CMP-7OHSA (0.94 ± 0.01 h–1; krel = 1), while introduction of two fluorine atoms resulted in a hundred fold decrease (0.0084 ± 0.0008 h–1; krel = 0.01).
We used a similar 1H NMR based assay to evaluate the effect of fluorination at the 7-position on rate constants for the spontaneous hydrolysis of the sialoside bond within the MU-containing sialyl–galactose conjugates 12 and 13. The degradation was measured using a high salt, dilute acid solution of each substrate and by monitoring the change in intensities of diagnostic protons. Since there are now two potential sugar linkages to be broken the position of hydrolysis was addressed by monitoring the changes in intensity of the anomeric proton of the galactose (H1′) and the H3-equatorial proton of the sialic acid (H3″eq). These protons are diagnostic since their chemical shifts are sensitive to changes in the anomeric configuration,40,41 and they will show a measurable change in chemical shift during hydrolysis. These products were identified as the free sialic acid and the β-arylgalactoside by the appearance of a second proton with a very small upfield chemical shift of H1′ and at a rate concurrent with the disappearance of H1′ of the substrate. A large change in chemical shift of this proton would indicate hydrolysis of the β-O-aryl bond, indicating that the aryl–galactoside bond remains intact. It was also observed that the intensity of the H3′′eq proton decreased without concurrent appearance of any other α-configured H3″eq protons,42 again indicating that the sialyl–galactose bond is being broken, rather than the aryl–galactose bond. This confirmed that the correct bond-breaking event is being observed and allowed the effect of 7-fluorination on the stability of the α-configured sialoside bonds to be addressed. This was achieved by monitoring the intensities of these protons over time (Figure S3), yielding a spontaneous hydrolysis rate constant of 1.45 (±0.08) h–1 for the parent sialyl galactoside substrate 12 and 0.29 (±0.01) h–1 for the 7-fluorosialyl galactoside 13, a 5-fold decrease.
These 3- and 5-fold decreases in rate constants upon monofluorination at the 7-position are consistent with the 3- and 4-fold decreases in rate constants for spontaneous hydrolysis of 6-deoxy-6-fluoropyranosides43 and 6-deoxy-6-fluoro-α-glucopyranosyl-1-phosphate observed previously.44 In all cases, a fluorine replaces a hydroxyl group on the carbon two bonds away from the ring oxygen. The much larger, 100-fold decrease in hydrolysis rate constants for the difluorinated β-CMP sialic acid derivative highlights the effect of additionally replacing a hydrogen substituent with a fluorine at this position, with much larger inductive consequences.
We moved on to evaluate the effect of 7-fluorination on sialidase activity for five neuraminidases across families GH33 and GH34 (Table 2). For this, we used the oNP and MU labeled disaccharides in a coupled enzymatic assay that has previously been used to monitor the activity of a trans-sialidase from Trypanosoma cruzi (see Figure S4).45−47 For the Micromonospora viridifaciens sialidase, NedA (GH33; catalytic domain) the kcat and Km values of the 7-fluorosialoside were, respectively ∼6-fold lower and ∼7-fold higher than for the parent substrate, resulting in a net 41-fold reduction in kcat/Km value. For two other GH33 enzymes, lack of saturation, even at high concentrations of 7F-sialoside 11, precluded determination of the individual parameters Km and kcat, but catalytic efficiency was reduced more than 100-fold for Trypanosoma rangeli TrSA48 and 200-fold for Clostridium perfringens NanI.49 A quantitative comparison for human Neu2 (GH33) proved impossible because turnover of 13 was too slow; a gratifying result. These substantial decreases in rates for GH33-catalyzed hydrolysis compared to the rates for spontaneous hydrolysis suggest that inductive effects alone are not responsible. Rather, much of the rate decrease for the enzymatic reactions must be due to loss of important noncovalent interactions at the active site that help stabilize the transition state.
Table 2. Kinetic Parameters for Sialidase-Catalyzed Cleavage of 7-Fluorosialosides vs Parent.
| enzyme | substrate | no. | Km (mM) | kcat (min–1) | kcat/Km (min–1 mM–1) | kcat/Km (relative to 7F substrate) |
|---|---|---|---|---|---|---|
| T. rangeli TrSA (GH33) | 7OH | 10 | 2.4 (±0.2) | 83 (±4) | 34 (±4) | 254 |
| 7F | 11 | not determined | not determined | 0.13 (±0.01) | 1 | |
| C. perfringens NanI (GH33) | 7OH | 10 | 0.80 (±0.04) | 78 (±2) | 97 (±7) | 133 |
| 7F | 11 | not determined | not determined | 0.73 (±0.03) | 1 | |
| M. viridifaciens NedA (GH33) | 7OH | 12 | 0.17 (±0.01) | 940 (±90) | 5500 (±900) | 41 |
| 7F | 13 | 1.2 (±0.15) | 170 (±50) | 135 (±60) | 1 | |
| hNEU2 (GH33) | 7OH | 12 | 3.5 (±0.3) | 3.0 (±0.1) | 0.9 (±0.1) | – |
| 7F | 13 | no activity detected | – | |||
| enzyme | substrate | no. | Km (mM) | Vmax rela (μM min–1) | Vmax/Km rela (min–1 × 10–3) | kcat/Km (relative to 7F substrate) |
|---|---|---|---|---|---|---|
| influenza N9 (GH34) | 7OH | 10 | 0.83 (±0.06) | 17.5 (±0.5) | 21 (±2.0) | 1.8 |
| 7F | 11 | 0.39 (±0.04) | 4.6 (±0.2) | 12 (±1.6) | 1 |
Values determined from a fixed volume of enzyme preparation.
