

Abstract
An iron(III) methoxide complex reacts with para-substituted triarylmethyl radicals to give iron(II) and methoxyether products. Second-order rate constants for the radical derivatives were obtained. Hammett and Marcus plots suggest the radical transfer reactions proceed via a concerted process. Calculations support the concerted nature of these reactions involving a single transition state with no initial charge transfer. These findings have implications for the radical “rebound” step invoked in nonheme iron oxygenases, halogenases, and related synthetic catalysts.
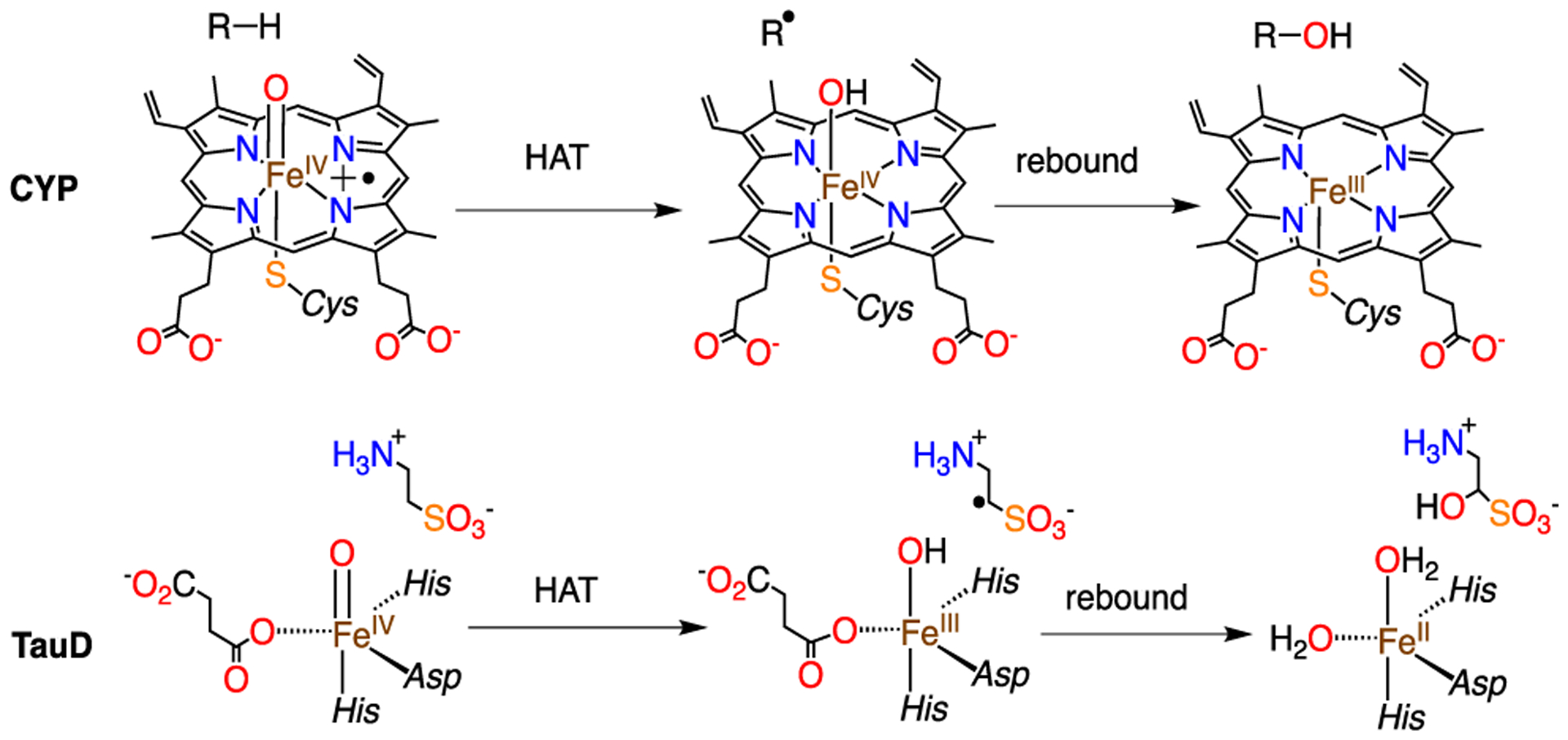
The preferential formation of C−O bonds by heme and nonheme iron enzymes typically occurs through C−H bond cleavage by a high-valent iron(IV)-oxo species, followed by a radical “rebound” step in which the newly formed carbon radical (R•) and Fe(OH) intermediate combine to give R−OH and a reduced Fe product.1–4 However, alternate outcomes are sometimes observed, including desaturation or decarboxylation (e.g., heme: Cytochrome P450 (CYP) OleT; nonheme: AsqJ, NapI, VioC, UndA).5–12 In the nonheme iron halogenases (e.g., CytC3, WelO5, SyrB2),13–17 the radical selectively combines with a halide ligand rather than an OH ligand, leading to halogenation. Interestingly, the nonheme iron enzyme isopenicillin N synthase (IPNS) may operate through a similar pathway, in which a CVal radical combines selectively with a coordinated thiolate ligand, instead of a bound OH group, to give the final thiazolidine ring.18 Studies on biomimetic, high-valent metal-oxo complexes showed that different outcomes can occur from the carbon radical stage, including conventional hydroxylation as well as simple radical dissociation away from the Fe(OH) species.4,19,20 The factors that control the rates and selectivities of these reactions remain poorly understood.
