

Abstract
Since the definition of the “12 Principles of Green Chemistry” more than 20 years ago, chemists have become increasingly mindful of the need to conserve natural resources and protect the environment through the judicious choice of synthetic routes and materials. The direct activation and functionalization of C–H bonds, bypassing intermediate functional group installation is, in abstracto, step and atom economic, but numerous factors still hinder the sustainability of large-scale applications. In this Outlook, we highlight the research areas seeking to overcome the sustainability challenges of C–H activation: the pursuit of abundant metal catalysts, the avoidance of static directing groups, the replacement of metal oxidants, and the introduction of bioderived solvents. We close by examining the progress made in the subfield of aryl C–H borylation from its origins, through highly efficient but precious Ir-based systems, to emerging 3d metal catalysts. The future growth of this field will depend on industrial uptake, and thus we urge researchers to strive toward sustainable C–H activation.
Short abstract
Applications of C−H activation face a sustainability challenge. This Outlook discusses progress in areas such as 3d metal catalysis, alternative oxidation, green solvents, and undirected systems.
1. Introduction
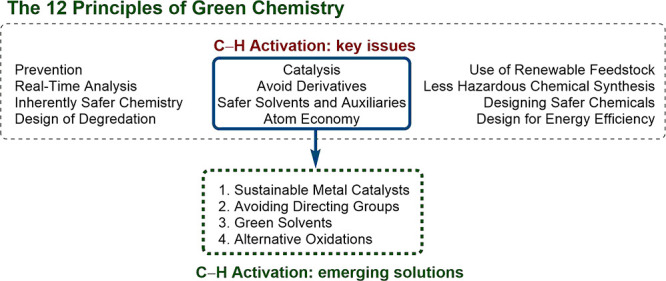
Sustainability is achieved when the needs of the present can be satisfied without compromising those of the future. In recent years, the ideals of sustainability have emerged as a dominant theme in organic chemistry, with a view to the technology transfer of novel reactions from the lab bench to process plant. The ideal sustainable transformation eliminates the use of scarce materials and materials which, through their production or disposal, represent an environmental hazard or burden. The design of “environmentally friendly” chemistry goes hand-in-hand with economic transformations.1,2 For instance, the use of catalytic reactions, mild conditions, and limiting step-count and waste typically results in cost savings and cycle-time reduction. As society seeks to counter resource scarcity and address the climate emergency, legislative restrictions on the use of materials perceived to be hazardous or highly polluting are increasing.3−5 In 1998, Anastas and Warner defined the “12 Principles of Green Chemistry” as “design rules” to make chemical processes more sustainable (Figure 1). Nowadays, transformations are evaluated numerically using parameters such as atom economy, E factor, process mass intensity, and many others.6−17 More recently, enormous efforts have been made to develop the science of “lifecycle assessment”—the quantitative metrics by which the total mass and energy inputs and waste outputs of a given chemical transformation are evaluated for their environmental impact.18−21 Since then, industrial chemists have led the charge by compiling important guides on reagent and solvent selection.22−28
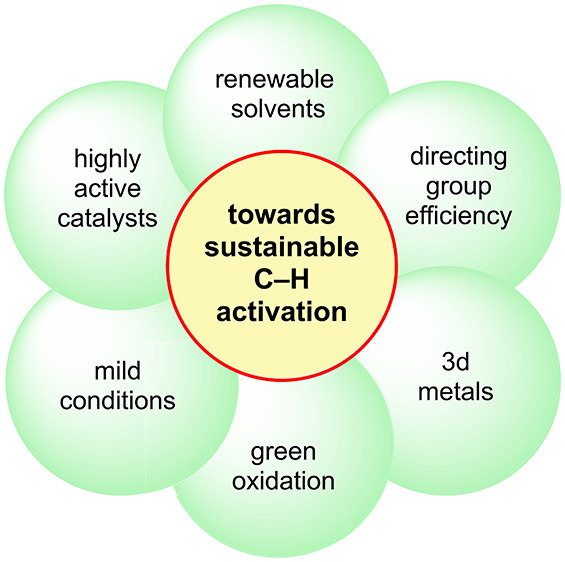
Figure 1.
C–H Activation: sustainability trends.
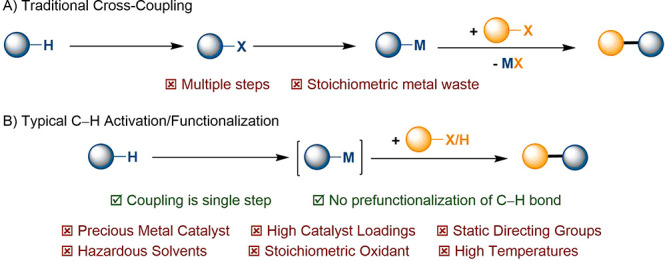
In comparison to the more established cross-coupling reactions, C–H activation removes the requirement for prefunctionalization of both partners; as such, C–H activation has long promised a means to decrease step-count and hence mass intensity of chemical processes (Scheme 1).29 That said, the selective cleavage of unactivated C–H bonds remains an active and comparatively difficult area of academic research. Consequently, the use of precious metal catalysts in high loadings, stoichiometric metal-based oxidants, high temperatures, and directing group manipulations are often required. Until now, practical and economic considerations have presented the major barriers to industrial application; sustainability considerations are likely to dominate in the years to come.
Scheme 1. C–H Activation: Intrinsic Opportunities.
A great body of work has already sought to address many of these limitations. Reviews on the subtopics of mild, efficient, and undirected C–H activation have been published, as well as those addressing the use of more sustainable solvents, nonprecious metals, and alternative oxidative systems, but to our knowledge there has yet to be a thematic overview from the perspective of sustainability.30−40 In this Outlook, we highlight noteworthy examples in areas where C–H activation faces its greatest sustainability challenges. Naturally, many transformations we have chosen span several such categories; most present at least one unsustainable aspect. The question is not whether these reactions are objectively green but rather in which ways do they represent an advance in sustainability. Our aim is to provide a cross-section that informs the reader of the fast-growing research lines in pursuit of sustainable, and thereby applicable, C–H activation.
2. Next Generation Metal Catalysts
C–H Activation and functionalization has relied heavily on the ability of precious transition metals to affect diverse catalytic steps. The sheer cost and price volatility of these elements disincentivizes large-scale applications. Even if resource scarcity does not inhibit future use, the supply of such materials may become subject to geopolitical risk.41 In addition, the low abundance of precious metals contributes to a significantly higher carbon footprint in their extraction.42 To initiate a move away from wasteful precious metal chemistry, many groups are now focused on developing alternative 3d metal systems and recoverable, heterogeneous catalysts.
2.1. Adopting 3d Metals
The 3d metals are generally viewed as inexpensive and less toxic, decreasing the impact of higher catalyst loadings.38 Promoting their uptake by the synthetic community depends heavily on expanding the range of chemistry which can be accomplished by these metals.
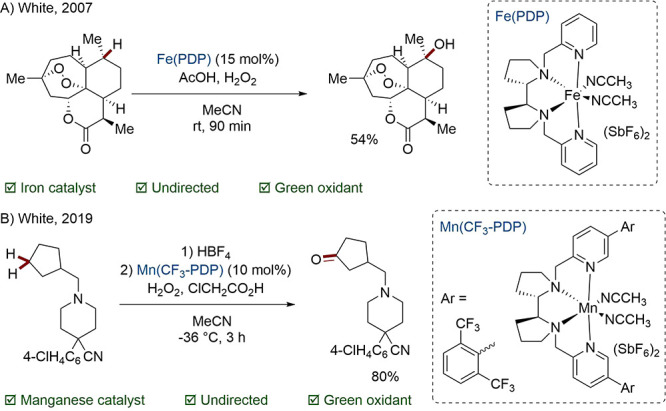
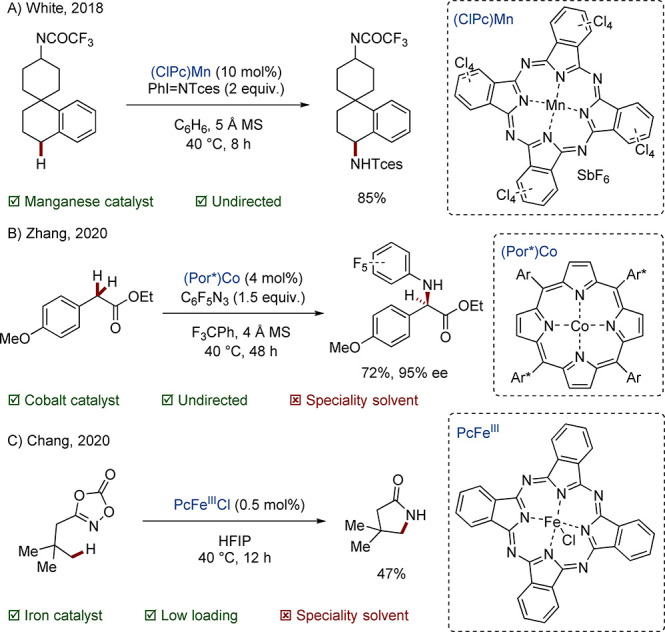
A well-known example of a 3d complex is the Fe-centered White–Chen catalyst, which is able to oxidize unactivated C(sp3)–H bonds. In the breakthrough work, it was discovered that the rigidity afforded by the pyrrolidine-pyridine (PDP) ligand conferred a high degree of regioselectivity based on subtle electronic differences. The original catalyst system favored oxidation of tertiary C–H bonds to alcohols over oxidation of methylene units to ketones (Scheme 2A).43,44 In a later study, a more substituted PDP complex was disclosed which reverses this trend by restricting substrate access to the metal center.45 Great efforts have been made to broaden the scope of this chemistry to molecules representative of biologically active compounds, for example, by coordinating problematic functionality with a Lewis acid.46−48 In one recent paper, interchange of the Fe center for Mn results in tolerance of basic nitrogen, halogens, and heterocyclic moieties (Scheme 2B).49
Scheme 2. PDP-Mediated Alkane Oxidations.
The related nitrenoid chemistry, based on electron-rich porphyrin (Por) and phthalocyanine (Pc) ligands, has likewise seen intense development.50,51 White’s 2012 FePc system was notable for selective, intramolecular allylic C–H amination over the aziridination favored by earlier Rh systems.52 The group of Che used an NHC-PorFe catalyst for the preparation of saturated nitrogen heterocycles from alkyl azides.53 Recently, a highly functional group tolerant PcMn analogue was disclosed for intermolecular, benzylic C–H amination; multiple late-stage derivatizations of biologically active compounds were exemplified (Scheme 3A).54 In parallel, the group of Zhang has led the development of the corresponding PorCo systems. Chiral amidoporphyrin ligands have enabled impressive enantioselective aminations, including the first intermolecular example in 2020 (Scheme 3B).55 In the same year, Chang extended the concept to intramolecular amidation for the preparation of γ-lactams from dioxazolones (Scheme 3C).56 The PcFe system is remarkable for its high activity, activating even primary alkyl C–H bonds at a catalyst loading of just 0.5 mol % (TON was 47 000 for benzylic C–H bonds).
Scheme 3. Porphyrin/Phthalocyanine-Mediated Amination and Amidation.
3d Metals are now well-established in reactions involving the insertion of C–C double and triple bonds. Early research on Mn-catalyzed C–H aromatic alkenylation was conducted by Wang and co-workers. High regio-, chemo-, and stereoselectivity were achieved, and anti-Markovnikov E-configured olefins were obtained in high yields using the simple MnBr(CO)5 catalyst (Scheme 4A).57 Shortly afterward, an imine directing group was harnessed to provide a Mn-catalyzed route to 3,4-disubstituted isoquinolines.58
Scheme 4. Mn- and Co-Catalyzed C–H Functionalizations.
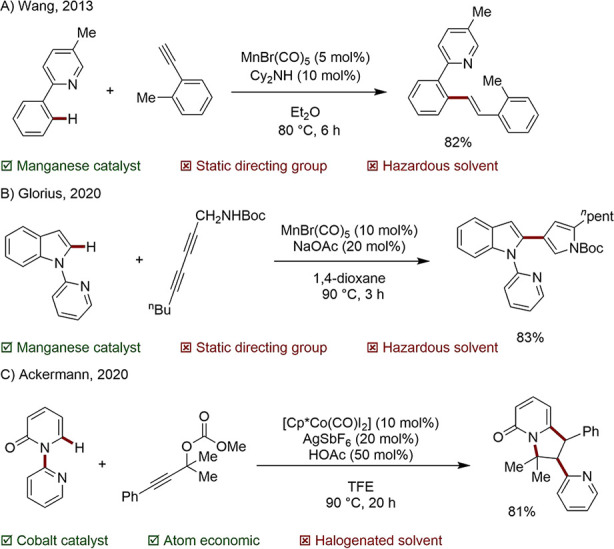
Glorius and co-workers later accomplished a highly selective synthesis of 1,3-enynes, pyrroles, and furans using MnBr(CO)5 as a catalyst (Scheme 4B).59 Significantly, Rh- and Ru-based catalysts normally used for coupling 1,3-diynes were not successful in this transformation. Co has likewise emerged as a competent metal in C–H addition chemistry. In a recent example, Ackermann and co-workers reported a selective domino C–H activation, pyridine migration-annulation sequence catalyzed by a pentamethyl cyclopentadienyl (Cp*) Co complex (Scheme 4C).60 Likewise, this reaction could not be accomplished by Rh or Ru, underlining the potential of 3d metals to serve not only as more sustainable alternatives, but in many cases offering contrasting reactivity. Until now, however, undirected Mn and Co examples of this type have been elusive.
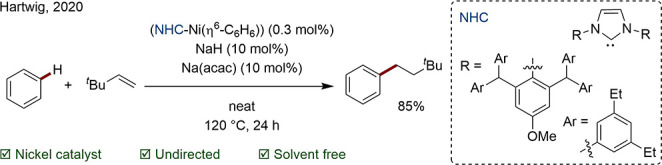
In contrast, Hartwig and Nakao have pioneered a Ni/NHC-catalyst system for the atypical, anti-Markovnikov hydroarylation of alkenes with arenes (Scheme 5). The authors demonstrate the presence of stabilizing, noncovalent interactions in the transition state between the bulky ligand and substrates. This chemistry is all the more remarkable for the high catalyst TON of 183, translating to a loading of just 0.3 mol %.61
Scheme 5. Ni-Catalyzed Undirected anti-Markovnikov Addition of Alkenes to Arenes.
The 3d metals have the potential to match and surpass the chemistry of the precious transition metals, but it is likely that further success will depend heavily on advanced ligand design so that these metal centers can participate in a wider range of elementary steps. The environmental impact of ligand syntheses can bear an outsize influence on the environmental burden of a process.9 Finding efficiencies in the multistep preparation of ligands represents an additional practical barrier to be overcome if such chemistry is to achieve mainstream appeal.
2.2. Heterogeneous Systems
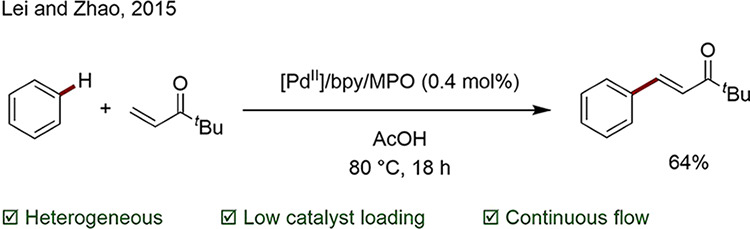
The use of heterogeneous catalysts has warranted attention owing to the ease of removal and recycling of the catalyst.62,63 Provided metal leaching is sufficiently low, such systems can render the use of precious metals more sustainable. The group of Glorius reported several examples of direct arylation of (hetero)aryls using simple Pd/C, followed by the first undirected C–H thiolation of electron-rich heteroarenes by Pd/Al2O3.64−67 In 2015, Lei and Zhao disclosed a system for the catalysis of the Fujiwara–Moritani reaction involving bipyridine bound Pd on mesoporous organosilica (MPO). Kinetic experiments demonstrated a superior catalytic activity relative to the corresponding homogeneous system, a longer lifetime of the catalyst owing to the prevention of Pd(0) aggregate formation, and the means to perform the reaction in continuous flow mode (Scheme 6).68
Scheme 6. Fujiwara–Moritani Reaction Catalyzed by Pd on Silica.
