

Abstract
Mutations to RAS proteins (H-, N-, and K-RAS) are among the most common oncogenic drivers, and tumors harboring these lesions are some of the most difficult to treat. Although covalent small molecules against KRASG12C have shown promising efficacy against lung cancers, traditional barriers remain for drugging the more prevalent KRASG12D and KRASG12V mutants. Targeted degradation has emerged as an attractive alternative approach, but for KRAS, identification of the required high-affinity ligands continues to be a challenge. Another significant hurdle is the discovery of a hybrid molecule that appends an E3 ligase-recruiting moiety in a manner that satisfies the precise geometries required for productive polyubiquitin transfer while maintaining favorable druglike properties. To gain insights into the advantages and feasibility of KRAS targeted degradation, we applied a protein-based degrader (biodegrader) approach. This workflow centers on the intracellular expression of a chimeric protein consisting of a high-affinity target-binding domain fused to an engineered E3 ligase adapter. A series of anti-RAS biodegraders spanning different RAS isoform/nucleotide-state specificities and leveraging different E3 ligases provided definitive evidence for RAS degradability. Further, these established that the functional consequences of KRAS degradation are context dependent. Of broader significance, using the exquisite degradation specificity that biodegraders can possess, we demonstrated how this technology can be applied to answer questions that other approaches cannot. Specifically, application of the GDP-state specific degrader uncovered the relative prevalence of the “off-state” of WT and various KRAS mutants in the cellular context. Finally, if delivery challenges can be addressed, anti-RAS biodegraders will be exciting candidates for clinical development.
Short abstract
Biodegraders, engineered proteins that fuse a high-affinity binder (e.g., RBD, K27, R11.1.6, and NS1) to an E3 ligase (e.g., SPOP), prompt degradation of RAS and reveal novel biological insights.
Introduction
Mutations to RAS proteins are among the most frequent drivers of human cancers with approximately 30% of all clinical malignancies containing an activating RAS mutation.1 KRAS is the most frequently mutated RAS isoform (86%), followed by NRAS (11%) and HRAS (3%).2 With a primary focus on KRAS, researchers have therapeutically pursued RAS oncogenes for nearly 40 years. Unfortunately, the intractability of this target to conventional approaches has impeded the identification of a clinically approved drug. However, recent advances are giving renewed hope that pharmacological inhibition of KRAS can finally be realized. In particular, recently discovered covalent inhibitors targeting the KRASG12C mutant protein are showing promising clinical efficacy,3,4 further validating mutant KRAS as a clinically relevant oncology target. In preclinical mouse models, these inhibitors have shown robust blockade of KRAS signaling and cell proliferation.3,4 Combinations with immunotherapy have led to increased efficacy and immune memory.3 More importantly, early Phase I clinical data with G12C inhibitor monotherapy has recorded responses in lung and, to a lesser degree, colon cancers.3,4 Despite these significant advances, the covalent strategy is thus far restricted to the relatively rare G12C mutation (found in 14% of nonsmall cell lung cancers, 5% of colorectal cancers, and 2% of pancreatic cancers).
For non-G12C mutations, traditional challenges for identifying therapeutic molecules remain. In particular, identification of high affinity noncovalent ligands against active KRAS has proven refractory–a consequence of the lack of appropriate small molecule-binding pockets. Removal of the covalent warhead and reinforcement of binding energies through noncovalent interactions is an approach worth considering. However, this binding pocket is occluded in the GTP-loaded state,5 and it remains unclear if non-G12C mutants cycle between nucleotide states rapidly enough for this approach to be effective. Overall, alternative strategies need to be considered. Among these, small molecule-based targeted degradation approaches have recently generated a great deal of excitement.6−10 These bifunctional molecules consist of a target-binding moiety linked to an E3-recruiting ligand. Successfully engineered small molecule degraders not only recruit the corresponding E3/E2 complex to the vicinity of the target-of-interest but also form productive ternary complexes that induce the transfer of polyubiquitin to the target to result in its proteasomal degradation.7 This strategy opens up new possibilities to tackle historically intractable targets since degradation is potentially achievable via engagement with a variety of binding sites–including but not restricted to those of functional consequence.8,11 Moreover, recent examples illustrate that targeted degradation offers better efficacy, potency, and selectivity.8,12 Finally, given the high intracellular concentration of KRAS13−15 (also Figure S1), achieving adequate target engagement with noncovalent stoichiometric inhibitors may be challenging.
As there are substantial challenges in identifying small molecule-based degraders, initial investigations aimed at assessing targeted degradation feasibility and providing insights on optimal design strategies are warranted. Key considerations include I) target degradability through engineered polyubiquitin transfer, II) “fitness” of E3 ligases recruited, III) interfaces on the target protein that can be bound yet remain amenable to polyubiquitination, and IV) functional consequences of target degradation. To resolve these questions, we have employed engineered fusion proteins herein termed biodegraders,16 also known in the literature as ubiquibodies,17 AdPROMs,18 and deGradFP.19 A biodegrader consists of a target-binding domain connected to an E3 ligase (E3). A variety of polypeptide scaffolds evolved to recognize the target with high affinity, and specificity can be selected as the target-binding domain.16 Indeed, active biodegraders have been generated with fusions between E3s and nanobodies, monobodies, alpha-reps, DARPins, and peptides.16,17 The choice of E3 is also flexible, with functional biodegraders having been engineered from both human and bacterial sequences.16,20
Cumulating evidence suggests that RAS is degradable. First, the natural turnover of RAS proteins was reported to be proteasome-dependent and regulated by the E3 ligases LTZR121−23 and βTrCP.24 Second, biodegrader equivalents consisting of the endogenous RAS-binding domain (RBD) fused to either VIF or CHIP E3 ligases have resulted in modest KRAS degradation.25,26 Third, the G12C covalent modifier fused to the Cereblon ligand was successful in degrading GFP-KRASG12C although the endogenous form was not affected.27 Here, we report the discovery of a panel of novel and potent KRAS-directed biodegraders that build on these earlier results and provide conclusive evidence for the degradability of various RAS isoforms and mutant proteins. By utilizing a variety of E3 ligases, our study unveils the possibility of engaging novel E3 ligases for a KRAS targeted degradation campaign beyond VHL and Cereblon. By exploring a variety of RAS-binding moieties, we shed light on KRAS interfaces that can be exploited for the design of small molecule degraders. We further demonstrate that while both GTP- and GDP-loaded forms of RAS proteins are amenable to targeted degradation, a biodegrader specific for GDP-loaded RAS (K27-SPOP) degraded wild-type and KRAS mutants (G12C, G12D, G12V, and Q61H) with different efficiencies, an observation that informs on the capacity of these mutants to cycle through nucleotide states in the cellular environment. Of broad significance, this KRAS-specific example illustrates the utility of biodegraders to remove subpopulations of a specific protein in ways that are not possible with RNAi or CRISPR approaches. Finally, we show that the delivery of biodegrader mRNA depleted endogenous KRASG12D and resulted in growth inhibition and apoptosis in AsPC-1, a KRAS-dependent cancer cell line. The stoichiometric inhibitor equivalent was ineffective, highlighting the superiority of a targeted degradation approach.
