

SUMMARY
Intrahepatic cholangiocarcinoma (ICC) is a highly malignant, heterogeneous cancer with poor treatment options. We found that mitochondrial dysfunction and oxidative stress trigger a niche favoring cholangiocellular overgrowth and tumorigenesis. Liver damage, reactive oxygen species (ROS) and paracrine tumor necrosis factor (Tnf) from Kupffer cells caused JNK-mediated cholangiocellular proliferation and oncogenic transformation. Anti-oxidant treatment, Kupffer cell depletion, Tnfr1 deletion, or JNK inhibition reduced cholangiocellular pre-neoplastic lesions. Liver-specific JNK1/2 deletion led to tumor reduction and enhanced survival in Akt/Notch- or p53/Kras-induced ICC models. In human ICC, high Tnf expression near ICC lesions, cholangiocellular JNK-phosphorylation, and ROS accumulation in surrounding hepatocytes are present. Thus, Kupffer cell-derived Tnf favors cholangiocellular proliferation/differentiation and carcinogenesis. Targeting the ROS/Tnf/JNK axis may provide opportunities for ICC therapy.
Graphical Abstract
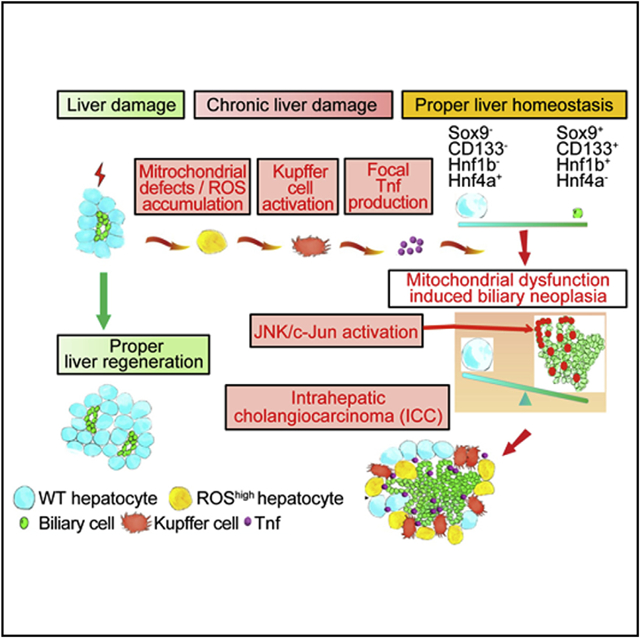
In Brief
Yuan et al. show that mitochondrial dysfunction and oxidative stress in the liver increase tumor necrosis factor (TNF) expression by Kupffer cells to cause JNK-mediated cholangiocellular proliferation and transformation. The ROS/TNF/JNK axis may be an effective target for intrahepatic cholangiocarcinoma therapy.
INTRODUCTION
Intrahepatic cholangiocarcinoma (ICC) is a liver cancer typically diagnosed at advanced stages, with poor prognosis and increasing incidence (Gatto and Alvaro, 2010). Cellular origins and molecular mechanisms underlying ICC progression are poorly understood (Zender et al., 2013; Zhang et al., 2008). ICC is observed in both diseases affecting biliary epithelial cells such as primary sclerosing cholangitis (Rizvi et al., 2015) and in diseases that cause chronic hepatocyte injury, such as chronic hepatitis C or B virus infection, chronic alcohol abuse, and nonalcoholic steatohepatitis (NASH) (Ariizumi and Yamamoto, 2014; Tyson and El-Serag, 2011). Of note, a common feature of these etiologies is mitochondrial dysfunction and high reactive oxygen species (ROS) levels.
Genomic and transcriptomic analyses revealed different mutational and transcriptome profiles between hepatocellular carcinoma (HCC) and ICC (Fujimoto et al., 2015; Jiao et al., 2013; Zou et al., 2014). A series of distinct developmental signals preferentially used by hepatocytes or cholangiocytes have been uncovered using rodent liver injury models (Boulter et al., 2012; Kang et al., 2012). However, the role of pro-inflammatory signaling pathways in ICC development under conditions of chronic liver damage remains elusive. Excessive remodeling of the inflammatory microenvironment is known to occur in chronic liver disease (Szabo and Petrasek, 2015). Given recent studies indicating a link between pro-inflammatory signaling pathways and cell plasticity in the intestine and breast epithelium (Scheeren et al., 2014; Taniguchi et al., 2015), it is likely that such pathways, in addition to their roles in immune regulation, may shape the cell plasticity of liver cells.
The pro-inflammatory cytokine tumor necrosis factor (Tnf) is mainly secreted by Kupffer cells in adult livers (Roberts et al., 2007), and has pro-survival/pro-growth effects on cells of specific lineages during development (Espin-Palazon et al., 2014; Liu et al., 2014). Malignant cells may hijack the Tnf-dependent pro-survival program to obtain a selective growth advantage. Indeed, Tnf has been implicated in cancer progression by sustaining expansion of neoplastic cells, including skin cancer, cervical carcinomas, and HCC (Arnott et al., 2004; Nakagawa et al., 2014; Pikarsky et al., 2004; Woodworth et al., 1995). However, the effects of persistent Tnf production on cholangiocytes under conditions of chronic liver injury and high ROS have remained elusive.
Thus, in this study we examined the role of mitochondrial dysfunction and ROS in ICC development.
RESULTS
Hepatic Mitochondrial Dysfunction Leads to Severe Liver Damage, Hepatocyte Proliferation, and Premalignant Cholangiocellular Lesions
To determine the effect of high ROS and mitochondrial dysfunction on ICC development, we analyzed ICC mouse models, including CRISPR/Cas9-induced ICC (Weber et al., 2015), constitutively active Akt-1 (Akt), together with Nras- (Akt/Nras) or Notch1-induced ICC (Akt/Notch) (Matter et al., 2016), and transposon-mediated in vivo delivery of KrasG12D-induced ICC (HDTV Kras) (M.S. and L.Z., unpublished data). 8-Hydroxy-2-deoxyguanosine (8-OHdG), an indicator of secondary metabolites due to oxidative DNA damage, was evaluated in tumors and adjacent tissues. All four ICC models exhibited extensive 8-OHdG staining in CK19+ neoplastic cells compared with normal cholangiocytes. Intriguingly, 8-OHdG positivity was not only seen in malignant cholangiocytes, but also in surrounding hepatocytes (Figure S1A), implying that oxidative stress in the liver microenvironment could correlate with ICC development.
We next performed 8-OHdG immunohistochemistry (IHC) in 121 human ICC samples. Nearly 80% of ICCs showed 8-OHdG+ malignant cholangiocytes as well as surrounding hepatocytes (Figure S1B), suggesting an association between oxidative stress and ICC development.
To mimic hepatic mitochondrial dysfunction, we generated knockin mice with loxP sites flanking exons 4 to 8 of Hspd1 (Figure S1C) (Berger et al., 2016) and crossed them with Alb-Cre transgenics (Postic et al., 1999) to produce mice with liver-specific Hspd1 deletion (Hspd1ΔLPC) (Figures S1D and S1E). qRT-PCR and Western blot revealed loss of Hspd1 transcript and protein in livers from 6-week-old mice (Figure S1F). IHC confirmed absence of Hspd1 in both hepatocytes and cholangiocytes (Figure S1G). Hepatic Hspd1 deletion induced severe mitochondrial defects. Ultrastructural analyses revealed fragmented, enlarged and swollen mitochondria, and mitochondria encapsulated in double-membrane autophagosomes or autolysosomes (mitophagy) in Hspd1ΔLPC livers (Figure 1A). Strong 8-OHdG staining in Hspd1ΔLPC liver cells and elevated levels of oxidized protein by oxyblot analysis further confirmed ROS accumulation (Figures 1A and S1H).
Figure 1. Lack of Hspd1 Causes Acute Liver Damage, Hepatic Proliferation, and Cholangiocellular Tumorigenesis.
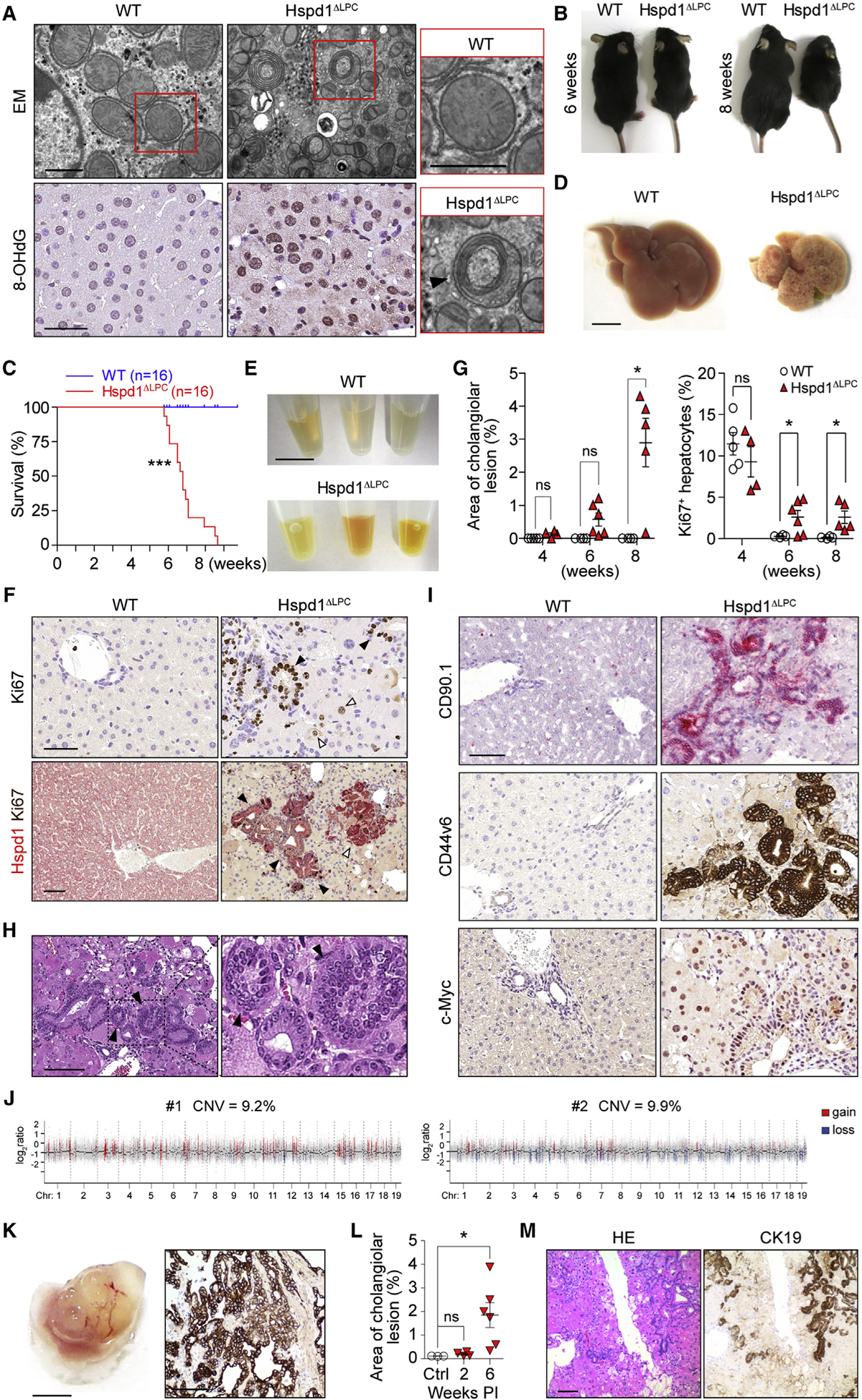
(A) Electron microscopy and 8-OHdG staining of WT and Hspd1ΔLPC livers. Arrowhead indicates a classic mitophagosome. Scale bars, 0.5 μm (upper), 50 μm (lower).
(B) Six- and 8-week-old Hspd1ΔLPC mice and WT littermates.
(C) Survival curve of Hspd1ΔLPC and WT mice.
(D) Eight-week-old Hspd1ΔLPC and WT littermate livers. Scale bar, 5 mm.
(E) Serum from 8-week-old Hspd1ΔLPC mice and WT littermates. Scale bar, 1 cm.
(F) Upper: Ki67 staining of 6-week-old Hspd1ΔLPC and WT livers. Black arrowheads, Ki67+ cholangiocytes; white arrowheads, Ki67+ hepatocytes. Lower: double staining of Hspd1 (red) and Ki67 (brown) of 8-week-old Hspd1ΔLPC and WT livers, indicating foci of hepatocyte (white arrowhead) and cholangiocellular (black arrowhead) proliferation. Scale bar, 50 μm.
(G) Quantification of cholangiocellular lesions and Ki67+ hepatocytes over time in Hspd1ΔLPC mice and WT littermates.
(H) H&E staining of Hspd1ΔLPC liver. Arrowheads indicate biliary intraepithelial neoplasia. Scale bar, 100 μm.
(I) IHC analysis in 8-week-old WT and Hspd1ΔLPC livers. Scale bar, 50 μm.
(J) Genomic hybridization profiles of CK19+ cholangiocellular neoplasia.
(K) Macroscopy (left) and CK19 staining (right) of tumor graft from severe combined immunodeficiency Beige liver. Scale bars, 5 mm (left), 100 μm (right).
(L) Quantification of cholangiocellular lesions over time in AAV8-Cre Hspd1loxP/loxP and control mice.
(M) H&E and CK19 staining of AAV8-Cre Hspd1loxP/loxP livers. Scale bar, 100 μm.
Data are represented as the mean ± SEM. *p < 0.05, ***p < 0.001. ns, not significant. See also Figures S1 and S2.
Postnatal day 2 (P2) and day 7 (P7) Hspd1ΔLPC livers showed no obvious histological or micro-architectural differences, excluding embryonic developmental defects (Figures S1I and S1J, data not shown). From the age of 4 to 6 weeks, Hspd1ΔLPC mice exhibited weakness and weight loss and had to be euthanized due to severe weight loss at 8–9 weeks of age (Figures 1B, 1C, and S1K). Macroscopic signs of focal fibrosis and necrosis were evident as early as P38 in Hspd1ΔLPC livers (Figure S1L). At 8 weeks, Hspd1ΔLPC mice had significantly higher liver-to-body weight ratio, pale livers, and yellow-colored serum, indicative of cholestasis (Figures 1D–1F and S1M). Serum analyses showed a progressive increase of ALT, AST, AP, and bilirubin levels over time (Figure S1N), indicating hepatocyte and bile duct damage. H&E and TUNEL staining revealed widespread hepatocyte cell death (Figure S1O), not attributable to caspase-3 activation (Figure S1P). Sirius Red staining at 8 weeks revealed severely altered liver architecture with concomitant collapse, regeneration, and fibrosis (Figure S1Q). Hepatocyte death and bile duct defects coincided with disturbed liver functions (Figure S1R). All of the above-described pathologic manifestations point to severe liver damage suggestive of liver failure, which most likely accounts for the premature lethality of the Hspd1ΔLPC mice.