Crystal structures of the GH33 enzymes have been obtained in ligand-free and inhibitor-bound states. In human Neu2, two hydrogen bonds between OH7 of the substrate and Glu111 have been suggested to help in substrate positioning.50 In the case of T. rangeli TrSA, OH7 is found to be part of a water-mediated hydrogen bonding network between the substrate and the enzyme, which includes interactions with Asn60, the acid/base catalyst and also the C5-N-acetyl oxygen.13,20 For C. perfringens NanI, OH7 is also involved in a tight water-mediated hydrogen bond network, this time to Asp291, also the acid/base catalyst.22 A significant restructuring of this hydrogen-bond network occurs upon ligand binding, along with a large displacement of Asn60 (T. rangeli) or Asp291 (C. perfringens). Loss of the hydrogen bond to OH7 would therefore be expected to result in significant deleterious effects on the enzyme-catalyzed hydrolysis reaction. The consequence of this is that 7-deoxy-7-fluorosialosides are only slowly hydrolyzed by these GH33 sialidases. Preliminary studies by Khedri et al. came to a similar conclusion.29
In addition, we examined influenza N9 neuraminidase as a representative of the GH34 family of viral sialidases and observed only minor effects of fluorination at the 7-position on catalytic efficiency (Table 2). This is markedly different from the ∼100-fold rate differences observed for the family GH33 enzymes. Interestingly the 4-fold decrease in maximum reaction rate between 7OH and the 7F substrate is very similar to the 5-fold decrease for the spontaneous hydrolysis above. Further, the modified substrate was bound 2-fold more tightly than the parent. The structure of the group 2 influenza neuraminidases, of which N9 is a member, solved in the presence of an inhibitor51 reveals only a very distant (3.8 Å) contact between the enzyme (Asp152) and the 7-position. By contrast, these enzymes interact strongly with both OH8 and OH9. These very weak interactions with OH7 thus explain the tolerance of the enzyme toward modifications at that position. This is in stark contrast with the family GH33 enzymes wherein the enzyme–substrate contacts around the 7-hydroxyl are important for enzyme activity.
Having established the ability of sialytransferases to transfer 7FSA and the increased resistance of 7FSA glycosides to spontaneous and enzymatic hydrolysis, we set out to investigate the possibility of installing hydrolysis-resistant 7FSA on proteins. For this, we explored one example of a therapeutic protein bearing an O-glycan and one bearing N-glycans to investigate whether both glycan classes can be engineered.
For O-glycans, we made use of a system that we have recently established52 allowing the expression of proteins carrying the core 1 disaccharide (β-Gal-1,3-α-GalNAc) in Escherichia coli. Using a purified sample of interferon-α2b (IFNα2b*, glycosylation-sequon optimized) bearing the T-antigen at Thr106 that we derived from this system we used the sialyltransferase ST3Gal1 to carry out an in vitro extension of the glycan with an α-2,3-linked sialic acid using, separately, CMP-7OHSA and CMP-7FSA as donor. Intact protein mass spectrometry (Figure 2) proved the high-efficiency transfer of both 7OHSA and 7FSA to the glycoprotein, as revealed by the correct mass differences compared to the precursor. This shows that the system works well at engineering O-glycans on proteins.
Figure 2.
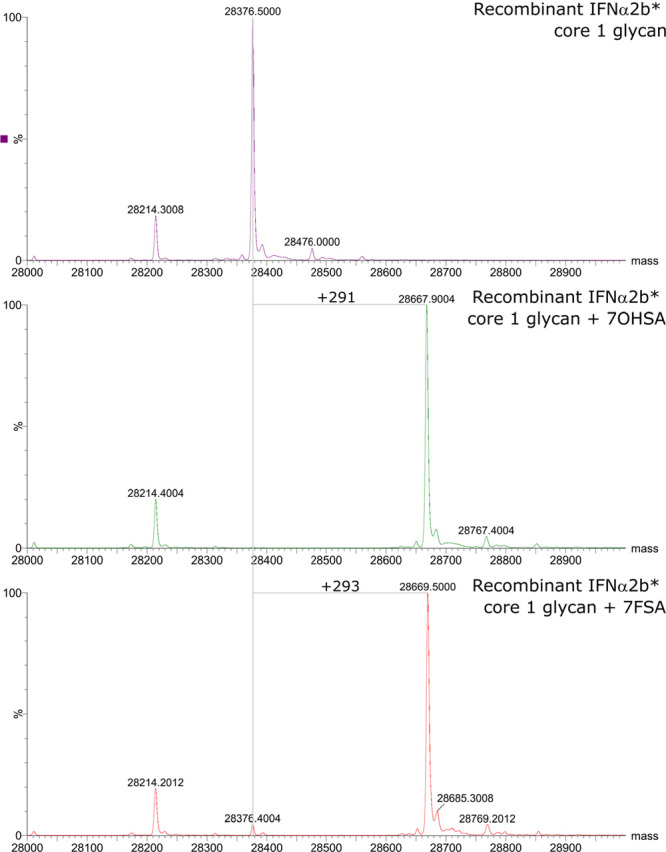
O-glycan engineering with 7FSA on IFNα2b* evaluated by intact protein mass spectrometry. The attachment of 7OHSA (control, middle panel) as well as the attachment of 7FSA (bottom panel) proceeded with high yields as seen by the shift of the protein peak representing the IFN carrying the core 1 glycan by the expected mass differences.