Results
GFP-KRAS Is Degraded by Multiple anti-GFP Biodegraders
As a starting point to determine if KRAS proteins can be targeted for ubiquitin-mediated proteasomal degradation, we applied our anti-GFP biodegrader platform,16 which features a panel of 10 representative Cullin-RING E3 ubiquitin ligase (CRL) family members fused to the GFP-binding nanobody vhhGFP428,29 (Figure 1a). By tagging KRAS with GFP, we sought to recruit an assortment of ubiquitination complexes to the vicinity of KRAS and evaluate its degradability. HEK293 stable cell lines with the constitutive expression of GFP or GFP-KRAS were established, and the panel of anti-GFP biodegraders was individually transfected with mCherry as an expression reporter. Flow cytometry was used to determine GFP levels in mCherry-positive (transfected) cells (Figure 1b). As noted previously,16 GFP alone was poorly degraded by our panel of anti-GFP biodegraders (Figure 1c left column). However, when fused to KRAS, GFP signal intensities were attenuated by 8 out of 10 biodegraders, with 6 of them (βTrCP, FBW7, SKP2, SPOP, SOCS2, and CHIP) having more than 70% of transfected cells in the GFP-negative quadrant (Q1) (Figure 1c right column) 24 h following transfection. Similar to observations against other targets,16 both CUL4-based (CRBN and DDB2) biodegraders failed to degrade GFP-KRAS; we speculate this is likely due to issues related to protein engineering rather than incompatibility of these E3 ligases. The depletion of GFP-KRAS, but not GFP, suggests that KRAS itself likely possesses the necessary traits for proteasomal degradation (i.e., solvent-exposed lysines for polyubiquitination and a structurally disordered segment that initiates unfolding at the 26S proteasome30).
Figure 1.


GFP-KRAS is degraded by multiple anti-GFP biodegraders. (a) Schematic of the anti-GFP biodegrader platform used to evaluate the degradability of a protein-of-interest (POI) fused to GFP. GFP is bound by vhhGFP4, a high-affinity anti-GFP nanobody, thereby bringing an E3 adaptor in close proximity to the POI. The collection of ten E3 adaptors spans representative members of the Cullin-RING E3 ligase (CRL) family. (b) Flow cytometry is used to determine the levels of the GFP-tagged protein. Transfected cells that express anti-GFP biodegrader will be mCherry-positive and therefore reside in quadrants 1 and 2 (Q1 and Q2). Successful degradation will reduce the GFP signal, and cells will cumulate in Q1. Cells with no degradation will be retained in Q2. (c) Flow cytometric analysis of HEK293 Tet-On 3G cells with stable integration of GFP or GFP-KRAS and transiently transfected with the panel of ten anti-GFP biodegraders. (d) Flow cytometric analysis of HEK293 Tet-On 3G cells with stable integration of GFP-KRAS and transiently transfected with vhhGFP4-SPOP or its controls. vhhGFP4mut lacks the complementarity determining region 3 (CDR3) and no longer recognizes GFP, whereas SPOPmut lacks the 3-box motif responsible for recruiting CUL3 and thus cannot assemble the ubiquitination machinery. (e) Confocal imaging analysis of HEK293 Tet-On 3G cells with stable integration of GFP-KRAS (green) and transiently transfected with the indicated anti-GFP biodegraders. mCherry (red) is a reporter of transfected cells.
For some of the active biodegraders such as vhhGFP4-SPOP, a characteristic hook effect was observed (Figure 1c and 1d). This is caused by excessively high concentrations which compromises degradation by decreasing the probability of ternary complex formation in favor of substrate:biodegrader and biodegrader:E3 binary complexes.31 Mutations to the binding domain (vhhGFP4mut) or the E3 ligase (SPOPmut) completely abrogated the downregulation of GFP-KRAS (Figure 1d), suggesting that both components of the chimeric protein are essential for activity. Targeted degradation of GFP-KRAS by anti-GFP biodegraders was further corroborated with confocal imaging. Like endogenous KRAS,32 the subcellular localization of GFP-KRAS was predominantly membrane-bound (Figure 1e). Transient expression of mCherry alone did not affect the levels and localization of GFP-KRAS (Figure 1e first column). However, when coexpressed with βTrCP-vhhGFP4, FBW7-vhhGFP4, or vhhGFP4-SPOP, the membrane-localized green fluorescence was specifically lost in mCherry positive (transfected) cells (Figure 1e middle 3 columns). DDB2-vhhGFP4 was identified as a nondegrader from the flow cytometric screen (Figure 1c). Interestingly, upon the expression of DDB2-vhhGFP4, GFP-KRAS was redistributed to the cytoplasm/nucleus (Figure 1e last column), suggesting that this biodegrader can bind GFP-KRAS but lacks the ability to induce its degradation. Overall, the anti-GFP biodegrader platform established GFP-KRAS as an amenable substrate and identified suitable E3s that can be employed to elicit its proteasomal degradation.
Leveraging High Affinity RAS Binders for Endogenous RAS Degradation
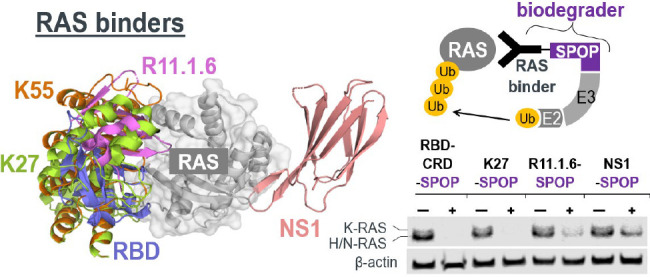
Having successfully demonstrated the degradability of GFP-KRAS, we were prompted to design anti-RAS biodegraders that can be used to directly engage and degrade endogenous KRAS. This involves the fusion of a KRAS binder to an appropriate E3 ligase. Based on published sources, we shortlisted five KRAS binders that interact at different interfaces (Figure 2a) and further validated their reported affinities and isoform/nucleotide-state specificities using Isothermal Titration Calorimetry (ITC). NS1 is a monobody that binds KRAS and HRAS but not NRAS33 (Figure S2a). The DARPins, K27 and K55, are specific for GDP- and GTP-loaded KRAS, respectively34 (Figure S2b and S2c). R11.1.6 is based on the ultrastable Sso7d scaffold and was described to be mutant KRAS-selective.35 Unfortunately, we were unable to purify sufficient quantities of recombinant R11.1.6 for biophysical analysis. We also tested the RAS-binding domain (RBD),36 a conserved region in RAS effector proteins (e.g., RAF, PI3K, and TIAM1) that interacts specifically with activated GTP-bound RAS. The RBD of RAF1 was made, and its affinity for GMPPCP-loaded KRASG12D was measured at 59 nM (Figure S2d).
Figure 2.

Leveraging high affinity binders for endogenous RAS degradation. (a) Overlay of KRAS binders from literature sources and a table summarizing their reported binding specificities and affinities. PDB structures used were as follows: 5E95 (NS1), 5O2S (K27), 5O2T (K55), 5UFQ (R11.1.6), and 4G0N (RBD). (b) Flow cytometric analysis of HEK293 Tet-On 3G cells with stable integration of GFP-KRAS and transiently transfected with anti-RAS biodegraders (in blue). Cells in Q1 represent successful GFP-KRAS depletion by the respective biodegrader. (c) Western blot analysis of HEK293 Tet-On 3G cells transiently transfected with the indicated anti-RAS biodegrader and sorted according to the levels of mCherry (a marker of transfected cells) using FACS. Gating was set such that mCherry (−) cells have the same signal intensities as untreated cells in the mCherry channel, and anything above this basal level was assigned mCherry (+). In the pan-RAS blot, the upper band corresponds to KRAS, while the lower band corresponds to HRAS and NRAS. Expression of the various anti-RAS biodegraders was detected using an anti-FLAG-tag antibody, and the expected molecular weight of each chimeric protein is indicated in kilodaltons (kDa). β-actin and HSP90 were used as loading controls.
