

Abstract
Resistant bacteria successfully evade the action of conventional antibiotic therapies during infection, often leading to significant illness and death. Our lab has discovered halogenated phenazine (HP) analogues which demonstrate potent antibacterial activities through a unique iron-starving mechanism. Herein, we describe synthetic efforts towards a stable cephalosporin-HP conjugate prodrug with the aim of translating HPs into useful clinical agents. Cephalosporin-antibiotic conjugates offer multiple advantages for antibacterial design, including the release of active agents through the targeting of intracellular cephalosporinase following selective ring-opening of the beta-lactam warhead. During these studies, carbonate-linked cephalosporin-HP conjugate 16 was synthesized; however, we were unable to successfully remove the ester group required for cephalosporinase processing. Cephalosporin-HP 16 was then utilized as a probe to investigate the stability of the carbonate linker in antibacterial assays and, as predicted, this compound proved to be inactive against Staphylococcus aureus (MIC > 100 μM). The lack of 16’s antibacterial activity can be attributed to the carbonate linker remaining intact throughout the MIC assay, thus not liberating the active HP moiety. These efforts have led to a more stable cephalosporin-HP conjugate joined through a carbonate linker compared to a highly unstable ether linked analogue we previously reported.
Keywords: Drug discovery, Drug design, Antibacterial prodrug, Halogenated phenazine, Cephalosporin-antibiotic conjugate
Pathogenic bacteria are able to effectively evade the action of conventional antibiotic therapies, leading to a significant number of resistant life-threatening infections.1–4 In the United States alone, there are nearly 2.8 million cases of antibiotic-resistant infections that result in > 35,000 deaths each year.4 Bacteria utilize several well-defined resistance mechanisms that enable them to escape the activities of conventional antibiotics, which include: (1) changes in membrane chemistry to reduce antibiotic penetration, (2) removal of antibiotics through efflux pumps, (3) mutations of antibacterial targets, and (4) enzymatic inactivation of antibiotics.2–10 In addition, very few new classes of antibiotics have entered the clinic over the past several decades further amplifying clinical challenges that result from resistance.3 Agents that operate through unique modes of action are desperately needed to address antibiotic resistance and select drug development strategies can enhance the likelihood of effectively treating multidrug resistant bacterial infections in the clinic.
Inspired by phenazine antibiotics, our group has discovered and explored an extensive series of halogenated phenazine (HP) small molecules that demonstrate excellent antibacterial activities against several antibiotic-resistant isolates of Staphylococcus aureus, S. epidermidis and Enterococcus faecium.11–18 In addition to targeting free-floating planktonic bacteria, HP analogues are able to eradicate surface-attached biofilms formed by methicillin-resistant S. aureus (MRSA), methicillin-resistant S. epidermidis (MRSE), and vancomycin-resistant E. faecium (VRE). Using RNA-seq technology, we recently demonstrated that HP-14 (a potent analogue) induces rapid iron starvation in established MRSA BAA-1707 biofilms as its mode of action.17
Our active halogenated phenazine agents require the 1-hydro-xyphenazine moiety to bind, or complex, to iron (coordination of the hydroxyl group’s oxygen atom and adjacent nitrogen forms a stable 5-membered ring when bound to iron) providing an excellent platform to functionalize for the development of prodrug analogues regarding translational efforts (Fig. 1).14,16–18 In previous studies, we have reported several carbonate-based prodrug versions of our most potent HP analogues (at the 1-hydroxyl position of the HP scaffold) and found these compounds to be stable and more soluble, yet very active in antibacterial assays.15,16 In addition, we reported the initial synthesis of an ether-linked cephalosporin-HP conjugate; however, this molecule proved highly unstable and fragmented during NMR experiments (Fig. S1).16
Fig. 1.
