

Abstract
Autophagy is a conserved quality-control pathway that degrades cytoplasmic contents in lysosomes. Autophagy degrades lipid droplets through a process termed lipophagy. Starvation and an acute lipid stimulus increase autophagic sequestration of lipid droplets and their degradation in lysosomes. Accordingly, liver-specific deletion of the autophagy gene Atg7 increases hepatic fat content, mimicking the human condition termed nonalcoholic fatty liver disease. In this review, we provide insights into the molecular regulation of lipophagy, discuss fundamental questions related to the mechanisms by which autophagosomes recognize lipid droplets and how ATG proteins regulate membrane curvature for lipid droplet sequestration, and comment on the possibility of cross talk between lipophagy and cytosolic lipases in lipid mobilization. Finally, we discuss the contribution of lipophagy to the pathophysiology of human fatty liver disease. Understanding how lipophagy clears hepatocellular lipid droplets could provide new ways to prevent fatty liver disease, a major epidemic in developed nations.
Keywords: lipophagy, lipases, lysosome, metabolism, steatosis
AN INTRODUCTION TO AUTOPHAGY
Autophagy is a cellular recycling pathway that is conserved across systems as diverse as yeast, flies, worms, and mammals (33). Autophagy, or “auto” and “phagos,” which translate to “eating one’s own self,” is a mechanism that delivers cytoplasmic cargo into acidic compartments of the cell known as lysosomes (19). The best-characterized functions of autophagy include maintenance of cellular quality control and provision of an alternate source of energy during starvation (33, 70, 107). Indeed, cells lacking autophagy are unable to clear dysfunctional and/or aged organelles and sustain themselves during nutrient insufficiency, which results in loss of function and cell death. It is therefore reasonable to conclude that cells have evolved multiple pathways to turn over their unwanted refuse in lysosomes. To that end, autophagy is classified in three distinct categories on the basis of the mechanism of lysosomal cargo delivery and the cargo type delivered to the lysosome: macroautophagy, chaperone-mediated autophagy (CMA) (13), and microautophagy (95, 97). Macroautophagy (33), a mechanism for in-bulk cargo degradation, requires a complex interplay of greater than ~30 autophagy proteins that orchestrate cargo delivery to lysosomes. In the basal state or under conditions of stress or starvation, macroautophagy entails de novo formation of double-membrane autophagosomes that sequester and deliver cargo to lysosomes. In contrast, CMA selectively degrades single soluble proteins with a specific amino acid signature, the KFERQ motif (66), which is recognized by cytosolic heat shock cognate protein 70 (Hsc70) and then delivered to lysosomes by the CMA receptor lysosome-associated membrane protein (LAMP)-2A. Microautophagy, the least characterized form of autophagy, involves direct sequestration of cytoplasmic cargo by invaginations of lysosomal and late endosomal membranes (95, 97), which is followed by the degradation of engulfed cargo. In this review, we focus on macroautophagy (hereafter referred to as autophagy) and its recently elucidated function in cellular lipid utilization, termed lipophagy (108).
REGULATION OF AUTOPHAGY
The Molecular Basis of Autophagosome Formation
Although the lysosome was identified as a distinct organelle in the 1950s through elegant electron microscopic work by Christian de Duve and colleagues (75), it is only through quite recent investigations that the molecular pathways regulating autophagy have begun to be understood (78). In fact, the cellular sources of autophagosomes and mechanisms of autophagosome biogenesis, how autophagy is regulated, and how cells identify and label cytoplasmic cargo for degradation currently are active areas of research.
It was through yeast genetic screens that autophagy gene (ATG) proteins were identified as molecular effectors of the macroautophagy pathway (31, 99, 122). Subsequent studies in higher organisms identified that induction of macroautophagy requires the release of the mammalian ortholog of ATG6, Beclin-1 (60, 129), from its binding partner Bcl-2, which allows Beclin-1 to form a complex with vps34 (34), ATG14/Barkor (39), and vps15/p150 (35) that together constitute class III phosphatidylinositol 3-kinase (PI3K) activity (Figure 1). Active class III PI3K generates phosphatidylinositol 3-phosphates (PI3Ps), which recruit additional ATG proteins to sites of phagophore assembly for de novo–limiting membrane formation (Figure 1) (33). Recruitment of a critical upstream regulator of autophagy, UNC51-like kinase 1 (Ulk1) (140), allows inclusion of additional ATGs, for instance ATG9, to the phagophore (62). ATG9 is the only transmembrane ATG protein that has been considered as a membrane donor for the growing autophagosome (128). PI3Ps also attract orthologs of ATG18, i.e., WD repeat domain phosphoinositide-interacting protein 1 (WIPI1) and WIPI2, to the phagophore that allows maturation of the autophagosome (88, 123). In recent years, great interest has been given to cellular sites of autophagosome biogenesis, and the subject remains under investigation (64, 121). A number of studies have now shown that autophagosomes originate from precursors derived from the plasma membrane (91), endoplasmic reticulum (32, 143), mitochondria-endoplasmic reticulum contact sites (28), and the Golgi apparatus (142). The expansion of limiting membranes into autophagosomes requires two ubiquitin-like conjugation cascades (33). ATG7 is a unique ubiquitin E1-like ligase that catalyzes binding of ATG12 with ATG5 (Figure 1) (116). In subsequent steps, the ATG12-ATG5 conjugate interacts with ATG16L1 to give rise to ATG12-ATG5-ATG16L1 in a reaction requiring the E2-like conjugating activity of ATG10 (Figure 1) (87, 94, 116). In the presence of ATG3, a protein with E2-like activity (139), and ATG7, ATG12-ATG5-ATG16L1 catalyzes the binding of soluble/cytosolic microtubule-associated protein 1 light chain 3 (LC3) to lipid moieties on the growing autophagic membranes (Figure 1) (29). Lipidated LC3, also known as LC3-II, is a molecular signature of the autophagosome (117) and is structurally and functionally essential for autophagosome formation and cargo recognition. LC3-II-positive autophagosomes recognize and capture cargo through the interaction of LC3 with p62/sequestosome-1 (SQSTM-1) (83), a cargo adaptor protein that is bound to polyubiquitinated cargo. Autophagosomes containing sequestered cargo traffic along microtubules to fuse with lysosomes in a manner that is soluble N-ethylmaleimide-sensitive factor–activating protein (SNARE) dependent, resulting in cargo degradation (40, 71). Although autophagy maintains a basal level of activity to fine-tune quality control and basal energetic needs, a wide variety of stressors rapidly activate autophagy, which allows the cell to survive through adverse conditions.
Figure 1.
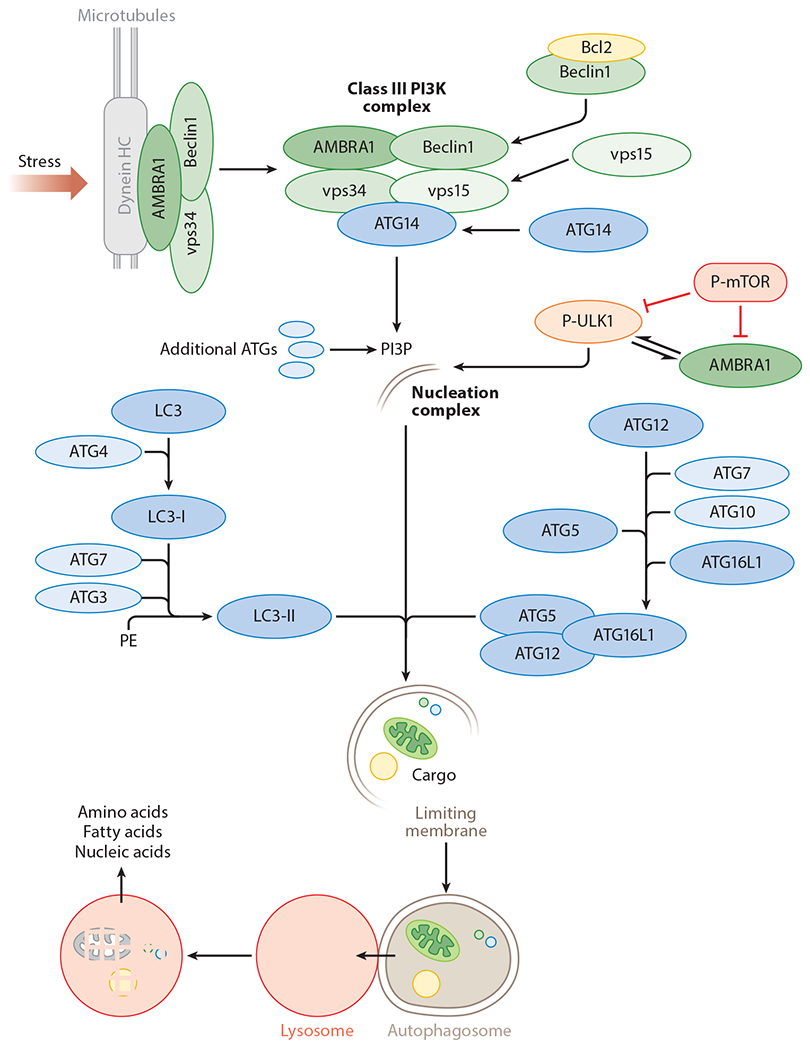
The autophagy pathway. Stress or starvation activates the class III phosphatidylinositol 3-kinases (PI3K) complex that generates phosphatidylinositol 3-phosphates (PI3Ps). PI3Ps recruit autophagy genes (ATGs) to the nucleation complex including UNC51-like kinase 1 (ULK1), an upstream regulator of autophagy. Two conjugation cascades, i.e., the ATG12-ATG5 and light chain 3 (LC3) conjugation cascades, are activated, which results in lipidation of cytosolic LC3-I into membrane-bound LC3-II. LC3-II-positive autophagosomes sequester cytoplasmic cargo that is delivered to the lysosomes, wherein a battery of acid hydrolases degrade engulfed cargo. Products of degradation—amino acids, fatty acids, and nucleic acids—are utilized by the starved cell. Abbreviations: AMBRA1, activating molecule in Beclin1-regulated autophagy; Beclin1, coiled-coil, myosin-like BCL2-interacting protein; Dynein HC, dynein heavy chain; P-mTOR, phospho-mammalian target of rapamycin; P-ULK1, phospho-UNC51-like kinase 1.
