

Abstract
Protein poly-ADP-ribosylation (PARylation) is a heterogeneous and dynamic posttranslational modification regulated by various writers, readers, and erasers. It participates in a variety of biological events and is involved in many human diseases. Currently, tools and technologies have yet to be developed for unambiguously defining readers and erasers of individual PARylated proteins or cognate PARylated proteins for known readers and erasers. Here, we report the generation of a bifunctional nicotinamide adenine dinucleotide (NAD+) characterized by diazirine-modified adenine and clickable ribose. By serving as an excellent substrate for poly-ADP-ribose polymerase 1 (PARP1)-catalyzed PARylation, the generated bifunctional NAD+ enables photocrosslinking and enrichment of PARylation-dependent interacting proteins for proteomic identification. This bifunctional NAD+ provides an important tool for mapping cellular interaction networks centered on protein PARylation, which are essential for elucidating the roles of PARylation-based signals or activities in physiological and pathophysiological processes.
Graphical Abstract
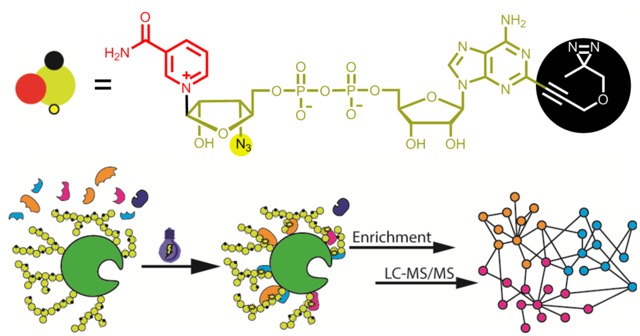
Introduction
Poly-ADP-ribose polymerases (PARPs) catalyze protein poly-ADP-ribosylation (PARylation) or mono-ADP-ribosylation (MARylation) with nicotinamide adenine dinucleotide (NAD+) as a cosubstrate. PARylated proteins can be specifically recognized by reader proteins, triggering downstream signaling cascades or effector functions1–6. The PARylation-dependent signals or activities can be modulated by eraser proteins that can remove covalently attached poly-ADP-ribose (PAR)5, 7–11. Protein PARylation plays vital roles in regulating genome stability, protein homeostasis, cell proliferation, differentiation and apoptosis12–16 and is strongly implicated in various human diseases17–22.
NAD+ with adenine modified by alkyne, biotin, and fluorescent groups have previously been generated for labeling and tracking cellular ADP-ribosylation23–29. Paired with PARP mutants, NAD+ analogues with alkyne-modified adenine facilitate identification of substrate proteins of PARPs30–32. Using agents that are known to bind to PAR, PARylated proteins and their associated protein complexes could be captured for proteomic identification33–36. However, such PAR-related non-covalent protein complexes could include candidate proteins that interact with PARylated proteins indirectly or in a PAR-independent manner. To date, no unambiguous approaches and technologies exist to identify reader(s) and eraser(s) for an individual PARylated protein or cognate PARylated protein(s) for a known reader or eraser protein, or to map PARylation-dependent interaction networks.
We recently generated NAD+ analogues functionalized at the ribose moiety with robust substrate activities for protein PARylation37. Here, we designed and synthesized novel NAD+ analogues that feature an azido group at ribosyl 3′-OH and a photocrosslinker at remote adenine moiety. The resulting bifunctional NAD+ with C2-diazirine not only displays excellent activity for protein PARylation but also enables photocrosslinking of reader and eraser proteins. Using the generated bifunctional NAD+, PARylation-dependent interacting proteins were identified for auto-PARylated PARP1 through proteomic analysis, which provides new knowledge for understanding the roles of protein PARylation in signal transduction and effector functions.
RESULTS AND DISCUSSION
Design, synthesis, and substrate activity of bifunctional NAD+.
Previous studies have shown chemical modifications at adenine N6 and C2 positions of NAD+ are well tolerated by PARPs23, 24, 27, 30, 31 and ribose-functionalized NAD+ with nicotinamide riboside (NR) 3′-azido displays strong activities for protein PARylation37. Thus, we envisioned that covalent attachment of a diazirine photocrosslinker to adenine N6 or C2 coupled with a 3′-azido NR could generate bifunctional NAD+ analogues with significant substrate activities. It is expected that by serving as excellent substrates for protein PARylation, the bifunctional NAD+ with diazirine-modified adenines and clickable riboses may enable photocrosslinking of PARylation-dependent interacting proteins, enrichments of covalently linked protein complexes, and subsequent proteomic identifications of protein “nodes” for PARylation-dependent interactome at both global and molecular levels (Figure 1).
Figure 1.
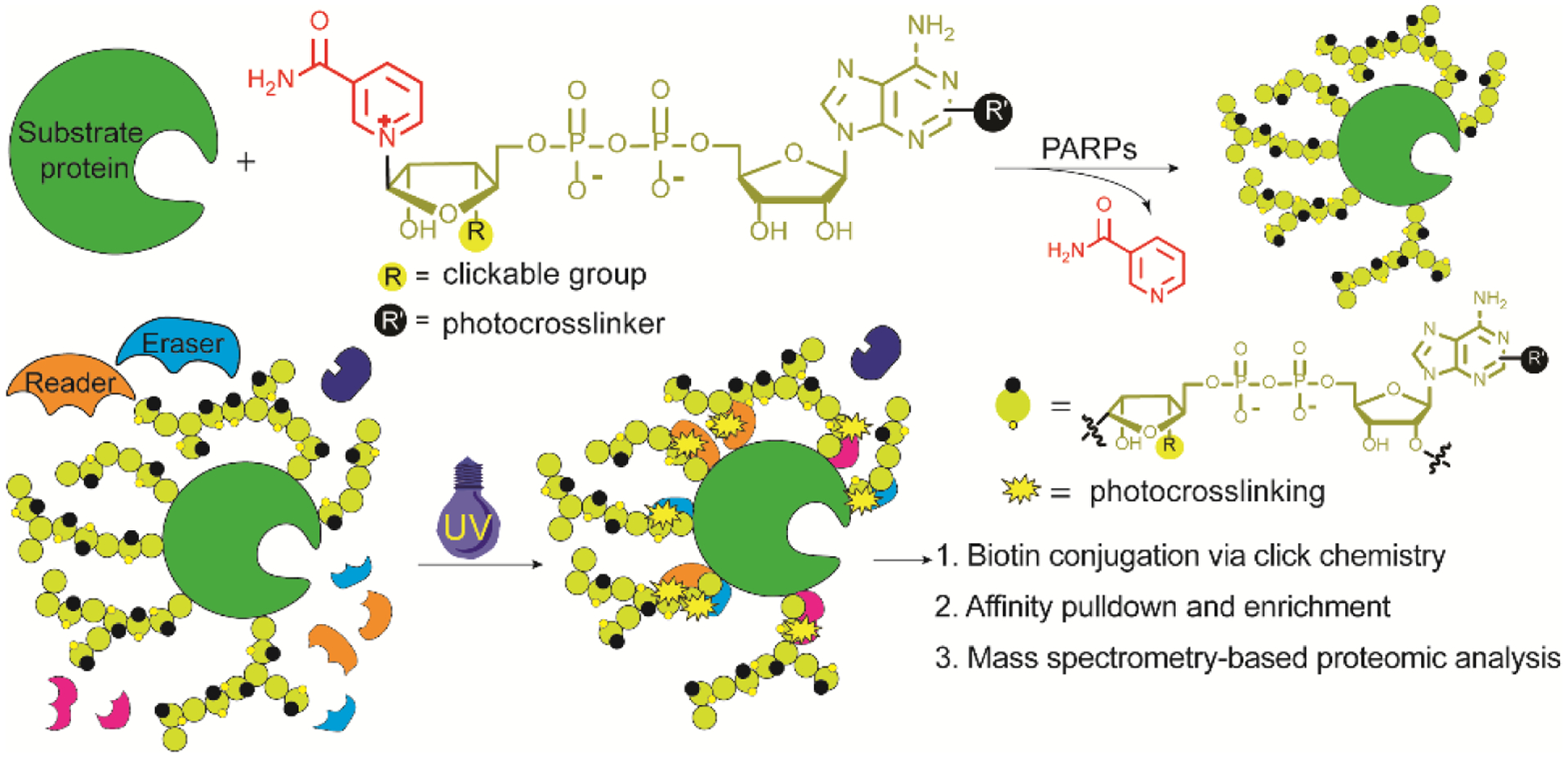
Profiling PARylation-dependent interacting proteins by a bifunctional NAD+.