The synthesis of a heme-like Fe(OH) corrole complex provided us with a platform to examine reactions with carbon radicals (trityl radical derivatives).21,22 These model reactions could be compared to the rebound hydroxylation process seen in CYP, involving protonated Compound II (Cpd-II, Scheme 1).21 The heme-based CYP undergoes reduction of a formal FeIV(OH)(porphyrin) to an FeIII(porphyrin). The analogous nonheme chemistry, exemplified by the hydroxylation performed by TauD shown in Scheme 1, occurs via the lower-valent FeIII(OH)-to-FeII transformation.1,23 Recently, we showed that a nonheme FeIII(OMe) complex reacts with trityl radical via homolytic cleavage of the Fe−OMe bond to give Ph3COMe and FeII.24 However, no kinetic data were obtained and the mechanism was not examined in detail. To our knowledge, experimentally determined rates of the rebound reaction in nonheme enzymes or models are not known.
Scheme 1.
Comparison of Heme and Nonheme Iron Enzymes
Herein, we show that [FeIII(N3PyO2Ph)(OCH3)](ClO4) (1) reacts with a series trityl radical derivatives, and a detailed kinetic study provides key insights regarding the mechanism of these reactions. Density functional theory (DFT) calculations help support the observed reactivity.
In our initial report, we employed triphenylmethyl radical (Ph3C•), a stable carbon radical, for reaction with 1. In the current work, we varied the electronic character of the radical derivatives by para substitution.25 A series of para-X-substituted radicals (X = OCH3, tBu, Ph, CN) were prepared. Addition of (4-tBu-C6H4)3C• (5 equiv) to 1 (Scheme 2) led to the conversion of dark purple 1 to yellow-orange within 5 min. Analysis by 1H NMR spectroscopy of the same reaction mixture in toluene-d8 showed formation of the methoxy group of (p-tBu-C6H4)3COCH3. Quantitation gave a 70% yield of methoxyether (see Figure S8 in the Supporting Information). The 57Fe-labeled 1 in THF/toluene revealed a broad doublet in the Mössbauer spectrum (δ = 0.5 mm s−1, |ΔEQ| = 1.29 mm s−1), which is indicative of an FeIII complex in an intermediate relaxation regime, as reported earlier.24 This doublet disappears following the addition of (4-tBu-C6H4)3C•, giving rise to two new high-spin FeII subcomponents: δ = 1.16 mm s−1, |ΔEQ| = 2.68 mm s−1 (80% of total fit) and δ = 1.34 mm s−1, |ΔEQ| = 3.29 mm s−1 (20% of total fit) (see Figure S10 in the Supporting Information). The latter component exhibits parameters that are similar to the FeII species formed after reaction with the unsubstituted Ph3C•,24 although this previous reaction was operated in pure THF. The solvent mixture employed here (toluene/THF 1:2) likely leads to different solvent binding equilibria that produces two, related high-spin FeII products, which, in turn, gives rise to the two overlapping Mössbauer signals. There is no evidence of any FeIII starting material remaining. When acetonitrile is added to the product mixture, the spectrum resolves to a single high-spin FeII component, δ = 1.16 mm s−1, ΔEQ = 2.66 mm s−1, matching that for [FeII(N3PyO2Ph)(CH3CN)]+,24 and indicating that 1 is quantitatively reduced to iron(II) via the radical reaction. The 1H NMR and Mössbauer spectra indicate that (4-tBu-C6H4)3C• reacts with 1 in good yield via the radical reaction shown in Scheme 2.
Scheme 2.