Our previous work16 and the screen described above (Figure 1c–1e) identified SPOP as a highly robust E3 ligase. Thus, we coupled each of the RAS binders to SPOP to generate anti-RAS biodegraders. To rapidly screen for activity, GFP-KRAS was picked as the initial substrate. Through their abilities to directly engage KRAS, NS1-SPOP, K27-SPOP, and R11.1.6-SPOP were all able to deplete the GFP signal (Figure 2b). Interestingly, while RBD-SPOP did not degrade GFP-KRAS, the addition of the cysteine-rich domain (CRD) that was reported to anchor RAF proteins on membrane patches and stabilize RAS-RAF interactions37,38 yielded an active biodegrader (RBD-CRD-SPOP) (Figure 2b). This exemplifies how increased avidity through membrane targeting could aid in the stabilization of ternary complex formation required for productive degradation of GTP-loaded KRAS. As KRAS switches to the “ON” state when bound to GTP, it engages in protein–protein interactions with a multitude of effector proteins, many of which are membrane localized. Hence, biodegraders that target GTP-loaded KRAS might benefit from increased membrane localization. This could explain why K55-SPOP was ineffective (Figure 2b) since it lacks membrane targeting. It is also worth noting that the affinity of K55 for GTP-loaded KRAS is 98 nM (Figure S2c), weaker than the endogenous RAS binder RBD, which is 59 nM (Figure S2d).
To probe for the degradation of endogenous RAS, we next transfected HEK293 cells with doxycycline-inducible DNA plasmids driving coexpression of anti-RAS biodegraders and mCherry reporter. Twenty-four-hours postinduction, cells were sorted into mCherry-negative (nontransfected) and mCherry-positive (transfected) populations and harvested for Western blot analysis. A pan-RAS antibody was used to probe for endogenous levels of RAS family proteins: KRAS, HRAS, and NRAS, which appeared as two bands in HEK293. A previous study using isoform-specific siRNAs demonstrated that the upper band corresponds to KRAS, whereas the lower band corresponds to HRAS and NRAS.39 In our experiments, the upper KRAS band was specifically lost with the expression of NS1-SPOP (Figure 2c lanes 6 and 8) but not with the nondegrading control NS1-SPOPmut (Figure 2c lane 10). These data suggest that it is possible to achieve selective degradation of closely related proteins if isoform-specificities are engineered into the binders. To understand if the degradation of RAS is affected by its guanine nucleotide status, we used K27 (pan-RAS, specific for the GDP-loaded state) and RBD-CRD (pan-RAS, specific for the GTP-loaded state) as the substrate-binding moieties. The expression of either K27-SPOP or RBD-CRD-SPOP led to complete disappearance of pan-RAS bands (Figure 2c lanes 16 and 18), suggesting that both nucleotide states across RAS isoforms are susceptible to degradation. Consistent with the results on GFP-KRAS (Figure 2b), K55-SPOP and RBD-SPOP failed to degrade endogenous RAS (Figure 2c lanes 12 and 20). R11.1.6-SPOP partially reduced pan-RAS band intensities (Figure 2c lane 4). The preferential binding of R11.1.6 to mutant KRAS35 could explain why there was incomplete degradation in HEK293 cells where the status of RAS is wild-type. All anti-RAS biodegraders were FLAG-tagged and expressed according to the expected sizes and at similar levels, with the exception of RBD-CRD-SPOP (Figure 2c lane 16). This biodegrader was also barely detectable in repeat experiments (Figure S3 lane 10). Using cell sorting, we were able to include the mCherry-negative (nontransfected) population as an internal control for RAS levels in all cases (Figure 2c lanes marked as mCherry−).
It is often challenging to achieve 100% efficiency with DNA transfection. In order to better characterize anti-RAS biodegraders and study the functional consequences of KRAS loss, we generated HEK293 stable cell lines with doxycycline-inducible expression of the various anti-RAS biodegraders. Pan-RAS deletion was achieved as early as 4-h postinduction of K27-SPOP. This effect persisted for up to 24 h (Figure 3a lanes 2–5, first panel) and coincided with inhibition of phospho-ERK1/2, a downstream effector of the mitogen-activated protein kinase (MAPK) pathway (Figure 3a lanes 2–5, second panel). With SPOP mutated, the E3 ligase activity of K27-SPOPmut was disabled such that pan-RAS protein levels were not affected (Figure 3a lanes 7–10, first panel). However, K27 on its own was reported to have inhibitory effects on the MAPK pathway,34 and indeed, phospho-ERK1/2 levels were reduced 4 h after the induction of K27-SPOPmut (Figure 3a lanes 7–8, second panel). Notably, this inhibitory effect could not be sustained, and phospho-ERK1/2 levels returned to baseline at 24 h (Figure 3a lanes 9 and 10, second panel), despite continued K27-SPOPmut expression (Figure 3a lanes 9 and 10, third panel). The nonbinder control K27mut-SPOP, wherein three RAS-binding residues were replaced by alanine,34 did not alter pan-RAS nor phospho-ERK1/2 levels as expected (Figure 3a lanes 11–15). Stable cell lines with doxycycline-inducible expression of other anti-RAS biodegraders, such as R11.1.6-SPOP, NS1-SPOP, and K27-VHL, were also generated (Figure S4), but K27-SPOP elicited the most complete RAS degradation and sustained phospho-ERK inhibition in HEK293 cells. Surprisingly, despite strong pan-RAS knockdown, HEK293 cells expressing K27-SPOP continued to proliferate at rates similar to controls (Figure 3b). Western blotting for pan-RAS confirmed that the cells proliferated in the absence of RAS proteins (Figure 3c). These data suggest that HEK293 cells are not dependent on RAS proteins for survival.
Figure 3.

Robust RAS degradation with doxycycline-inducible anti-RAS biodegraders. (a) Western blot analysis of T-REx-293 cells with stable integration of K27-SPOP (or its controls) under the control of a Tet-responsive promoter. Various concentrations of doxycycline (1 or 10 ng/mL) were added to the culture media for the indicated length of time (4 or 24 h), and protein lysates were collected. Degradation of RAS was detected using a pan-RAS antibody, and disruption to the MAPK pathway was measured using the levels of phospho-ERK1/2. Expression of K27-SPOP (or its controls) was detected using an anti-FLAG-tag antibody. HSP90 was used as a loading control. (b) Incucyte confluency measurements of T-REx-293 cells with stable integration of K27-SPOP (or its controls) under the control of a Tet-responsive promoter. Various concentrations of doxycycline (0.1 to 100 ng/mL) were added to the culture media, and the percentage confluency of the cells was tracked continuously over 4 days. (c) Western blot analysis as in (a) on protein lysates collected at 1, 2, or 4 days after treatment with 1 ng/mL doxycycline.