Focal areas of hepato- and cholangiocellular regeneration were found from 6 weeks onwards (Figures 1G and 1H), accompanied by upregulation of Sox9, cell-cycle-related proteins Pcna and Cyclin D1, and p-Akt (Figures S1S and S1T). Ki67+ hepatocytes forming regenerative nodules were Hspd1+ (Figure S1U), while Hspd1− hepatocytes were rarely Ki67+ in 8-week-old Hspd1ΔLPC livers (Figure S1V). PCR and qRT-PCR of regenerative nodules and surrounding Hspd1− hepatocytes revealed that the Hspd1loxP/loxP alleles were largely intact in regenerative hepatocytes, in contrast to surrounding Hspd1− regions (Figures S1W and S1X). Next, we crossed Hspd1ΔLPC mice with the stochastic multicolor Cre-recombinase reporter line, Confetti (Livet et al., 2007). Hspd1− hepatocytes were labeled with multi-colored fluorescent proteins, while Hspd1+ hepatocytes remained unlabeled in 8-week-old Hspd1ΔLPC mice, supporting the lack of Cre-mediated recombination in regenerating Hspd1+ hepatoctyes (Figure S2A).
In parallel with proliferating hepatocytes, marked expansion of cholangiocytes was observed, including glands or trabecular structures of cells with scant cytoplasm and hyperchromatic nuclei by 7–8 weeks (Figures 1F, 1G, S2B, and S2C). Cholangiocellular lesions, characterized by irregular glands, loss of polarity, multi-layering of cells, and frequent mitosis, were evident in 8-week-old Hspd1ΔLPC livers (Figure 1H). These foci, assessed by board-certified pathologists, resembled human biliary intraepithelial neoplasia, a precursor cancerous ICC lesion (Rougemont et al., 2010). Cholangiocellular lesions were positive for pan-CK, A6, CK19, and Erbb2, and negative for α-fetoprotein (Afp) and intestinal lineage markers, underlining the biliary fate of these cells (Figures S2D–S2F). Furthermore, these lesions co-stained for validated cancer stem cell (CSC) markers CD44v6 and CD90.1, c-Myc, and for collagen IV-positive stromal reaction as seen in human ICC (Figures 1I and S2D). Microarray-based comparative genomic hybridization analysis of laser-dissected CK19+ cholangiocellular lesions from independent Hspd1ΔLPC livers revealed copy-number changes of >9% across the genome compared with controls (Figure 1J). Synteny analyses revealed that 95% of chromosomal losses and 53% of chromosomal gains in Hspd1ΔLPC livers were congruent with loci in the human cholangiocarcinoma cohort from The Cancer Genome Atlas (p < 0.0001, Fisher’s exact test) (Figure S2G). Transplantation of liver pieces from one out of three 8-week-old Hspd1ΔLPC donor mice resulted in tumor formation with ICC, but not HCC characteristics, including glandular structures and CK19+ cells in three out of four transplantation experiments in severe combined immunodeficiency Beige recipient mice (Figures 1K and S2H), supporting their malignant potential. Comparable cholangiocellular lesions were absent in several other liver injury models investigated (Figure S2I). Similar to regenerating hepatocytes, proliferating cholangiocytes were almost exclusively Hspd1+ (Figures 1F and S2J). Tracing with Confetti-Hspd1ΔLPC mice further showed lack of Cre-recombinase activity in these cells (Figure S2K).
Using an adeno-associated virus (AAV) vector Cre expression system (Figure S2L) (Malato et al., 2011), we reproduced this phenotype in Hspd1loxP/loxP mice. Progressive postnatal deletion of Hspd1 was restricted to hepatocytes from 2 weeks post-AAV8 injection on (AAV8-Cre Hspd1loxP/loxP) (Figure S2M), including progressive liver damage, cholestasis, and cholangiocellular proliferation at 6–8 weeks post injection (Figures 1L, 1M, S2N, and S2O). Hspd1 expression in these cholangiocellular lesions was observed by IHC (Figure S2P). This confirms the non-cell-autonomous mode of ICC development upon Hspd1 deficiency, and suggests that the phenotype does not result from developmental defects due to Cre induction.
Mitochondrial Dysfunction Induces an Oxidative and Pro-carcinogenic Environment in Hspd1ΔLPC Livers
Genes involved in mitochondria homeostasis, the mitochondrial respiratory chain, and molecules downstream of CHOP (encoded by Ddit3) capable of regulating cell death were upregulated in Hspd1ΔLPC livers (Figures S3A and S3B). In contrast, most ER unfolded protein response associated genes remained unchanged, indicating that mitochondrial UPR (mtUPR) is primarily responsive to Hspd1 deficiency. A large panel of anti-oxidant enzymes was also strongly upregulated in Hspd1-deficient livers (Figures 2A and S3C). 8-OHdG extensively labeled Hspd1− but not Hspd1+ hepatocytes (Figure 2B). Early increase of phosphorylated Eif2a (p-Eif2a) and CHOP was confirmed in Hspd1ΔLPC livers from 4 weeks by Western blot (Figure 2C). Increased phosphatidylethanolamine-conjugated LC3 (LC3-II) was also observed, providing ultrastructural evidence of mitophagy in Hspd1ΔLPC hepatocytes (Figures 1A and 2C).
Figure 2. Oxidative and Pro-carcinogenic Liver Microenvironment in Hspd1ΔLPC Mice.
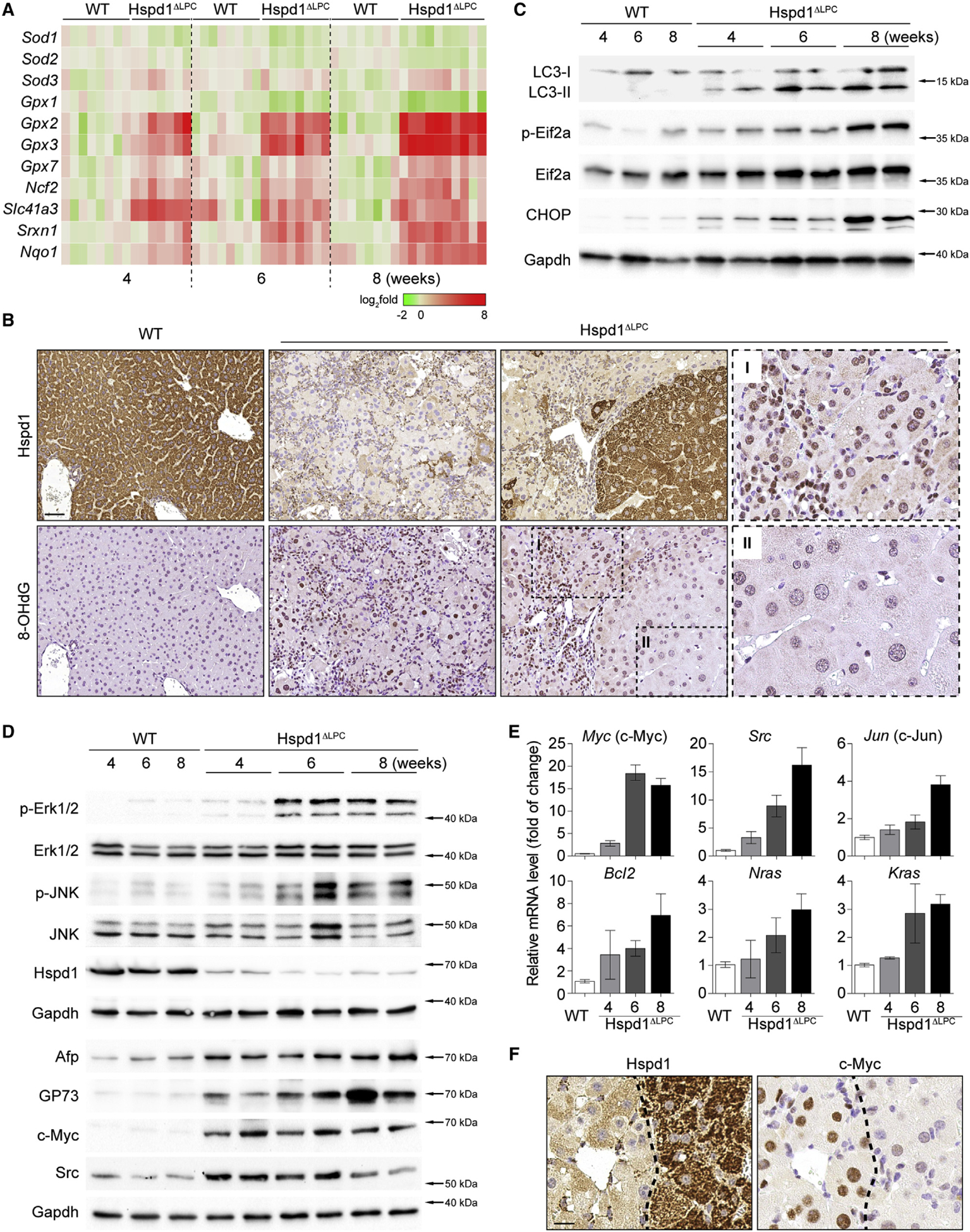
(A) Heatmap of the expression of anti-oxidant genes in Hspd1ΔLPC livers compared with WT over time. Red, upregulated; green, downregulated.
(B) Hspd1 and 8-OHdG staining of 8-week-old Hspd1ΔLPC and WT livers. Scale bar, 50 μm.
(C) Western blot analysis of Hspd1ΔLPC and WT livers from the indicated ages.
(D) Western blot analysis of Hspd1ΔLPC and WT livers from the indicated ages.
(E) qRT-PCR for the top upregulated pro-survival genes in Hspd1ΔLPC liver compared with 8-week-old WT liver. Data are represented as the mean ± SEM.
(F) Hspd1 and c-Myc IHC in consecutive liver sections from 8-week-old WT and Hspd1ΔLPC mice. The dashed lines indicate the border between the Hspd1+ and Hspd1− areas. Scale bar, 20 μm.
See also Figure S3.
We next investigated which signaling pathways were activated downstream of ROS: phosphorylation of JNK and Erk1/2, along with HCC markers, were elevated in Hspd1ΔLPC livers from week 4 (Figure 2D) but not p-p38 MAPK (Figure S3D). Staining consecutive sections revealed that most p-JNK+ cells were Hspd1− (Figure S3E). JNK activation was corroborated by nuclear translocation of c-Jun (Figure S3E). Screening also identified c-Myc, Kras, and Src as the top upregulated pro-survival genes in Hspd1ΔLPC livers (Figures 2D, 2E, and S3F), the expression of which was mainly restricted to Hspd1− areas by IHC (Figures 2F and S3G–S3I). Thus, mitochondrial defects driven by Hspd1 loss may induce an oxidative and pro-survival/oncogenic microenvironment stimulating growth of surrounding wild-type (WT) cells.
Anti-oxidant BHA Attenuates ROS Accumulation and Formation of Premalignant Cholangiocellular Lesions
To examine the role of ROS in Hspd1ΔLPC liver pathology, Hspd1ΔLPC mice were fed chow containing the anti-oxidant butylated hydroxyanisole (BHA) starting at the age of 4 weeks. BHA reduced 8-OHdG staining, expression of ROS-scavenging enzymes, and Afp level in Hspd1ΔLPC livers (Figures S4A and S4B), indicating efficient ROS blockade. Remarkably, treating Hspd1ΔLPC mice with BHA strongly attenuated macroscopically visible liver damage, weight loss, and prevented early lethality (Figures 3A, 3B, and S4C). At 8 weeks, hepatocyte necrosis and serum liver enzymes were significantly reduced (Figures 3C, 3D, and S4D). Intriguingly, BHA almost completely blocked cholangiocyte proliferation and development of cholangiocellular lesions (Figures 3E and S4E). In contrast, no effect on hepatocyte proliferation or cyclin D1 expression was found (Figure S4F). Accordingly, CK19 expression was suppressed and hepatocyte nuclear factor 4α (Hnf4a) and Aldh2 levels were partially rescued at 8 weeks (Figures 3F and S4G). Compensatory proliferation of WT hepatocytes reversed weight loss in Hspd1ΔLPC mice around weeks 18 to 20 (Figures S4C and S4H).
Figure 3. Anti-oxidants Attenuate ROS Accumulation and Premalignant Cholangiocellular Lesions in Hspd1ΔLPC Mice.
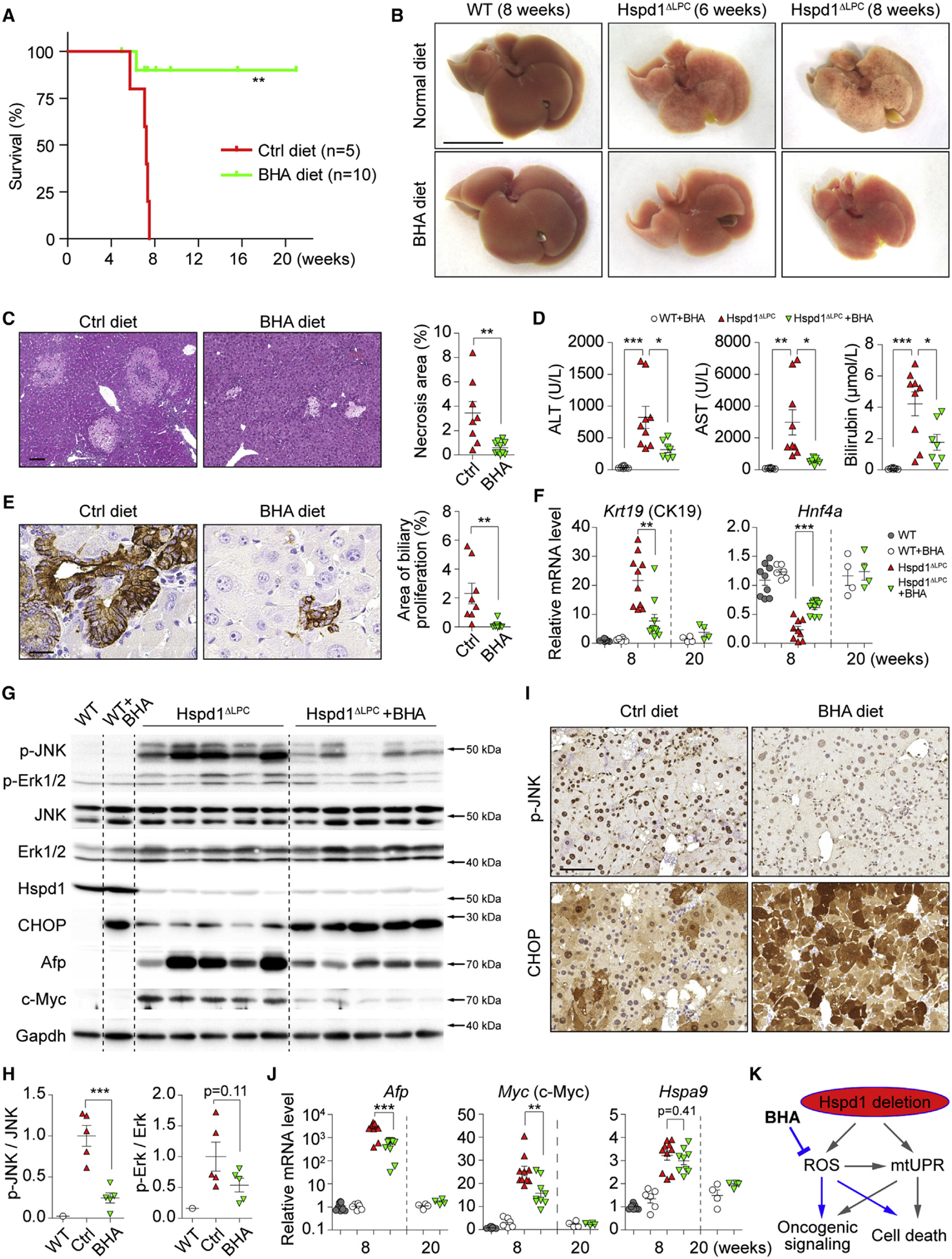
(A) Survival plots for Hspd1ΔLPC mice on normal or BHA diet.