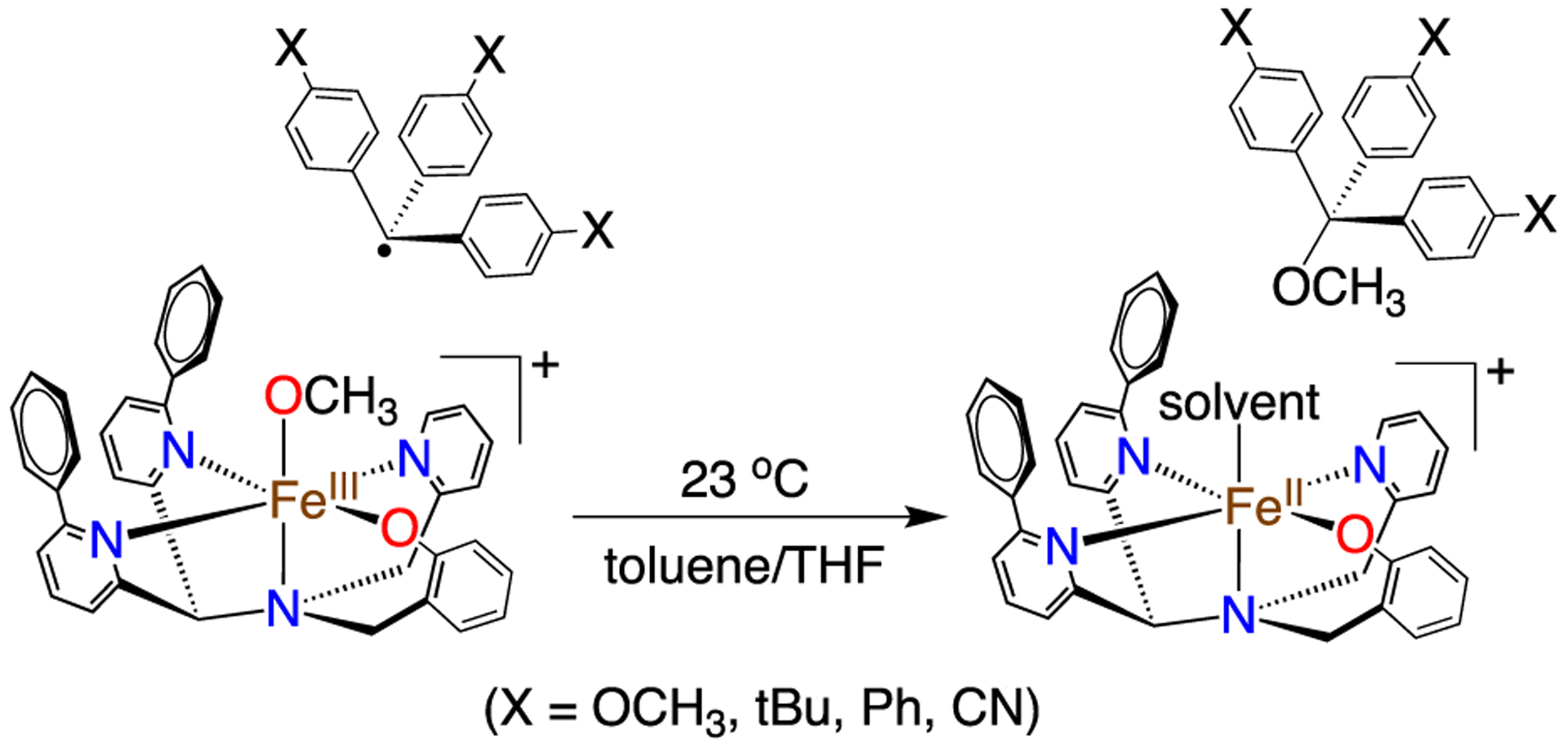
Reaction of 1 with para-X-Substituted Triphenylmethyl Radicals
Mechanistic information was obtained from kinetic studies. Complex 1 was reacted with (p-X-C6H4)3C• derivatives under pseudo-first-order conditions (excess radical) in toluene at 23 °C. The consumption of 1 could be followed by UV-vis spectroscopy, leading to decay curves that give a pseudo-first-order rate constant (kobs) (see Figures S2–S5 in the Supporting Information). Measuring the kobs values at different radical concentrations led to second-order rate constants (k2) (Figure 1A) with a range of k2 values between 2–25 M−1 s−1 (X = CN < Ph < tBu < OMe), which increased with the electron-donating ability of the para-substituent.
Figure 1.
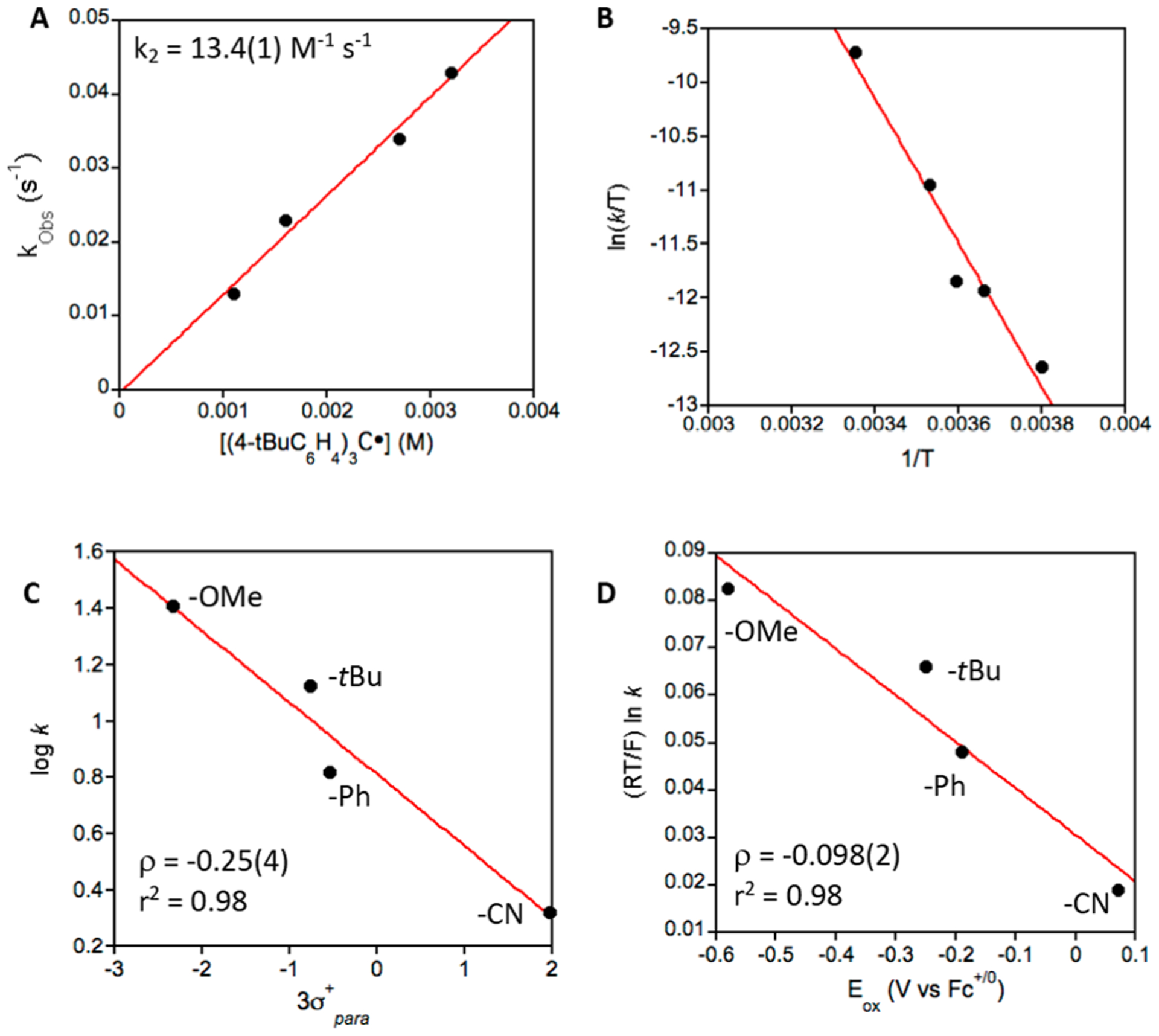
(A) Plot of kobs versus [(p-tBu-C6H4)3C•], where the slope of the best fit (red line) gives k2 = 13.4(1) M−1 s−1. (B) Eyring plot of (ln(kobs/T) vs 1/T for the reaction of 1 and (p-tBu-C6H4)3-C• from −10 °C to 25 °C. (C) Hammett plot. (D) Marcus plot.
Although there are no other analogous rate constants available for nonheme iron complexes for comparison, we can compare these rate constants to those measured previously for a heme-like corrole complex. The iron corrole Fe(OH)(ttppc) (ttppc = tris(2,4,6-triphenylphenyl corrole) undergoes similar radical reactivity with (p-X-C6H4)3C• (X = −OCH3, −tBu, −Ph, Cl) to give (p-X-C6H4)3COH and FeIII (ttppc), with rate constants ranging from 12.6(1) to 357(4) M−1 s−1.21 The ttppc complex is sterically encumbered by large triphenylphenyl groups, but appears to react significantly faster than 1; e.g., Fe(OH)(ttppc) reacts 14 times faster than 1 with the para-OMe derivative. The physical oxidation state of the iron center in the ttppc complex is not easily assigned, because of the possible noninnocent behavior of the corrole ligand, but the overall redox level is one unit above the nonheme system (formally FeIV(OH)), and this difference in redox levels