To this end, we synthesized three dually modified NAD+ with NR 3′-azido and N6- or C2-diazirine with linkers varying in length (Figure 2A and Schemes S1 and S2). To evaluate their substrate activities, full-length human PARP1 was expressed and purified from Escherichia coli (Figure S1). The activities of dually modified NAD+ were examined through PARP1 auto-PARylation (Figure 2). Immunoblot analysis indicated that the activity of 3 is higher than those of 1 and 2 and comparable to that of 3′-N3-NAD+ which has shown similar activity to NAD+ for protein PARylation previously37. Treatment with olaparib (100 μM), a PARP inhibitor, resulted in suppression of PARP1 auto-PARylation by 1–3 and 3′-N3-NAD+. Of note, the purified PARP1 is highly sensitive to proteolysis, resulting in clevaged fragments during auto-PARylation reactions as observed in the PARP1 loading controls of immunoblots throughout this work. HPLC-based activity assays were then performed to determine kinetic parameters of NAD+, 3′-N3-NAD+, and 3 for PARP1-catalyzed auto-PARylation (PARP activity) and hydrolysis (NADase activity) (Table 1). The kcat and Km of 3 for PARP1 automodification are 25.8 ± 2.7 min−1 and 407.7 ± 83.2 μM, respectively, at comparable levels to those of NAD+ and 3′-N3-NAD+. These results support 3 as an excellent substrate for PARP1-catalyzed PARylation.
Figure 2.
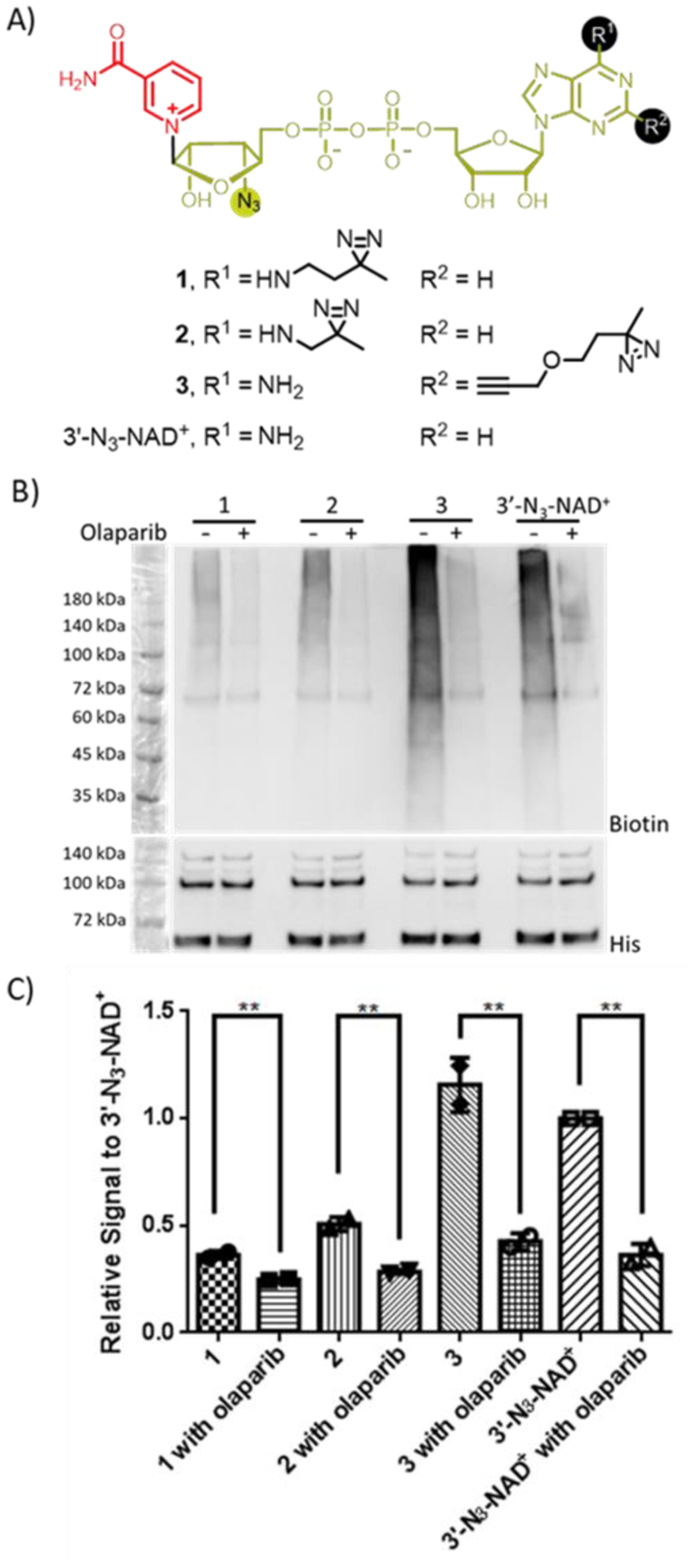
Substrate activities of NAD+ analogues for human PARP1. (A) Chemical structures of NAD+ analogues. (B) Immunoblot analysis of auto-PARylation of PARP1 with NAD+ analogues using a streptavidin-HRP conjugate following biotin conjugation via click chemistry. Auto-PARylation reactions were performed in the absence or presence of the PARP inhibitor olaparib. Lower panel: PARP1 loading controls detected using an anti-His6 antibody. The observed bands between 60 and 72 kDa were proteolyzed PARP1 formed during auto-modification reactions. (C) Relative densitometric analysis of PARylation levels. Error bars represent standard deviation of two replicates.