(B) Macroscopy of the liver of 6- and 8-week-old Hspd1ΔLPC or 8-week-old WT mice on normal or BHA diet. Scale bar, 1 cm.
(C) H&E of the liver of 8-week-old Hspd1ΔLPC mice on normal versus BHA diet and quantification of necrotic areas. Scale bar, 100 μm.
(D) Serum ALT, AST, and bilirubin in 8-week-old Hspd1ΔLPC mice on normal and BHA diet.
(E) A6 IHC in 8-week-old normal versus BHA diet Hspd1ΔLPC livers and quantification of cholangiocellular overgrowth. Scale bar, 20 μm.
(F) qRT-PCR of livers from 8- and 20-week-old normal versus BHA diet Hspd1ΔLPC mice for CK19 and Hnf4α.
(G and H) Western blot of liver lysates from 8-week-old normal versus BHA diet Hspd1ΔLPC mice (G) and quantifications the p-JNK to total JNK and the p-Erk1/2 to total Erk1/2 ratio (H).
(I) IHC for p-JNK and CHOP in consecutive liver sections from normal versus BHA diet 8-week-old Hspd1ΔLPC mice. Scale bar, 50 μm.
(J) qRT-PCR of livers from 8- and 20-week-old normal versus BHA diet Hspd1ΔLPC mice for genes indicated.
(K) A schematic depicting the action of BHA in Hspd1ΔLPC mice.
Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significant. See also Figure S4.
Analysis of downstream signaling revealed that p-JNK was dampened by BHA (Figures 3G and 3H), while Erk1/2, p38 MAPK, and Eif2a were unaffected (Figures 3G, 3H, and S4I). Moreover, BHA normalized expression of pro-survival genes (Figures 3G, 3J, and S4G). However, CHOP expression was further elevated by BHA (Figures 3G and 3I). This was possibly due to BHA itself, as BHA-fed WT mice also had elevated CHOP (Figures 3G and S4J). BHA did not attenuate Hspa9, p-Eif2a, or LC3-II (Figures 3J and S4I). Thus, anti-oxidant treatment appeared to protect Hspd1ΔLPC mice by inhibiting ROS and its downstream pro-carcinogenic signaling pathways rather than attenuating mtUPR signaling (Figure 3K).
Kupffer Cell-Derived Tnf Promotes Premalignant Cholangiocellular Lesions
Next, we investigated the propagating factors that link high ROS levels and the pro-carcinogenic niche with the neoplastic phenotype. We measured mRNA expression of cytokines/ligands in Hspd1ΔLPC and WT livers (Figure 4A). Strikingly, rapid and robust induction of Tnf mRNA and protein levels, far exceeding other candidates, was observed (Figures 4A, 4B, and S5A). By contrast, Il6 mRNA levels were not changed and even reduced at protein level (Figures 4A and S5A), suggesting a niche favoring cholangiocellular proliferation. Consistent with the ameliorated Hspd1ΔLPC phenotype, Tnf expression was strongly attenuated upon BHA treatment (Figures 4B and 4C).
Figure 4. Kupffer Cells Promote Cholangiocellular Tumorigenesis.
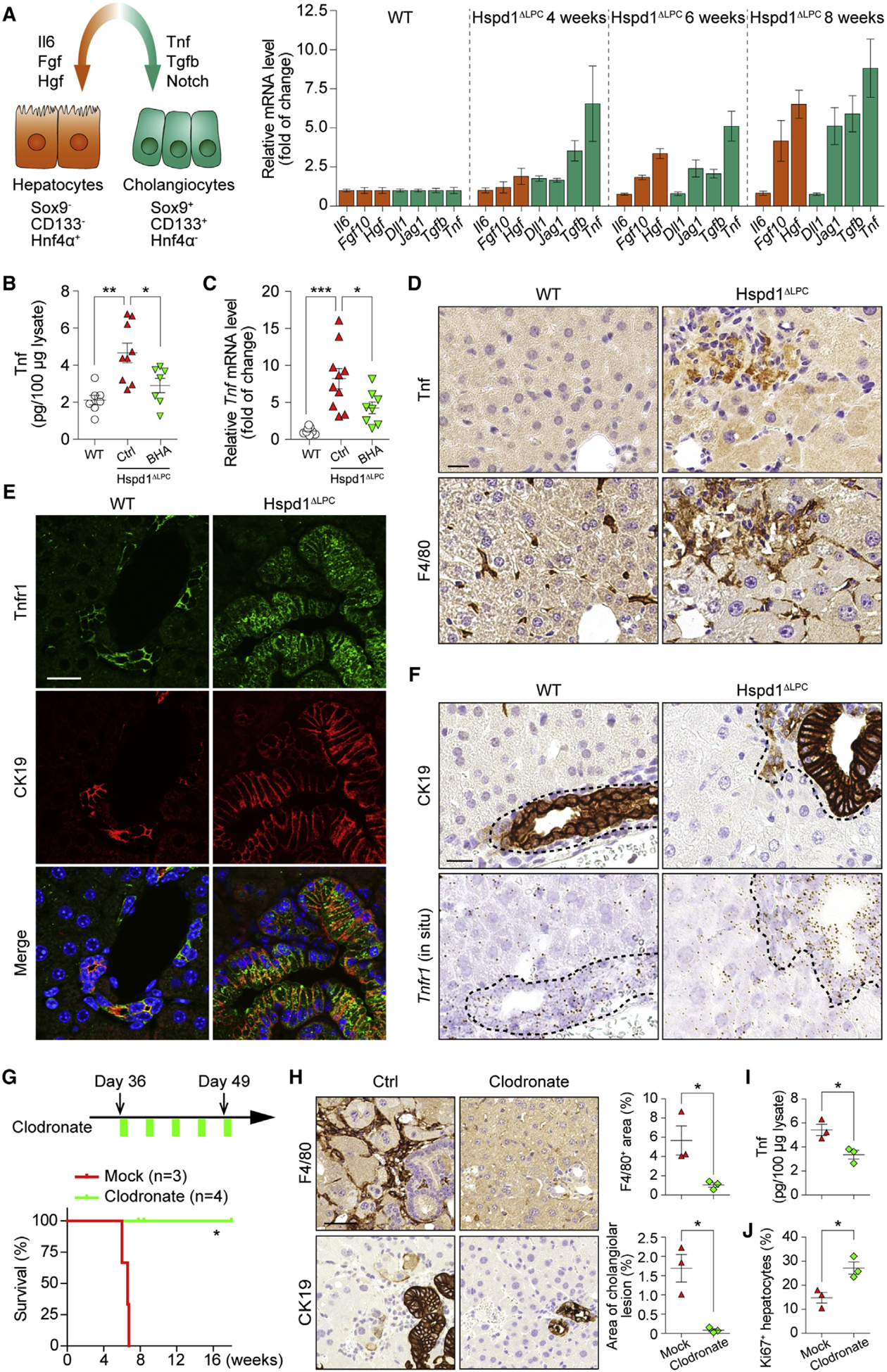
(A) Left: schematic of key signals regulating hepato- or cholangiocellular proliferation. Right: qRT-PCR of Hspd1ΔLPC livers at different ages for the expression of key ligands.
(B and C) Tnf ELISA (B) and qRT-PCR (C) of WT and normal versus BHA diet 8-week-old Hspd1ΔLPC livers.
(D) Tnf and F4/80 IHC in consecutive sections of 8-week-old Hspd1ΔLPC and WT livers. Scale bar, 20 μm.
(E) Immunofluorescence (IF) of Tnfr1 and CK19 in 8-week-old Hspd1ΔLPC and WT livers. Scale bar, 20 μm.
(F) CK19 IHC and Tnfr1 in situ hybridization in consecutive sections from 8-week-old Hspd1ΔLPC and WT livers. The dashed lines indicate the border between bilary cells and hepatocytes. Scale bar, 20 μm.
(G) Upper: timeline of clodronate administration on Hspd1ΔLPC mice. Lower: survival plot for Hspd1ΔLPC mice treated with mock or clodronate.
(H) F4/80 and CK19 IHC in consecutive sections from 8-week-old Hspd1ΔLPC livers treated with mock or clodronate. Quantification of each staining was shown in the right panels. Scale bar, 50 μm.
(I) Tnf ELISA from 8-week-old Hspd1ΔLPC livers treated with mock or clodronate.
(J) Quantification of Ki67+ hepatocytes in Hspd1ΔLPC livers treated with mock or clodronate.
Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significant. See also Figure S5.
Cells under stress conditions release pro-inflammatory cytokines into the environment as “endogenous danger signals” (Shi et al., 2003). Indeed, cytokines known to activate Kupffer cells, including Ccl2, Ccl5, and interleukin 1β (IL-1β), were upregulated in Hspd1ΔLPC livers and reduced by BHA treatment (Figure S5B). IHC and in situ hybridization revealed that focal Tnf expression mainly localized to F4/80+ Kupffer cells (Figures 4D and S5C), the numbers of which were more extensively increased in Hspd1ΔLPC livers compared with T and B lymphocytes and neutrophils (Figure S5D). qRT-PCR analysis of Tnf mRNA in purified liver cells confirmed that Tnf is mainly produced by Kupffer cells (Figure S5E). Treating primary Kupffer cells or a murine myeloid cell line (RAW 264.7) with supernatant of primary hepatocytes isolated from Hspd1ΔLPC mice or H2O2-treated primary WT hepatocytes increased Tnf expression (Figure S5F). Tnf expression was inhibited by the inflammasome inhibitor isoliquiritigenin (Huang et al., 2013), but not by inhibitors of several other pathways (Figure S5G). In addition, less activated Kupffer cells in BHA-treated Hspd1ΔLPC mice correlated with attenuated Tnf expression (Figures 4B, 4C, and S5H). Furthermore, immunofluorescence and in situ hybridization for Tnfr1 showed enriched expression in biliary epithelial cells and cholangiocellular lesions when compared with surrounding hepatocytes (Figures 4E, 4F, and S5I). Therefore, Tnf from Kupffer cells could link ROS production to cholangiocellular tumorigenesis in Hspd1ΔLPC mice.
Next, we depleted Kupffer cells by liposomal clodronate injection in Hspd1ΔLPC mice once every 3 days from weeks 6 to 8 (Figure 4G). One day after the final injection, depletion efficacy in liver and spleen was confirmed by F4/80 IHC (Figures 4H and S5J). Clodronate significantly reduced Tnf levels in Hspd1ΔLPC livers (Figure 4I), and cholangiocyte proliferation and development of cholangiocellular lesions were almost entirely abolished (Figures 4H and S5K), while Ki67+ hepatocytes increased up to 1.84-fold (Figures 4J and S5L). Clodronate-treated Hspd1ΔLPC mice began to restore body weight from day 42 (Figure S5M), and 8-week-old mice had rescued liver architecture and decreased liver enzymes (Figures S5N and S5O). Clodronate-treated Hspd1ΔLPC mice displayed no abnormalities up to 4 months (Figure S5N), indicating that cholangiocellular overgrowth in Hspd1ΔLPC mice is triggered by Tnf from Kupffer cells in response to hepatic mitochondrial dysfunction and oxidative stress.
Tnfr1 Signaling Promotes Formation of Premalignant Cholangiocellular Lesions
Next, we treated primary cholangiocytes isolated from WT or Tnfr1−/− mice with Tnf. After 5 days, WT cholangiocytes showed a 2.5-fold increase in bromodeoxyuridine incorporation, which was comparable with the mitogenic effect of Egf together with forskolin (Figure 5A). Tnfr1 deficiency altered the response of cholangiocytes to Tnf, but not to Egf and forskolin (Figure 5A), confirming that the mitogenic effect of Tnf is mediated by Tnfr1. We then tested the effect of Tnf on bipotential hepatoblasts isolated from E14.5 livers (Nitou et al., 2002). To induce differentiation, hepatoblasts were maintained in basal medium with DMSO for 7 days. Biliary lineage markers A6 and CK19 were downregulated, while hepatocyte lineage markers Hnf4α and Afp were highly expressed (Figures 5B and S5P). Strikingly, when hepatoblasts were treated with Tnf, cells adopted a biliary morphology (Figures 5B and S5P). IHC indicated high A6 and strongly reduced Hnf4α expression, suggesting that Tnf can bias toward a cholangiocellular cell fate in hepatoblasts (Figures 5B and S5P). These data were also reproduced in bipotential HepaRG cells (Figure S5Q), strongly suggesting that Tnf can promote cholangiocellular differentiation and proliferation of bipotential hepatic cells in vitro.
Figure 5. Tnfr1 Signaling Promotes Premalignant Cholangiocellular Lesions.
