

Abstract
Purpose:
Simultaneously targeting the tumor and tumor microenvironment (TME) may hold promise in treating children with refractory solid tumors. Pexidartinib, an oral inhibitor of tyrosine kinases including Colony Stimulating Factor 1 Receptor (CSF-1R), KIT, and FLT3, is FDA approved in adults with tenosynovial giant cell tumor (TGCT). A phase I trial was conducted in pediatric and young adult patients (pts) with refractory leukemias or solid tumors including neurofibromatosis type 1 (NF1) related plexiform neurofibromas (PN).
Materials and Methods:
A rolling-six design with dose levels (DL) of 400 mg/m2, 600 mg/m2, and 800 mg/m2 once daily for 28 day cycles (C) was used. Response was assessed at regular intervals. PK and population PK were analyzed during C1.
Results:
Twelve pts (4 per DL, 9 evaluable) enrolled on the dose escalation phase and four patients enrolled in the expansion cohort: median (lower, upper quartile) age 16 (14, 16.5) years. No dose-limiting toxicities (DLT) were observed. PK appeared linear over three DLs. PK modeling and simulation determined a weight based recommended phase 2 dose (RP2D). Two pts had stable disease and 1 pt with peritoneal mesothelioma (C49+) had a sustained partial response 67% RECIST reduction. PD markers included a rise in plasma macrophage colony stimulating factor levels and a decrease in absolute monocyte count.
Conclusions:
Pexidartinib in pediatric pts was well tolerated at all DL tested, achieved target inhibition and resulted in a weight based RPD2 dose.
INTRODUCTION
Despite improvement in therapies for children and young adults with cancer many patients develop metastasis and progressive disease and there is a growing appreciation for the tumor cell extrinsic regulation in this process1–3. Targeting microenvironment dependencies may be a promising direction for improving survival in pediatric solid tumor patients. Agents that have both a direct anti-tumor effect and reform the tumor microenvironment are especially attractive.
Pexidartinib (PLX3397) is an oral small molecule inhibitor of class III protein tyrosine kinases including CSF-1R, KIT, and oncogenic FLT3 kinase4,5. In addition to direct tumor targeting, pexidartinib acts on solid tumors by inhibiting FLT3 kinase and KIT on myeloid progenitor cells and CSF-1R signaling which is important in the mobilization, migration, survival, and proliferation of monocytes and macrophages. Myeloid cells are the most abundant immune cell within many tumors and often increase in number during metastatic progression6,7.
The microenvironment of many pediatric solid tumors is rich in immune suppressive tumor associated macrophages (TAMs)8–10, and inhibition of CSF-1R may interfere with their development or function11–13. Although the full picture of the diversity and differential function of myeloid cells in pediatric malignancies is incomplete, there is growing evidence that a heterogeneous population of myeloid cells regulate progression of many diverse pediatric cancers including myeloid cells that promote cancer growth progression, and regulate immune suppression in osteosarcoma, soft tissue sarcomas and rhabdoid tumors 14–18. The myeloid cell populations within the tumor microenvironment in pediatric cancers can hold both pro-tumorigenic and anti-tumor functions19. Polarization of monocytes and neutrophils to an immune suppressive phenotype or to antigen presentation and phagocytic roles is seen in different myeloid cell populations and may vary in myeloid cell populations in different cell states depending on the signals within the local microenvironment. Evolving evidence suggests that classical monocytes or monocytic myeloid derived suppressor cells that express high levels of CSF1R can establish a supportive environment that promotes cancer cell survival, therapeutic resistance in pediatric leukemia, gliomas and neuroblastoma similar to findings in adult carcinomas14,19,20. Targeting any one particular signaling axis may not be sufficient to dramatically alter the myeloid component of the tumor microenvironment, but inhibition of the CSF1-CSF1R axis holds promise to limit the CSF1R high expressing M2 macrophage and monocytic myeloid derived suppressor cells (MDSCs) which are associated with enhanced inflammation and angiogenesis, diminished tumor specific T cell responses and increased tumor invasion and metastasis11,15,20. Diminishing CSF-CSF1R signaling may tip the balance in favor of M1 macrophages that can induce anti-tumor T cell responses and phagocytosis of stressed and dying tumor cells. Neurofibromatosis type 1 (NF1) related plexiform neurofibromas (PN) contain abundant TAMs, mast cells and NF1 -/- Schwann cells and this microenvironment produces high levels of stem cell factor I (scf-1) and IL34, the ligands for KIT and CSF-1R respectively21–24. Inhibition of CSF-1R and KIT in NF1 related PN may decrease tumor progression25.
In refractory leukemias, FLT3 and KIT inhibition may be beneficial through a direct effect on neoplastic cells. KIT is overexpressed in up to 80% of acute myelogenous leukemia (AML) 26–28 and FLT3 and FLT3 ligand are increased in several pediatric leukemias, with aberrant expression in more than 90% of AML including leukemia stem cells29and nearly 100% of B-cell acute lymphoblastic leukemia30. In regard to both acute myeloid leukemia and acute lymphoblastic leukemia, especially in the recurrent setting, the leukemia cells may be regulated by a myeloid immune suppressive bone marrow microenvironment31–33. Given the role of CSF1R in myeloid biology and the ability to create an immune suppressive myeloid microenvironment in the bone marrow, suggests the utility of CSF1R targeting for patients with leukemia. Therefore, CSF1R targeting in leukemia can hold both direct tumor targeting effects as well as targeting of signaling in the bone marrow microenvironment that can regulate leukemia progression.
Central nervous system (CNS) tumors were included in this study as recent published work indicates that CSF1R is highly expressed on microglial cells as well as bone marrow-derived myeloid cells infiltrating pediatric brain tumors34. Microglial cells have been shown to express CSF1R and require CSF1/CSF1R signaling for their maintenance35. A recent study of PK and CSF1R targeting with Pexidartinib in a non-human primate model showed low but detectable levels of the drug in the cerebral spinal fluid36. This study was in a nontumor setting and notably there may be significant differences with enhanced blood brain barrier permeability in tumor bearing hosts. PK and PD studies performed in patients with recurrent glioblastoma showed plasma PK levels that lead to decreased circulating CD14+CD16+ monocytes also lead to a decrease in macrophages in brain tissue after Pexidartinib therapy. Comparison of CSF1R targeting in tumor tissue with PK bioanalysis of plasma taken at the time of surgery revealed a median ratio of tumor/plasma PLX3397 concentrations of 70%5. These studies suggest Pexidartinib may hold efficacy in penetrating into brain tissue and impacting the myeloid cell populations in the tumor microenvironment.
