

Abstract
Background
Epigenetic alterations may contribute to early detection of cancer. We evaluated the association of blood DNA methylation with lymphatic–hematopoietic cancers and, for comparison, with solid cancers. We also evaluated the predictive ability of DNA methylation for lymphatic–hematopoietic cancers.
Methods
Blood DNA methylation was measured using the Illumina Infinium methylationEPIC array in 2324 Strong Heart Study participants (41.4% men, mean age 56 years). 788,368 CpG sites were available for differential DNA methylation analysis for lymphatic–hematopoietic, solid and overall cancers using elastic-net and Cox regression models. We conducted replication in an independent population: the Framingham Heart Study. We also analyzed differential variability and conducted bioinformatic analyses to assess for potential biological mechanisms.
Results
Over a follow-up of up to 28 years (mean 15), we identified 41 lymphatic–hematopoietic and 394 solid cancer cases. A total of 126 CpGs for lymphatic–hematopoietic cancers, 396 for solid cancers, and 414 for overall cancers were selected as predictors by the elastic-net model. For lymphatic–hematopoietic cancers, the predictive ability (C index) increased from 0.58 to 0.87 when adding these 126 CpGs to the risk factor model in the discovery set. The association was replicated with hazard ratios in the same direction in 28 CpGs in the Framingham Heart Study. When considering the association of variability, rather than mean differences, we found 432 differentially variable regions for lymphatic–hematopoietic cancers.
Conclusions
This study suggests that differential methylation and differential variability in blood DNA methylation are associated with lymphatic–hematopoietic cancer risk. DNA methylation data may contribute to early detection of lymphatic–hematopoietic cancers.
Keywords: Lymphatic cancers, Hematopoietic cancers, DNA methylation, Epigenetics, American Indians
Introduction
Epigenetic modifications—heritable and reversible changes in the genome without changes in the DNA sequence—are involved in tumorigenesis, potentially enabling early cancer detection. Modifications in DNA methylation, the most established epigenetic measure, occur in early stages of tumor development [1] and have been associated with cancer-related biological processes including oxidative stress [2] and apoptosis [3]. Many types of human cancers show hypermethylation of regulatory regions of certain tumor-suppressor genes [4]. DNA methylation-based biomarkers have been a target for early detection of cancer [5] due to their early and frequent emergence in tumors, their high quality measurement by well-established methods, their stability over time, their presence in different body fluids, and their cell type specificity [6]. However, only two DNA methylation-based tests have received FDA approval to date, both of them for colorectal cancer screening protocols [6].
Lymphatic and hematopoietic cancers affect the blood, bone marrow, lymph, and lymphatic system tissues. They are classified as myeloid (affecting mainly blood, including leukemia) and lymphoid (affecting mainly lymph nodes) neoplasms [7]. In 2019, they were expected to account for 10% of new cancer cases diagnosed in the United States [8].
For most cancers, early detection using DNA methylation is limited by the need for biopsy and access to the target tissue. For lymphatic and hematopoietic neoplasms, blood is a much more readily available biospecimen, providing a ready opportunity to identify markers that can detect cancer in early stages of development. Global DNA hypomethylation has been associated with better clinical outcomes in acute lymphoblastic leukemia [9] and acute myeloid leukemia [10, 11], and has also been used to conduct genetic characterization for stratification of acute myeloid leukemia risk groups [12]. In addition, site-specific differential blood DNA methylation in humans has been identified in several epigenome-wide association studies for multiple myeloma [13], B-cell lymphoma [14] and chronic lymphocytic leukemia [15], and in vitro for T-acute lymphoblastic leukemia [16]. Those studies, however, compared prevalent cases to controls and lacked follow-up, which is critical both for prediction and association purposes. In addition, the number of samples or the number of CpGs included in prior studies was small.
Because blood represents the relevant target tissue for lymphatic–hematopoietic tumors, we hypothesized that DNA methylation changes in blood may have a better ability to predict these compared to solid tumors. The objective of this study was to investigate the association of blood DNA methylation with lymphatic–hematopoietic and non-lymphatic–hematopoietic (solid) tumors in the Strong Heart Study (SHS), a prospective cohort study that has followed adult men and women since 1989–1991. In addition to estimating Differentially Methylated Positions (DMPs) and Differentially Methylated Regions (DMRs), we also tested for Differentially Variable Positions (DVPs) and regions (DVRs), which are underexplored but increasingly recognized as predictors of field defects (tissue transformations that predate tumor development). We assessed replication in an independent population: the Framingham Heart Study (FHS), a prospective cohort study of adults of European ancestry in Framingham, MA followed for health outcomes for decades [17].
Methods
Main study population: the Strong Heart Study
The SHS is a prospective cohort study funded by the National Heart, Lung and Blood Institute to investigate cardiovascular diseases and risk factors in American Indian adults [18]. In 1989–1991, 4549 men and women aged 45–75 years members of 13 tribes from Arizona, Oklahoma, and North and South Dakota agreed to participate. To analyze blood DNA methylation, we had a series of exclusion criteria that were not related to the cancer outcome (Fig. 1): (1) Due to tribal request, samples from one of the tribes were not selected for DNA methylation analyses, leaving 4091 participants. (2) As we needed to use metal data to answer other research questions, participants without sufficient urine for metal determinations were excluded, leaving 3515 participants. (3) Cardiovascular disease was a primary aim for the methylation data, so participants who were free of cardiovascular disease and were not missing other variables of interest at baseline (1989–1991) were eligible for blood DNA methylation analyses (N = 2730). (4) Sufficient genomic DNA was available for DNA methylation analyses in 2350 participants. (5) After laboratory analyses, data from individuals without classical bimodal distribution in DNA methylation levels and from individuals with low median intensity levels were removed, leaving a total of 2324 participants for this study. These participants were similar by sociodemographic and anthropometric characteristics to the eligible participants (Table 1).
Fig. 1.
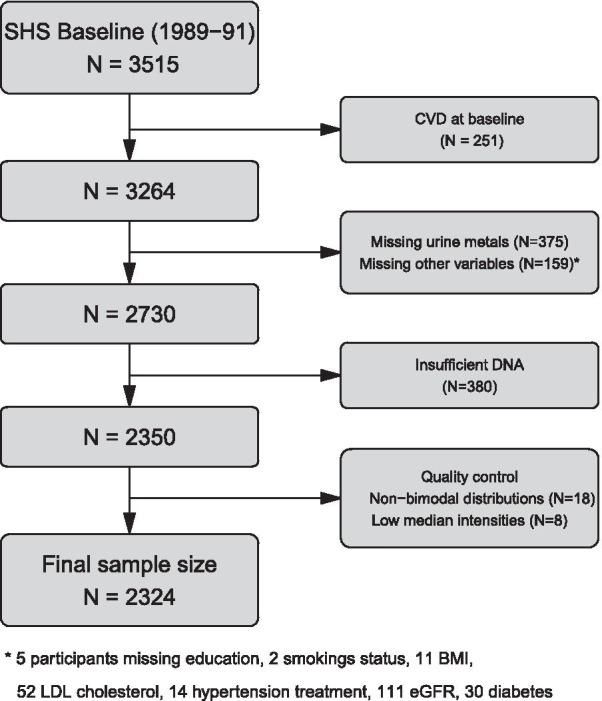
Flowchart of the included participants from the Strong Heart Study
Table 1.
Descriptive characteristics for eligible participants versus finally selected participants
| Included (N = 2324) | Eligible (N = 2730) | |
|---|---|---|
| Age (years), median (IQR) | 55.0 (49.2, 62.0) | 54.9 (49.2, 62.0) |
| Sex (% male) | 41.4 | 40.7 |
| Smoking status | ||
| % Current | 38.4 | 37.7 |
| % Former | 32.2 | 33.0 |
| BMI, median (IQR) | 29.6 (26.3, 33.6) | 29.7 (26.3, 33.7) |
| Education | ||
| % High | 58.6 | 59.4 |
| % Medium | 23.9 | 23.5 |
| Alcohol consumption | ||
| % Current | 43.1 | 42.9 |
| % Former | 42.2 | 42.2 |
IQR interquartile range
Participant characteristics
Trained and certified personnel collected information on sociodemographic factors, medical history, smoking status and alcohol consumption in a personal interview. Participants having smoked < 100 cigarettes in their lifetime were considered never smokers. Participants having smoked ≥ 100 cigarettes in their lifetime and smoking at the time of the interview were considered current smokers. Participants having smoked ≥ 100 cigarettes in their lifetime but currently not smoking were classified as former smokers. Current alcohol consumption was defined as any alcohol use within the past year. Former alcohol consumption was defined as no use of any alcohol during the last year but previous use of > 12 drinks of alcohol. The physical exam included anthropometric measures (height and weight), and collected fasting blood and spot urine samples.