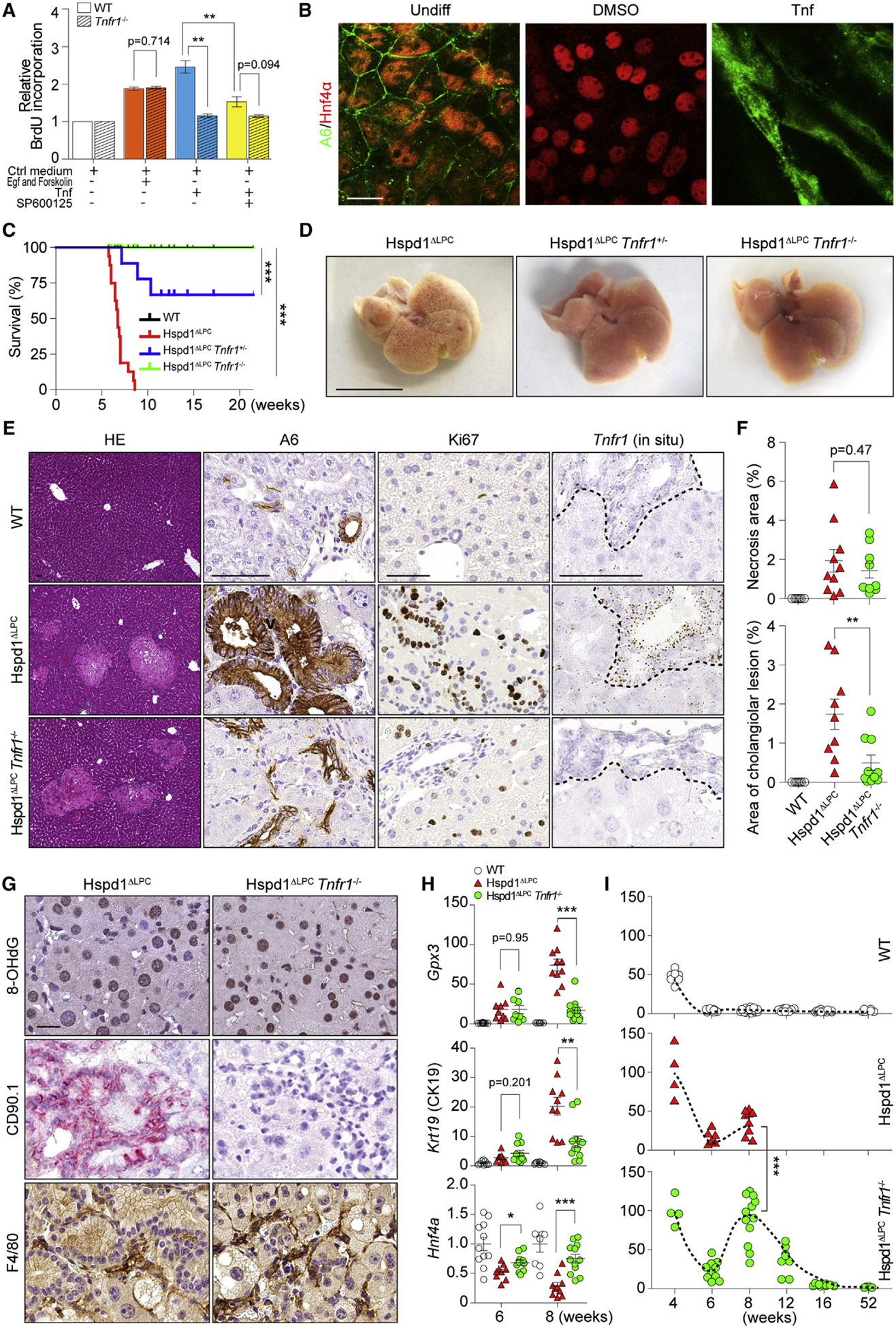
(A) Cell proliferation of WT or Tnfr1−/− cholangiocytes was analyzed by bromodeoxyuridine (BrdU) incorporation.
(B) Hepatoblasts were kept in basal medium or basal medium supplemented with DMSO or Tnf. IF of A6 and Hnf4α was performed. Scale bar, 20 μm.
(C) Survival analysis for Hspd1ΔLPC, Hspd1ΔLPC Tnfr1+/−, and Hspd1ΔLPC Tnfr1−/− mice.
(D) Macroscopic appearance of 8-week-old Hspd1ΔLPC, Hspd1ΔLPC Tnfr1+/−, and Hspd1ΔLPC Tnfr1−/− livers. Scale bar, 1 cm.
(E and F) H&E, A6 and Ki67 IHC, and Tnfr1 in situ hybridization of 8-week-old WT, Hspd1ΔLPC, and Hspd1ΔLPC Tnfr1−/− livers (E), as well as quantification of necrosis and cholangiocellular overgrowth (F). The dashed lines indicate the border between bilary cells and hepatocytes. Scale bar, 50 μm.
(G) IHC of 8-week-old Hspd1ΔLPC and Hspd1ΔLPC Tnfr1−/− livers. Scale bar, 20 μm.
(H) qRT-PCR of 6- and 8-week-old WT, Hspd1ΔLPC, and Hspd1ΔLPC Tnfr1−/− livers for indicated genes.
(I) Percentage of Ki67+ hepatocytes in WT, Hspd1ΔLPC, and Hspd1ΔLPC Tnfr1−/− livers.
Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significant. See also Figure S5.
Next, we generated Hspd1ΔLPC Tnfr1−/− double knockout mice. Loss of Tnfr1 in Hspd1ΔLPC mice prevented early lethality and significantly reduced liver damage at 8 weeks (Figures 5C, 5D, S5R, and S5S). Interestingly, while in most Hspd1ΔLPC Tnfr1+/− mice lethality was prevented, 4 out of 18 were not, indicating the dose dependence of Tnfr1 (Figures 5C and S5S). Importantly, the prevention of cholangiocellular overgrowth was unlikely to be dependent on the pro-survival or pro-apoptotic function of Tnfr1: equal levels of necrosis were observed in Hspd1ΔLPC as well as in 6-week-old double knockout mice (Figures 5E and 5F). Likewise, ROS accumulation, as well as the resulting induction of Gpx3, Kupffer cell activation, and Tnf mRNA levels remained unchanged in Hspd1ΔLPC Tnfr1−/− when compared with Hspd1ΔLPC mice, suggesting that Kupffer cell activation and Tnf production were not affected by Tnfr1 deletion (Figures 5G, 5H, and S5T). No difference in liver enzyme levels between Hspd1ΔLPC Tnfr1−/− and Hspd1ΔLPC mice was found by week 6 but ALT, AST, AP, and bilirubin were significantly reduced in double knockout mice by week 8 (Figure S5U). Consistent with the idea that Tnf promotes biliary proliferation, cholangiocellular overgrowth and CSC expansion was apparently blocked (Figures 5E–5H and S5V). Importantly, Ki67+ hepatocytes increased significantly in 8-week-old livers in double knockout mice at the expense of cholangiocytes (Figures 5E and 5I). One-year-old double knockout mice did not show any abnormalities (Figure S5W). Thus, Tnfr1 signaling favors biliary proliferation and neoplasia over hepatocyte proliferation under the conditions of mitochondrial dysfunction, ROS, and liver damage.
JNK/c-Jun Activation Is Required for Cholangiocellular Hyperproliferation
Tnf/Tnfr1 signaling activates a diverse set of downstream molecules, including Rela, Stat3, and JNK. We observed strong nuclear p-JNK staining in cholangiocytes, whereas nuclear Rela and Stat3 were mainly present in non-parenchymal cells (Figure 6A). Nuclear c-Jun was also observed in cholangiocytes of Hspd1ΔLPC livers (Figure S6A). Similarly, primary hepatoblasts cultured with Tnf showed nuclear staining of c-Jun but not Rela (Figure 6B). Notably, nuclear p-JNK was specifically attenuated in cholangiocytes but not in hepatocytes after in vivo clodronate treatment (Figure S6B), indicating that JNK is activated downstream of Tnf.
Figure 6. JNK/c-Jun Activation Is Required for Premalignant Cholangiocellular Lesions.
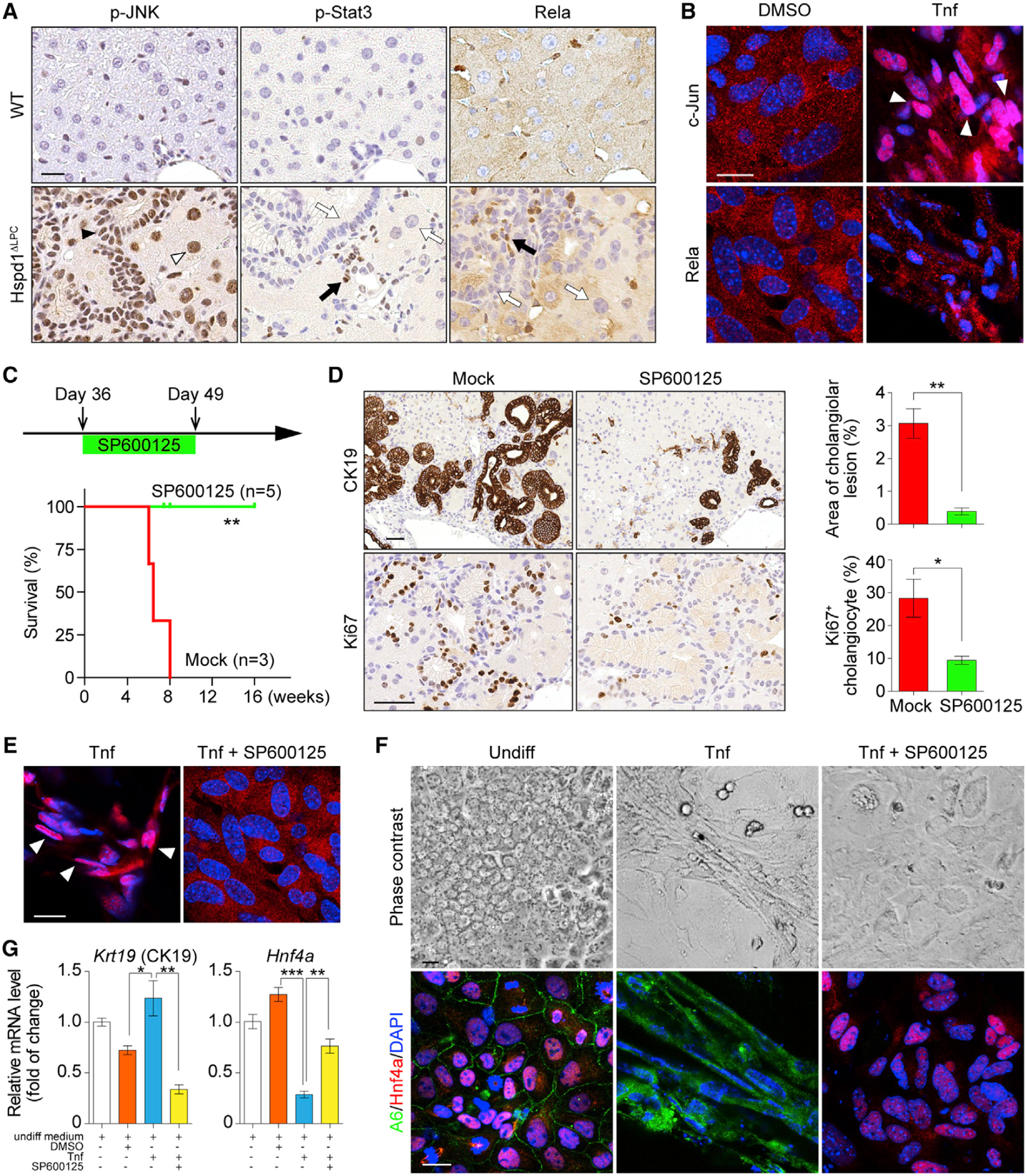
(A) p-JNK, p-Stat3, and Rela IHC in 8-week-old WT and Hspd1ΔLPC livers. The black arrowhead indicates a p-JNK+ cholangiocyte, the white arrowhead indicates a p-JNK+ hepatocyte. Black arrows indicate positive staining in non-parenchymal cells, while white arrows indicate negative staining in liver parenchymal cells. Scale bar, 20 μm.
(B) Rela and c-Jun IF in hepatoblasts cultured in basal medium supplemented with DMSO or Tnf. White arrowheads indicate c-Jun nuclear staining. Scale bar, 20 μm.
(C) Timeline of SP600125 administration on Hspd1ΔLPC mice and survival of Hspd1ΔLPC mice not treated (mock) or treated with SP600125.
(D) CK19 and Ki67 IHC in 8-week-old Hspd1ΔLPC livers treated with mock or SP600125, and quantification of cholangiolar cancerous lesion areas and Ki67+ cholangiocytes relative to total cholangiocytes. Scale bar, 50 μm.
(E) c-Jun IF in hepatoblasts cultured in Tnf-containing medium with or without SP600125. White arrowheads indicate the c-Jun nuclear staining. Scale bar, 20 μm.
(F and G) Phase contrast and A6 and Hnf4α IF images (F) and qRT-PCR (G) of hepatoblasts in basal or Tnf-containing medium with or without SP600125. Scale bar, 20 μm.
Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. ns, not significant. See also Figure S6.
To test the therapeutic potential of JNK inhibition on cholangiocellular overgrowth, the JNK inhibitor SP600125 was administered intraperitoneally (i.p.) daily for 2 weeks beginning at P36 in Hspd1ΔLPC mice (Figure 6C). JNK inhibition reduced cholangiocellular lesions, resulting in proper regeneration and eventually functional livers with WT hepatocytes at week 16 (Figures 6D, S6C, and S6D). Cholangiocellular proliferation was also strongly reduced (Figure 6D), whereas hepatocyte proliferation was not influenced (Figure S6E). Since p-JNK was also found in Hspd1− hepatocytes in untreated Hspd1ΔLPC mice, SP600125 might also exert its role by attenuating hepatocyte injury and/or the carcinogenic signals in Hspd1− hepatocytes. Indeed, we observed decreased nuclear p-JNK in both hepatocytes and cholangiocytes due to SP600125 treatment, accompanied by reduced necrosis and liver enzymes and abolished c-Myc upregulation (Figures S6F–S6I). Expression of UPR-related and redox signaling-related genes also declined in livers of SP600125-treated Hspd1ΔLPC mice, concomitant with reduced 8-OHdG staining (Figures S6J and S6K). In addition, less F4/80+ staining and lower levels of Tnf mRNA were evident (Figures S6J and S6K), indicating that Kupffer cell activation and Tnf production was attenuated. In contrast, Kupffer cell depletion strongly reduced p-JNK in cholangiocytes but not surrounding hepatocytes (Figure S6B), suggesting that JNK activation in cholangiocytes was dependent on paracrine Tnf.
To directly assess the role of JNK on biliary proliferation, isolated cholangiocytes were cultured with SP600125 in the presence of Tnf. Tnf-enhanced proliferation of WT cholangiocytes was abrogated by SP600125 treatment (Figure 5A). This result was confirmed with two additional JNK inhibitors (JNK-IN-8 and JNK6o; Figure S6L).
Hepatoblasts were also treated with SP600125 during in vitro differentiation. SP600125-treated hepatoblasts lacked c-Jun activation (Figures 6E and S6M). As a result, biliary differentiation of hepatoblasts was suppressed, with reduced ductular morphogenesis and decreased expression of A6 and CK19 (Figures 6F and 6G). Meanwhile, SP600125 treatment restored expression of hepatocyte markers Hnf4α and Afp comparable with controls (Figures 6F, 6G, and S6M). Therefore, JNK/c-Jun signaling appears to be activated by Tnf and required for biliary differentiation and proliferation.