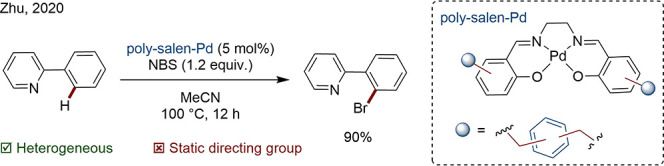
Zhu and co-workers developed a salen-based, hyper-cross-linked polymer-supported Pd catalyst to carry out C–H bromination and chlorination. The catalyst exhibited superior activity in comparison to homogeneous Pd(OAc)2 under the same conditions. Only trace Pd leaching was detected, confirming the suitability of poly salen as support material (Scheme 7).69
Scheme 7. C–H Bromination Catalyzed by Pd Supported on a Salen-Based, Hyper-Cross-Linked Polymer.
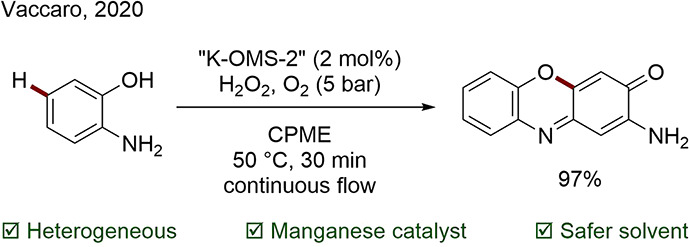
Vaccaro and co-workers reported the use of a Mn based heterogeneous catalyst for the oxidative coupling reaction of 2-aminophenols, O-phenylenediamines, and pyrogallol in continuous flow (Scheme 8).70 Notably, this catalyst showed minimal leaching and low contamination of the product, which was readily purified by crystallization upon cooling of the CPME reaction solution, avoiding mass and solvent inefficient chromatography. (This solvent is regarded as process-friendly owing to its hydrophobicity, high boiling point, and low tendency to form peroxides.) The solvent could be recovered by distillation and reused, contributing to a very small E factor of 1.4 under continuous flow conditions versus 19 in batch.
Scheme 8. Oxidative Coupling of Aminophenol Catalyzed by Heterogeneous Mn-Based K-OMS-2.
3. Avoiding and Replacing Static Directing Groups
Most of the time, C–H bond activation is enabled by directing groups (DGs) that are covalently linked to the substrate (Scheme 9). A wide variety of functional groups with Lewis basic properties, for instance, monodentate amides, pyridines, esters, or imines, as well as bidentate DGs, have been utilized.71 This coordination expedites C–H activation since it increases the local concentration of substrate in the proximity of the catalyst as well as controlling regioselectivity.
Scheme 9. Directing Group Mode of Action.

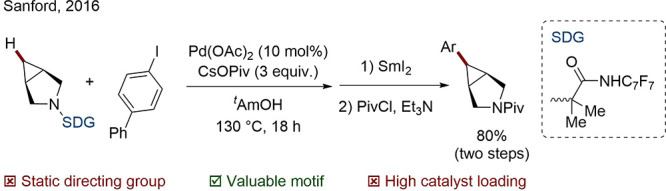
Unfortunately, for most valuable substrates, existing functionality is not suitable for the facilitation of directed C–H functionalization, and an existing functional group must be converted to a DG. Removal of the DG is often less trivial than is commonly admitted, but is crucial to obtain products of genuine utility. For example, Sanford was able to demonstrate a Pd-catalyzed C(sp3)–H arylation of bicyclic amines, which represent an important class of medicinally relevant compounds, with successful and efficient DG removal (Scheme 10).72 In later work, it was found that the addition of picolinic or quinaldic acid as ligands improved the reactivity of the system and expanded the scope to the valuable tropane and homotropane cores.73
Scheme 10. Arylation of Bicyclic Amines with Removable DG.
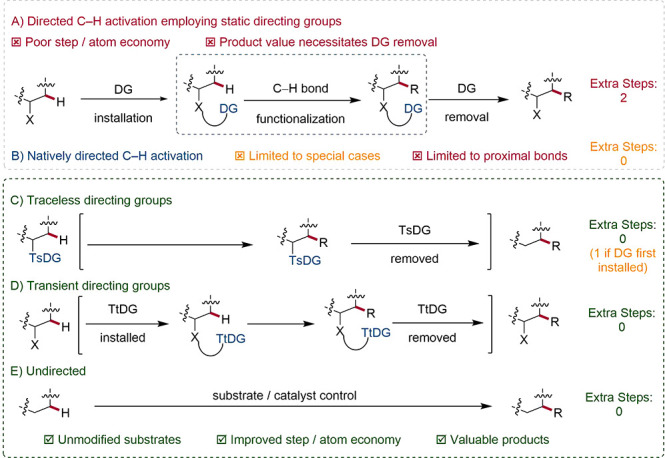
For high value-structures, it is perhaps possible to make an exceptional case for so-called static directing groups (SDGs). At best, the installation and removal of covalent SDGs negate the step and atom economy potential of C–H activation—at worst, final removal of the DG is either not possible, requires harsh conditions, or offers low yields. Since SDG removal is likely to be the final step, this represents a disproportionate resource burden on a synthesis. SDGs arguably represent the single largest barrier to the uptake of C–H activation by industry, conflicting with the Green Chemistry principle of avoiding derivatives. For fundamental research, SDGs are a necessary evil; realistically, chemistry of this design is unlikely to find applications unless high product value and a lack of alternative methodology are evident.
3.1. Native Directing Groups
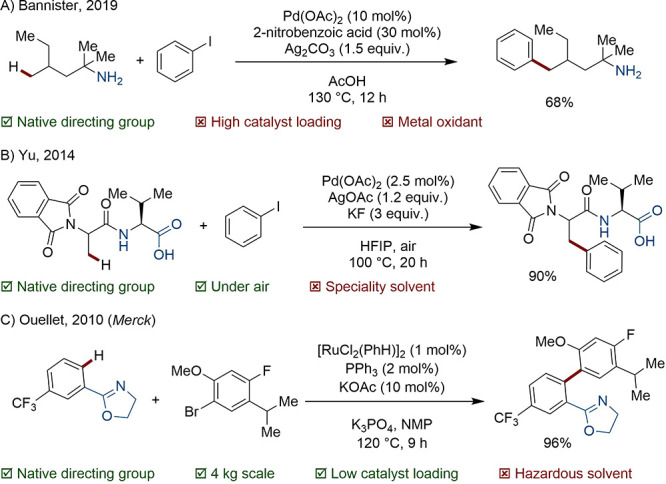
In many cases, so-called native functionality, where a Lewis basic site is already present, can be harnessed as a DG. For instance, Bannister and co-workers reported the utilization of primary amines as a native directing group (Scheme 11A).74 Yu accomplished the site-selective C(sp3)–H functionalization of N-protected di-, tri-, and tetrapeptides by making use of bidentate N,O coordination (Scheme 11B).75 In Merck’s kilo-scale synthesis of Anacetrapib, Ru ortho-coordination is helpfully provided by a neighboring oxazoline which forms part of the final active pharmaceutical ingredient (Scheme 11C).76
Scheme 11. Native Heteroatom Directed C(sp3)–H Arylation.
In a high-profile example of native functionality exploitation, Ackermann recently published a Co-catalyzed methylation of a number of bioactive compounds.77 The addition of a methyl group is widely recognized to have an outsize influence on biological activity and physicochemical properties. In this work, a catalytic system was identified that could transfer a methyl group to C(sp2) and C(sp3) centers coordinated by a range of Lewis basic moieties including N-containing heterocycles, amides, amines, ketones, and aldehydes. The reaction was then applied successfully to the late-stage functionalization of 22 biologically active compounds, although several required isomeric separation. A highlight of this work was the selective monomethylation of paclitaxel, which contains some 47 C–H bonds (Scheme 12).
Scheme 12. Co-Catalyzed C–H Methylation of Paclitaxel.
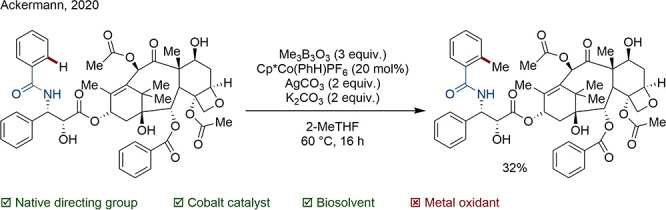
Of course, natively directed C–H functionalizations are highly substrate specific and cannot offer a general strategy for functionalization of each and every C–H bond, especially for advanced synthetic intermediates.
3.2. Traceless Directing Groups
Recently, attention has been drawn to the replacement of preinstalled SDGs by more step-economic alternatives. These strategies allow for the use of less functionalized substrates. Consequently, a rapid increase in value and diversity from simple substrates can be achieved. As a special case of SDGs, traceless directing groups (TsDGs) are commonly pre-existing coordinating groups that can be engineered in the C–H bond functionalization of the substrate and subsequently removed from the product without an additional step.78
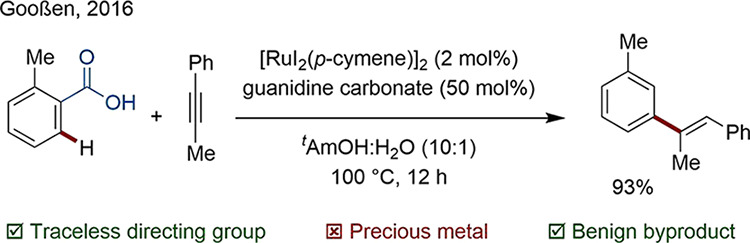
Among the most used TsDGs are carboxylic acids. In particular, the use of benzoic acid derivatives is convenient, since they are often inexpensive and commercially available. In 2016, Gooßen accomplished a regioselective C–H hydroarylation of internal alkynes with benzoic acid derivatives using a Ru-based catalyst (Scheme 13).79 Facile liberation of carbon dioxide (CO2) revealed the product. In the same year, Zhao published a similar method to convert lignin-derived 4-hydroxybenzoic acid into the corresponding meta-substituted alkenyl arene.80
Scheme 13. Regioselective C–H Hydroarylation with Internal Asymmetric Alkynes.
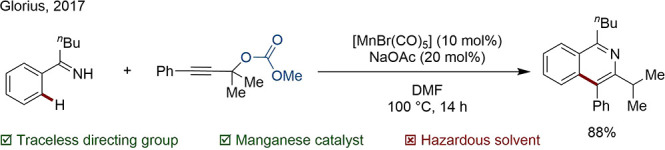
Although removal of the DG can still be achieved in one pot, often synthesis of the starting material is required. For instance, Glorius accomplished a Mn-catalyzed annulation with perfect regioselectivity owing to the presence of a carbonate-based TsDG on the alkyne (Scheme 14).81
Scheme 14. Regioselective Annulation Using a Carbonate TsDG.
3.3. Transient Directing Groups
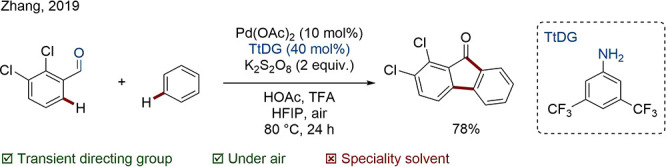
In the transient directing group (TtDG) strategy, DGs are installed as well as removed in situ. Often the TtDG can be added in catalytic amounts, rendering this process in theory more resource and step economic.82 The most common TtDGs are imines formed by the condensation of amines and carbonyls; examples of phosphonites and enamines are also known. Zhang exploited the monodentate and commercially available TtDG 3,5-bis(trifluoromethyl)aniline for the synthesis of 9-fluorenones in a cross-dehydrogenative coupling (CDC).83
Scheme 15. Pd-Catalyzed CDC Using an Aniline TtDG.
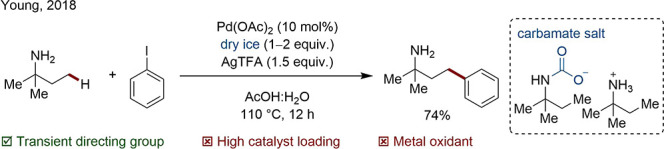
Young and co-workers have reported protocols in which cheap and abundant CO2 was used as a TtDG. The group applied this strategy to the C(sp3)–H γ-arylation of primary and secondary aliphatic amines (Scheme 16) and later to the C(sp2)–H arylation of primary and secondary benzylamines.84,85 The formation of a carbamate TtDG was suggested based on mechanistic investigations in which the corresponding carbamate salt, prepared from a reaction of the amine with dry ice, was converted to the coupled product without additional CO2.
Scheme 16. C(sp3)–H Arylation Using a Carbamate TtDG.
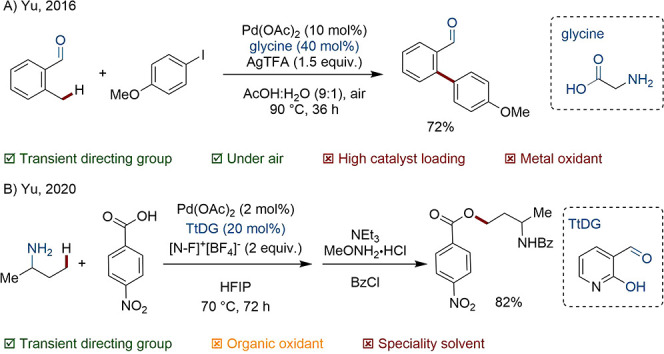
Bidentate TtDGs are now common. Yu utilized unfunctionalized glycine for the C(sp3)–H β-arylation of aliphatic ketones and C(sp3)–H γ-arylation of benzylic aldehydes (Scheme 17A).86
Scheme 17. Bidentate TtDG Mediated C(sp3)–H Arylation and Alkylation.
The same group likewise utilized 2-hydroxynicotinaldehyde as a TtDG group for the oxygenation of free amines (Scheme 17B).87 In comparison to earlier protocols, the protection and deprotection of the amine were not necessary, and a one-step coupling was thus enabled. The same TtDG was also applied in the fluorination of free amines.88 Sorensen and co-workers utilized a commercially available orthanilinic acid as a TtDG for the C–H ortho-methylation or fluorination of benzaldehydes.89 [N–F]+ salts were used either as the oxidant or as an electrophilic fluorine source.
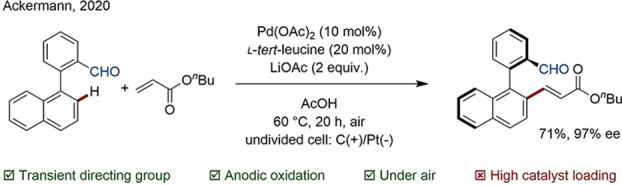
Expansion of the concept to chiral TtDGs gives access to comparatively step and atom economic enantioselective C–H functionalizations. Among common chiral DGs are amino acids, amino amides, or chiral amines that form imine intermediates. Chiral amino acids are often utilized in combination with Pd; examples for the generation of molecules with central, axial, and planar chirality have been reported. Recently, Ackermann accomplished the synthesis of enantioenriched chiral biaryl and N-aryl pyrroles using simple l-tert-leucine as a chiral TtDG (Scheme 18).90
Scheme 18. Atroposelective C–H Activation.
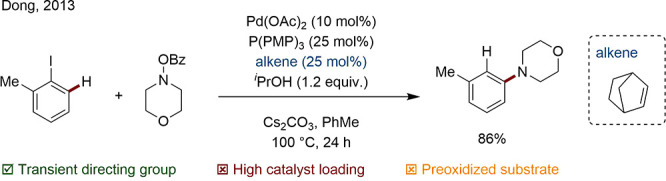
Finally, Catellani reaction variants represent noncondensation examples of TtDG mediated processes. ortho-Directed amination of aryl iodides was achieved by Dong following an oxidative addition-carbopalladation directed C–H activation process (Scheme 19).91
Scheme 19. Transient Norbornene-Directed Amination.
Nevertheless, a common criticism of TtDG reactions is the requirement for high metal catalyst and organic cocatalyst loadings, which mainstream applications will need to address.