In adult clinical trials, pexidartinib has been evaluated for safety and tolerability both alone and in combination with other agents including immune checkpoint therapy (NCT02777710, NCT0245242), BRAF/MEK inhibition (NCT03158103, NCT01826448), mitotic inhibitors (NCT01525602), and alkylating agents combined with radiation (NCT01790503). The recommended phase 2 dose (RP2D) of single agent pexidartinib in adults with tenosynovial giant cell tumor is 1000 mg/day given as a split dose on a continuous dosing schedule4 with higher doses of 1200–3000 mg given alone or in combination in patients with recurrent, progressive leukemias or solid tumors37. Dosing was not based on maximum tolerated dose (MTD). PK studies of Pexidartinib show increased plasma concentrations and exposure with increased dose and evidence of potential saturation at around the 1200 mg dose. Common adverse effects included fatigue, nausea, dysgeusia, periorbital edema, hair color lightening, anorexia, and elevated aspartate transaminase (AST) and alanine transaminase (ALT)4,5. In a phase I trial pexidartinib showed activity in patients with tenosynovial giant cell tumor (TGCT)4, and in a subsequent phase III trial demonstrated clinical benefit and led to FDA approval for this indication at a dose of 400 mg twice daily on a continuous dosing schedule(NCT02371369)38. While the majority of patients experienced minimal toxicity, idiosyncratic, serious (grade 4–5) liver toxicity was observed in a small subset of patients39,40.
We report the first pediatric phase I trial of pexidartinib in patients with refractory solid tumors including NF1 PN and with refractory leukemias.
MATERIALS AND METHODS
Patients
Patients age 3 to 21 years with recurrent or refractory acute leukemias or solid tumors including primary neoplasms of the CNS and patients with NF1 and inoperable PN that cause morbidity were eligible for phase I trial. The phase I study protocol conformed to the Declaration of Helsinki, Good Clinical Practice guidelines, and was approved by the National Cancer Institute Institutional Review Board and the US Food and Drug Administration. All patients or their legal guardians signed a document of informed consent indicating their understanding of the investigational nature and risks of this study. Assent was obtained per institutional guidelines. The written informed consent was obtained for patients who met eligibility requirements, including performance status and organ function parameters, prior to pexidartinib (NCT ClinicalTrials.gov Identifier: NCT02390752, Supplemental Appendix 1 with elibility criteria).
Study Design and Objectives
This is a phase I, single-center, investigator-initiated, open-label study of single agent pexidartinib conducted by the NCI Pediatric Oncology Branch. The primary objectives were to evaluate safety and tolerability of pexidartinib in pediatric patients and to determine the MTD and/or recommended phase II dose (RP2D). Secondary objectives included assessment of plasma pharmacokinetics (PK), preliminary clinical activity, and the pharmacodynamics (PD) of pexidartinib in peripheral blood immune cells and circulating biomarkers of KIT and CSF-1R inhibition. Pexidartinib was supplied by Plexxikon Inc./Daiichi-Sankyo, Inc., and administered orally once daily in 200 mg capsules (1 cycle =28 days) on a continuous schedule (C). Dosing was based on body surface area (BSA) with the total weekly dose rounded to within 10% of the calculated dose using a dosing nomogram. Patients were enrolled into one of three dose levels (DL) at 400 mg/m2 (70% of the adult RP2D based on an average adult BSA of 1.8 m2), 600 mg/m2, and 800 mg/m2 dose daily using a rolling six design41. We also performed population PK modeling to determine weight based dosing to achieve a similar drug exposure to adults receiving the active FDA approved dose of 800 mg daily.
Pharmacokinetic Studies
PK samples were collected from peripheral blood on C1 day 1 (pre-dose and 0.5, 1, 2, 4, 6, 10, 12, 19, 22, 23 and 24 hours post-dose (pre day 2 dose), day 15 (pre-dose and 1, 2, 4, and 6 hours post-dose), and day 16 (pre-dose) for patients in the dose escalation phase. Plasma PK analysis was performed by Plexxikon Inc., with interpretation and analysis by Daiichi-Sankyo and the investigators at the NCI. The Pexidartinib concentration was quantified using a validated liquid chromatography/tandem mass spectrometry method with a lower limit of quantification of 10 nmol/L and interday reproducibility variability less than 5%40. The Pexidartinib concentration-time data and PK parameters (maximal plasma concentration -Cmax-, time to maximal concentration -Tmax-, drug exposure -AUC-,and apparent Clearance -CLF ) were analyzed using noncompartmental methods using Phoenix WINNONLIN 6.3 (Certara, L.P. St Louis, MO).
Additionally, a population PK analysis was performed by using nonlinear mixed effects modeling (NONMEM). A two-compartment model with sequential zero- and first-order absorption with a lag time and linear elimination was used. Body weight was included as a covariate on the disposition parameters using allometric scaling. Based on the PK parameter estimates from the final model, simulations were subsequently conducted to evaluate doses in pediatric patients ages 6 to 18 years that would provide similar steady-state pexidartinib exposure to that in adult TGCT patients receiving pexidartinib 800 mg daily.
Safety and Response Evaluation
Safety evaluations including history, vital signs, physical exam, performance status and laboratory tests (complete blood count, T cell, B cell, NK cell profile, comprehensive profile with liver function tests, creatine phosphokinase, gamma-glutamyl transferase and blood urea nitrogen and coagulation studies) were conducted pre treatment, mid-C1, prior to C2-C9 then every other C, and at the end of therapy. ECG and echocardiogram were performed on C1 day 1 and day 15, prior to C3, and then every 4C. Toxicities were graded according to the NCI Common Toxicity Criteria (CTCAE v4.0) and dose limiting toxicities (DLT) observed during C1 (28 days) were used to determine the MTD. Patients were considered evaluable for determination of the MTD if they completed at least 24 of 28 doses in C1 (approximately 85%) or experienced DLT prior to the 24th dose.