The Tnf/JNK Signaling Axis Is Activated in Distinct Mouse ICC Models
Since both HCCs and ICCs were found in the multiplexed CRISPR/Cas9-induced liver cancer model (Weber et al., 2015), we were able to directly compare the molecular traits of HCC versus ICC. IHC revealed that JNK activation, Tnfr1 expression, and high levels of CK19 and Sox9 were exclusively present in ICC, but not in HCC or unaffected tissue (Figure 7A and S7A–S7C). Kupffer cell activation and Tnf production were strongly enriched in ICC samples, surrounding the malignant cholangiocytes, regardless of their distinct genetic signature (Figures 7B and S7D–S7F). Moreover, nuclear p-JNK was strongly enriched in all ICC models examined (Figures 7A and S7G), but absent from HCC models induced by chronic inflammation (Haybaeck et al., 2009) or NASH (Wolf et al., 2014) (Figure S7H).
Figure 7. Kupffer Cell-Derived Tnf/JNK Axis Is Activated in Different ICC Models.
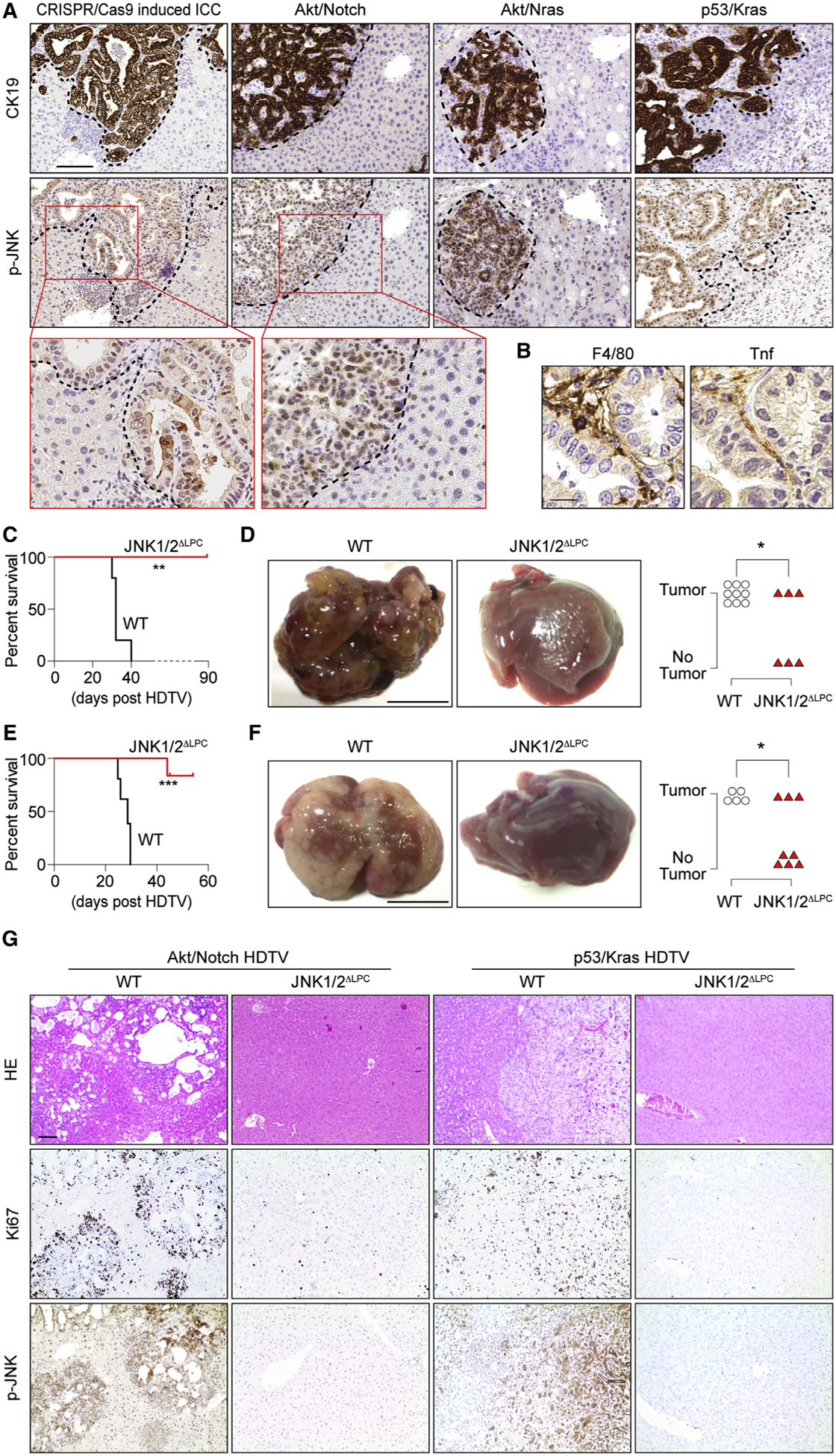
(A) IHC of CK19 and p-JNK in liver sections from four ICC models as indicated. The dashed lines indicate the border between ICC and the surrounding liver tissues. Scale bar, 100 μm.
(B) F4/80 and Tnf IHC in consecutive sections from CRISPR/cas9-mediated multiplex mutagenesis livers. Scale bar, 20 μm.
(C–F) Survival plot (C and E) and representative macroscopic view of ICC and quantification of tumor incidence (D and F) for JNK1/2ΔLPC and control C57BL/6 mice in HDTV-Akt/Notch-induced (C and D) or HDTV-p53/Kras-induced (E and F) ICC model. Scale bar, 1 cm.
(G) H&E and IHC staining of Ki67 and p-JNK in livers as indicated in (C and E). Scale bar, 100 μm.
*p < 0.05, **p < 0.01, ***p < 0.001. ns, not significant. See also Figure S7.
JNK inhibition was tested on two aggressive ICC models by hydrodynamic tail vein injection of different sets of constructs (Akt/Notch; p53/Kras) into WT or liver-specific JNK1/2 knockout mice (JNK1/2ΔLPC). JNK1/2 knockout in liver parenchymal cells provided a strong survival benefit (e.g., >40 days in Akt/Notch; >25 days in p53/Kras) and significantly reduced tumor incidence in both models (Figures 7C–7F). IHC showed lack or reduction of tumors (Figure 7G). Tumors in JNK1/2ΔLPC mice found at later time points displayed features of ICC (Figure S7I), suggesting the existence of uncharacterized JNK bypass signaling.
Next, we established an ICC cell line (mICC), derived from a multiplexed CRISPR/Cas9 ICC. mICC cells had reduced p-JNK and severe growth defects in the presence of SP600125 (Figures S7J and S7K). mICC cells were subcutaneously injected into mice, resulting in A6+ and p-JNK+ tumors with histological features similar to primary tumors of human ICC patients (Figure S7L). SP600125 was administered i.p. daily starting at day 1 post-inoculation for 7 days, which resulted in significantly reduced tumor formation (Figure S7M).
p-JNK is Strongly Increased in ICC Patient Specimens and Cell Lines
Next, we analyzed human HCC and ICC for Tnf and p-JNK by IHC (Figures 8A and S8A). Only 3.2% of HCC were Tnf+, as opposed to 55.2% of ICC (Figure 8B). p-JNK was found in approximately 20% of HCC, as opposed to 80% in ICC (Figure 8C). We also found 8-OHdG+ ICC predominantly surrounding hepatocytes but not in malignant cholangiocytes (Figure 8A), supporting our notion that mitochondrial dysfunction and oxidative stress can also drive JNK activation non-automatously.
Figure 8. Tnf and p-JKN Are Strongly Increased in ICC Patient Specimens.
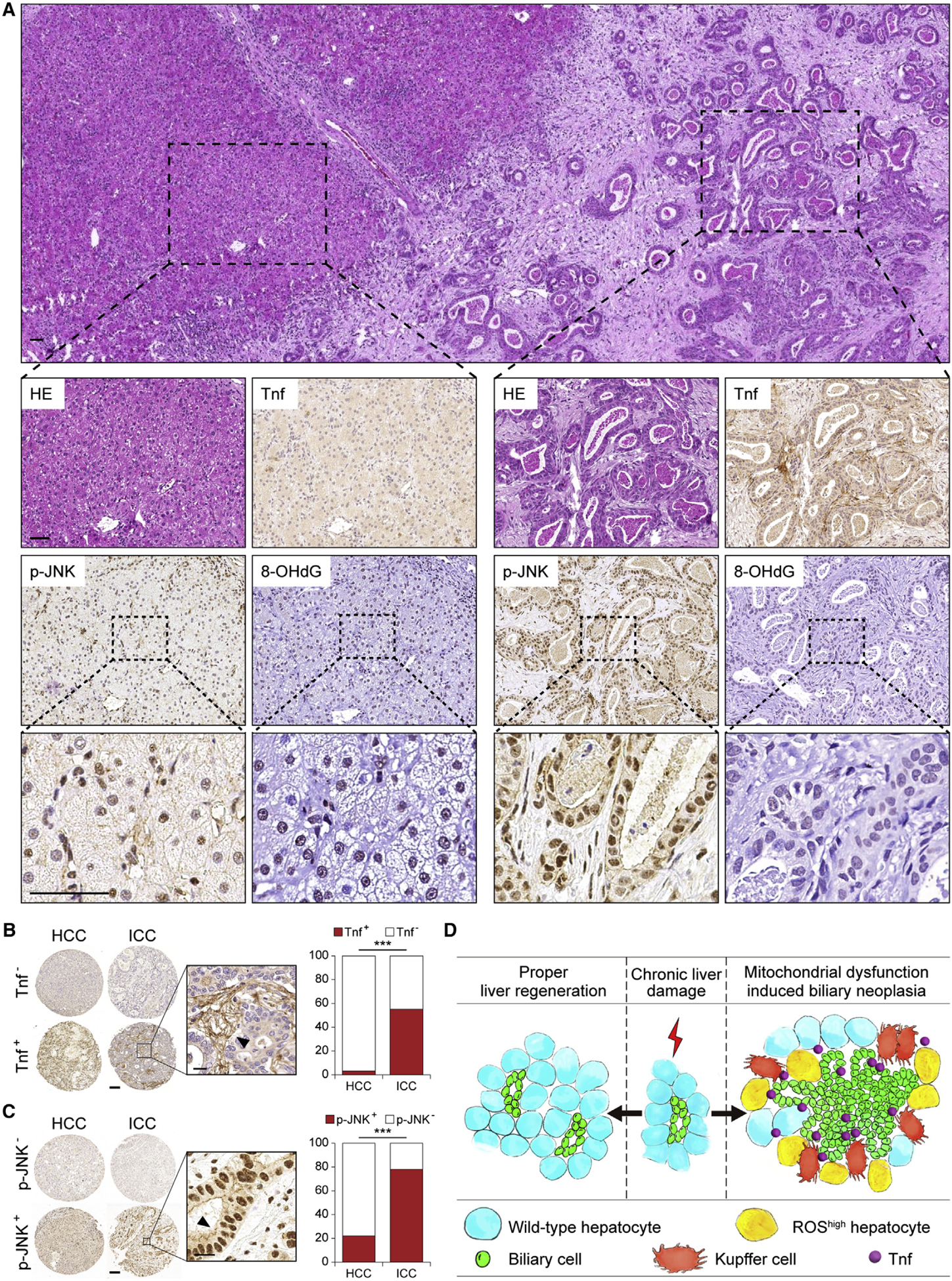
(A) H&E and IHC staining of Tnf, p-JNK, and 8-OHdG in a representative human ICC sample. Scale bar, 50 μm.
(B and C) Representative IHC images and quantification of Tnf (B) and p-JNK (C) in human ICC and HCC. Arrowheads indicate positive staining. Scale bars, 100 μm (left), 20 μm (right).
(D) Model depicting a non-cell-autonomous mechanism of hepatocyte mitochondrial dysfunction in regulating cholangiocellular tumorigenesis.
***p < 0.001. ns, not significant. See also Figure S8.
Finally, we tested the response of well-characterized human ICC (ETK-1 and HuCCT-1) and HCC (Huh-7 and HepG2) cell lines to JNK inhibition. ICC cells had much higher p-JNK than HCC cells, consistent with our histopathological studies (Figure S8B). Inhibiting JNK activity impaired cell proliferation in a dose-dependent manner (Figures S8C–S8E). Both human ICC cell lines showed a higher sensitivity to SP600125 compared with HCC cell lines (p < 0.001, Figures S8D and S8E). ICC cell lines also showed a higher degree to which maximal growth inhibition was achieved by JNK inhibition (Figure S8E). Similar effects were seen with additional JNK inhibitors (p < 0.01, Figures S8F and S8G). This suggests that targeted inhibition of JNK signaling in ICC may have therapeutic value.
DISCUSSION
Mitochondrial dysfunction and ROS are common features of liver diseases (Kamata et al., 2005; Maeda et al., 2005; Szabo and Petrasek, 2015). A recent genetic screen in Drosophila indicated that mitochondrial defects and ROS activate oncogenic signaling in neighboring cells by forming a tumorigenic niche (Ohsawa et al., 2012). Here, we demonstrate that mitochondrial defects in hepatocytes trigger ROS accumulation leading to focal recruitment of Tnf-producing Kupffer cells, which create a favorable niche for biliary proliferation via JNK signaling. This suggests that the liver microenvironment could be pharmacologically manipulated to improve liver function in advanced liver disease.
Tnf/JNK/c-Jun signaling appears to induce a biliary proliferation program in hepatocytes or liver bipotential cells (Nishikawa et al., 2013), implicating this pathway in the pathogenesis of human cholangiocellular tumors. Examination of human specimens showed that Tnf and p-JNK were found far more frequently in ICC than in HCC, consistent with findings from others, showing that p-c-Jun emanates from infiltrating inflammatory cells in human HCC (Hefetz-Sela et al., 2014). Chemotherapeutic options for ICC are lacking due to unidentified ICC-specific targets. Our results with human HCC and ICC reveal distinct molecular traits of these two pathological entities, indicating that HCC and ICC may respond differently to the same treatment. Accordingly, cancer cells isolated from human ICC are more sensitive to JNK inhibition compared with cells from HCC. Further investigation of the link between p-JNK level and clinicopathological features may provide important clues regarding disease mechanisms.