Table 1.
Kinetic parameters of NAD+ and its analogues with human PARP1.
| Substrate | kcat (min−1) | Km (μM) | kcat/Km (min−1 M−1) | |
|---|---|---|---|---|
| PARP activity | NAD+ | 15.4 ± 1.2 | 162.1 ± 33.9 | 9.5 × 104 |
| 3′-N3-NAD+ | 17.7 ± 3.6 | 557.7 ± 193.7 | 3.2 × 104 | |
| 3 | 25.8 ± 2.7 | 407.7 ± 83.2 | 6.3 × 104 | |
| NADase activity | NAD+ | 3.4 ± 1.1 | 346.1 ± 223.9 | 9.8 × 103 |
| 3′-N3-NAD+ | < 0.2 | N/A | N/A | |
| 3 | < 0.017 | N/A | N/A |
Photocrosslinking capability of bifunctional NAD+.
Next, 3 was evaluated for its capability of photocrosslinking PARylation-dependent interacting proteins. An active, truncated human poly-ADP-ribose glycohydrolase (tPARG) was expressed and purified from E. coli (Figures S1 and S2)38. Although tPARG could not efficiently degrade automodified PARP1 by 3 (data not shown), ELISA confirmed its recognition of 3-modified PARP1 (Figure S3A). Immunoblotting against tPARG indicated that UV irradiation resulted in crosslinking of tPARG with 3-modified PARP1 as revealed by the smearing pattern due to PARylation (Figures 3A, S4A, and S5A), which is sensitive to the addition of NAD+-modified PARP1 to compete for tPARG binding.
Figure 3.
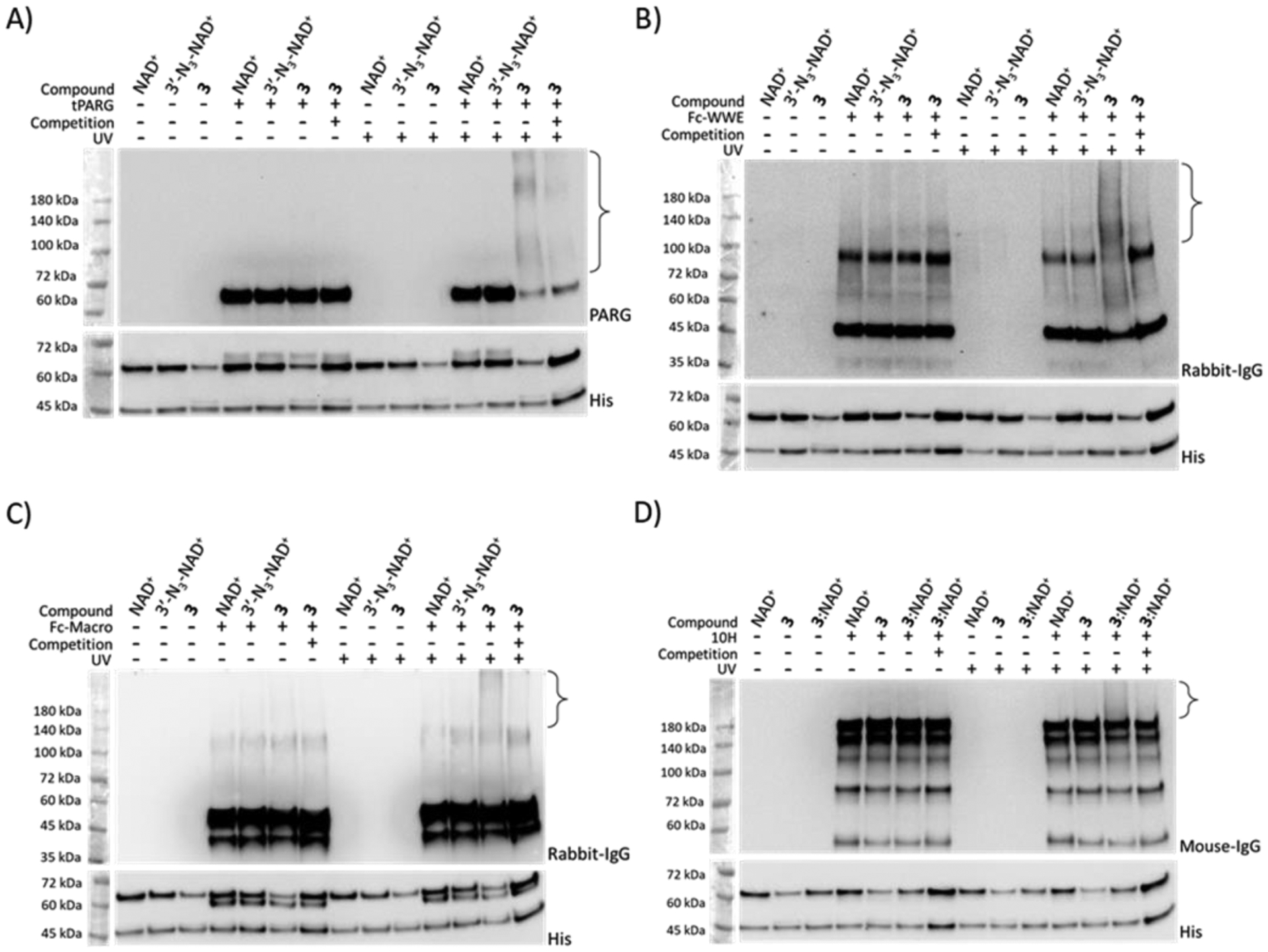
Photocrosslinking of automodified PARP1 with model interacting proteins. PARP1 PARylated by NAD+, 3′-N3-NAD+, 3, or a mixture of 3:NAD+ at a 1:1 molar ratio was incubated with (A) tPARG, (B) Fc-WWE, (C) Fc-Macro, and (D) antibody 10H in the absence or presence of 365-nm UV irradiation and automodified PARP1 by NAD+ for binding competition, followed by immunoblot analysis using an anti-PARG antibody for (A) tPARG, an anti-rabbit IgG antibody for (B) Fc-WWE and (C) Fc-Macro, and an anti-mouse IgG antibody for (D) antibody 10H. Lower panels: PARP1 loading controls detected using an anti-His6 antibody. The observed bands between 45 and 72 kDa were proteolyzed PARP1 formed during auto-modification reactions. Brackets indicate regions of crosslinked protein complexes. Full-sized anti-His6 immunoblots are shown in Figure S5.