Non hematologic dose limiting toxicities (DLTs) were defined as any grade 4 toxicity related to PLX3397, any grade 3 PLX3397 related toxicity which failed to recover to Grade ≤1 toxicity or to baseline toxicity after interruption of PLX3397 for 72 hours, with the exception of the following grade 3 toxicities: constipation managed with bowel regimen, tumor lysis syndrome, correctable and asymptomatic electrolyte abnormalities, hypoalbuminemic hypercalcemia not fully corrected, grade 3 neutronpenic fever resolved to grade 1 within seven days, grade 3 hypertriglyceridemia or hypercholesterolemia, grade 2 transaminase, alkaline phosphatase, bilirubin or other liver function test elevation; provided there was resolution to grade 1 within 7 days of interrupting treatment with PLX3397. Persistent (lasting longer than 7 days) grade 2 toxicities were considered dose-limiting if they were intolerable to the patient or if, in the opinion of the principal investigator, they posed a continued risk to the patient. In patients with normal hematological function at enrollment, hematologic DLT was defined as, any grade 4 neutropenia, anemia or thrombocytopenia. Lymphopenia was not considered in the definition of DLT.
Evaluation of measurable or evaluable tumors was performed at baseline within 14 days prior to starting treatment and then after every even C (with consistent use of imaging modality). For patients with leukemia, peripheral blood and bone marrow analysis was conducted every C and as clinically indicated. Response criteria was RECISTv1.1 for refractory solid tumors, WHO for brain tumors, and volumetric MRI analysis criteria for NF1 PN42. In patients with NF1 PN response was assessed at baseline, every 4th C for the first year then every 6 C in year 2 and onward42. Patients were allowed to continue on therapy until evidence of unacceptable toxicity, progression of disease, or for a maximum of two years in the absence of clinical or imaging response.
Biology Studies and Statistical Analysis
Whole blood was obtained prior to starting therapy, on C1 (day 7, day 15, end of C1 plus or minus one day), and every other C for analysis of changes in peripheral blood immune cells and serum cytokines (IL-10, IL-12p70, IL-6, MCP-1, M-CSF, MIF and IL-34). See supplemental statistical section for statistical analysis.
RESULTS
Patients, Disease, and Treatment Characteristics
16 patients were enrolled including 4 patients on the phase I expansion from April 2015 to October 2017 (Table 1). All patients were eligible. Patients with solid refractory tumors were heavily pretreated with most having received multiple previous therapies (Table 1). Patients were primarily teenagers or young adults, with a median age of 16 years and two patients under the age of 13 years. Seven patients were being treated for sarcomas, and no patients with leukemia were enrolled in the dose escalation portion of the trial. In the dose escalation phase, four patients were enrolled onto each DL, and nine of twelve patients completed at least 24 of 28 doses in C1 and were fully evaluable for toxicity. The patients who were not evaluable included two patients with rapidly progressive disease and one patient with NF1 PN who withdrew from the study due to patient preference.
Table 1:
Demographics and baseline characteristics of patients treated with pexidartinib
| Characteristic | Number of patients (n=16) | |
|---|---|---|
| Age (years) | Min., Quartiles, Max. | 4, 14.5, 16, 20, 21 |
| Sex | Female/Male | 7/9 |
| Race | White | 10 |
| African American | 3 | |
| Asian | 1 | |
| Hispanic | 2 | |
| Performance Status (%) | Min., Quartiles, Max. | 60, 80, 90, 90, 90 |
| Tumor Type | Sarcomas (Osteosarcoma, Ewing Sarcoma, Rhabdomyosarcoma, Malignant Peripheral Nerve Sheath Tumor) | 8 |
| Neurofibromatosis type 1 (NF1) Plexiform Neurofibroma | 3 | |
| Central Nervous System tumors | 3 | |
| Acute myeloid leukemia | 1 | |
| Peritoneal mesothelioma | 1 | |
| Prior Therapies | Surgery | 13 |
| Chemotherapy | 12 | |
| Radiation | 9 | |
| Immunotherapy | 5 | |
| Targeted therapy | 5 | |
| None | 1 |
Safety and Tolerability
No DLTs were seen across the three DLs in 16 patients enrolled (9 evaluable for determination of the MTD). Pexidartinib related toxicities during C1 are listed in Table 2. Common non-DLT toxicities included fatigue, headache, proteinuria, decreases in WBC and lymphocyte counts, and asymptomatic increases in creatine phosphokinase and serum amylase. Toxicities observed after cycle 1 are listed in supplemental Table 1 and included skin hypopigmentation in several patients. A MTD was not reached and the RP2D was defined as 800 mg/m2/day, equivalent to 130% of the adult FDA approved dose of 800 mg daily (400 mg BID) based on an adult BSA of 1.8 m2. Four additional patients were enrolled on this dose level in the expansion cohort and did not experience DLT. The RP2D based on body weight targeting a drug exposure equivalent to that in adults receiving 800 mg daily using population PK is described below.
Table 2:
Adverse events with at least possible attribution to pexidartinib during cycle 1 by the highest grade per patient
| Toxicity Grade CTCAE v4 | Dose level and patients enrolled | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| DL 1 (n = 4) | DL2 n =4) | DL3 (n=8)* | |||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| Hematologic | |||||||||
| Anemia | 2 | 1 | 1 | 1 | |||||
| White blood cell count decreased | 2 | 1 | 2 | 2 | |||||
| Lymphocyte count decreased | 1 | 1 | 1 | 2 | 1 | 2 | 1 | ||
| Neutrophil count decreased | 1 | 1 | 1 | ||||||
| Platelet count decreased | 3 | 1 | 1 | ||||||
| Prolonged APTT | 1 | ||||||||
| Constitutional | |||||||||
| Anorexia | 3 | 1 | |||||||
| Fatigue | 3 | 3 | 1 | 3 | |||||
| Cough | 1 | ||||||||
| Gastrointestinal | |||||||||
| Diarrhea | 1 | 2 | 1 | ||||||
| Constipation | 1 | ||||||||
| Nausea | 3 | 2 | |||||||
| Vomiting | 3 | ||||||||
| Hepatic | |||||||||
| ALT increased | 2 | 1 | |||||||
| AST increased | 1 | 2 | |||||||
| Metabolism | |||||||||
| CPK increased | 3 | 2 | 4 | 1 | |||||
| Alkaline phosphatase increased | 2 | ||||||||
| Hypoalbuminemia | 1 | 2 | |||||||
| Hypocalcemia | 1 | 1 | |||||||
| Hypercalcemia | 2 | ||||||||
| Hypoglycemia | 1 | ||||||||
| Hyperglycemia | 1 | 2 | |||||||
| Hypokalemia | 1 | ||||||||
| Hyponatremia | 1 | 2 | |||||||
| Hypophosphatemia | 1 | 2 | |||||||
| Serum lipase increased | 1 | ||||||||
| Serum amylase increased | 1 | 1 | 1 | 1 | 2 | ||||
| Neurologic/Psychiatric | |||||||||
| Anxiety | 2 | ||||||||
| Dizziness | 4 | ||||||||
| Headache | 1 | 2 | 3 | ||||||
| Non-cardiac chest pain | 1 | ||||||||
| Pain | 1 | ||||||||
| Restlessness | 1 | ||||||||
| Renal | |||||||||
| Creatinine increased | 1 | 1 | |||||||
| Glycosuria | 1 | ||||||||
| Proteinuria | 1 | 1 | 1 | 1 | 1 | ||||
| Dermatologic | |||||||||
| Bruise | 1 | 1 | |||||||
| Hair depigmentation | 1 | 1 | |||||||
| Petechiae | 1 | ||||||||
| Rash | 1 | 2 | 1 | ||||||
| Edema Face | 1 | 1 | |||||||
| Oral/ENT | |||||||||
| Dysgeusia | 2 | ||||||||
| Epistaxis | 1 | ||||||||
| Mucositis | 1 | ||||||||
| Oral Thrush | 1 | ||||||||
Pharmacokinetics
Pharmacokinetic parameters for patients in the dose escalation cohort (n=12) are shown in Supplemental Table 2. Pexidartinib reached peak plasma concentrations (Cmax) between 2 and 12 hours across dose levels. The AUC0–24 and the Cmax rose with increasing dose and were highest in DL3 (Figures 1A and 1B). Concentration x time curves on the first day of pexidartinib dosing showed slow clearance (Figure 1C). Plasma half-life could not be calculated due to timing of samples only until 24 hours after the first dose of pexidartinib. Due to the long half-life of pexidartinib, the sampling period did not allow for full characterization of the terminal half-life. The median terminal half-life in adults was 16.8 hours with lower and upper quartiles of 12.7 hours to 24.2 hours4. The accumulation ratio decreased with increasing DL and the median was 1.22 (n=3) for day 15 vs day 1 in DL3 (Supplemental Table 2).