We then moved on to explore the potential for engineering the larger and more structurally complex N-glycans on a more densely glycosylated protein with 7FSA. This sample would then be used to test the hypothesis that the hydrolysis-resistant 7FSA should withstand spontaneous or enzymatic hydrolysis longer than the natural version, thereby keeping underlying galactose residues from interacting with lectin receptors and inhibiting the associated clearance pathway. We selected α1-antitrypsin (A1AT), an abundant serpin-type protease inhibitor as our target. A1AT is a 52 kDa glycoprotein that protects tissue, mainly lung, from destruction by dead-end inhibition of the inflammation-related protease neutrophil elastase.53 Clinically, purified A1AT is used to treat the lung pathology of α-1 antitrypsin deficiency, which is a genetic disease caused by single nucleotide point mutations or preliminary stop codons. These can lead to enzyme variants with reduced or no activity or misfolded variants that form plaques in the liver causing progressive dysfunction.54 Patients present with mild to severe COPD-like symptoms and currently the only effective therapy is A1AT supplementation by weekly intravenous injection.55 Stabilized proteins may prevent premature clearance from circulation and extend circulation lifetimes, with clear benefits in terms of patient compliance and costs. A1AT possesses no O-glycosylations, but has N-glycosylation at three sites. These are mainly occupied by biantennary N-glycans terminating with α-2,6-sialic acids; triantennary N-glycans (two α-2,6 and one α-2,3 linkage for the terminal sialic acids) are the second most abundant species.56 This very homogeneous and well-studied N-glycan pattern allowed us to install 7FSA units globally across the glycosylation sites and to track all steps of remodelling. We decided to work with the marketed formulation Prolastin C to ensure highest possible protein quality.
Replacement of surface sialic acids by the 7-fluoro analogue was done in a two-step desialylation–resialylation protocol. Using the catalytic domain of M. viridfaciens NedA, we stripped off all natural sialic acids followed by extensive purification to ensure complete neuraminidase removal to avoid compromising the resialylation step. Sialidase activity was monitored throughout the process by kinetic analysis or analytical TLC.
We screened different recombinantly expressed sialyltransferases for their ability to resialylate the protein with either 7OHSA or 7FSA (Figure 3). To assess sialic acid attachment efficiency, N-glycans were released from the protein by PNGase F treatment and analyzed by high performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD, Figure 3 and Figure S1). Monosialylated N-glycans were the major sialylated products for two enzymes, namely CjST1 and a recombinant human α-2,6-sialyltransferase ST6Gal1 (rhST6Gal1). CjST1slowly converted nonsialylated to mono- and disialylated glycans, but the failure to detect significant amounts of CMP-7FSA, used in excess, after prolonged incubation suggests unavoidable background hydrolysis (Figure 3). rhST6Gal1 yielded only monosialylated glycan species within the first 8 h, with no further glycosylation even after 48 h, suggesting a prohibitive mechanism in the given context. We also tested wildtype and mutant PmST1 variants, but the high background hydrolysis of either CMP-7FSA or the resulting sialosides excluded these enzymes from further investigation. In contrast, high levels of sialylation were observed for PsST with essentially complete formation of at least the predominant species, the biantennary glycan. Non- and monosialylated glycans were essentially absent, but some triantennary structures were modified with only two 7FSA moieties, leaving one arm untouched. The results were confirmed by LC-MS, but the question of whether the same or different sites on the triantennary glycans remain unmodified was unanswered.
Figure 3.
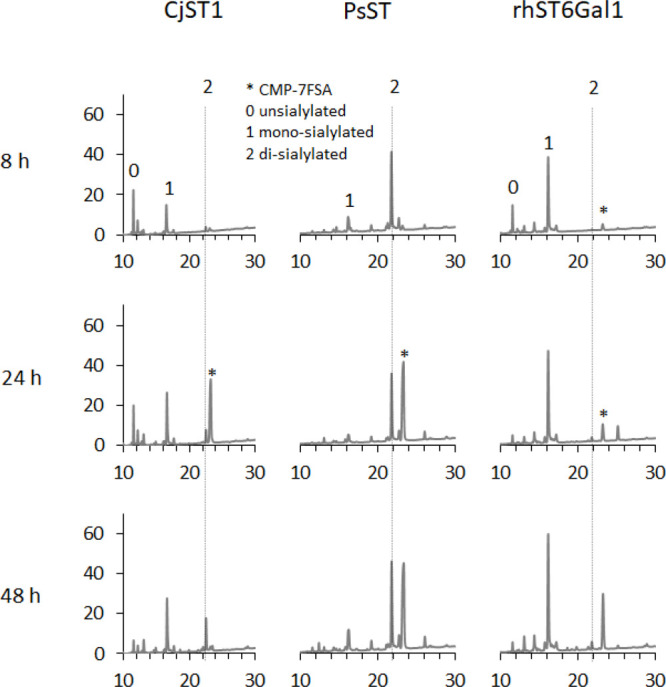
Evaluation of different recombinant sialyltransferases for their ability to attach 7FSA to asialo-A1AT. N-glycans were released from the substrate protein after 8, 24, and 48 h of reaction and were analyzed by HPAEC-PAD to separate glycans with different 7FSA attachment levels.