The Signaling Cascades Regulating Autophagy
In light of the fact that autophagy is activated in response to diverse environmental cues, it is likely that multiple signaling cascades are engaged to tightly regulate the activity of this pathway, and in fact, this has been shown to be the case. For instance, the cellular nutrient sensor, mechanistic target of rapamycin (mTOR), is a critical inhibitor of autophagy (92). Nutrient availability and growth factors activate mTOR, which blocks autophagy by inhibiting upstream autophagy activator Ulk1 through its phosphorylation at Serine-757 (Figure 2) (49). Suppression of autophagy ensures that this pathway does not inadvertently degrade cellular components while nutrients are abundant. By contrast, starvation-associated increases in cellular adenosine monophosphate (AMP) activate adenosine monophosphate-activated protein kinase (AMPK) (Figure 2) (30). Active AMPK induces autophagy by phosphorylating Ulk1 at several residues (22, 49), for instance, Serine-555 and Serine-777, which are distinct from those phosphorylated by the nutrient-activated mTOR target Serine-757 (49). As noted previously, the activation of Ulk1 initiates membrane trafficking events that include localization of transmembrane protein ATG9 to sites of de novo autophagosome biogenesis and induction of autophagy (144). Elegant studies have identified complex regulatory loops that sense changes in nutrient availability and modify autophagy (22, 49, 73); for instance, interplay between mTOR and a recently described autophagy regulator AMBRA1 (25) and Ulk1 has recently been shown to regulate autophagy (73).
Figure 2.
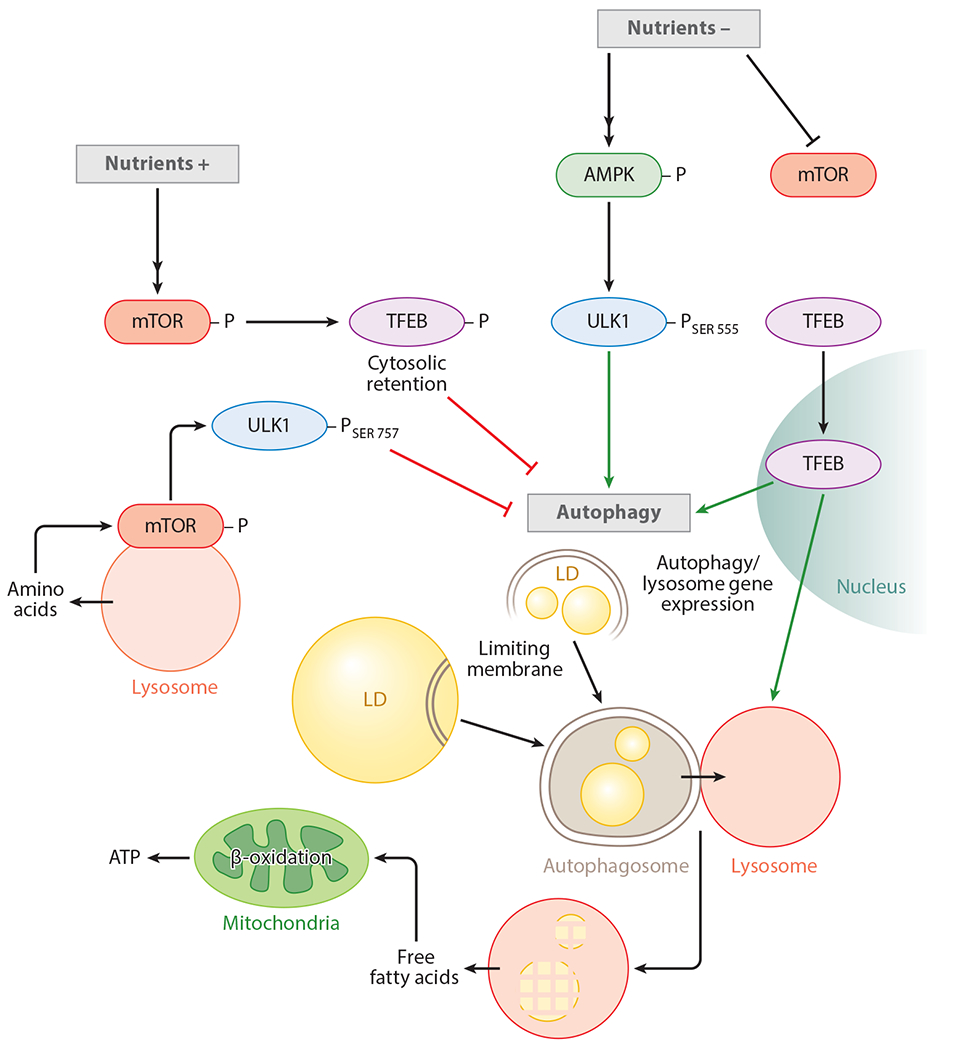
The regulation of lipophagy. The availability of nutrients phosphorylates and activates the mechanistic target of rapamycin (mTOR). It is likely that lysosomal efflux of amino acids also activates mTOR. Active mTOR phosphorylates transcription factor EB (TFEB) and retains it in the cytoplasm, and it decreases autophagy and lysosomal gene expression. Active mTOR also suppresses UNC51-like kinase 1 (ULK1) activity by phosphorylating ULK1 at SER 757. During nutrient depletion, mTOR is inactivated while adenosine monophosphate-activated protein kinase (AMPK) is activated. These events allow ULK1 phosphorylation at SER 555, which results in ULK1 activation. Suppression of mTOR activity also allows TFEB to localize to the nucleus to drive autophagy and lysosomal gene expression. Activation of autophagy begins to sequester and degrade lipid droplets (LDs) in lysosomes. Lysosome-derived free fatty acids are oxidized in the mitochondria for production of adenosine triphosphate (ATP).
Although maintenance of active autophagy was not traditionally considered to require de novo ATG protein synthesis because ATG proteins were not considered rate limiting, recent studies have identified a gene regulatory network that activates autophagy by increasing lysosomal biogenesis (104). Indeed, a basic HLH-leucine zipper transcription factor EB (TFEB) has been identified as a master regulator of the CLEAR (coordinated lysosomal enhancement and regulation) network that directly regulates Atg and lysosomal gene expression (82, 98, 104). TFEB is regulated by its nuclear exclusion, and studies exploring the regulation of TFEB have identified two kinases—ERK2 and mTOR—that control autophagy activity by modulating TFEB phosphorylation and its cellular localization (104, 105). It has been shown that nutrient and growth factors stimulate ERK2 and mTOR activation that phosphorylates TFEB at Serine-142 and increases its cytoplasmic accumulation (104). Work from the same group has shown that amino acids recruit mTOR to the lysosomal surface by action of a protein complex that includes v-ATPase, Rag GTPases, and Ragulator (96). At the lysosomal surface, active mTOR interacts and phosphorylates TFEB and retains it in the cytosol, which effectively reduces macroautophagy activity. In contrast, fasting decreases lysosomal v-ATPase activity, which in turn blocks Rags and thus releases mTOR from the lysosomes. Consequently, in the absence of active mTOR, TFEB remains in the dephosphorylated state, allowing it to traverse to the nucleus and turn on the CLEAR network of genes and hence macroautophagy (Figure 2). Given the intricate control of macroautophagy at several steps by a plethora of regulatory proteins, it is conceivable that disruptions in any of the regulatory steps would contribute to an age-related decline in macroautophagy and cell function. Indeed, a large body of work supports the concept of reduced autophagy with age (11, 43, 86, 89, 100, 145) and forms the basis of much of the ongoing work that is aimed at identifying ways to restore autophagy during aging.
LIPID DROPLET TURNOVER BY LIPOPHAGY
Introduction to Lipophagy
Lysosomes contain a battery of hydrolases, including proteases, lipases, nucleases, and glycases, that allow rapid turnover of unwanted portions of the cell (9). In fact, autophagy can be classified on the basis of the type of cargo it degrades; for instance, autophagy degrades mitochondria via mitophagy (47), peroxisomes by pexophagy (48), endoplasmic reticulum via reticulophagy (118), and ribosomes through ribophagy (55); in addition, autophagy has been shown to degrade RNA (37, 130). Thus, the autophagic apparatus eliminates cargo in bulk yet maintains remarkable selectivity in its ability to distinguish damaged organelles marked for degradation from those that are healthy and functional.
