

Abstract
Therapeutic immunotoxins composed of antibodies and bacterial toxins provide potent activity against malignant cells, but joining them with a defined covalent bond while maintaining the desired function is challenging. Here, we develop novel immunotoxins by dovetailing full-length immunoglobulin G (IgG) antibodies and nontoxic anthrax proteins, in which the C terminus of the IgG heavy chain is connected to the side chain of anthrax toxin protective antigen. This strategy enabled efficient conjugation of protective antigen variants to trastuzumab (Tmab) and cetuximab (Cmab) antibodies. The conjugates effectively perform intracellular delivery of edema factor and N terminus of lethal factor (LFN) fused with diphtheria toxin and Ras/Rap1-specific endopeptidase. Each conjugate shows high specificity for cells expressing human epidermal growth factor receptor 2 (HER2) and epidermal growth factor receptor (EGFR), respectively, and potent activity across six Tmab- and Cmab-resistant cell lines. The conjugates also exhibit increased pharmacokinetics and pronounced in vivo safety, which shows promise for further therapeutic development.
Short abstract
Using tools from protein engineering and chemical biology, we combine antibodies and nontoxic anthrax proteins to develop new immunotoxins that target cancer cells and deliver therapeutic proteins.
Introduction
Harnessing delivery systems from nature may offer the key to achieving antibody-directed protein delivery into mammalian cells. Immunotoxins are a class of therapeutic delivery systems comprising a bacterial toxin and receptor-binding component, which mediate cytosolic delivery of the toxin upon binding to the target receptor. Most immunotoxins consist of a truncated form of either exotoxin A from Pseudomonas aeruginosa, ricin toxin from Ricinus communis, or the A chain of diphtheria toxin (DTA) from Corynebacterium diphtheria.1,2 These toxins are combined with proteins, antibodies, or antibody fragments that target receptors on malignant cells,3,4 including the CD22 receptor on hairy cell leukemia cells, the CD25 receptor on chronic lymphocytic leukemia and cutaneous T-cell lymphoma cells, and the mesothelin receptor on mesothelioma,5 pancreatic,6 and ovarian7 cancer cells. A notable example of an immunotoxin is moxetumomab pasudotox, which is a fusion protein of an anti-CD22 single-chain antibody fragment and a truncated form of Pseudomonas exotoxin A. This immunotoxin was FDA approved in 2018 for the treatment of hairy cell leukemia8,9 and has paved the way for other immunotoxins in the clinic. Recently, oportuzumab monatox has generated exciting clinical results for the treatment of bladder cancer, which is an immunotoxin composed of an anti-EpCAM single-chain antibody fragment conjugated to Pseudomonas aeruginosa exotoxin A fragment.10
Although a few immunotoxins have been successful in the clinic, improved designs are needed that exhibit enhanced efficacy and target-recognition affinity, tunable in vivo lifetimes, and reduced immunogenic and other adverse effects.11 Unfortunately, developing new variants is particularly challenging. Insufficient selectivity of receptor-binding proteins has been associated with off-target toxicity.1 Full-length antibodies can provide increased antigen affinity through avidity, a wider therapeutic window, and attractive in vivo properties, but few methods have been developed for site-selective ligation of toxins to antibodies.2,12 Continued development of immunotoxins is therefore needed for these therapeutics to reach their full potential.
Anthrax is a potent toxin that comprises two main proteins: protective antigen (PA) and lethal factor (LF).13,14 In nature, wild-type (WT) PA ultimately forms a pore to translocate toxins LF or edema factor (EF) into the cell cytosol, inducing rapid cell death (Figure 1A).15 Translocation proceeds by the following mechanism: (1) The 83 kDa protein, PA83, binds to one of two cell receptors, TEM8 or CMG2.16,17 (2) PA83 is proteolytically cleaved by a furin protease into two fragments, PA63 and PA20.18 (3) The PA63 fragment oligomerizes into heptameric19 and octameric20 prepores. (4) Three or four LF (or EF) molecules bind to PA63 with nanomolar affinity.21,22 (5) The entire complex is endocytosed, where acidification of the endosome promotes PA to conformationally change from prepores to active transmembrane pores for translocating LF or EF into the cytosol.23−25
Figure 1.

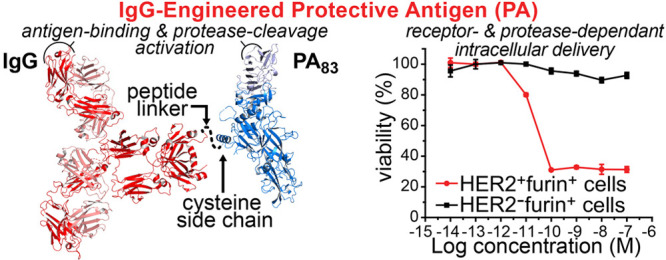
Protective antigen (PA) mediates cytosolic protein delivery into cells. (A) PA translocation mechanism of lethal factor (LF) or edema factor (EF), targeting TEM8 or CMG2 receptors. (B) Envisioned IgG-directed targeting to HER2 or EGFR receptors, followed by PA-mediated translocation of the N-terminal LF domain (LFN) with cargo. (C) X-ray crystallographic structures of full-length PA (PDB: 1ACC) with PA20 and PA63; and IgG antibody (PDB ID: 1HZH) with heavy (HC) and light (LC) chains. The linker peptide is illustrated as a dotted line. N and C termini are indicated by closed and open face arrows, respectively.
Engineered PA and LF variants provide effective tools for delivering non-native cargo into cells.26 The nontoxic N-terminal region of LF, called LFN, provides the basis for harnessing this machinery. LFN retains nanomolar binding affinity to PA prepores and permits conjugation and delivery of beta-lactamase,27Pseudomonas exotoxin A,28 Ras/Rap1-specific endopeptidase (RRSP),29 DTA,30 cytotoxic T lymphocyte epitopes from Listeria monocytogenes listeriolysin O and ovalbumin,31,32 peptide nucleic acids,33,34 and other non-native cargo.35 Targeting mutant PA variants to specific cell types has recently been achieved by combining them with a receptor-binding protein36−40 or by altering the protease cleavage site between PA63 and PA20.41,42 These retargeted PA variants have provided a glimpse of the therapeutic potential for delivering effector proteins into specific cells but have not yet generated sufficient in vivo selectivity for translation to the clinic.
Here, we introduce an immunotoxin platform that combines full-length antibodies with nontoxic anthrax proteins. We envisioned that this platform would provide enhanced in vivo properties and targeting to mammalian cell receptors and, upon binding, would maintain the PA translocation mechanism (Figure 1B). In practice, however, combining an antibody with PA is challenging. Simply fusing PA to an antibody N or C terminus either would obstruct the antibody binding region or, upon proteolytic cleavage of PA20, would separate the antibody from PA63, respectively. To develop this platform, we carefully designed a bioconjugation strategy to connect a side chain on mutant PA to the C terminus of an immunoglobulin G (IgG) antibody (Figure 1C). This strategy enabled successful preparation of two classes of PA conjugates: one with trastuzumab (Tmab) for targeting human epidermal growth factor receptor 2 (HER2)43 and the other with cetuximab (Cmab) for targeting epidermal growth factor receptor (EGFR).44
In vitro studies show that these Tmab- and Cmab-directed PA conjugates selectively deliver DTA into HER2- and EGFR-positive cells, respectively. These studies also show that DTA delivery provides potent toxicity across six antibody-resistant cancer cell lines, including one HER2-positive cell line and five EGFR-positive cell lines. Further in vitro studies show that the conjugates efficiently deliver EF and RRSP into target cells. Also, two additional Cmab-mPAC conjugates with dual antibody- and protease-specific cleavage site-targeting conjugates provide effective translocation into target cells. In vivo studies show that these dual-targeting conjugates exhibit enhanced pharmacokinetic properties and pronounced in vivo safety, relative to unconjugated PA, which shows promise for further therapeutic development.
Results
Design and Preparation of Antibody-Directed Protective Antigen
We designed and prepared two main classes of antibody-directed PA conjugates, which each comprise a full-length IgG antibody and PA. One class exhibits Tmab-directed targeting of HER2-positive cells; the other class exhibits Cmab-directed targeting of EGFR-positive cells.
To prepare these conjugates, we designed a mutant PA, called mPAC, that contains two sets of previously reported mutations (Figure 2A): (1) a pair of mutations associated with mPA, N682A and D683A, which ablate binding to native anthrax receptors;45 and (2) a single mutation associated with PAC, K563C, which permits cysteine conjugation.46 These mutations were combined for preparing mPAC and a translocation-deficient mPAC [F427A] mutant, called mPACA (Table S1).47,48 Both mutants were expressed and purified by anion-exchange chromatography (AEX), followed by SDS-PAGE (Figure S1) and LC/MS analysis (Figure S2).
Figure 2.