Figure 1.

Right Top Panel 1A, Left Top Panel 1B
Right Bottom Panel 1C, Left Bottom Panel 1D
Population PK: The PK modeling resulted in weight based dosing aligned with the adult single agent FDA approved dose: To achieve similar steady-state pexidartinib AUC (median: 145000 ng*h/mL; 5th to 95th percentile: 96000 to 255000 ng*h/mL) in adult TGCT patients receiving 800 mg daily, the recommended dose regimen is 800 mg daily for body weight ≥ 40 kg, 600 mg daily for body weight ≥ 30 & < 40 kg; and 400 mg daily for body weight < 30 kg (Figure 1D).
Pharmacodynamics
Changes in plasma PD markers are presented in Tables 3, 4 and Supplemental Table 3, 4 and 5. Results showed a decrease in circulating monocytes within the first C with a median fold change (95% CI) for absolute monocyte count (AMC) at C1 day 6–8, day 14–16, and day 27–29 were respectively 0.58 (0.35, 0.81), 0.56 (0.40, 0.89), and 0.64 (0.26, 0.88) (Table 3). Similarly, the percentage of monocytes was decreased with median fold change in percent monocytes at C1 day 6–8, day 14–16, and day 27–29 were respectively 0.60 (0.40, 0.87), 0.67 (0.52, 0.89), and 0.84 (0.26, 1.06) (Table 3). Serum cytokine analysis showed substantial increases in M-CSF (CSF-1) levels on treatment with pexidartinib, with both a linear and weak curvilinear response over time with weak evidence for a dosage effect (Table 4, Supplemental Table 3). M-CSF median fold change at C1 day 14–15 was 3.70 (2.31, 5.24) and at C1 day 27–29 was 4.52 (2.40, 13.3). Additionally, monocyte chemoattractant protein-1 (MCP-1) increased on treatment, with median fold change of 1.28 (1.01, 1.56) at C1 days 14–15 and 1.27 (1.11, 1.54) at C1 days 27–29 (Table 4, Supplemental Table 3). Flow cytometric analysis of PBMC trended toward a decrease in CD11b+ cells at C1 days 6–8 with median fold change of 0.47 (0.21, 1.31) but levels did not continue to decrease throughout C1 (Supplemental Table 4). No significant changes were observed in CD14dimCD16+ nonclassical, pro-inflammatory monocytes in C1(Supplemental Table 4). There were no notable changes seen in peripheral blood lymphocyte subset analysis during C1 (Supplemental Table 5).
Table 3:
Pexidartinib pharmacodynamics: Fold change in peripheral blood markers compared to baseline
| 6–8 Days from C1 | 14–16 Days from C1 | 27–29 Days from C1 | Repeated Measures ANCOVA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CBC Parameter (Fold Change from Day 1) | N | P* | Med. Fold Change (95% CI) | N | P* | Med. Fold Change (95% CI) | N | P* | Med. Fold Change (95% CI) | Time Linear | Dose Linear |
| White blood cell (WBC) | 13 | 0.15 | 0.89 (0.78, 1.07) | 13 | 0.04 | 0.85 (0.65, 1.06) | 10 | 0.19 | 0.81 (0.58, 1.28) | 0.007 | 0.10 |
| Hemoglobin (Hgb) | 13 | 0.08 | 1.03 (0.97, 1.08) | 13 | 0.02 | 0.97 (0.91, 1.00) | 10 | 0.37 | 1.03 (0.95, 1.13) | 0.91 | 0.71 |
| Platelet Count | 13 | 0.24 | 1.10 (0.84, 1.17) | 13 | 0.64 | 0.97 (0.83, 1.13) | 10 | 0.43 | 0.94 (0.79, 1.18) | 0.59 | 0.002 |
| Neutrophils % + bands | 13 | 0.13 | 1.08 (0.96, 1.13) | 13 | 0.07 | 1.08 (0.96, 1.13) | 10 | 0.85 | 0.97 (0.78, 1.30) | 0.91 | 0.26 |
| Immature granulocytes | 9 | 0.01 | 0.50 (0.08, 0.67) | 9 | 0.01 | 0.46 (0.30, 0.67) | 6 | 0.44 | 0.24 (0.00, 2.00) | ||
| Lymphocytes % | 13 | 0.79 | 0.96 (0.81, 1.29) | 13 | 0.95 | 0.92 (0.87, 1.25) | 10 | 0.28 | 1.28 (0.47, 1.95) | 0.53 | 0.29 |
| Monocytes % | 13 | 0.002 | 0.60 (0.40, 0.87) | 13 | 0.002 | 0.67 (0.52, 0.89) | 10 | 0.05 | 0.84 (0.26, 1.06) | 0.03 | 0.10 |
| Eosinophils % | 10 | 0.03 | 0.78 (0.55, 1.04) | 11 | 0.70 | 0.97 (0.92, 1.53) | 8 | 0.46 | 0.76 (0.48, 4.19) | 0.87 | 0.79 |