Compound 3 was further examined for photocrosslinking two model readers with distinct structural preference, the WWE domain of human RNF146 and macrodomain of AF152139. As shown by ELISA, Fc fusions of WWE and macrodomain (designated as Fc-WWE and Fc-Macro) could specifically bind to NAD+-, 3′-N3-NAD+-, and 3-modified PARP1 (Figure S3B–C). Immunoblot analysis indicated that UV treatment led to crosslinking of WWE domain and macrodomain with 3-modified PARP1 in a competition-sensitive manner (Figures 3B–C, S4B–C, and S5B–C).
We also tested 3 for photocrosslinking an anti-PAR monoclonal antibody 10H. Based on ELISA, the antibody 10H showed weak binding to 3-modified PARP1 (Figure S3D). Immunoblot analysis revealed little crosslinking of antibody 10H with PARylated PARP1 by 3 (Figure 3D). To improve the photocrosslinking with antibody 10H, a mixture of 3:NAD+ (1:1) was used to generate auto-modified PARP1 with chimeric PAR polymers, which may facilitate capturing of reader and eraser proteins by mimicking native PARylation. Immunoblotting showed that UV irradiation induced significant crosslinking of antibody 10H with PARylated PARP1 by the mixture of 3:NAD+ (Figures 3D, S4D, and S5D). Taken together, these results indicate that 3 allows efficient photocrosslinking with PARylation-dependent interacting proteins.
Identification of PARylation-dependent interacting proteins.
To demonstrate its utility, 3 was applied to profile proteins potentially interacting with PARylated PARP1 in a cellular context. PARP1 automodified by the mixture of 3:NAD+ was incubated with HEK293T cell lysates in the absence or presence of NAD+-modified PARP1 for binding competition. Upon UV irradiation, photocrosslinked protein complexes were labeled with biotin for enrichment and proteomic identification. Comparative analysis was then performed for the lists of proteins identified without and with NAD+-modified PARP1. Protein hits that were present in the group without binding competition but disappeared in the presence of NAD+-modified PARP1 as a binding competitor were considered as potential PARylation-dependent interacting proteins. In total, 247 potential binding proteins were identified (Figure 4A and Table S1), of which several hits were known PARylation-dependent interacting proteins from previous studies, such as DNA-dependent protein kinase catalytic subunit (PRKDC) and mitotic checkpoint protein BUB3 (BUB3)4, 6, 40.
Figure 4.
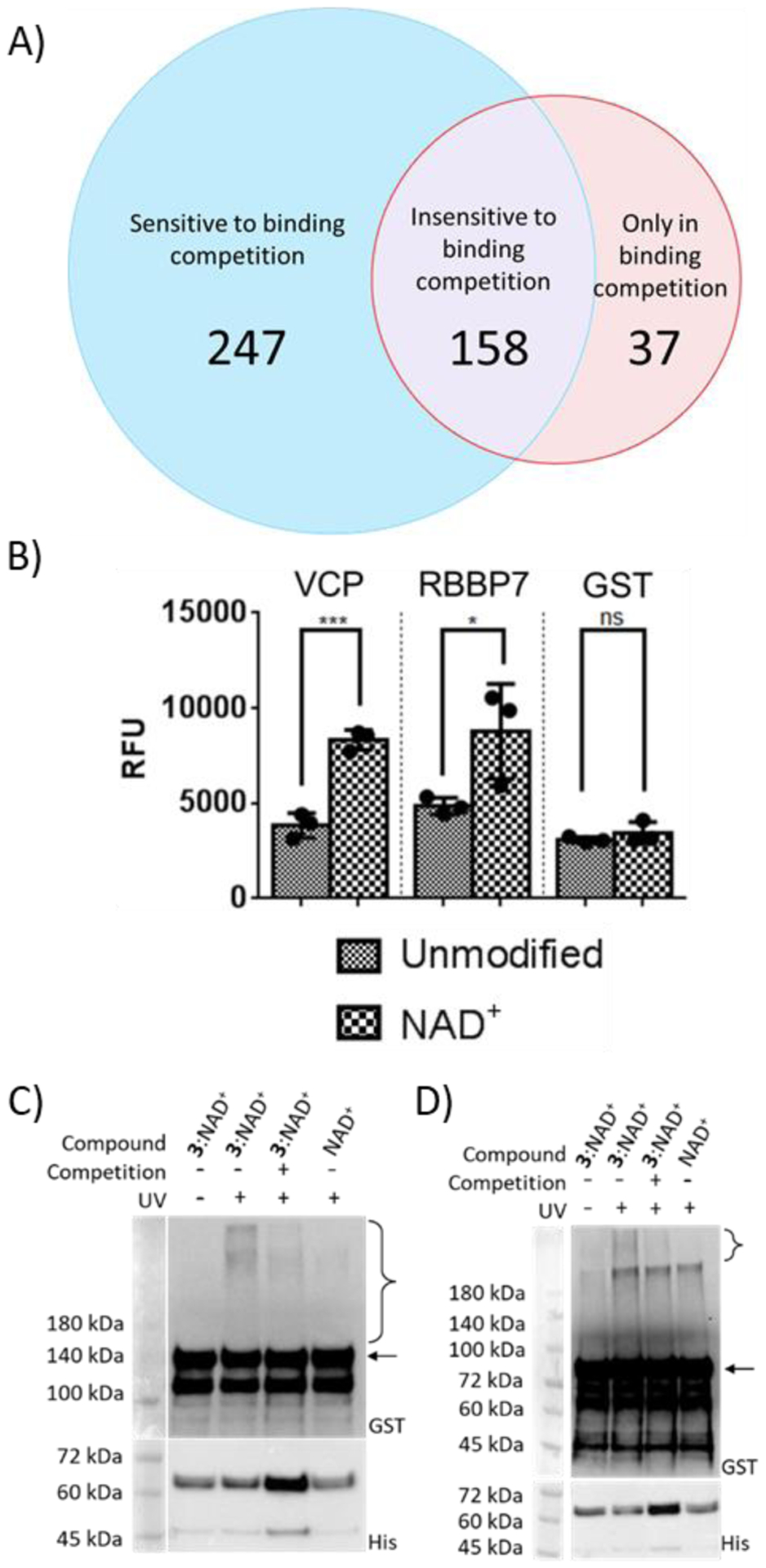
Verification of novel PARylation-dependent interacting proteins. (A) Venn diagram of protein hits identified from photocrosslinking in cell lysates. (B) Binding of GST-VCP and GST-RBBP7 for unmodified PARP1 and automodified PARP1 by NAD+. Unmodified PARP1 and NAD+-modified PARP1 were coated on ELISA plates for binding analysis with two identified protein hits. ns: not significant, *P < 0.05, *** P < 0.001 by one-tailed unpaired t-test. Error bars represent standard deviation of three replicates. RFU: relative fluorescence unit. (C) and (D) Photocrosslinking of automodified PARP1 with (C) VCP and (D) RBBP7. PARP1 PARylated by NAD+ or a mixture of 3:NAD+ (molar ratio 1:1) was incubated with GST-VCP or GST-RBBP7 without and with 365-nm UV and NAD+-modified PARP1 for binding competition, followed by immunoblot analysis using an anti-GST antibody. Expected sizes for VCP and RBBP7 are indicated by the arrow for (C) and (D) respectively. Lower panels: PARP1 loading controls detected using an anti-His6 antibody. The observed bands between 45 and 72 kDa were proteolyzed PARP1 formed during auto-modification reactions. Brackets indicate regions of crosslinked protein complexes.