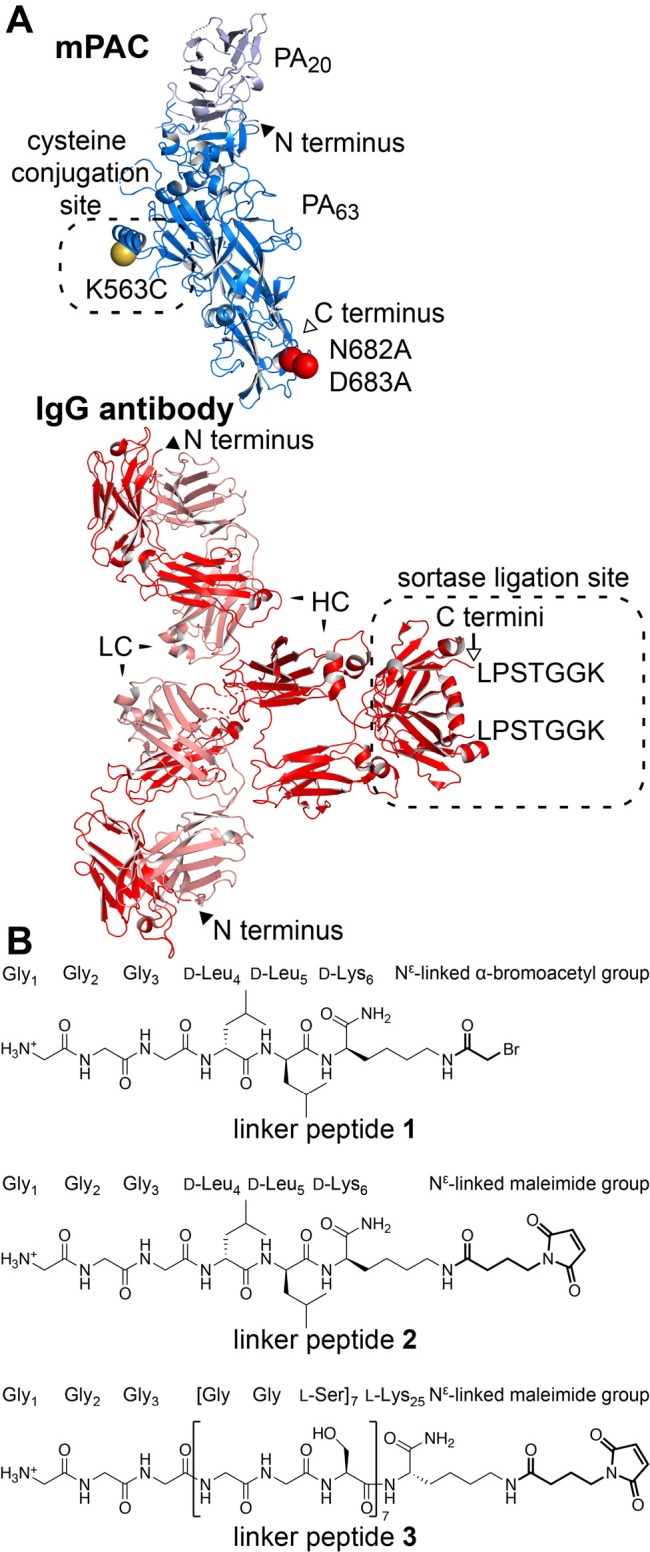
Designs of IgG, PA, and linker peptides are critical for creating an anthrax-based immunotoxin. (A) mPAC, with mutated residues indicated by spheres: N682A and D683A (red); K563C (yellow). IgG antibody, with the sortase-recognition sequence indicated by LPSTGGK. N and C termini are indicated by closed and open face arrows, respectively. (B) Linker peptides 1–3, illustrating the three N-terminal Gly residues for sortase-mediated ligation and the C-terminal group for conjugation to a cysteine side chain.
We also prepared two mutant IgG antibodies of Tmab and Cmab, which each contain residues LPSTGGK for recognition by the sortase enzyme (Figure 2A).49 These residues were incorporated at the C terminus of the antibody heavy chain (HC) to provide distance between the PA translocase and the antibody-binding region. The antibodies were prepared by incorporating the sortase-recognition sequence into plasmids for Tmab (Table S2) and Cmab (Table S3), followed by expression in mammalian cells, purification by protein A resin, and LC/MS analysis (Figure S3).
We also designed and prepared linker peptides 1–3 to combine IgG antibodies with mPAC (Figure 2B). The peptides each contain three Gly residues at the N terminus for sortase-mediated ligation but differ at the C terminus. Peptide 1 contains residues d-Leu4, d-Leu5, and d-Lys6 to impart proteolytic stability and also contains an Nε-linked α-bromoacetyl group for conjugation to a cysteine side chain. Peptide 2 is similar to peptide 1, but 2 contains an Nε-linked maleimide group for conjugation to a cysteine side chain. Peptide 3 also contains an Nε-linked maleimide group but contains a long chain of l-amino acids, [GGS]7K, to provide an extended linkage (ca. 80 Å) between the IgG antibody and mPAC. The three peptides were synthesized by solid-phase peptide synthesis on Rink-amide resin, followed by cleavage of the peptide, RP-HPLC purification, lyophilization, and LC/MS analysis (Figure S4).
We developed a two-step procedure for preparing these antibody-directed PA conjugates from the mutant IgG and PA variants, which enabled modular assembly of these variants. In the first step, mPAC was incubated with linker peptide 1, 2, or 3 at pH 8.5 for 60 min to give the corresponding G3-mPAC (Figure 3A, Figure S5). In the second step, after removal of excess peptide, G3-mPAC was incubated with an IgG antibody (IgG-LPSTGGK) and sortase enzyme (SrtA*) for 60 min (Figure 3B).49 The proteins were analyzed by SDS-PAGE, before and after adding SrtA*, which revealed a mixture of IgG-mPAC and IgG-(mPAC)2 conjugates (Figure 3C). These mono- and disubstituted conjugates were separated by size-exclusion chromatography (SEC), followed by AEX chromatography. SDS-PAGE analysis showed that purification gave clean IgG-mPAC1 conjugates for both Tmab-mPAC (Figure 3 D,E) and Cmab-mPAC (Figure S6).
Figure 3.
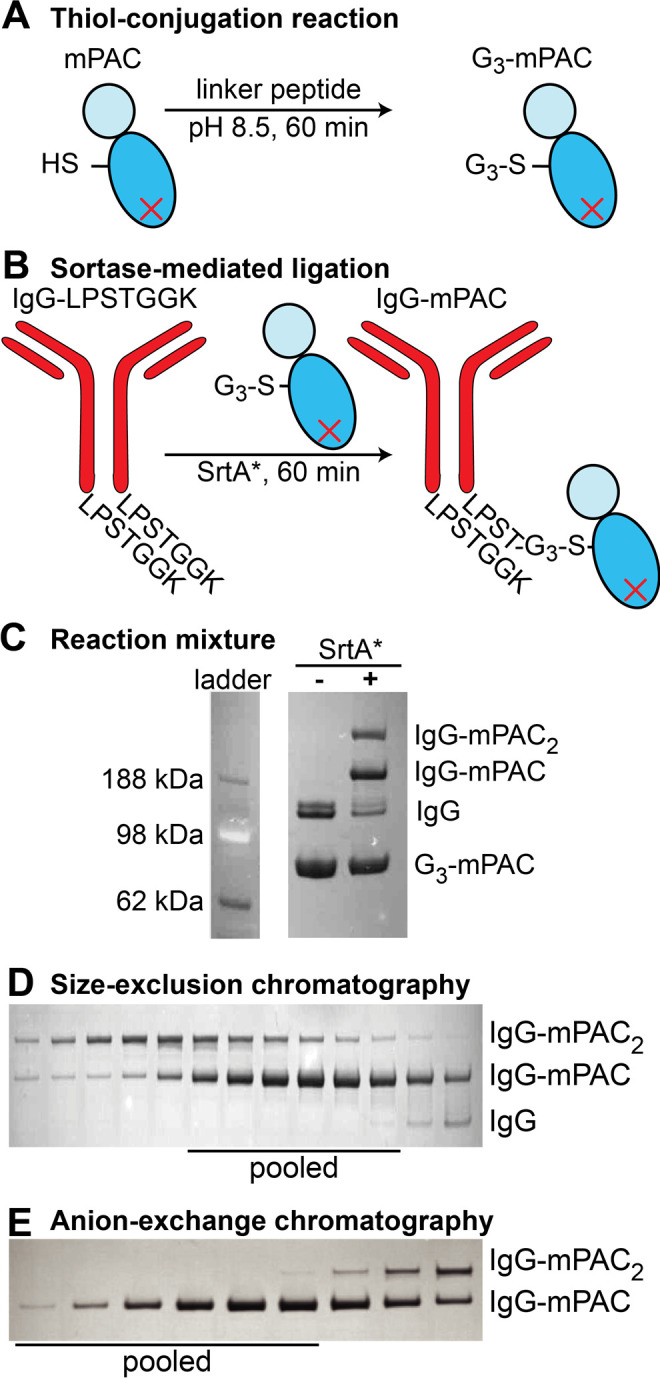
A two-step procedure enables bioconjugation of IgG to mPAC. (A) The thiol-conjugation reaction with mPAC and linker peptide affords G3-mPAC. (B) Sortase-mediated ligation with IgG antibody (IgG-LPSTGGK) and G3-mPAC affords a mixture of IgG-mPAC and IgG-(mPAC)2 conjugates. Coomassie-visualized SDS-PAGE gel analysis of mixtures containing: (C) Tmab and G3-mPAC (linker peptide 1), before and after adding SrtA*; and Tmab-mPAC conjugate after (D) size-exclusion chromatography (SEC, HiLoad 16/600 Superdex 200) and (E) anion-exchange (AEX) chromatography (HiTrap Q HP).