| Basophils % | 12 | 0.16 | 0.95 (0.50, 1.13) | 11 | 0.04 | 0.83 (0.50, 1.13) | 9 | 0.01 | 0.22 (0.00, 0.83) | 0.001 | 0.28 |
| Absolute Neutrophil Count | 13 | 0.74 | 0.90 (0.77, 1.26) | 13 | 0.41 | 0.96 (0.64, 1.15) | 10 | 0.49 | 0.70 (0.46, 1.66) | 0.035 | 0.13 |
| Absolute Immature Granulocyte | 9 | 0.09 | 0.50 (0.10, 1.00) | 9 | 0.02 | 0.50 (0.19, 1.00) | 6 | 0.69 | 0.20 (0.00, 2.50) | ||
| Absolute Lymphocyte Count | 13 | 0.49 | 0.97 (0.68, 1.20) | 13 | 0.05 | 0.88 (0.73, 1.03) | 10 | 0.36 | 0.86 (0.60, 1.21) | 0.075 | 0.56 |
| Absolute Monocyte Count | 13 | 0.001 | 0.58 (0.35, 0.81) | 13 | 0.0002 | 0.56 (0.40, 0.89) | 10 | 0.002 | 0.64 (0.26, 0.88) | 0.0003 | 0.95 |
| Absolute Eosinophil Count | 10 | 0.01 | 0.76 (0.50, 1.07) | 11 | 0.92 | 1.00 (0.81, 1.27) | 8 | 0.25 | 0.63 (0.31, 5.37) | 0.73 | 0.97 |
| Absolute Basophil Count | 12 | 0.06 | 0.92 (0.50, 1.00) | 11 | 0.01 | 0.67 (0.50, 1.00) | 9 | 0.01 | 0.33 (0.00, 0.67) | 0.0006 | 0.057 |
Two-tailed unadjusted signed rank test p-value for Mu=1
Table 4:
Pexidartinib pharmacodynamics: Serum cytokine analysis fold change from Day 1
| 14–15 Days from C1 | 27–29 Days from C1 | |||||
|---|---|---|---|---|---|---|
| Cytokine | N | P* | Med. Fold Change (95% CI) | N | P* | Med. Fold Change (95% CI) |
| IL-10 | 12 | 0.021 | 1.25 (1.11, 1.44) | 8 | 0.64 | 1.22 (0.81, 1.30) |
| IL-12p70 | 12 | 0.79 | 0.92 (0.80, 1.25) | 8 | 0.023 | 0.86 (0.66, 1.05) |
| IL-6 | 12 | 0.85 | 0.91 (0.78, 1.34) | 8 | 0.95 | 0.98 (0.64, 2.39) |
| MCP-1 | 12 | 0.003 | 1.28 (1.01, 1.56) | 8 | 0.023 | 1.27 (1.11, 1.54) |
| M-CSF | 12 | 0.0005 | 3.70 (2.31, 5.24) | 8 | 0.008 | 4.52 (2.40, 13.3) |
Two-tailed unadjusted signed rank test p-value for Mu=1
Treatment Duration and Response:
Patients received a median of 1 C of treatment (range less than 1 C to >45 C). Patient enrollment is detailed in Table 5. There was one sustained partial response 67% decrease in longest diameter of target lesion in a patient with peritoneal mesothelioma on the 400mg/m2/day DL (Figure 2). Stable disease was seen in three patients. One patient with metastatic, rapidly progressive osteosarcoma had stable disease for 4 C prior to disease progression. Two patients with NF1 PN with stable disease and one with improvement in pain discontinued therapy to initate MEK inhibitor therapy with established benefit for patients with NF1 PN44.
Table 5:
Patient enrollment and duration of treatment
| Patient | Diagnosis | Dose Level | Time-point Off Treatment | Reason Off Treatment |
|---|---|---|---|---|
| 1 | Peritoneal mesothelioma | 1 | On Cycle 45 | Remains on Study |
| 2 | Osteosarcoma | 1 | Cycle 1 Day 23 | Progressive Disease |
| 3 | Ewing Sarcoma | 1 | Cycle 1 Day 28 | Progressive Disease |
| 4 | NF1 plexiform neurofibroma | 1 | After Cycle 4 | Best Interest of Patient |
| 5 | NF1 plexiform neurofibroma | 2 | After Cycle 1 (took 23 doses) | Withdrawal of Consent |
| 6 | NF1 plexiform neurofibroma | 2 | After Cycle 6 | Best Interest of Patient |
| 7 | Central Nervous System PNET | 2 | After Cycle 1 | Progressive Disease |
| 8 | Primary brain tumor | 2 | After Cycle 1 | Progressive Disease |
| 9 | NF1 malignant peripheral nerve sheath tumor | 3 | Cycle 2 Day 28 | Progressive Disease |
| 10 | Osteosarcoma | 3 | Cycle 4 Day 6 | Progressive Disease |
| 11 | Osteosarcoma | 3 | Cycle 3 Day 1 | Progressive Disease |
| 12 | Embryonal rhabdomyosarcoma | 3 | Cycle 1 Day 12 | Progressive Disease |
| 13 | Acute myeloid leukemia | 3 | Cycle 1 Day 15 | Progressive Disease |
| 14 | Spindle cell carcinoma | 3 | Cycle 1 Day 23 | Progressive Disease |
| 15 | Aneurysmal fibrous histiocytoma | 3 | Cycle 3 Day 16 | Progressive Disease |
| 16 | Glioblastoma | 3 | Cycle 2 Day 5 | Progressive Disease |
Figure 2.