Mutant KRAS Degradation, Inhibition of Proliferation, and Induction of Apoptosis with mRNA-Mediated Expression of anti-RAS Biodegraders
To extend our study to KRAS-dependent cancer cell lines, we employed mRNA transfection to yield higher transfection rates. As an example, in AsPC-1 cells (pancreatic adenocarcinoma cell line, homozygous KRASG12D), transfection efficiencies of a GFP-encoding DNA plasmid versus GFP mRNA were 1% and 90%, respectively, after 12 h (Figure S5). High mRNA transfection efficiency was also recapitulated in a panel of 14 cancer cell lines, wherein 9 of the lines were more than 80% transfected at 24 h (Figure S6). Leveraging this workflow, we transfected AsPC-1 cells with K27-SPOP mRNA and observed pan-RAS degradation and corresponding phospho-ERK1/2 inhibition within 4 h (Figure 4a). This effect persisted for up to 24 h and ultimately resulted in growth inhibition of AsPC-1 cells at all three mRNA concentrations tested (Figure 4b). These data suggest that the KRASG12D mutant protein retains adequate intrinsic hydrolysis to cycle back to the GDP-loaded state, where it can be effectively targeted by a GDP-specific biodegrader such as K27-SPOP. On the contrary, although the stoichiometric inhibitor K27-SPOPmut was initially successful at disrupting ERK1/2 phosphorylation, the effects were not sustained (Figure 4a), and cells expressing K27-SPOPmut showed similar proliferation rates as the nonbinding control K27mut-SPOP (Figure 4b). Morphologically, AsPC-1 cells transfected with the K27-SPOP biodegrader appeared rounded up (Figure 4c) and increased cleaved caspase-3 levels confirmed that they were undergoing apoptosis (Figure 4d). Overall, our data highlights the superiority of employing an event-driven strategy (such as targeted degradation)40 for inhibiting KRAS rather than an occupancy-driven stoichiometric inhibitor approach.
Figure 4.

Mutant KRAS degradation, inhibition of proliferation, and induction of apoptosis in AsPC-1 cells. (a) Western blot analysis of AsPC-1 cells transfected with mRNA encoding K27-SPOP (or its controls). Protein lysates were collected 4-, 8-, 12-, 24-, and 48-h post-transfection. Degradation of RAS was detected using a pan-RAS antibody, and disruption to the MAPK pathway was measured using the levels of phospho-ERK1/2. Expression of K27-SPOP (or its controls) was detected using an anti-FLAG-tag antibody. HSP90 was used as a loading control. (b) Incucyte confluency measurements of AsPC-1 cells transfected with mRNA as in (a) and tracked continuously over 5 days. (c) Phase-contrast images acquired 5-days post-transfection of AsPC-1 cells with 2 nM K27-SPOP mRNA (or its controls). (d) Immunostaining for the levels of cleaved caspase-3, an indicator of apoptosis, 4-days post-transfection of AsPC-1 cells with 2 nM K27-SPOP mRNA (or its control). Treatment with 400 nM staurosporine was used as a positive control for apoptotic cells.
Establishment of the NanoLuc Assay to Inform on Degradation Selectivity and Quantify Degradation Rates
To better characterize isoform specificities and degradation efficacies of the anti-RAS biodegraders, we established a panel of stable cell lines with doxycycline-inducible expression of a RAS protein tagged with Nanoluc luciferase at its N-terminus (Figure 5a). Compared to the published HiBiT-LgBiT platform,41 which tags endogenous RAS protein with HiBiT through CRISPR and is thus time-consuming to generate, the NanoLuc approach can be established rapidly, enabling one to track NanoLuc levels real-time in live cells and report quantitative metrics of degradation efficiencies for any RAS isoform or mutant protein in the same genetic background.
Figure 5.



Real-time quantitative measurements of RAS degradation efficiency and selectivity using the NanoLuc assay. (a) Illustration of the NanoLuc degradation assay. T-REx-293 cells with stable integration of NanoLuc-tagged RAS proteins under the control of a Tet-responsive promoter were generated. Expression was induced through a transient pulse of doxycycline, after which anti-RAS biodegraders were introduced through mRNA transfection. If successfully ubiquitinated and targeted for proteasomal degradation, the NanoLuc protein would not be available to react with its substrate (produced from slow ester hydrolysis of Endurazine), and the level of luminescence will drop. The rate of decline in luminescence decline reflects the effectiveness of the transfected biodegrader. (b) Western blot analysis of T-REx-293 stable cell lines as described in (a). Various concentrations of doxycycline (1, 10, and 100 ng/mL) were added to the culture media for 4 h, and protein lysates were collected. Fusion of a 19.7 kDa NanoLuc-tag to the RAS protein results in a slower migrating band when probed with pan-RAS antibodies. Activation of the MAPK pathway was determined using the levels of phospho-MEK1/2 and phospho-ERK1/2. HSP90 was used as a loading control. (c) T-REx-293 cells with doxycycline-induced expression of NanoLuc-HaloTag, NanoLuc-KRAS, NanoLuc-HRAS, and NanoLuc-NRAS were transfected with a 10-point 2-fold dose titration of the indicated biodegrader mRNA at time 0. Luminescence (RLU) was measured continuously every hour over a period of 40 h. Profiles were plotted as fractional RLU by normalizing to values of doxycycline induction with a transfection reagent only (MAX) and no doxycycline (MIN). (d) Degradation rate calculated from (c) plotted against biodegrader mRNA amount in nanograms (ng). (e) Degradation profile and degradation rate calculated from T-REx-293 cells with doxycycline-induced expression of NanoLuc-KRAS and NanoLuc-KRASR135K and transfected with a 10-point 2-fold dose titration of NS1-SPOP mRNA at time 0. (f) Degradation rate calculated from T-REx-293 cells with doxycycline-induced expression of NanoLuc-KRASG12C, pretreated with the indicated concentration of AMG510, and transfected with a 10-point 2-fold dose titration of the indicated biodegrader mRNA at time 0. (g) Degradation rate calculated from T-REx-293 cells with doxycycline-induced expression of various NanoLuc-tagged mutant KRAS and transfected with a 10-point 2-fold dose titration of the indicated biodegrader mRNA at time 0. (h) Fractional RLU specifically retrieved for the 24-h time-point from (g) and expressed as a percentage to represent the residual protein compared to transfection reagent only control.
HEK293 cells with stable integration of different NanoLuc-tagged RAS proteins were selected, and varying concentrations of doxycycline were added to induce expression (Figure 5b). Using a pan-RAS antibody, we noted that the overexpression of NanoLuc-tagged RAS proteins was significantly higher compared to endogenous levels (Figure 5b). Interestingly, the overexpression of NanoLuc-KRASG12D was sufficient to stimulate the MAPK pathway and increase the phosphorylation of MEK and ERK (Figure 5b). This was not observed with the overexpression of the wild-type NanoLuc-RAS proteins (Figure 5b), validating NanoLuc-KRASG12D as a functional and activating mutant protein.