Antibody-Directed Protective Antigen Variants Oligomerize and Perform Translocation
We used three key experiments to establish IgG-mPAC oligomerization, serum stability, and translocation activity.
First, we established oligomer-forming ability by SDS-PAGE analysis. Tmab-mPAC prepared with linker peptide 1 was incubated with furin protease to cleave PA20, which was then removed by AEX chromatography. Upon subjecting Tmab-mPAC63 to acidic conditions, SDS-PAGE analysis revealed the appearance of a single, high-molecular-weight band consistent with the formation of SDS-stable oligomers (Figure S7). This result indicates that IgG-mPAC conjugates can oligomerize under acidic conditions and may also perform protein translocation into mammalian cells.
Second, we evaluated the serum stability of the linker peptides. Two Tmab-mPAC variants prepared from linker peptides 1 and 2 were incubated with 10% fetal bovine serum at 37 °C for 168 h. SDS-PAGE gels visualized by Western blot analysis showed that Tmab-mPAC remains largely intact with linker peptide 1 but substantially degrades after 1 h with linker peptide 2 (Figure S8).
Third, we evaluated the translocation activity of the IgG-mPAC conjugates. Three Tmab-mPAC conjugates prepared from linker peptides 1–3 were incubated with HER2-positive cells (BT474), using 10-fold serial dilutions of each conjugate and 10 nM of the fusion protein LFN-DTA. LFN-DTA served as a reporter of translocation activity by decreasing cell viability, in which the DTA component ribosylates eukaryotic elongation factor 2 in the cytosol and inhibits protein synthesis.50 This experiment showed that all three IgG-mPAC conjugates exhibit comparable protein synthesis inhibition activity (Figure S9), indicating that varying the linker length does not alter translocation activity. In addition, Tmab-mPAC and Tmab-(mPAC)2 also showed comparable activity (Figure S10), indicating that increasing the mPAC substituents does not alter translocation activity.
These three experiments established that linker peptide 1 provides a robust, serum-stable linkage between IgG antibodies and mPAC. The experiments also establish that efficient translocation activity is achieved through a 1:1 and 1:2 IgG/mPAC ratio for the IgG-mPAC conjugates. All IgG-mPAC conjugates mentioned hereafter were prepared using linker peptide 1, followed by isolation of fractions containing IgG-mPAC1 conjugates (fractions containing IgG-mPAC2 conjugates were discarded).
Antibody-Directed Cytosolic Protein Delivery into HER2- and EGFR-Positive Cells
In vitro cell assays revealed that both Tmab-mPAC and Cmab-mPAC conjugates act on cells bearing the corresponding target receptors. These experiments also showed that the IgG-mPAC conjugates deliver cargo by a translocation mechanism. We established this activity against HER2- and EGFR-positive cells with the IgG-mPAC conjugates and appropriate controls.
HER2-positive cells (BT474) were incubated with 10 nM LFN-DTA and 10-fold serial dilutions of Tmab-mPAC (Figure 4A). This treatment showed decreased cell viability at Tmab-mPAC concentrations as low as 100 pM, indicating potent DTA delivery into the cytosol. The cells were also incubated with 10 nM LFN-DTA and a 1:1 mixture of Tmab + mPAC, which did not decrease cell viability. In addition, incubation with either the unconjugated Tmab alone or conjugated Tmab-mPAC alone did not decrease cell viability. The cells were also incubated with two translocation-deficient PA variants: (1) Tmab-mPACA (Table S1 and Figure S2) and LFN-DTA; and (2) Tmab conjugated directly to LFN-DTA, called Tmab-LFN-DTA (Figure S11). Neither treatment decreased viability, indicating that free LFN-DTA and a functional IgG-mPAC conjugate are important for achieving antibody-directed cytosolic protein delivery.
Figure 4.

Antibody-Targeting Provides High Selectivity for Target Cells. Relative cell viability after 72 h of incubation with or without 10 nM LFN-DTA, and with 10-fold serial dilutions of an IgG or IgG-mPAC conjugate: (A) HER2-positive cells (BT474) with Tmab, Tmab-mPAC, Tmab + mPAC, Tmab-mPACA, or Tmab-LFN-DTA; (B) EGFR-positive cells (A431) with Cmab, Cmab-mPAC, Cmab + mPAC, or Cmab-mPACA; (C) HER2-positive cells (BT474) and HER2-negative cells (BT549) with Tmab-mPAC; (D) EGFR-positive cells (A431) and EGFR-negative cells (Jurkat) with Cmab-mPAC. mPACA represents the mPAC [F427A] variant, which is translocation-deficient. Cell viability was determined by the relative luminescence from a CellTiter-Glo assay, which was normalized to untreated cells. Data represent the mean of three replicate wells ± the standard deviation (±s.d.).
Also, Cmab-mPAC delivers LFN-DTA into EGFR-positive cells by a translocation mechanism (Figure 4B). EGFR-positive cells (A431) were incubated with Cmab-mPAC and LFN-DTA. These treatments decreased viability at Cmab-mPAC concentrations as low as 100 pM. No other treatment decreased cell viability, indicating that both Cmab-mPAC and LFN-DTA are required for cytotoxic protein delivery into EGFR-positive cells.
Targeted Delivery of Bacterial Toxins for Receptor-Specific Toxicity against Cancer Cells
We established that the IgG-mPAC conjugates only perform translocation on cells with the target receptor (Figure 4C,D).51 HER2-positive (BT474) and HER2-negative (BT549) cells were incubated with serial dilutions of Tmab-mPAC and 10 nM of LFN-DTA. These treatments decreased viability of the HER2-positive cells but had no effect on the HER2-negative cells (Figure 4C). EGFR-positive (A431) and EGFR-negative (Jurkat) cells were incubated with Cmab-mPAC and LFN-DTA. These treatments decreased viability of the EGFR-positive cells at concentrations as low as 100 pM but had no effect on the EGFR-negative cells (Figure 4D). To show that LFN-DTA is toxic to BT549 and Jurkat cells, which do not display the target receptors, these cells were incubated with WT PA and LFN-DTA. For both cell lines, the treatments decreased cell viability at PA concentrations as low as 100 pM (Figure S12).
Since the parent antibodies are toxic to several HER2- and EGFR-positive cell lines, we performed additional experiments to compare antibody-specific activity with the additional toxicity from delivering LFN-DTA. We also evaluated background activity that may occur through Fc binding to cell receptors. Two cell lines were used for these studies: (1) HER2-positive (BT474) and (2) EGFR-positive (A431) cells. These cells were selected because they each display only one of the two receptors, HER2 or EGFR, and are sensitive to Tmab and Cmab, respectively. For these experiments, cells were either incubated with 10 nM IgG-mPAC alone or coincubated with serial dilutions of LFN-DTA. To demonstrate toxicity from PA and LFN-DTA in parallel, cells were also treated with 10 nM WT PA and with serial dilutions of LFN-DTA. The studies showed that Tmab-mPAC alone had no effect on HER2-negative (A431) cells but partially decreased viability of the HER2-positive (BT474) cells (Figure S13). In addition, coincubation of Tmab-mPAC + LFN-DTA further decreased cell viability (EC50 = 3.9 ± 3.5 pM). Alternatively, Cmab-mPAC had no effect on the EGFR-negative (BT474) cells but partially decreased viability of the EGFR-positive (A431) cells (Figure S14). In addition, coincubation of Cmab-mPAC + LFN-DTA further decreased cell viability of the EGFR-positive cells (EC50 = 4.3 ± 1.5 pM). These studies show that the IgG-PA conjugates alone exhibit partial toxicity to cells bearing the target receptor, and exhibit increased toxicity through delivering LFN-DTA. Furthermore, the absence of toxicity to cells lacking the target receptor suggests that limited activity is exhibited through binding of the Fc region.
To further evaluate selectivity of the IgG-mPAC conjugates, two HER2-negative EGFR-positive cells (A549 and HCT116) were incubated with LFN-DTA in the presence of either Tmab-mPAC or Cmab-mPAC. For both cell lines, the Cmab-mPAC showed toxicity at concentrations as low as 100 pM, while the Tmab-mPAC treatments showed no toxicity (Figure S15).
Targeted Delivery of Bacterial Toxins into Drug-Resistant Cancer Cells
Tmab- and Cmab-directed DTA delivery is also toxic to cells that are resistant to the parent Tmab52 and Cmab53 antibodies. We established this activity across six drug-resistant cell lines, including one with HER2 receptors and five with EGFR receptors. HER2-positive ovarian cancer cells (SKOV-3) were incubated with serial dilutions of Tmab-mPAC and 10 nM LFN-DTA (Figure 5A). This treatment decreased cell viability at Tmab-mPAC concentrations as low as 100 pM. In contrast, cells incubated with Tmab alone did not decrease viability.
Figure 5.

Antibody-directed PA conjugates overcome resistance to Tmab and Cmab therapeutic antibodies. Relative cell viability after 72 h of incubation with or without 10 nM LFN-DTA, and with 10-fold serial dilutions of IgG, IgG-mPAC conjugate, or gefitinib: (A) ovarian cancer cells (SKOV-3); (B, C) lung cancer cells (A549, H441); (D) colon cancer cells (HCT-116); and (E, F) triple-negative breast cancer cells (BT549, MDA-MB-231). Cell viability was determined by the relative luminescence from a CellTiter-Glo assay, which was normalized to untreated cells. Data represent the mean ± s.d. of three replicate wells.