In recent years, much attention has been focused on understanding the physiology behind the regulation of cellular fat stores within lipid droplets (133). During nutrient sufficiency, cells store their energy reserves as fat within lipid droplets—which are large stores of neutral lipid esters, triglycerides, and cholesteryl esters (46)—surrounded by a phospholipid monolayer and lipid droplet coat proteins that are classified into a family of perilipins (PLINs) (50). Indeed, due to the dynamic cross talk of lipid droplets with additional organelles, and due to the fact that lipid droplets have been shown to buffer misfolded proteins (12) or infectious agents (24), these seemingly inert fat stores have often been considered as distinct organelles (17).
The synthesis of lipid droplets occurs at the endoplasmic reticulum (27, 46), and most cells generate lipid droplets in the size range of 0.1 to 10 μm, although cells dedicated for fat storage—adipocytes—contain much larger lipid droplets that grow up to 100 μm in size. During starvation, lipid droplets undergo lipolysis, a process well studied in adipocytes, wherein lipolytic stimuli activate protein kinase A, which phosphorylates lipid droplet coat protein PLIN1 (112). This results in PLIN1 degradation via the proteasome and in turn facilitates recruitment of cytosolic lipases to the lipid droplet surface (112). Given that starvation activates autophagy and that starvation-induced autophagy generates nutrients through lysosomal degradation of unwanted cytoplasmic contents, it was proposed that the autophagolysosomal system played a role in breaking down lipid droplets to provide free fatty acids to the starved cell.
To test the role of autophagy in the degradation of cellular lipid stores, hepatocytes were exposed to the pharmacological autophagy inhibitor 3-methyladenine, which blocks class III PI3K activity, or were treated with small hairpin (sh) RNA against the autophagy gene Atg5. Inhibition of autophagy remarkably increased lipid droplet content in hepatocytes cultured in the basal state or when cells were exposed to free fatty acids (108). Mouse embryonic fibroblasts knocked out for Atg5 also displayed increased triglyceride levels (108). Increased lipid droplet content in autophagy-deficient cells did not result from increased triglyceride synthesis but occurred due to a decrease in lipid turnover as indicated by reduced rates of triglyceride decay and mitochondrial β-oxidation (108). To determine whether autophagosomes and lysosomes interact with lipid droplets, fed and serum-starved cells were subjected to fractionation and imaging studies. To that end, immunogold electron microscopy revealed sequestration of lipid droplets by LC3-labeled membranes, and accordingly, isolated lipid droplets from fasted livers displayed enrichment of LC3 (108). Conversely, autophagosomes and lysosomes isolated from fasted livers revealed enrichment of lipid droplet markers PLIN1 and PLIN2 (108). These studies indicated that dynamic interactions occurred between these seemingly distinct cellular compartments. In a second experimental approach, indirect immunofluorescence of cultured hepatocytes showed colocalization of BODIPY 493/503, a lipid droplet marker, with markers of autophagosomes and lysosomes, LC3 and LAMP1, respectively (108). The inclusion of a lipid stimulus increased associations of lipid droplets with LAMP1-positive structures; blocking lysosomal function further increased BODIPY 493/503/LAMP1 colocalization, indicating the accumulation of lipids within lysosomes. By contrast, interfering with autophagosome-lysosome fusion led to decreased BODIPY 493/503/LAMP1 colocalization, indicating the failure of delivery of lipids to lysosomes (108). Although it remains undetermined whether neutral cytosolic lipases contribute, in part, to this lysosomal consumption of lipid, a ubiquitous effect of dissipating lysosomal pH, which suppresses lysosomal degradation but leaves intact neutral lipase activity, was blocked lipolysis (44, 108). These findings demonstrate a fundamental role of lysosomes in lipid droplet turnover. Validation of the role for autophagy in lipid droplet mobilization in vivo came from mice that selectively lacked the autophagy gene Atg7 in livers. Consistent with findings of increased lipid accumulation in autophagy-deficient hepatocytes and Atg5−/− mouse embryonic fibroblasts, liver-specific Atg7−/− mice displayed remarkable lipid accumulation that histologically mimicked the commonly observed human condition—nonalcoholic fatty liver disease (108).
Intriguingly, we find that providing a lipid stimulus acutely, for instance treating hepatocytes with a physiological fatty acid such as oleic acid, activates lipophagy (108). This activation occurs possibly as a mechanism to consume excessive lipid influx into the hepatocyte. Oleic acid–induced autophagy has also been reported in cultured hypothalamic neurons (44), which suggests that this is perhaps a ubiquitous effect observed in diverse cell types. In contrast, chronic lipid availability, such as prolonged high-fat-diet feeding, has been shown to suppress lipophagy (108). Indeed, in models of obesity, such as high-fat-diet-fed mice and the leptin-deficient ob/ob mice, it has been reported that chronic overnutrition decreases hepatic ATG7 levels and decreased autophagy activity (141). Furthermore, overnutrition-induced autophagy suppression has been associated with hepatic inflammation and endoplasmic reticulum stress (141). In our electron microscopy studies, high-fat-diet feeding led to decreased LC3 positivity in liver lipid droplets (108), indicating decreased autophagic sequestration of lipid droplets. In an attempt to analyze whether restoring ATG7 levels can reverse hepatic fat accumulation in obese mice, Yang and colleagues (141) utilized an adenoviral approach to overexpress ATG7 in livers of genetic and diet-induced models of obesity. Liver-specific overexpression of ATG7 was sufficient to prevent hepatic steatosis in ob/ob mice, supporting the central role of autophagy in controlling hepatic fat content (141). However, the question of why autophagy flux decreases in models of chronic overnutrition remains. Although it is possible that multiple mechanisms contribute to decreased autophagy during obesity, a study using an in vitro assay that measured rates of fusion between isolated autophagosome and lysosomal compartments revealed that feeding mice a high-fat diet decreased autophagosome-lysosome fusion by up to 70% (52). In this context, ethanol-induced hepatotoxicity has also been associated with decreased autophagolysosomal fusion (10). In addition to lysosomal pH and ATP availability, which are predominant factors controlling autophagolysosomal fusion, it is likely that dietary factors alter lysosomal membrane lipid composition (52, 53), which in turn determines autophagolysosomal fusion events. As previously noted, in recent years SNARE proteins have been shown to regulate autophagy by recruiting ATG proteins to sites of autophagosome formation (40, 71). Detailed work from the Mizushima laboratory (40) has demonstrated a role for SNARE protein syntaxin 17 in regulating the fusion of autophagosomes with acidic organelles; however, whether changes in SNARE protein function are behind decreased autophagolysosomal fusion and lipophagy during obesity states remains to be determined.
Since the original finding of autophagic lipid droplet turnover in hepatocytes, a plethora of studies have now identified lipophagy as a means to mobilize cellular fat stores in diverse cell types, including orexigenic hypothalamic neurons (44), primary striatal neurons (65), glial cells (65), lymphocytes (38), macrophage foam cells (80, 81), adipose-resident macrophages (137), cultured adipocytes (59), gastrointestinal epithelial cells (45), and prostate cancer cells (41); in nonmammalian systems such as the yeast Saccharomyces cerevisiae (124, 126), Caenorhabditis elegans (57, 76), and the fungus Fusarium graminearum (74); in experimental plant systems such as rice (56); and in phyllosphere microorganisms (79). Taken together, these studies are a testament to a conserved role for autophagy in maintaining lipid homeostasis across multiple organisms, which supports the use of the term macrolipophagy or lipophagy (108) as a ubiquitous pathway for in-bulk mobilization of cellular lipid droplets.
Transcriptional Regulators of Lipophagy
A growing interest in the field of lipid metabolism is the identification of the regulatory networks linking the activation of autophagy to catabolism of cellular lipid droplets. To that end, a study by Lettieri Barbato (59) and colleagues revealed that starvation-induced activation of transcription factor forkhead box protein O1 (FoxO1) modulates adipose mass through the transcriptional upregulation of lysosomal acid lipase-mediated autophagy. More recent studies have revealed that activation of TFEB, a master regulator of autophagy and its nuclear localization, has been linked to activation of autophagy-mediated lipid utilization. Indeed, in a study performed in C. elegans, the nutrient status in a given state orchestrates the coordinated regulation of transcription factors HLH-30 (the TFEB ortholog in worms) andMXL-3 that, in turn, modulate lysosomal acidic lipase LIPL1- and LIPL3-dependent lipid droplet breakdown through lipophagy (76). In addition to this study in worms, a study conducted in mammals revealed that TFEB exerts a central role in hepatic lipid metabolism by orchestrating a transcriptional program in response to starvation via the PGC1α-PPARα-lipophagy axis that mediates lipid catabolism (103). Related work by Seok and colleagues (102) has provided evidence that the cAMP response element-binding protein (CREB) promotes lipophagy in the fasted state via direct transcriptional activation of TFEB and autophagy genes; in contrast, nutrient availability suppresses this effect by activation of the nuclear receptor farnesoid X receptor (FXR). Thus, the CREB-FXR axis represents a major nutrient-sensing network that tightly regulates lipophagy during periods of fasting and refeeding. Lee and colleagues (58) have described a similar regulatory interplay between FXR and peroxisome proliferator-activated receptor alpha (PPARα), wherein starvation-induced PPARα activation suppresses feeding-induced FXR-driven inhibition of autophagy. These findings indicate that, contrary to previous belief, lipid utilization via lipophagy requires complex regulatory networks that entail transcriptional regulation of autophagy genes.