To run the NanoLuc assay in a high-throughput 384-well format to accommodate a full dose titration of biodegrader mRNAs, we first performed a series of optimization to select 1) type of live-cell substrate, 2) cell seeding densities, and 3) doxycycline concentrations and length of induction (Figure S7). With these conditions established, we chose K27-SPOP and NS1-SPOP as tools to evaluate if the NanoLuc assay can inform on the selectivity of biodegrader-mediated degradation. A previous report indicated that while K27 is specific for the GDP-loaded form of RAS (Figure S2b), it does not discriminate between RAS isoforms.34 Accordingly, K27-SPOP degraded all RAS isoforms (NanoLuc-KRAS, NanoLuc-HRAS, and NanoLuc-NRAS) in a dose-dependent manner but not a control substrate NanoLuc-HaloTag (Figure 5c first panel). Neither K27-SPOPmut nor K27mut-SPOP degraded any of the NanoLuc-tagged proteins tested (Figure 5c second and third panel). This suggested that the decline in luminescence is specific to the binding of the NanoLuc-tagged substrate by an active biodegrader, which then induces its proteasomal turnover. The degradation rate, as described previously,41 was calculated for each concentration and plotted (Figure 5d). K27-SPOP was the most effective at degrading NanoLuc-KRAS, followed by NanoLuc-NRAS and finally NanoLuc-HRAS.
NS1 is a monobody that binds KRAS and HRAS but not NRAS33 (Figure S2a). Using conventional Western blotting, the upper band corresponding to KRAS was preferentially lost in cells transfected with NS1-SPOP (Figure 2c lanes 6 and 8). However, it was difficult to establish if other RAS isoforms were also affected since isoform-specific antibodies are lacking. Using the NanoLuc assay, it was clear that NS1-SPOP degraded NanoLuc-KRAS and NanoLuc-HRAS but not NanoLuc-NRAS (Figure 5c fourth panel), in line with its reported binding specificities33 (Figure S2a). When the substrate-binding domain of NS1-SPOP was replaced by the fibronectin type III domain (FN3), which forms the basis of the monobody scaffold, degradation was lost (Figure 5c fifth panel). Interestingly, degradation rate constants suggested that NS1-SPOP degraded NanoLuc-KRAS more efficiently than NanoLuc-HRAS (Figure 5d fourth panel), despite the stronger affinity of NS1 for HRAS than for KRAS33 as determined from in vitro biophysical assays (Figure S2a). This result is consistent with the reported activity of the NS1 monobody in the cellular context, where it disrupted plasma membrane localization and RAF engagement for KRAS but not for HRAS.33 Overall, we have demonstrated that the NanoLuc assay is a useful tool to (1) inform on the specificity of degradation among closely related proteins and (2) provide quantitative measurements of degradation efficiencies inside live cells.
To further validate the NanoLuc assay, we generated a NanoLuc-KRASR135K stable cell line. R135 is a conserved residue in KRAS and HRAS but not NRAS, where it is instead a lysine. R135 makes extensive contacts with NS1 and is a major specificity determinant since its mutation to lysine greatly diminished NS1 binding.33 Likewise, NS1-SPOP degraded NanoLuc-KRAS but was ineffective against NanoLuc-KRASR135K (Figure 5e). Conversely, when K135 on NRAS was mutated to arginine, NRAS is now recognized and degraded by NS1-SPOP (Figure S8a). Taken together, these results clearly establish how degradation specificity can be tightly controlled through precise molecular recognition of a POI by the biodegrader substrate-binding domain. By extension, biodegraders represent exciting tools for uncovering biological insights in the cellular context (vida infra).
While we have shown that the GDP-selective biodegrader K27-SPOP is able to degrade KRASG12D and reduce the viability of AsPC-1 cells (Figure 4), it is not known if the same can be achieved with other oncogenic KRAS mutations. Specifically, it was reported that intrinsic GTP hydrolysis rates are highly variable between KRAS mutants, and therefore, the pool of GDP-loaded forms available at any given time is expected to differ.42 If this can be validated in the cellular context, it will have important implications for drug development strategies beyond KRASG12C. We sought to use K27-SPOP as a probe to investigate the relative abundance of GDP-loaded forms across various KRAS mutations. To first validate that K27-SPOP is indeed GDP-specific, we measured degradation rates in the presence of the covalent G12C inhibitor, AMG510.3 AMG510 skews the population of KRASG12C toward the inactive GDP-bound state.3 In accordance with these literature findings, K27-SPOP increased degradation rates in a dose-dependent manner (Figure 5f). Conversely, in the presence of AMG510, degradation of KRASG12C by the GTP-specific biodegrader RBD-CRD-SPOP was completely blocked but was unaffected by the nucleotide-agnostic biodegrader NS1-SPOP (Figure 5f). These data strongly suggest K27-SPOP only recognizes and degrades RAS if it is in the GDP-bound state.
To determine if mutant KRAS does indeed cycle between the nucleotide states at different rates, we generated NanoLuc-tagged lines of the most common KRAS mutations (G12C, G12D, G12V, and Q61H) and compared their degradability by K27-SPOP (Figures 5g and S9). We expect that the higher the intrinsic hydrolysis rate, the greater the proportion of GDP-loaded mutant KRAS, and consequently, the better the rate of degradation by K27-SPOP. NS1-SPOP was used as a normalizing comparator since it binds both the GTP- and GDP-loaded forms equally.33 FN3-SPOP was used as a nondegrading control. Consistent with the nucleotide-state agnostic nature of NS1, the corresponding biodegrader NS1-SPOP degraded all five NanoLuc-tagged proteins with similar efficiencies (Figure 5g black lines). However, for K27-SPOP, a prominent difference in the rate of degradation was observed for each mutant (Figure 5g red lines). In accordance with the reported intrinsic hydrolysis rates,42 K27-SPOP was the most effective against wild-type KRAS (even exceeding NS1-SPOP), followed by KRASG12C, KRASG12D, and finally KRASG12V. The same trend was reproduced when we plotted the percentage of NanoLuc-tagged proteins remaining at 24-h post-transfection of respective biodegrader mRNAs (Figure 5h red lines). KRASG12V was barely degraded by K27-SPOP, while it was degraded by NS1-SPOP to a similar extent as the other mutants. One notable exception was KRASQ61H. Although it was reported that Q61L and Q61H mutants exhibit the lowest intrinsic hydrolysis rates,42 NanoLuc-KRASQ61H continued to be degraded by K27-SPOP (Figure 5g and 5h last column). It is currently unclear what accounts for this discrepancy. Finally, when the activating G12C, G12D, or G12V mutation was combined with the A59G substitution, KRAS becomes locked in the GTP state,5,43 and degradation by K27-SPOP was blocked in all cases (Figure S10). This further underscores the GDP-state specificity of K27-SPOP.
During the preparation of this manuscript, there was a report of a KRAS-specific DARPin, K1944 (Figure S11). Specificity was conferred through extensive interactions with histidine 95, a residue that is unique to KRAS. We generated the K19-SPOP biodegrader and confirmed that it was only able to degrade NanoLuc-KRAS (and KRASG12D) but not NanoLuc-HRAS and NanoLuc-NRAS (Figure 6a). By replacing histidine at position 95 with glutamine that is found in HRAS or leucine that is found in NRAS, K19-SPOP was no longer able to bind to and therefore degrade NanoLuc-KRASH95Q and NanoLuc-KRASH95L, while its counterpart K27-SPOP continued to degrade all proteins (Figures 6b, 6c, and S12). Degradation by K19-SPOP can be imparted to HRAS by mutating Q95 to histidine. On the contrary, a single point mutation of L95 to histidine on NRAS was insufficient to induce its degradation by K19-SPOP (Figure S8b). Since K19 interacted with KRAS independently of the nucleotide state,44 K19-SPOP degraded the various NanoLuc-tagged KRAS mutants to a similar extent (Figures 6d and S12). This result highlights how biodegraders that specifically degrade KRAS can be rapidly generated by engineering KRAS selectivity in the substrate-binding domain.