Five EGFR-positive cells were incubated with unconjugated Cmab alone or with the Cmab-mPAC conjugate and 10 nM LFN-DTA. Cells were also incubated with gefitinib, which is a small-molecule drug inhibitor of EGFR.54 The cell lines evaluated include two derived from lung cancer (A549 and H441), one derived from colorectal cancer (HCT-116), and two derived from triple negative breast cancer (BT549 and MDA-MB-231). All five cell lines decreased in viability after incubation with Cmab-mPAC and LFN-DTA, but not after incubation with Cmab or gefitinib alone (Figure 5B–F).55,56 These results show that immunotoxins can overcome resistance associated with the parent antibody and gefitinib.
Targeted Enzyme Delivery Enables Cell-Specific Control of Intracellular Processes
Delivering effector proteins into cells, other than DTA, shows that antibody-directed PA variants enable control of cell processes without promoting cell death. For these experiments, we used Cmab-mPAC to deliver two enzymes, EF and RRSP, and evaluated the corresponding activity.
EF is a native effector protein of anthrax lethal toxin that functions by increasing intracellular concentrations of cyclic AMP (cAMP), through calmodulin- and Ca2+-dependent adenylate cyclase activity.57 This activity is important, because cAMP is a key intracellular signaling molecule that modulates the immune system and is associated with inflammatory disease.58,59 Here, we show that delivering EF increases cAMP concentrations selectively in EGFR-positive cells. To establish this activity, we measured cAMP levels with an ELISA-based competition assay after incubating EGFR-positive cells with EF and IgG-mPAC constructs.
MDA-MB-231 cells incubated with 20 nM EF and 100 nM Cmab-mPAC showed increased intracellular cAMP levels (Figure 6A). In contrast, cells incubated with either EF alone or Cmab-mPAC alone did not exhibit increased cAMP levels. Cells also did not exhibit increased cAMP levels after incubation with EF in the presence of 100 nM Cmab-mPACA, Tmab-mPAC, or a 1:1 mixture of Cmab + mPAC. In addition, cells did not exhibit increased cAMP levels after incubation with LFN-DTA, rather than EF, in the presence of Cmab-mPAC. These studies show that the observed increase of cAMP is associated with cytosolic EF delivery by a functional Cmab-mPAC.
Figure 6.
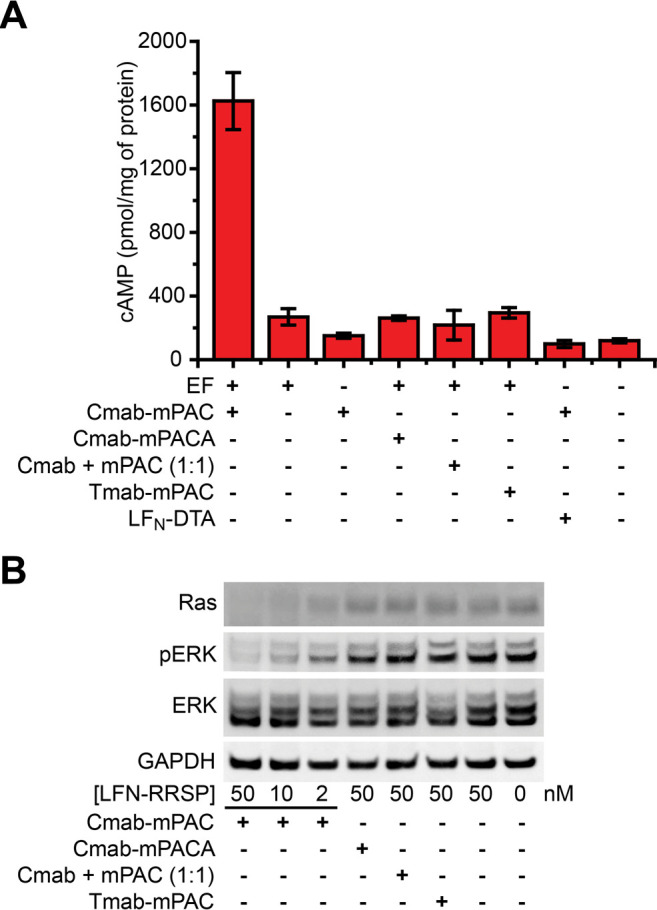
Antibody-directed PA conjugates deliver EF and RRSP into EGFR-positive cells. (A) Intracellular cAMP concentrations of MDA-MB-231 cells after 2 h of incubation with 20 nM EF or LFN-DTA and with 100 nM Cmab-mPAC or Tmab-mPAC. Concentrations of cAMP were measured with an ELISA-based competition assay. Data represent the mean ± s.d. of three replicate wells. (B) Western blot analysis of intracellular protein amounts in MDA-MB-231 cells after 24 h of incubation of LFN-RRSP and with 50 nM Cmab-mPAC, Cmab-mPACA, Tmab-mPAC, or a 1:1 mixture of Cmab + mPAC.
Intracellular RRSP delivery by the Cmab-mPAC conjugate enables cell-specific disruption of Ras signaling. Ras is an important oncoprotein involved in cancer development that is particularly difficult to target. Protease-mediated cleavage of Ras with RRSP has recently emerged as a promising strategy to rapidly cleave this protein inside of cancer cells, which interferes with downstream signaling of the MAPK pathway and promotes cancer cell death.29
To evaluate Cmab-mPAC-mediated Ras cleavage, MDA-MB-231 cells were incubated with LFN-RRSP in the presence of 50 nM Cmab-mPAC. After 24 h, Western blot analysis showed that bands from the Ras protein decreased in a dose-dependent fashion from LFN-RRSP and Cmab-mPAC (Figure 6B). We also compared the relative amount of ERK and phospho-ERK (pERK), which is influenced by Ras signaling. The blot showed that delivery of LFN-RRSP facilitated a dose-dependent decrease in pERK levels, while total ERK remained constant. LFN-RRSP did not exhibit any apparent activity when combined with translocation-deficient Cmab-mPACA, Tmab-mPAC, or a 1:1 mixture of Cmab + mPAC. These studies show that Cmab-mPAC enables cell-selective delivery of LFN-RRSP, and disruption of Ras signaling.
Protease-Specific Targeting of Cancer Cells
Protease-specific PA mutants are compatible with IgG-mPAC conjugates, which enables further targeting to cells. Two mutant PA variants developed by Leppla and co-workers have enabled proteolytic activation by uPA and MMP-9 proteases, rather than the native furin protease.41,42 We prepared Cmab-mPAC variants with each of these two protease cleavage sites (Figure 7A, Table S4): (1) Cmab-mPAC-uPA, which is only activated by the uPA protease; and (1) Cmab-mPAC-MMP, which is only activated by the MMP-9 protease. We generated these variants in a similar fashion as WT PA, using recombinant expression and AEX chromatography purification (Figures S16 and S17). We then combined these variants with linker peptide 1, followed by sortase-mediated ligation with Cmab (Figures S18–S20).
Figure 7.

Protease- and antibody-directed PA conjugates provide dual-targeting selectivity. (A) Cartoon illustrations of IgG-mPAC conjugates cleaved by proteases uPA and MMP-9. (B) SDS-PAGE gel analysis with Western blot visualization of the protease-treated conjugates: Cmab-mPAC, Cmab-mPAC-uPA (Cmab-uPA), and Cmab-mPAC-MMP (Cmab-MMP). (C–F) Relative viability of EGFR-positive cells expressing the uPA and MMP proteases (MDA-MB-231, BT549) after incubation with or without 10 nM LFN-DTA, and with 10-fold serial dilutions of (C, D) Cmab-mPAC-uPA or (E, F) Cmab-mPAC-MMP. Cell viability was determined by the relative luminescence from a CellTiter-Glo assay, which was normalized to untreated cells. Data represent the mean ± s.d. of three replicate wells.
We established the protease-cleavage selectivity of Cmab-mPAC-uPA and Cmab-mPAC-MMP against the MMP-9, uPA, and furin proteases. The three proteases were incubated with each conjugate for 3 h; then, the reaction mixtures were analyzed by SDS-PAGE under reducing conditions.60 At t = 0, Western blot analysis showed bands associated with the LC, HC, and heavy chain–mPAC conjugate (HC-mPAC). Over time, protease cleavage was indicated by the disappearance of bands for HC-mPAC83 and by the appearance of two new bands: HC-mPAC63 and PA20. Based on this pattern, the blots showed that the MMP-9 and uPA proteases cleaved only the corresponding Cmab-mPAC-MMP or Cmab-mPAC-uPA conjugate (Figure 7B), rather than Cmab-mPAC (Figure S21).