With the α-2,6-resialylated proteins in hand, we wanted to ensure that sialic acid fluorination does not interfere with the ability of the sialic acid to protect the glycoprotein from being recognized by hepatocyte-residing galactose-binding lectins, mainly the asialoglycoprotein receptor. These receptors recognize terminal Gal and GalNAc residues, but also, with lower affinity and species-dependently, α-2,6-SiaGal(NAc).57−59 The different resialylated Prolastin C samples were nonspecifically labeled with the amine-reactive fluorophore BODIPY-FL-NHS, incubated with HepG2 cells for 3 h, and uptake was quantified by fluorescence microscopy (Figure 4). Asialo-A1AT exhibited the highest uptake as expected from the large number of Gal residues that were exposed to interaction with lectins. For all sialylated samples, uptake was low with no significant differences between the unmodified A1AT and the protein resialylated with either the natural or the 7-fluorosialic acid. This demonstrates that reattachment of both sialic acid variants efficiently blocked receptor interaction at least with the major receptors. Other (lectin) receptors might also be involved, but our results clearly demonstrate that the 7-hydroxyl to 7-fluorine replacement does not seem to change the recognition pattern of relevant lectin receptors on liver cells. The results of the uptake assay therefore indicate preservation of biological properties.
Figure 4.

Cellular uptake of A1AT glycoforms by the hepatocyte cell line HepG2. (a) Fluorescent image of SDS-PAGE to analyze fluorescently labeled A1AT. (b) Quantification of cellular fluorescence. (c) Representative images showing BODIPY-FL-labeled A1AT (green) and nuclear staining with DAPI (blue).
Encouraged by those results, we moved on to evaluate the effects of 7F-fluorination on circulatory half-life in vivo in a proof-of-principle study. A1AT (Prolastin C) was again desialylated and decorated with either 7OHSA or 7FSA. The two proteins exhibited very similar glycoforms in which relative abundances were slightly shifted from the unmodified protein as seen by HPAEC-PAD analysis of released glycans (see Figure S5). The major difference is an increased peak around 16 min (17 min for 7FSA as 7-fluorination caused a shift in retention times) and fewer peaks at later retention times. LC-MS analysis indicates that this difference might represent incomplete resialylation of triantennary N-glycans leading to some structures terminating with galactose residues (Figures S6–S11). This peak was present to the same extent in A1AT modified with 7OHSA and with 7FSA. Samples were labeled with a near-infrared fluorophore (CF770), injected into CD-1 mice, and protein concentration in blood was determined for up to 72 h by measurement of fluorescence in collected samples (Figure 5).60 All sialylated A1AT versions showed longer retention in the circulatory system than the asialo-A1AT, which was rapidly cleared from circulation. Direct comparison of the two resialylated A1AT preparations showed clear advantages for 7FSA over the natural 7OHSA with the half-life being extended from 4.6 ± 0.8 h (mean ± SD) to 13.8 ± 1.1 h (see Table S1 for all calculated values). The area under the curve, which reflects the actual body exposure to the drug after administration, increased from 116 ± 24 to 245 ± 27 μg h/mL. These differences do not reside in altered levels of glycosylation since the N-glycan profiles of these proteins are basically identical (Figure S5). The results therefore strongly support the original hypothesis that the increased hydrolytic stability of the 7-fluorosialoside in vivo leads to longer retention of the glycoprotein in the circulatory system.
Figure 5.
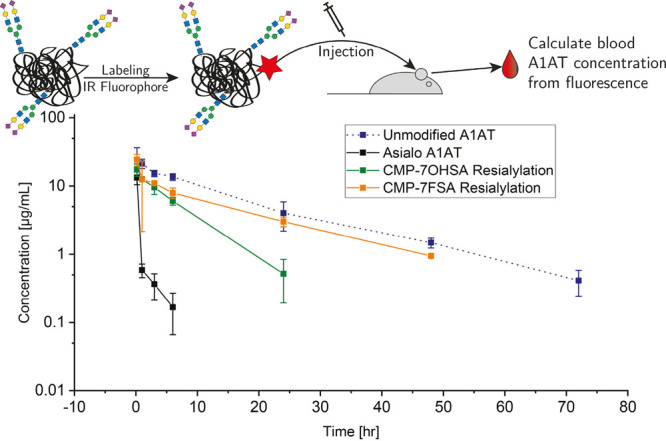
Pharmacokinetic profile of unmodified and modified glycoproteins labeled with CF770 fluorophore. Mice were intravenously injected with 6 mg/kg of CF770 labeled protein and blood was collected at various time points. Data are mean ± SEM for three mice per group. Levels of the CF770-labeled proteins in blood samples were measured by an IVIS Lumina III (excitation/emission: 740/790 nm) and their concentrations interpolated from a standard curve of known concentrations of the labeled protein diluted in control blood.