may, in part, help to explain the enhanced reactivity of the corrole complex. Alternatively, the difference in reactivity may be due to the steric demands of the axial OMe versus OH ligand, or may arise from an inherent difference in Fe−OMe versus Fe−OH homolytic bond strengths. Further work is needed on both heme and nonheme systems to resolve these fundamental questions.
A Hammett plot (Figure 1C) consisting of log k2 vs 3σ +, where σ + is the Hammett parameter for the para-X substituents in (p-X-C6H4)3C•. The rates decrease linearly with the σ + values, indicating that 1 is behaving as an electrophile, as expected. However, the slope is small (ρ = −0.25). In fact, it is less than half the slope seen for the Hammett plot for Fe(OH)(ttppc) (ρ = −0.55) with the same trityl radical derivatives. Both ρ values suggest little charge separation in the transition state,21,26 and 1 is even less sensitive to the electrophilicity of the radical derivatives.
The reaction for 1 with the carbon radicals could occur as a concerted process in which C−O bond formation occurs with concomitant Fe−O bond cleavage and reduction of FeIII to FeII, or it could proceed following a stepwise electron-transfer/cation transfer (ET/CT) pathway shown in Scheme 3. The possibility of an ET/CT mechanism was addressed by a Marcus plot (Figure 1). The plot shows reasonable linearity, but the slope (ρ = −0.098) is quite small, showing only a weak dependence on the redox potentials of the radical substrates. In comparison, a rate-limiting, outer-sphere ET process gives typical slopes on the order of −0.5.21,27 The same plot for the reaction of Fe(OH)(ttppc) also gave a small slope (ρ = −0.15). This analysis indicates the mechanism for 1 is best-described as a concerted process (i.e., the diagonal path in Scheme 3). The same Marcus analysis has also been applied to metal-mediated H atom transfer reactions, with small slopes indicating a concerted proton electron-transfer (CPET) process.28,29
Scheme 3.