Cancer incidence follow-up
The SHS used tribal records, death certificates, medical records, and direct annual contact with participants and their families to assess health outcomes and vital status over time. Cancer incidence was assessed by interviews, death certificates and/or chart reviews. For these analyses, we evaluated total cancer incidence, lymphatic and hematopoietic cancer incidence (codes 200–208), and non-lymphatic and hematopoietic cancer incidence (all cancer codes minus codes 200–208, for simplicity called solid cancers). Participants with any prior history of cancer before baseline were excluded (136 for solid and 1 for lymphatic–hematopoietic cancers). We calculated follow-up from the date of baseline examination to the date of the cancer diagnosis or 31 December 2017, whichever occurred first.
Microarray DNA methylation measurements
Details of microarray DNA methylation measurements at the baseline visit of the SHS (1989–1991) have been published elsewhere [19]. Briefly, buffy coats from fasting blood samples were collected in 1989–1991 and stored at − 70 °C. DNA from white blood cells was extracted and stored at the Penn Medical Laboratory, MedStar Health Research Institute under a strict quality-control system. In 2015, blood DNA was shipped with dry ice to the analytical laboratory at the Texas Biomedical Research Institute for DNA methylation analysis. DNA was bisulfite-converted with the EZ DNAm kit (Zymo Research) according to the manufacturer’s instructions. Bisulfite converted DNA methylation from white blood cells was measured using the Illumina MethylationEPIC BeadChip (850 K). Individuals with low detection p-values, cross-hybridizing probes, probes located in sex chromosomes and SNPs (Single Nucleotide Polymorphisms) with minor allele frequency > 0.05 were excluded. Single sample noob normalization and regression on correlated probes normalization were conducted following Illumina’s recommendations for preprocessing [20]. Blood cell proportions (CD8T, CD4T, NK cells, B cells, monocytes and neutrophils) were estimated using the R package FlowSorted.Blood.EPIC. The preprocessing resulted in data from 2324 individuals and 788,368 CpG sites in our analyses.
Replication population: the Framingham Heart Study
The FHS is a community-based study [17]. In this study, participants from the FHS Offspring cohort, participants who attended exam cycle 8 (2005–2008, N = 2202) and Third Generation cohort participants who attended exam cycle 2 (2008–2011, N = 1455) were eligible. The study protocol was approved by the Institutional Review Board at Boston University Medical Center (Boston, MA).
Cancer was defined as the occurrence of any type of malignant tumor excluding non-melanoma skin neoplasms. Diagnoses were confirmed from pathology and laboratory reports and clinical notes. Age-specific incidence rates were compared with Connecticut Surveillance, Epidemiology, and End Results (SEER) data [21]. Participants with any prior history of cancer before the blood draw for DNA methylation measurements were excluded. Participants were followed from the time of blood collection to the time of cancer incidence (N = 376), which extended to December 31, 2016. These included hematological cancers (N = 28) and other (solid tumor) cancers (N = 348). Body Mass Index (BMI) was calculated as weight (kg) divided by height squared (m2). Current smoking (yes/no) was defined as smoking on average at least one cigarette per day during the past 12 months. Smoking pack-years was computed by multiplying the average number of cigarettes smoked per day by the number of years smoked, divided by 20. Cell type fractions of CD4T, CD8T, NK cells, monocytes and eosinophils were estimated from DNA methylation data using the Houseman method [22].
DNA samples were extracted from whole blood buffy coat samples using the Gentra Puregene DNA extraction kit (Qiagen, Venlo, Netherland) and subsequently underwent bisulfite conversion using the EZ DNA methylation kit (Zymo Research, Irvine, CA). DNA methylation levels were measured using the Illumina Infinium Human Methylation450 BeadChip (450 K). FHS Offspring cohort samples were run in two laboratory batches (batch #1 and #2). The Third Generation samples were run in batch #3. For each separate lab batch, DNA methylation beta values from Illumina GenomeStudio were further normalized using the DASEN methodology implemented in the wateRmelon R package. We used surrogate variable analyses to eliminate unwanted variation in the DNA methylation data. Beta values were regressed on batch-specific surrogate variables, and the DNA methylation residual was taken forward. The three lab batches were merged for analyses. For sample quality control, we excluded samples with a missing DNA methylation value (detection p > 0.01) for > 1% CpGs, poor matching of SNPs between the 65 SNPs on the Illumina 450 K array and the GWAS array, or outliers at the multi-dimensional scaling plot. For quality control at the CpG level, we excluded CpGs with methylation values missing (detection p value > 0.01) for > 20% of samples, as well as CpGs previously identified to map to multiple locations on the sex chromosomes, or to have an underlying SNP (minor allele frequency > 5% in European ancestry in the 1000 Genomes Project data) at the CpG site or within 10 bp of the single base extension. A total of 415,318 CpGs were retained for analyses.
Statistical methods
Differentially Methylated Positions (DMPs)
Standard Cox Proportional Hazard Regression models are limited in accounting for large numbers of predictors or correlated data. Thus, we used GLMnet penalized regression, a mix between Ridge and Lasso regression in an elastic-net framework [23] which tests all CpG sites simultaneously. This approach has shown to be successful for high-dimensional methylation data [24] as well as genome-wide association studies of SNPs [25, 26]. The elastic-net penalty is controlled by the α parameter, where the default would be α = 1 (Lasso regression) and Ridge regression would be α = 0. Importantly, the Lasso penalty tends to select only one variable among the set of correlated variables, whereas the Ridge penalty offers more flexibility and could introduce more than one predictor from a correlated set in the models. We selected α = 0.05 based on the performance of the model after trying different values on the range between 0 and 1. This level of α, which is close to Ridge regression, has been a popular choice and has shown to work well for methylation data. The regularization path is computed for the selected penalty at a set of values as specified by the regularization parameter λ, which was selected using 10-folds cross-validation in our study. This model is thus also useful for avoiding genomic inflation, which is a concern in all Epigenome-Wide and Genome-Wide Association Studies. DNA methylation proportions at a given CpG (beta values) were used as predictors with age as time scale and individual entry times (age at baseline) treated as staggered entries for lymphatic–hematopoietic, solid and overall cancers. Models were adjusted for biologically relevant variables (sex, smoking status (never, former, current), BMI, blood cell counts (CD8T, CD4T, NK cells, monocytes and B cells), study region (Arizona, Oklahoma, North Dakota and South Dakota) and five genetic PCs [27]. Predictive ability was evaluated using Harrell’s concordance or C index. For replication, we ran elastic-net in the SHS restricting the CpGs to those present in 450 K (as no data from the EPIC array were available in the FHS) and we fitted an elastic-net model in the FHS population introducing the CpGs that the model selected in the SHS.
Since statistical inference based on the coefficients from the elastic-net model is unreliable given the shrinkage of the coefficients, we ran Cox proportional hazards models comparing the 90th versus the 10th percentile of DNA methylation with the CpGs selected by the elastic-net in order to report hazard ratios (HRs).
For comparison with approaches commonly used in the literature, we ran Cox proportional hazard models comparing the 90th versus the 10th percentile of DNA methylation epigenome-wide (i.e. including all CpG sites) for lymphatic–hematopoietic, solid and all cancers.
Protein–protein interaction network
We created lists of unique protein-coding genes from the CpGs selected by elastic-net for lymphatic–hematopoietic and solid tumors, respectively. We constructed a protein interaction network using the STRING database v11.0 [28], which provides a confidence score (from 0 to 1) to indicate the estimated likelihood that the annotated interaction between a given pair of proteins is biologically meaningful, specific and reproducible, according to the evidence derived from in-house predictions, homology transfers and externally maintained databases. We displayed a protein interaction network with Cytoscape v. 3.8.0 [29] using the yfiles Organic layout. In the resultant network, we only kept connections obtained from experimental studies, publicly available databases and text mining with a minimum confidence score of 0.3. Nodes that had no connections were excluded.