We then established the translocation activity of Cmab-mPAC-MMP and Cmab-mPAC-uPA on EGFR-positive cells expressing the MMP-9 and uPA proteases. We began by evaluating these variants on BT549 and MDA-MB-231 cells, which express both proteases.61,62 These cells were incubated with serial dilutions of Cmab-mPAC-MMP and 10 nM LFN-DTA, which decreased cell viability (Figure 7C,D). These cells were similarly treated with Cmab-mPAC-uPA, which also decreased cell viability (Figure 7E,F). These results show that the modified cleavage sites enable receptor- and protease-specific cell targeting, without affecting the translocase activity.63
We further evaluated these variants on H2030 cells, which express furin proteases but not MMP and uPA proteases.64 H2030 cells were incubated with LFN-DTA and with either Cmab-mPAC, Cmab-mPAC-MMP, or Cmab-mPAC-uPA. The results showed that neither Cmab-mPAC-MMP nor Cmab-mPAC-uPA decreased cell viability, but Cmab-mPAC decreased viability at concentrations as low as 100 pM (Figure S21). We also assessed the activity against normal human endothelial cells (HMEC-1), which express MMP and uPA proteases but fewer copies of the EGFR receptor. HMEC-1 cells were incubated with LFN-DTA, and with either WT PA, Cmab-mPAC, Cmab-mPAC-MMP, or Cmab-mPAC-uPA. The results showed that only the treatments with WT PA decreased cell viability (Figure S21). These results establish that the dual-activation mechanism provided by the antibody and the protease cleavage sites provides enhanced control for targeting cancer cells over healthy cells.
Antibody-Directed PA Variants Increase Pharmacokinetic Profiles
We evaluated pharmacokinetics and biodistribution with a series of in vivo studies in tumor-free animals. These studies showed that IgG-mPAC conjugates exhibit an increased clearance time and an altered biodistribution pattern, relative to unconjugated mPAC. Radioactive 89Zr was incorporated onto the Cmab antibody, mPAC-uPA, and the Cmab-mPAC-uPA conjugate, followed by i.v. administration into mice at 1 mg/kg (Figure 8A). In vivo properties of each construct were monitored over time by measuring the radioactivity of blood samples and collecting whole-body animal PET images (Figure 8B).
Figure 8.
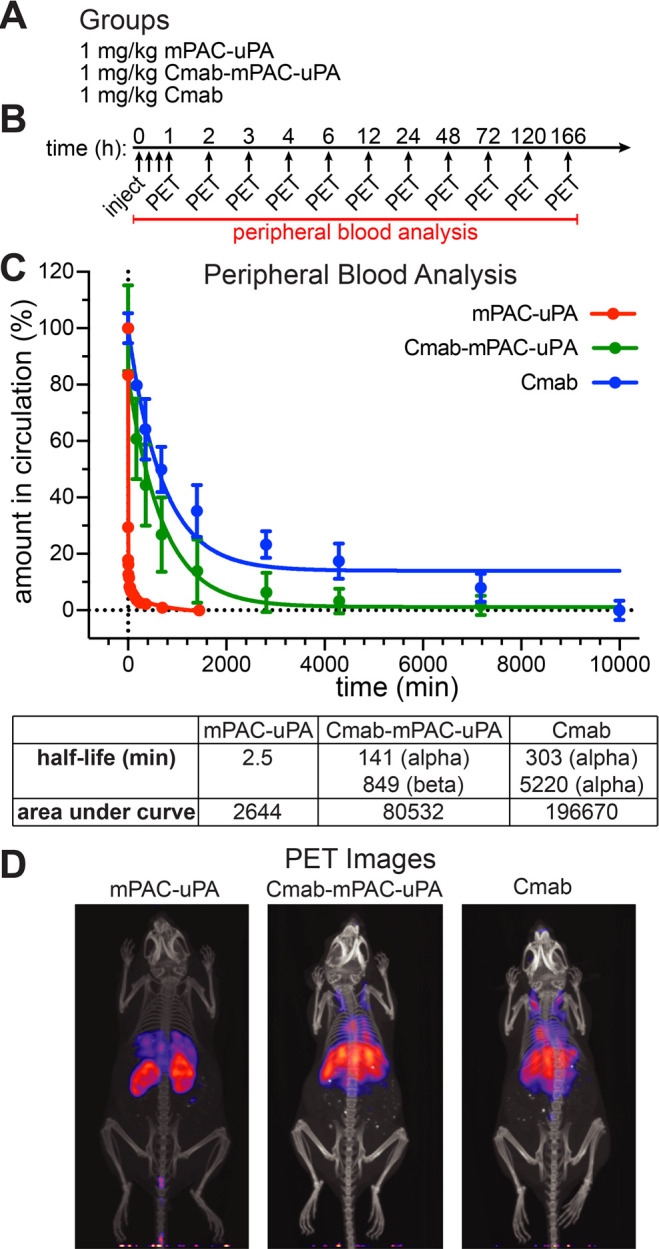
Antibody-directed PA conjugates enhance biodistribution and pharmacokinetics. (A) Groups of female nude mice i.v. injected with 1 mg/kg 89Zr-labeled mPAC-uPA (n = 1), Cmab-mPAC-uPA (n = 4), or Cmab (n = 4). (B) Whole-body PET images and peripheral blood analysis at the indicated time points. (C) Amount in circulation (%) of mPAC-uPA, Cmab-mPAC-uPA, and Cmab based on blood sample radiation measurements using a gamma counter. Data represent the mean ± s.d. of injected dose per gram of tissue (%ID/g), which was normalized to t = 0. The curves represent a two-phase decay model fitted to the data and were used to determine the values for half-life and area under curve. (D) Representative whole-body animal PET images collected at 6 h after injection.
The amount in circulation (%) was measured from blood-sample radioactivity using a gamma counter, which showed an exponential decrease of the constructs over time (Figure 8C). Pharmacokinetics was analyzed based on a two-compartment model: with a fast (α) clearance phase due to equilibration with the central compartment and a slower (β) clearance phase due to absorption by other tissue.65,66 This model revealed that the Cmab-mPAC-uPA conjugate exhibited biexponential clearance, in which the α phase half-life (t1/2) = 140 min, and β phase t1/2 = 850 min. The Cmab antibody also exhibited biexponential clearance, but with a longer α phase t1/2 = 303 min and β phase t1/2 = 5220 min. The mPAC-uPA exhibited monocompartment clearance and a single half-life of t1/2 = 2.5 min, indicating rapid removal in a first-order fashion.67,68 Analyzing the area under each curve showed the in vivo lifetime markedly increased for Cmab-mPAC-uPA relative to mPAC-uPA, but decreased relative to the Cmab antibody alone. These results indicate that the antibody component on Cmab-mPAC-uPA enhances the pharmacokinetic properties, which is an important feature for generating enhanced clinical responses.
The biodistribution images also show that Cmab and Cmab-mPAC-uPA exhibit comparable biodistribution patterns (Figure 8D, Figure S22). Further analysis of the animal tissue showed that both Cmab and Cmab-mPAC-uPA accumulate in the liver and spleen, which may serve as reservoirs to extend the clearance time (Figure S23). For mPAC-uPA, substantial accumulation was observed in the kidneys, and some accumulation was observed in the liver and spleen (Figure S24). These results further show that the IgG component, rather than mPAC, directs distribution and increases clearance time.
Antibody-Directed PA Conjugates Exhibit Pronounced in Vivo Safety
We also established the in vivo safety of IgG-mPAC conjugates in tumor-free animals. Groups of female nude mice (n = 3) were i.v. administered with 1 mg/kg LFN-DTA alone or coadministered with 3 or 15 mg/kg of Cmab-mPAC-uPA or Cmab-mPAC-MMP (Figure 9A). The mice were monitored over 48 h for outward signs of toxicity; then, tissue samples from the kidney and liver were subjected to microscopic examination (Figure 9B). The animals showed no outward signs or histological features associated with toxicity from Cmab-mPAC-uPA or Cmab-mPAC-MMP at doses of 3 mg/kg (Figure S25) and up to 15 mg/kg (Figure 9C). These results indicate that Cmab-mPAC-uPA and Cmab-mPAC-MMP exhibit pronounced in vivo safety up to 15 mg/kg, which is a 5-fold higher dosage than previously studied PA variants (e.g., PA-MMP up to 2.25 mg/kg).69−71
Figure 9.
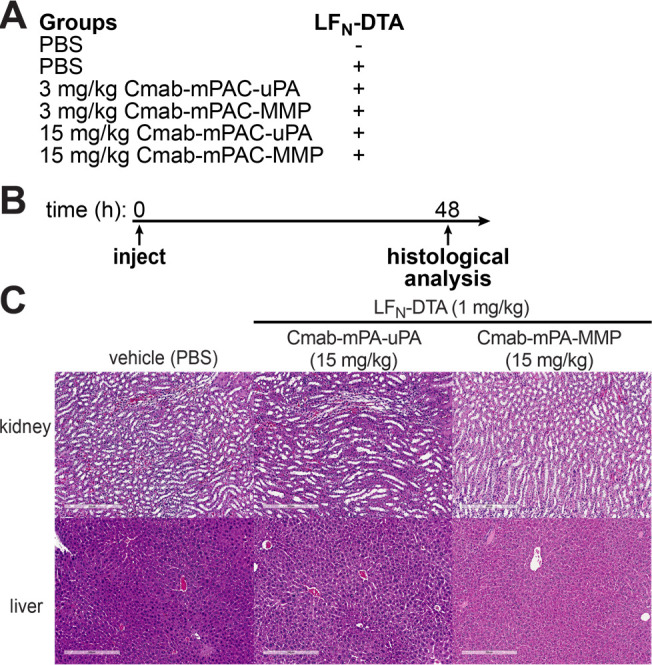
Antibody-directed PA conjugates exhibit pronounced in vivo safety. (A) Groups of nude mice (n = 3) i.v. administered with Cmab-mPAC-uPA or Cmab-mPAC-MMP, either with or without 1 mg/kg LFN-DTA. (B) Animals were monitored over 48 h for outward signs of toxicity; then, tissue samples from the kidney and liver were harvested and subjected to histological analysis. (C) Representative histological images of kidney and liver sections visualized with hematoxylin and eosin (H&E).