Conflicting roles for Kupffer cells in hepatocyte regeneration, cholangiocellular growth (Boulter et al., 2012, 2015), and HCC development (Maeda et al., 2005; Zhu et al., 2000) have been reported. The role of Kupffer cells likely depends on the type of tissue injury. Indeed, the cytokine signatures of Kupffer cells isolated from CDE or DDC diet models are different (Boulter et al., 2012). In addition, the spatial regulation of the regenerative niche also differs in distinct contexts. In DDC-induced biliary regeneration, Kupffer cells are separated from ductular reactive cells (Boulter et al., 2012), whereas in Hspd1ΔLPC and multiplexed CRISPR/Cas9 mice, Kupffer cells closely associate with cholangiocellular tumor cells. Thus, immune components appear to be highly adaptable to various stimuli. In addition, it remains to be determined how the crosstalk between Tnf and other signaling pathways (e.g., Wnt, Tgfb, Notch, and Hippo signaling) coordinates liver cell plasticity in different models of murine liver cancer (Boulter et al., 2015; Yimlamai et al., 2014; Zender et al., 2013).
Our analysis on human HCC and ICC revealed that a subgroup of HCC are strongly p-JNK+. p-JNK/p-c-Jun positivity in HCC varies greatly across studies, from 11% (Hefetz-Sela et al., 2014) to 56% (Hui et al., 2008), probably due to sample heterogeneity. JNK activation has also been implicated in HCC development in mouse models. For instance, liver-specific Mapk14 deletion leads to JNK hyperactivation and HCC development in diethylnitrosamine (DEN)-treated mice (Hui et al., 2007). JNK1-deficient mice show impaired HCC formation in a DEN-phenobarbital-induced mouse liver cancer model (Hui et al., 2008). JNK1 disruption also reverses increased susceptibility to DEN-induced HCC formation in IkkbΔHep mice (Sakurai et al., 2006). However, lack of JNK1 plus JNK2 in hepatocytes increases tumor burden (Das et al., 2011), whereas the pro-tumorigenic effects of JNK on HCC are associated with inflammation and require JNK function in non-parenchymal cells such as myeloid cells (Han et al., 2016) and hematopoietic cells (Cubero et al., 2015). Thus, the role of JNK in HCC might depend on the subtype and cellular context. Retrospective analysis of patient disease histories found no correlation of JNK activity in HCC with pathophysiological features (Hui et al., 2008). Moreover, continuous efforts with next-generation sequencing (NGS) have shown that the JNK pathway is not recurrently amplified in virus-, alcohol-, and other risk-factor-associated HCCs (Guichard et al., 2012; Huang et al., 2012; Jhunjhunwala et al., 2014; Schulze et al., 2015). Interestingly, recent NGS characterization of human pediatric liver cancers induced by intrahepatic cholestasis has shown massive gene amplification and rearrangements frequently targeting direct regulators of JNK (Iannelli et al., 2014). This suggests that the mechanistic involvement of JNK in the development of liver cancers is largely context dependent. Supporting this notion, HCCs from Mdr2-KO mice, a model of defects in hepatocyte biliary transporter, show similar genetic profiles affecting JNK activity as human pediatric liver cancers (Iannelli et al., 2014).
Our model of chronic cholestasis induced by oxidative stress further highlights that different disease etiologies have distinct impacts on signaling reliance and tumor phenotypes. In addition to the reported roles of JNK in cell proliferation, survival, apoptosis, and necrosis, as well as a lineage determinant role of JNK in liver parenchymal cells may also exist, where it not only favors proliferation of biliary cells but also directly biases biliary cell fate decisions in bipotential hepatic cells (Figure 8D). Importantly, we and others observed that JNK inhibition retards ICC progression. Therefore, ICC patients and HCC patients with hepatobiliary abnormalities may preferentially benefit from JNK inhibition. Thus, stratifying liver cancers by disease etiologies may have great implications for preclinical studies with JNK inhibitors for targeted therapy.
One remaining question is the origin of the Cre-recombinase−/Hspd1+ escaper cells. Using different lineage-tracing systems, several groups have failed to demonstrate a significant contribution of adult biliary-derived liver progenitor cells (LPCs) to hepatocyte neogenesis in diet or injury-induced mouse models (Jors et al., 2015; Tarlow et al., 2014; Yanger et al., 2014). However, LPC-mediated liver regeneration may require complete hepatocyte growth arrest of the hepatocyte and biliary compartment, as recently proposed (Lu et al., 2015). In our model, it remains unclear whether Hspd1+ ICC cells arise from LPCs, from dedifferentiated Sox9+ hepatocytes, and/or cholangiocytes. Indeed, recent studies propose that either of the latter two cell types can potentially contribute to ICC development (Fan et al., 2012; Guest et al., 2014; Saha et al., 2014; Tschaharganeh et al., 2014; Yimlamai et al., 2014; Zender et al., 2013). Further lineage-tracing studies should clarify this open question.
In conclusion, we show that Tnf is a key molecule driving cholangiocellular overgrowth and cancer development. In addition to the known signaling molecules implicated in ICC growth, such as Notch and Wnt, non-parenchymal cell-derived inflammatory signals can be added. As a reciprocal supportive interplay between cancer cells and their niche may be critical to promote cancer initiation and progression, our finding not only provides an example of a mechanism in which a pro-inflammatory microenvironment can function as pre-carcinogenic niche but also suggests the ROS/Tnf/JNK axis as an attractive ICC therapeutic target.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mathias Heikenwalder (heikenwaelder@helmholtz-muenchen.de or m.heikenwaelder@dkfz-heidelberg.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were maintained under SPF conditions. Experiments were conducted in accordance with the guidelines of the Germany Animal Protection Law and were approved by local committees of government of Bavaria (license number: 55.2–1-54–2532-134–14; license number: 55.2–1-54–2532-118–2016) and the government of Baden-Würrtemberg (license number: 35–9185.81/G178/16). C57BL/6J mice were purchased from Harlan or bred in-house. R26R-Confetti mice and Tnfr1KO mice were purchased from The Jackson Laboratory. Conditional Hspd1 knockout mice were generated by Taconic-Artemis (Cologne, Germany) in close collaboration with the Chair of Nutrition and Immunology. Both male and female mice were used in all mouse experiments and no gender disparity was found when it came to the phenotype of Hspd1ΔLPC mice or intercrossings of the latter. Diet-induced liver injury models and BDL model were provided by Prof. Geisler (Jors et al., 2015). HDTV induced ICC models (license number: G178/16) were generated as previously described (Matter et al., 2016). AAV mediated Cre expression in hepatocytes were done by i.v. injection of 4 × 1011 viral genomes of AAV8-Ttr-Cre, purchased from VectorBiolabs, into 6–8-week-old male mice. We analyzed between 3 and 6 mice per condition in each experiment.
Human Material
Formalin-fixed, paraffin-embedded human liver tissue samples were retrieved from the archives and the biobank of the Institute of Surgical Pathology, University Hospital Zurich, Northwestern and Central Switzerland (EKNZ), and Faculty of Medicine, University Hospital RWTH Aachen, for morphological, immunohistological and molecular analyses. The study was approved by the local ethics committee (“Kantonale Ethikkommission Zürich”, application number StV26/2005 and KEK-ZH-Nr. 2013–0382; EKNZ, application number 310/13; University Hospital RWTH Aachen, application number EK122/16).
Cell Lines and Primary Cultures
Isolation, Culture and Differentiation of Hepatoblasts
Isolation of fetal livers (C57BL/6J) at gestation stage E14.5 was done by a modified MACS based immune-isolation protocol (Nitou et al., 2002). Briefly, fetal livers were dissected, freed of adherent tissue, shredded and subsequently incubated with dispase II (1.6 U/ml) (Sigma) for 1hour at 37°C. Liver specimen were further dispersed by pipetting and filtered three times through a 100 μm Nylon cell strainer (Falcon). The cell suspension was further centrifuges for 10 min at 800 rpm, washed twice using Hepes-buffered (20 mM) (Sigma) DMEM (Dulbecco’s Modified Eagle Medium; Gibco) with 0.01% DNase (Roche), and subsequently re-suspended in Hepes-buffered (20 mM) (Sigma) DMEM (Dulbecco’s Modified Eagle Medium; Gibco) supplemented with 10% FBS (Biochrom) and 1x PenStrep (Life Technologies). Purification of E-Cadherin+ hepatoblasts was performed using the rat anti-mouse E-Cadherin antibody (clone Decma-1, Merck Millipore). Briefly, anti-rat IgG microbeads were incubated with Decma-1 antibody for 45 min at room temperature on a Intelli-mixer (LTF Labortechnik) and the complexes of primary and secondary antibody were subsequently washed twice times with DMEM (Dulbecco’s Modified Eagle Medium; Gibco) supplemented with 10% FBS (Biochrom) and 1x PenStrep (Life Technologies) using a table centrifuge for 10min at 13 rpm. Following the incubation of the cell suspension with the complexed microbeads, the cells were purified using the MACS columns (MS, Milteny Biotec). Cells were washed and afterwards eluded in Hepes-buffered (20 mM) (Sigma) DMEM (Dulbecco’s Modified Eagle Medium; Gibco).
Isolated hepatocytes were plated onto rat tail type I collagen (Sigma) coated plates. Cells were kept in an undifferentiated state in basal medium composed of Williams’ Medium E supplemented with 10% FCS, 10mM nicotinamide, 20 ng/ml mouse Egf (Roche Diagnostics), and 40 IU / ml insulin (Sigma Chemical Company). After 3 days, 0.5% DMSO or 10 ng/ml of recombinant murine Tnf was added into the basal medium to induce differentiation. For JNK inhibition in presence of Tnf, 10 μM of SP600125 (Abcam) was added. Cells were kept in basal medium or differentiation medium for another 7 days until harvest, changing the culture medium daily.
Isolation and Cultivation of Cholangiocytes
Ducts were isolated from 8–10 weeks old mice by microdissection (Keitel et al., 2010). The isolated bile ducts were cut into 3–4 mm segments, placed in a collagen matrix and covered with growth factor-containing medium. After 10–14 days cholangiocytes grew out of the ducts forming a monolayer. Cells were passaged once 80% confluent and cells were used in passages 4–9.
Isolation and Culture of Primary Hepatocytes
Male 8–12-week old C57Bl/6 mice were anesthetized by i.p. injection of 100 mg kg−1 body weight ketamine hydrochloride and 5 mg kg−1 body weight xylazine hydrochloride. After opening the abdominal cavity, the liver was perfused at 37 °C with HANKS buffer, supplemented with 1mM EDTA, via the portal vein for 5 min and subsequently with HANKS buffer, supplemented with 0,3 mg/ml Collagenase IV (Roche) and 10% glucose, for 5–7 min until disintegration of the liver structure was observed. The liver capsule was removed and the cell suspension was filtered through a 100 μm mesh. The cells were washed and, subsequently, viability of cells was determined by trypan blue staining. 1 × 105 living cells per well were seeded on collagen I-coated 12-well plates and cultured in Williams E medium (Life Technologies, Glutamax) supplemented with 2% fetal calf serum, 100 μg/ml penicillin/streptomycin.
Isolation and Culture of Primary Kupffer Cells
C57Bl/6 mice were heart perfused with HBSS. Liver lobes were collected and processed for flow cytometry. To obtain single-cell suspensions, organs were homogenized and incubated for 45 min in RPMI containing 60U Collagenase IV (Sigma) and 25 μg/ml DNase I (Sigma). Cell suspensions were washed with HBSS and layered upon 25%/50% percoll for gradient centrifugation (1800g/15min). Middle layers containing leukocytes were collected and washed twice in HBSS. Fc receptors were blocked by incubating cells in 5% FCS with purified mouse IgG (500 mg/ml, Jackson ImmunoResearch Laboratories). Stainings were performed in 5% FCS on ice for 30 min with optimal dilutions of commercially-prepared Lin (CD4 BV421 (GK1.5), CD8 BV421 (53–6.7), CD19 BV421 (6d5), Ter119 BV421 (Ter119)), F4/80 BV711 (BM8) (BioLegend), CD11b PE-Cy7 (M1/70), CD45 Alexa700 (30-F11) (eBioscience), Ly6G-PerCP5.5 (1A8) (BD Pharmingen). Lin−Ly6G−Ly6C−CD45+CD11bintF4/80hi Kupffer cells were sorted on a BD FacsARIA III. After sortimg, 3×103 Kupffer cells were seeded on a 96-well plate and cultured in DMEM medium supplemented with 10% fetal calf serum, 1% non-essential amino acid and 100 μg/ml penicillin/streptomycin.
Isolation of Murine Liver Cells
Liver cells were isolated from 6–8 weeks old mice. For isolation, animals were sacrificed and liver was perfused via the inferior vena cava with EGTA containing buffer followed by a buffer containing collagenase (0,12 U/ml type NB 4G collagenase (Serva, Germany)). Liver was removed and mechanically disrupted. Cells suspension was centrifuged at 20g for 5 min. Pellet was washed and subjected to Percoll gradient (GE healthcare, Sweden). Hepatocytes were cultured on collagen-coated (Serva, Germany) plates in Williams’E medium supplemented with Pen/Strep, Gentamycin, 0,22 mM Glutamin, 0,02M HEPES, Insulin, 110nM Hydrocortison and 1,6% DMSO. The supernatant was subjected to Nycodenz gradient and the cells used for fluorescence-activated cell sorting.
Cell Lines and Drug Treatment
Human ICC cell lines, HuCCT-1 and ETK-1, and Mouse ICC cell line were generously provided by Dr. Roland Rad. Human HCC cell lines, HepG2 and Huh-7 were routinely maintained in our lab. The three ICC cell lines were cultured in RPMI medium supplemented with 10% fetal calf serum, 1% non-essential amino acid and 100 μg/ml penicillin/streptomycin. The two HCC cell lines were maintained in DMEM medium supplemented with 10% fetal calf serum, 1% non-essential amino acid and 100 μg/ml penicillin/streptomycin. For treatment with SP600125 (Abcam), JNK-IN-8 (Merck) or JNK6o (Bio-techne), 800 ~ 1,000 cells/well were seeded in 96-well plates. SP600125 (10, 20, 30 and 60 μM), JNK-IN-8 (1, 5, and 10 μM) or JNK6o (1, 5, and 10 μM) was added into the culture medium from the second day for 72 hours. Cell viability assay was performed using a CellTiter-Blue Cell Viability Assay (Promega) following the manufacturer’s instruction. Primary Hepatocytes were treated with 2μM H2O2 for 6h. Subsequently, supernatant was collected and used for further treatment of primary Kupffer cells and Raw 246.7 cells. Primary Kupffer cells/well were treated with respective hepatocyte supernatants for 16h. Raw 246.7 cells were cultured in DMEM medium supplemented with 10% fetal calf serum, 1% non-essential amino acid and 100 μg/ml penicillin/streptomycin. 3 × 105 Raw 246.7 cells were seeded in 12-well plates and were either left untreated or were pretreated for 4h with the Inhibitors S3I-201 (working conc. 80μM, Biovision), LY-294002 (working conc. 50μM, Sigma), Isoliquiritigenin (working conc. 10μM, Sigma), TPCA-1 (working conc. 5μM, Sigma) or for 24h with MYD88 Inhibitor Peptide Set (NBP2–29328, working conc. 100μM, Novus Biologicals) respectively, before treatment with respective hepatocyte supernatants for 16h. Raw 246.7 cells were well tolerated under the treatment of the inhibitions without showing major cell death.