Differentially Methylated Regions (DMRs)
Testing differential methylation at the regional level might have several advantages as compared to the single position approach. DMRs can remove spatial redundancy by reducing the dimensionality of the often spatially correlated methylation levels and might offer increased robustness [30]. In addition, some studies have argued that DMRs might be more biologically relevant than DMPs [31, 32]. We used the R package DMRcate, which computes a kernel estimate against a null comparison to identify Differentially Methylated Regions, and ranks the DMRs by Stouffer p value [33]. DMRs were calculated based on the combination of the Cox regression results for individual CpGs. CpGs were annotated to the closest gene based on hg19 notation.
Differentially Variable Positions (DVPs) and Regions (DVRs)
We used the R package missMethyl for the DVP analysis between cases and non-cases (no survival method is available to date). The function varFit calculates a measure of variability (absolute deviation) for each CpG site and then fits a linear model to the deviations. Empirical Bayes shrinkage is applied to the residuals of the linear model to obtain robust moderated t statistics [34]. Multiple comparisons were accounted for using the Benjamini and Hochberg method to control for the false discovery rate (FDR) [35]. We report Log Var Ratios, which are defined as the natural log of the ratio of the absolute deviations of cancers versus non-cancers. A Log Var Ratio of log(2) would mean that the variance of one group is twice that of the second group. For the regional analysis, we used the DMRcate package.
Sensitivity analyses
We further adjusted the cancer models for a family history of cancer in first-degree relatives and for alcohol consumption (never, former, current) to see if the predictive ability changed. Additionally, we excluded all cases diagnosed in the first 5 years of follow-up (before 1995) to evaluate if DNA methylation could predict better cases in the near future. We analyzed lymphatic cancers (lymphomas) and hematopoietic cancers (myelomas and leukemias) separately to see if we could observe differences. Last, among the CpG sites that were selected by the elastic-net model, we repeated the Cox models adjusting for epigenetic aging instead of chronological age, using three different epigenetic aging biomarkers: the Hannum clock [36], the Horvath clock [37] and the PhenoAge [38]. The aim was to explore if some of the methylation changes might be reflecting aging.
Results
Descriptive analysis
Participants with incident cancer were older and more likely to be current smokers than non-cases (Table 2). Participants with incident lymphatic–hematopoietic cancers had higher BMI at baseline than solid cancers and non-cases. During follow up there were 420 new-onset cancer cases including 41 lymphatic–hematopoietic tumor cases. The mean follow-up time among participants who did not develop cancer was 26.8 years. The mean time from blood samples collection to cancer diagnosis was 14.7 years for lymphatic–hematopoietic cancers and 15.1 years for solid cancers and overall cancer. Solid cancers included 85 lung cancers, 49 breast cancers, 44 colorectal cancers, 24 kidney cancers, 23 pancreatic cancers, 22 stomach-esophagus cancers, 21 liver cancers, 15 ovarian cancers, 15 gallbladder cancers, 4 endometrial cancers, 2 thyroid cancers, and 214 other solid neoplasms (one individual might have several types of cancers).
Table 2.
Participants’ characteristics by cancer status
| Lymphatic–hematopoietic cancer (N = 41) | Solid cancers (N = 394) | Overall cancer (N = 420) | No cancer (N = 1904) | |
|---|---|---|---|---|
| Age (years), median (IQR) | 53.2 (49.8, 59.9) | 56.4 (50.5, 64.0) | 56.2 (50.4, 63.7) | 54.7 (49.0, 61.7) |
| Sex, % male | 36.6 | 46.2 | 46.0 | 40.5 |
| Smoking status, % | ||||
| Former | 22.0 | 30.2 | 22.6 | 30.9 |
| Current | 46.3 | 47.2 | 46.4 | 36.7 |
| BMI, median (IQR) | 31.5 (26.9, 36.5) | 29.0 (25.5, 33.8) | 29.2 (25.7, 33.9) | 29.7 (26.3, 33.5) |
Medians (IQR) or percentages are shown for continuous or categorical variables, respectively
IQR interquartile range
Differentially Methylated Positions
The elastic-net model for lymphatic–hematopoietic cancer selected 126 CpG sites as relevant. Among them, 10 were annotated to the gene FAM65B. The C index comparing the model that only included risk factors (age, sex, smoking status, BMI, blood cell counts, study region and five genetic PCs) to the model that further included DNA methylation increased from 0.5 to 0.87 (Table 3). The results from the Cox proportional hazards model for the selected CpGs by elastic-net are shown in Table S1 (Additional file 1). When considering each CpG separately, 12,342 DMPs were epigenome-wide significant at FDR < 0.05. The genomic inflation factor was 1.41 (41% of false positives, data not shown).
Table 3.
Predictive ability of DNA methylation for lymphatic–hematopoietic, solid and overall cancers in the Strong Heart Study from the elastic-net model
| N predictors | Lymphatic–hematopoietic cancer | Solid cancers | Overall cancer | ||||
|---|---|---|---|---|---|---|---|
| C index | N coef > 0b | C index | N coef > 0b | C index | N coef > 0b | ||
| Risk factorsa | 5 | 0.50 | 0 | 0.66 | 5 | 0.65 | 5 |
| Risk factorsa + cell counts + genetic PCs | 15 | 0.50 | 0 | 0.67 | 15 | 0.66 | 14 |
| Risk factorsa + cell counts + genetic PCs + DMPs | 788,383 | 0.87 | 126 | 0.79 | 396 | 0.79 | 414 |
coef coefficient, PCs principal components, DMPs Differentially Methylated Positions
aAge (years), smoking status (current/former/never), sex (men/women), BMI (kg/m2) and study center (AZ, OK, ND/SD)
bVariables with coef 0 are considered not to play any role in prediction
For solid cancers, the elastic-net model selected 396 CpG sites including one CpG annotated to the oncogene LMO2 and seven CpGs annotated to smoking-related genes (AHRR, F2RL3, PRSS23 and GFI1). All the CpGs annotated to smoking-related genes were inversely associated with incident lung cancer in our population (data not shown), meaning that hypomethylation in those genes would increase lung cancer risk. The C index comparing the model that only included risk factors to the model that further included DNA methylation increased from 0.66 to 0.79 (Table 3). The results from the Cox proportional hazards model for those CpGs are shown in Table S2 (Additional file 1). No DMPs were found by the traditional epigenome-wide association study (EWAS) approach at 0.05 FDR significance level.
For overall cancer, the elastic-net model selected 414 CpG sites of which 250 were also selected for solid tumors and two for lymphatic–hematopoietic cancers. The C index increased from 0.66 to 0.79 after including DNA methylation in the model (Table 3). The results from the Cox proportional hazards model for those CpGs are shown in Table S3 (Additional file 1). No DMPs were found by the traditional epigenome-wide association study (EWAS) approach at 0.05 FDR significance level.
Replication
Replication results of DNA methylation and cancer in the FHS are shown in Table 4. For lymphatic–hematopoietic cancers, the C index for a model including only risk factors in the FHS (age, sex, BMI and smoking status) was 0.76, and it increased to 0.89 when further including CpG sites selected by the SHS model as well as cell counts (Table 4). For solid tumors, the C index for a model including only risk factors in the FHS was 0.69, and it increased to 0.75 when further including the CpGs selected by the SHS model and cell counts (Table 4). For overall cancers, the C index when only including risk factors in the FHS was 0.69, and it increased to 0.74 when further including the CpGs selected by the SHS model and cell counts (Table 4). The results from the Cox proportional hazards model for those CpGs for lymphatic–hematopoietic, solid and overall cancers are show in Additional file 1 (Tables S1, S2 and S3, respectively). 28 CpGs for lymphatic–hematopoietic, 54 for solid and 37 for overall cancers had HRs in the same direction as in the SHS.
Table 4.