3.4. Undirected C–H Activation
The area of undirected C–H activation, in which electronic and steric factors determine regioselectivity, is an exciting but incredibly challenging subfield. Unfortunately, in the absence of highly active catalysts, harsh conditions and high metal loadings are typical. Problematically, reactions often do not run to completion or give product mixtures which may require challenging separations.32
The group of White was instrumental in developing the direct functionalization of allylic C(sp3)–H bonds, presenting an alternative to the Tsuji–Trost reaction, which requires an allylic leaving group. Initial work in 2004 revealed that under Wacker oxidation conditions the presence of DMSO led to linear allylic acetates from terminal olefins. The adduct of Pd(OAc)2 and a simple disulfoxide ligand, which gives the alternative branched products, became known as White’s catalyst (Scheme 20A).92,93 Since then, White and others have intensely developed this subfield, with related systems enabling intermolecular allylic amination, alkylation, and Heck-type arylation, and applied them to the synthesis of natural products.94−98 Furthermore, an array of methods now exist for highly regio- and stereoselective transformations.99−102 In one example, medicinally relevant anti- and syn-1,3 amino alcohols were prepared using complementary aryl-sulfoxide oxazoline (ArSOX) ligands (Scheme 20B).103 Unfortunately, most systems of this type require high Pd loadings and multiple equivalents of benzoquinone (BQ) type oxidants, presenting considerable scope for sustainability improvements.
Scheme 20. Pd-Catalyzed Allylic Functionalization.
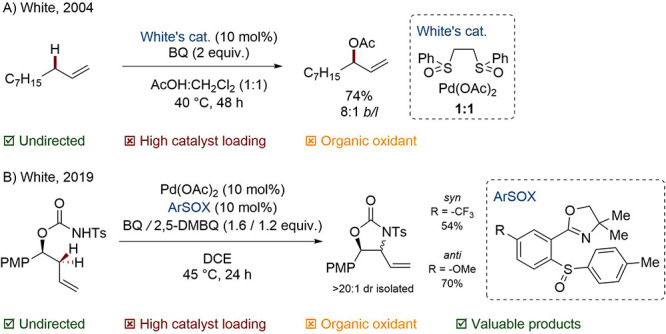
Cp* complexes of the Group IX metals were later harnessed by Tanaka, Cossy, Blakey, Glorius, and others to accomplish related allylic C–H arylations, aminations, and arylations.104−111 A common weakness of these transformations is found in the product regioselectivity in the absence of nearby coordinating groups or strong electronic bias. For this reason, Rovis’ 2020 work, in which allylic amination is guided by very subtle electronic effects via the σ-framework, is all the more remarkable (Scheme 21).112 Despite the value of the products obtained, once again such chemistry is blighted by the high loading of transition metals required as catalyst and oxidant as well as frequently environmentally hazardous solvents, such as 1,2-dichloroethane (DCE).
Scheme 21. Rh-Catalyzed Allylic Functionalization.
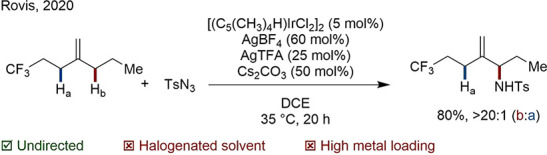
Other precious metal systems are known with significantly better efficiency. In 1999, Fujiwara published an oxidative Heck reaction giving styrene-type products with catalyst loadings as low as 0.2% (Scheme 22A).113 Since then, Stahl was able to show that the addition of weakly coordinating ligands increases the activity of a Pd system used for CDC, enabling improvements in the industrial synthesis of important polymers (Scheme 22B).114,115 In Hong’s direct arylation, the rigid, planar diimine ligand is thought to allow the reaction to proceed with a TON of up to 290 (Scheme 22C).116
Scheme 22. Low-Loading Pd-Catalyzed Examples of Undirected C–H Activation.
Although exciting developments in undirected C–H activation are forthcoming, it is likely that SDGs will still dominate in the short-to-medium term. In the meantime, more and more groups are exploring the possibilities of traceless and transient DGs. A summary of directing strategies, including a comparison of their relative step-counts, is given below (Scheme 23).
Scheme 23. Greener Alternatives to Static DGs.
4. Modern Oxidation Strategies
The coupling of C–H bonds and nucleophiles is among the most common of C–H functionalizations. A general such catalytic cycle consists of four steps (Scheme 24): C–H activation (1), functionalization of the carbometallic intermediate (2), reductive elimination (3), and finally reoxidation of the metal center (4). Usually, this reoxidation is carried out using (super)stoichiometric oxidants, often outweighing the inherent virtue of catalysis.
Scheme 24. General C–H Activation Mechanism Using Stoichiometric Oxidants.
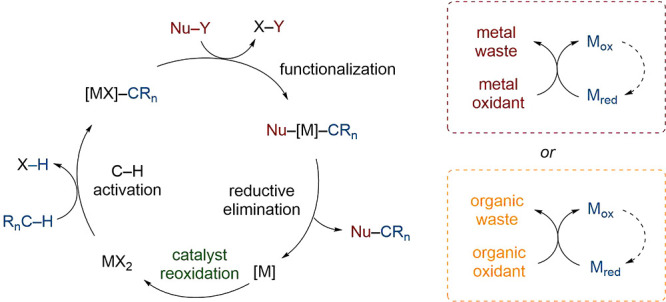
Ag(I) and Cu(II) salts are the most commonly used transition metal oxidants. These salts are typically expensive, possess high molecular weights, and may pose safety hazards (Table 1). Their consumption also produces quantitative, potentially toxic metal waste. Organic oxidants, such as BQ, may be less expensive but still contribute significantly to the E factor. There is therefore high interest in avoiding stoichiometric oxidants.
Table 1. Properties of Common C–H Activation Oxidants.
| oxidant | Mr | cost/mol (US $)a | major issue |
|---|---|---|---|
| NFSI | 315.14 | 4223 | atom economy |
| PhI(TFA)2 | 430.04 | 1450 | atom economy |
| Ag2O | 231.74 | 973 | precious metal |
| AgOAc | 166.91 | 883 | precious metal |
| PhI(OAc)2 | 322.10 | 412 | atom economy |
| Cu(OAc)2 | 181.63 | 303 | metal waste |
| oxone | 307.38 | 163 | waste |
| K2S2O8 | 270.32 | 128 | waste |
| tBuOOH | 90.12 | 51 | waste |
| BQ | 108.09 | 39 | waste |
Source: Sigma-Aldrich, accessed October 2020. Batch size chosen was closest to 100 g; reagent grade or most similar.
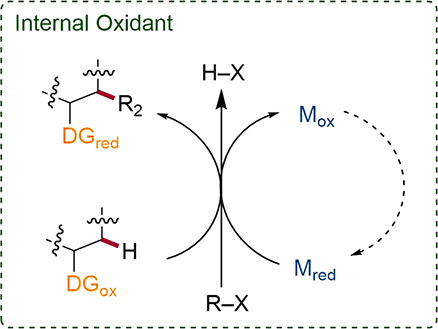
4.1. Internal Oxidants
One strategy involves the use of a pre-installed, internal oxidant that also serves as a DG (Scheme 25).117,118 Often these moieties consist of cleavable N–O and N–N bonds, originating from hydroxylamine and hydrazine derivatives produced on ton scale via efficient, established processes. Transformations using these moieties are often milder than their metal oxidant driven alternatives.
Scheme 25. General C–H Activation Mechanism Using a Preoxidized DG.
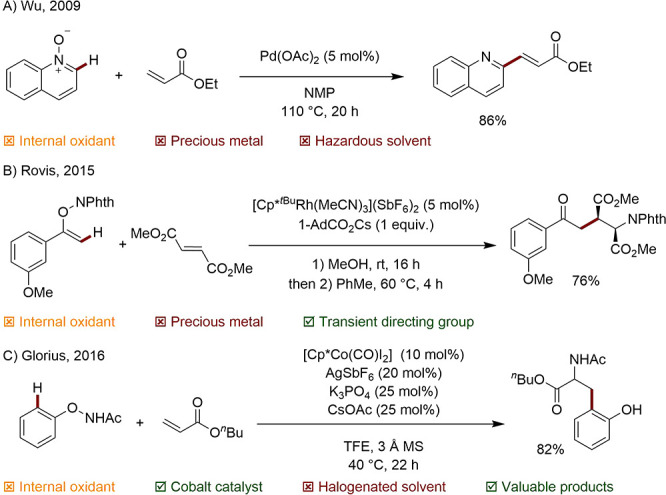
In 2009, the group of Wu revealed that N-oxidized quinolines could successfully direct Pd-catalyzed C2 alkenylation with acrylates (Scheme 26A).119 The following year, Hartwig published an efficient, Pd-catalyzed, oxime ester-directed synthesis of indoles.120 Later, Rovis was able to apply the concept to the syn-carboamination of alkenes using a bulky Cp*-type Rh complex. After undergoing solvolytic ring-opening, the N-eneoxy phthalimide starting material is proposed to act as a bidentate directing group as well as offering an oxidizable N–O bond for closure of the catalytic cycle (Scheme 26B).121 In 2016, the group of Glorius disclosed a synthesis of amino acid esters mediated by a Cp*Co system (Scheme 26C).122
Scheme 26. C–H Functionalizations Using Preoxidized DGs.
4.2. Molecular Oxygen
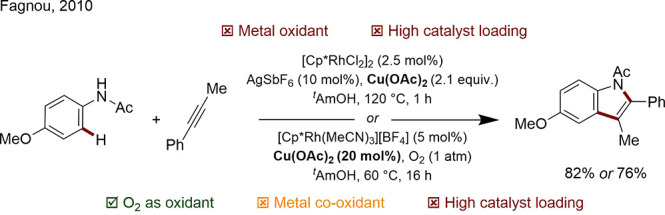
The use of molecular oxygen (O2) is attractive for obvious reasons: O2 or air is readily available, nontoxic, and inexpensive; water is the only byproduct.123 Unfortunately, this method is normally limited to metal systems with the appropriate redox potential. In 2009, Yu demonstrated that a bulky 2,6-substituted pyridine ligand both promoted meta-regioselectivity in a Fujiwara–Moritani reaction and facilitated the reoxidation of Pd(0) to Pd(II) within the catalytic cycle.124 Fagnou acknowledged the problematic use of stoichiometric Cu(OAc)2 in an early Cp*Rh mediated indole synthesis and demonstrated the reaction could become cocatalytic in Cu using O2 as the terminal oxidant with a minor modification of the reaction conditions (Scheme 27).125
Scheme 27. Indole Syntheses with Stoichiometric and Catalytic Cu Oxidant.
Cheng and co-workers later disclosed a synthesis of isoquinolinones requiring only air as the O2 source. Furthermore, the products precipitated readily from solution, avoiding resource-intensive purification. The authors applied this method to preparation of the pharmaceutical agent ISQ-1 in 82% yield (Scheme 28A).126 Ackermann reported a twofold C–H functionalization between benzoic acids and alkenes. C–H Activation was mediated by a Ru biscarboxylate catalyst with O2 as the terminal oxidant (Scheme 28B).127
Scheme 28. Rh-Catalyzed C–H Alkenylation with Air as the Terminal Oxidant.
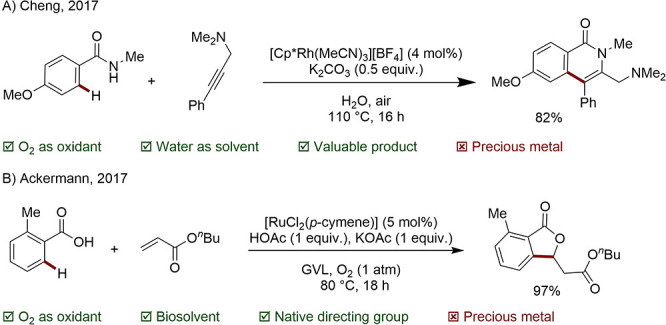
4.3. Photoredox Catalysis
A further method that has drawn attention is the use of photoredox chemistry. In this case, electrons are transferred to a photoredox catalyst, thus reoxidizing the C–H activating metal center. The photoredox catalyst is then regenerated by a terminal oxidant. Although the use of organic terminal oxidants is preferable to the generation of metal waste, the best examples involve the reduction of O2 to a superoxide anion and ultimately water (Scheme 29).
Scheme 29. General C–H Activation Involving Catalyst Photooxidation, with O2 as Terminal Oxidant.
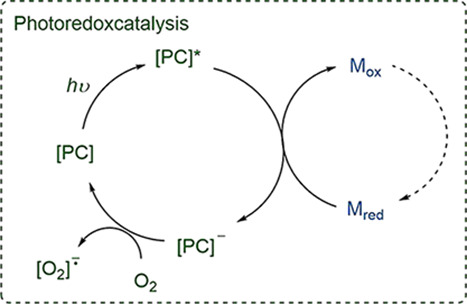
Van d’Eycken and co-workers established a procedure for a selective C2-acylation of indoles using a visible-light photoredox catalyst for the reoxidation of Pd. The reaction procedure was compatible with number of functional groups and was applied to aromatic, primary, and secondary aliphatic aldehydes. The combination of continuous flow and photochemistry allowed a significant decrease in the reaction time and photocatalyst loading (Scheme 30).128
Scheme 30. Photoredox Enabled C2-Acylation of Indoles.
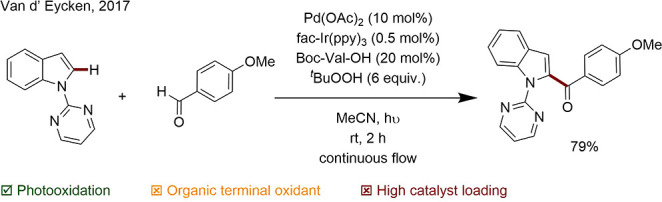
Sundararaju and Rueping developed a mild protocol for C–H/N–H annulation using a dual catalytic approach and O2 as the terminal oxidant. Co(acac)2 mediated the C–H activation, while Na2[Eosin Y] functioned as the electron transfer agent (Scheme 31).129
Scheme 31. Sequential C–C and C–N Bond Formation Catalyzed by a Co and a Photoredox Catalyst.
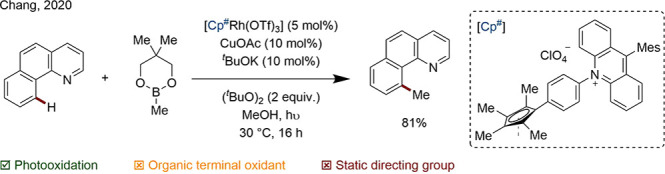
In an exciting recent development, Chang and co-workers detailed a bifunctional catalyst containing both a Cp*Rh center and an acridinium moiety. Internal oxidation of the metal center by the photosensitizing module expedites the reductive elimination step. A cocatalytic Cu salt and an organic terminal oxidant complete the catalytic cycle (Scheme 32).130 Developments beyond this proof-of-concept may offer a further alternative to stoichiometric metal oxidants.
Scheme 32. An Integrated Catalyst for C–H Activation and Photooxidation.
4.4. Metallaelectrocatalysis
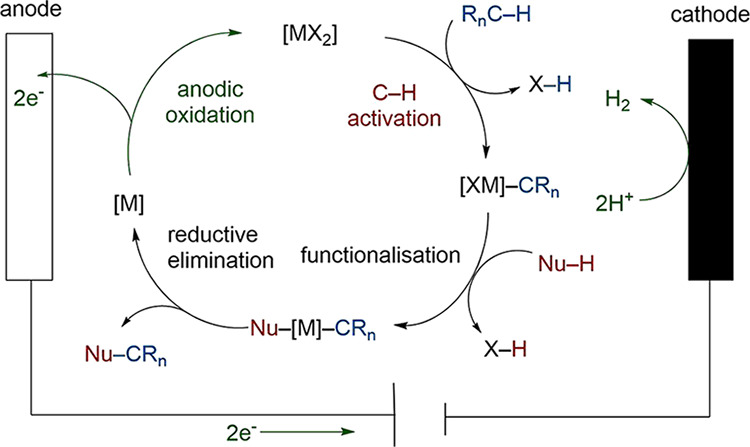
The upscaling of photochemical reactions is often challenging. Another drawback of photoredox catalysts is the use of mostly precious metals, such as Ir, as photocatalysts, as well as their discrete redox states. To access differing redox potentials, chemical modifications via resource-demanding, multistep synthesis are necessary. Surpassing these shortcomings, the field of metallaelectrocatalysis has emerged.36,40,131−133 Redox potentials can be adjusted continuously, for instance, by a potentiostat. This allows broader functional group tolerance and a decreased need for complex ligand systems. Molecular hydrogen is generated as a potentially useful byproduct (Scheme 33).