METHOD DETAILS
Subcutaneous Transplantation of Liver Tissue
The donor liver tissue of Hspd1ΔLPC mice for the subcutaneous transplantation was prepared under sterile conditions. The recipient SCID Beige mice at the age of 4–8 weeks were anesthetized with Ketamin (100 mg/kg) and Rompun (10 mg/kg) under sterile conditions as well. For transplantation, a small incision into the previous disinfected skin of the back of the recipient mouse was used to place the minced donor tissue under the skin. For each of both flanks one tissue sample (3–4 mm in diameter) was transplanted. In total 12 transplantations on 6 mice were set up from 3 independent donors. Afterwards the incision was closed and disinfected again. Tumors were harvested 4–8 weeks after transplantation and fixed in 4% PFA in PBS, followed by immune-histochemical analysis.
Immunofluorescence Microscopy
Deparaffinized and rehydrated slides were subjected to microwave antigen retrieval in 25 mM citrate buffer (pH 6.0) and allowed to cool to room temperature. Antigen retrieval for F4/80 was performed by incubation with 1mg/ml Pronase XXIV (Sigma) for 10 minutes at room temperature. Slides were blocked in TBS containing 0.1% Triton X-100 and 2% serum from the species that the secondary antibody was raised in at room temperature for two hours. After blocking, primary antibodies diluted in TBS with 0.1% Triton X-100 and 2% blocking serum were added overnight at 4°C. After extensive washing, slides were incubated with the secondary antibody in TBS with 0.1% Triton X-100 and 2% blocking serum for 2 hours at room temperature in dark and counterstaining with DAPI. Slides were mounted with Fluoromount-G (SouthernBiotech, Birmingham, Alabama, USA) and images were captured with Olympus FV10i confocal Microscope (Olympus, Germany).
Electron Microscopy
Liver tissues were fixed with 4% glutaraldehyde in PBS for less than 24 h and embedded in Epon. The tissues were then trimmed and reoriented so that ultrathin sections could be cut and treated with 1% uranyl acetate and lead citrate. Electron micrographs were obtained using a Philips CM 100 electron microscope.
RNA In-Situ Hybridization
Probes for Tnf, Tnfr1, the housekeeping gene ubiquitin C (positive control), the bacterial gene dapB (negative control), and the hybridization kit (RNAscope 2.0 FFPE Assay) were purchased from Advanced Cell Diagnostics. RNA in-situ hybridization was performed using the RNAscope 2.0 technology according to the manufacturer instructions. Briefly, 2 μm paraffin sections were pretreated with heat at 75°C in EZprep buffer for 20 minutes and protease at 37°C for 30 minutes. Tnfr1 specific probe pairs (Advanced Cell Diagnostics) were then hybridized at 48°C for 2 hours, followed by a series of signal amplification and washing steps. Hybridization signals were detected by DAB staining, followed by counterstaining with hematoxylin. For fluorescent in situ, 4 μm sections were pretreated by boiling with pretreat 2 for 30 minutes followed by incubation with pretreat 4 at 40°C for 35 minutes. Tnf specific probe pairs were then hybridized at 40°C for 2 hours. Stained slides were scanned into digital images using a Leica SCN400 scanner (Leica) at ×40 magnification.
BrdU Assay
For proliferation experiments cholangiocytes were seeded on collagen-coated 96 well plates (confluence of the cells: 30–40%). After 24 hours cells were washed 2 times with starvation medium and stimulated with various substances for another 36 hr. 24 hr prior to end of the experiment the cells were additionally treated with BrdU. The BrdU-assay was carried out according to the manufacturer’s protocol (BrdU Roche). SP600125, JNK-IN-8 or JNK6o was added 30 min prior to the stimulation with Tnf.
Histology and Immunohistochemistry
Liver tissue samples were fixed overnight in 4% paraformaldehyde, embedded in paraffin. Paraffin sections were subjected to hematoxylin-eosin or immunohistochemistry staining as previously described (Haybaeck et al., 2009). Briefly, paraffin sections (2 μm) and frozen sections (5 μm) of livers were used for automated staining performed on Bond MAX (Leica) using the DAB detection kit (bornmax). The following primary antibodies were used: mouse anti-8-OHdG (1:1000 dilution, Abcam); goat anti-Afp (1:1000 dilution, R&D); rabbit anti-CHOP (1:1000 dilution, Cell Signaling); rabbit anti-c-Jun (1:1000 dilution, Cell Signaling); p-c-Jun (1:1000 dilution, Cell Signaling); rabbit anti-cleaved Caspase-3 (1:1000 dilution, Cell Signaling); rabbit anti-Collagen type IV (1:1000 dilution, Cedarlane); rabbit anti-Cytokeratin (1:1000 dilution, Dako); rat anti-F4/80 (1:1000 dilution, BioLegend); rabbit anti-Glutamine Synthetase (1:1000 dilution, Abcam); goat anti-Hspd1 (1:4000 dilution, Santa Cruz); rabbit anti-Ki67 (1:200 dilution, NeoMarkers); rat anti-Ly6G (1:1000 dilution, BD); rabbit anti-RelA (1:200 dilution NeoMarkers); rabbit anti-Tnf (1:150 dilution, Abcam); rabbit anti-c-Myc (1:1000 dilution, Abcam); rat anti-CK19 (1:100 dilution, Developmental Studies Hybridoma Bank). Paraffin-embedded tissue sections were used for staining with 0.1% Sirius red dissolved in saturated picric acid. Oval cell proliferation was detected by A6 staining (1:50), kindly provided by Dr. Valentina Factor. TUNEL assays were performed using the Fluorescein cell death detection kit (Clontech).
Measurement of Serum Parameters
The analysis for AST, ALT, AP, bilirubin, cholesterol, triglycerides and bile acid was performed with mouse serum on a Roche Modular System (Roche Diagnostics) with a commercially available automated colorimetric system using a Hitachi P-Modul (Roche).
ELISA
Tnf protein levels from liver homogenates were measured using a Quantikine-Elisa-Kit from R&D Systems (Oxon, UK). IL6 protein levels were measured using a mouse IL6 Elisa kit from RayBiotech (GA, US). The procedures closely followed the manufacturer’s instructions. The detection limit was 16 pg/ml for Tnf and 2 pg/ml for IL6 respectively. The homogenization buffer was tested as a negative control.
Immunoblot Analysis
Liver homogenates were prepared by mechanical grinding in RIPA buffer (50 mM Tris; 1% NP40; 0.25% Deoxycholic acid sodium salt; 150 mM NaCl; 1 mM EGTA) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific), and quantified with a BCA protein assay kit (Thermo Scientific) according to the manufacturer’s manual. 50 μg protein were denatured in Laemmli buffer containing 5% β-mercaptoethanol and separated by gel electrophoresis and blotted by wet blotting onto nitrocellulose membranes (Bio Rad). Membranes were blocked in 5% milk/TBS-T for at least 1hr at RT. Primary antibodies against Hspd1 (Santa Cruz), Pcna (Santa Cruz), Gp73 (Santa Cruz), c-Jun (Abcam), p-c-Jun (Cell Signaling), p-JNK, JNK, p-Erk1/2, Erk1/2, CHOP, p-p38 MAPK, p38 MAPK, p-Akt, Akt and Gapdh (all Cell Signaling) and were incubated at 4°C overnight under shaking conditions. Incubation with the secondary antibody (HRP-anti rabbit IgG, 1:10000, HRP-anti mouse IgG, 1:10000, Jackson; HRP-anti Goat IgG, 1:10000, Dako) was performed under shaking conditions for 1 hr. Detection was achieved with Clarity Western ECL Substrate (Bio Rad) using Stella 3200 imaging system (Bio Rad). To assure equal loading, membranes reprobed with anti-Gapdh antibody (Cell Signaling) and detected as described above.
RNA Isolation from Liver Tissue
Total RNA from mouse liver samples or cultured cells was isolated using RNeasy Mini kit (Qiagen). The quantity and quality of the RNA was determined spectroscopically using a nanodrop (Thermo Scientific). Purified RNA was reversely transcribed into cDNA using Quantitect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions.
Array-based Comparative Genomic Hybridization (aCGH)
200 ng of isolated DNA were amplified using the REPLI-g Whole Genome Amplification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Efficiency of amplification of high molecular weight DNA was controlled by agarose gel electrophoresis. 3 ug of amplified DNA were digested by Rsa I and Alu I for 2h at 37°C. Digested DNA was subsequently purified using QIAprep Spin Miniprep Columns (Qiagen, Hilden, Germany). Briefly, the digestion reaction was mixed with 5 volumes buffer PB and loaded onto the column. The column was washed once with buffer PE and DNA fragments eluted in buffer EB. 1 μg amplified sample DNA and sex-matched reference DNA was labeled with Cy3 or Cy5, respectively, using the SureTag DNA Labeling Kit (Agilent, Santa Clara, CA) according to the manufacturer’s instructions (protocol version 7.3). Labeled DNA was subsequently hybridized on Mouse Genome CGH 1 × 244K Oligo Microarrays (Agilent, Santa Clara, CA) for 40h. After washing slides were processed on an Agilent G2565BA scanner. Agilent Technologies’ Feature Extraction (version 10.7.3.1) was used for data quality control and extraction. aCGH analysis and plotting was performed as described in (Vucur et al., 2013). In order to reduce GC wave patterns of the resulting profiles the NoWaves approach was used. For this purpose a reference data set consisting of 10 normal-tissue profiles was generated. The synteny analysis between mouse and human was conducted as described in Wolf et al. (2014). Matching of mouse copy number changes to syntenic human regions that were also changed in copy number was tested for random occurrence using Fisher’s exact test. Copy number profiles of human intrahepatic cholangiocarcinomas were generated from level 3 segmented data downloaded from The Cancer Genome Atlas (TCGA http://cancergenome.nih.gov/) CHOL dataset. R packages used for aCGH and synteny analysis were as follows: CGHcall, CGHregions and RCircos.
Real-time PCR
For mRNA expression analysis quantitative real-time PCR was performed in triplicates in 384-well plates using Fast Start SYBR Green Master Rox (Roche) on a 7900 HT qPCR system (Applied Biosystems, Life Technologies Darmstadt, Germany). Relative mRNA levels were calculated according to the ΔΔCt relative quantification method and were normalized to at least two of the examined house-keeping genes (Hprt; Rhot2; Gapdh) levels.
QUANTIFICATION AND STATISTICAL ANALYSIS
Counting of Proliferating Hepatocytes
For quantification of Ki67 staining, slides were scanned using a SCN400 slide scanner (Leica). The total number of Ki67+ hepatocytes was counted at the screen (20 x). For each mouse 5 fields were counted.
Statistical Analysis
Statistical analyses were performed with Prism software (Graphpad Prism version 5.0a). The standard error of the mean was calculated from the average of at least 3 independent samples per condition. To evaluate statistical significance, data was subjected to Student’s t-test (unpaired, two-tailed test). Human specimens were compared using the chi-square test. A p value of less than 0.05 was considered significant.
DATA AND SOFTWARE AVAILABILITY
The accession number for the aCGH data reported in this paper is array express: E-MTAB-4624.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Hspd1 | Santa Cruz | Cat# sc-1052; RRID: AB_631683 |
| Goat anti-GP73 | Santa Cruz | sc-48011; RRID: AB_2113325 |
| Rabbit anti-c-Jun | Abcam | Cat# ab31367; RRID: AB_731606 |
| p-c-Jun | Cell Signaling | Cat# 9164L; RRID: AB_330893 |
| Rabbit anti-p-JNK | Abcam | Cat# ab131499; RRID: AB_11157976 |
| Rabbit anti-JNK | Cell Signaling | Cat# 9252; RRID: AB_2250373 |
| Rabbit anti-p-Erk1/2 | Cell Signaling | Cat# 4370; RRID: AB_2315112 |
| Erk1/2 | Cell Signaling | Cat# 9107; RRID: AB_2235073 |
| Rabbit anti-CHOP | Cell Signaling | Cat# 5554S; RRID: AB_10694399 |
| Rabbit anti-p-p38 MAPK | Cell Signaling | Cat# 4511; RRID: AB_2139682 |
| Rabbit anti-p38 MAPK | Cell Signaling | Cat# 9212; RRID: AB_330713 |
| Rabbit anti-p-Akt | Cell Signaling | Cat# 4060; RRID: AB_2315049 |
| Rabbit anti-Akt | Cell Signaling | Cat# 2938; RRID: AB_915788 |
| Gapdh | Cell Signaling | Cat# 2118; RRID: AB_561053 |
| Rabbit anti-Pcna | Santa Cruz | Cat# sc-7907; RRID: AB_2160375 |
| HRP-anti rabbit IgG | Jackson | Cat# 111-055-045; RRID: AB_2337950 |
| HRP-anti mouse IgG | Jackson | Cat# 115-035-003; RRID: AB_10015289 |
| HRP-anti Goat | Dako | Cat# P0449; RRID: AB_2617143 |
| Mouse anti-8-ohdg | Abcam | Cat# ab48508; RRID: AB_867461 |
| Goat anti-Afp | R&D | Cat# AF5369; RRID: AB_2258018 |
| Rabbit anti-Collagen Type IV | Cedarlane | Cat# CL50451AP; RRID: AB_10059811 |
| Rabbit anti-cleaved Caspase-3 | Cell Signaling | Cat# 9661; RRID: AB_2341188 |
| Rabbit anti-Cytokeratin | Dako | Cat# Z0622; RRID: AB_2650434 |
| Rat anti-F4/80 | BioLegend | Cat# 123105; RRID: AB_893499 |
| Rabbit anti-Glutamine Synthetase | Abcam | Cat# ab16802; RRID: AB_302521 |
| Rabbit anti-Ki67 | NeoMarkers | Cat# RM-9106-S0; RRID: AB_2341197 |
| Rat anti-Ly6G | BD | Cat# 551459; RRID: AB_394206 |
| Rabbit anti-Rela | NeoMarkers | Cat# RB-1638-P0; RRID: AB_59726 |
| Rabbit anti-Tnf | Abcam | Cat# ab6671; RRID: AB_305641 |
| Rabbit anti-Tnfr1 | Abcam | Cat# ab19139; RRID: AB_2204128 |
| Rabbit anti-c-Myc | Abcam | Cat# ab32072; RRID: AB_731658 |
| Rat anti-CK19 | Developmental Studies Hybridoma Bank | Cat# TROMA-III; RRID: AB_2133570 |
| Bacterial and Virus Strains | ||
| AAV8-Cre | VectorBiolabs | Cat#7060 |
| Biological Samples | ||
| Liver tissues from mice after bile duct ligation | Jors et al., 2015 | N/A |
| Liver tissues from mice fed with CDE | Jors et al., 2015 | N/A |
| Liver tissues from mice fed with DDC | Jors et al., 2015 | N/A |
| Liver tissues from CRISPR/Cas9 mulitplex mutagenesis model | Weber et al., 2015 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SP600125 | Abcam | CAS: 129-56-6 |
| JNK-IN-8 | Merck | CAS: 1410880-22-6 |
| JNK6o | Bio-techne | CAS: 894804-07-0 |
| S3I-201 | Biovision | CAS: 501919-59-1 |
| LY-294002 | Sigma | CAS: 934389-88-5 |
| Isoliquiritigenin | Sigma | CAS: 961-29-5 |
| TPCA-1 | Sigma | CAS: 507475-17-4 |
| NBP2–29328 | Novus Biologicals | Cat#NBP2–29328 |
| Critical Commercial Assays | ||
| Mouse Genome CGH Oligo Microarrays | Agilent | Cat#4839A |
| REPLI-g Whole Genome Amplification Kit | Qiagen | Cat#150025 |
| TNF Quantikine-Elisa-Kit | R&D | Cat#SMAT00B |
| Mouse Il6 Elisa kit | RayBiotech | Cat#ELM-IL6 |
| RNAscope 2.0 in situ hybridization kit | Advanced Cell Diagnostics | Cat#310033 |
| Deposited Data | ||
| aCGH data | This paper | The accession number for the aCGH data reported in this paper is arrayexpress: E-MTAB-4624 |
| Intrahepatic cholangiocarcinomas data set in TCGA | TCGA | http://cancergenome.nih.gov/ |
| Experimental Models: Cell Lines | ||
| HuCCT-1 | Roland Rad | Cellbankaustralia, JCRB0425 |
| ETK-1 | Roland Rad | N/A |
| HepG2 | ATCC | HB-8065 |
| Huh7 | Ulrike Protzer | N/A |
| HepRG | Ulrike Protzer | N/A |
| mICC | Roland Rad | N/A |
| Experimental Models: Organisms/Strains | ||
| Hspd1flox/flox | Taconic-Artemis; Dirk Haller (Berger et al., 2016) | N/A |
| R26R-Confetti | Jackson | JAX: 013731 |
| Tnfr1−/− | Jackson | JAX: 003242 |
| Alb-Cre | Jackson | JAX: 003574 |
| JNK1/2flox/flox | Roger Davis | N/A |
| Oligonucleotides | ||
| qRT-PCR primers | This paper | Please see Table S1. |
| Recombinant DNA | ||
| pT3 EF1a-KrasG12D-IRES-GFP | Darjus F. Tschaharganeh | N/A |
| pT3 EF1a-NICD-IRES-cherry | Darjus F. Tschaharganeh | N/A |
| pT3 EF1a-myrAKT | Xin Chen (Fan et al., 2012) | N/A |
| pX330 sgp53 | Darjus T. | N/A |
| Software and Algorithms | ||
| Agilent Technologies’ Feature Extraction (version 10.7.3.1) | Agilent | http://www.agilent.com |
| RCircos | http://bioconductor.org | |
| CGHcall | http://bioconductor.org | |
| CGHregions | http://bioconductor.org | |
Highlights.