Replication: predictive ability in the Framingham Heart Study (450 K) of the CpGs selected in the Strong Heart Study for lymphatic–hematopoietic, solid and overall cancers
| N predictors | Lymphatic–hematopoietic cancer | Solid cancers | Overall cancer | ||||
|---|---|---|---|---|---|---|---|
| C index | N coef > 0b | C index | N coef > 0b | C index | N coef > 0b | ||
| Risk factorsa | 5 | 0.76 | 5 | 0.69 | 4 | 0.69 | 5 |
| Risk factorsa + cell counts | 11 | 0.79 | 10 | 0.69 | 11 | 0.69 | 10 |
| Risk factorsa + cell counts + DMPs | 132/379/399c | 0.89 | 62 | 0.75 | 34 | 0.74 | 32 |
coef coefficient, PCs principal components, DMPs Differentially Methylated Positions
aAge (years), smoking status (current/former/never), sex (men/women) and BMI (kg/m2)
bVariables with coef 0 are considered not to play any role in prediction
cFor lymphatic–hematopoietic and overall cancers, among the 125 CpGs selected by elastic-net in the Strong Heart Study (restricting to CpGs included in 450 K), 123 were present in the Framingham Heart Study. In addition, the four risk factor variables and the five cell count variables were included in the elastic-net model: a total of 132 variables. For solid cancers, 373 CpGs were selected in the Strong Heart Study, 370 being present in the Framingham Heart Study. With the four risk factor variables and the five cell count variables, a total of 379 variables were included. For overall cancers, 395 CpGs were selected in the Strong Heart Study, 390 being present in the Framingham Heart Study. With the four risk factor variables and the five cell count variables, a total of 399 variables were included
Protein–protein interaction network
When restricting the SHS analyses to 450 K, 126 and 373 CpGs were selected for lymphatic–hematopoietic and solid tumors, respectively, which included 442 unique genes. Among those, 218 were ncRNA genes or non-connected nodes. Thus, a network with 224 nodes and 398 interactions was obtained (Fig. 2). From 57 lymphatic–hematopoietic nodes identified in the SHS, 26 were also identified in the FHS population, being GATA4, SOX1 and PPARGC1A the most connected (11, 9 and 9 interaction, respectively). For 162 solid cancer nodes identified in the SHS, 50 nodes were also identified in the FHS population, being MYC, NOTCH1 and SHH the most connected nodes in the network (> 20 connections). The remaining 5 nodes (PRDM16, GALNT9, PACRG, PDLIM1 and ZMIZ1) were reported in both lymphatic–hematopoietic and solid tumors. Details of the network are included in Additional file 2.
Fig. 2.

Protein–Protein interaction network of Differentially Methylated Positions in lymphatic–hematopoietic cancer. The circle nodes indicate Differentially Methylated Positions in the Strong Heart Study and the square nodes those replicated in the Framingham Heart Study. The red nodes indicate Differentially Methylated Positions for lymphatic–hematopoietic cancers, the blue nodes for solid cancers and the yellow nodes for all cancers. The size of the nodes is proportional to the number of connections. The edges indicate confidence scores for interactions from 0 to 1
Differentially Methylated Regions (DMRs)
We found 159 DMRs for lymphatic–hematopoietic cancers. The top 15 are shown in Table 5. No DMRs were found for overall or solid tumors. The number of CpGs included in the DMRs for lymphatic–hematopoietic cancers ranged from 4 to 41. The region 24910562: 24912385 (chromosome 6), annotated to the gene FAM65B, was the top DMR, including 20 CpGs. The top two DMR, reflecting 41 CpG sites, was annotated to the gene WT1. Figure 3 shows the tendency of the associations of the individual CpGs within this DMR; a bump of highly hypermethylated CpG sites followed by a flat area with no significant sites and another hypermethylation bump is observed.
Table 5.
Top 15 Differentially Methylated Regions for lymphatic–hematopoietic cancers
| Positions | Chr | Width | N CpGs | Stouffer p value | Gene | Function | Overlapping promoters |
|---|---|---|---|---|---|---|---|
| 24910562: 24912385 | chr6 | 1824 | 20 | 2.30E−28 | FAM65B | Inhibits the proliferation of human leukemic T cells | FAM65B-001 |
| 32447944: 32455735 | chr11 | 7792 | 41 | 2.28E−23 | WT1 | Oncogene in Acute Myeloid Leukemia | WT1-001, WT1-005, WT1-002, WT1-AS-001, WT1-AS-201, WT1-AS-005, WT1-009, WT1-004, WT1-AS-003, WT1-AS-004, WT1-AS-002, WT1-AS-006, WT1-006, WT1-003 |
| 31159558: 31160393 | chr16 | 836 | 6 | 5.77E−16 | PRSS36 | Serine protease (cleaves peptide bonds in proteins) | PRSS36 |
| 73565440: 73565966 | chr10 | 527 | 5 | 4.22E−09 | CDH23 | Encodes calcium dependent cell–cell adhesion glycoproteins. Might be associated with breast cancer | CDH23, SNORA71, SNORA17 |
| 159869223: 159870915 | chr1 | 1693 | 10 | 1.28E−08 | CFAP45 | Uncharacterized function | snoU13, Y_RNA, SCARNA16, SNORD112, SNORA63, U3, SNORA51, SNORA25, SNORD59, SCARNA20, SNORA67, U6, SNORA70, SNORA77, SNORA26, SNORA72, U8, SNORA31, SNORA40, CCDC19, hsa-mir-4259, ACA64, SNORD78, snoU109, SNORD60, SNORD116 |
| 52995053: 52995634 | chr12 | 582 | 7 | 1.56E−08 | KRT72 | Structural integrity of epithelial cells | RP11-641A6.2, KRT72, snoMe28S-Am2634 |
| 30476089: 30477270 | chr22 | 1182 | 15 | 3.62E−08 | HORMAD2 | Meiotic prophase quality control. Associated with lung cancer | HORMAD2, CTA-85E5.10 |
| 149112318: 149113196 | chr7 | 879 | 6 | 5.09E−08 | ZNF777 | Zinc finger protein. Nucleic acid binding | None |
| 42951711: 42952369 | chr5 | 659 | 4 | 1.21E−07 | LOC648987 | Uncharacterized function | SNORA27, SNORA68, RPS23P5, SNORA57, SNORA76, 7SK, SNORD45 |
| 157164556: 157165335 | chr1 | 780 | 5 | 2.26E−07 | ETV3 | Transcriptional repressor associated to dendritic cell tumor | snoU13, Y_RNA, SCARNA16, SNORD112, SNORA63, U3, SNORA51, SNORA25, SNORD59, SCARNA20, SNORA67, U6, SNORA70, SNORA77, SNORA26, SNORA72, U8, SNORA31, SNORA40, SNORD64, ACA64, SNORD78, snoU109, SNORD60, SNORD116 |
| 49726500: 49727110 | chr12 | 611 | 5 | 2.28E−07 | C1QL4 | Tumor necrosis factor | C1QL4, snoMe28S-Am2634 |
| 375248: 375830 | chr10 | 583 | 5 | 2.83E-07 | DIP2C | Mutations in breast and lung cancer (potential diagnosis target) | DIP2C |
| 157182707: 157187341 | chr2 | 4635 | 22 | 3.92E−07 | NR4A2 | Transcription factor. Potential therapeutic target for gastrointestinal cancer | 5S_rRNA, SNORA4, SNORD11, SNORD51, SNORA41, SCARNA6, SNORD39, SNORA75, ACA59, SNORA48, NR4A2, SNORA43, SNORA1, Vault |
| 76803270: 76803925 | chr10 | 656 | 5 | 6.10E−07 | DUPD1 | Dual specificity phosphatase | SNORA71, SNORA17, DUPD1 |
| 64253534: 64253818 | chr3 | 285 | 6 | 1.19E−06 | PRICKLE2 | Related to WNT signaling pathways | U7, SNORD77, SNORA33, SNORA81, SNORD66, SNORD2, SNORD5, SNORD38, SNORD63, PRICKLE2, Metazoa_SRP, SNORA18 |
Model adjusted for age, smoking status (never, former, current), sex (male/female), BMI (kg/m2) Houseman cell proportions (CD8T, CD4T, NK, B cells and monocytes), five genetic PCs and study center (Arizona, Oklahoma or Dakota)
Fig. 3.