Scheme 33. A Generalized Mechanism of C–H Activation Involving Anodic Oxidation.
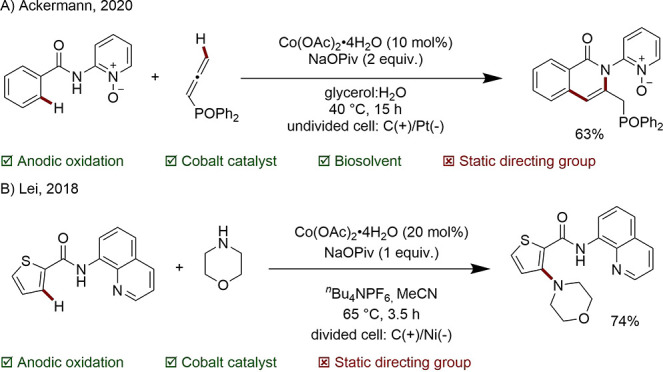
The applicability of Co, Ni, and Cu electrochemistry to C–H functionalization has been demonstrated extensively by Ackermann since 2017.90,134−139 In a recent protocol, a challenging C–C/C–N bond formation with a potentially sensitive allene was accomplished under mild conditions in the biosolvent glycerol using simple Co(OAc)2 salt (Scheme 34A).140 Furthermore, electricity generated in house from wind and solar energy was exploited. Building on Ackermann’s earlier work, Lei and co-workers used electrochemistry for the amination of (hetero)arenes including aryls, furans, and thiophenes, up to gram scale (Scheme 34B).141,142
Scheme 34. Cobalta-Electrocatalyzed C–C/C–N Bond Formation.
The replacement of metal-based oxidants by greener methods has accelerated in recent years; intensive work in this field has established applicable and sustainable alternatives (Figure 2). While internal and organic oxidants avoid the use of transition metals, these strategies are still responsible for stoichiometric waste. Meanwhile, the use of O2, photoredox catalysis and electrochemistry offer sustainable alternatives with benign byproducts. These methods have already been applied to a range of substrates and proven compatible with 3d metals and biosolvents.
Figure 2.
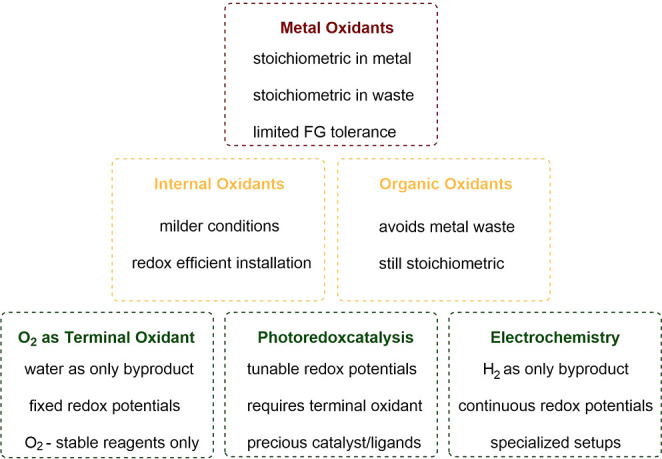
Pyramid of oxidation method sustainability.
5. Choosing Greener Solvents
Organic solvents are almost always the largest weight component of reactive chemistry and purification. The environmental impact of solvents has been the topic of extensive industrial interest. Indeed, the CHEM21 consortium of major pharmaceutical companies have released multiple solvent selection guides to aid sustainable process development, which quantify solvent attributes such as carbon footprint, reactive hazards, and human health impact.22,23,25−28 Choice of solvent (or mixture) is often dictated by the mechanism of the chemistry at hand and by coupling partner solubility or compatibility. In many cases, greener solvents are too quickly overlooked. For example, in C–H activation chemistry involving heterocycle preparation, unsustainable solvents, such as 1,2-DCE, HFIP, and TCE are common (Table 2); many greener alternatives have been shown to be practical under the right conditions.143
Table 2. Common Solvents used in C–H Activation, Associated Hazards, and Environmental Burdensa.
| solvent | cost/L (US $)b | hazardb | environment |
|---|---|---|---|
| HFIP | 1170 | a, e | f, g |
| TFA | 492 | a | g |
| THF | 119 | a, b, c | g |
| DCE | 116 | a, b, c, d | e, f, g |
| DMF | 109 | a, b, c, d | |
| NMP | 104 | a, c, d | |
| 1,4-dioxane | 92 | a, b, c | g |
| CH2Cl2 | 67 | a, b | e, f |
a. Toxicity. b. Cancer risk. c. Flammable. d. Fertility risk. e. Severe greenhouse gas. f. Ozone depletory. g. Environmentally persistent.
Source: Sigma-Aldrich, accessed October 2020. Batch size chosen was closest to 1 L; solvent grade.
It is appreciated that the environmental impact of solvents is more limited in terms of scale, and receives less attention, within academia than in industry; novel reactions are optimized for yield (or selectivity). Likewise, academic researchers likely do not have ready access to the many emerging biosolvents screened in industry. Nevertheless, the inclusion of sustainable alternatives in deviation tables, even if they do not provide the most optimal results, would provide starting points for applied researchers, including for emerging machine learning-based reaction optimization.144,145 Principle component analysis (PCA) is a frequently used method to find compatible alternative solvents based on physicochemical properties.146 The investigation of the properties of solvent mixtures remains an under-researched area.
5.1. Biosolvents
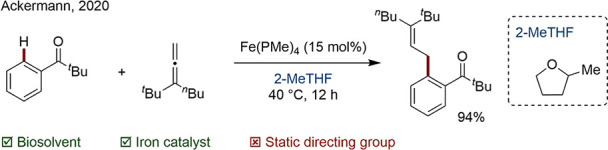
Biomass-derived solvents, commonly referred to as biosolvents, are increasing in popularity. In comparison to traditional solvents, they typically have lower toxicity and show higher biodegradability. 2-MeTHF is now a commonly adopted green alternative and has been used as a substitute for THF, 1,4-dioxane, DCE, and others.147 Its low miscibility in water facilitates purification by organic-water phase separations. Ackermann and co-workers chose 2-MeTHF in their mild C–H activation of allenes using a simple Fe phosphine catalyst (Scheme 35).148
Scheme 35. Iron-Catalyzed C–H Activation of Allenes in 2-MeTHF.
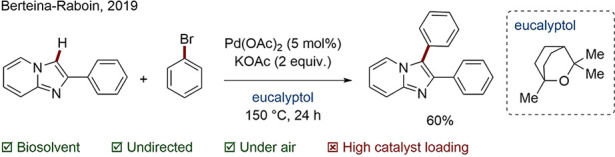
Berteina-Raboin and co-workers published the first C–H activation protocol in which eucalyptol was used as a green solvent (Scheme 36).149 Eucalyptol is the main constituent of eucalyptus essential oil (up to 90%), is immiscible with water, and exhibits low toxicity. On the basis of physical properties, such as its polarity, the authors postulate that eucalyptol may rival 2-MeTHF; it has a comparable cost and shows a lower tendency to form radicals. As a byproduct of the global paper industry, the potential supply of this solvent is estimated to be in the millions of tons; it is also a precursor for the hydrocarbon substitute cymene.150
Scheme 36. Direct C–H Arylation in Eucalyptol.
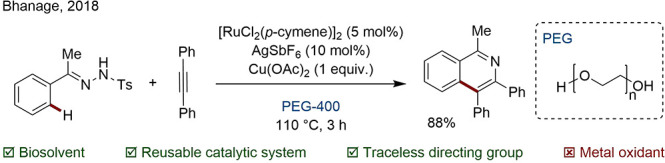
Polyethylene glycols (PEGs) are sugar-derived, low volatility, polar liquid polymers with diverse physical properties. Molecular weights ranging from 300 to 10 000 000 g/mol are commercially available. Bhanage and co-workers performed an alkyne annulation via directed C–H/N–H activation using a homogeneous Ru(II)/PEG-400 catalytic system. Remarkably, the catalyst was recovered by extraction during product isolation and reused four times with negligible impact on yield (Scheme 37).151
Scheme 37. Ru-Catalyzed Annulation Reaction in PEG-400.
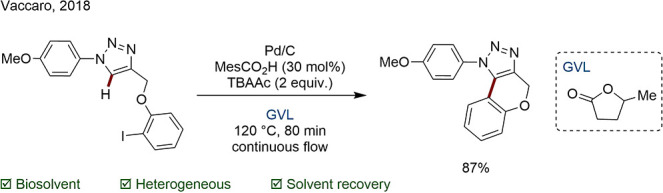
γ-Valerolactone (GVL) is another sugar-derived solvent that is often considered as an alternative to polar aprotics like DMF or MeCN. Vaccaro and co-workers accomplished a regioselective C–H functionalization of 1,2,3-triazoles in GVL under continuous flow (Scheme 38).152 Remarkably, the solvent could be readily recovered by distillation and reused. The product was purified by recrystallization in acetone and water, contributing to a low overall E factor of 23.9. This value was significantly smaller than for previously reported protocols for the synthesis of triazoles. Though the exact figures have to be taken with care, this example underlines the importance of solvent in the mass efficiency of chemical processes: significant improvements can be made by recycling the solvent or by operating under highly concentrated conditions.
Scheme 38. Intramolecular C–H Functionalization of 1,2,3-Triazoles in GVL.
Although many are skeptical regarding the cost and supply of biosolvents, with increasing demand the wholesale cost is expected to decrease, especially for large-scale applications (Table 3). We do not discount the role of the more recognized green solvents, which may or may not be obtained from biological sources, such as ethanol and ethyl acetate.
Table 3. Common Biosolvents, Miscibility, Costs, and Sources.
| solvent | cost/L (US $)a | water miscible | substitutes | source153 |
|---|---|---|---|---|
| GVL | 571 | yes | polar aprotics | sugars |
| 2-MeTHF | 235 | no | ethers, chlorinated | sugars |
| eucalyptol | 218 | no | various | paper industry |
| cyrene | 191 | yes | polar aprotics | cellulose |
| limonene | 144 | no | alkanes | citrus waste |
| ethyl acetate | 71 | no | esters | sugars or petrochem |
| ethyl lactate | 68 | yes | esters | starch |
| cymene | 57 | no | alkanes, aryls | paper industry |
| PEG-400 | 43 | yes | various | sugars |
| ethanolb | 40 | yes | alcohols | sugars or petrochem |
Source: Sigma-Aldrich, accessed October 2020.
Denatured. Batch size chosen was closest to 1 L; solvent grade.
5.2. Aqueous and Solvent-Free Reactions
Provided solvent is not required for heat-transfer purposes, neat chemistry has the potential to offer a sustainability advantage by lowering the E factor, as well as aiding low activity catalytic systems. For example, in 2017 a Mn-catalyzed aryl allylation was accomplished by Glorius and co-workers under solvent-free conditions.154 Sequential C–H and C–C/C–X bond activation led to the synthesis of diverse, valuable products.
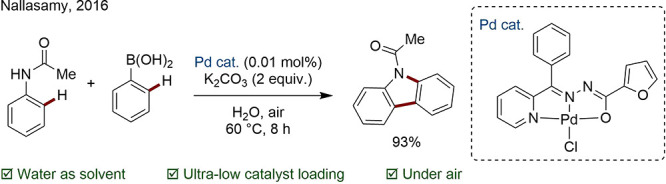
The use of water appears attractive for industrial purposes, since it is inherently safe, poses no health hazard, and is of course highly abundant. An example by Nallasamy is the tandem C–H/N–H activation of acetanilide in water with an active Pd pincer complex (Scheme 39).155 In another preparation of an Anacetrapib intermediate (recall Scheme 11C), a water-soluble Ru formate based system “MCAT-53” was used to carry out the directed arylation in 73% yield with precipitation of the product from solution. Following an organic wash, the aqueous layer could be reused.156
Scheme 39. Aqueous Tandem C–H/N–H Activation.
Notwithstanding, a significant note of caution must be attached to claims that the use of water is always sustainable. Following isolation of the products, the aqueous waste stream must be safely disposed of. If this cannot be remediated by other means, incineration of the waste is an endergonic process.
C–H Activation is not unique in its hitherto reliance on a narrow range of fossil-fuel derived and chlorinated solvents; as organic chemistry adopts bioderived materials more widely, it is likely that the application of green solvents will become commonplace within this field.
6. Case Study: Arene C–H Borylation
6.1. Origins
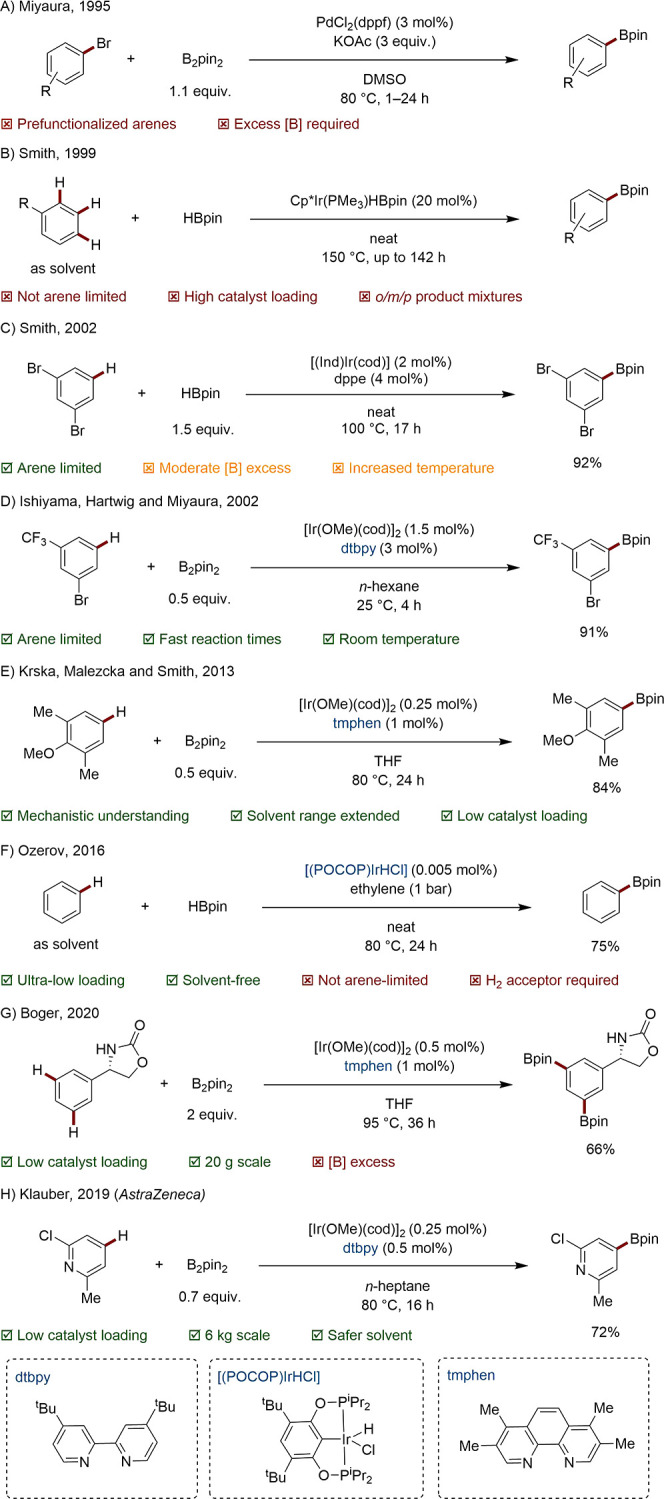
In the absence of general and direct systems for many important C–H transformations (e.g., arylations, alkylations, oxidations, halogenations), aryl boron species are highly valued synthetic intermediates. Before the establishment of C–H borylation, such compounds could only be prepared by stoichiometric organometallic chemistry or the Miyaura borylation of aryl halides (Scheme 40A).157 The group of Smith pioneered the first undirected Ir-catalyzed C–H borylations for installation of the highly versatile bis(pinacolato)borane (Bpin) handle (Scheme 40B).158,159 Although this early work suffered from high metal loading and poor regioselectivity, this was shortly followed a much improved system involving bulkier, more stable bidentate phosphine ligands such as 1,2-bis(diphenylphosphino)ethane (dppe) (Scheme 40C).160 Simultaneously, the eponymous Ishiyama–Miyaura–Hartwig (IMH) catalyst, consisting of an Ir(I) cyclooctadiene precatalyst and the 4,4′-di-tert-butylbipyridine ligand (dtbpy), was shown to be capable of installing the Bpin unit under mild conditions with unrivalled functional group tolerance.161 The somewhat bulky ligand is responsible for the steric control of the reaction, selectively delivering the products from a 1:1 ratio of boron units and starting arenes (Scheme 40D).162 From an early stage, it was clear the novel system could offer a clear step and atom economy advantage over established methods for aryl boronic ester synthesis, in addition to new product substitution patterns.