Figure 6.

Characterization of a KRAS-specific biodegrader, K19-SPOP. (a–d) Degradation profile and degradation rate calculated from T-REx-293 cells with doxycycline-induced expression of the indicated NanoLuc-tagged RAS protein and transfected with a 10-point 2-fold dose titration of K19-SPOP (a, b, and d) or K27-SPOP (c) mRNA at time 0.
Discussion
The work described herein advances our understanding of KRAS degradability and provides a compelling example of applying biodegraders as novel biological tools.
Specificity of anti-RAS Biodegraders
Prior to discussing how this work informs on (i) the degradability of KRAS proteins and (ii) KRAS biology, it is important to establish the specificity of the anti-KRAS biodegrader tools. As noted, the library of anti-RAS biodegraders was discovered using previously reported RAS binders spanning different affinities, isoform specificities, and nucleotide-state selectivities (Figures 2a and S2). Remarkably, all constructs, except for the K55-based biodegrader, resulted in functional degradation of endogenous RAS proteins (Figure 2c). The expected specificities of these biodegraders were also observed, with the clearest examples coming from the NanoLuc-RAS panel of cell lines. For example, NS1-SPOP was only able to degrade KRAS and HRAS but not NRAS (Figure 5c and 5d). When a single specificity-determining residue on KRAS was mutated to the corresponding NRAS residue (KRASR135K, Figure 5e), the loss of KRAS molecular recognition resulted in a lack of degradation by NS1-SPOP. The converse is also true–a K153R mutation in NRAS imparted recognition and degradation of NRAS by NS1-SPOP (Figure S8a). These observations highlight how biodegrader-mediated degradation is driven by precise biomolecular interactions. This point was further underscored using K19-SPOP, which was able to degrade KRAS but not HRAS nor NRAS (Figure 6a), as expected based on K19-binding specificities44 (Figure S11). X-ray structures have shown that the KRAS specificity of K19 is governed by its interaction with histidine 95, a residue where the equivalent amino acid is glutamine and leucine in HRAS and NRAS, respectively. As predicted, K19-SPOP failed to degrade the KRASH95Q and KRASH95L point mutants (Figure 6b). The ability to engineer exquisite specificities, coupled with their ease of discovery, makes biodegraders valuable research tools.
RAS Degradability
To gain rapid insights into KRAS degradation, we used GFP-KRAS and the toolbox of anti-GFP biodegraders we developed in previous work.16 Robust degradation was seen with most constructs (Figure 1c). Among the E3 ligases achieving significant degradation was VHL, an important result as VHL ligands have been used extensively for small molecule-based targeted degradation7 and therefore implies that they could be leveraged for degrading KRAS as well. Indeed, during the preparation of this manuscript, three relevant peer-reviewed reports were released.45−47 First, a biodegrader equivalent was constructed employing full-length VHL fused to NS1 (which the authors termed VHL-aHRAS).45 This so-called Affinity-directed PROtein Missile (AdPROM) achieved some knockdown in A549GFPKRAS cells but unfortunately did not yield significant growth inhibition in the three cancer cell lines tested–A549, HT29, and SW620. The Rabbitts group also reported a KRAS-specific biodegrader consisting of K19 fused to VHL.46 In the present study, we specifically removed the natural substrate-binding domain of VHL and demonstrated that it was highly effective at degrading both GFP-KRAS (when fused to vhhGFP4, Figure 1c) and endogenous KRAS (when fused to K27 and R11.1.6, Figure S3, lanes 16 and 18). Notably, the other two RAS binders, NS1 and RBD-CRD, that had worked in combination with SPOP (Figure S3, lanes 4 and 10) failed to degrade endogenous KRAS when conjugated to truncated VHL (Figure S3, lanes 14 and 20), suggesting that not all binder and E3 ligase combinations will produce active biodegraders. A publication from the Crews group appears to confirm that small molecule-based degraders which couple G12C covalent inhibitors to VHL ligands can achieve KRASG12C degradation.47 The current DC50 value (concentration to achieve 50% maximal degradation) stands at the micromolar range. Indeed, employment of the VHL E3 ligase in a degradation strategy is a convenient starting point as compatible ligands are available. However, our study also uncovered other E3 ligases that gave superior GFP-KRAS degradation (Figure 1), suggesting that time spent generating ligands to alternative E3 ligases could potentially yield more effective small molecule degraders.
While the case for converting an irreversible covalent inhibitor into a degrader molecule may not be immediately compelling, seminal work by the Crews lab47 provides solid evidence for the degradation of oncogenic KRASG12C through a targeted degradation approach and paves the way for future exploration in this direction. However, it is paramount to understand if the same can be applied to other KRAS mutants as they behave quite differently, both in terms of protein dynamics48 and, ultimately, in vivo tumorgenicity.49,50 Specifically, it was reported that the intrinsic GTP hydrolysis of various KRAS mutants differs in magnitude with the G12C mutant protein retaining the highest capacity to convert from the GTP-bound to the GDP-bound state.42 The two nucleotide states adopt distinct conformations and interact differently with the lipid bilayer,51 which may impact ligand accessibility and ternary complex formation. More importantly, the binding pocket used by covalent inhibitors is only accessible in the GDP-loaded state. Thus, targeted degradation strategies that aim to (noncovalently) exploit this pocket might be limited to KRAS mutant proteins that retain sufficiently high GTPase activity. In this study, we further investigated the degradability of KRAS under different nucleotide states and containing different oncogenic mutations. By applying biodegraders that are either GDP-specific (K27-SPOP) or GTP-specific (RBD-CRD-SPOP), we have demonstrated that both nucleotide states of K-, N-, and H-RAS are degradable substrates (Figures 2c and 5c). We have also shown that wild-type and a spectrum of KRAS mutants (G12D, G12C, G12V, and Q61H) are degradable (Figure 5g and 5h).
Cellular Prevalence of the GDP-Loaded State
Biodegraders represent novel cell-based tools for advancing biological understanding in entirely new ways. Here, we applied the GDP-specific biodegrader, K27-SPOP, to probe the prevalence of the inactive state in individual KRAS mutants. The corresponding data adds to a growing body of literature challenging the dogma that oncogenic RAS proteins are “locked” in the GTP state.52 Instead, a more nuanced view is one where the oncogenic mutations bias RAS to the GTP state. In particular, biochemical studies have suggested that while phenotypic RAS mutations greatly compromised GAP-mediated hydrolysis of GTP, low levels of both GAP-mediated and intrinsic hydrolysis still occur, albeit with a range of rate constants across the different mutations.42 Among them, KRASG12C had the highest intrinsic hydrolysis rate implying that a significant proportion of this protein may be present in the GDP-loaded (inactive) state. Indeed, the robust cellular activity demonstrated by G12C covalent inhibitors supports this notion since the corresponding binding pocket is only accessible in the GDP state. In fact, the covalent inhibitors were able to capture more than 90% of KRASG12C proteins within 1 h of treatment,4 attesting to the significant rate of GTP hydrolysis of the G12C mutant. We investigated the capacity of other KRAS mutants to cycle through the GDP/GTP states in the cellular context by using K27-SPOP as a gauge of the prevalence of the GDP-loaded state. K27-SPOP-induced degradability was WT > G12C > G12D > Q61H > G12V (from highest to lowest). Except for the Q61H mutant protein, this rank-order matches that determined previously.42 Our study has thus corroborated the biochemical data with physiologically relevant cell-based readouts.