The observation that the circulatory half-life of the 7FSA protein is very similar to that of the unmodified A1AT (13.0 ± 1.4 h) was initially disappointing and must reside in the slight differences in sialylation level previously discussed, as seen in the peaks at 16/17 min (Figure S5). Triantennary glycans constitute about 20% of the total N-glycans on native A1AT56 but are found at only one of the three glycosylation sites per protein molecule, Asn-107. This means that more than half of the A1AT molecules have a triantennary N-glycan at this position. As a consequence, incomplete sialylation of this glycan type would result in a substantial fraction of protein molecules missing a sialic acid at the time of injection. This will be a disadvantage not only just after injection but also during the whole time course due to multivalent recognition of glycoconjugates by liver lectins.61 Whereas hydrolysis of one sialic acid from a fully sialylated protein would yield a protein that interacts in a weak monovalent fashion, equivalent loss of a sialic acid from a protein already missing one sialic acid will lead to a strong divalent interaction and more rapid clearance. More extensive evaluation of sialyltransferases and conditions will likely solve this issue for A1AT, which may well not be a problem for other therapeutic proteins. Most importantly, these results confirm the validity of the approach.
Conclusions
By exploiting differences in transition state structure and substrate recognition between sialyltransferases and sialidases, we have established a practical methodology for increasing circulatory half-lives of therapeutic glycoproteins by incorporating terminal 7-fluorosialic acids. Kinetic studies with a series of sialyltransferases confirmed that the 7-fluoro substitution has little impact on sialyltransfer, while having dramatic effects on rates of enzymatic hydrolysis of the resultant sialosides, as desired. The enzymatic synthesis of the requisite CMP-7-fluorosialic acid donor is achieved on a gram scale from readily available 4-deoxy-4-fluoro-N-acetylglucosamine. Use of this donor in conjunction with sialyltransferases allows efficient sialylation of the asialoglycoproteins, recombinant IFNα2b* for O-glycans and the in vitro desialylated therapeutic glycoprotein alpha-1-antitrypsin for N-glycans. Glycan-remodelled α-1-antitrypsin exhibits essentially equivalent cellular uptake into HepG2 cells as the equivalent protein bearing sialic acid. Most importantly the half-life for elimination of the 7-fluorosialylated protein from the circulation is increased by 3-fold. This serves as a proof of principle for the concept of 7-fluorosialylation and opens the door to optimization of resialylation protocols. This choice of substitution strikes a balance between ease of synthesis and degree of circulatory lifetime extension and is readily transferable to a range of therapeutic glycoproteins.
Acknowledgments
We thank the Canadian Institutes of Health Research (CIHR) and the Canadian Glycomics Network, GlycoNet for financial support of this work and Dr. Hong-ming Chen for assistance with synthesis. T.J.M. was supported by a Leverhulme Trust fellowship, L.B. was funded by a DFG fellowship, J.R.R. was supported by an NSERC fellowship and A.G. by a Leopoldina Fellowship. CMP-N-acetyl neuraminic acid was a kind gift from Neose Technologies. We thank Dr. Andrew G Watts for earlier donation of a sample of epimerase and Dr. Nicholas McGregor for support with the HPAEC-PAD and MS measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01589.
Detailed experimental procedures including synthesis and characterization of fluorinated donors and transfer products, protein production, determination of kinetic parameters, glycan engineering and analysis for O-linked and N-linked glycoproteins, cell-based protein uptake assays, and pharmacokinetic profile determination in CD-1 mice; figures showing TLC analysis of 7FSia transfer, NMR-based time course of CMP7F Sia hydrolysis, overview of coupled assay for 7FSia-Lac hydrolysis, PAGE and HPLC analysis of protein resialylation, and PGC-LC-MS analyses of released glycans; and a table of pharmacokinetic parameter estimates (PDF)
Author Contributions
# A.G., L.B, and T.J.M. contributed equally
The authors declare the following competing financial interest(s): We hold an issued patent on the technology.