INSIGHTS INTO THE MOLECULAR REGULATION OF LIPOPHAGY
Cytosolic lipases mobilize lipid droplets by breaking down one triglyceride molecule into three molecules of fatty acids and a molecule of glycerol via tandem reactions catalyzed by adipose triglyceride lipase (ATGL) (147), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (125). By contrast, autophagosomes sequester portions of larger lipid droplets or possibly consume smaller lipid droplets in toto for their delivery to lysosomes (108). Within lysosomes, the lysosomal acid lipase degrades triglycerides into fatty acids and glycerol (14, 108). In this regard, the autophagy pathway can be viewed as an attractive mechanism that rapidly eliminates cellular lipid stores in bulk.
How Does the Autophagy Machinery Consume Lipid Droplets?
Autophagosomes and lysosomes range from 0.5 to 1.5 μm and 50 to 100 nm in size, respectively; lipid droplets are much larger in size and can be up to 10 μm in size in nonadipocytes. Given these differences in organelle size, it is likely that autophagosome assembly occurs at the surface of the lipid droplet. Indeed, electron microscopy data suggest that limiting membranes generate at the lipid droplet surface and expand into the core of the lipid droplets (108). The limiting membrane then seals to generate an autophagosome, which pinches off portions of the lipid droplet for lysosomal targeting (108). In fact, electron microscopic analyses revealed that lipid droplets were less often consumed in totality and observed more often to be consumed piecemeal (108). In support of this notion, we detected enrichment of soluble LC3-I and the upstream regulators of autophagy, ATG7, ATG5, and Beclin 1, on the lipid droplet surface (unpublished data). In addition, mice lacking ATG7 in the liver displayed accumulation of LC3-I in lipid droplet fractions (108), which would suggest that although the E1-like ligase ATG7 is necessary for sequestration of lipid droplets by LC3-II-positive autophagosomes, ATG7 was not required for the recruitment of cytosolic LC3-I to the lipid droplet surface. These findings have opened up several new questions; for instance, how might ATG proteins be recruited to the lipid droplet, and how might lipid droplets be recognized as an autophagic cargo? How do autophagosomes attain the appropriate curvature and dimensions to sequester a lipid droplet? Although these questions currently remain unanswered, possible solutions are being considered.
How Does the Autophagy Machinery Recognize Lipid Droplets?
The mechanism of sequestration of cytoplasmic cargo by the autophagosomes is an exciting area of research, and the current paradigm, as revealed through studies of sequestration of intracellular protein aggregates, suggests that polyubiquitinated chains of specific lysine linkages serve as biological labels that identify cargo destined for degradation and spare those that are considered essential for the cell. To that end, protein polyubiquitination patterns have served as biological tags that allow recognition of bacteria invading a host cell as well as damaged organelles. Whereas organelles such as mitochondria display a dedicated tagging system requiring Pink1, a serine/threonine kinase that localizes to the mitochondria via its mitochondrial localization sequence, and Parkin, an E3 ubiquitin ligase, it remains unknown whether such a dedicated tagging system exists for lipid droplet degradation by autophagy. Whether ubiquitination of lipid-droplet-associated proteins per se targets lipid droplets for consumption by autophagy also is not known. Quite interestingly, ancient ubiquitous protein 1 (AUP1), a protein that contains a domain at the C-terminus that binds E2 ubiquitin conjugases, is recruited to lipid droplets. It has been shown that the C-terminus of AUP1 binds to an E2 ubiquitin-conjugating enzyme, Ube2g2 (113). These findings suggest that recruitment of the AUP1-Ube2g2 molecular complex to the lipid droplet surface could be a potential mechanism that initiates the labeling of the lipid droplet components. However, in light of these considerations, a second set of questions comes to mind: If ubiquitination is the signal that allows elimination of the PLIN coat protein layer of the lipid droplet, then is the process coordinated by cross talk between multiple proteolytic systems? Indeed, both the lysosomal and proteasomal proteolytic systems have been shown to turn over lipid-droplet-associated coat proteins (54, 67, 77, 135, 136), although the precise mechanism for their degradation via autophagy remains unclear. Furthermore, what might be the biological tag that labels lipids for lysosomal degradation? Is it possible that the autophagy apparatus identifies a distinct lipid tag and converges onto the exposed lipid surface following the removal of the PLIN layer on lipid droplets?
A unified theme in the mechanisms described to mediate cargo recognition is the binding of the biological tag, i.e., polyubiquitinated proteins, to cargo adaptors such as SQSTM1/p62 (8). SQSTM1/p62 is a unique protein that is able to bind to a wide variety of polyubiquitinated proteins through a UBA domain at its C-terminus (90). SQSTM1/p62 also polymerizes via its Phox and Bem1 (PB1) domain at the N-terminus, and this ability to polymerize with itself is considered to favor its binding to diverse polyubiquitinated substrates (7). SQSTM1/p62 also binds to the autophagosome marker LC3-II, which allows it to serve as a cargo adaptor for autophagy (115). Additional autophagy cargo adaptors have been identified, for instance, NBR1, NDP52, and optineurin, each of which regulates the recognition of diverse cytoplasmic material for autophagic degradation (18, 120, 132). Whether any of these autophagy receptors serve as the candidate molecule that targets lipid droplets for autophagic degradation remains to be investigated. Interestingly, recent work has linked mutations in huntingtin to accumulation of cytoplasmic lipid droplets in multiple cell types (65). Huntingtin is a widely expressed protein whose mutations result in development of Huntington’s disease, a severely debilitating neurological condition. Huntingtin has been found to associate with membranes of various organelles (4), and mutations in huntingtin result in the accumulation of large autophagosomes that are unable to sequester cargo and appear to be devoid of organelles (65). These “empty” autophagosomes fail to sequester and target cargo for lysosomal degradation, and it has been proposed that mutations in huntingtin lead to a defective association with SQSTM1/p62, which impairs cargo recognition (65). Interestingly, as reported by the Cuervo group (65), cells that express mutant huntingtin display remarkable lipid accumulation, demonstrating a role of this protein in regulating lipid droplet breakdown via autophagy. These intriguing findings beg a number of questions, including whether huntingtin associates with lipid droplets and whether huntingtin could be the cargo recognition molecule that tethers lipid droplets to the forming autophagosome. Needless to say, additional studies will be necessary to dissect the potential roles for huntingtin—and for that matter, SQSTM1/p62, NBR1, NDP52, and optineurin—in lipid droplet turnover through lipophagy.
Autophagy Genes, Membrane Curvature, and Lipid Droplets
This brings us to questions pertaining to the development of autophagic membrane curvature, which is likely to be a critical determinant for efficient sequestration of lipid droplets of diverging sizes. Elegant work from Fan and colleagues (23) has identified a domain at the C-terminus of ATG14, which is termed Barkor/Atg14 autophagosome targeting sequence (BATS). Mutational analyses indicate that the ATG14 BATS domain binds directly to autophagic membranes via a hydrophobic region on its amphipathic alpha helix (23). Furthermore, the BATS domain of ATG14 binds to areas of curvature in the early autophagic structures. The authors also show that the BATS domain senses membrane curvature by selectively binding to curved membranes containing phosphatidylinositol 3-phosphate but not phosphatidylinositol 4,5-biphosphate (23). Given the ability of ATG14 to sense and maintain curvature, it is possible that ATG14 is a critical molecule that governs recruitment of ATGs to strategic sites on the lipid droplet surface for de novo autophagosome formation. A more recent study has highlighted that the E2-like enzyme ATG3, which serves to regulate lipidation of LC3, binds to regions of the autophagic membrane that contain local lipid-packing defects. Similar to ATG14, the hydrophobicity of the ATG3 N-terminal amphipathic helix determines the degree of binding of ATG3 to membranes, which is followed by lipidation of LC3 (72). On the basis of these findings, Nath and colleagues (72) suggest that the E2-like enzyme ATG3 is perhaps suitable to work at highly curved membranes, and it is possible that the unique hydrophobic domains of ATG14 and ATG3 are critical determinants that allow recruitment and expansion of the autophagic machinery at the lipid droplet surface. In keeping with these concepts, modifying ATG14 has been shown to disrupt hepatocyte lipid metabolism (134). However, additional studies will be required to determine the precise role of ATG14 and ATG3 in targeting to regions of membrane curvature, which in turn could be important in understanding how autophagic sequestration of lipid droplets occurs.
Autophagy and Cross Talk with Lipases
An important issue that needs to be considered is the possibility of cooperativity between lipases and ATG proteins in the regulation of lipolysis. To the authors’ understanding, no information is currently available on ATG protein–cytosolic lipase cross talk in the control of lipolysis. White adipocytes have traditionally been the most optimal system to study lipolysis, largely because their unique architecture and function are geared toward fat storage and breakdown. White adipocytes are characterized by miniscule amounts of cytoplasm, a single large lipid droplet that essentially flattens the nucleus; most importantly, adipocytes exhibit abundant levels and activity of cytosolic lipases. Why then would autophagy be required to mobilize fat in the adipose tissue?