Differentially methylated region for lymphatic–hematopoietic cancer. Hazard ratios (95% confidence intervals) and genomic location of the top 2 differentially methylated region for lymphatic–hematopoietic cancers including 41 CpG sites. Orange bars represent overlapping promoters. Locations of CpGs of the differentially methylated region in the chromosome are represented by blue vertical bars above the overlapping promoters. The grey area in the plot represents the differentially methylated region
Differentially Variable Positions (DVPs) and Regions (DVRs)
At a 0.05 FDR significance level, we found 12,967 DVPs for lymphatic–hematopoietic (Table 6 shows top 15), 7 for solid (Table 7), and 9 for all cancers (data not shown). There were five common DVPs for overall and solid tumors annotated to CCDC92, AQP12B, GFI1, XIRP2 and SPRY2 genes. Other DVPs associated to solid neoplasms (Table 7) were annotated to TBC1D12 and MTOR genes. The violin plots in Fig. 4 show the distribution of the methylation proportions for lymphatic–hematopoietic cancer cases versus non-cases for the top 4 DVPs. The Log Var Ratios of the top 15 DVPs for lymphatic–hematopoietic cancers range between 1.57 and 2.22, indicating the group variance is between 5 and 9 times higher (log(5) = 1.6, log(9) = 2.2) in lymphatic–hematopoietic cancer cases compared to non-cases (Table 6). 106 of the 152 CpGs selected by elastic-net were DVPs as well. We found 432 DVRs for lymphatic–hematopoietic cancers (Table 8 shows top 15); 78 were DMRs as well.
Table 6.
Top 15 Differentially Variable Positions for lymphatic–hematopoietic cancers
| CpG | Chr | Gene | Function | In 450 k | Log Var Ratio | p value | FDR |
|---|---|---|---|---|---|---|---|
| cg03098814 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 2.22 | 1.31E−22 | 1.03E−16 |
| cg11083276 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 1.97 | 3.61E−21 | 1.42E−15 |
| cg17090968 | chr12 | SLC38A1 | Sodium-dependent amino acid transporter. Mediates the saturable, pH-sensitive and electrogenic cotransport of glutamine and sodium ions | Yes | 1.67 | 1.08E−19 | 2.85E−14 |
| cg18761994 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 2.15 | 1.78E−19 | 3.52E−14 |
| cg17757602 | chr5 | Intergenic | Uncharacterized | Yes | 2.02 | 4.34E−19 | 6.84E−14 |
| cg11211942 | chr15 | Intergenic | Uncharacterized | No | 1.92 | 1.26E−17 | 1.66E−12 |
| cg19936032 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 2.04 | 2.34E−17 | 2.64E−12 |
| cg02915015 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 2.04 | 1.15E−16 | 1.13E−11 |
| cg14536812 | chr12 | Intergenic | Uncharacterized | 1.84 | 3.60E−16 | 3.15E−11 | |
| cg08576643 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 2.15 | 4.26E−16 | 3.36E−11 |
| cg17896599 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | No | 2.15 | 6.59E−16 | 4.73E−11 |
| cg01726103 | chr6 | FAM65B | Inhibits the proliferation of human leukemic T cells | Yes | 1.91 | 8.37E−16 | 5.50E−11 |
| cg24698979 | chr17 | ARHGAP23 | Increases p53 proto-oncogene’s transactivity | No | 1.57 | 1.09E−15 | 6.58E−11 |
| cg18368658 | chr15 | CHST14 | Regulates proliferation and neurogenesis of neural progenitor cells | No | 1.60 | 1.62E−15 | 9.12E−11 |
| cg14216285 | chr6 | LINC01623 | Uncharacterized | Yes | 1.79 | 2.20E−15 | 1.16E−10 |
Log Var Ratios: Natural log of the ratio of the absolute deviations of cancers versus non-cancers. A Log Var Ratio of log(2) would mean that the variance of one group is twice that of the second group
Model adjusted for age, smoking status (never, former, current), sex (male/female), BMI (kg/m2), Houseman cell proportions (CD8T, CD4T, NK, B cells and monocytes), five genetic PCs and study center (Arizona, Oklahoma or Dakota)
Table 7.
Differentially Variable Positions for solid cancers
| CpG | Chr | Gene | Function | In 450 k | Log Var Ratio | p value | FDR |
|---|---|---|---|---|---|---|---|
| cg21902846 | 12 | CCDC92 | DNA repair and reduction/oxidation reactions | No | 0.91 | 1.22E−11 | 9.65E−06 |
| cg23070169 | 2 | XIRP2 | Associated to Alzheimer’s disease and Down syndrome | No | − 0.64 | 5.90E−09 | 0.0022 |
| cg26292116 | 2 | AQP12B | Migration, invasion and proliferation of human breast tumor cells | No | − 0.33 | 8.44E−09 | 0.0022 |
| cg13911116 | 1 | MTOR | Its activation promotes tumor growth and metastasis, many MTOR inhibitors have been approved to treat human cancers | Yes | 0.28 | 1.05E−07 | 0.021 |
| cg21383151 | 10 | TBC1D12 | Mutations suggested to be related to bladder cancer | Yes | 0.41 | 1.55E−07 | 0.023 |
| cg08598861 | 13 | SPRY2 | Regulates metastatic potential and differentiation in several cancers | No | − 0.59 | 1.74E−07 | 0.023 |
| cg18146737 | 1 | GFI1 | Significant role in development of lung cancer and prostate cancer and tumor suppressor gene in colorectal cancer | Yes | 0.53 | 3.37E−07 | 0.038 |
Log Var Ratios: Natural log of the ratio of the absolute deviations of cancers versus non-cancers. A Log Var Ratio of log(2) would mean that the variance of one group is twice that of the second group
Models adjusted for age, smoking status (never, former, current), sex (male/female), BMI (kg/m2), cell proportions (CD8T, CD4T, NK, B cells and monocytes), five genetic PCs and study center (Arizona, Oklahoma or Dakota)
Fig. 4.

Violin plots for lymphatic–hematopoietic cancer. Distribution of the methylation proportions of lymphatic–hematopoietic cancers versus non-cases for the top four Differentially Variable Positions
Table 8.
Top 15 Differentially Variable Regions for lymphatic–hematopoietic cancers
| Positions | Chr | Width | N CpGs | Stouffer p value | Gene | Function | Overlapping promoters |
|---|---|---|---|---|---|---|---|
| 24910562: 24912896 | chr6 | 2335 | 21 | 1.55E−51 | FAM65B | Inhibits the proliferation of human leukemic T cells | FAM65B-001 |
| 32447944: 32456069 | chr11 | 8126 | 42 | 7.89E−33 | WT1 | Oncogene in Acute Myeloid Leukemia | WT1-001, WT1-005, WT1-002, WT1-AS-001, WT1-AS-201, WT1-AS-005, WT1-009, WT1-004, WT1-AS-003, WT1-AS-004, WT1-AS-002, WT1-AS-006, WT1-006, WT1-003 |
| 748992: 749620 | chr20 | 629 | 9 | 1.66E−23 | SLC52A3 | Predictive and prognostic biomarker in esophageal cancer | SLC52A3-001, SLC52A3-004, SLC52A3-003 |
| 36147197: 36150135 | chr20 | 2939 | 48 | 1.08E−17 | BLCAP | Regulates cell proliferation and coordinate apoptosis and cell cycle progression. Associated to bladder and cervical cancers | NNAT-001, NNAT-002, BLCAP-009, BLCAP-010, BLCAP-005 |
| 27141388: 27144595 | chr7 | 3208 | 28 | 2.55E−17 | HOXA2 | Aberrant expression associated to several cancers | HOXA2-001 |
| 16829666: 16830859 | chr19 | 1194 | 10 | 8.58E−12 | NWD1 | Suggested causal role in dysregulation of androgen receptor signaling during prostate cancer progression | NWD1-002, NWD1-001, NWD1-006 |
| 42950995: 42952369 | chr5 | 1375 | 5 | 6.85E−11 | LOC648987 | Uncharacterized function | - |
| 157164556:157165335 | chr1 | 780 | 5 | 1.72E−10 | ETV3 | Transcriptional repressor associated to dendritic cell tumor | - |
| 13120555: 13123217 | chr19 | 2663 | 7 | 3.50E−10 | NFIX | DNA-binding transcription factor activity | NFIX-009, NFIX-008 |
| 121624862: 121625735 | chr2 | 874 | 6 | 2.07E−09 | GLI2 | Zinc finger protein thought to play a role in embryogenesis | RP11-297J22.1-001 |
| 36665826: 36667782 | chr17 | 1957 | 10 | 2.23E−09 | ARHGAP23 | Increases p53 proto-oncogene’s transactivity | - |
| 46997630: 46999840 | chr19 | 2211 | 19 | 1.03E−08 | PPP5D1 | MAPK signaling pathway. Abnormal MAPK signaling may lead to uncontrolled cell proliferation and resistance to apoptosis | AC011484.1-201, PNMAL2-201, PNMAL2-004, PNMAL2-001 |
| 42431109: 42433041 | chr17 | 1933 | 8 | 1.68E−08 | FAM171A2 | Associated to ceroid lipofuscinosis | GRN-022 |
| 78526804: 78527410 | chr15 | 607 | 5 | 3.19E−08 | ACSBG1 | Associated to malignant ovarian surface epithelial-stromal neoplasm and ovary epithelial cancer | ACSBG1-001, ACSBG1-201, ACSBG1-012, ACSBG1-014, ACSBG1-002, ACSBG1-008, ACSBG1-003 |
| 17603531: 17604184 | chr17 | 654 | 6 | 3.55E−08 | RAI1 | Transcriptional regulator of the circadian clock components. Chromatin remodeling | - |
Models adjusted for age, smoking status (never, former, current), sex (male/female), BMI (kg/m2), Houseman cell proportions (CD8T, CD4T, NK, B cells and monocytes), five genetic PCs and study center (Arizona, Oklahoma or Dakota)
Sensitivity analyses
Adjustment for cancer family history or alcohol consumption made no changes in the C index of the predictive models. After excluding five cases of lymphatic–hematopoietic cancers diagnosed before 1995, the C index dropped from 0.85 to 0.75. The C index did not change when excluding 33 cases of solid cancers that were diagnosed before 1995. A model including 19 cases of lymphatic cancers had a C index of 0.83, with seven CpGs being selected. A model including 20 cases of hematopoietic cancers had a C index of 0.94, with 184 CpGs being selected (including the gene FAM65B selected several times). Adjustment for any of the three epigenetic aging biomarkers did not change the results as compared to the adjustment for chronological aging (data not shown).