Among other identified hits, two were chosen for validation, valosin-containing protein (VCP) and retinoblastoma binding protein 7 (RBBP7). VCP participates in a myriad of cellular functions and was found to localize at DNA damage sites and interact with different chromatin remodeling complexes41, 42. RBBP7 is involved with transcriptional repression by binding to histones as part of several histone deacetylase complexes43–45. Using GST-VCP and GST-RBBP7 fusions, ELISA-based binding assays indicated both proteins displayed tighter binding to PARylated PARP1 by NAD+ relative to unmodified PARP1 as well as significant binding to PAR (Figures 4B and S6). Furthermore, immunoblot analysis revealed that upon UV irradiation both VCP and RBBP7 could be crosslinked with PARP1 automodified by the mixture of 3:NAD+ in a competition-sensitive fashion (Figures 4C–D and S7). These results confirmed VCP and RBBP7 as novel proteins interacting with PARylated PARP1.
This work reports the synthesis and characterization of a novel bifunctional NAD+. Upon modifying the previously generated 3′-N3-NAD+ with a diazirine at the adenine C2 position, the resulting dually modified 3 exhibits surprisingly high activity for PARP1-catalyzed PARylation. As a robust substrate of PARP1, this NAD+ analogue thus enables simultaneous incorporation of photocrosslinker and clickable groups into PAR polymers for capturing and identifying PARylation-dependent interacting proteins. This study, for the first time, discovers a bifunctional NAD+ molecule with excellent substrate activity for protein PARylation. Notably, these findings suggest adequate tolerance of PARP1 active site to the modifications at NR 3′-OH and adenine C2 positions, possibly allowing for the creation of other types of dually modified NAD+ with distinct functions and properties. In addition to PARP1, the substrate activity of 1–3 need to be assessed for other PARP enzymes responsible for cellular protein PARylation and mono-ADP-ribosylation.
In comparison to native PAR polymers, the 3-derived PAR polymers are likely to display altered binding affinity and specificity for PARylation-dependent interacting proteins due to modifications at the adenine and ribose moieties of NAD+. Use of 3 for protein PARylation may thus cause low efficiency in photocrosslinking certain types of readers and erasers of PARylated proteins or lead to biased results. Chimeric PAR polymers with limited chemical modifications could be generated through using mixtures of NAD+ and 3, which, by mimicking native PARylation, may facilitate faithful and unbiased mapping of PARylation-dependent interaction networks.
It needs to be noted that in the proof-of-concept study to demonstrate the utility of 3, lysates of non-stressed cells were used for incubation with the automodified PARP1 by the mixture of 3:NAD+. The identified proteins potentially interacting with PARylated PARP1 may not truly reflect PARylation-based interactome present in stressed cells. For this purpose, future proteomic studies need to be performed in physiologically relevant conditions and quantitative analysis will provide insights into the binding affinity of identified proteins for PARylated PARP1. Using PARP1 auto-PARylated by the mixture of 3:NAD+, VCP and RBBP7 were identified and confirmed as novel proteins interacting with PARylated PARP1. The nucleotide binding domain of VCP and WD40 domain of RBBP7 may involve in binding to PARylated PARP1. Future studies are required to characterize their binding sites, binding specificity, contribution of PARP1 to PARylation-dependent interactions, and effects of the PARylation-dependent interactions on related biological functions.
In summary, 3 is a bifunctional NAD+ molecule with excellent substrate activity for protein PARylation and strong capability of photocrosslinking PARylation-dependent interacting proteins for proteomic identifications. This molecule serves as a valuable tool for discovering novel readers and erasers for individual PARylated proteins and dissecting cellular interaction networks centered on protein PARylation, which will advance the understanding of the roles of protein PARylation in physiology and pathophysiology.
METHODS
Synthetic procedures and characterization of NAD+ analogues.
The experimental details and results for synthesis of the NAD+ analogues 1–3 are provided in the Supporting Information.
Protein expression, purification, and enzymatic activity with NAD+ analogues.
The experimental details for expression and purification of human PARP1 and tPARG and characterization of their activities with NAD+ and its analogues 1–3 are described in the Supporting Information.
Preparation of automodified PARP1 for photocrosslinking and ELISA assays.
Automodification of PARP1 was performed at 30°C overnight using 3 μM purified PARP1 without NAD+, with 200 μM NAD+, or with NAD+ analogues in PARP reaction buffer (100 mM Tris-HCl pH 8.0, 10 mM MgCl2, 50 mM NaCl, 1mM DTT, and 100 ng μL−1 activated DNA). Reactions were stopped by the additions of 100 μM of olaparib. In the case of mixtures of 3:NAD+ (1:1), 100 μM of each was added.
Photocrosslinking with interacting proteins.
1.5 μg of automodified PARP1 was added to 12 μL final volume of PBS with 100 μM olaparib plus 150 ng Fc-WWE, 100 ng tPARG, 150 ng monoclonal anti-PAR antibody (clone: 10H, Santa Cruz Biotechnology: sc-56198), 150 ng Fc-Macro (Millipore: MABE1016), or PBS. Reactions were incubated at room temperature for 10 minutes before UV irradiation with 365-nm wavelength light with a handheld UV lamp placed 2 cm away from reactions in PCR strips for 15 minutes. Reactions with competition contained 1.5 μg of PARP1 automodified with NAD+ added at a final volume of 12 μL. Reactions were separated on PAGE gels for immunoblot detection Blocking was done for 1 hour using 5% non-fat milk (w/v) in PBS with 0.1% Tween-20 (v/v) (PBST). Fc-WWE and Fc-Macro were detected with 1:3000 goat anti-rabbit secondary antibody in PBST + 1% non-fat milk (w/v). Anti-PAR antibody 10H was detected using 1:3000 goat anti-mouse secondary antibody conjugated to HRP (ThermoFisher: G-21040) in PBST. tPARG was detected using 1:300 mouse anti-PARG antibody (Santa Cruz Biotechnology: sc-398563) in PBST followed by goat anti-mouse secondary antibody at 1:3000 in PBST. Images were analyzed and quantified using ImageJ. Regions above expected size of the binding protein were quantified and normalized to the lane with NAD+ and the respective binding proteins without UV irradiation.