To determine the contribution of autophagy in lipid metabolism in white adipose tissue, two independent groups deleted Atg7 in adipocytes using aP2/fatty acid–binding protein 4 as the promoter to drive Cre recombination and deletion of Atg7. Quite surprisingly, both studies reported that selective loss of autophagy in white adipocytes resulted in mice exhibiting decreased fat mass and heightened insulin sensitivity. Molecular analyses of Atg7−/− adipocytes revealed a remarkable reduction in levels of markers of adipocyte differentiation, indicating a failure of white fat differentiation, which likely contributed to decreased white fat mass in these mice. It is possible that in the absence of white fat differentiation, a subset of unique adipose progenitors is activated and repopulates the adipose tissue with brown adipocyte-like cells, enhancing energy expenditure and insulin sensitivity in these mice. In the context of the role of autophagy in lipid mobilization, it is important to note that the failure to observe accumulation of lipid droplets in Atg7−/− adipocytes suggests that autophagy plays a predominant role in white fat development and is perhaps dispensable in the steps regulating adipocyte lipid droplet turnover. It should also be noted that because the loss of Atg7 from birth triggers developmental changes, it is likely that an accurate analysis of lipophagy and its cross talk with lipases in white fat lipid metabolism would require in vivo models wherein Atg7 can be deleted conditionally in the postdevelopmental state.
It is currently thought that stimulation of lipolysis in adipocytes activates protein kinase A, which phosphorylates the lipid droplet coat protein PLIN1 (formerly called perilipin). Phosphorylation of PLIN1 results in its degradation, which allows neutral lipases to gain access to the lipid core of lipid droplets (69). It has also been shown that persistent lipolytic stimulation leads to fragmentation of larger perinuclear lipid droplets into much smaller lipid droplets that are coated with PLIN1 (63). A likely effect of this fragmentation is an increase in the available surface area upon which lipases can efficiently act to mobilize triglycerides. Intriguingly, preliminary data from our laboratory indicate that activating autophagy in adipocytes contributes to the breakup of larger lipid droplets into smaller lipid droplets, possibly through the ability of autophagy to degrade large-sized lipid droplets in bulk. Conversely, we also examined the consequence of an acute loss of autophagy on lipid droplet size, and our findings suggest that acutely inhibiting autophagy in adipocytes depletes small-sized adipocytes and leaves behind abnormally large and dysmorphic lipid droplets. Although we appreciate this to be an effect restricted to brown adipocytes, these results allow us to consider a possible paradigm wherein activation of autophagy breaks down larger lipid droplets into smaller droplets, which in turn increases the net lipid droplet surface area upon which cytosolic lipases can act effectively. It is thus possible that autophagy facilitates adipose lipolysis by increasing cytosolic lipase activity merely by remodeling lipid droplet size. Whether these events occur in white adipocytes remains to be seen. In addition, whether activation of autophagy translates to increased lipase activity also needs detailed investigation.
Additional Autophagy Pathways and Lipid Metabolism
Finally, we consider the question of whether additional forms of autophagy participate in lysosomal lipid turnover. To date no studies in mammalian systems have linked CMA or the microautophagy pathway to lysosomal delivery and turnover of lipid droplets. However, in a recent study in the yeast S. cerevisiae, lysosomal lipid droplet breakdown was shown to occur via microautophagy. As briefly discussed in the introduction, microautophagy refers to a form of autophagy wherein lysosomal or late endosomal membranes directly engulf cytoplasmic cargo that is then degraded within these acidic compartments (95).
In the study performed in yeast, van Zutphen and colleagues (124) determined that the yeast vacuole directly engulfs lipid droplets in a mechanism that resembles microautophagy in mammals. When yeast cells were cultured in the basal state or exposed to fatty acids, the authors observed lipid droplets within the vacuole, although providing fatty acids led to further increases in vacuolar lipid droplet content. Electron microscopic analyses indicated that lipid droplets associated closely with invaginations in the lysosomal membrane, and no additional membranes were detected in the vicinity of lipid droplets, which suggested that cargo delivery occurred via direct lysosomal engulfment. Intriguingly, using several strains mutated for various ATG proteins, the authors revealed that much of the core autophagic machinery, except ATG11 and ATG20 or Shp1, an ATG8 cofactor, is required for direct vacuolar lipid consumption (124). In contrast, the authors revealed that this process requires a unique set of proteins, in particular, ATG17 and ATG15. Using Atg15 mutants, the authors demonstrated that loss of ATG15 results in marked increases in vacuolar lipid accumulation. These findings suggest that ATG15 is the vacuolar triglyceride lipase and support the previously proposed function of ATG15 as the triglyceride lipase in organisms such as F. graminearum (74). These results also suggest that factors inherent to lysosomes and/or late endosomes could affect rates of lysosomal lipid consumption. For instance, recent work from the McNiven laboratory (101) has shown a new role for Dynamin 2 in the vesiculation of lysosomal tubules and demonstrated the importance of these vesiculations in lipid droplet turnover via lipophagy. The function of Dynamin 2 is to regulate vesicle budding from late endosomes by binding to endocytic adaptors. In this work, Schulze and colleagues (101) show that after lysosomal consumption of lipid droplets, nascent lysosomal tubules originate from autolysosomes in an attempt to generate new lysosomes. Dynamin 2 regulates the scission events necessary for production of these nascent lysosomes. Accordingly, depleting Dynamin 2 reduces production of nascent lysosomes and promotes accumulation of larger incompetent lysosomes that in unison compromise lysosomal lipid degradation. These important results suggest that factors independent of the mechanisms regulating autophagosome formation could also impact lipophagy. It is thus tempting to speculate that age-associated changes in lysosomal vesiculation and lysosomal regeneration could contribute to the increased lipid accumulation observed in these conditions.
In contrast to roles of lipophagy in breaking down lipid droplets, Shibata and colleagues (106) have previously shown that the LC3 lipidation system is required for the generation of lipid droplets and that starvation-induced lipid accumulation fails to occur in autophagy-deficient mice. Unlike the findings by Shibata et al. (106) and in keeping with the degradative role of lipophagy, van Zutphen and colleagues have shown that mutants for Atg1, Atg8, and Atg15 are comparable to wild-type strains in their abilities to generate cytosolic lipid droplets when maintained in growth medium. Given the dynamic nature of the interaction of autophagy with its substrates across diverging physiological conditions, i.e., feeding or fasting, it is not surprising that although autophagosomes originate from plasma membranes (6, 91), endoplasmic reticulum (118), and mitochondria, autophagosomes also serve to devour the very cellular components and organelles (endoplasmic reticulum and mitochondria) that generate them (5, 28, 32, 143). Furthermore, although autophagy degrades lipid droplets via lipophagy, a recent study reveals that lipid-droplet-localized neutral lipase, PNPLA5, and lipid utilization via PNPLA5 are required for the optimal activation of autophagy by providing the lipid fuel needed for the generation of autophagosomes (21).
WHY IS LIPOPHAGY REQUIRED IN THE LIVER?
Lipophagy in the Pathophysiology of Liver Disease
So, why is lipophagy necessary in the liver? As discussed previously, in adipocytes a significant proportion of cytosolic triglyceride hydrolysis occurs via lipases ATGL and HSL. Although ATGL displays highest activity in fat tissue, several nonadipose tissues express low amounts of ATGL. Accordingly, ATGL-deficient mice have shown lipid accumulation in adipose tissue, heart, and liver. However, ATGL does not hydrolyze cholesteryl esters, and furthermore, ATGL contributes to variable degrees of phospholipid hydrolysis, at least as observed in different experimental conditions. In addition, under normal physiological states, the liver expresses very low levels of HSL, and no studies have associated HSL activity to lipolysis in the liver. In contrast, under experimental conditions, overexpression of HSL has been shown to reverse hepatic steatosis (93). These results would allow one to consider that lipophagy perhaps evolved as a mechanism for in-bulk removal of hepatic lipid droplets generated in response to free fatty acids delivered to liver in the fasted state. Because excessive lipid accumulation generates oxidant stress, and given the role of autophagy as a prosurvival factor, it is not unreasonable to consider that the autophagic system plays an important role in buffering fatty acid toxicity and maintaining hepatic lipohomeostasis in the background of enhanced adipose lipolysis. Consistent with this notion, Wang and colleagues (127) have shown that autophagy-deficient hepatocytes are more sensitive to menadione-induced oxidant stress when compared to those with intact autophagy. Of particular note, blocking β-oxidation led to decreased viability in hepatocytes exposed to oxidant stress; in contrast, simply providing free fatty acids was sufficient to bypass oxidant stress–induced cell death in autophagy-deficient cells (127). These findings suggest that lipophagy potentially supersedes hepatotoxicity by generating ATP that would allow the cells to sustain an antioxidant response. Alternatively, lipophagy could facilitate the clearance of toxic lipid species through their degradation in lysosomes. In accordance with this possibility, Alexaki et al. (2) have linked defective autophagy to the accumulation of diverse lipid species in the liver, for instance, sphingolipids and their bioactive derivative ceramide, which have been shown to contribute to cellular toxicity. In keeping with this notion, it is not surprising that diet-induced and genetic models of obesity reveal inhibition of autophagy that occurs in parallel with tissue inflammation (108, 141) and cell death.