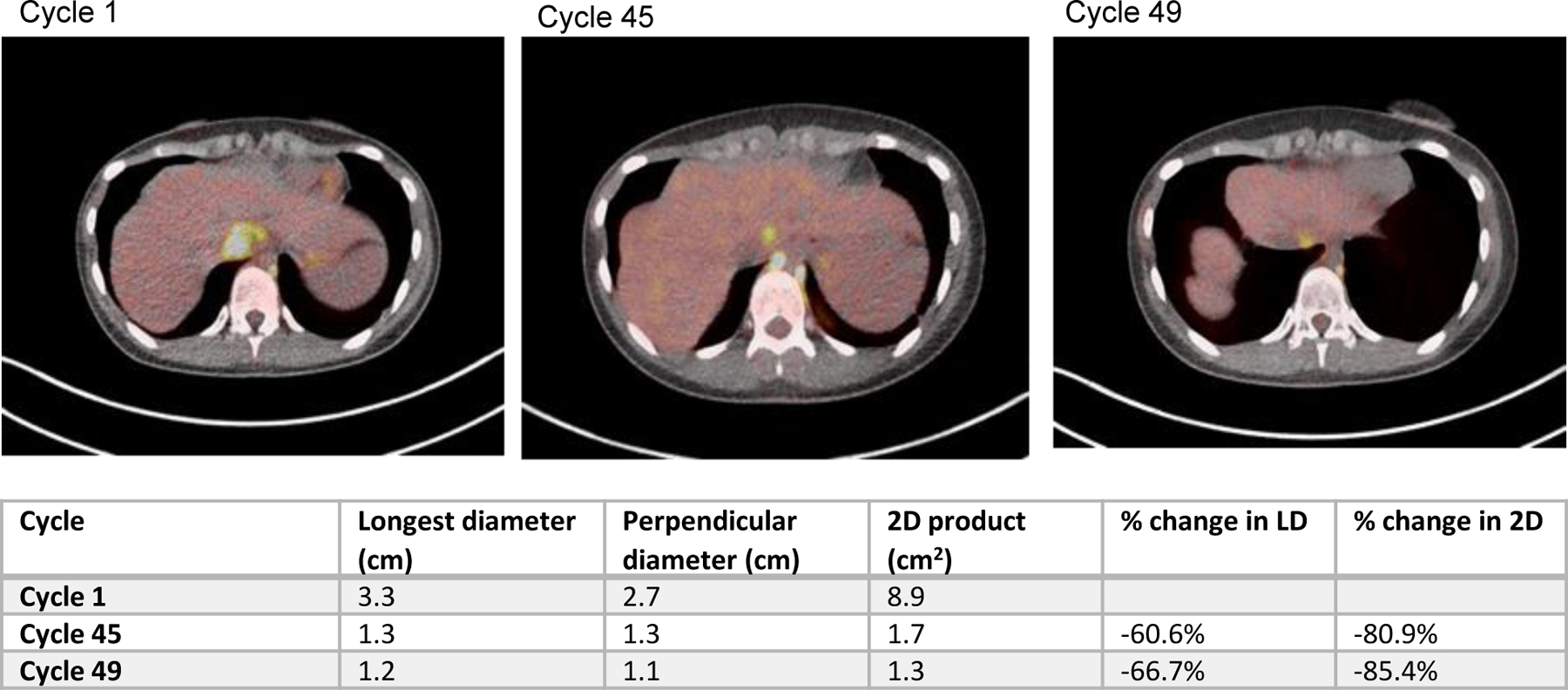
FDG PET Imaging top three panels
Table measurement of lesion of over time for indicated cycle of therapy
DISCUSSION
This first-in-pediatric phase I dose-escalation study demonstrated that pexidartinib administered on an oral continuous daily dosing schedule was well tolerated in pediatric patients with solid tumors including patients with NF1 PN as well as heavily pretreated patients.
The toxicity profile was mild and no DLTs were seen including at the highest DL of 800mg/m2/dose which is approximately 130 percent of the single agent adult RP2D for patients with TCGT4,5. The MTD was therefore not reached. Although elevations in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) and creatinine phosphokinase were seen in our patients, these were mild compared to the more substantial idosyncratic liver abnormalities observed in a subset of adult patients treated with pexidartinib 4,5 as well as with other CSF-1R inhibitors45. The rise in AST and ALT has been suggested to be secondary to a decrease in CSF-1R positive Kupffer cells, which are responsible for physiologic clearance of these enzymes 46. Other adverse events included fatigue, pain, and changes in hair color and hematologic labs such as mild decrease in WBC, lymphocyte, and platelet counts. We elected to determine the RP2D using population PK and modeling based on doses needed to achieve equivalent drug exposure to that in adults with TCGT tumor receiving the FDA approved dose of 800 mg daily. The calculated RP2D is 800 mg daily for body weight ≥ 40 kg, 600 mg daily for body weight ≥ 30 & < 40 kg; and 400 mg daily for body weight < 30 kg. Pexidartinib target inhibition was seen at all DLs with CSF1 levels elevated (Table 4) and AMC levels decreased at each DL (Table 3). Target inhibition was achieved at doses below 800mg/m2/dose and PK modeling established pediatric dosing that avoids use of a nomogram and limits dosing above what is required for target inhibition.
Preliminary activity was observed in several patients. Three patients had stable disease for four or more C, with one patient with peritoneal mesothelioma with a deep partial response now on C 47. There were no other partial or complete responders and the majority of patients discontinued treatment due to progressive disease.
Although this study is limited by small sample size and incomplete pharmacodynamic data, our biology studies focused on circulating markers of effect in the tumor microenvironment (TME) such as peripheral blood immune cells and cytokines may reflect some aspects of the TME13. Monocytes receive signals through circulating chemokines to differentiate into TAMs, and this signal is regulated primarily through CSF-1R 13. In a preclinical model pexidartinib was associated with decreases in circulating monocytes and regulatory T cells and TAMs43. Similarly, CSF-1R targeting in pancreatic adenocarcinoma models and, in combination with chemotherapy, in breast cancer models suggest that limiting CSF-1R dependent macrophage infiltration improves anti-tumor immunity and overall survival 47,48.
In this study, we chose to focus on circulating monocytes including classical monocytes as these cells most often develop into macrophages in tissue. CD14dimCD16+ nonclassical, pro-inflammatory monocytes decreased with CSF-1R inhibition in other trials43 but did not show similar changes in C1 in this trial (Supplemental Table 4). However, in order to explore an easier and potentially less biased approach to examining the total peripheral myeloid population that can become tumor associated macrophages and other tissue infiltrating myeloid cells we examined absolute monocyte count from the complete blood count. Absolute monocyte count captures multiple monocyte populations including circulating myeloid progenitors and immature myeloid derived suppressor cells and could provide a widely available, standardized and reliable approach to examine the impact of CSF1R targeted therapy. Given the challenges of obtaining tumor biopsies in children, a main limitation of this current study is the lack of tissue sampling to compare to the peripheral blood measurements to validate the utility of one particular myeloid population over another and to assess if the circulating populations reflect cell populations within the local tumor microenvironment.