Scheme 40. Evolution of Precious-Metal-Catalyzed Aryl Borylation.
6.2. Development and Applications
Guided by increasing mechanistic understanding,163−165 further exploration of the IMH system led to adoption of the ligand 3,4,7,8-tetramethylphenanthroline (tmphen), scope-expansion, and sustainability improvements in the reaction conditions.166−170 For example, in 2013 the groups of Krska, Maleczka, and Smith conducted extensive high-throughput screening-optimization from which they elucidated subtle relationships between order-of-addition, precatalyst choice, temperature, and solvent. Using this knowledge, they were able to affect competent borylation systems requiring just 0.25 mol % Ir loadings with simultaneously high boron economy. It was shown that polar solvents, including the biosolvent 2-MeTHF, are amenable to this chemistry (Scheme 40E).171 In 2019, Hartwig and co-workers elucidated that the often superior performance of phenanthroline-based systems over the classic IMH dtbpy catalyst is owing to greater binding stability and hence catalyst lifetime.172 In a similar vein, Ozerov and co-workers revealed a strikingly active catalyst based on a POCOP ligand, exhibiting a turnover number (TON) in excess of 20 000 (Scheme 40F).173
The compatibility of this chemistry with downstream chemistry in “telescoped” sequences, avoiding potentially wasteful purification, has facilitated its take-up in total syntheses. In 2011, Hartwig harnessed a two-step, one-pot borylation-bromination protocol developed in his laboratory to prepare a key intermediate in his route toward (−)-Taiwaniaquinone H and (−)-Taiwaniaquinol B.174,175 In Baran’s total syntheses of Verruculogen and Fumitremorgin A, use of the simple, unsubstituted phenanthroline ligand was shown to be optimal for the 10 g borylation of a key indole intermediate.176 Boger affected a dual 3,5-diborylation on 20 g scale in his preparation of the critically important antibiotic vancomycin (Scheme 40G).177 There are now multiple process chemistry examples of C–H borylation in the public domain, revealing the industrial uptake of this technology. In Merck’s preparation of Dorivirine, a 75 kg scale borylation-oxidation sequence rendered key intermediate 3-bromo-5-iodophenol in 94% yield over two steps (simple bipyridine was used as an adequately performing, low-cost ligand).178 Pfizer used the tmphen ligand to obtain 19 kg of a borylated nicotine intermediate.179 AstraZeneca carried out a 6 kg borylation of a key pyridine intermediate using just 0.25% Ir loading to supply their Phase IIA trial of the A2AR agonist AZD4635 (Scheme 40H).180,181
6.3. Future Directions
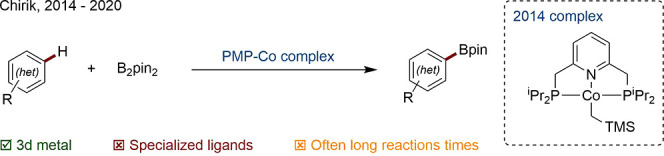
The accumulated knowledge of the past 20 years, in terms of increased catalyst activity and solvent range, suggests there are likely still gains to be found in the effectiveness of this system. This said, one hugely valid criticism remains: the high cost and scarcity of Ir, which may be offset in the cases of highly efficient or heterogeneous, recyclable systems.182−187 The development of undirected 3d metal-based systems, which may replicate and complement Ir borylation chemistry, is therefore an exciting area of growth. Ni,188−190 Fe,191 and dual-metal192 systems have been developed, but the most general of the new catalysts seem to be Co based, of which the work of Chirik is best known (Scheme 41).193−198 Use of the bis(phosphomethyl)pyridine (PNP) family of ligands was shown to act by providing a suitably electron-rich environment for the Co center to emulate Ir-like behavior. A more user-friendly, air-stable terpyridine-based precatalyst has been disclosed.199 The discovery of yet more active, faster 3d systems which can be deployed industrially is a highly anticipated development.
Scheme 41. PMP-Co Complex-Mediated Borylation.
7. Summary and Conclusion
In this Outlook, we have sought to highlight active and growing branches of research in C–H activation which exemplify aspects of sustainability. We have showcased the emergence of abundant 3d metal systems, such as Co, Mn, Fe, Ni, and heterogeneous systems as competent and complementary catalysts. We have discussed elegant transformations that do not require coordinating functionality, or can exploit native DGs. We have highlighted exciting, modern alternatives to transition metal-based oxidants including photoredox and electrochemical methods, and reiterated that the selection of green reaction media contributes substantially toward the mass intensity of a transformation. We hope that researchers will be motivated to explore and understand the sustainability potential of new C–H transformations. With the intention of making environmental considerations from the beginning, we pose ourselves the following questions:
-
(1)
What is the efficiency of my catalyst? Can I provide an alternative, low-loading optimization with an acceptable yield? Is there a high-abundancy metal capable of the elementary steps required by my transformation?
-
(2)
What is the environmental impact of my solvent choice? Are there commercially available green solvents (or mixtures) available which I may not have considered, that may give similar results? Can I simply increase the reaction concentration?
-
(3)
Can I minimize or substitute metal-based oxidants with a modern alternative?
-
(4)
Can I avoid using a DG? Is it genuinely straightforward to remove an SDG; are my products useful if not? Would a novel directed reaction offer a real advantage over an established classical method?
-
(5)
How robust is my chemistry? Are strictly dried solvent and inert atmosphere really necessary? Can I use air-stable catalysts and additives? Would exploring the sensitivity of my conditions lead to better reproducibility and fewer wasted resources?200
-
(6)
Can I quantitatively estimate the environmental burden of my transformation, for comparison and targeted improvement?
We envisage that in the future, by combining several green design elements, newly published reactions will offer promising starting points for applied research. Until now, “greenness” has historically taken a back seat to reaction novelty. Nevertheless, we are convinced that cumulative advances in sustainability will finally enable the industrial potential of C–H activation to be fulfilled.
Acknowledgments
We earnestly thank Malte Schrader, Tobias Pinkert, Shobhan Mondal, Peter Bellotti, Daniel Moock, and Tiffany Paulisch for constructive and insightful discussions as well as the reviewer for their constructive feedback.
Generous financial support by the Deutsche Forschungsgemeinschaft (Leibniz Award, CRC 1459) is gratefully acknowledged.
The authors declare no competing financial interest.
References
- Hayler J. D.; Leahy D. K.; Simmons E. M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2019, 38, 36–46. 10.1021/acs.organomet.8b00566. [DOI] [Google Scholar]
- Laird T. Green Chemistry Is Good Process Chemistry. Org. Process Res. Dev. 2012, 16, 1–2. 10.1021/op200366y. [DOI] [Google Scholar]
- Gilding P. Why I Welcome a Climate Emergency. Nature 2019, 573, 311. 10.1038/d41586-019-02735-w. [DOI] [PubMed] [Google Scholar]
- Communication from the Commission to the European Parliament, The Council, The European Economic and Social Committee and the Committee of the Regions: Chemical Strategy for Sustainability Towards a Toxic-Free Environment; Brussels, 2020.
- Regulation (EC) No. 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), Establishing a European Chemicals Agency, Amending Directive 1999/4. Off. J. Eur. Union, 2006.
- Anastas P. T.; Warner J. C.. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998. [Google Scholar]
- Trost B. M. The Atom Economy - A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- Trost B. M. Atom Economy—A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. 10.1002/anie.199502591. [DOI] [Google Scholar]
- Constable D. J. C.; Curzons A. D.; Cunningham V. L. Metrics to ‘Green’ Chemistry–Which Are the Best?. Green Chem. 2002, 4, 521–527. 10.1039/B206169B. [DOI] [Google Scholar]
- Sheldon R. A. The E Factor: Fifteen Years On. Green Chem. 2007, 9, 1261–1273. 10.1039/b713736m. [DOI] [Google Scholar]
- Sheldon R. A. E Factors, Green Chemistry and Catalysis: An Odyssey. Chem. Commun. 2008, 3352–3365. 10.1039/b803584a. [DOI] [PubMed] [Google Scholar]
- Anastas P.; Eghbali N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. 10.1039/B918763B. [DOI] [PubMed] [Google Scholar]
- Jimenez-Gonzalez C.; Ponder C. S.; Broxterman Q. B.; Manley J. B. Using the Right Green Yardstick: Why Process Mass Intensity Is Used in the Pharmaceutical Industry To Drive More Sustainable Processes. Org. Process Res. Dev. 2011, 15, 912–917. 10.1021/op200097d. [DOI] [Google Scholar]
- McGonagle F. I.; Sneddon H. F.; Jamieson C.; Watson A. J. B. Molar Efficiency: A Useful Metric To Gauge Relative Reaction Efficiency in Discovery Medicinal Chemistry. ACS Sustainable Chem. Eng. 2014, 2, 523–532. 10.1021/sc4004532. [DOI] [Google Scholar]
- Li J.; Albrecht J.; Borovika A.; Eastgate M. D. Evolving Green Chemistry Metrics into Predictive Tools for Decision Making and Benchmarking Analytics. ACS Sustainable Chem. Eng. 2018, 6, 1121–1132. 10.1021/acssuschemeng.7b03407. [DOI] [Google Scholar]
- Sheldon R. A. Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustainable Chem. Eng. 2018, 6, 32–48. 10.1021/acssuschemeng.7b03505. [DOI] [Google Scholar]
- Monteith E. R.; Mampuys P.; Summerton L.; Clark J. H.; Maes B. U. W.; McElroy C. R. Why We Might Be Misusing Process Mass Intensity (PMI) and a Methodology to Apply It Effectively as a Discovery Level Metric. Green Chem. 2020, 22, 123–135. 10.1039/C9GC01537J. [DOI] [Google Scholar]
- Lankey R. L.; Anastas P. T. Life-Cycle Approaches for Assessing Green Chemistry Technologies. Ind. Eng. Chem. Res. 2002, 41, 4498–4502. 10.1021/ie0108191. [DOI] [Google Scholar]
- Capello C.; Fischer U.; Hungerbühler K. What Is a Green Solvent? A Comprehensive Framework for the Environmental Assessment of Solvents. Green Chem. 2007, 9, 927–934. 10.1039/b617536h. [DOI] [Google Scholar]
- McElroy C. R.; Constantinou A.; Jones L. C.; Summerton L.; Clark J. H. Towards a Holistic Approach to Metrics for the 21st Century Pharmaceutical Industry. Green Chem. 2015, 17, 3111–3121. 10.1039/C5GC00340G. [DOI] [Google Scholar]
- Kralisch D.; Ott D.; Gericke D. Rules and Benefits of Life Cycle Assessment in Green Chemical Process and Synthesis Design: A Tutorial Review. Green Chem. 2015, 17, 123–145. 10.1039/C4GC01153H. [DOI] [Google Scholar]
- Henderson R. K.; Jiménez-González C.; Constable D. J. C.; Alston S. R.; Inglis G. G. A.; Fisher G.; Sherwood J.; Binks S. P.; Curzons A. D. Expanding GSK’s Solvent Selection Guide - Embedding Sustainability into Solvent Selection Starting at Medicinal Chemistry. Green Chem. 2011, 13, 854–862. 10.1039/c0gc00918k. [DOI] [Google Scholar]
- Prat D.; Pardigon O.; Flemming H.-W.; Letestu S.; Ducandas V.; Isnard P.; Guntrum E.; Senac T.; Ruisseau S.; Cruciani P.; Hosek P. Sanofi’s Solvent Selection Guide: A Step Toward More Sustainable Processes. Org. Process Res. Dev. 2013, 17, 1517–1525. 10.1021/op4002565. [DOI] [Google Scholar]
- Adams J. P.; Alder C. M.; Andrews I.; Bullion A. M.; Campbell-Crawford M.; Darcy M. G.; Hayler J. D.; Henderson R. K.; Oare C. A.; Pendrak I.; Redman A. M.; Shuster L. E.; Sneddon H. F.; Walker M. D. Development of GSK’s Reagent Guides - Embedding Sustainability into Reagent Selection. Green Chem. 2013, 15, 1542–1549. 10.1039/c3gc40225h. [DOI] [Google Scholar]
- Prat D.; Hayler J.; Wells A. A Survey of Solvent Selection Guides. Green Chem. 2014, 16, 4546–4551. 10.1039/C4GC01149J. [DOI] [Google Scholar]
- Prat D.; Wells A.; Hayler J.; Sneddon H.; McElroy C. R.; Abou-Shehada S.; Dunn P. J. CHEM21 Selection Guide of Classical-and Less Classical-Solvents. Green Chem. 2016, 18, 288–296. 10.1039/C5GC01008J. [DOI] [Google Scholar]
- Byrne F. P.; Jin S.; Paggiola G.; Petchey T. H. M.; Clark J. H.; Farmer T. J.; Hunt A. J.; McElroy R. C.; Sherwood J.. Tools and Techniques for Solvent Selection: Green Solvent Selection Guides. Sustainable Chem. Processes 2016, 4.7. 10.1186/s40508-016-0051-z [DOI] [Google Scholar]
- Diorazio L. J.; Hose D. R. J. J.; Adlington N. K. Toward a More Holistic Framework for Solvent Selection. Org. Process Res. Dev. 2016, 20, 760–773. 10.1021/acs.oprd.6b00015. [DOI] [Google Scholar]
- Wencel-Delord J.; Glorius F. C–H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem. 2013, 5, 369–375. 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- Wencel-Delord J.; Dröge T.; Liu F.; Glorius F. Towards Mild Metal-Catalyzed C–H Bond Activation. Chem. Soc. Rev. 2011, 40, 4740–4761. 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Beyond Directing Groups: Transition-Metal-Catalyzed C–H Activation of Simple Arenes. Angew. Chem., Int. Ed. 2012, 51, 10236–10254. 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.; Larsen M. A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensch T.; Hopkinson M. N.; Glorius F.; Wencel-Delord J. Mild Metal-Catalyzed C–H Activation: Examples and Concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. 10.1039/C6CS00075D. [DOI] [PubMed] [Google Scholar]