Furthermore, an extended safety study was performed with repeated dosing to the animals, while monitoring survival, body weight, inflammation, liver function, and histological toxicity (Figure S26). In this study, treatment dosage and intervals were chosen based on previous studies performed by Leppla and co-workers.72 Tumor-free animals (C57BL/6, F, n = 5) were treated six times over 21 days with 5 μg (0.25 mg/kg) of LFN-DTA and 15 μg (0.75 mg/kg) of WT PA, Cmab, Cmab-mPAC, Cmab-mPAC-uPA, or mPAC. During this time, the mice were monitored twice daily for signs of outward toxicity and changes in body weight. Sera samples from peripheral blood were collected on days 9, 17, and 21 for analysis of inflammatory cytokine levels (Figure S27), aspartate aminotransferase (AST) activity (Figure S28), and alanine transaminase (ALT) activity (Figure S28). On day 21, animal organs were harvested and processed to analyze toxicity across five major organs: liver, lung, heart, kidney, and spleen.
This study demonstrated that signs of off-target toxicity either presented after 48 h or were absent. In addition, the study further showed that LFN-DTA administered with Cmab-mPAC-uPA does not exhibit toxicity to healthy tissue. In contrast, LFN-DTA administered with either Cmab-mPAC or mPAC exhibited substantial toxicity after 48 h. These findings illustrate that IgG-targeting to specific cell types remains a major challenge but also show that this challenge can be overcome with IgG-PA conjugates engineered with specific protease-activated linkers. The combined effect of these antibody-directed and protease-activated delivery systems enables precise targeting to tumor cells without background toxicity to healthy cells, while also extending in vivo lifetimes, increasing receptor binding avidity, and providing robust payload delivery. Harnessing this dual-targeting approach holds promise for enhancing immunotoxin safety and treatment responses in clinical settings.
Discussion
In vivo pharmacokinetics and safety have critical roles in generating therapeutic responses. Typically, immunotoxins exhibit a half-life of several minutes and are undetectable after 24 h.66,73,74 The in vivo half-life for the Cmab-mPAC-uPA conjugate is 140 min, which is 50-fold longer than unconjugated PA and 5-fold longer than other immunotoxins undergoing preclinical development.66 Off-target toxicity is another challenge that has prevented the use of immunotoxins in the clinic. Although Cmab-mPAC showed off-target toxicity in animals, both Cmab-mPAC-uPA and Cmab-mPAC-MMP were tolerated up to a dose of 15 mg/kg. These findings show that protease-activated targeting can eliminate off-target toxicity at 15 mg/kg, which is 5-fold higher than previously studied PA variants.69−71 In addition, these findings of prolonged clearance time and pronounced in vivo safety demonstrate an attractive therapeutic profile for the dual-targeted IgG-mPAC conjugates and show promise for developing additional IgG-mPAC delivery systems.
The design of retargeted PA variants is critical to achieving target-binding and PA-translocase activity. Previous designs contain a receptor-binding protein at the C terminus of a native-receptor-ablated mPA (Figure 10A), which were combined by recombinant expression as a fusion protein36,37 or by conjugation using sortase.38 These strategies have been compatible with a variety of receptor-binding proteins, including native receptor-binding proteins,36 engineered binding proteins,37,38 and single-chain variable fragments,38,75 but have not been successful with antibodies. Although C- to C-terminal ligation strategies are possible using the sortase enzyme, “LPSTG”-containing IgG76 and mPA38 mutants were primarily designed for efficient N- to C-terminal ligation strategies. As a result, C- to C-terminal ligation strategies are prohibitively cumbersome approaches that rely on incorporating a linker peptide with two different amide-bond forming enzymes,77 and/or on a multistep reaction and purification procedure.78
Figure 10.

Antibody conjugation to the side chain of mutant PA is critical for enabling anthrax-based immunotoxins. (A) Design of previous retargeted-PA variants, in which a receptor-binding protein is either fused or conjugated to the C terminus of mPA. (B) Design of antibody-directed PA variants, in which an antibody is conjugated to a side chain of mPAC. (C, D) Design of antibody-directed PA variants, in which an antibody is fused to PA at either the N or C terminus of mPA.
We anticipated that conjugation would successfully retain antibody-binding and PA-mediated translocation activity, particularly upon connecting the IgG to a side chain on mPAC (Figure 10B). We were attracted to this strategy for several reasons: (1) Remote placement of mPAC at the C terminus of the IgG, distant from the binding region, helps preserve IgG binding activity. (2) Conjugation enables rapid assembly of different IgG and mPAC components, without re-expressing an entire IgG-mPAC fusion protein. (3) Separately preparing the mPAC and IgG antibody enables bacterial PA expression and mammalian IgG expression, which are the standard expression conditions for each component. (4) Tuning of the final IgG/mPAC ratio (IgG-mPAC1 vs. IgG-mPAC2), if necessary, is achievable by altering the conjugation conditions, followed by chromatographic separation of the desired conjugate.
We further envisioned that recombinantly fusing the IgG to mPA or mPAC could present challenges. The preparation of IgG-mPA fusion proteins would generally give a 1:2 antibody to mPA ratio. If the IgG was placed at the C terminus of mPA, binding may be reduced from obstruction by the mPA translocase (Figure 10C). If placed at the N terminus of mPA, proteases would cleave PA63 from the antibody–receptor complex and lead to PA63 dissociation from the cell surface (Figure 10D). These challenges may limit the function of either the IgG or PA component and may also preclude further development of the delivery platform.
Conclusion
The anthrax delivery platform described here is part of a larger body of ongoing work in our laboratory on antibody-directed cytosolic delivery of effector proteins into cells. Preparing the IgG-mPAC constructs with sortase-mediated ligation is a simple two-step procedure and is reproducible among different IgG antibody and PA variants. The conjugates enable robust protein delivery, including for EF, LFN-DTA, and LFN-RRSP. In vitro cell assays show that Tmab- and Cmab-mPAC conjugates deliver these proteins into HER2- and EGFR-positive cells, respectively, to promote the death of cancer and drug-resistant cancer cells at picomolar concentrations. These conjugates are also compatible with the cleavage sites for MMP-9 and uPA proteases, which further enables cell-specific targeting control. Combining these two targeting strategies provides a promising general platform for achieving in vivo therapeutic delivery, without unwanted off-target activity or toxicity.
The immunogenic response against PA and LFN has limited their use in clinical settings, but new avenues of therapeutic development are beginning to overcome this challenge. Immunosuppressive regimens with pentostatin and cyclophosphamide (PC) have been shown to mitigate this response by depleting lymphocytes, particularly B cells, in immunocompetent C57BL/6J mice.70,71 In addition, Pastan and co-workers have reported approaches for promoting antigen-specific tolerance79 and for reducing off-target toxicity.74 These strategies, in addition to the current work, offer the promise of administering repeat doses of PA and LFN, which will enable drug delivery applications for a wide range of diseases.
Acknowledgments
This work was funded by MIT start-up funds, the MIT Reed Fund, a Damon Runyon Cancer Research Foundation Innovation Award, a National Science Foundation (NSF) CAREER Award (CHE-1351807), and the Bridge Project between the Koch Institute and Dana-Farber/Harvard Cancer Center to B.L.P. This work was also funded by a National Institutes of Health Postdoctoral Fellowship (F32-CA239362) to N.L.T. In addition, D.J.I. is an investigator of the Howard Hughes Medical Institute. This work was supported in part by the NERCE facility (Grant: U54-AI057159) for expression of toxin proteins and by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. The authors thank R. J. Collier (Harvard University) for his contribution of laboratory equipment, S. H. Leppla (NIH) for providing the MMP-9 and uPA proteases, and R. T. Bronson (Harvard Medical School) for assistance with analyzing rodent pathology slides. The authors also thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically Preclinical Modeling, Imaging and Testing and Histology facilities. The authors also thank A. Loas, A. R. Loftis, and C. Hanna for critically reading this manuscript and providing detailed feedback.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01670.
Experimental methods for the synthesis and purification of peptide linkers 1–3, protein expression and purification, CellTiter-Glo luminescent cell viability assays, and Western blot analysis (PDF)
Author Contributions
◆ Z.L. and N.L.T. contributed equally to this work.
The authors declare the following competing financial interest(s): B.L.P. is a founder of Amide Technologies and Resolute Bio.