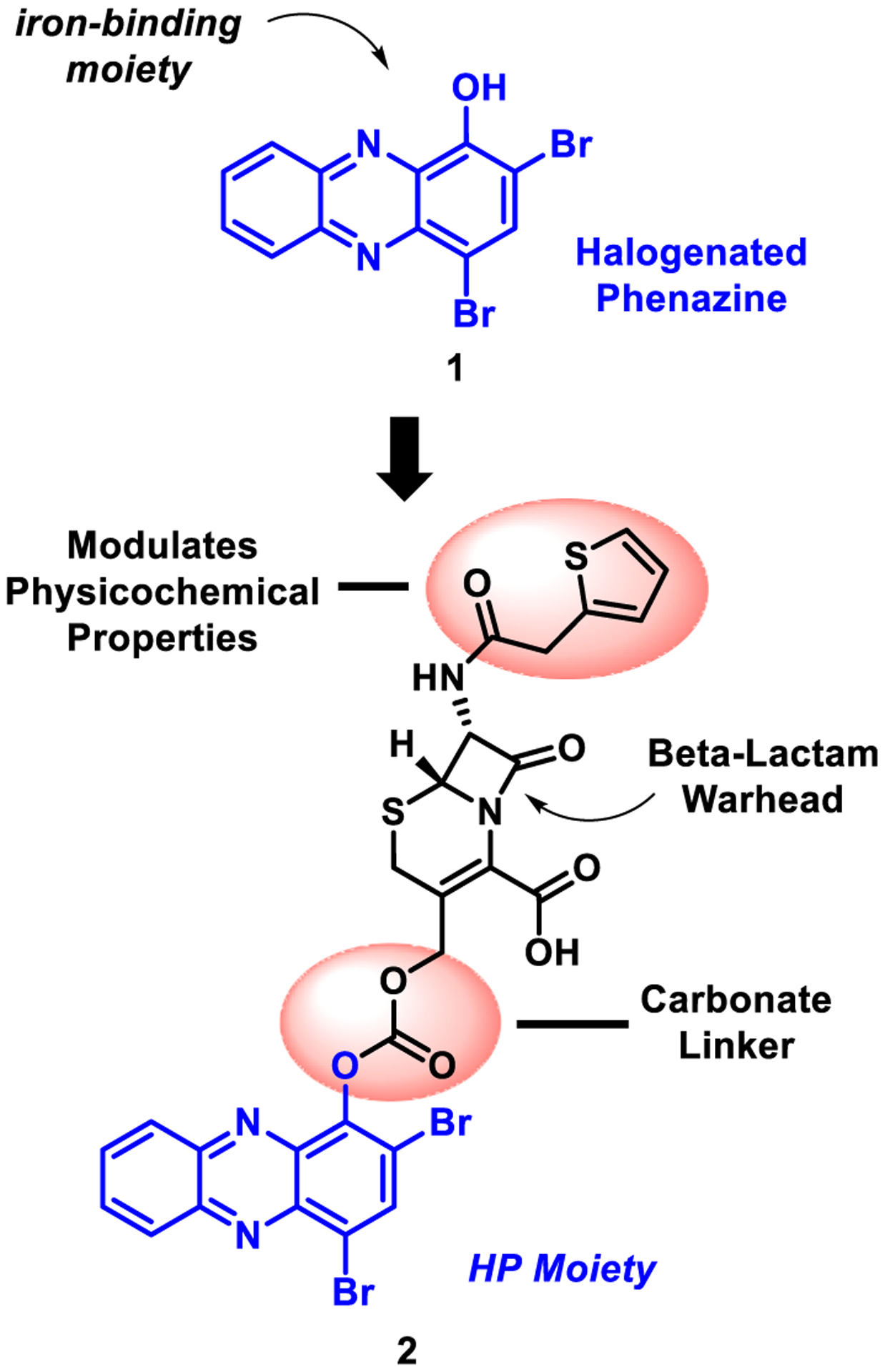
Design of cephalosporin-halogenated phenazine conjugate prodrug 2.
Herein, we describe our efforts towards developing a more stable cephalosporin-halogenated phenazine conjugate prodrug using a synthetic approach that enabled the rapid assembly of a carbonate linker. The beta-lactam warhead of the cephalosporin scaffold is designed for intracellular bacterial targeting of cephalosporinase enzymes involved in cell wall synthesis (Fig. 2).19–27 Following entry into a bacterial cell, a key serine residue of a cephalosporinase (or beta-lactamase) enzyme will undergo nucleophilic attack with the electrophilic beta-lactam motif of the cephalosporin scaffold leading to covalent modification of the target enzyme and subsequent fragmentation to release carbon dioxide and the halogenated phenazine. As such, the HP prodrug system is designed to stay intact by keeping the HP-linked to the cephalosporin scaffold until the beta-lactam moiety is first processed (see Fig. 3).