- Santoro S.; Ferlin F.; Luciani L.; Ackermann L.; Vaccaro L. Biomass-Derived Solvents as Effective Media for Cross-Coupling Reactions and C–H Functionalization Processes. Green Chem. 2017, 19, 1601–1612. 10.1039/C7GC00067G. [DOI] [Google Scholar]
- Gensch T.; James M. J. M. J.; Dalton T.; Glorius F. Increasing Catalyst Efficiency in C–H Activation Catalysis. Angew. Chem., Int. Ed. 2018, 57, 2296–2306. 10.1002/anie.201710377. [DOI] [PubMed] [Google Scholar]
- Meyer T. H.; Finger L. H.; Gandeepan P.; Ackermann L. Resource Economy by Metallaelectrocatalysis: Merging Electrochemistry and C–H Activation. Trends Chem. 2019, 1, 63–76. 10.1016/j.trechm.2019.01.011. [DOI] [Google Scholar]
- Gandeepan P.; Kaplaneris N.; Santoro S.; Vaccaro L.; Ackermann L. Biomass-Derived Solvents for Sustainable Transition Metal-Catalyzed C–H Activation. ACS Sustainable Chem. Eng. 2019, 7, 8023–8040. 10.1021/acssuschemeng.9b00226. [DOI] [Google Scholar]
- Gandeepan P.; Müller T.; Zell D.; Cera G.; Warratz S.; Ackermann L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]
- Yu C.; Sanjosé-Orduna J.; Patureau F. W.; Pérez-Temprano M. H. Emerging Unconventional Organic Solvents for C–H Bond and Related Functionalization Reactions. Chem. Soc. Rev. 2020, 49, 1643–1652. 10.1039/C8CS00883C. [DOI] [PubMed] [Google Scholar]
- Gandeepan P.; Finger L. H.; Meyer T. H.; Ackermann L. 3d Metallaelectrocatalysis for Resource Economical Syntheses. Chem. Soc. Rev. 2020, 49, 4254–4272. 10.1039/D0CS00149J. [DOI] [PubMed] [Google Scholar]
- Graedel T. E.; Harper E. M.; Nassar N. T.; Nuss P.; Reck B. K. Criticality of Metals and Metalloids. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4257–4262. 10.1073/pnas.1500415112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss P.; Eckelman M. J. Life Cycle Assessment of Metals: A Scientific Synthesis. PLoS One 2014, 9, e101298. 10.1371/journal.pone.0101298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- Gormisky P. E.; White M. C. Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- Howell J. M.; Feng K.; Clark J. R.; Trzepkowski L. J.; White M. C. Remote Oxidation of Aliphatic C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2015, 137, 14590–14593. 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osberger T. J.; Rogness D. C.; Kohrt J. T.; Stepan A. F.; White M. C. Oxidative Diversification of Amino Acids and Peptides by Small-Molecule Iron Catalysis. Nature 2016, 537, 214–219. 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanjo T.; de Lucca E. C. Jr.; White M. C. Remote, Late-Stage Oxidation of Aliphatic C–H Bonds in Amide-Containing Molecules. J. Am. Chem. Soc. 2017, 139, 14586–14591. 10.1021/jacs.7b07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Nanjo T.; de Lucca E. C. Jr.; White M. C. Chemoselective Methylene Oxidation in Aromatic Molecules. Nat. Chem. 2019, 11, 213–221. 10.1038/s41557-018-0175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Zhang X. P. Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev. 2011, 40, 1899–1909. 10.1039/C0CS00070A. [DOI] [PubMed] [Google Scholar]
- Liu Y.; You T.; Wang H.-X.; Tang Z.; Zhou Y.; Che C.-M. Iron-and Cobalt-Catalyzed C(sp3)–H Bond Functionalization Reactions and Their Application in Organic Synthesis. Chem. Soc. Rev. 2020, 49, 5310–5358. 10.1039/D0CS00340A. [DOI] [PubMed] [Google Scholar]
- Paradine S. M.; White M. C. Iron-Catalyzed Intramolecular Allylic C–H Amination. J. Am. Chem. Soc. 2012, 134, 2036–2039. 10.1021/ja211600g. [DOI] [PubMed] [Google Scholar]
- Shing K.-P.; Liu Y.; Cao B.; Chang X.-Y.; You T.; Che C.-M. N-Heterocyclic Carbene Iron(III) Porphyrin-Catalyzed Intramolecular C(sp3)–H Amination of Alkyl Azides. Angew. Chem., Int. Ed. 2018, 57, 11947–11951. 10.1002/anie.201806059. [DOI] [PubMed] [Google Scholar]
- Clark J. R.; Feng K.; Sookezian A.; White M. C. Manganese-Catalysed Benzylic C(sp3)–H Amination for Late-Stage Functionalization. Nat. Chem. 2018, 10, 583–591. 10.1038/s41557-018-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L.-M.; Xu P.; Xie J.; Zhang X. P. Enantioselective Intermolecular Radical C–H Amination. J. Am. Chem. Soc. 2020, 142, 20828–20836. 10.1021/jacs.0c10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.; Kweon J. Highly Robust Iron Catalyst System for Intramolecular C(sp3)–H Amidation Leading to γ-Lactams. Angew. Chem., Int. Ed. 2020, anie.202013499. 10.1002/anie.202013499. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Chen H.; Wang C. Mn-Catalyzed Aromatic C–H Alkenylation with Terminal Alkynes. J. Am. Chem. Soc. 2013, 135, 1264–1267. 10.1021/ja311689k. [DOI] [PubMed] [Google Scholar]
- He R.; Huang Z. T.; Zheng Q. Y.; Wang C. Manganese-Catalyzed Dehydrogenative [4 + 2] Annulation of N–H Imines and Alkynes by C–H/N–H Activation. Angew. Chem., Int. Ed. 2014, 53, 4950–4953. 10.1002/anie.201402575. [DOI] [PubMed] [Google Scholar]
- Cembellín S.; Dalton T.; Pinkert T.; Schäfers F.; Glorius F. Highly Selective Synthesis of 1,3-Enynes, Pyrroles, and Furans by Manganese(I)-Catalyzed C–H Activation. ACS Catal. 2020, 10, 197–202. 10.1021/acscatal.9b03965. [DOI] [Google Scholar]
- Zhu C.; Kuniyil R.; Jei B. B.; Ackermann L. Domino C–H Activation/Directing Group Migration/Alkyne Annulation: Unique Selectivity by D6-Cobalt(III) Catalysts. ACS Catal. 2020, 10, 4444–4450. 10.1021/acscatal.9b05413. [DOI] [Google Scholar]
- Saper N. I.; Ohgi A.; Small D. W.; Semba K.; Nakao Y.; Hartwig J. F. Nickel-Catalysed Anti-Markovnikov Hydroarylation of Unactivated Alkenes with Unactivated Arenes Facilitated by Non-Covalent Interactions. Nat. Chem. 2020, 12, 276–283. 10.1038/s41557-019-0409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro S.; Kozhushkov S. I.; Ackermann L.; Vaccaro L. Heterogeneous Catalytic Approaches in C–H Activation Reactions. Green Chem. 2016, 18, 3471–3493. 10.1039/C6GC00385K. [DOI] [Google Scholar]
- Hübner S.; De Vries J. G.; Farina V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts?. Adv. Synth. Catal. 2016, 358, 3–25. 10.1002/adsc.201500846. [DOI] [Google Scholar]
- Tang D.-T. D.; Collins K. D.; Glorius F. Completely Regioselective Direct C–H Functionalization of Benzo[b]Thiophenes Using a Simple Heterogeneous Catalyst. J. Am. Chem. Soc. 2013, 135, 7450–7453. 10.1021/ja403130g. [DOI] [PubMed] [Google Scholar]
- Tang D.-T. T. D.; Collins K. D.; Ernst J. B.; Glorius F. Pd/C as a Catalyst for Completely Regioselective C–H Functionalization of Thiophenes under Mild Conditions. Angew. Chem., Int. Ed. 2014, 53, 1809–1813. 10.1002/anie.201309305. [DOI] [PubMed] [Google Scholar]
- Collins K. D.; Honeker R.; Vásquez-Céspedes S. V.; Tang D.-T. D.; Glorius F. C–H Arylation of Triphenylene, Naphthalene and Related Arenes Using Pd/C. Chem. Sci. 2015, 6, 1816–1824. 10.1039/C4SC03051F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vásquez-Céspedes S.; Ferry A.; Candish L.; Glorius F. Heterogeneously Catalyzed Direct C–H Thiolation of Heteroarenes. Angew. Chem., Int. Ed. 2015, 54, 5772–5776. 10.1002/anie.201411997. [DOI] [PubMed] [Google Scholar]
- Duan H.; Li M.; Zhang G.; Gallagher J. R.; Huang Z.; Sun Y.; Luo Z.; Chen H.; Miller J. T.; Zou R.; Lei A.; Zhao Y. Single-Site Palladium(II) Catalyst for Oxidative Heck Reaction: Catalytic Performance and Kinetic Investigations. ACS Catal. 2015, 5, 3752–3759. 10.1021/acscatal.5b00569. [DOI] [Google Scholar]
- Meng D.; Bi J.; Dong Y.; Hao B.; Qin K.; Li T.; Zhu D. Salen-Based Hypercrosslinked Polymer-Supported Pd as an Efficient and Recyclable Catalyst for C–H Halogenation. Chem. Commun. 2020, 56, 2889–2892. 10.1039/C9CC09781C. [DOI] [PubMed] [Google Scholar]
- Ferlin F.; Luque Navarro P. M.; Gu Y.; Lanari D.; Vaccaro L. Waste Minimized Synthesis of Pharmaceutically Active Compounds via Heterogeneous Manganese Catalysed C–H Oxidation in Flow. Green Chem. 2020, 22, 397–403. 10.1039/C9GC02961C. [DOI] [Google Scholar]
- Sambiagio C.; Schönbauer D.; Blieck R.; Dao-Huy T.; Pototschnig G.; Schaaf P.; Wiesinger T.; Zia M. F.; Wencel-Delord J.; Besset T.; Maes B. U. W.; Schnürch M. A Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C–H Functionalisation Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. 10.1039/C8CS00201K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topczewski J. J.; Cabrera P. J.; Saper N. I.; Sanford M. S. Palladium-Catalysed Transannular C–H Functionalization of Alicyclic Amines. Nature 2016, 531, 220–224. 10.1038/nature16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera P. J.; Lee M.; Sanford M. S. Second-Generation Palladium Catalyst System for Transannular C–H Functionalization of Azabicycloalkanes. J. Am. Chem. Soc. 2018, 140, 5599–5606. 10.1021/jacs.8b02142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Pan X.; Barsamian A. L.; Kamenecka T. M.; Bannister T. D. Native Directed Site-Selective δ-C(sp3)-H and δ-C(sp2)-H Arylation of Primary Amines. ACS Catal. 2019, 9, 4887–4891. 10.1021/acscatal.8b04927. [DOI] [Google Scholar]
- Gong W.; Zhang G.; Liu T.; Giri R.; Yu J. Q. Site-Selective C(sp3)–H Functionalization of Di-, Tri-, and Tetrapeptides at the N-Terminus. J. Am. Chem. Soc. 2014, 136, 16940–16946. 10.1021/ja510233h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet S. G.; Roy A.; Molinaro C.; Angelaud R.; Marcoux J.-F.; O’Shea P. D.; Davies I. W. Preparative Scale Synthesis of the Biaryl Core of Anacetrapib via a Ruthenium-Catalyzed Direct Arylation Reaction: Unexpected Effect of Solvent Impurity on the Arylation Reaction. J. Org. Chem. 2011, 76, 1436–1439. 10.1021/jo1018574. [DOI] [PubMed] [Google Scholar]
- Friis S. D.; Johansson M. J.; Ackermann L. Cobalt-Catalysed C–H Methylation for Late-Stage Drug Diversification. Nat. Chem. 2020, 12, 511–519. 10.1038/s41557-020-0475-7. [DOI] [PubMed] [Google Scholar]
- Paul K. D.; Luxami V.; Rani G. Traceless Directing Group: A Novel Strategy in Regiodivergent C–H Functionalization. Chem. Commun. 2020, 56, 12479–12521. 10.1039/D0CC04863A. [DOI] [PubMed] [Google Scholar]
- Huang L.; Biafora A.; Zhang G.; Bragoni V.; Gooßen L. J. Regioselective C–H Hydroarylation of Internal Alkynes with Arenecarboxylates: Carboxylates as Deciduous Directing Groups. Angew. Chem., Int. Ed. 2016, 55, 6933–6937. 10.1002/anie.201600894. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Shrestha R.; Hartwig J. F.; Zhao P. A Decarboxylative Approach for Regioselective Hydroarylation of Alkynes. Nat. Chem. 2016, 8, 1144–1151. 10.1038/nchem.2602. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Greßies S.; Cembellín S.; Klauck F. J. R.; Daniliuc C. G.; Glorius F. Redox-Neutral Manganese(I)-Catalyzed C–H Activation: Traceless Directing Group Enabled Regioselective Annulation. Angew. Chem., Int. Ed. 2017, 56, 12778–12782. 10.1002/anie.201707396. [DOI] [PubMed] [Google Scholar]
- Gandeepan P.; Ackermann L. Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem. 2018, 4, 199–222. 10.1016/j.chempr.2017.11.002. [DOI] [Google Scholar]
- Wang Y. F.; Xu W. G.; Sun B.; Yu Q. Q.; Li T. J.; Zhang F. L. Monodentate Transient Directing Group Assisted Pd-Catalyzed Direct Dehydrogenative Cross-Coupling of Benzaldehydes with Arenes toward 9-Fluorenones. J. Org. Chem. 2019, 84, 13104–13111. 10.1021/acs.joc.9b02139. [DOI] [PubMed] [Google Scholar]
- Kapoor M.; Chand-Thakuri P.; Young M. C. Carbon Dioxide-Mediated C(sp2)–H Arylation of Primary and Secondary Benzylamines. J. Am. Chem. Soc. 2019, 141, 7980–7989. 10.1021/jacs.9b03375. [DOI] [PubMed] [Google Scholar]
- Kapoor M.; Liu D.; Young M. C. Carbon Dioxide-Mediated C(sp3)–H Arylation of Amine Substrates. J. Am. Chem. Soc. 2018, 140, 6818–6822. 10.1021/jacs.8b05061. [DOI] [PubMed] [Google Scholar]
- Zhang F. L.; Hong K.; Li T. J.; Park H.; Yu J. Q. Functionalization of C(sp3)–H Bonds Using a Transient Directing Group. Science 2016, 351, 252–256. 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Q.; Wu Y.; Wang Z.; Qiao J. X.; Yu J. Q. Transient Directing Group Enabled Pd-Catalyzed γ-C(sp3)–H Oxygenation of Alkyl Amines. ACS Catal. 2020, 10, 5657–5662. 10.1021/acscatal.0c01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Q.; Singh S.; Wu Y.; Wang Z.; Hao W.; Verma P.; Qiao J. X.; Sunoj R. B.; Yu J. Q. Pd-Catalyzed γ-C(sp3)-H Fluorination of Free Amines. J. Am. Chem. Soc. 2020, 142, 9966–9974. 10.1021/jacs.9b13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Y.; Sorensen E. J. Pd-Catalyzed, ortho C–H Methylation and Fluorination of Benzaldehydes Using Orthanilic Acids as Transient Directing Groups. J. Am. Chem. Soc. 2018, 140, 2789–2792. 10.1021/jacs.8b00048. [DOI] [PubMed] [Google Scholar]