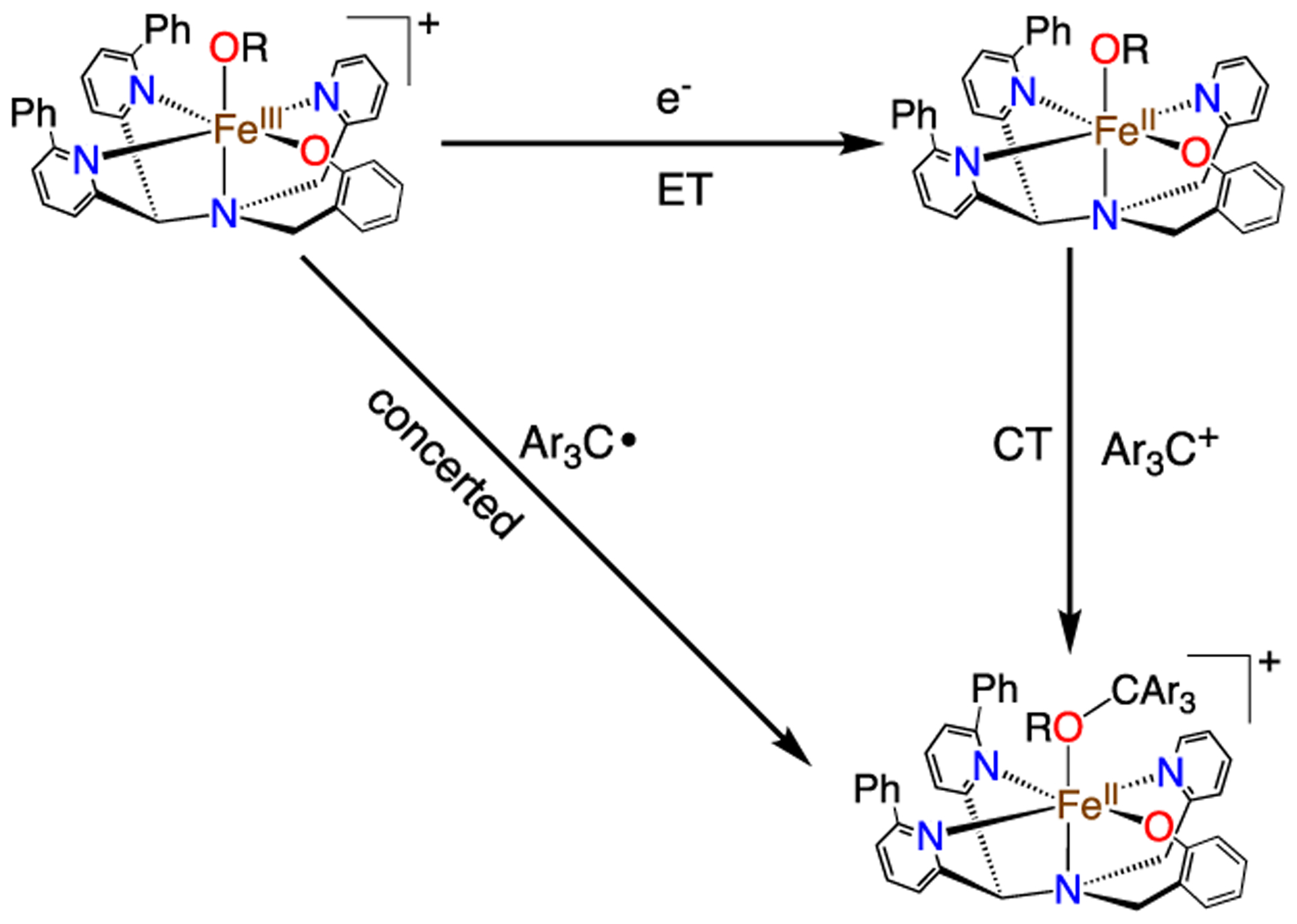
Concerted versus ET/CT Pathways
The pseudo-first-order rate constants (kobs) were measured between −10 °C and 25 °C, and a plot of ln(k/T) versus 1/T (Figure 1) gave activation parameters: ΔH‡ = 13.2(1.6) kcal mol−1 and ΔS‡ = −22.1(0.4) cal mol−1 K−1, and ΔG‡ = 19.7(1.7) kcal mol−1 at 298 K. These values are consistent with a bimolecular rate-determining step.
Reaction of 1 and trityl cation, Ph3C+, gives very different results. Combining 1 and (Ph3C)BF4 (4 equiv) in THF at 23 °C led to a reaction that was over within seconds, as seen by UV-vis. The ether product (Ph3COCH3) was formed in 80% yield (Figure S9 in the Supporting Information), and the iron complex did not change oxidation state, as seen by UV-vis (Figure S7 in the Supporting Information) and Mössbauer spectroscopy (see Figure S10 in the Supporting Information). The nucleophilic reactivity of 1 with trityl cation is consistent with the reactivity observed for other metal-alkoxide complexes.30 It appears as though heterolytic cleavage of the Fe−O bond in 1 is facile, compared to homolytic cleavage, although the origins of this difference are not well understood at this time.