Supplementary Material
References
- Pierpont T. M.; Limper C. B.; Richards K. L. Past, Present, and Future of Rituximab—The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. 10.3389/fonc.2018.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch I. B. Insulin Analogues. N. Engl. J. Med. 2005, 352, 174–183. 10.1056/NEJMra040832. [DOI] [PubMed] [Google Scholar]
- Kiss Z.; Elliott S.; Jedynasty K.; Tesar V.; Szegedi J. Discovery and basic pharmacology of erythropoiesis-stimulating agents (ESAs), including the hyperglycosylated ESA, darbepoetin alfa: an update of the rationale and clinical impact. Eur. J. Clin. Pharmacol. 2010, 66, 331–340. 10.1007/s00228-009-0780-y. [DOI] [PubMed] [Google Scholar]
- Rustgi V. K. Albinterferon alfa-2b, a novel fusion protein of human albumin and human interferon alfa-2b, for chronic hepatitis C. Curr. Med. Res. Opin. 2009, 25, 991–1002. 10.1185/03007990902779186. [DOI] [PubMed] [Google Scholar]
- Kesik-Brodacka M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. 10.1002/bab.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. 10.1038/nbt.4305. [DOI] [PubMed] [Google Scholar]
- Sauerborn M.; Brinks V.; Jiskoot W.; Schellekens H. Immunological mechanism underlying the immune response to recombinant human protein therapeutics. Trends Pharmacol. Sci. 2010, 31, 53–59. 10.1016/j.tips.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Gamblin D. P.; Scanlan E. M.; Davis B. G. Glycoprotein Synthesis: An Update. Chem. Rev. 2009, 109, 131–163. 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- Palaniappan K. K.; Bertozzi C. R. Chemical Glycoproteomics. Chem. Rev. 2016, 116, 14277–14306. 10.1021/acs.chemrev.6b00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solá R. J.; Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharm. Sci. 2009, 98, 1223–45. 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A. R.; Rodrigues M. E.; Henriques M.; Oliveira R.; Azeredo J. Glycosylation: impact, control and improvement during therapeutic protein production. Crit. Rev. Biotechnol. 2014, 34, 281–299. 10.3109/07388551.2013.793649. [DOI] [PubMed] [Google Scholar]
- Morell A. G.; Gregoriadis G.; Scheinberg I. H.; Hickman J.; Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J. Biol. Chem. 1971, 246, 1461–7. 10.1016/S0021-9258(19)76994-4. [DOI] [PubMed] [Google Scholar]
- Buschiazzo A.; Alzari P. M. Structural insights into sialic acid enzymology. Curr. Opin. Chem. Biol. 2008, 12, 565–572. 10.1016/j.cbpa.2008.06.017. [DOI] [PubMed] [Google Scholar]
- Yang W. H.; et al. An intrinsic mechanism of secreted protein aging and turnover. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 13657–13662. 10.1073/pnas.1515464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Y. Functional Consequences of Mannose and Asialoglycoprotein Receptor Ablation. J. Biol. Chem. 2016, 291, 18700. 10.1074/jbc.M116.738948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozawa R.; et al. Asialoglycoprotein Receptor Deficiency in Mice Lacking the Major Receptor Subunit. J. Biol. Chem. 2001, 276, 12624–12628. 10.1074/jbc.M011063200. [DOI] [PubMed] [Google Scholar]
- Powell L. D.; Jain R. K.; Matta K. L.; Sabesan S.; Varki A. Characterization of Sialyloligosaccharide Binding by Recombinant Soluble and Native Cell-associated CD22. J. Biol. Chem. 1995, 270, 7523–7532. 10.1074/jbc.270.13.7523. [DOI] [PubMed] [Google Scholar]
- Kale R. R.; et al. Detection of Intact Influenza Viruses using Biotinylated Biantennary S -Sialosides. J. Am. Chem. Soc. 2008, 130, 8169–8171. 10.1021/ja800842v. [DOI] [PubMed] [Google Scholar]
- Rich J. R.; Wakarchuk W. W.; Bundle D. R. Chemical and Chemoenzymatic Synthesis of S-Linked Ganglioside Analogues and Their Protein Conjugates for Use as Immunogens. Chem. - Eur. J. 2006, 12, 845–858. 10.1002/chem.200500518. [DOI] [PubMed] [Google Scholar]
- Watts A. G.; Oppezzo P.; Withers S. G.; Alzari P. M.; Buschiazzo A. Structural and Kinetic Analysis of Two Covalent Sialosyl-Enzyme Intermediates on Trypanosoma rangeli Sialidase. J. Biol. Chem. 2006, 281, 4149–4155. 10.1074/jbc.M510677200. [DOI] [PubMed] [Google Scholar]
- Watts A. G.; Withers S. G. The synthesis of some mechanistic probes for sialic acid processing enzymes and the labeling of a sialidase from Trypanosoma rangeli.. Can. J. Chem. 2004, 82, 1581–1588. 10.1139/v04-125. [DOI] [Google Scholar]