Previous studies have suggested that ≥75% KRAS occupancy is needed to achieve therapeutic efficacy in tumor models.53 Irreversible inhibitory mechanisms have demonstrated the capacity to attain and sustain these levels despite the high intracellular concentration of KRAS (0.3 to 1.5 μM, Figure S1). However, for other KRAS mutants where a noncovalent inhibitor approach is required, achieving sufficient intracellular concentrations such that ≥75% stoichiometric target engagement is maintained will be challenging. A KRAS degradation approach is an attractive solution since these chimeric molecules can potentially be recycled to catalyze multiple rounds of target degradation at substoichiometric concentrations.6 The binding pocket that is available in the GDP state and bound by the G12C covalent inhibitors is an obvious starting point for the discovery of degraders against other KRAS mutations. However, considering our current data and previous work,42 leveraging this binding pocket for KRAS mutations with slower intrinsic hydrolysis may be challenging. As alternatives, our study has uncovered at least two additional RAS interfaces that might be leveraged for small molecule target degradation strategies. The regions bound by NS1-SPOP and K19-SPOP are especially attractive since we have shown that degradation efficiencies are comparable regardless of KRAS mutational status (Figure 5g and 6d). Although the path toward the identification of small molecule ligands that bind to these sites remains challenging, our study has nonetheless shown definitively that degraders occupying these spaces do not obstruct polyubiquitination sites and proteasomal degradation of KRAS.
Probing RAS Dependency/Superiority of a Degradation Strategy
Biodegraders can be used as novel tools to probe for RAS dependency, with examples herein of (1) lack of dependency (despite complete pan-RAS degradation, HEK293 cells, Figure 3) and (2) robust dependency (AsPC-1 cells, Figure 4). Compared to protein knockdown using conventional siRNA where effects only occur after turnover of the pre-existing pool of proteins (for KRASG12C, the reported half-life is ∼24 to 48 h4), targeted degradation by biodegraders can be achieved within 4 h following transfection (Figures 3a and 4a). The present study also shows that a degradation modality outperforms the stoichiometric equivalent. For example, the biodegrader K27-SPOP demonstrated sustained pERK inhibition up to 24-h postdoxycycline induction, whereas pERK levels rebounded at this time point with the stoichiometric inhibitor K27-SPOPmut despite its continued expression (Figure 3a). It is likely that feedback mechanisms related to RAS reactivation are at play as have been reported elsewhere with inhibitors of the RAS-signaling pathway.54−56 The superior effects of biodegraders were also recapitulated in functional assays where K27-SPOP resulted in complete growth arrest (Figure 4b) and induction of apoptosis (Figure 4d), whereas K27-SPOPmut and the nonbinding control K27mut-SPOP had no impact on AsPC-1 cells. Collectively, our study suggests that a degradation strategy can elicit a more comprehensive and durable inhibition of KRAS-dependent signaling compared to a stoichiometric approach, a finding that may have important implications for the treatment of KRAS mutant tumors.
Concluding Remarks
Perhaps the most significant impact from this work is the clear demonstration that the exquisite specificity of biodegraders for their targets allows them to answer questions that other approaches cannot. Although RNAi and CRISPR techniques can deplete/remove a protein from cellular environments, they lack the capacity to differentiate subpopulations that differ in conformations, post-translational modifications, or subcellular localizations. The application of the GDP-specific biodegrader to inform on nucleotide-loaded states represents a KRAS-specific example to what should be a generally applicable approach. This work has immediate impacts as it firmly establishes the degradability of KRAS and informs on the design of small molecule-based degraders for KRAS mutants, important oncogenic drivers. The superiority of a targeted degradation approach was also demonstrated. Beyond their use as tool molecules, this work highlights the potential therapeutic application of biodegraders and related intracellular biologics. Indeed, using an engineered overexpression system, the Rabbitts group recently demonstrated that biodegrader equivalents have the potential to degrade KRAS and achieve tumor growth inhibition in vivo.46 Moving forward, obtaining sufficient delivery and intracellular expression of macromolecules will be among the most important challenges in therapeutic settings. Encouragingly, recent success of mRNA-based COVID-19 vaccines in nonhuman primates and in clinical trials57−59 suggest that mRNA therapy may be a potential approach for addressing these hurdles. Although outside the scope of this work, recent advances have started to chart road maps for optimizing mRNA molecules with 1) improved cellular half-lives,60,61 2) improved translation efficiencies,62 and 3) greatly reduced innate immune system activation.63−65 As well, developments in lipid nanoparticle design are improving mRNA cell entry while minimizing toxicity.66 Such advances have contributed–in part–to the pioneering successes that are starting to be realized for in vivo delivery of therapeutic mRNA outside of the vaccine arena.67−72
Acknowledgments
We thank Tomi K. Sawyer, Kaustav Biswas, Nicolas Boyer, Chunhui Huang, Alexander Stoeck, Nicole Boo, Jeff Chang, Sybil M. G. Williams, Payal Sheth, Jason E. Imbriglio, Uyen Phan, Ruban Mangadu, Mohammed Selman, C. M. Hsieh, Veronica Juan, Sara Zarnowski, Li Ding, Lei Chen, Amy C. Doty, Lauren A. Austin, Jeffrey S. Smith, Nicolas Solban, David P. Lane, Christopher J. Brown, Charles W. Johannes, Tsz Ying Yuen, Chandra Verma, Srinivasaraghavan Kannan, and all members of the Quantitative Biosciences team for helpful discussions and comments on the manuscript. The authors acknowledge support from the MRL Postdoctoral Research Program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01337.