Hepatic mitochondrial dysfunction induces premalignant cholangiocellular lesions
Hepatocytes under oxidative stress activate Tnf-producing Kupffer cells
Kupffer cell-derived Tnf triggers cholangiocyte overgrowth through JNK
JNK inhibition impairs intrahepatic cholangiocarcinoma development in several models
Significance.
HCC and ICC are malignant, heterogeneous cancers with rising incidence, poor prognosis, and limited treatment options. The liver microenvironment might favor the development of either HCC or ICC. However, mechanistic underpinnings remain elusive. Analyses of distinct murine and human ICC and HCC specimens revealed especially high ROS levels within ICC and surrounding hepatocytes. Using a mouse model mimicking severe liver damage caused by mitochondrial dysfunction and high ROS levels, we induced profound compensatory proliferation of cholangiocytes and high, focal Tnf levels favoring cholangiocellular overgrowth and premalignant transformation. Our results indicate that Tnf fuels ICC through activation of Tnfr1/JNK in cholangiocytes. Deletion of JNK dampened ICC formation in several aggressive ICC models. Thus, targeting the ROS/Tnf/JNK axis might be promising for ICC therapy.
ACKNOWLEDGMENTS
We thank Ruth Hillermann, Olga Seelbach, Robert Baier, Reiner Dunkl, Tjeerd Sijmonsma, Danijela Heide, Jenny Hetzer, Daniel Kull, and Fiona Mayer for technical support. We thank Jan Tchorz for constructive suggestions. This work was supported by the Horizon 2020 HEPCAR, the ERC Consolidator grant “HepatoMetaboPath”, the SFB Transregio SFB/TR36, the SFB/TR179, the SFB/TR209, and the Graduiertenkolleg (GRK482) to M.H. and D.H., DFG grant (GE 2289/1) to F.G., and NIH grant DK107220 to R.J.D. A.W. was supported by grants from the Hartmann Müller Stiftung and the Swiss National Science Foundation (SNF) (grant SNF 310030_146940).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2017.05.006.
REFERENCES
- Ariizumi SI, and Yamamoto M (2014). Intrahepatic cholangiocarcinoma and cholangiolocellular carcinoma in cirrhosis and chronic viral hepatitis. Surg. Today 45, 682–687. [DOI] [PubMed] [Google Scholar]
- Arnott CH, Scott KA, Moore RJ, Robinson SC, Thompson RG, and Balkwill FR (2004). Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene 23, 1902–1910. [DOI] [PubMed] [Google Scholar]
- Berger E, Yuan D, Waldschmitt N, Rath E, Allgäuer M, Staszewski O, Kober O, Lobner E, Schöttl T, Prinz M, et al. (2016). Mitochondrial function controls epithelial stemness and proliferation in the intestine independent of unfolded protein response signaling mechanisms. Nat. Commun 7, 13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, Ridgway RA, Seo SS, Spee B, Van Rooijen N, et al. (2012). Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med 18, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, et al. (2015). WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Invest 125, 1269–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubero FJ, Zhao G, Nevzorova YA, Hatting M, Al Masaoudi M, Verdier J, Peng J, Schaefer FM, Hermanns N, Boekschoten MV, et al. (2015). Haematopoietic cell-derived Jnk1 is crucial for chronic inflammation and carcinogenesis in an experimental model of liver injury. J. Hepatol 62, 140–149. [DOI] [PubMed] [Google Scholar]
- Das M, Garlick DS, Greiner DL, and Davis RJ (2011). The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 25, 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espin-Palazon R, Stachura DL, Campbell CA, Garcia-Moreno D, Del Cid N, Kim AD, Candel S, Meseguer J, Mulero V, and Traver D (2014). Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 159, 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, Dombrowski F, Evert M, Chen X, et al. (2012). Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest 122, 2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Furuta M, Shiraishi Y, Gotoh K, Kawakami Y, Arihiro K, Nakamura T, Ueno M, Ariizumi S, Hai Nguyen H, et al. (2015). Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun 6, 6120. [DOI] [PubMed] [Google Scholar]
- Gatto M, and Alvaro D (2010). New insights on cholangiocarcinoma. World J. Gastrointest. Oncol 2, 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest RV, Boulter L, Kendall TJ, Minnis-Lyons SE, Walker R, Wigmore SJ, Sansom OJ, and Forbes SJ (2014). Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 74, 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. (2012). Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet 44, 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Barrett T, Brehm MA, and Davis RJ (2016). Inflammation mediated by JNK in myeloid cells promotes the development of hepatitis and hepatocellular carcinoma. Cell Rep. 15, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, Bremer J, Iezzi G, Graf R, Clavien PA, et al. (2009). A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell 16, 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefetz-Sela S, Stein I, Klieger Y, Porat R, Sade-Feldman M, Zreik F, Nagler A, Pappo O, Quagliata L, Dazert E, et al. (2014). Acquisition of an immunosuppressive protumorigenic macrophage phenotype depending on c-Jun phosphorylation. Proc. Natl. Acad. Sci. USA 111, 17582–17587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, et al. (2012). Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet 44, 1117–1121. [DOI] [PubMed] [Google Scholar]
- Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, Ding Q, Loughran P, Beer-Stolz D, Billiar TR, et al. (2013). Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J. Immunol 191, 2665–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H, and Wagner EF (2007). p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet 39, 741–749. [DOI] [PubMed] [Google Scholar]
- Hui L, Zatloukal K, Scheuch H, Stepniak E, and Wagner EF (2008). Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Invest 118, 3943–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannelli F, Collino A, Sinha S, Radaelli E, Nicoli P, D’Antiga L, Sonzogni A, Faivre J, Buendia MA, Sturm E, et al. (2014). Massive gene amplification drives paediatric hepatocellular carcinoma caused by bile salt export pump deficiency. Nat. Commun 5, 3850. [DOI] [PubMed] [Google Scholar]
- Jhunjhunwala S, Jiang Z, Stawiski EW, Gnad F, Liu J, Mayba O, Du P, Diao J, Johnson S, Wong KF, et al. (2014). Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 15, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. (2013). Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet 45, 1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jors S, Jeliazkova P, Ringelhan M, Thalhammer J, Durl S, Ferrer J, Sander M, Heikenwalder M, Schmid RM, Siveke JT, et al. (2015). Lineage fate of ductular reactions in liver injury and carcinogenesis. J. Clin. Invest 125, 2445–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, and Karin M (2005). Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661. [DOI] [PubMed] [Google Scholar]
- Kang LI, Mars WM, and Michalopoulos GK (2012). Signals and cells involved in regulating liver regeneration. Cells 1, 1261–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keitel V, Ullmer C, and Haussinger D (2010). The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol. Chem 391, 785–789. [DOI] [PubMed] [Google Scholar]
- Liu B, Zupan B, Laird E, Klein S, Gleason G, Bozinoski M, Gal Toth J, and Toth M (2014). Maternal hematopoietic TNF, via milk chemokines, programs hippocampal development and memory. Nat. Neurosci 17, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, and Lichtman JW (2007). Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62. [DOI] [PubMed] [Google Scholar]
- Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, Guest RV, Wojtacha D, Man TY, Mackinnon A, et al. (2015). Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol 17, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, and Karin M (2005). IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121, 977–990. [DOI] [PubMed] [Google Scholar]
- Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J, Kay MA, Grimm D, and Willenbring H (2011). Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest 121, 4850–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter MS, Marquardt JU, Andersen JB, Quintavalle C, Korokhov N, Stauffer JK, Kaji K, Decaens T, Quagliata L, Elloumi F, et al. (2016). Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology 63, 1888–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress co-operates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa Y, Sone M, Nagahama Y, Kumagai E, Doi Y, Omori Y, Yoshioka T, Tokairin T, Yoshida M, Yamamoto Y, et al. (2013). Tumor necrosis factor-alpha promotes bile ductular transdifferentiation of mature rat hepatocytes in vitro. J. Cell Biochem 114, 831–843. [DOI] [PubMed] [Google Scholar]
- Nitou M, Sugiyama Y, Ishikawa K, and Shiojiri N (2002). Purification of fetal mouse hepatoblasts by magnetic beads coated with monoclonal anti-e-cadherin antibodies and their in vitro culture. Exp. Cell Res 279, 330–343. [DOI] [PubMed] [Google Scholar]
- Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, and Igaki T (2012). Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 490, 547–551. [DOI] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, and Ben-Neriah Y (2004). NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431, 461–466. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, and Magnuson MA (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem 274, 305–315. [DOI] [PubMed] [Google Scholar]
- Rizvi S, Eaton JE, and Gores GJ (2015). Primary sclerosing cholangitis as a premalignant biliary tract disease: surveillance and management. Clin. Gastroenterol. Hepatol 13, 2152–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, and Klaunig JE (2007). Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol. Sci 96, 2–15. [DOI] [PubMed] [Google Scholar]
- Rougemont AL, Genevay M, McKee TA, Gremaud M, Mentha G, and Rubbia-Brandt L (2010). Extensive biliary intraepithelial neoplasia (BilIN) and multifocal early intrahepatic cholangiocarcinoma in non-biliary cirrhosis. Virchows Arch. 456, 711–717. [DOI] [PubMed] [Google Scholar]
- Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, et al. (2014). Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Maeda S, Chang L, and Karin M (2006). Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 103, 10544–10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheeren FA, Kuo AH, van Weele LJ, Cai S, Glykofridis I, Sikandar SS, Zabala M, Qian D, Lam JS, Johnston D, et al. (2014). A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat. Cell Biol 16, 1238–1248. [DOI] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet 47, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Evans JE, and Rock KL (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521. [DOI] [PubMed] [Google Scholar]
- Szabo G, and Petrasek J (2015). Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol 12, 387–400. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. (2015). A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlow BD, Pelz C, Naugler WE, Wakefield L, Wilson EM, Finegold MJ, and Grompe M (2014). Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 15, 605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, Katz SF, Kastenhuber ER, Weissmueller S, et al. (2014). p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson GL, and El-Serag HB (2011). Risk factors for cholangiocarcinoma. Hepatology 54, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, Kreggenwinkel K, Koppe C, Hammerich L, Hakem R, et al. (2013). RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep 4, 776–790. [DOI] [PubMed] [Google Scholar]
- Weber J, Ollinger R, Friedrich M, Ehmer U, Barenboim M, Steiger K, Heid I, Mueller S, Maresch R, Engleitner T, et al. (2015). CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc. Natl. Acad. Sci. USA 112, 13982–13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, et al. (2014). Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26, 549–564. [DOI] [PubMed] [Google Scholar]
- Woodworth CD, McMullin E, Iglesias M, and Plowman GD (1995). Interleukin 1 alpha and tumor necrosis factor alpha stimulate autocrine amphiregulin expression and proliferation of human papillomavirus-immortalized and carcinoma-derived cervical epithelial cells. Proc. Natl. Acad. Sci. USA 92, 2840–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanger K, Knigin D, Zong Y, Maggs L, Gu G, Akiyama H, Pikarsky E, and Stanger BZ (2014). Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 15, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, and Camargo FD (2014). Hippo pathway activity influences liver cell fate. Cell 157, 1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, El-Khatib M, Geffers R, Bektas H, Manns MP, et al. (2013). A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 23, 784–795. [DOI] [PubMed] [Google Scholar]
- Zhang F, Chen XP, Zhang W, Dong HH, Xiang S, Zhang WG, and Zhang BX (2008). Combined hepatocellular cholangiocarcinoma originating from hepatic progenitor cells: immunohistochemical and double-fluorescence immunostaining evidence. Histopathology 52, 224–232. [DOI] [PubMed] [Google Scholar]
- Zhu HZ, Ruan YB, Wu ZB, and Zhang CM (2000). Kupffer cell and apoptosis in experimental HCC. World J. Gastroenterol 6, 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS, Zhao X, Li Y, Li Q, Wang H, et al. (2014). Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun 5, 5696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the aCGH data reported in this paper is array express: E-MTAB-4624.