Fig. 2.
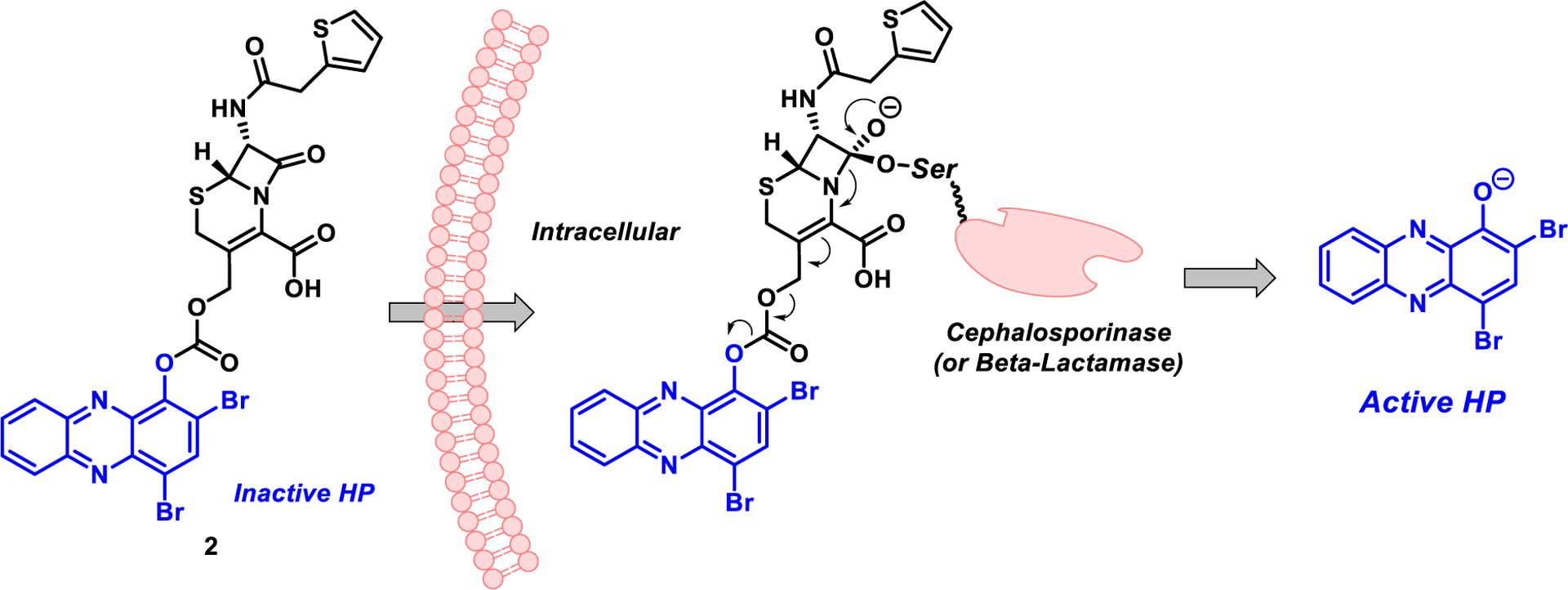
Cephalosporin-halogenated phenazine conjugate 2 and cephalosporinase-triggered release of the active halogenated phenazine moiety.
Fig. 3.