For VCP (Abnova: H00007415-P02) photocrosslinking, 750 ng of PARP1 automodified with a mixture of 3:NAD+ or NAD+ was added to 200 ng of VCP in PBS with 100 μM olaparib in a final volume of 14 μL. Reactions with competition contained 3 μg of PARP1 automodified with NAD+ added to a final volume of 14 μL. Reactions were incubated, subjected to UV irradiation, and run on precast PAGE gels for immunoblot detection. Membranes were blocked with PBST + 5% non-fat milk (w/v) for 1 hour, followed by detection with primary antibody 1:2000 rabbit anti-GST antibody (Cell Signaling: 2625S) primary antibody in PBST for 1 hour. 1:3000 goat anti-rabbit secondary antibody in PBST + 1% (w/v) non-fat milk for 1 hour was used afterwards. Detection, analysis, and quantification were done as described above but with normalization to the lane with NAD+ and the binding proteins with UV irradiation. For RBBP7 (Abnova: H00005931-P01) photocrosslinking, the final volume was 12 μL and 160 ng of RBBP7 was used in place of VCP. For samples with competitions, 1.5 μg of PARP1 automodified with NAD+ was used in a final volume of 12 μL. The samples were otherwise treated the same as VCP photocrosslinking.
ELISA binding assays.
To examine Fc-WWE binding toward PARylated PARP1, 10 ng of PARP1 automodified with NAD+ or NAD+ analogues were diluted into 100 μL of PBS and plated into 96-well high-binding plates (Grenier Bio-One: 655077) and incubated overnight at 4°C. Following 3 washes using 400 μL of PBS with 0.05% Tween-20 (v/v) (PBS-0.05T), wells were blocked with 300 μL 3% BSA (w/v) in PBS-0.05T for 2 hours at room temperature. This was followed by another 3 washes with PBS-0.05T before adding 100 μL of Fc-WWE at 1:5000 in PBS-0.05T supplemented with 0.1% BSA (w/v) for 2 hours at room temperature. Anti-rabbit IgG antibody conjugated to HRP (System Biosciences) was used as the secondary antibody at a dilution factor of 1:3000 in PBS-0.05T with 0.1% BSA (w/v) for 1 hour at room temperature. Detection was done using QuantBlu fluorogenic peroxidase substrate (ThermoFisher: 15169) with an excitation wavelength of 325 nm and an emission wavelength of 420 nm. Results and graphs were generated using Graphpad Prism.
To analyze Fc-Macro binding for PARylated PARP1, 50 ng of automodified PARP1 was diluted into 100 μL and plated as before. In place of Fc-WWE at 1:5000, Fc-Macro was used at 1:3000. All other steps were done as for Fc-WWE.
To evaluate binding of anti-PAR antibody 10H for PARylated PARP1, 1 μg of automodified PARP1 was diluted into 100 μL and plated as before. In place of Fc-WWE, antibody 10H was used at 1:250. Anti-mouse secondary antibody was used at 1:3000 in PBS-0.05T with 0.1% BSA (w/v) for 1 hour at room temperature. Detection was done as described above.
To assess binding of tPARG against PARylated PARP1, 200 ng of automodified PARP1 was seeded onto plates overnight in 100 μL at 4°C. Blocking was done as described before. 100 μL of recombinant tPARG was incubated in the plates at a concentration of 1 μg mL−1 for 2 hours before washing. Incubation with 100 μL of anti-PARG antibody diluted at 1:100 was done for 2 hours. Anti-mouse secondary antibody was used at 1:3000 in PBS-0.05T with 0.1% BSA (w/v) for 1 hour at room temperature. Detection was done as described above.
To test binding of VCP, RBBP7, and GST (Sigma-Aldrich: SRP5348) for PARylated PARP1, 50 ng of automodified PARP1 was seeded onto plates overnight in 100 μL at 4°C. Blocking and washing were done as described before. 27 nM of VCP, 27 nM of RBBP7, or 400 nM of GST in PBS-0.05T with 0.1% BSA (w/v) was added for 2 hours. After washing, 100 μL of 1:1000 anti-GST antibody in PBS-0.05T with 0.1% BSA (w/v) was added for 2 hours before washing the wells. 1:3000 goat anti-rabbit conjugated to HRP was used at 1:3000 in PBS-0.05T with 0.1% BSA (w/v) as the secondary antibody for 1 hour at room temperature. Detection was done as described above.
To study protein binding for PAR, VCP (7.8 nM), RBBP7 (10.8 nM), and GST (20 nM) were seeded onto plates overnight in 100 μL at 4°C. Blocking and washing were done as described before. PAR (200 nM) (R&D Systems: 4336-100-01) was incubated in the wells for 2 hours at room temperature before washing the wells. 10H antibody (1:500) was added to the wells for 2 hours to detect PAR. Anti-mouse HRP secondary antibody was used at 1:3000 in PBS-0.05T with 0.1% BSA (w/v) for 1 hour at room temperature. Detection was done as described above.
Preparation of HEK293T cell lysates.
HEK293T cell lysate preparations for photocrosslinking for proteomic analysis were done by growing HEK293T cells in T75 flasks to 80% confluency and then collected using trypsin-EDTA solution to detach the cells from the flasks before spinning down and washing the cell pellets with PBS and spinning down again. 200 μL of 293T lysis buffer (25 mM Tris-HCl pH 7.5, 50 mM NaCl, 10% glycerol (v/v), 1% Nonidet P-40 (v/v), and Halt Protease Inhibitor Cocktail (ThermoFisher: 78430)) was used to resuspend the cell pellets. Cells were shaken for 10 minutes at room temperature before spinning down cellular debris at 14,000×g at 4°C for 15 minutes. Cell lysates were collected, and the cellular debris was resuspended with another 100 μL of 293T lysis buffer, and the cell lysis was repeated. Both fractions of cell lysates were combined. Protein concentrations of cell lysates were determined using Bradford reagent (ThermoFisher: 23236).
Photocrosslinking of PARylated PARP1 in cell lysates for mass spectrometry.
60 μg of automodified PARP1 was prepared as described above using the mixtures of 3:NAD+. 1 mg of HEK293T lysate was added to automodified PARP1 in 400 μL total. For samples with competitions, 300 μg of PARP1 was automodified with 200 μM NAD+ and concentrated to 100 μL using Amicron 10 kDa filters before additions to PARP1 automodified with the mixtures of 3:NAD+ prior to the additions of 293T lysates. Reactions without and with competitions were preincubated for 10 minutes before UV irradiation with 365-nm wavelength light for 15 minutes in PCR tubes placed 2 cm away from the light source. Samples were then treated with 3× CuAAC buffer (4.5 mM THPTA, 2.25 mM CuSO4, 900 μM biotin-PEG4-alkyne, 22.5 mM sodium ascorbate) for 1 hour at room temperature. Excess alkyne-biotin was removed by buffer exchange into PBS using Amicron 10 kDa MWCO Ultra-4 Centrifugal Filter Units (Millipore: UFC801024) by over 1000-fold dilution. Samples were added to 5 mL of PBS with 1% NP-40 (v/v), 100 mM NaCl, and 100 μL of NeutrAvidin beads (ThermoFisher: 29200) and incubated with head-over-end rotation overnight at room temperature. Beads were spun down at 2,000×g for 5 minutes and the supernatant was discarded. Beads were resuspended in 500 μL PBS with 4 M urea pH 7.4 and incubated with head-over-end rotation for 10 minutes. Beads were spun down, and the supernatant was discarded. This was repeated one additional time. Beads were then incubated in PBS with 1% NP-40 (v/v and incubated the same way for 3 times, 50 mM ammonium bicarbonate for 2 times, PBS for 2 times, 20% acetonitrile (v/v) 2 times, PBS for two times, and then 50 mM ammonium bicarbonate for two times.