A time-tested model for the development and progression to nonalcoholic steatohepatitis (NASH) is the “two-hit theory” (15, 16). In this theory, the first hit includes the development of a fatty liver disease, and the second hit entails development of oxidative stress, mitochondrial deficits, and hepatocellular inflammation that cumulatively lead to the development of NASH. How autophagy might fit in with the two-hit theory in development of NASH is unclear. First, it was proposed and conclusively shown that lipophagy plays a protective role against the first hit by preventing excessive fat accumulation (108). Second, in all likelihood, autophagy prevents the second hit through its ability to eliminate damaged reactive oxygen species–generating mitochondria and to maintain hepatocellular ATP levels. A common mediator of liver injury is the inflammatory molecule tumor necrosis factor (TNF)-α, and Amir and colleagues (3) have shown that deleting Atg7 in the liver led to hepatocellular injury when mice were exposed to known liver toxins, i.e., galactosamine combined with lipopolysaccharide or galactosamine administered with TNF-α. The authors demonstrated that loss of autophagy increases liver cell death through the activation of the mitochondrial death pathway and that hepatotoxicity is reversed upon overexpression of Beclin-1 in mice (3). Autophagy/lipophagy has also been shown to contribute to hepatic fibrosis through effects on hepatic stellate cell function. Indeed, autophagy induction was detected in activated hepatic stellate cells, and autophagy inhibition suppressed hepatic stellate cell activation (119). A second group has shown that autophagy is necessary for stellate cell activation in vitro and in vivo, and that lipophagy-driven fatty acid β-oxidation is necessary for stellate cell activation (36). Accordingly, replacing free fatty acids led to stellate cell activation in autophagy-deficient cells (36). These findings indicate that autophagy plays an important part in preventing steatosis and in the progression to hepatocellular toxicity in the background of fatty liver disease; quite surprisingly, autophagy also plays a regulatory role in the development of hepatic fibrosis.
In addition to protection against lipid-driven hepatotoxicity, the physiological function of lipophagy is perhaps to regulate the availability of free fatty acids, which is required for generation of very-low-density lipoproteins (VLDL) particles. It has been shown that in hepatocytes cultured in the absence of free fatty acids, a significant proportion (~70%) of the secreted VLDL is generated through the intracellular breakdown of triglycerides (131); however, how free triglycerides were catabolized to generate VLDL was unclear. Studies in mice lacking Atg7 in the liver displayed a reduction in the secretion of triglycerides that were packaged in VLDL particles (108), which indicates that lipophagy plays an important part in the turnover of cellular triglycerides to generate free fatty acids that are utilized for VLDL production. In keeping with these findings, studies employing a pharmacological inhibition of autophagy revealed decreased fatty acid oxidation and VLDL production, whereas in contrast, activating autophagy resulted in increased VLDL production (84, 111). In addition to modulating liver disease through control of hepatic fat content, lipophagy could also modify the infectious pathology of hepatotropic agents. For instance, hepatitis B and hepatitis C viruses activate autophagy, and this autophagy activation supports replication of these viruses (1, 20, 110). It is also possible that breakdown of lipid droplets provides the fuel required for their replication. In fact, hepatitis C virus particles have been shown to dock to lipid droplets that, in turn, allow viral assembly (68). Consistent with this possibility, blocking autophagy inhibited viral replication, and in contrast, providing free fatty acids rescued viral replication in autophagy-deficient cells. Although it remains unknown how viruses utilize cellular resources for their replication, these emerging studies build a clear case for the involvement of lipophagy in fueling viral replication.
Lipophagy and Human NAFLD
Autophagy analysis in human subjects has remained a major limitation in terms of understanding the degree to which loss of autophagy contributes to human NAFLD. Moreover, analyzing autophagy activity relies exclusively on measuring autophagy flux, which requires pretreatment of samples with lysosomal inhibitors (51, 138). In spite of these limitations, correlative studies in human subjects have provided the proof of principle that autophagy indeed plays a significant part in preventing the development of NAFLD in humans. For instance, liver sections obtained from deceased subjects and liver biopsies from adult subjects revealed accumulation of the autophagy cargo p62/SQSTM1 in individuals with severe steatosis (26, 42). These findings support that autophagy activity, as one would expect, correlates inversely with total liver fat content. Further links between lipophagy and steatosis originate from studies wherein pharmaceutical agents commonly known to generate steatosis have now been shown to suppress autophagy. For instance, thymidine analogs used as antiretroviral agents, particularly stavudine and zidovudine, have now been shown to inhibit autophagy activity, which in turn could be the reason for the increased hepatic lipid content observed in individuals treated with these agents (114). Conversely, the anticonvulsant carbamazepine has been shown to prevent development of steatosis through its ability to activate autophagy (61). More recently, Park and colleagues (85) have shown that obesity and lipotoxic states lead to autophagy blockage, and this inhibition of autophagy occurs from increased cytosolic calcium levels. They further showed that increases in hepatocellular calcium leads to impaired autophagosome-lysosome fusion (85). On the basis of these findings, the authors administered the commonly used calcium channel blocker verapamil to obese mice. Interestingly, verapamil therapy activated autophagy and eliminated lipid accumulation in livers from obese mice, which in turn enhanced insulin sensitivity (85). Given that calcium channel blockers have been used for treatment of hypertension for decades, it is likely that verapamil will be rapidly available for use to prevent metabolic complications stemming from decreased autophagy. Furthermore, two commonly used beverages, green tea and coffee, have active biological ingredients, epigallocatechin-3-gallate (146) and caffeine (109), respectively, that have been shown to prevent hepatic steatosis by activating lipophagy. In sum, these findings support a major role for lipophagy in maintaining hepatic fat levels in mice models and in humans and form the basis for understanding the molecular mechanisms regulating autophagy-mediated lipid metabolism.
CONCLUSIONS AND FUTURE DIRECTIONS
Lipophagy degrades hepatocellular lipid droplets within lysosomes. Several studies have now identified the functionality of lipophagy in diverse cell types as well as in organisms such as yeast, worms, and fungal structures. Whereas blocking autophagy promotes hepatocellular lipid accumulation, activating autophagy leads to the clearance of hepatocellular lipid droplets. These findings suggest that activating lipophagy is an attractive strategy to rapidly mobilize hepatocellular lipids and prevent human fatty liver disease. However, several questions remain unanswered. For instance, it remains unknown how autophagy proteins are recruited to lipid droplets. How does the autophagic machinery recognize lipid droplets? How do autophagosomes sense the curvature and size of lipid droplets? It also remains unknown whether lipophagy and cytosolic lipases act in concert to eliminate lipid droplets. The answers to many of these questions would help better explain how autophagy, an in-bulk degradative pathway, eliminates cellular lipid stores. Knowing how lipophagy functions could be helpful in designing new strategies for the prevention of hepatic steatosis, a major health condition in developed countries.
SUMMARY POINTS.
Lipophagy entails the sequestration of lipid droplets by autophagosomes and their degradation within lysosomes.
Blocking lipophagy promotes hepatic lipid accumulation and mimics the human condition termed nonalcoholic fatty liver disease.
Starvation and an acute lipid stimulus activate lipophagy; refeeding or prolonged overnutrition suppresses autophagy.
More than 30 autophagy (Atg) genes orchestrate the formation of autophagosomes.
Lipophagy requires complex regulatory networks that include transcriptional control of autophagy genes.
In addition to the liver, lipophagy degrades lipid droplets in diverse cell types as well as in yeast, worms, and fungal structures.
How the autophagic machinery recognizes lipid droplets remains unknown.
Activating hepatic lipophagy prevents excessive fat accumulation in the liver.
FUTURE ISSUES.
How are ATG proteins recruited to lipid droplets?
How might lipid droplets be recognized as an autophagic cargo?
Do recognition tags for lipophagy differ from those meant for recognition of other organelles?
How does an autophagosome assess the curvature and dimension of a lipid droplet?
Does lipophagy control lipid droplet fission events to generate smaller droplets?
Is there cross talk between lipophagy and cytosolic lipases in regulating lysosomal lipid turnover?
Are additional autophagy pathways involved in lysosomal lipid degradation?
Can activation of lipophagy prevent obesity-associated steatosis?