Supplementary Material
References
- Allahyari H.; Heidari S.; Ghamgosha M.; Saffarian P.; Amani J. Immunotoxin: A new tool for cancer therapy. Tumor Biol. 2017, 39, 101042831769222. 10.1177/1010428317692226. [DOI] [PubMed] [Google Scholar]
- Akbari B.; Farajnia S.; Ahdi Khosroshahi S.; Safari F.; Yousefi M.; Dariushnejad H.; Rahbarnia L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. 10.1080/08830185.2017.1284211. [DOI] [PubMed] [Google Scholar]
- Pastan I.; Hassan R.; Fitzgerald D. J.; Kreitman R. J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- Ghetie V.; Vitetta E. S. Chemical Construction of Immunotoxins. Mol. Biotechnol. 2001, 18, 251–268. 10.1385/MB:18:3:251. [DOI] [PubMed] [Google Scholar]
- Chang K.; Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 136–40. 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani P.; Iacobuzio-Donahue C.; Ryu B.; Rosty C.; Goggins M.; Wilentz R. E.; Murugesan S. R.; Leach S. D.; Jaffee E.; Yeo C. J.; Cameron J. L.; Kern S. E.; Hruban R. H. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 2001, 7 (12), 3862–3868. [PubMed] [Google Scholar]
- Hassan R.; Kindler H. L.; Jahan T.; Bazhenova L.; Reck M.; Thomas A.; Pastan I.; Parno J.; O’Shannessy D. J.; Fatato P.; Maltzman J. D.; Wallin B. A. Phase II clinical trial of amatuximab, a chimeric antimesothelin antibody with pemetrexed and cisplatin in advanced unresectable pleural mesothelioma. Clin. Cancer Res. 2014, 20, 5927–36. 10.1158/1078-0432.CCR-14-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. 10.1007/s40265-018-1000-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon H.; Reichert J. M. Antibodies to watch in 2018. MAbs 2018, 10, 183–203. 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon H.; Reichert J. M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S.; Fukuhara T.; Hattori N. Immunotoxin Screening System: A Rapid and Direct Approach to Obtain Functional Antibodies with Internalization Capacities. Toxins 2020, 12 (10), 658. 10.3390/toxins12100658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirzer T.; Becher K.-S.; Rieker M.; Meckel T.; Mootz H. D.; Kolmar H. Generation of Potent Anti-HER1/2 Immunotoxins by Protein Ligation Using Split Inteins. ACS Chem. Biol. 2018, 13, 2058–2066. 10.1021/acschembio.8b00222. [DOI] [PubMed] [Google Scholar]
- Young J. A.; Collier R. J. Anthrax Toxin: Receptor Binding, Internalization, Pore Formation, and Translocation. Annu. Rev. Biochem. 2007, 76, 243–265. 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- Collier R. J. Membrane Translocation by Anthrax Toxin. Mol. Aspects Med. 2009, 30, 413–422. 10.1016/j.mam.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Pentelute B. L.; Collier R. J.; Zhou Z. H. Atomic Structure of Anthrax Protective Antigen Pore Elucidates Toxin Translocation. Nature 2015, 521, 545–549. 10.1038/nature14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley K. A.; Mogridge J.; Mourez M.; Collier R. J.; Young J. A. Identification of the Cellular Receptor for Anthrax Toxin. Nature 2001, 414, 225–229. 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- Scobie H. M.; Rainey G. J. A.; Bradley K. A.; Young J. A. T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5170–5174. 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimpel K. R.; Molloy S. S.; Thomas G.; Leppla S. H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 10277–81. 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne J. C.; Furlong D.; Hanna P. C.; Wall J. S.; Collier R. J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–12. 10.1016/S0021-9258(17)32036-7. [DOI] [PubMed] [Google Scholar]
- Kintzer A. F.; Thoren K. L.; Sterling H. J.; Dong K. C.; Feld G. K.; Tang II; Zhang T. T.; Williams E. R.; Berger J. M.; Krantz B. A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld G. K.; Thoren K. L.; Kintzer A. F.; Sterling H. J.; Tang II; Greenberg S. G.; Williams E. R.; Krantz B. A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–90. 10.1038/nsmb.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge J.; Cunningham K.; Collier R. J. Stoichiometry of anthrax toxin complexes. Biochemistry 2002, 41, 1079–82. 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- Nassi S.; Collier R. J.; Finkelstein A. PA63 channel of anthrax toxin: an extended beta-barrel. Biochemistry 2002, 41, 1445–50. 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- Lacy D. B.; Wigelsworth D. J.; Melnyk R. A.; Harrison S. C.; Collier R. J. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13147–51. 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. J.; Elliott J. L.; Collier R. J. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 1999, 38, 10432–10441. 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- Verdurmen W. P. R.; Mazlami M.; Pluckthun A. A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci. Rep. 2017, 7, 13194. 10.1038/s41598-017-13469-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Leppla S. H. Anthrax toxin uptake by primary immune cells as determined with a lethal factor-beta-lactamase fusion protein. PLoS One 2009, 4, e7946 10.1371/journal.pone.0007946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora N.; Klimpel K. R.; Singh Y.; Leppla S. H. Fusions of Anthrax Toxin Lethal Factor to the Adp-Ribosylation Domain of Pseudomonas Exotoxin-a Are Potent Cytotoxins Which Are Translocated to the Cytosol of Mammalian-Cells. J. Biol. Chem. 1992, 267, 15542–15548. 10.1016/S0021-9258(19)49569-0. [DOI] [PubMed] [Google Scholar]
- Antic I.; Biancucci M.; Zhu Y.; Gius D. R.; Satchell K. J. F. Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat. Commun. 2015, 6, 7396. 10.1038/ncomms8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne J. C.; Blanke S. R.; Hanna P. C.; Collier R. J. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 1995, 15, 661–6. 10.1111/j.1365-2958.1995.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Ballard J. D.; Collier R. J.; Starnbach M. N. Anthrax Toxin-Mediated Delivery of a Cytotoxic T-Cell Epitope in vivo. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 12531–12534. 10.1073/pnas.93.22.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard J. D.; Doling A. M.; Beauregard K.; Collier R. J.; Starnbach M. N. Anthrax Toxin-Mediated Delivery In Vivo and In Vitro of a Cytotoxic T-Lymphocyte Epitope from Ovalbumin. Infect. Immun. 1998, 66, 615–619. 10.1128/IAI.66.2.615-619.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright D. G.; Zhang Y.; Murphy J. R. Effective delivery of antisense peptide nucleic acid oligomers into cells by anthrax protective antigen. Biochem. Biophys. Res. Commun. 2008, 376, 200–205. 10.1016/j.bbrc.2008.08.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Paolella B. R.; Truex N. L.; Loftis A. R.; Liao X.; Rabideau A. E.; Brown M. S.; Busanovich J.; Beroukhim R.; Pentelute B. L. Targeting Cancer Gene Dependencies with Anthrax-Mediated Delivery of Peptide Nucleic Acids. ACS Chem. Biol. 2020, 15, 1358–1369. 10.1021/acschembio.9b01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabideau A. E.; Pentelute B. L. Delivery of Non-Native Cargo into Mammalian Cells Using Anthrax Lethal Toxin. ACS Chem. Biol. 2016, 11, 1490–1501. 10.1021/acschembio.6b00169. [DOI] [PubMed] [Google Scholar]
- Mechaly A.; McCluskey A. J.; Collier R. J. Changing the receptor specificity of anthrax toxin. mBio 2012, 3, e00088-12. 10.1128/mBio.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey A. J.; Olive A. J.; Starnbach M. N.; Collier R. J. Targeting HER2-Positive Cancer Cells with Receptor-Redirected Anthrax Protective Antigen. Mol. Oncol. 2013, 7, 440–451. 10.1016/j.molonc.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey A. J.; Collier R. J. Receptor-directed chimeric toxins created by sortase-mediated protein fusion. Mol. Cancer Ther. 2013, 12, 2273–81. 10.1158/1535-7163.MCT-13-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahaf N. I.; Lang A. E.; Kaiser L.; Fichter C. D.; Lassmann S.; McCluskey A.; Augspach A.; Aktories K.; Schmidt G. Targeted delivery of an ADP-ribosylating bacterial toxin into cancer cells. Sci. Rep. 2017, 7, 41252. 10.1038/srep41252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack S.; Madhivanan K.; Ramadesikan S.; Subramanian S.; Edwards D. F.; Elzey B. D.; Dhawan D.; McCluskey A.; Kischuk E. M.; Loftis A. R.; Truex N.; Santos M.; Lu M.; Rabideau A.; Pentelute B.; Collier J.; Kaimakliotis H.; Koch M.; Ratliff T. L.; Knapp D. W.; Aguilar R. C. A novel, safe, fast and efficient treatment for Her2-positive and negative bladder cancer utilizing an EGF-anthrax toxin chimera. Int. J. Cancer 2020, 146, 449–460. 10.1002/ijc.32719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Netzel-Arnett S.; Birkedal-Hansen H.; Leppla S. H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [PubMed] [Google Scholar]
- Liu S.; Bugge T. H.; Leppla S. H. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 2001, 276, 17976–17984. 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- Hortobagyi G. N. Trastuzumab in the treatment of breast cancer. N. Engl. J. Med. 2005, 353, 1734–6. 10.1056/NEJMe058196. [DOI] [PubMed] [Google Scholar]
- Graham J.; Muhsin M.; Kirkpatrick P. Cetuximab. Nat. Rev. Drug Discovery 2004, 3, 549–50. 10.1038/nrd1445. [DOI] [PubMed] [Google Scholar]
- Rosovitz M. J.; Schuck P.; Varughese M.; Chopra A. P.; Mehra V.; Singh Y.; McGinnis L. M.; Leppla S. H. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J. Biol. Chem. 2003, 278, 30936–44. 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]
- Mourez M.; Yan M.; Lacy D. B.; Dillon L.; Bentsen L.; Marpoe A.; Maurin C.; Hotze E.; Wigelsworth D.; Pimental R. A.; Ballard J. D.; Collier R. J.; Tweten R. K. Mapping Dominant-Negative Mutations of Anthrax Protective Antigen by Scanning Mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13803–13808. 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The design of mPACA is based on the well-characterized PA [427A] mutant, which oligomerizes in the presence of cells but does not perform translocation. We prepared this mutant as a control to reveal whether intracellular delivery occurs through PA-mediated oligomerization and translocation, which cannot occur with the mPACA mutant, or through another mechanism, which may occur with the mPACA mutant.