Discussion
Differential methylation at a number of CpGs and regions was associated with the incidence of lymphatic–hematopoietic, solid, and overall cancers. The strongest epigenetic signals were apparent for lymphatic–hematopoietic cancers, and the increase in prediction ability was substantially higher for lymphatic–hematopoietic cancers compared to the other cancers. Of note, improvement in event prediction for lymphatic–hematopoietic cancer cases was due to cases occurring during early follow up and may reflect blood DNA methylation predicting subclinical disease. The improvement in predictive ability for lymphatic–hematopoietic cancers as well as the direction of association for several CpGs was replicated in the FHS, an independent population of white men and women from Framingham, MA. Whereas several signals showed to be robust across both populations, other CpGs were not replicated in the FHS and some of them had opposite directions of association. Given that DNA methylation is highly influenced by environmental and genetic factors, population-specific effects for methylation sites might exist [39]. Our results support stronger and more robust signals for hematopoietic than for lymphatic cancers. This might be related to the specificity of the blood tissue.
The issue of genomic inflation and the spatial redundancy among correlated CpGs may make DMRs a more appropriate and robust approach than DMPs calculated by individual models for each CpG [30]. DMR approaches, however, remain spatially defined and do not include non-contiguous CpG sets [40]. For this reason, studying all CpG sites together in the same model might be more appropriate than studying them separately. When introducing all the CpG sites in the elastic-net model for lymphatic–hematopoietic cancers, only 126 were selected, in contrast to the 12,342 sites identified in the traditional EWAS DMP modeling. One possible reason for this large drop in the number of CpGs is the reduction in redundancy among correlated methylation across multiple CpGs, either due to spatial correlation or to methylation-level interactions on disease risk.
Our results are consistent with those from a case–control study in a population from three different cities in the US [15] that studied genome-wide DNA methylation changes in chronic lymphocytic leukemia. They found cancer-related hypermethylation in HOX gene clusters. Two of our DVRs and a DMR for lymphatic–hematopoietic cancers were annotated to genes HOXA2 and HOXA-AS3 and overlapped with promoters of the HOX family, whose aberrant expression levels have been related to several cancers [41–45]. The second top both DMR and DVR in our study (including 41 CpG sites) was annotated to WT1, an oncogene in acute myeloid leukemia. Another top DMR was annotated to PRICKLE2. WT1 and PRICKLE2 genes are part of the WNT signaling pathway. Hypermethylation in genes related to WNT signaling pathway was also found in the aforementioned case–control study [15]. Moreover, mutations in WT1 have been recurrently identified in acute myeloid leukemia and associated with poor prognosis and chemotherapy resistance [46, 47]. The DMRs annotated to HOXA2 and WT1 in our study were hypermethylated, consistently with the case–control study [15].
Despite limitations in methods for prospective analyses, DVPs have previously been shown to be valuable for early cancer detection [30]. Differential variability detected field defects (tissue transformations that may predate cancer) in breast [48] and cervical [49] cancers. In our study, differential variability was associated with lymphatic–hematopoietic cancer with an extremely large number of DVPs identified. In addition, 96 of the 126 CpGs selected by the elastic-net models for lymphatic–hematopoietic cancers were also DVPs, reflecting the importance of variability in methylation for the occurrence of these tumors. An example of the aforementioned spatial redundancy can be seen in our DVP results (Table 6), where most of the top CpGs are annotated to FAM65B. These DVPs are encompassed into a single DVR in chromosome 6 annotated to FAM65B in Table 8. The gene FAM65B is repeatedly showing as differentially methylated and differentially variable in our study; furthermore, seven of the selected CpGs by the elastic-net model were annotated to this gene, suggesting its importance for lymphatic–hematopoietic cancers. FAM65B’s function is to control the proliferation of transformed and primary T cells [50]. In transformed T lymphocytes, forced expression of FAM65B blocks their mitosis, leading to G2 cell cycle arrest and apoptosis. In a public database including 75,000 individuals with methylation and cancer data [51], the CpG sites from chromosome 6 annotated to gene FAM65B had more variability in acute myeloid leukemia cases than in controls, which is consistent with our results. Research is needed to understand the potential role of this gene in lymphatic–hematopoietic cancers. Other genes to which DVRs were annotated were also related to the lymphatic or hematopoietic systems such as the gene ETV3, associated to dendritic cell tumor, which develops from cells of the immune system, typically beginning in the lymph system [52].
Differential variability might also be relevant for solid cancers. We found a DVP annotated to MTOR, which regulates cell growth, survival, metabolism and immunity. Activation of MTOR promotes tumor growth and metastasis, and many MTOR inhibitors have been developed to treat cancer [53]. Some of them have already been approved and are being used with modest success, while others are still being evaluated in clinical trials [54]. Other DVPs for solid cancers were annotated to genes related to bladder (TBC1D12), breast (AQP12B) or lung, prostate and colorectal (GFI1) cancers. GFI1 has been identified as a potential therapeutic target for interfering with inflammation-induced colorectal cancer progression and spread [55]. Of note, several CpGs annotated to smoking-associated genes were identified as predictive of solid cancers in both the SHS and the FHS (AHRR and F2RL3) or only in the SHS (PRSS23 and GFI1). These genes were individually associated with lung cancer in the SHS and might be predictive of other specific solid smoking-related cancers as well.
In addition, the protein interaction network showed highly connected nodes in both populations that have previously been related to cancer. For instance, the hub nodes MYC, NOTCH1 and SHH have been associated to different types of cancer [56]. The GATA4 gene encodes a member of a zinc-finger transcription factors family and alterations in gene expression in this gene have been associated with cancer [57]. Methylation in PPARGC1A gene was reported to predict cancer incidence [58]. The common nodes for solid and lymphatic–hematopoietic cancers have also been previously associated to cancer, for instance PRDM16 was related to acute myeloblastic leukemia [59]. Those highly connected nodes could be key factors for lymphatic–hematopoietic cancers development. Additional experimental research is needed to confirm the biological relevance of the findings.