Materials and Methods; coding sequences of all constructs (Table S1); quantitation of intracellular RAS concentration in panel of KRAS mutant cell lines, isothermal titration calorimetric analysis (ITC), degradation of endogenous RAS by SPOP- and VHL-based biodegraders, degradation of endogenous RAS by doxycycline-inducible anti-RAS biodegraders, comparison of DNA and mRNA transfection efficiencies in AsPC-1 cells, determination of mRNA transfection efficiencies in panel of KRAS mutant cells, optimization of NanoLuc degradation assay, specificity determinants in biodegrader-mediated degradation, real-time monitoring of the degradation of various KRAS mutant proteins, introduction of A59G mutation abolishes degradation by K27-SPOP, and characterization of KRAS-specific biodegrader K19-SPOP (Figures S1–S12) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Khan I.; Rhett J. M.; O’Bryan J. P. Therapeutic targeting of RAS: New hope for drugging the “undruggable. Biochim. Biophys. Acta, Mol. Cell Res. 2020, 1867, 118570. 10.1016/j.bbamcr.2019.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick F. Progress in targeting RAS with small molecule drugs. Biochem. J. 2019, 476, 365–374. 10.1042/BCJ20170441. [DOI] [PubMed] [Google Scholar]
- Canon J.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Hallin J.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery 2020, 10, 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P.; Solomon M.; Li L. S.; Hansen R.; Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18, 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. Proteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discovery Today: Technol. 2019, 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidou M.; et al. PROTACs- a game-changing technology. Expert Opin. Drug Discovery 2019, 14, 1255–1268. 10.1080/17460441.2019.1659242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt G. F.; Scott-Stevens P.; Gaohua L. Targeted protein degradation in vivo with Proteolysis Targeting Chimeras: Current status and future considerations. Drug Discovery Today: Technol. 2019, 31, 69–80. 10.1016/j.ddtec.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Chessum N. E. A.; et al. Demonstrating In-Cell Target Engagement Using a Pirin Protein Degradation Probe (CCT367766). J. Med. Chem. 2018, 61, 918–933. 10.1021/acs.jmedchem.7b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines J.; Lartigue S.; Dong H.; Qian Y.; Crews C. M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. 10.1158/0008-5472.CAN-18-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka A.; et al. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem. 2006, 281, 8917–8926. 10.1074/jbc.M509344200. [DOI] [PubMed] [Google Scholar]
- Mageean C. J.; Griffiths J. R.; Smith D. L.; Clague M. J.; Prior I. A. Absolute Quantification of Endogenous Ras Isoform Abundance. PLoS One 2015, 10, e0142674 10.1371/journal.pone.0142674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B.; et al. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Lim S.; et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 5791–5800. 10.1073/pnas.1920251117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoff A. D.; Stephens E. A.; Varner J. D.; DeLisa M. P. Ubiquibodies, synthetic E3 ubiquitin ligases endowed with unnatural substrate specificity for targeted protein silencing. J. Biol. Chem. 2014, 289, 7844–7855. 10.1074/jbc.M113.544825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher L. J.; Hutchinson L. D.; Macartney T. J.; Turnbull C.; Sapkota G. P. Targeting endogenous proteins for degradation through the affinity-directed protein missile system. Open Biol. 2017, 7, 170066. 10.1098/rsob.170066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E.; Kanca O.; Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 2012, 19, 117–121. 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- Ludwicki M. B.; et al. Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci. 2019, 5, 852–866. 10.1021/acscentsci.9b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigenzahn J. W.; et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362, 1171–1177. 10.1126/science.aap8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steklov M.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. 10.1126/science.aap7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T.; et al. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 2020, 27, 1023–1035. 10.1038/s41418-019-0395-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. K. beta-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK3beta. EMBO Rep 2018, 19, e46060. 10.15252/embr.201846060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; et al. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 286–294. 10.1158/1535-7163.MCT-12-0650. [DOI] [PubMed] [Google Scholar]
- Pan T.; et al. A recombinant chimeric protein specifically induces mutant KRAS degradation and potently inhibits pancreatic tumor growth. Oncotarget 2016, 7, 44299–44309. 10.18632/oncotarget.9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 2020, 27, 19–31.e16. 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Saerens D.; et al. Identification of a universal VHH framework to graft non-canonical antigen-binding loops of camel single-domain antibodies. J. Mol. Biol. 2005, 352, 597–607. 10.1016/j.jmb.2005.07.038. [DOI] [PubMed] [Google Scholar]
- Rothbauer U.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- Guharoy M.; Bhowmick P.; Sallam M.; Tompa P. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat. Commun. 2016, 7, 10239. 10.1038/ncomms10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J.; Poganik J. R.; Aye Y. On-Demand Targeting: Investigating Biology with Proximity-Directed Chemistry. J. Am. Chem. Soc. 2016, 138, 3610–3622. 10.1021/jacs.5b12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmick M.; et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. 10.1016/j.cell.2014.02.051. [DOI] [PubMed] [Google Scholar]
- Spencer-Smith R.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillard S.; et al. Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat. Commun. 2017, 8, 16111. 10.1038/ncomms16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauke M. J.; et al. An engineered protein antagonist of K-Ras/B-Raf interaction. Sci. Rep. 2017, 7, 5831. 10.1038/s41598-017-05889-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C. J. Ras effectors. Curr. Opin. Cell Biol. 1996, 8, 197–204. 10.1016/S0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- Travers T.; et al. Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci. Rep. 2018, 8, 8461. 10.1038/s41598-018-26832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Jang H.; Zhang J.; Nussinov R. Raf-1 Cysteine-Rich Domain Increases the Affinity of K-Ras/Raf at the Membrane, Promoting MAPK Signaling. Structure 2018, 26, 513–525. 10.1016/j.str.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omerovic J.; Hammond D. E.; Clague M. J.; Prior I. A. Ras isoform abundance and signalling in human cancer cell lines. Oncogene 2008, 27, 2754–2762. 10.1038/sj.onc.1210925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; et al. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Hunter J. C.; et al. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- Lu J.; Bera A. K.; Gondi S.; Westover K. D. KRAS Switch Mutants D33E and A59G Crystallize in the State 1 Conformation. Biochemistry 2018, 57, 324–333. 10.1021/acs.biochem.7b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bery N.; et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 2019, 10, 2607. 10.1038/s41467-019-10419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S. Targeting Endogenous K-RAS for Degradation through the Affinity-Directed Protein Missile System. Cell Chem. Biol. 2020, 27, 1151. 10.1016/j.chembiol.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bery N.; Miller A.; Rabbitts T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020, 11, 3233. 10.1038/s41467-020-17022-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M. J.; Chu L.; Nalawansha D. A.; Li K.; Crews C. M. Targeted Degradation of Oncogenic KRASG12C by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantsar T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. 10.1016/j.csbj.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters I. P.; et al. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat. Commun. 2017, 8, 2053. 10.1038/s41467-017-01519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis K. M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. 10.1016/j.trecan.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhab-Jafari M. T.; et al. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6625–6630. 10.1073/pnas.1419895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs G. A.; Wittinghofer A.; Der C. J. Selective Targeting of the KRAS G12C Mutant: Kicking KRAS When It’s Down. Cancer Cell 2016, 29, 251–253. 10.1016/j.ccell.2016.02.015. [DOI] [PubMed] [Google Scholar]
- Janes M. R.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e517. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Turke A. B.; et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A.; et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Villanueva J.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett K. S.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. 10.1056/NEJMoa2024671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. J. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N Engl J. Med. 2020, 383, 2427. 10.1056/NEJMoa2028436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. 10.1038/s41586-020-2814-7. [DOI] [PubMed] [Google Scholar]
- Mauger D. M.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 24075–24083. 10.1073/pnas.1908052116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandini von Niessen A. G.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. 10.1016/j.ymthe.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample P. J.; et al. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809. 10.1038/s41587-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S.; Kataoka K.; Itaka K. Screening of mRNA Chemical Modification to Maximize Protein Expression with Reduced Immunogenicity. Pharmaceutics 2015, 7, 137–151. 10.3390/pharmaceutics7030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Luo X.; Dong Y. Effects of Chemically Modified Messenger RNA on Protein Expression. Bioconjugate Chem. 2016, 27, 849–853. 10.1021/acs.bioconjchem.6b00090. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan S.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther.--Nucleic Acids 2018, 12, 530–542. 10.1016/j.omtn.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis S.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong N. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl Med. 2019, 11, eaaw1565. 10.1126/scitranslmed.aaw1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt S. L. Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci. Transl Med. 2019, 11, eaat9143. 10.1126/scitranslmed.aat9143. [DOI] [PubMed] [Google Scholar]
- Gan L. M.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. 10.1038/s41467-019-08852-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. 10.1038/s41591-018-0199-z. [DOI] [PubMed] [Google Scholar]
- Stadler C. R.; et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817. 10.1038/nm.4356. [DOI] [PubMed] [Google Scholar]
- Hauser S.; et al. mRNA as a Novel Treatment Strategy for Hereditary Spastic Paraplegia Type 5. Mol. Ther.--Methods Clin. Dev. 2019, 15, 359–370. 10.1016/j.omtm.2019.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.