- Sellman B. R.; Nassi S.; Collier R. J. Point Mutations in Anthrax Protective Antigen that Block Translocation. J. Biol. Chem. 2001, 276, 8371–8376. 10.1074/jbc.M008309200. [DOI] [PubMed] [Google Scholar]
- Ling J. J.; Policarpo R. L.; Rabideau A. E.; Liao X.; Pentelute B. L. Protein Thioester Synthesis Enabled by Sortase. J. Am. Chem. Soc. 2012, 134, 10749–10752. 10.1021/ja302354v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. A.; Collier R. J. Diphtheria-Toxin and Pseudomonas-Aeruginosa Exotoxin-a - Active-Site Structure and Enzymatic Mechanism. Curr. Top. Microbiol. Immunol. 1992, 175, 27–41. 10.1007/978-3-642-76966-5_2. [DOI] [PubMed] [Google Scholar]
- For clarity, the toxicity data from panels A and B with Tmab-mPAC (BT474) and Cmab-mPAC (A431) were overlaid with the data in panels C and D.
- Pohlmann P. R.; Mayer I. A.; Mernaugh R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T. M.; Iida M.; Wheeler D. L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–92. 10.4161/cbt.11.9.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukohara T.; Engelman J. A.; Hanna N. H.; Yeap B. Y.; Kobayashi S.; Lindeman N.; Halmos B.; Pearlberg J.; Tsuchihashi Z.; Cantley L. C.; Tenen D. G.; Johnson B. E.; Janne P. A. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J. Natl. Cancer Inst 2005, 97, 1185–94. 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- Corkery B.; Crown J.; Clynes M.; O’Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol 2009, 20, 862–7. 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- Napolitano S.; Martini G.; Rinaldi B.; Martinelli E.; Donniacuo M.; Berrino L.; Vitagliano D.; Morgillo F.; Barra G.; De Palma R.; Merolla F.; Ciardiello F.; Troiani T. Primary and Acquired Resistance of Colorectal Cancer to Anti-EGFR Monoclonal Antibody Can Be Overcome by Combined Treatment of Regorafenib with Cetuximab. Clin. Cancer Res. 2015, 21, 2975–83. 10.1158/1078-0432.CCR-15-0020. [DOI] [PubMed] [Google Scholar]
- Leppla S. H. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 3162–6. 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serezani C. H.; Ballinger M. N.; Aronoff D. M.; Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–32. 10.1165/rcmb.2008-0091TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raker V. K.; Becker C.; Steinbrink K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. 10.3389/fimmu.2016.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The protease cleavage experiments are designed to illustrate the differences in the peptide substrates cleaved by furin, MMP-9, and uPA proteases. These experiments are not intended to assess or replicate protease expression levels by various mammalian cell types. The conditions for each experiment are described in the Materials and Methods section of the Supporting Information. Enzyme concentration was based on the units listed on the certificate of analysis from the manufacturer (units/mL or μg).
- Mehner C.; Hockla A.; Miller E.; Ran S.; Radisky D. C.; Radisky E. S. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 2014, 5, 2736–49. 10.18632/oncotarget.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.; New L.; Pan Z.; Han J.; Nemerow G. R. Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38alpha mitogen-activated protein kinase activity. J. Biol. Chem. 2000, 275, 12266–72. 10.1074/jbc.275.16.12266. [DOI] [PubMed] [Google Scholar]
- After 72 h, the parent Cmab-mPAC conjugate is toxic to BT549 cells at concentrations as low as 100 pM in the presence of 10 nM LFN-DTA (shown in Figure 5E). Cmab-mPAC-MMP and Cmab-mPAC-uPA exhibit nearly identical activity to BT549 cells after the same 72 h incubation period (Figure 7), which suggests that the altered protease activation sites neither decrease translocase activity nor reduce proteolytic stability.
- Abi-Habib R. J.; Singh R.; Liu S. H.; Bugge T. H.; Leppla S. H.; Frankel A. E. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol. Cancer Ther. 2006, 5, 2556–2562. 10.1158/1535-7163.MCT-06-0315. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Zia-Amirhosseini P. Basic principles of pharmacokinetics. Toxicol. Pathol. 1995, 23, 115–23. 10.1177/019262339502300203. [DOI] [PubMed] [Google Scholar]
- Shancer Z.; Liu X. F.; Nagata S.; Zhou Q.; Bera T. K.; Pastan I. Anti-BCMA immunotoxins produce durable complete remissions in two mouse myeloma models. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 4592–4598. 10.1073/pnas.1821733116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The short residence time of mPAC-uPA is comparable to other unmodified proteins administered intravenously, which typically results in faster protein clearance over other injection methods (e.g., intraperitoneal and subcutaneous).
- Sumbria R. K.; Zhou Q.-H.; Hui E. K.-W.; Lu J. Z.; Boado R. J.; Pardridge W. M. Pharmacokinetics and Brain Uptake of an IgG-TNF Decoy Receptor Fusion Protein Following Intravenous, Intraperitoneal, and Subcutaneous Administration in Mice. Mol. Pharmaceutics 2013, 10, 1425–1431. 10.1021/mp400004a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Redeye V.; Kuremsky J. G.; Kuhnen M.; Molinolo A.; Bugge T. H.; Leppla S. H. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol. 2005, 23, 725–730. 10.1038/nbt1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Liu J.; Ma Q.; Cao L.; Fattah R. J.; Yu Z.; Bugge T. H.; Finkel T.; Leppla S. H. Solid tumor therapy by selectively targeting stromal endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4079–E4087. 10.1073/pnas.1600982113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Ma Q.; Fattah R.; Bugge T. H.; Leppla S. H. Anti-tumor activity of anthrax toxin variants that form a functional translocation pore by intermolecular complementation. Oncotarget 2017, 8, 65123–65131. 10.18632/oncotarget.17729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Wang H.; Currie B. M.; Molinolo A.; Leung H. J.; Moayeri M.; Basile J. R.; Alfano R. W.; Gutkind J. S.; Frankel A. E.; Bugge T. H.; Leppla S. H. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J. Biol. Chem. 2008, 283, 529–540. 10.1074/jbc.M707419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F.; Cunningham T.; Stookey S.; Tai C. H.; Burkett S.; Jailwala P.; Stetler Stevenson M.; Cam M. C.; Wayne A. S.; Pastan I. 5-Azacytidine prevents relapse and produces long-term complete remissions in leukemia xenografts treated with Moxetumomab pasudotox. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E1867–E1875. 10.1073/pnas.1714512115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. F.; Wei J.; Zhou Q.; Molitoris B. A.; Sandoval R.; Kobayashi H.; Okada R.; Nagaya T.; Karim B.; Butcher D.; Pastan I. Immunotoxin SS1P is rapidly removed by proximal tubule cells of kidney, whose damage contributes to albumin loss in urine. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 6086–6091. 10.1073/pnas.1919038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis A. R.; Santos M. S.; Truex N. L.; Biancucci M.; Satchell K. J. F.; Pentelute B. L. Anthrax protective antigen retargeted with single-chain variable fragments delivers enzymes to pancreatic cancer cells. ChemBioChem 2020, 21, 2772 10.1002/cbic.202000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehlmann S.; Mahlert C.; Greven S.; Scholz P.; Harrenga A. In vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem 2011, 12, 1774–1780. 10.1002/cbic.201100002. [DOI] [PubMed] [Google Scholar]
- Harmand T. J.; Bousbaine D.; Chan A.; Zhang X.; Liu D. R.; Tam J. P.; Ploegh H. L. One-Pot Dual Labeling of IgG 1 and Preparation of C-to-C Fusion Proteins Through a Combination of Sortase A and Butelase 1. Bioconjugate Chem. 2018, 29, 3245–3249. 10.1021/acs.bioconjchem.8b00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.; Kwakkenbos M. J.; Claassen Y. B.; Maijoor K.; Bohne M.; van der Sluijs K. F.; Witte M. D.; van Zoelen D. J.; Cornelissen L. A.; Beaumont T.; Bakker A. Q.; Ploegh H. L.; Spits H. Bispecific antibody generated with sortase and click chemistry has broad antiinfluenza virus activity. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 16820–16825. 10.1073/pnas.1408605111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor R.; King E. M.; Onda M.; Cuburu N.; Addissie S.; Crown D.; Liu X. F.; Kishimoto T. K.; Pastan I. Tolerogenic nanoparticles restore the antitumor activity of recombinant immunotoxins by mitigating immunogenicity. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E733–E742. 10.1073/pnas.1717063115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.