This study has several limitations. First, we only have 41 cases of lymphatic–hematopoietic cancers, and we might lack power to detect signals for lymphatic and hematopoietic cancers separately. Second, we might not have been able to capture all risk factors associated with some of these tumors (e.g., data on Epstein–Barr virus infection, a risk factor for Hodgkin lymphoma). Also, the C index measure has shown to be problematic in some settings. Training a new model different to that of the discovery set might overestimate C index in replication sets [60]. At the same time, using the model trained on the discovery set on the replication set might lead to underestimation of the C index due to differences in biological factors between cohorts [60]. The development of more appropriate predictive accuracy methods for replication sets needs further investigation. Non-fatal cancer data in the SHS might be incomplete, as no linkage between the SHS cancer data and cancer registry data has been conducted to date. However, the lymphatic–hematopoietic cancer diagnosis is very specific and it is unlikely that the reported cases are incorrectly classified. On the other hand, this study has several strengths which include having comprehensive methylation in one of the largest microarrays available nowadays (Infinium methylationEPIC), the high quality of the study protocols, the availability of data to account for potential confounders, the innovative statistical methods and the replication in an independent population with a large sample size. Moreover, this is the first prospective study evaluating DNA methylation in lymphatic–hematopoietic cancers (including almost 30 years of follow-up).
Conclusions
In conclusion, this study supports that differential methylation and differential variability in methylation are associated with lymphatic–hematopoietic cancers. Blood DNA methylation data could improve early detection of cancer beyond known risk factors. The identified DNA methylation markers may not only constitute a precision medicine tool for the early identification of blood cancers in adults, but may also help elucidate mechanisms that can inform prevention and treatment.
Supplementary Information
Additional file 1. Hazard ratios (95% CIs) for lymphatic-hematopoietic, solid and overall cancers in the Strong Heart Study and the Framingham Heart Study.
Additional file 2. Network nodes and network edges for the protein-protein interaction network.
Acknowledgements
We thank the dedication of the SHS and the FHS participants, investigators and staff, without whom this work would not have been possible.
Abbreviations
- CpG
Cytosine–guanine dinucleotide
- SHS
Strong Heart Study
- FHS
Framingham Heart Study
- DMP
Differentially methylated position
- DMR
Differentially methylated region
- DVP
Differentially variable position
- DVR
Differentially variable region
- SNPs
Single nucleotide polymorphism
- HRs
Hazard ratio
Authors’ contributions
ADR conducted data analysis, planning and writing of the article. TH and ALRC conducted data analysis. KH and SC obtained and processed the blood DNA methylation data in the Strong Heart Study. DL, MDF, MBT, DAR, EGE and MTP helped with the planning of the study and provided feedback, suggestions and careful review. YZ was responsible for the development of the database of the Strong Heart Study and the study protocol for cancer outcomes assessment. MH was responsible for the data analysis platform and resources. ANA was responsible for the design and planning of the study and conducted writing and review. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (NHLBI) (Contract Numbers 75N92019D00027, 75N92019D00028, 75N92019D00029 and 75N92019D00030) and previous Grants (R01HL090863, R01HL109315, R01HL109301, R01HL109284, R01HL109282, and R01HL109319 and Cooperative Agreements: U01HL41642, U01HL41652, U01HL41654, U01HL65520 and U01HL65521); by the National Institute of Environmental Health Sciences (Grant Numbers R01ES021367, R01ES025216, P42ES010349, P30ES009089); by the Chilean CONICYT/FONDECYT-POSTDOCTORADO Nº3180486 and by a fellowship from “la Caixa” Foundation (ID 100010434) (fellowship code “LCF/BQ/DR19/11740016”). The funders had no role in the planning, conducting, analysis, interpretation, or writing of this study. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (United States) or the National Health Institute Carlos III (Spain).
Availability of data and materials
The data underlying this article cannot be shared publicly in an unrestricted manner due to limitations in the consent forms and in the agreements between the Strong Heart Study tribal communities and the Strong Heart Study investigators. The data can be shared to external investigators following the procedures established by the Strong Heart Study, available at https://strongheartstudy.org/. All analyses were conducted in R version 3.6.2 and all packages used are freely available in the CRAN repository.
Ethics approval and consent to participate
This study was approved by Institution Review Boards of the academic organizations, tribal communities, and the Indian Health Service for the Strong Heart Study and by the Institutional Review Board at Boston University Medical Center (Boston, MA) for the Framingham Heart Study.
Consent for publication
Informed consent from participants was obtained for both the Strong Heart Study and the Framingham Heart Study.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Arce Domingo-Relloso, Email: ad3531@cumc.columbia.edu.
Ana Navas-Acien, Email: an2737@cumc.columbia.edu.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13148-021-01030-8.
References
- 1.Paska AV, Hudler P. Aberrant methylation patterns in cancer: a clinical view. Biochem Med. 2015;25(2):161–176. doi: 10.11613/BM.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barciszewska AM, Giel-Pietraszuk M, Perrigue PM, Naskręt-Barciszewska M. Total DNA methylation changes reflect random oxidative DNA damage in gliomas. Cells. 2019;8(9):1065. doi: 10.3390/cells8091065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gopisetty G, Ramachandran K, Singal R. DNA methylation and apoptosis. Mol Immunol. 2006;43:1729–1740. doi: 10.1016/j.molimm.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 5.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3(4):253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 6.Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15(7):459–466. doi: 10.1038/s41571-018-0004-4. [DOI] [PubMed] [Google Scholar]
- 7.Swerdlow SH, International Agency for Research on Cancer., World Health Organization . WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2008. [Google Scholar]
- 8.Leukemia and Lymphoma Society. Facts and statistics. https://www.lls.org/facts-and-statistics/facts-and-statistics-overview/facts-and-statistics. Accessed 8 Jan 2021.
- 9.Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, Garate L, et al. Promoter hypermethylation and global hypomethylation are independent epigenetic events in lymphoid leukemogenesis with opposing effects on clinical outcome. Leukemia. 2006;20:1445–1448. doi: 10.1038/sj.leu.2404257. [DOI] [PubMed] [Google Scholar]
- 10.Zhang LY, Yuan YQ, Zhou DM, Wang ZY, Ju SG, Sun Y, et al. Impact of global and gene-specific DNA methylation in de novo or relapsed acute myeloid leukemia patients treated with decitabine. Asian Pac J Cancer Prev. 2016;17(1):431–437. doi: 10.7314/APJCP.2016.17.1.431. [DOI] [PubMed] [Google Scholar]
- 11.Deneberg S, Grövdal M, Karimi M, Jansson M, Nahi H, Corbacioglu A, et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia. 2010;24(5):932–941. doi: 10.1038/leu.2010.41. [DOI] [PubMed] [Google Scholar]
- 12.Yang X, Wong MPM, Ng RK. Aberrant DNA methylation in acute myeloid leukemia and its clinical implications. Int J Mol Sci. 2019;20:4576. doi: 10.3390/ijms20184576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salhia B, Baker A, Ahmann G, Auclair D, Fonseca R, Carpten JD. DNA methylation analysis determines the high frequency of genic hypomethylation and low frequency of hypermethylation events in plasma cell tumors. Cancer Res. 2010;70(17):6934–6944. doi: 10.1158/0008-5472.CAN-10-0282. [DOI] [PubMed] [Google Scholar]
- 14.Shaknovich R, Geng H, Johnson NA, Tsikitas L, Cerchietti L, Greally JM, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood. 2010;116(20):e81–e89. doi: 10.1182/blood-2010-05-285320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei L, Choi J-H, Liu J, Lee E-J, McCarthy B, Wilson JM, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7(6):567–578. doi: 10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tejedor JR, Bueno C, Cobo I, Bayón GF, Prieto C, Mangas C, et al. Epigenome-wide analysis reveals specific DNA hypermethylation of T cells during human hematopoietic differentiation. Epigenomics. 2018;10(7):903–923. doi: 10.2217/epi-2017-0163. [DOI] [PubMed] [Google Scholar]
- 17.Dawber TR, Meadors GF, Moore FE. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health. 1951;41(3):279–281. doi: 10.2105/AJPH.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee ET, Welty TK, Fabsitz R, Cowan LD, Le NA, Oopik AJ, et al. The Strong Heart Study. A study of cardiovascular disease in American Indians: design and methods. Am J Epidemiol. 1990;132(6):1141–1155. doi: 10.1093/oxfordjournals.aje.a115757. [DOI] [PubMed] [Google Scholar]
- 19.Domingo-Relloso A, Riffo-Campos AL, Haack K, Rentero-Garrido P, Ladd-Acosta C, Fallin DM, et al. Cadmium, smoking, and human blood DNA methylation profiles in adults from the strong heart study. Environ Health Perspect. 2020;128(6):067005. doi: 10.1289/EHP6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fortin J-P, Triche TJ, Hansen KD, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33(4):558–560. doi: 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreger BE, Splansky GL, Schatzkin A. The cancer experience In the Framingham Heart Study cohort. Cancer. 1991;67(1):1–6. doi: 10.1002/1097-0142(19910101)67:1<1::AID-CNCR2820670102>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 22.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13(1):86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman J, Hastie T, Tibshirani R, Narasimhan B, Simon N, Qian J, et al. Package “glmnet”. CRAN repository. 2020.