DFT calculations on [Fe(OCH3)(N3PyO2Ph)]+, and the related hypothetical hydroxide complex [Fe(OH)(N3PyO2Ph)]+, led to optimized geometries for the sextet spin ground states of these species (see Figure S11 in the Supporting Information). The metrical parameters for 6[Fe(OCH3)(N3PyO2Ph)]+ match reasonably well with the reported crystal structure, except for Fe−N1(py), which is elongated by ~0.10 Å in the DFT structure. The initial reactant complex (Re) involves the association of the FeIII complex with a radical substrate whose unpaired spin is aligned parallel (septet) or antiparallel (quintet) to the Fe d electrons. The quintet states were uniformly lower in energy. No charge transfer in the Re was observed. Attempts to interchange molecular orbitals to generate a charge-transfer state (e.g., 5[Fe(OH)(N3PyO2Ph)]0−(4-Cl-C6H4)3C+) for 5ReOH,Cl) always converged back to the structure with a radical on the carbon moiety. The [FeII(N3PyO2Ph)+ + ROCAr3)] (R = H, Me) products (Pr) were also calculated, and their relative energies showed similar exothermicities (5Pr(ΔE + ZPE) − (5Re(ΔE + ZPE)) = −7 to −8 kcal mol−1) for the FeIII(OMe) and FeIII(OH) complexes, suggesting similar reaction pathways for these species. Transition state (TS) structures were located for the FeIII(OMe) and FeIII(OH) complexes and the p-Cl− radical, and free energies of activation with the Wertz entropy correction give values of 23.9 and 25.0 kcal mol−1, respectively. The calculated enthalpy values (see Table S9 in the Supporting Information) are relatively close to the experimental value obtained from the Eyring analysis.
Transition state (TS) structures were located for the FeIII(OMe) and FeIII(OH) complexes and the p-Cl− radical, and they are indeed similar in both geometry and relative energies, although one of the py donors appears dissociated from the metal in both structures (Figure 2) due to steric clash, as a result of substrate approach. However, this Fe-py bond is restored during product formation, giving the expected FeII product. The structures shown in Figure 2 indicate relatively early transition states, consistent with facile, exothermic reactions. The DFT results show that a concerted, radical group transfer mechanism is favored for both the FeIII(OMe) and FeIII(OH) complexes, and the electronic changes are similar for both species. Calculations for other heme and nonheme iron-hydroxo complexes have also shown a single transition state, implicating a concerted process.31–33
Figure 2.
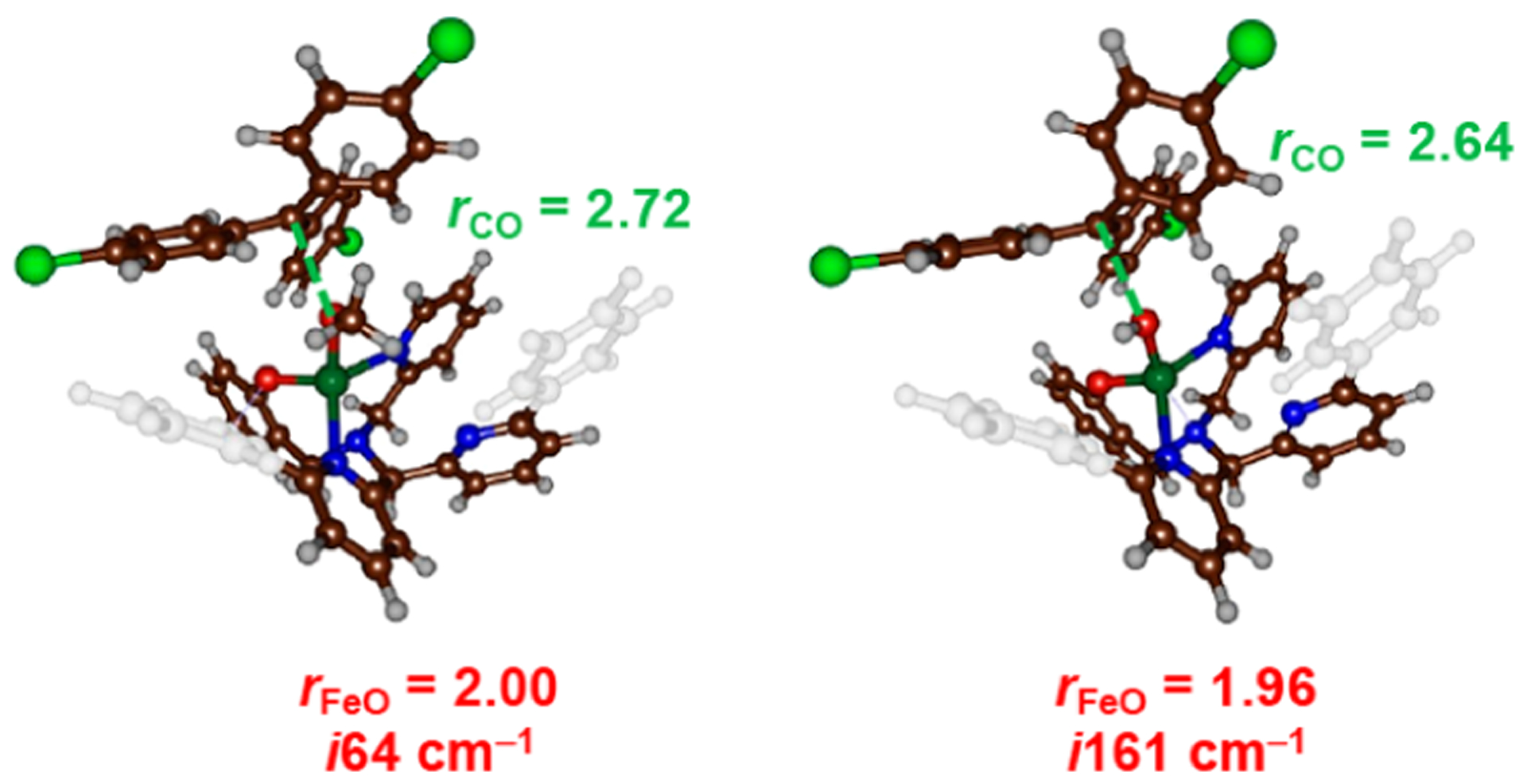
Transition-state structures for [FeIII(OCH3)(N3PyO2Ph)]+ (left) and [FeIII(OH)(N3PyO2Ph)]+ (right) with (4-Cl-C6H4)3C•. The C−O and Fe−O distances (in Å), and imaginary frequencies, are listed for each structure.