- Newstead S. L.; et al. The Structure of Clostridium perfringens NanI Sialidase and Its Catalytic Intermediates. J. Biol. Chem. 2008, 283, 9080–9088. 10.1074/jbc.M710247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C. P. C.; et al. Structural analysis of the α-2,3-sialyltransferase Cst-I from Campylobacter jejuni in apo and substrate-analogue bound forms. Biochemistry 2007, 46, 7196–7204. 10.1021/bi602543d. [DOI] [PubMed] [Google Scholar]
- Chiu C. P. C.; et al. Structural analysis of the sialyltransferase CstII from Campylobacter jejuni in complex with a substrate analog. Nat. Struct. Mol. Biol. 2004, 11, 163–170. 10.1038/nsmb720. [DOI] [PubMed] [Google Scholar]
- Ni L.; et al. Crystal Structures of Pasteurella multocida Sialyltransferase Complexes with Acceptor and Donor Analogues Reveal Substrate Binding Sites and Catalytic Mechanism † ‡. Biochemistry 2007, 46, 6288–6298. 10.1021/bi700346w. [DOI] [PubMed] [Google Scholar]
- Burkart M. D.; Vincent S. P.; Wong C.-H. An efficient synthesis of CMP-3-fluoroneuraminic acid. Chem. Commun. 1999, 1525–1526. 10.1039/a903362i. [DOI] [Google Scholar]
- Chokhawala H. A.; Cao H.; Yu H.; Chen X. Enzymatic synthesis of fluorinated mechanistic probes for sialidases and sialyltransferases. J. Am. Chem. Soc. 2007, 129, 10630–10631. 10.1021/ja072687u. [DOI] [PubMed] [Google Scholar]
- Lo H.-J.; et al. Synthesis of Sialidase-Resistant Oligosaccharide and Antibody Glycoform Containing α2,6-Linked 3F ax -Neu5Ac. J. Am. Chem. Soc. 2019, 141, 6484. 10.1021/jacs.9b01991. [DOI] [PubMed] [Google Scholar]
- Khedri Z.; et al. Chemoenzymatic synthesis of sialosides containing C7-modified sialic acids and their application in sialidase substrate specificity studies. Carbohydr. Res. 2014, 389, 100–111. 10.1016/j.carres.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Bernacki R. J.; Paul B.; Korytnyk W. Fluorinated carbohydrates as potential plasma membrane modifiers. Synthesis of 4- and 6-fluoro derivatives of 2-acetamido-2-deoxy-D-hexopyranoses. Carbohydr. Res. 1990, 198, 205–21. 10.1016/0008-6215(90)84293-4. [DOI] [PubMed] [Google Scholar]
- Karwaski M.-F.; Wakarchuk W. W.; Gilbert M. High-level expression of recombinant Neisseria CMP-sialic acid synthetase in Escherichia coli. Protein Expression Purif. 2002, 25, 237–40. 10.1016/S1046-5928(02)00004-9. [DOI] [PubMed] [Google Scholar]
- Nahálka J.; Pätoprstý V. Enzymatic synthesis of sialylation substrates powered by a novel polyphosphate kinase (PPK3). Org. Biomol. Chem. 2009, 7, 1778. 10.1039/b822549b. [DOI] [PubMed] [Google Scholar]
- Mizanur R. M.; Pohl N. L. Bacterial CMP-sialic acid synthetases: production, properties, and applications. Appl. Microbiol. Biotechnol. 2008, 80, 757–765. 10.1007/s00253-008-1643-7. [DOI] [PubMed] [Google Scholar]
- Hartlieb S.; et al. Chemoenzymatic synthesis of CMP-N-acetyl-7-fluoro-7-deoxy-neuraminic acid. Carbohydr. Res. 2008, 343, 2075–2082. 10.1016/j.carres.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Morley T. J.; Withers S. G. Chemoenzymatic Synthesis and Enzymatic Analysis of 8-Modified Cytidine Monophosphate-Sialic Acid and Sialyl Lactose Derivatives. J. Am. Chem. Soc. 2010, 132, 9430–9437. 10.1021/ja102644a. [DOI] [PubMed] [Google Scholar]
- Yu H.; et al. A multifunctional Pasteurella multocida sialyltransferase: A powerful tool for the synthesis of sialoside libraries. J. Am. Chem. Soc. 2005, 127, 17618–17619. 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- Gosselin S.; Alhussaini M.; Streiff M. B.; Takabayashi K.; Palcic M. M. A Continuous Spectrophotometric Assay for Glycosyltransferases. Anal. Biochem. 1994, 220, 92–97. 10.1006/abio.1994.1303. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H.; Takakura Y.; Mine T.; Yamamoto T. Photobacterium sp. JT-ISH-224 Produces Two Sialyltransferases, -/ -Galactoside 2,3-Sialyltransferase and -Galactoside 2,6-Sialyltransferase. J. Biochem. 2008, 143, 187–197. 10.1093/jb/mvm208. [DOI] [PubMed] [Google Scholar]
- Morley T. J.; Willis L. M.; Whitfield C.; Wakarchuk W. W.; Withers S. G. A New Sialidase Mechanism. J. Biol. Chem. 2009, 284, 17404–17410. 10.1074/jbc.M109.003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski U.; Friebolin H.; Brossmer R.; Supp M. 1H-NMR studies at N-acetyl-D-neuraminic acid ketosides for the determination of the anomeric configuration II. Tetrahedron Lett. 1979, 20, 4637–4640. 10.1016/S0040-4039(01)86670-3. [DOI] [Google Scholar]
- Lemieux R. U.; Stevens J. D. Substitutional and configurational effects on chemical shift in pyranoid carbohydrate derivatives. Can. J. Chem. 1965, 43, 2059–2070. 10.1139/v65-276. [DOI] [Google Scholar]