- 24.Benton MC, Sutherland HG, Macartney-Coxson D, Haupt LM, Lea RA, Griffiths LR. Methylome-wide association study of whole blood DNA in the Norfolk Island isolate identifies robust loci associated with age. Aging (Albany NY) 2017;9(3):753–768. doi: 10.18632/aging.101187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abraham G, Kowalczyk A, Zobel J, Inouye M. SparSNP: fast and memory-efficient analysis of all SNPs for phenotype prediction. BMC Bioinform. 2012;13(1):88. doi: 10.1186/1471-2105-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waldmann P, Mészáros G, Gredler B, Fuerst C, Sölkner J. Evaluation of the lasso and the elastic net in genome-wide association studies. Front Genet. 2013 doi: 10.3389/fgene.2013.00270/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol. 2014;38(3):231. doi: 10.1002/gepi.21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teschendorff AE, Relton CL. Statistical and integrative system-level analysis of DNA methylation data. Nat Rev Genet. 2017;19(3):129–147. doi: 10.1038/nrg.2017.86. [DOI] [PubMed] [Google Scholar]
- 31.Schlosberg CE, VanderKraats ND, Edwards JR. Modeling complex patterns of differential DNA methylation that associate with gene expression changes. Nucleic Acids Res. 2017;45(9):5100–5111. doi: 10.1093/nar/gkx078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.VanderKraats ND, Hiken JF, Decker KF, Edwards JR. Discovering high-resolution patterns of differential DNA methylation that correlate with gene expression changes. Nucleic Acids Res. 2013;41(14):6816–6827. doi: 10.1093/nar/gkt482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heard NA. Choosing between methods of combining p-values. Biometrika. 2017;105(1):239–246. doi: 10.1093/biomet/asx076. [DOI] [Google Scholar]
- 34.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 35.Chen S-Y, Feng Z, Yi X. A general introduction to adjustment for multiple comparisons. J Thorac Dis. 2017;9(6):1725–1729. doi: 10.21037/jtd.2017.05.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda SV, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 2018;10(4):573–591. doi: 10.18632/aging.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husquin LT, Rotival M, Fagny M, Quach H, Zidane N, McEwen LM, et al. Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation 06 biological sciences 0604 genetics. Genome Biol. 2018;19(1):1–17. doi: 10.1186/s13059-018-1601-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tj P, Mj B. The DMRcate package user’s guide. 2019. https://www.bioconductor.org/packages/release/bioc/vignettes/DMRcate/inst/doc/DMRcate.pdf.
- 41.Makiyama K, Hamada JI, Takada M, Murakawa K, Takahashi Y, Tada M, et al. Aberrant expression of HOX genes in human invasive breast carcinoma. Oncol Rep. 2005;13(4):673–679. [PubMed] [Google Scholar]
- 42.Mustafa M, Lee JY, Kim MH. CTCF negatively regulates HOXA10 expression in breast cancer cells. Biochem Biophys Res Commun. 2015;467(4):828–834. doi: 10.1016/j.bbrc.2015.10.058. [DOI] [PubMed] [Google Scholar]
- 43.Hur H, Lee JY, Yun HJ, Park BW, Kim MH. Analysis of HOX gene expression patterns in human breast cancer. Mol Biotechnol. 2014;56(1):64–71. doi: 10.1007/s12033-013-9682-4. [DOI] [PubMed] [Google Scholar]
- 44.Carrera M, Bitu CC, de Oliveira CE, Cervigne NK, Graner E, Manninen A, et al. HOXA10 controls proliferation, migration and invasion in oral squamous cell carcinoma. Int J Clin Exp Pathol. 2015;8(4):3613–3623. [PMC free article] [PubMed] [Google Scholar]
- 45.Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med. 2014;92:811–823. doi: 10.1007/s00109-014-1181-y. [DOI] [PubMed] [Google Scholar]
- 46.Pandey S, Moazam M, Eisermann K, Hord J, Fraizer G, Kuerbitz SJ. The importance of WT1 in leukemia. Blood. 2011;118(21):4645–4645. doi: 10.1182/blood.V118.21.4645.4645. [DOI] [Google Scholar]
- 47.Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, Miwa H, et al. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood. 1994;84(9):3071–3079. doi: 10.1182/blood.V84.9.3071.3071. [DOI] [PubMed] [Google Scholar]
- 48.Teschendorff AE, Gao Y, Jones A, Ruebner M, Beckmann MW, Wachter DL, et al. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun. 2016;7(1):10478. doi: 10.1038/ncomms10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, et al. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012;4(3):24. doi: 10.1186/gm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Froehlich J, Versapuech M, Megrelis L, Largeteau Q, Meunier S, Tanchot C, et al. FAM65B controls the proliferation of transformed and primary T cells. Oncotarget. 2016;7(39):63215–63225. doi: 10.18632/oncotarget.11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiong Z, Li M, Yang F, Ma Y, Sang J, Li R, et al. EWAS Data Hub: a resource of DNA methylation array data and metadata. Nucleic Acids Res. 2019;48(D1):D890–D895. doi: 10.1093/nar/gkz840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davick JJ, Kim J, Wick MR, Gru AA. Indeterminate dendritic cell tumor: a report of two new cases lacking the ETV3-NCOA2 translocation and a literature review. Am J Dermatopathol. 2018;40(10):736–748. doi: 10.1097/DAD.0000000000001191. [DOI] [PubMed] [Google Scholar]
- 53.Zhou H, Luo Y, Huang S. Updates of mTOR Inhibitors. Anticancer Agents Med Chem. 2012;10(7):571–581. doi: 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12:71. doi: 10.1186/s13045-019-0754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xing W, Xiao Y, Lu X, Zhu H, He X, Huang W, et al. GFI1 downregulation promotes inflammation-linked metastasis of colorectal cancer. Cell Death Differ. 2017;24(5):929–943. doi: 10.1038/cdd.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolós V, Grego-Bessa J, De La Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- 57.Lentjes MHFM, Niessen HEC, Akiyama Y, Bruïne DAP, Melotte V, Engeland MVAN. The emerging role of GATA transcription factors in development and disease. Expert Rev Mol Med. 2016;18:e3. [DOI] [PMC free article] [PubMed]
- 58.Kresovich JK, Joyce BT, Gao T, Zheng Y, Zhang Z, Achenbach CJ, et al. Promoter methylation of PGC1A and PGC1B predicts cancer incidence in a veteran cohort. Epigenomics. 2018;10(6):733–743. doi: 10.2217/epi-2017-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corrigan DJ, Luchsinger LL, De Almeida MJ, Williams LJ, Strikoudis A, Snoeck HW. PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J Clin Investig. 2018;128(8):3250–3264. doi: 10.1172/JCI99862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hattab MW, Clark SL, van den Oord EJCG. Overestimation of the classification accuracy of a biomarker for assessing heavy alcohol use. Mol Psychiatry. 2018;23:2114–2115. doi: 10.1038/mp.2017.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Hazard ratios (95% CIs) for lymphatic-hematopoietic, solid and overall cancers in the Strong Heart Study and the Framingham Heart Study.
Additional file 2. Network nodes and network edges for the protein-protein interaction network.
Data Availability Statement
The data underlying this article cannot be shared publicly in an unrestricted manner due to limitations in the consent forms and in the agreements between the Strong Heart Study tribal communities and the Strong Heart Study investigators. The data can be shared to external investigators following the procedures established by the Strong Heart Study, available at https://strongheartstudy.org/. All analyses were conducted in R version 3.6.2 and all packages used are freely available in the CRAN repository.