ACKNOWLEDGMENTS
This work was funded by grants from the National Institutes of Health DK087776 (R.S.), AG043517 (R.S.), DK020541 (Einstein Diabetes Research Center), and AG031782 (R.S.); an Ellison Medical Foundation New Scholar Award (R.S.); and institutional funds from the Albert Einstein College of Medicine (R.S.). We apologize to those whose work could not be cited due to space constraints.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, et al. 2008. Hepatitis C virus genotype 1 a growth and induction of autophagy. J. Virol 82:2241–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexaki A, Gupta SD, Majumder S, Kono M, Tuymetova G, et al. 2014. Autophagy regulates sphingolipid levels in the liver. J. Lipid Res 55:2521–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amir M, Zhao E, Fontana L, Rosenberg H, Tanaka K, et al. 2013. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 20:878–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, et al. 2007. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet 16:2600–15 [DOI] [PubMed] [Google Scholar]
- 5.Bejarano E, Girao H, Yuste A, Patel B, Marques C, et al. 2012. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol. Biol. Cell 23:2156–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bejarano E, Yuste A, Patel B, Stout RF Jr, Spray DC, et al. 2014. Connexins modulate autophagosome biogenesis. Nat. Cell Biol 16:401–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, et al. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol 171:603–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjorkoy G, Lamark T, Johansen T. 2006. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2:138–39 [DOI] [PubMed] [Google Scholar]
- 9.Bonten EJ, Annunziata I, d’Azzo A. 2014. Lysosomal multienzyme complex: pros and cons of working together. Cell Mol. Life Sci 71:2017–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho HI, Choi JW, Lee SM. 2014. Impairment of autophagosome-lysosome fusion contributes to chronic ethanol-induced liver injury. Alcohol 48:717–25 [DOI] [PubMed] [Google Scholar]
- 11.Choi AM, Ryter SW, Levine B. 2013. Autophagy in human health and disease. N. Engl. J. Med 368:651–62 [DOI] [PubMed] [Google Scholar]
- 12.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, et al. 2002. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem 277:6344–52 [DOI] [PubMed] [Google Scholar]
- 13.Cuervo AM, Wong E. 2014. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24:92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czaja MJ, Cuervo AM. 2009. Lipases in lysosomes, what for? Autophagy 5:866–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Day CP. 2002. Non-alcoholic steatohepatitis (NASH): Where are we now and where are we going? Gut 50:585–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Day CP, James OF. 1998. Steatohepatitis: a tale of two “hits”? Gastroenterology 114:842–45 [DOI] [PubMed] [Google Scholar]
- 17.de Mattos KA, Sarno EN, Pessolani MC, Bozza PT. 2012. Deciphering the contribution of lipid droplets in leprosy: multifunctional organelles with roles in Mycobacterium leprae pathogenesis. Mem. Inst. Oswaldo Cruz 107(Suppl. 1):156–66 [DOI] [PubMed] [Google Scholar]
- 18.Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, et al. 2013. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci 126:939–52 [DOI] [PubMed] [Google Scholar]
- 19.Dice JF, Terlecky SR, Chiang HL, Olson TS, Isenman LD, et al. 1990. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin. Cell Biol 1:449–55 [PubMed] [Google Scholar]
- 20.Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. PNAS 106:14046–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, et al. 2014. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol 24:609–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, et al. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan W, Nassiri A, Zhong Q. 2011. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). PNAS 108:7769–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filipe A, McLauchlan J. 2014. Hepatitis C virus and lipid droplets: finding a niche. Trends Mol. Med 21:34–42 [DOI] [PubMed] [Google Scholar]
- 25.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, et al. 2007. Ambra1 regulates autophagy and development of the nervous system. Nature 447:1121–25 [DOI] [PubMed] [Google Scholar]
- 26.Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, et al. 2014. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol. Res 44:1026–36 [DOI] [PubMed] [Google Scholar]
- 27.Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, et al. 2011. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Invest 121:2102–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, et al. 2013. Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–93 [DOI] [PubMed] [Google Scholar]
- 29.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, et al. 2007. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem 282:37298–302 [DOI] [PubMed] [Google Scholar]
- 30.Hardie DG, Carling D, Halford N. 1994. Roles of the Snf1/Rkin1/AMP-activated protein kinase family in the response to environmental and nutritional stress. Semin. Cell Biol 5:409–16 [DOI] [PubMed] [Google Scholar]
- 31.Harding TM, Hefner-Gravink A, Thumm M, Klionsky DJ. 1996. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem 271:17621–24 [DOI] [PubMed] [Google Scholar]
- 32.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, et al. 2009. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol 11:1433–37 [DOI] [PubMed] [Google Scholar]
- 33.He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet 43:67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herman PK, Emr SD. 1990. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol. Cell Biol 10:6742–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herman PK, Stack JH, Emr SD. 1991. A genetic and structural analysis of the yeast Vps15 protein kinase: evidence for a direct role of Vps15p in vacuolar protein delivery. EMBO J. 10:4049–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, et al. 2012. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142:938–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang H, Kawamata T, Horie T, Tsugawa H, Nakayama Y, et al. 2015. Bulk RNA degradation by nitrogen starvation-induced autophagy in yeast. EMBO J. 34:154–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, et al. 2010. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol 185:7349–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itakura E, Kishi C, Inoue K, Mizushima N. 2008. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalianAtg14 and UVRAG. Mol. Biol. Cell 19:5360–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Itakura E, Kishi-Itakura C, Mizushima N. 2012. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151:1256–69 [DOI] [PubMed] [Google Scholar]
- 41.Kaini RR, Sillerud LO, Zhaorigetu S, Hu CA. 2012. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate 72:1412–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kashima J, Shintani-Ishida K, Nakajima M, Maeda H, Unuma K, et al. 2014. Immunohistochemical study of the autophagy marker microtubule-associated protein 1 light chain 3 in normal and steatotic human livers. Hepatol. Res 44:779–87 [DOI] [PubMed] [Google Scholar]
- 43.Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, et al. 2012. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 13:258–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, et al. 2011. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 14:173–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khaldoun SA, Emond-Boisjoly MA, Chateau D, Carriere V, Lacasa M, et al. 2014. Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol. Biol. Cell 25:118–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khor VK, Shen WJ, Kraemer FB. 2013. Lipid droplet metabolism. Curr. Opin. Clin. Nutr. Metab. Care 16:632–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim I, Rodriguez-Enriquez S, Lemasters JJ. 2007. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys 462:245–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J, Klionsky DJ. 2000. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem 69:303–42 [DOI] [PubMed] [Google Scholar]
- 49.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13:132–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C. 2010. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J. Lipid Res 51:468–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koga H, Kaushik S, Cuervo AM. 2010. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 24:3052–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koga H, Kaushik S, Cuervo AM. 2010. Inhibitory effect of intracellular lipid load on macroautophagy. Autophagy 6:825–27 [DOI] [PubMed] [Google Scholar]
- 54.Kovsan J, Ben-Romano R, Souza SC, Greenberg AS, Rudich A. 2007. Regulation of adipocyte lipolysis by degradation of the perilipin protein: nelfinavir enhances lysosome-mediated perilipin proteolysis. J. Biol. Chem 282:21704–11 [DOI] [PubMed] [Google Scholar]
- 55.Kraft C, Deplazes A, Sohrmann M, Peter M. 2008. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol 10:602–10 [DOI] [PubMed] [Google Scholar]
- 56.Kurusu T, Koyano T, Hanamata S, Kubo T, Noguchi Y, et al. 2014. OsATG7 is required for autophagy-dependent lipid metabolism in rice postmeiotic anther development. Autophagy 10:878–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lapierre LR, Gelino S, Melendez A, Hansen M. 2011. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans.Curr. Biol 21:1507–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, et al. 2014. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516:112–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lettieri Barbato D, Tatulli G, Aquilano K, Ciriolo MR. 2013. FoxO1 controls lysosomal acid lipase in adipocytes: implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 4:e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, et al. 1999. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–76 [DOI] [PubMed] [Google Scholar]
- 61.Lin CW, Zhang H, Li M, Xiong X, Chen X, et al. 2013. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol 58:993–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mack HI, Zheng B, Asara JM, Thomas SM. 2012. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 8:1197–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marcinkiewicz A, Gauthier D, Garcia A, Brasaemle DL. 2006. The phosphorylation of serine 492 of perilipin A directs lipid droplet fragmentation and dispersion. J. Biol. Chem 281:11901–9 [DOI] [PubMed] [Google Scholar]
- 64.Mari M, Tooze SA, Reggiori F. 2011. The puzzling origin of the autophagosomal membrane. F1000 Biol. Rep 3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, et al. 2010. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci 13:567–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Massey AC, Zhang C, Cuervo AM. 2006. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol 73:205–35 [DOI] [PubMed] [Google Scholar]
- 67.Masuda Y, Itabe H, Odaki M, Hama K, Fujimoto Y, et al. 2006. ADRP/adipophilin is degraded through the proteasome-dependent pathway during regression of lipid-storing cells. J. Lipid Res 47:87–98 [DOI] [PubMed] [Google Scholar]
- 68.McLauchlan J 2009. Lipid droplets and hepatitis C virus infection. Biochim. Biophys. Acta 1791:552–59 [DOI] [PubMed] [Google Scholar]
- 69.Moore HP, Silver RB, Mottillo EP, Bernlohr DA, Granneman JG. 2005. Perilipin targets a novel pool of lipid droplets for lipolytic attack by hormone-sensitive lipase. J. Biol. Chem 280:43109–20 [DOI] [PubMed] [Google Scholar]
- 70.Murrow L, Debnath J. 2013. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu. Rev. Pathol 8:105–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, et al. 2011. SNARE proteins are required for macroautophagy. Cell 146:290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, et al. 2014. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat. Cell Biol 16:415–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, et al. 2013. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function throughAMBRA1 and TRAF6. Nat. Cell Biol 15:406–16 [DOI] [PubMed] [Google Scholar]