The combined experimental and computational data suggest that the radical rebound step in nonheme iron enzymes or synthetic catalysts may proceed by a concerted process, with little or no charge transfer prior to C−O bond formation. The sizable reaction barrier for the rebound process for complex 1 is significantly different from the almost-barrierless rebound processes predicted for nonheme iron enzymes. However, the model complex reacts via a bimolecular process, whereas the radicals in the enzymes are trapped in the active site pocket and react directly via a first-order process. In addition, the trityl radical derivatives are significantly more stable than typical primary or secondary alkyl radicals generated in the biological systems. The conclusion that a concerted “rebound” pathway is operative for 1 suggests that radical rebound in nonheme iron enzymes also may be concerted, which could help determine product selectivity. The substrate radical must be held in a close, appropriate orientation for a concerted pathway to lead to a productive reaction. Obtaining such information from model systems should further our understanding of the selectivity of rebound processes in nonheme iron enzymes such as the α-KG halogenases, IPNS, and related systems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH through No. GM119374 to D.P.G., S.P.d.V., and E.F.G. thank the BBSRC for a studentship (under Grant No. BB/M011208/1).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.9b01208.
Experimental procedures, UV-vis data, kinetics, Mössbauer data, and DFT methods (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Huang X; Groves JT Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C-H activation. JBIC, J. Biol. Inorg. Chem 2017, 22, 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nam W; Lee Y-M; Fukuzumi S Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res 2014, 47, 1146–1154. [DOI] [PubMed] [Google Scholar]
- (3).Bollinger JM Jr.; Price JC; Hoffart LM; Barr EW; Krebs C Mechanism of Taurine: α-Ketoglutarate Dioxygenase (TauD) from Escherechia coli. Eur. J. Inorg. Chem 2005, 2005, 4245–4254. [DOI] [PubMed] [Google Scholar]
- (4).Cho KB; Hirao H; Shaik S; Nam W To rebound or dissociate? This is the mechanistic question in C-H hydroxylation by heme and nonheme metal-oxo complexes. Chem. Soc. Rev 2016, 45, 1197–210. [DOI] [PubMed] [Google Scholar]
- (5).Liao H-J; Li J; Huang J-L; Davidson M; Kurnikov I; Lin T-S; Lee JL; Kurnikova M; Guo Y; Chan N-L; Chang W.-c . Insights into the Desaturation of Cyclopeptin and its C3 Epimer Catalyzed by a non-Heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chem., Int. Ed 2018, 57, 1831− 1835. [DOI] [PubMed] [Google Scholar]
- (6).Dunham NP; Chang W.-c.; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Two Distinct Mechanisms for C-C Desaturation by Iron(II)- and 2-(Oxo)glutarate-Dependent Oxygenases: Importance of α-Heteroatom Assistance. J. Am. Chem. Soc 2018, 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hsieh CH; Huang X; Amaya JA; Rutland CD; Keys CL; Groves JT; Austin RN; Makris TM The Enigmatic P450 Decarboxylase OleT is Capable of, but Evolved to Frustrate, Oxygen Rebound Chemistry. Biochemistry 2017, 56, 3347–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rude MA; Baron TS; Brubaker S; Alibhai M; Del Cardayre SB; Schirmer A Terminal Olefin (1-Alkene) Biosynthesis by a Novel P450 Fatty Acid Decarboxylase from Jeotgalicoccus Species. Appl. Environ. Microbiol 2011, 77, 1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu Y; Wang C; Yan J; Zhang W; Guan W; Lu X; Li S Hydrogen peroxide-independent production of α-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol. Biofuels 2014, 7, 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dennig A; Kuhn M; Tassoti S; Thiessenhusen A; Gilch S; Bülter T; Haas T; Hall M; Faber K Oxidative Decarboxylation of Short-Chain Fatty Acids to 1-Alkenes. Angew. Chem., Int. Ed 2015, 54, 8819–8822. [DOI] [PubMed] [Google Scholar]