Despite limitations of this study due to small sample sizes and potential bias due to non-random missing data (i.e. sicker patients coming off study earlier), an increase in serum M-CSF (CSF-1) levels and decrease in both monocyte percentage and AMC were found to correlate with dose suggesting pexidartinib targets the myeloid compartment as anticipated. While it is interesting to consider the potential of CSF-1 and AMC as prognostic markers in response to CSF1R inhibition, larger studies will be required to determine if this is the case. As such, all statistical analysis should be considered in this context. High AMC is a marker of poor prognosis in several cancers including Hodgkin’s lymphoma, diffuse large B-cell lymphoma, breast, lung and GI malignancies49–51. Elevated serum M-CSF (CSF-1) was also observed and has been reported as a reproducible PD marker of CSF-1R inhibition5. Immature granulocytes can also be seen in the immune suppressive microenvironment 52, and these cells decreased after pexidartinib in a small sample of the patients. Targeting monocyte/macrophage mediated immune suppression via M-CSF (CSF-1)/CSF-1R axis holds promise for improving efficacy of conventional chemotherapy or potentially in combination with other immune modulatory therapy to enhance anti-tumor immunity. Overall, plasma M-CSF (CSF-1) levels and AMC are consistent PD markers for CSF-1R targeting, and linking to biopsy samples may determine the utility of blood markers for monitoring local tissue effects53. Preclinical work suggests that the M-CSF (CSF-1)/CSF-1R axis can play an important role in the recruitment of immune suppressive myeloid populations to the TME, and along with other immunomodulatory approaches, its inhibition may be an effective anti-metastatic strategy 11,43,54.
Given its favorable toxicity profile in children and young adults, pexidartinib may be tolerated well in combination with other agents. The TME may be rendered less immune suppressive after CSF-1R inhibition, and treatment with cytotoxic agents or immune activating agents could be more effective in decreasing tumor burden when used in combination. There may also be a role for CSF-1R inhibition as maintenance therapy in the setting of minimal residual disease to decrease metastasis through inhibition of myeloid derived suppressor cells associated with metastasis of disseminated tumor cells55,56. The aggressive nature of pediatric leukemia in the relapsed setting makes detailed investigations in the role of myeloid mediated immune suppression and leukemia resistance a challenge. However, the combination of leukemia directed cytotoxic chemotherapy and pexidartinib in disease with high CSF1 expression may warrant further clinical investigation. Further investigation using pexidartinib either as a single agent or in combination with other cytotoxic drugs, targeted agents, or immunotherapy treatments is underway in adult trials and merits further study in pediatric oncology populations.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE:
Despite aggressive multimodal therapy, many high risk pediatric solid tumor patients develop metastatic progression and succumb to their disease. Novel treatment approaches targeting both tumor intrinsic pathways as well as elements of the metastatic microenvironment including immune suppressive myeloid cells such as tumor associated macrophages (TAMs) may be a promising strategy for improving outcomes. Colony Stimulating Factor 1 Receptor (CSF-1R) signaling is important in macrophage biology including impacting myeloid cell mobilization, migration, survival, and proliferation. Inhibition of CSF-1R through pexidartinib, an oral inhibitor of tyrosine kinases including CSF-1R, KIT, and FLT3, may decrease tumor progression by suppressing the effects of TAMs and other monocyte-derived populations in the tumor microenvironment. We conducted the first pediatric phase I trial of pexidartinib to study its safety profile, pharmacokinetics, and recommended pediatric phase II dose. The trial resulted in establishment of a safe, and feasible phase 2 dosing regimen and potential biomarkers for pediatric patients.
ACKNOWLEDGEMENTS
This research was supported in part by the Intramural Research Program of the NIH, NCI, Center for Cancer Research. The drug manufacturer, Plexxikon, Inc/ Daiichi Sanyko, Inc, provided the study drug and performed PK assays and PK modeling.
Footnotes
Conflict of Interest Disclosures:
The authors have no conflicts of interest to disclose
REFERENCES
- 1.Quail DF, Joyce JA: Microenvironmental regulation of tumor progression and metastasis. Nat Med 19:1423–37, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altorki NK, Markowitz GJ, Gao D, et al. : The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer 19:9–31, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Kenawi A, Hanggi K, Ruffell B: The Immune Microenvironment and Cancer Metastasis. Cold Spring Harb Perspect Med 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tap WD, Wainberg ZA, Anthony SP, et al. : Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med 373:428–37, 2015 [DOI] [PubMed] [Google Scholar]
- 5.Butowski N, Colman H, De Groot JF, et al. : Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol 18:557–64, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrilovich DI: Myeloid-Derived Suppressor Cells. Cancer Immunol Res 5:3–8, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giles AJ, Reid CM, Evans JD, et al. : Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre-metastatic Niche. Cancer Res, 2015 [DOI] [PMC free article] [PubMed]
- 8.Endo-Munoz L, Evdokiou A, Saunders NA: The role of osteoclasts and tumour-associated macrophages in osteosarcoma metastasis. Biochim Biophys Acta 1826:434–42, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Hadjidaniel MD, Muthugounder S, Hung LT, et al. : Tumor-associated macrophages promote neuroblastoma via STAT3 phosphorylation and up-regulation of c-MYC. Oncotarget 8:91516–91529, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liou P, Bader L, Wang A, et al. : Correlation of tumor-associated macrophages and clinicopathological factors in Wilms tumor. Vasc Cell 5:5, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannarile MA, Weisser M, Jacob W, et al. : Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer 5:53, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hume DA, MacDonald KP: Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119:1810–20, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Zhang Y: Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol 10:58, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leruste A, Tosello J, Ramos RN, et al. : Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 36:597–612 e8, 2019 [DOI] [PubMed] [Google Scholar]
- 15.Webb MW, Sun J, Sheard MA, et al. : Colony stimulating factor 1 receptor blockade improves the efficacy of chemotherapy against human neuroblastoma in the absence of T lymphocytes. Int J Cancer 143:1483–1493, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kather JN, Horner C, Weis CA, et al. : CD163+ immune cell infiltrates and presence of CD54+ microvessels are prognostic markers for patients with embryonal rhabdomyosarcoma. Sci Rep 9:9211, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kansara M, Thomson K, Pang P, et al. : Infiltrating Myeloid Cells Drive Osteosarcoma Progression via GRM4 Regulation of IL23. Cancer Discov 9:1511–1519, 2019 [DOI] [PubMed] [Google Scholar]