- Dhawa U.; Tian C.; Wdowik T.; Oliveira J. C. A.; Hao J.; Ackermann L. Enantioselective Pallada-Electrocatalyzed C–H Activation by Transient Directing Groups: Expedient Access to Helicenes. Angew. Chem., Int. Ed. 2020, 59, 13451–13457. 10.1002/anie.202003826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z.; Dong G. Ortho vsIpso: Site-Selective Pd and Norbornene-Catalyzed Arene C–H Amination Using Aryl Halides. J. Am. Chem. Soc. 2013, 135, 18350–18353. 10.1021/ja410823e. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. A Sulfoxide-Promoted, Catalytic Method for the Regioselective Synthesis of Allylic Acetates from Monosubstituted Olefins via C–H Oxidation. J. Am. Chem. Soc. 2004, 126, 1346–1347. 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; Prabagaran N.; Labenz N. A.; White M. C. Serial Ligand Catalysis: A Highly Selective Allylic C–H Oxidation. J. Am. Chem. Soc. 2005, 127, 6970–6971. 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]
- Reed S. A.; Mazzotti A. R.; White M. C. A Catalytic, Brønsted Base Strategy for Intermolecular Allylic C–H Amination. J. Am. Chem. Soc. 2009, 131, 11701–11706. 10.1021/ja903939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraunhoffer K. J.; White M. C. syn-1,2-Amino Alcohols via Diastereoselective Allylic C–H Amination. J. Am. Chem. Soc. 2007, 129, 7274–7276. 10.1021/ja071905g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A. J.; White M. C. Catalytic Intermolecular Allylic C–H Alkylation. J. Am. Chem. Soc. 2008, 130, 14090–14091. 10.1021/ja806867p. [DOI] [PubMed] [Google Scholar]
- Delcamp J. H.; Brucks A. P.; White M. C. A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction. J. Am. Chem. Soc. 2008, 130, 11270–11271. 10.1021/ja804120r. [DOI] [PubMed] [Google Scholar]
- Strambeanu I. I.; White M. C. Catalyst-Controlled C–O versus C–N Allylic Functionalization of Terminal Olefins. J. Am. Chem. Soc. 2013, 135, 12032–12037. 10.1021/ja405394v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R.; White M. C. C–H to C–N Cross-Coupling of Sulfonamides with Olefins. J. Am. Chem. Soc. 2018, 140, 3202–3205. 10.1021/jacs.7b13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Li C.-J. Catalytic Allylic Alkylation via the Cross-Dehydrogenative-Coupling Reaction between Allylic sp3 C–H and Methylenic sp3 C–H Bonds. J. Am. Chem. Soc. 2006, 128, 56–57. 10.1021/ja056541b. [DOI] [PubMed] [Google Scholar]
- Ammann S. E.; Liu W.; White M. C. Enantioselective Allylic C–H Oxidation of Terminal Olefins to Isochromans by Palladium(II)/Chiral Sulfoxide Catalysis. Angew. Chem., Int. Ed. 2016, 55, 9571–9575. 10.1002/anie.201603576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Ali S. Z.; Ammann S. E.; White M. C. Asymmetric Allylic C–H Alkylation via Palladium(II)/cis-ArSOX Catalysis. J. Am. Chem. Soc. 2018, 140, 10658–10662. 10.1021/jacs.8b05668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R.; Young J.; Promontorio R.; Dannheim F. M.; Pattillo C. C.; White M. C. Synthesis of anti-1,3 Amino Alcohol Motifs via Pd(II)/SOX Catalysis with the Capacity for Stereodivergence. J. Am. Chem. Soc. 2019, 141, 9468–9473. 10.1021/jacs.9b02690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazerouni A. M.; McKoy Q. A.; Blakey S. B. Recent Advances in Oxidative Allylic C–H Functionalization via Group IX-Metal Catalysis. Chem. Commun. 2020, 56, 13287–13300. 10.1039/D0CC05554A. [DOI] [PubMed] [Google Scholar]
- Cochet T.; Bellosta V.; Roche D.; Ortholand J. Y.; Greiner A.; Cossy J. Rhodium(III)-Catalyzed Allylic C–H Bond Amination. Synthesis of Cyclic Amines from ω-Unsaturated N-Sulfonylamines. Chem. Commun. 2012, 48, 10745–10747. 10.1039/c2cc36067e. [DOI] [PubMed] [Google Scholar]
- Archambeau A.; Rovis T. Rhodium(III)-Catalyzed Allylic C(sp3)–H Activation of Alkenyl Sulfonamides: Unexpected Formation of Azabicycles. Angew. Chem., Int. Ed. 2015, 54, 13337–13340. 10.1002/anie.201504150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y.; Kudo E.; Sugiyama H.; Uekusa H.; Tanaka K. Facile Generation and Isolation of π-Allyl Complexes from Aliphatic Alkenes and an Electron-Deficient Rh(III) Complex: Key Intermediates of Allylic C–H Functionalization. Organometallics 2016, 35, 1547–1552. 10.1021/acs.organomet.6b00143. [DOI] [Google Scholar]
- Lerchen A.; Knecht T.; Koy M.; Ernst J. B.; Bergander K.; Daniliuc C. G.; Glorius F. Non-Directed Cross-Dehydrogenative (Hetero)Arylation of Allylic C(sp3)–H Bonds Enabled by C–H Activation. Angew. Chem., Int. Ed. 2018, 57, 15248–15252. 10.1002/anie.201807047. [DOI] [PubMed] [Google Scholar]
- Knecht T.; Mondal S.; Ye J.; Das M.; Glorius F. Intermolecular, Branch-Selective, and Redox-Neutral Cp*IrIII-Catalyzed Allylic C–H Amidation. Angew. Chem., Int. Ed. 2019, 58, 7117–7121. 10.1002/anie.201901733. [DOI] [PubMed] [Google Scholar]
- Knecht T.; Pinkert T.; Dalton T.; Lerchen A.; Glorius F. Cp*RhIII-Catalyzed Allyl-Aryl Coupling of Olefins and Arylboron Reagents Enabled by C(sp3)–H Activation. ACS Catal. 2019, 9, 1253–1257. 10.1021/acscatal.8b04677. [DOI] [Google Scholar]
- Lei H.; Rovis T. Ir-Catalyzed Intermolecular Branch-Selective Allylic C–H Amidation of Unactivated Terminal Olefins. J. Am. Chem. Soc. 2019, 141, 2268–2273. 10.1021/jacs.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H.; Rovis T. A Site-Selective Amination Catalyst Discriminates between Nearly Identical C–H Bonds of Unsymmetrical Disubstituted Alkenes. Nat. Chem. 2020, 12, 725–731. 10.1038/s41557-020-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C.; Lu W.; Kitamura T.; Fujiwara Y. Highly Efficient Pd-Catalyzed Coupling of Arenes with Olefins in the Presence of tert-Butyl Hydroperoxide as Oxidant. Org. Lett. 1999, 1, 2097–2100. 10.1021/ol991148u. [DOI] [Google Scholar]
- Wang D.; Izawa Y.; Stahl S. S. Pd-Catalyzed Aerobic Oxidative Coupling of Arenes: Evidence for Transmetalation between Two Pd(II)-Aryl Intermediates. J. Am. Chem. Soc. 2014, 136, 9914–9917. 10.1021/ja505405u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Stahl S. S. Pd-Catalyzed Aerobic Oxidative Biaryl Coupling: Non-Redox Cocatalysis by Cu(OTf)2 and Discovery of Fe(OTf)3 as a Highly Effective Cocatalyst. J. Am. Chem. Soc. 2017, 139, 5704–5707. 10.1021/jacs.7b01970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Hong S. H. Ligand-Promoted Direct C–H Arylation of Simple Arenes: Evidence for a Cooperative Bimetallic Mechanism. ACS Catal. 2017, 7, 3336–3343. 10.1021/acscatal.7b00397. [DOI] [Google Scholar]
- Huang H.; Ji X.; Wu W.; Jiang H. Transition Metal-Catalyzed C–H Functionalization of N-Oxyenamine Internal Oxidants. Chem. Soc. Rev. 2015, 44, 1155–1171. 10.1039/C4CS00288A. [DOI] [PubMed] [Google Scholar]
- Patureau F. W.; Glorius F. Oxidizing Directing Groups Enable Efficient and Innovative C–H Activation Reactions. Angew. Chem., Int. Ed. 2011, 50, 1977–1979. 10.1002/anie.201007241. [DOI] [PubMed] [Google Scholar]
- Wu J.; Cui X.; Chen L.; Jiang G.; Wu Y. Palladium-Catalyzed Alkenylation of Quinoline-N-Oxides via C–H Activation under External-Oxidant-Free Conditions. J. Am. Chem. Soc. 2009, 131, 13888–13889. 10.1021/ja902762a. [DOI] [PubMed] [Google Scholar]
- Tan Y.; Hartwig J. F. Palladium-Catalyzed Amination of Aromatic C–H Bonds with Oxime Esters. J. Am. Chem. Soc. 2010, 132, 3676–3677. 10.1021/ja100676r. [DOI] [PubMed] [Google Scholar]
- Piou T.; Rovis T. Rhodium-Catalysed syn-Carboamination of Alkenes via a Transient Directing Group. Nature 2015, 527, 86–90. 10.1038/nature15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerchen A.; Knecht T.; Daniliuc C. G.; Glorius F. Unnatural Amino Acid Synthesis Enabled by the Regioselective Cobalt(III)-Catalyzed Intermolecular Carboamination of Alkenes. Angew. Chem., Int. Ed. 2016, 55, 15166–15170. 10.1002/anie.201608729. [DOI] [PubMed] [Google Scholar]
- Campbell A. N.; Stahl S. S. Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C–H Oxidation Reactions Catalyzed by Pd (and Cu). Acc. Chem. Res. 2012, 45, 851–863. 10.1021/ar2002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-H.; Shi B.-F.; Yu J.-Q. Pd(II)-Catalyzed Olefination of Electron-Deficient Arenes Using 2,6-Dialkylpyridine Ligands. J. Am. Chem. Soc. 2009, 131, 5072–5074. 10.1021/ja900327e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart D. R.; Alsabeh P.; Kuhn M.; Fagnou K. Rhodium(III)-Catalyzed Arene and Alkene C–H Bond Functionalization Leading to Indoles and Pyrroles. J. Am. Chem. Soc. 2010, 132, 18326–18339. 10.1021/ja1082624. [DOI] [PubMed] [Google Scholar]
- Upadhyay N. S.; Thorat V. H.; Sato R.; Annamalai P.; Chuang S. C.; Cheng C. H. Synthesis of Isoquinolones via Rh-Catalyzed C–H Activation of Substituted Benzamides Using Air as the Sole Oxidant in Water. Green Chem. 2017, 19, 3219–3224. 10.1039/C7GC01221G. [DOI] [Google Scholar]
- Bechtoldt A.; Baumert M. E.; Vaccaro L.; Ackermann L. Ruthenium(II) Oxidase Catalysis for C–H Alkenylations in Biomass-Derived γ-Valerolactone. Green Chem. 2018, 20, 398–402. 10.1039/C7GC03353B. [DOI] [Google Scholar]
- Sharma U. K.; Gemoets H. P. L.; Schröder F.; Noël T.; Van Der Eycken E. V. Merger of Visible-Light Photoredox Catalysis and C–H Activation for the Room-Temperature C-2 Acylation of Indoles in Batch and Flow. ACS Catal. 2017, 7, 3818–3823. 10.1021/acscatal.7b00840. [DOI] [Google Scholar]
- Kalsi D.; Dutta S.; Barsu N.; Rueping M.; Sundararaju B. Room-Temperature C–H Bond Functionalization by Merging Cobalt and Photoredox Catalysis. ACS Catal. 2018, 8, 8115–8120. 10.1021/acscatal.8b02118. [DOI] [Google Scholar]
- Kim J.; Kim D.; Chang S. Merging Two Functions in a Single Rh Catalyst System: Bimodular Conjugate for Light-Induced Oxidative Coupling. J. Am. Chem. Soc. 2020, 142, 19052–19057. 10.1021/jacs.0c09982. [DOI] [PubMed] [Google Scholar]
- Sauermann N.; Meyer T. H.; Qiu Y.; Ackermann L. Electrocatalytic C–H Activation. ACS Catal. 2018, 8, 7086–7103. 10.1021/acscatal.8b01682. [DOI] [Google Scholar]
- Meyer T. H.; Choi I.; Tian C.; Ackermann L. Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis?. Chem. 2020, 6, 2484–2496. 10.1016/j.chempr.2020.08.025. [DOI] [Google Scholar]
- Samanta R. C.; Meyer T. H.; Siewert I.; Ackermann L. Renewable Resources for Sustainable Metallaelectro-Catalysed C–H Activation. Chem. Sci. 2020, 11, 8657–8670. 10.1039/D0SC03578E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C.; Massignan L.; Meyer T. H.; Ackermann L. Electrochemical C–H/N–H Activation by Water-Tolerant Cobalt Catalysis at Room Temperature. Angew. Chem., Int. Ed. 2018, 57, 2383–2387. 10.1002/anie.201712647. [DOI] [PubMed] [Google Scholar]
- Sauermann N.; Mei R.; Ackermann L. Electrochemical C–H Amination by Cobalt Catalysis in a Renewable Solvent. Angew. Chem., Int. Ed. 2018, 57, 5090–5094. 10.1002/anie.201802206. [DOI] [PubMed] [Google Scholar]
- Tian C.; Dhawa U.; Scheremetjew A.; Ackermann L. Cupraelectro-Catalyzed Alkyne Annulation: Evidence for Distinct C–H Alkynylation and Decarboxylative C–H/C–C Manifolds. ACS Catal. 2019, 9, 7690–7696. 10.1021/acscatal.9b02348. [DOI] [Google Scholar]
- Zhang S.; Struwe J.; Hu L.; Ackermann L. Nickela-electrocatalyzed C–H Alkoxylation with Secondary Alcohols: Oxidation-Induced Reductive Elimination at Nickel(III). Angew. Chem., Int. Ed. 2020, 59, 3178–3183. 10.1002/anie.201913930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C.; Meyer T. H.; Stangier M.; Dhawa U.; Rauch K.; Finger L. H.; Ackermann L. Cobaltaelectro-Catalyzed C–H Activation for Resource-Economical Molecular Syntheses. Nat. Protoc. 2020, 15, 1760–1774. 10.1038/s41596-020-0306-8. [DOI] [PubMed] [Google Scholar]
- Meyer T. H.; Samanta R. C.; Del Vecchio A.; Ackermann L. Mangana(III/IV)electro-catalyzed C(sp3)–H azidation. Chem. Sci. 2021, 10.1039/d0sc05924b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T. H.; Chesnokov G. A.; Ackermann L. Cobalta-Electrocatalyzed C–H Activation in Biomass-Derived Glycerol: Powered by Renewable Wind and Solar Energy. ChemSusChem 2020, 13, 668–671. 10.1002/cssc.202000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauermann N.; Meyer T. H.; Tian C.; Ackermann L. Electrochemical Cobalt-Catalyzed C–H Oxygenation at Room Temperature. J. Am. Chem. Soc. 2017, 139, 18452–18455. 10.1021/jacs.7b11025. [DOI] [PubMed] [Google Scholar]
- Gao X.; Wang P.; Zeng L.; Tang S.; Lei A. Cobalt(II)-Catalyzed Electrooxidative C–H Amination of Arenes with Alkylamines. J. Am. Chem. Soc. 2018, 140, 4195–4199. 10.1021/jacs.7b13049. [DOI] [PubMed] [Google Scholar]
- Santhoshkumar R.; Cheng C. H. Reaching Green: Heterocycle Synthesis by Transition Metal-Catalyzed C–H Functionalization in Sustainable Medium. Chem. - Eur. J. 2019, 25, 9366–9384. 10.1002/chem.201901026. [DOI] [PubMed] [Google Scholar]
- Strieth-Kalthoff F.; Sandfort F.; Segler M. H. S.; Glorius F. Machine Learning the Ropes: Principles, Applications and Directions in Synthetic Chemistry. Chem. Soc. Rev. 2020, 49, 6154–6168. 10.1039/C9CS00786E. [DOI] [PubMed] [Google Scholar]
- Pflüger P. M.; Glorius F. Molecular Machine Learning: The Future of Synthetic Chemistry?. Angew. Chem., Int. Ed. 2020, 59, 18860–18865. 10.1002/anie.202008366. [DOI] [PubMed] [Google Scholar]
- Murray P. M.; Sheppard T. D.; Bellany F.; Benhamou L.; Bučar D.-K.; Tabor A. B. The Application of Design of Experiments (DoE) Reaction Optimisation and Solvent Selection in the Development of New Synthetic Chemistry. Org. Biomol. Chem. 2016, 14, 2373–2384. 10.1039/C5OB01892G. [DOI] [PubMed] [Google Scholar]
- Aycock D. F. Solvent Applications of 2-Methyltetrahydrofuran in Organometallic and Biphasic Reactions. Org. Process Res. Dev. 2007, 11, 156–159. 10.1021/op060155c. [DOI] [Google Scholar]