- (11).Matthews S; Belcher JD; Tee KL; Girvan HM; McLean KJ; Rigby SEJ; Levy CW; Leys D; Parker DA; Blankley RT; Munro AW Catalytic Determinants of Alkene Production by the Cytochrome P450 Peroxygenase OleTJE. J. Biol.Chem 2017, 292, 5128–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rui Z; Li X; Zhu X; Liu J; Domigan B; Barr I; Cate JHD; Zhang W Microbial biosynthesis of medium-chain 1-alkenes by a nonheme iron oxidase. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 18237–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mitchell AJ; Zhu Q; Maggiolo AO; Ananth NR; Hillwig ML; Liu X; Boal AK Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat. Chem. Biol 2016, 12, 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Martinie RJ; Livada J; Chang W; Green MT; Krebs C; Bollinger JM Jr.; Silakov A Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J. Am. Chem. Soc 2015, 137, 6912–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wong C; Fujimori DG; Walsh CT; Drennan CL Structural Analysis of an Open Active Site Conformation of Nonheme Iron Halogenase CytC3. J. Am. Chem. Soc 2009, 131, 4872–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Puri M; Biswas AN; Fan R; Guo Y; Que L Jr. Modeling Non-Heme Iron Halogenases: High-Spin Oxoiron(IV)-Halide Complexes That Halogenate C-H Bonds. J. Am. Chem. Soc 2016, 138, 2484–2487. [DOI] [PubMed] [Google Scholar]
- (18).Tamanaha E; Zhang B; Guo Y; Chang W.-c.; Barr EW; Xing G; St. Clair J; Ye S; Neese F; Bollinger JM; Krebs C Spectroscopic Evidence for the Two C-H Cleaving Intermediates of Aspergillis nidulans Isopenicillin N Synthase. J. Am. Chem. Soc 2016, 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Planas O; Clemancey M; Latour J-M; Company A; Costas M Structural modeling of iron halogenases: synthesis and reactivity of halide-iron(IV)-oxo compounds. Chem. Commun 2014, 50, 10887− 10890. [DOI] [PubMed] [Google Scholar]
- (20).Rana S; Dey A; Maiti D Mechanistic elucidation of C-H oxidation by electron rich non-heme iron(IV)-oxo at room temperature. Chem. Commun 2015, 51, 14469–14472. [DOI] [PubMed] [Google Scholar]
- (21).Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc 2017, 139, 13640–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Baglia RA; Zaragoza JPT; Goldberg DP Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev 2017, 117, 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Price JC; Barr EW; Hoffart LM; Krebs C; Bollinger JMJ Kinetic Dissection of the Catalytic Mechanism of Taurine: α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2005, 44, 8138–8147. [DOI] [PubMed] [Google Scholar]
- (24).Pangia TM; Davies CG; Prendergast JR; Gordon JB; Siegler MA; Jameson GNL; Goldberg DP Observation of Radical Rebound in a Mononuclear Nonheme Iron Model Complex. J. Am. Chem. Soc 2018, 140, 4191–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co-H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]
- (26).Colclough N; Smith JRL A Mechanistic Study of the Oxidation of Phenols in Aqueous Solution by Oxoiron(IV) Tetra(N-methylpyridyl)porphyrins. A Model for Horseradish Peroxidase Compound II? J. Chem. Soc., Perkin Trans 2 1994, 2, 1139–1149. [Google Scholar]
- (27).Marcus RA; Sutin N Electron transfers in chemistry and biology. Biochim. Biophys. Acta, Rev. Bioenerg 1985, 811, 265–322. [Google Scholar]
- (28).Osako T; Ohkubo K; Taki M; Tachi Y; Fukuzumi S; Itoh S Oxidation Mechanism of Phenols by Dicopper-Dioxygen (Cu2/O2) Complexes. J. Inorg. Biochem 2003, 96, 207. [DOI] [PubMed] [Google Scholar]
- (29).Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric-Superoxo Complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Blanchard J; In M; Schaudel B; Sanchez C Hydrolysis and Condensation Reactions of Transition Metal Alkoxides: Calorimetric Study and Evaluation of the Extent of Reaction. Eur. J. Inorg. Chem 1998, 1998, 1115–1127. [Google Scholar]
- (31).Shaik S; Cohen S; de Visser SP; Sharma PK; Kumar D; Kozuch S; Ogliaro F; Danovich D The “Rebound Controversy”: An Overview and Theoretical Modeling of the Rebound Step in C-H Hydroxylation by Cytochrome P450. Eur. J. Inorg. Chem 2004, 2004, 207–226. [Google Scholar]
- (32).Li X-X; Postils V; Sun W; Faponle AS; Solà M; Wang Y; Nam W; de Visser SP Reactivity Patterns of (Protonated) Compound II and Compound I of Cytochrome P450: Which is the Better Oxidant? Chem. - Eur. J 2017, 23, 6406–6418. [DOI] [PubMed] [Google Scholar]
- (33).Timmins A; Quesne MG; Borowski T; de Visser SP Group Transfer to an Aliphatic Bond: A Biomimetic Study Inspired by Nonheme Iron Halogenases. ACS Catal. 2018, 8, 8685–8698. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.