- Kao Y.-H.; Lerner L.; Warner T. G. Stereoselectivity of the Chinese hamster ovary cell sialidase: sialoside hydrolysis with overall retention of configuration. Glycobiology 1997, 7, 559–563. 10.1093/glycob/7.4.559. [DOI] [PubMed] [Google Scholar]
- Namchuk M. N.; McCarter J. D.; Becalski A.; Andrews T.; Withers S. G. The role of sugar substituents in glycoside hydrolysis. J. Am. Chem. Soc. 2000, 122, 1270–1277. 10.1021/ja992044h. [DOI] [Google Scholar]
- Withers S. G.; MacLennan D. J.; Street I. P. The synthesis and hydrolysis of a series of deoxyfluoro-D-glucopyranosyl phosphates. Carbohydr. Res. 1986, 154, 127–144. 10.1016/S0008-6215(00)90028-4. [DOI] [Google Scholar]
- Harrison J. A.; et al. Hydrolase and sialyltransferase activities of trypanosoma cruzi trans-sialidase towards NeuAc-alpha-2,3-gal-Gal-beta-O-PNP. Bioorg. Med. Chem. Lett. 2001, 11, 141–4. 10.1016/S0960-894X(00)00611-9. [DOI] [PubMed] [Google Scholar]
- Cao H.; et al. Sialidase substrate specificity studies using chemoenzymatically synthesized sialosides containing C5-modified sialic acids. Org. Biomol. Chem. 2009, 7, 5137. 10.1039/b916305k. [DOI] [PubMed] [Google Scholar]
- Indurugalla D.; Watson J. N.; Bennet A. J. Natural sialoside analogues for the determination of enzymatic rate constants. Org. Biomol. Chem. 2006, 4, 4453. 10.1039/b613909d. [DOI] [PubMed] [Google Scholar]
- Reuter G.; Schauer R.; Prioli R.; Pereira M. E. A. Isolation and properties of a sialidase fromTrypanosoma rangeli. Glycoconjugate J. 1987, 4, 339–348. 10.1007/BF01048367. [DOI] [Google Scholar]
- Traving C.; Schauer R.; Roggentin P. Gene structure of the ‘large’ sialidase isoenzyme from Clostridium perfringens A99 and its relationship with other clostridial nanH proteins. Glycoconjugate J. 1994, 11, 141–51. 10.1007/BF00731154. [DOI] [PubMed] [Google Scholar]
- Chavas L. M. G.; et al. Crystal structure of the human cytosolic sialidase Neu2: Evidence for the dynamic nature of substrate recognition. J. Biol. Chem. 2005, 280, 469–475. 10.1074/jbc.M411506200. [DOI] [PubMed] [Google Scholar]
- Bossart-Whitaker P.; et al. Three-dimensional Structure of Influenza A N9 Neuraminidase and Its Complex with the Inhibitor 2-Deoxy 2,3-Dehydro-N-Acetyl Neuraminic Acid. J. Mol. Biol. 1993, 232, 1069–1083. 10.1006/jmbi.1993.1461. [DOI] [PubMed] [Google Scholar]
- Du T.; et al. A Bacterial Expression Platform for Production of Therapeutic Proteins Containing Human-like O-Linked Glycans. Cell Chem. Biol. 2019, 26, 203–212. 10.1016/j.chembiol.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Stockley R. A. Alpha1-antitrypsin review. Clinics in Chest Medicine 2014, 35, 39–50. 10.1016/j.ccm.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Greene C. M.; et al. α1-Antitrypsin deficiency. Nat. Rev. Dis. Prim. 2016, 2, 1–17. 10.1038/nrdp.2016.51. [DOI] [PubMed] [Google Scholar]
- Strange C. Anti-proteases and alpha-1 antitrypsin augmentation therapy. Respir. Care 2018, 63, 690–698. 10.4187/respcare.05933. [DOI] [PubMed] [Google Scholar]
- Kolarich D.; Weber A.; Turecek P. L.; Schwarz H.-P.; Altmann F. Comprehensive glyco-proteomic analysis of human α1-antitrypsin and its charge isoforms. Proteomics 2006, 6, 3369–3380. 10.1002/pmic.200500751. [DOI] [PubMed] [Google Scholar]
- Park E. I.; Mi Y.; Unverzagt C.; Gabius H. J.; Baenziger J. U. The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acidα2,6GalNAc. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17125–17129. 10.1073/pnas.0508537102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusch A.; et al. Development and Analysis of Alpha 1-Antitrypsin Neoglycoproteins: The Impact of Additional N -Glycosylation Sites on Serum Half-Life. Mol. Pharmaceutics 2013, 10, 2616–2629. 10.1021/mp400043r. [DOI] [PubMed] [Google Scholar]
- Park E. I.; Baenziger J. U. Closely related mammals have distinct asialoglycoprotein receptor carbohydrate specificities. J. Biol. Chem. 2004, 279, 40954–40959. 10.1074/jbc.M406647200. [DOI] [PubMed] [Google Scholar]
- Lindhout T.; et al. Site-specific enzymatic polysialylation of therapeutic proteins using bacterial enzymes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7397–402. 10.1073/pnas.1019266108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. E.; Paulson J. C. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr. Opin. Chem. Biol. 2004, 8, 617–625. 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.