For elution, beads were then resuspended in 100 μL of 0.1% Rapigest (w/v) (Waters: 186001861) and the slurry was boiled at 98°C for 10 minutes. The beads were spun down and the supernatant was collected. An additional 50 μL of 0.5% Rapigest (w/v) was added to the boiled beads and the beads were subjected to another round of boiling. Supernatant was collected and combined with the previous Rapigest fraction.
Eluted samples were first reduced by incubating in 10 mM DTT at 56°C for 20 minutes. The resulting thiols were alkylated with 55 mM iodoacetamide in the dark for 15 minutes. Proteins were digested with trypsin (Promega: V5111) at a 1:50 (w:w) ratio (trypsin:protein) overnight at 37°C. Peptides were acidified to 1% trifluoroacetic acid (v/v) and then desalted using C18 ZipTip (Millipore: ZTC18 5096). Dried peptides were resuspended in 10 μL of 0.1% TFA (v/v) in water.
1 μg of sample was injected onto an UltiMate 3000 UHP liquid chromatography system (Thermo Fisher Scientific). Peptides were separated using a μPAC C18 Trapping column (PharmaFluidics) in-line with a 50 cm μPAC column (PharmaFluidics). Peptides were eluted with a 90-min gradient (0–30% acetonitrile with 0.1% formic acid (v/v) for 60 min then 30–60% for 30 min) at a flow rate of 1 μL min−1 and electrosprayed into an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) with a Nanospray Flex ion source (Thermo Fisher Scientific). The source voltage was set to 2.5 kV, and the S-Lens RF level was set to 30%. The instrument method consisted of one survey (full) scan from m/z 375 to 1500 at a resolution of 120,000 in the Orbitrap mass analyzer, followed by data-dependent MS/MS scans of selected precursor ions in the linear quadrupole ion trap (LTQ) using the topN method. The AGC target value was set to 4e05, and the maximum injection time was set to 50 ms in the Orbitrap. The parameters were set to 2e04 and 120 ms in the LTQ with an isolation width of 1.2 Da and normalized collision energy of 28 for precursor isolation and MS/MS scanning. Precursor dynamic exclusion was enabled for a duration 40 s.
Thermo.Raw files were imported into Proteome Discoverer 2.2 (Thermo Fisher Scientific) using default parameters. The search engine Sequest HT was used. Parameters for protein searching were defined as follows: database—Uniprot human protein database (updated October 2018); precursor mass tolerance—10 ppm; fragment mass tolerance—0.6 Da; digestion—trypsin with two missed cleavages allowed; fixed modification: carbamidomethylation (C); variable modification: oxidation (M) and N-terminal protein acetylation. The Percolator node was used for peptide validation based on the PEP score. For protein identification a cut-off value of at least two unique high confidence peptide per protein with at 1% false discovery rate (FDR) was used.
Comparative analysis was performed for the lists of identified protein hits from the samples without and with added PARP1 automodified with NAD+ for binding competition. Only hits with at least 2 unique peptides were included in the analysis. Identified proteins that were found in the group without binding competition but disappeared in the presence of competition control were considered potential PARylation-dependent interacting proteins.
Supplementary Material
Acknowledgements
This work was supported by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, Sharon L. Cockrell Cancer Research Fund, The V Foundation for Cancer Research V Scholar PLUS Grant (to Y.Z.) National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) grant R35GM137901 (to Y. Z.), and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the NIH grant P30DK048522 (to USC Research Center for Liver Diseases).
Footnotes
Conflict of Interest Disclosure
The authors declare no competing financial interests.
Supporting Information
The Supporting Information includes supplementary methods and results. Supporting Information Available: This material is available free of charge via the Internet.
References
- [1].Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z, West AB, Koehler RC, Poirier GG, Dawson TM, and Dawson VL (2011) Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death, Nat. Med 17, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Han W, Li X, and Fu X (2011) The macro domain protein family: structure, functions, and their potential therapeutic implications, Mutat. Res 727, 86–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kalisch T, Ame JC, Dantzer F, and Schreiber V (2012) New readers and interpretations of poly(ADP-ribosyl)ation, Trends Biochem. Sci 37, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pleschke JM, Kleczkowska HE, Strohm M, and Althaus FR (2000) Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins, J. Biol. Chem 275, 40974–40980. [DOI] [PubMed] [Google Scholar]
- [5].Rack JG, Perina D, and Ahel I (2016) Macrodomains: Structure, Function, Evolution, and Catalytic Activities, Annu. Rev. Biochem 85, 431–454. [DOI] [PubMed] [Google Scholar]
- [6].Teloni F, and Altmeyer M (2016) Readers of poly(ADP-ribose): designed to be fit for purpose, Nucleic Acids Res. 44, 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fontana P, Bonfiglio JJ, Palazzo L, Bartlett E, Matic I, and Ahel I (2017) Serine ADP-ribosylation reversal by the hydrolase ARH3, eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Leung AKL, McPherson RL, and Griffin DE (2018) Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases, PLoS Pathog 14, e1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li C, Debing Y, Jankevicius G, Neyts J, Ahel I, Coutard B, and Canard B (2016) Viral Macro Domains Reverse Protein ADP-Ribosylation, J. Virol 90, 8478–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, Schellenberg MJ, Weston R, Williams JG, Rossi MN, Galehdari H, Krahn J, Wan A, Trembath RC, Crosby AH, Ahel D, Hay R, Ladurner AG, Timinszky G, Williams RS, and Ahel I (2013) Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease, EMBO J. 32, 1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, and Ahel I (2011) The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase, Nature 477, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gibson BA, and Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs, Nature reviews. Molecular cell biology 13, 411–424. [DOI] [PubMed] [Google Scholar]
- [13].Hassa PO, Haenni SS, Elser M, and Hottiger MO (2006) Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going?, Microbiol. Mol. Biol. Rev 70, 789–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hottiger MO (2015) Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics, Annu. Rev. Biochem 84, 227–263. [DOI] [PubMed] [Google Scholar]