- Messinis A. M.; Finger L. H.; Hu L.; Ackermann L. Allenes for Versatile Iron-Catalyzed C–H Activation by Weak O-Coordination: Mechanistic Insights by Kinetics, Intermediate Isolation, and Computation. J. Am. Chem. Soc. 2020, 142, 13102–13111. 10.1021/jacs.0c04837. [DOI] [PubMed] [Google Scholar]
- Campos J. F.; Scherrmann M. C.; Berteina-Raboin S. Eucalyptol: A New Solvent for the Synthesis of Heterocycles Containing Oxygen, Sulfur and Nitrogen. Green Chem. 2019, 21, 1531–1539. 10.1039/C8GC04016H. [DOI] [Google Scholar]
- Leita B. A.; Warden A. C.; Burke N.; O’Shea M. S.; Trimm D. Production of p-Cymene and Hydrogen from a Bio-Renewable Feedstock-1,8-Cineole (Eucalyptus Oil). Green Chem. 2010, 12, 70–76. 10.1039/B916460J. [DOI] [Google Scholar]
- Deshmukh D. S.; Bhanage B. M. N-Tosylhydrazone Directed Annulation via C–H/N–N Bond Activation in Ru(II)/PEG-400 as Homogeneous Recyclable Catalytic System: A Green Synthesis of Isoquinolines. Org. Biomol. Chem. 2018, 16, 4864–4873. 10.1039/C8OB01082J. [DOI] [PubMed] [Google Scholar]
- Ferlin F.; Luciani L.; Santoro S.; Marrocchi A.; Lanari D.; Bechtoldt A.; Ackermann L.; Vaccaro L. A Continuous Flow Approach for the C–H Functionalization of 1,2,3-Triazoles in γ-Valerolactone as a Biomass-Derived Medium. Green Chem. 2018, 20, 2888–2893. 10.1039/C8GC01115J. [DOI] [Google Scholar]
- Clarke C. J.; Tu W.-C.; Levers O.; Bröhl A.; Hallett J. P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. 10.1021/acs.chemrev.7b00571. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Klauck F. J. R.; Glorius F. Manganese-Catalyzed Allylation via Sequential C–H and C–C/C–Het Bond Activation. Chem. Sci. 2017, 8, 3379–3383. 10.1039/C7SC00230K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam V.; Kaminsky W.; Nallasamy D. Pd(II) Pincer Type Complex Catalyzed Tandem C–H and N–H Activation of Acetanilide in Aqueous Media: A Concise Access to Functionalized Carbazoles in a Single Step. Green Chem. 2016, 18, 3295–3301. 10.1039/C5GC02937F. [DOI] [Google Scholar]
- Mehta A.; Saha B.; Koohang A. A.; Chorghade M. S. Arene Ruthenium Catalyst MCAT-53 for the Synthesis of Heterobiaryl Compounds in Water through Aromatic C–H Bond Activation. Org. Process Res. Dev. 2018, 22, 1119–1130. 10.1021/acs.oprd.8b00141. [DOI] [Google Scholar]
- Ishiyama T.; Murata M.; Miyaura N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. 10.1021/jo00128a024. [DOI] [Google Scholar]
- Iverson C. N.; Smith M. R. III Stoichiometric and Catalytic B–C Bond Formation from Unactivated Hydrocarbons and Boranes. J. Am. Chem. Soc. 1999, 121, 7696–7697. 10.1021/ja991258w. [DOI] [Google Scholar]
- Cho J.-Y.; Iverson C. N.; Smith M. R. III Steric and Chelate Directing Effects in Aromatic Borylation. J. Am. Chem. Soc. 2000, 122, 12868–12869. 10.1021/ja0013069. [DOI] [Google Scholar]
- Cho J.-Y.; Kin Tse M.; Holmes D.; Maleczka R. E. Jr.; Smith M. R. III Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C–H Bonds. Science 2002, 295, 305–308. 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; Takagi J.; Ishida K.; Miyaura N.; Anastasi N. R.; Hartwig J. F. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; Takagi J.; Hartwig J. F.; Miyaura N. A Stoichiometric Aromatic C–H Borylation Catalyzed by Iridium(I)/2,2′-Bipyridine Complexes at Room Temperature. Angew. Chem., Int. Ed. 2002, 41, 3056–3058. 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Tamura H.; Yamazaki H.; Sato H.; Sakaki S. Iridium-Catalyzed Borylation of Benzene with Diboron. Theoretical Elucidation of Catalytic Cycle Including Unusual Iridium(V) Intermediate. J. Am. Chem. Soc. 2003, 125, 16114–16126. 10.1021/ja0302937. [DOI] [PubMed] [Google Scholar]
- Boller T. M.; Murphy J. M.; Hapke M.; Ishiyama T.; Miyaura N.; Hartwig J. F. Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc. 2005, 127, 14263–14278. 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- Larsen M. A.; Oeschger R. J.; Hartwig J. F. Effect of Ligand Structure on the Electron Density and Activity of Iridium Catalysts for the Borylation of Alkanes. ACS Catal. 2020, 10, 3415–3424. 10.1021/acscatal.0c00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. A.; Hartwig J. F. Iridium-Catalyzed C–H Borylation of Heteroarenes: Scope, Regioselectivity, Application to Late-Stage Functionalization, and Mechanism. J. Am. Chem. Soc. 2014, 136, 4287–4299. 10.1021/ja412563e. [DOI] [PubMed] [Google Scholar]
- Seechurn C. C. C. J.; Sivakumar V.; Satoskar D.; Colacot T. J. Iridium-Catalyzed C–H Borylation of Heterocycles Using an Overlooked 1,10-Phenanthroline Ligand: Reinventing the Catalytic Activity by Understanding the Solvent-Assisted Neutral to Cationic Switch. Organometallics 2014, 33, 3514–3522. 10.1021/om500420d. [DOI] [Google Scholar]
- Saito Y.; Segawa Y.; Itami K. para-C–H Borylation of Benzene Derivatives by a Bulky Iridium Catalyst. J. Am. Chem. Soc. 2015, 137, 5193–5198. 10.1021/jacs.5b02052. [DOI] [PubMed] [Google Scholar]
- Wang G.; Xu L.; Li P. Double N,B-Type Bidentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation. J. Am. Chem. Soc. 2015, 137, 8058–8061. 10.1021/jacs.5b05252. [DOI] [PubMed] [Google Scholar]
- Haines B. E.; Saito Y.; Segawa Y.; Itami K.; Musaev D. G. Flexible Reaction Pocket on Bulky Diphosphine–Ir Complex Controls Regioselectivity in para-Selective C–H Borylation of Arenes. ACS Catal. 2016, 6, 7536–7546. 10.1021/acscatal.6b02317. [DOI] [Google Scholar]
- Preshlock S. M.; Ghaffari B.; Maligres P. E.; Krska S. W.; Maleczka R. E. Jr.; Smith M. R. III High-Throughput Optimization of Ir-Catalyzed C–H Borylation: A Tutorial for Practical Applications. J. Am. Chem. Soc. 2013, 135, 7572–7582. 10.1021/ja400295v. [DOI] [PubMed] [Google Scholar]
- Oeschger R. J.; Larsen M. A.; Bismuto A.; Hartwig J. F. Origin of the Difference in Reactivity between Ir Catalysts for the Borylation of C–H Bonds. J. Am. Chem. Soc. 2019, 141, 16479–16485. 10.1021/jacs.9b08920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press L. P.; Kosanovich A. J.; McCulloch B. J.; Ozerov O. V. High-Turnover Aromatic C–H Borylation Catalyzed by POCOP-Type Pincer Complexes of Iridium. J. Am. Chem. Soc. 2016, 138, 9487–9497. 10.1021/jacs.6b03656. [DOI] [PubMed] [Google Scholar]
- Murphy J. M.; Liao X.; Hartwig J. F. Meta Halogenation of 1,3-Disubstituted Arenes via Iridium-Catalyzed Arene Borylation. J. Am. Chem. Soc. 2007, 129, 15434–15435. 10.1021/ja076498n. [DOI] [PubMed] [Google Scholar]
- Liao X.; Stanley L. M.; Hartwig J. F. Enantioselective Total Syntheses of (−)-Taiwaniaquinone H and (−)-Taiwaniaquinol B by Iridium-Catalyzed Borylation and Palladium-Catalyzed Asymmetric α-Arylation. J. Am. Chem. Soc. 2011, 133, 2088–2091. 10.1021/ja110215b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Holte D.; Zoller J.; Umemiya S.; Simke L. R.; Baran P. S. Total Synthesis of Verruculogen and Fumitremorgin a Enabled by Ligand-Controlled C–H Borylation. J. Am. Chem. Soc. 2015, 137, 10160–10163. 10.1021/jacs.5b07154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J.; Qu S.; Tan C.; Cai Y.; Mogi Y.; Keith D. J.; Boger D. L. Next-Generation Total Synthesis of Vancomycin. J. Am. Chem. Soc. 2020, 142, 16039–16050. 10.1021/jacs.0c07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau L. C.; Chen Q.; Gauvreau D.; Girardin M.; Belyk K.; Maligres P.; Zhou G.; Gu C.; Zhang W.; Tan L.; O’Shea P. D. A Robust Kilo-Scale Synthesis of Doravirine. Org. Process Res. Dev. 2016, 20, 1476–1481. 10.1021/acs.oprd.6b00163. [DOI] [Google Scholar]
- Sieser J. E.; Maloney M. T.; Chisowa E.; Brenek S. J.; Monfette S.; Salisbury J. J.; Do N. M.; Singer R. A. Ir-Catalyzed Borylation as an Efficient Route to a Nicotine Hapten. Org. Process Res. Dev. 2018, 22, 527–534. 10.1021/acs.oprd.8b00053. [DOI] [Google Scholar]
- Littleson M. M.; Campbell A. D.; Clarke A.; Dow M.; Ensor G.; Evans M. C.; Herring A.; Jackson B. A.; Jackson L. V.; Karlsson S.; Klauber D. J.; Legg D. H.; Leslie K. W.; Moravčík Š.; Parsons C. D.; Ronson T. O.; Meadows R. E. Synthetic Route Design of AZD4635, an A2AR Antagonist. Org. Process Res. Dev. 2019, 23, 1407–1419. 10.1021/acs.oprd.9b00171. [DOI] [Google Scholar]
- Douglas J. J.; Adams B. W. V.; Benson H.; Broberg K.; Gillespie P. M.; Hoult O.; Ibraheem A. K.; Janbon S.; Janin G.; Parsons C. D.; Sigerson R. C.; Klauber D. J. Multikilogram-Scale Preparation of AZD4635 via C–H Borylation and Bromination: The Corrosion of Tantalum by a Bromine/Methanol Mixture. Org. Process Res. Dev. 2019, 23, 62–68. 10.1021/acs.oprd.8b00342. [DOI] [Google Scholar]
- Kawamorita S.; Ohmiya H.; Hara K.; Fukuoka A.; Sawamura M. Directed Ortho Borylation of Functionalized Arenes Catalyzed by a Silica-Supported Compact Phosphine–Iridium System. J. Am. Chem. Soc. 2009, 131, 5058–5059. 10.1021/ja9008419. [DOI] [PubMed] [Google Scholar]
- Tagata T.; Nishida M.; Nishida A. Continuous-Flow C–H Borylation of Arene Derivatives. Adv. Synth. Catal. 2010, 352, 1662–1666. 10.1002/adsc.201000160. [DOI] [Google Scholar]
- Iwai T.; Murakami R.; Harada T.; Kawamorita S.; Sawamura M. Silica-Supported Tripod Triarylphosphane: Application to Transition Metal-Catalyzed C(sp3)–H Borylations. Adv. Synth. Catal. 2014, 356, 1563–1570. 10.1002/adsc.201301147. [DOI] [Google Scholar]
- Grüning W. R.; Siddiqi G.; Safonova O. V.; Copéret C. Bipyridine Periodic Mesoporous Organosilica: A Solid Ligand for the Iridium-Catalyzed Borylation of C–H Bonds. Adv. Synth. Catal. 2014, 356, 673–679. 10.1002/adsc.201400125. [DOI] [Google Scholar]
- Waki M.; Maegawa Y.; Hara K.; Goto Y.; Shirai S.; Yamada Y.; Mizoshita N.; Tani T.; Chun W.-J.; Muratsugu S.; Tada M.; Fukuoka A.; Inagaki S. A Solid Chelating Ligand: Periodic Mesoporous Organosilica Containing 2,2′-Bipyridine within the Pore Walls. J. Am. Chem. Soc. 2014, 136, 4003–4011. 10.1021/ja4131609. [DOI] [PubMed] [Google Scholar]
- Wu F.; Feng Y.; Jones C. W. Recyclable Silica-Supported Iridium Bipyridine Catalyst for Aromatic C–H Borylation. ACS Catal. 2014, 4, 1365–1375. 10.1021/cs4009539. [DOI] [Google Scholar]
- Das A.; Hota P. K.; Mandal S. K. Nickel-Catalyzed C(sp2)–H Borylation of Arenes. Organometallics 2019, 38, 3286–3293. 10.1021/acs.organomet.9b00364. [DOI] [Google Scholar]
- Khake S. M.; Chatani N. Nickel-Catalyzed C–H Functionalization Using A Non-Directed Strategy. Chem. 2020, 6, 1056–1081. 10.1016/j.chempr.2020.04.005. [DOI] [Google Scholar]
- Tian Y. M.; Guo X. N.; Wu Z.; Friedrich A.; Westcott S. A.; Braunschweig H.; Radius U.; Marder T. B. Ni-Catalyzed Traceless, Directed C3-Selective C–H Borylation of Indoles. J. Am. Chem. Soc. 2020, 142, 13136–13144. 10.1021/jacs.0c05434. [DOI] [PubMed] [Google Scholar]
- Dombray T.; Werncke C. G.; Jiang S.; Grellier M.; Vendier L.; Bontemps S.; Sortais J. B.; Sabo-Etienne S.; Darcel C. Iron-Catalyzed C–H Borylation of Arenes. J. Am. Chem. Soc. 2015, 137, 4062–4065. 10.1021/jacs.5b00895. [DOI] [PubMed] [Google Scholar]
- Parmelee S. R.; Mazzacano T. J.; Zhu Y.; Mankad N. P.; Keith J. A. A Heterobimetallic Mechanism for C–H Borylation Elucidated from Experimental and Computational Data. ACS Catal. 2015, 5, 3689–3699. 10.1021/acscatal.5b00275. [DOI] [Google Scholar]
- Obligacion J. V.; Semproni S. P.; Chirik P. J. Cobalt-Catalyzed C–H Borylation. J. Am. Chem. Soc. 2014, 136, 4133–4136. 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]
- Obligacion J. V.; Semproni S. P.; Pappas I.; Chirik P. J. Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design. J. Am. Chem. Soc. 2016, 138, 10645–10653. 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]
- Obligacion J. V.; Chirik P. J. Mechanistic Studies of Cobalt-Catalyzed C(sp2)–H Borylation of Five-Membered Heteroarenes with Pinacolborane. ACS Catal. 2017, 7, 4366–4371. 10.1021/acscatal.7b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obligacion J. V.; Bezdek M. J.; Chirik P. J. C(sp2)-H Borylation of Fluorinated Arenes Using an Air-Stable Cobalt Precatalyst: Electronically Enhanced Site Selectivity Enables Synthetic Opportunities. J. Am. Chem. Soc. 2017, 139, 2825–2832. 10.1021/jacs.6b13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Obligacion J. V.; Chirik P. J.; Hall M. B. Cobalt Pincer Complexes in Catalytic C–H Borylation: The Pincer Ligand Flips Rather Than Dearomatizes. ACS Catal. 2018, 8, 10606–10618. 10.1021/acscatal.8b03146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst T. P.; Obligacion J. V.; Rochette É.; Pappas I.; Chirik P. J. Cobalt-Catalyzed Borylation of Fluorinated Arenes: Thermodynamic Control of C(sp2)–H Oxidative Addition Results in ortho-to-Fluorine Selectivity. J. Am. Chem. Soc. 2019, 141, 15378–15389. 10.1021/jacs.9b07984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léonard N. G.; Bezdek M. J.; Chirik P. J. Cobalt-Catalyzed C(sp2)–H Borylation with an Air-Stable, Readily Prepared Terpyridine Cobalt(II) Bis(Acetate) Precatalyst. Organometallics 2017, 36, 142–150. 10.1021/acs.organomet.6b00630. [DOI] [Google Scholar]
- Pitzer L.; Schäfers F.; Glorius F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem., Int. Ed. 2019, 58, 8572–8576. 10.1002/anie.201901935. [DOI] [PubMed] [Google Scholar]