- 18.Mantovani A, Marchesi F, Malesci A, et al. : Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14:399–416, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xin C, Zhu J, Gu S, et al. : CD200 is overexpressed in neuroblastoma and regulates tumor immune microenvironment. Cancer Immunol Immunother, 2020 [DOI] [PMC free article] [PubMed]
- 20.Edwards DKt, Watanabe-Smith K, Rofelty A, et al. : CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood 133:588–599, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutmann DH, Ferner RE, Listernick RH, et al. : Neurofibromatosis type 1. Nat Rev Dis Primers 3:17004, 2017 [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Szretter KJ, Vermi W, et al. : IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13:753–60, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei S, Nandi S, Chitu V, et al. : Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol 88:495–505, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Vries WM, Briaire-de Bruijn IH, van Benthem PPG, et al. : M-CSF and IL-34 expression as indicators for growth in sporadic vestibular schwannoma. Virchows Arch 474:375–381, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson KA, Nalepa G, Yang FC, et al. : Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol 13:1218–24, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heo SK, Noh EK, Kim JY, et al. : Radotinib induces high cytotoxicity in c-KIT positive acute myeloid leukemia cells. Eur J Pharmacol 804:52–56, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Ikeda H, Kanakura Y, Tamaki T, et al. : Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood 78:2962–8, 1991 [PubMed] [Google Scholar]
- 28.Pollard JA, Alonzo TA, Gerbing RB, et al. : Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 115:2372–9, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aikawa Y, Katsumoto T, Zhang P, et al. : PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat Med 16:580–5, 1p following 585, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown P, Small D: FLT3 inhibitors: a paradigm for the development of targeted therapeutics for paediatric cancer. Eur J Cancer 40:707–21, discussion 722–4, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Witkowski MT, Lasry A, Carroll WL, et al. : Immune-Based Therapies in Acute Leukemia. Trends Cancer 5:604–618, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epperly R, Gottschalk S, Velasquez MP: A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front Oncol 10:262, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medyouf H: The microenvironment in human myeloid malignancies: emerging concepts and therapeutic implications. Blood 129:1617–1626, 2017 [DOI] [PubMed] [Google Scholar]
- 34.Haage V, Semtner M, Vidal RO, et al. : Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol Commun 7:20, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oosterhof N, Kuil LE, van der Linde HC, et al. : Colony-Stimulating Factor 1 Receptor (CSF1R) Regulates Microglia Density and Distribution, but Not Microglia Differentiation In Vivo. Cell Rep 24:1203–1217 e6, 2018 [DOI] [PubMed] [Google Scholar]
- 36.Shankarappa PS, Peer CJ, Odabas A, et al. : Cerebrospinal fluid penetration of the colony-stimulating factor-1 receptor (CSF-1R) inhibitor, pexidartinib. Cancer Chemother Pharmacol 85:1003–1007, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wesolowski R, Sharma N, Reebel L, et al. : Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol 11:1758835919854238, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tap WD: Multidisciplinary care in tenosynovial giant cell tumours. Lancet Oncol 20:755–756, 2019 [DOI] [PubMed] [Google Scholar]
- 39.William D. Tap HG, Stacchiotti Silvia, Palmerini Emanuela, Ferrari Stefano, Desai Jayesh, Bauer Sebastian, Blay Jean-Yves, Alcindor Thierry, Ganjoo Kristen N., Broto Javier Martin, Ryan Christopher W., Shuster Dale Edward, Zhang Ling, Wang Qiang, Hsu Henry, Lin Paul S., Tong Sandra, Wagner Andrew J.: Final results of ENLIVEN: A global, double-blind, randomized, placebo-controlled, phase 3 study of pexidartinib in advanced tenosynovial giant cell tumor (TGCT). ASCO Annual Meeting 2018, 2018, pp Abstract 11502 [Google Scholar]
- 40.Tap WD, Gelderblom H, Palmerini E, et al. : Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet, 2019 [DOI] [PMC free article] [PubMed]
- 41.Skolnik JM, Barrett JS, Jayaraman B, et al. : Shortening the timeline of pediatric phase I trials: the rolling six design. J Clin Oncol 26:190–5, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Dombi E, Ardern-Holmes SL, Babovic-Vuksanovic D, et al. : Recommendations for imaging tumor response in neurofibromatosis clinical trials. Neurology 81:S33–40, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dammeijer F, Lievense LA, Kaijen-Lambers ME, et al. : Depletion of Tumor-Associated Macrophages with a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol Res 5:535–546, 2017 [DOI] [PubMed] [Google Scholar]
- 44.Dombi E, Baldwin A, Marcus LJ, et al. : Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med 375:2550–2560, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papadopoulos KP, Gluck L, Martin LP, et al. : First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res 23:5703–5710, 2017 [DOI] [PubMed] [Google Scholar]
- 46.Radi ZA, Koza-Taylor PH, Bell RR, et al. : Increased serum enzyme levels associated with kupffer cell reduction with no signs of hepatic or skeletal muscle injury. Am J Pathol 179:240–7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Candido JB, Morton JP, Bailey P, et al. : CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep 23:1448–1460, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeNardo DG, Brennan DJ, Rexhepaj E, et al. : Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1:54–67, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishijima TF, Muss HB, Shachar SS, et al. : Prognostic value of lymphocyte-to-monocyte ratio in patients with solid tumors: A systematic review and meta-analysis. Cancer Treat Rev 41:971–8, 2015 [DOI] [PubMed] [Google Scholar]
- 50.Wilcox RA, Ristow K, Habermann TM, et al. : The absolute monocyte and lymphocyte prognostic score predicts survival and identifies high-risk patients in diffuse large-B-cell lymphoma. Leukemia 25:1502–9, 2011 [DOI] [PubMed] [Google Scholar]
- 51.Gu L, Li H, Chen L, et al. : Prognostic role of lymphocyte to monocyte ratio for patients with cancer: evidence from a systematic review and meta-analysis. Oncotarget 7:31926–42, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singel KL, Segal BH: Neutrophils in the tumor microenvironment: trying to heal the wound that cannot heal. Immunol Rev 273:329–43, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quandt D, Dieter Zucht H, Amann A, et al. : Implementing liquid biopsies into clinical decision making for cancer immunotherapy. Oncotarget 8:48507–48520, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holmgaard RB, Zamarin D, Lesokhin A, et al. : Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine 6:50–58, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Umansky V, Blattner C, Gebhardt C, et al. : The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) 4, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaplan RN, Rafii S, Lyden D: Preparing the “soil”: the premetastatic niche. Cancer Res 66:11089–93, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.