- [15].Ryu KW, Kim DS, and Kraus WL (2015) New Facets in the Regulation of Gene Expression by ADP-Ribosylation and Poly(ADP-ribose) Polymerases, Chem. Rev [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhen Y, and Yu Y (2018) Proteomic Analysis of the Downstream Signaling Network of PARP1, Biochemistry 57, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cardinale A, Paldino E, Giampa C, Bernardi G, and Fusco FR (2015) PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington’s Disease, PloS one 10, e0134482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gariani K, Ryu D, Menzies KJ, Yi HS, Stein S, Zhang H, Perino A, Lemos V, Katsyuba E, Jha P, Vijgen S, Rubbia-Brandt L, Kim YK, Kim JT, Kim KS, Shong M, Schoonjans K, and Auwerx J (2017) Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease, J. Hepatol 66, 132–141. [DOI] [PubMed] [Google Scholar]
- [19].Martire S, Fuso A, Mosca L, Forte E, Correani V, Fontana M, Scarpa S, Maras B, and d’Erme M (2016) Bioenergetic Impairment in Animal and Cellular Models of Alzheimer’s Disease: PARP-1 Inhibition Rescues Metabolic Dysfunctions, Journal of Alzheimer’s disease : JAD 54, 307–324. [DOI] [PubMed] [Google Scholar]
- [20].Riffell JL, Lord CJ, and Ashworth A (2012) Tankyrase-targeted therapeutics: expanding opportunities in the PARP family, Nature reviews. Drug discovery 11, 923–936. [DOI] [PubMed] [Google Scholar]
- [21].Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Canto C, Conley KE, Walter L, Lovering RM, Chin ER, Jasmin BJ, Marcinek DJ, Menzies KJ, and Auwerx J (2016) NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation, Sci Transl Med 8, 361ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sahaboglu A, Tanimoto N, Kaur J, Sancho-Pelluz J, Huber G, Fahl E, Arango-Gonzalez B, Zrenner E, Ekstrom P, Lowenheim H, Seeliger M, and Paquet-Durand F (2010) PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function, PloS one 5, e15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Buntz A, Wallrodt S, Gwosch E, Schmalz M, Beneke S, Ferrando-May E, Marx A, and Zumbusch A (2016) Real-Time Cellular Imaging of Protein Poly(ADP-ribos)ylation, Angew. Chem. Int. Ed. Engl 55, 11256–11260. [DOI] [PubMed] [Google Scholar]
- [24].Jiang H, Kim JH, Frizzell KM, Kraus WL, and Lin H (2010) Clickable NAD analogues for labeling substrate proteins of poly(ADP-ribose) polymerases, J. Am. Chem. Soc 132, 9363–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Krebs C, Koestner W, Nissen M, Welge V, Parusel I, Malavasi F, Leiter EH, Santella RM, Haag F, and Koch-Nolte F (2003) Flow cytometric and immunoblot assays for cell surface ADP-ribosylation using a monoclonal antibody specific for ethenoadenosine, Anal. Biochem 314, 108–115. [DOI] [PubMed] [Google Scholar]
- [26].Laing S, Unger M, Koch-Nolte F, and Haag F (2011) ADP-ribosylation of arginine, Amino Acids 41, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wallrodt S, Buntz A, Wang Y, Zumbusch A, and Marx A (2016) Bioorthogonally Functionalized NAD(+) Analogues for In-Cell Visualization of Poly(ADP-Ribose) Formation, Angew. Chem. Int. Ed. Engl 55, 7660–7664. [DOI] [PubMed] [Google Scholar]
- [28].Kalesh K, Lukauskas S, Borg AJ, Snijders AP, Ayyappan V, Leung AKL, Haskard DO, and DiMaggio PA (2019) An Integrated Chemical Proteomics Approach for Quantitative Profiling of Intracellular ADP-Ribosylation, Scientific reports 9, 6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ando Y, Elkayam E, McPherson RL, Dasovich M, Cheng SJ, Voorneveld J, Filippov DV, Ong SE, Joshua-Tor L, and Leung AKL (2019) ELTA: Enzymatic Labeling of Terminal ADP-Ribose, Mol Cell 73, 845–856.e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carter-O’Connell I, Jin H, Morgan RK, David LL, and Cohen MS (2014) Engineering the substrate specificity of ADP-ribosyltransferases for identifying direct protein targets, J Am Chem Soc 136, 5201–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carter-O’Connell I, Jin H, Morgan RK, Zaja R, David LL, Ahel I, and Cohen MS (2016) Identifying Family-Member-Specific Targets of Mono-ARTDs by Using a Chemical Genetics Approach, Cell Rep 14, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, and Kraus WL (2016) Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation, Science 353, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dani N, Stilla A, Marchegiani A, Tamburro A, Till S, Ladurner AG, Corda D, and Di Girolamo M (2009) Combining affinity purification by ADP-ribose-binding macro domains with mass spectrometry to define the mammalian ADP-ribosyl proteome, Proc. Natl. Acad. Sci. U.S.A 106, 4243–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, and Poirier GG (2008) Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes, Nucleic Acids Res. 36, 6959–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gagne JP, Pic E, Isabelle M, Krietsch J, Ethier C, Paquet E, Kelly I, Boutin M, Moon KM, Foster LJ, and Poirier GG (2012) Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress, Nucleic Acids Res. 40, 7788–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, and Nielsen ML (2013) Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses, Mol. Cell 52, 272–285. [DOI] [PubMed] [Google Scholar]
- [37].Zhang X-N, Cheng Q, Chen J, Lam AT, Lu Y, Dai Z, Pei H, Evdokimov NM, Louie SG, and Zhang Y (2019) A ribose-functionalized NAD+ with unexpected high activity and selectivity for protein poly-ADP-ribosylation, Nature communications 10, 4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tucker JA, Bennett N, Brassington C, Durant ST, Hassall G, Holdgate G, McAlister M, Nissink JW, Truman C, and Watson M (2012) Structures of the human poly (ADP-ribose) glycohydrolase catalytic domain confirm catalytic mechanism and explain inhibition by ADP-HPD derivatives, PloS one 7, e50889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gibson BA, Conrad LB, Huang D, and Kraus WL (2017) Generation and Characterization of Recombinant Antibody-like ADP-Ribose Binding Proteins, Biochemistry 56, 6305–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Saxena A, Saffery R, Wong LH, Kalitsis P, and Choo KH (2002) Centromere proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly(ADP-ribosyl)ated, J. Biol. Chem 277, 26921–26926. [DOI] [PubMed] [Google Scholar]
- [41].Torrecilla I, Oehler J, and Ramadan K (2017) The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks, Philos. Trans. R. Soc. Lond. B. Biol. Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].van den Boom J, and Meyer H (2018) VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling, Mol. Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- [43].Yang J, Kiefer S, and Rauchman M (2002) Characterization of the gene encoding mouse retinoblastoma binding protein-7, a component of chromatin-remodeling complexes, Genomics 80, 407–415. [DOI] [PubMed] [Google Scholar]
- [44].Li GC, and Wang ZY (2006) Retinoblastoma suppressor associated protein 46 (RbAp46) attenuates the beta-catenin/TCF signaling through up-regulation of GSK-3beta expression, Anticancer Res. 26, 4511–4518. [PubMed] [Google Scholar]
- [45].Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, and Reinberg D (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation, Genes Dev. 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.