- 74.Nguyen LN, Bormann J, Le GT, Starkel C, Olsson S, et al. 2011.Autophagy-related lipase FgATG15 of Fusarium graminearum is important for lipid turnover and plant infection. Fungal Genet. Biol 48:217–24 [DOI] [PubMed] [Google Scholar]
- 75.Novikoff AB, Beaufay H, de Duve C. 1956. Electron microscopy of lysosome-rich fractions from rat liver. J. Biophys. Biochem. Cytol 2:179–84 [PMC free article] [PubMed] [Google Scholar]
- 76.O’Rourke EJ, Ruvkun G. 2013. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol 15:668–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ogasawara J, Kitadate K, Nishioka H, Fujii H, Sakurai T, et al. 2012. Oligonol-induced degradation of perilipin 1 is regulated through lysosomal degradation machinery. Natural Prod. Commun 7:1193–96 [PubMed] [Google Scholar]
- 78.Ohsumi Y 2014. Historical landmarks of autophagy research. Cell Res. 24:9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oku M, Takano Y, Sakai Y. 2014. The emerging role of autophagy in peroxisome dynamics and lipid metabolism of phyllosphere microorganisms. Front. Plant Sci 5:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, et al. 2011. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 13:655–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ouimet M, Marcel YL. 2012. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol 32:575–81 [DOI] [PubMed] [Google Scholar]
- 82.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, et al. 2011. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet 20:3852–66 [DOI] [PubMed] [Google Scholar]
- 83.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, et al. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem 282:24131–45 [DOI] [PubMed] [Google Scholar]
- 84.Papackova Z, Dankova H, Palenickova E, Kazdova L, Cahova M. 2012. Effect of short- and long-term high-fat feeding on autophagy flux and lysosomal activity in rat liver. Physiol. Res 61(Suppl. 2):S67–76 [DOI] [PubMed] [Google Scholar]
- 85.Park HW, Park H, Semple IA, Jang I, Ro SH, et al. 2014. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun 5:4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phadwal K, Watson AS, Simon AK. 2013. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol. Life Sci 70:89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Phillips AR, Suttangkakul A, Vierstra RD. 2008. The ATG12-conjugating enzyme ATG10 is essential for autophagic vesicle formation in Arabidopsis thaliana. Genetics 178:1339–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Polson HE, deLartigue J, Rigden DJ, Reedijk M, Urbe S, et al. 2010. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6:506–22 [DOI] [PubMed] [Google Scholar]
- 89.Pyo JO, Yoo SM, Jung YK. 2013.The interplay between autophagy and aging. Diabetes Metab. J 37:333–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Raasi S, Varadan R, Fushman D, Pickart CM. 2005. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat. Struct. Mol. Biol 12:708–14 [DOI] [PubMed] [Google Scholar]
- 91.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. 2010. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol 12:747–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, et al. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet 36:585–95 [DOI] [PubMed] [Google Scholar]
- 93.Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, et al. 2008. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem 283:13087–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, et al. 2012. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 31:4304–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, et al. 2011. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20:131–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, et al. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Santambrogio L, Cuervo AM. 2011. Chasing the elusive mammalian microautophagy. Autophagy 7:652–54 [DOI] [PubMed] [Google Scholar]
- 98.Sardiello M, Ballabio A. 2009. Lysosomal enhancement: a CLEAR answer to cellular degradative needs. Cell Cycle 8:4021–22 [DOI] [PubMed] [Google Scholar]
- 99.Schlumpberger M, Schaeffeler E, Straub M, Bredschneider M, Wolf DH, et al. 1997. AUT1, a gene essential for autophagocytosis in the yeast Saccharomyces cerevisiae. J. Bacteriol 179:1068–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schneider JL, Cuervo AM. 2014. Liver autophagy: much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol 11:187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, et al. 2013. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J. Cell Biol 203:315–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Seok S, Fu T, Choi SE, Li Y, Zhu R, et al. 2014. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 516:108–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, et al. 2013. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol 15:647–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, et al. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332:1429–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, et al. 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31:1095–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, et al. 2009. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun 382:419–23 [DOI] [PubMed] [Google Scholar]
- 107.Singh R, Cuervo AM. 2011. Autophagy in the cellular energetic balance. Cell Metab. 13:495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, et al. 2009. Autophagy regulates lipid metabolism. Nature 458:1131–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, et al. 2014. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 59:1366–80 [DOI] [PubMed] [Google Scholar]
- 110.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, et al. 2010. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. PNAS 107:4383–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Skop V, Cahova M, Papackova Z, Palenickova E, Dankova H, et al. 2012. Autophagy-lysosomal pathway is involved in lipid degradation in rat liver. Physiol. Res 61:287–97 [DOI] [PubMed] [Google Scholar]
- 112.Souza SC, Muliro KV, Liscum L, Lien P, Yamamoto MT, et al. 2002. Modulation of hormone-sensitive lipase and protein kinase A-mediated lipolysis by perilipin A in an adenoviral reconstituted system. J. Biol. Chem 277:8267–72 [DOI] [PubMed] [Google Scholar]
- 113.Spandl J, Lohmann D, Kuerschner L, Moessinger C, Thiele C. 2011. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J. Biol. Chem 286:5599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stankov MV, Panayotova-Dimitrova D, Leverkus M, Vondran FW, Bauerfeind R, et al. 2012. Autophagy inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS 26:1995–2006 [DOI] [PubMed] [Google Scholar]
- 115.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, et al. 2005. A human protein-protein interaction network: a resource for annotating the proteome. Cell 122:957–68 [DOI] [PubMed] [Google Scholar]
- 116.Tanida I, Tanida-Miyake E, Ueno T, Kominami E. 2001. The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem 276:1701–6 [DOI] [PubMed] [Google Scholar]
- 117.Tanida I, Ueno T, Kominami E. 2004. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol 36:2503–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, et al. 2007. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 6:2263–67 [DOI] [PubMed] [Google Scholar]
- 119.Thoen LF, Guimaraes EL, Dolle L, Mannaerts I, Najimi M, et al. 2011. A role for autophagy during hepatic stellate cell activation. J. Hepatol 55:1353–60 [DOI] [PubMed] [Google Scholar]
- 120.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. 2009. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol 10:1215–21 [DOI] [PubMed] [Google Scholar]
- 121.Tooze SA, Yoshimori T. 2010. The origin of the autophagosomal membrane. Nat. Cell Biol 12:831–35 [DOI] [PubMed] [Google Scholar]
- 122.Tsukada M, Ohsumi Y. 1993. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333:169–74 [DOI] [PubMed] [Google Scholar]
- 123.Tsuyuki S, Takabayashi M, Kawazu M, Kudo K, Watanabe A, et al. 2014. Detection of WIPI1 mRNA as an indicator of autophagosome formation. Autophagy 10:497–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van Zutphen T, Todde V, de Boer R, Kreim M, Hofbauer HF, et al. 2014. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 25:290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vaughan M, Berger JE, Steinberg D. 1964. Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. J. Biol. Chem 239:401–9 [PubMed] [Google Scholar]
- 126.Wang CW, Miao YH, Chang YS. 2014. A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J. Cell Biol 206:357–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang Y, Singh R, Xiang Y, Czaja MJ. 2010. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 52:266–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Webber JL, Young AR, Tooze SA. 2007. Atg9 trafficking in mammalian cells. Autophagy 3:54–56 [DOI] [PubMed] [Google Scholar]
- 129.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30:678–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Welter E, Elazar Z. 2015. Autophagy mediates nonselective RNA degradation in starving yeast. EMBO J. 34:131–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wiggins D, Gibbons GF. 1992. The lipolysis/esterification cycle of hepatic triacylglycerol. Its role in the secretion of very-low-density lipoprotein and its response to hormones and sulphonylureas. Biochem. J 284(Part 2):457–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, et al. 2011. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333:228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wilfling F, Haas JT, Walther TC, Farese RV Jr. 2014. Lipid droplet biogenesis. Curr. Opin. Cell Biol 29:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiong X, Tao R, DePinho RA, Dong XC. 2012. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem 287:39107–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xu G, Sztalryd C, Londos C. 2006. Degradation of perilipin is mediated through ubiquitination-proteasome pathway. Biochim. Biophys. Acta 1761:83–90 [DOI] [PubMed] [Google Scholar]
- 136.Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, et al. 2005. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J. Biol. Chem 280:42841–47 [DOI] [PubMed] [Google Scholar]
- 137.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, et al. 2013. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 18:816–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yamada E, Singh R. 2012. Mapping autophagy on to your metabolic radar. Diabetes 61:272–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yamada Y, Suzuki NN, Hanada T, Ichimura Y, Kumeta H, et al. 2007. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J. Biol. Chem 282:8036–43 [DOI] [PubMed] [Google Scholar]
- 140.Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, et al. 1998. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegarn UNC-51. Biochem. Biophys. Res. Commun 246:222–27 [DOI] [PubMed] [Google Scholar]
- 141.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. 2010. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11:467–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yen WL, Shintani T, Nair U, Cao Y, Richardson BC, et al. 2010. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell. Biol 188:101–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 2009. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5:1180–85 [DOI] [PubMed] [Google Scholar]
- 144.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, et al. 2006. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci 119:3888–900 [DOI] [PubMed] [Google Scholar]
- 145.Zhang C, Cuervo AM. 2008. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med 14:959–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhou J, Farah BL, Sinha RA, Wu Y, Singh BK, et al. 2014. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLOS ONE 9:e87161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, et al. 2004. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383–86 [DOI] [PubMed] [Google Scholar]