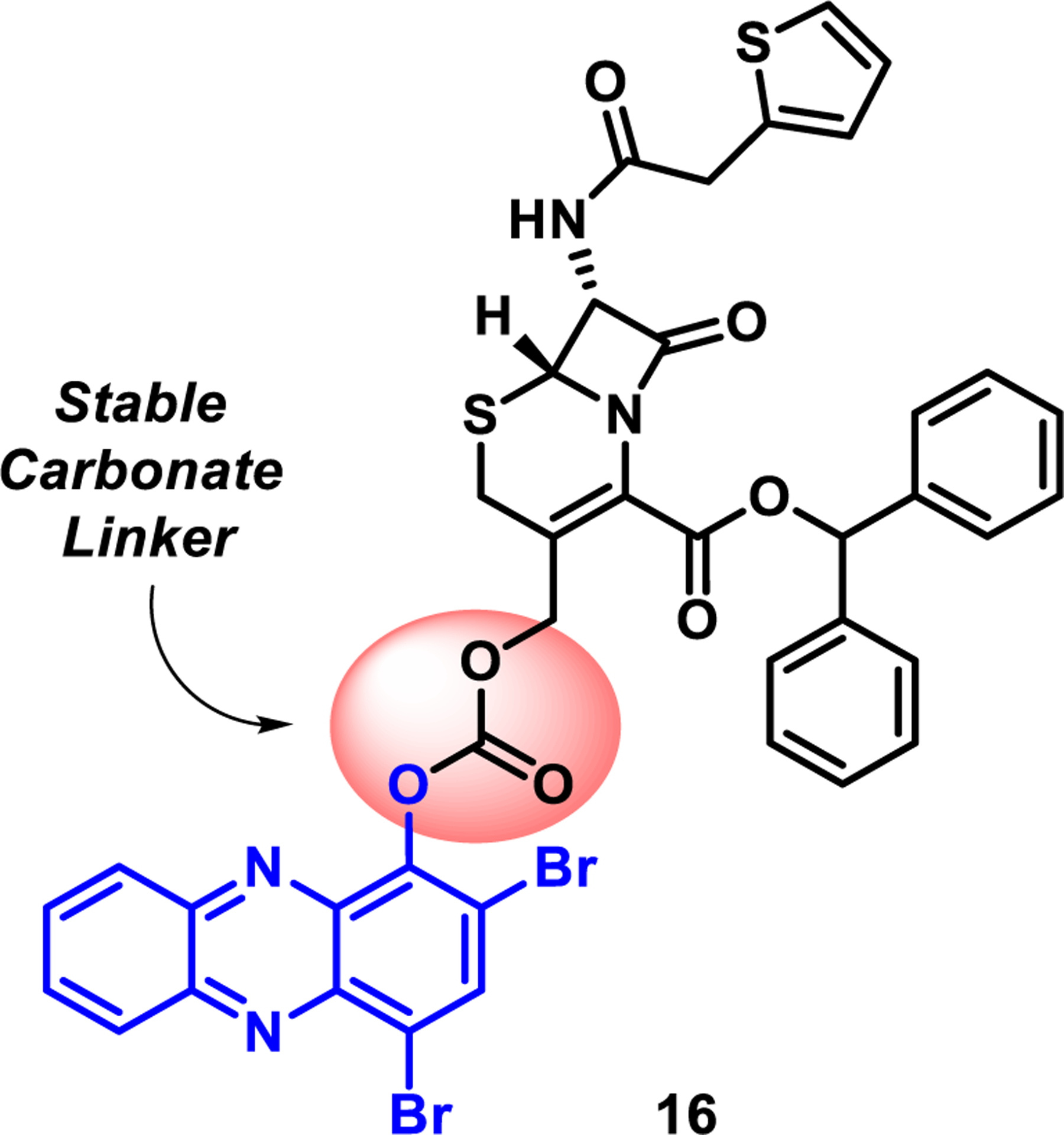
Progress to identify a stable carbonate linker regarding HP-cephalosporin conjugate prodrugs and ongoing work.
During these investigations, our initial synthesis goal was to convert 7-aminocephalosporanic acid (7-ACA, 3; commercially available) into target compound 7 (Scheme 1A). 7-ACA (3) underwent smooth esterification to 4 using thionyl chloride in methanol (90% yield)28; however, we were unable to advance 4 to amide 5 despite several attempts. In a second route, 7-ACA was subjected to an amide-forming reaction upon treatment with thiophene-2-acetyl chloride to yield 6 in 85% yield.29 The carboxylic acid in 6 was readily converted to its corresponding methyl ester 5 in 98% yield following treatment with (trimethylsilyl)diazomethane; however, base-promoted hydrolysis of the acetate group with potassium carbonate in methanol/water led to undesired lactone 8 as the sole product in 62% yield. Despite several attempts, we were unable to produce our initial target cephalosporin compound 7.
Scheme 1.
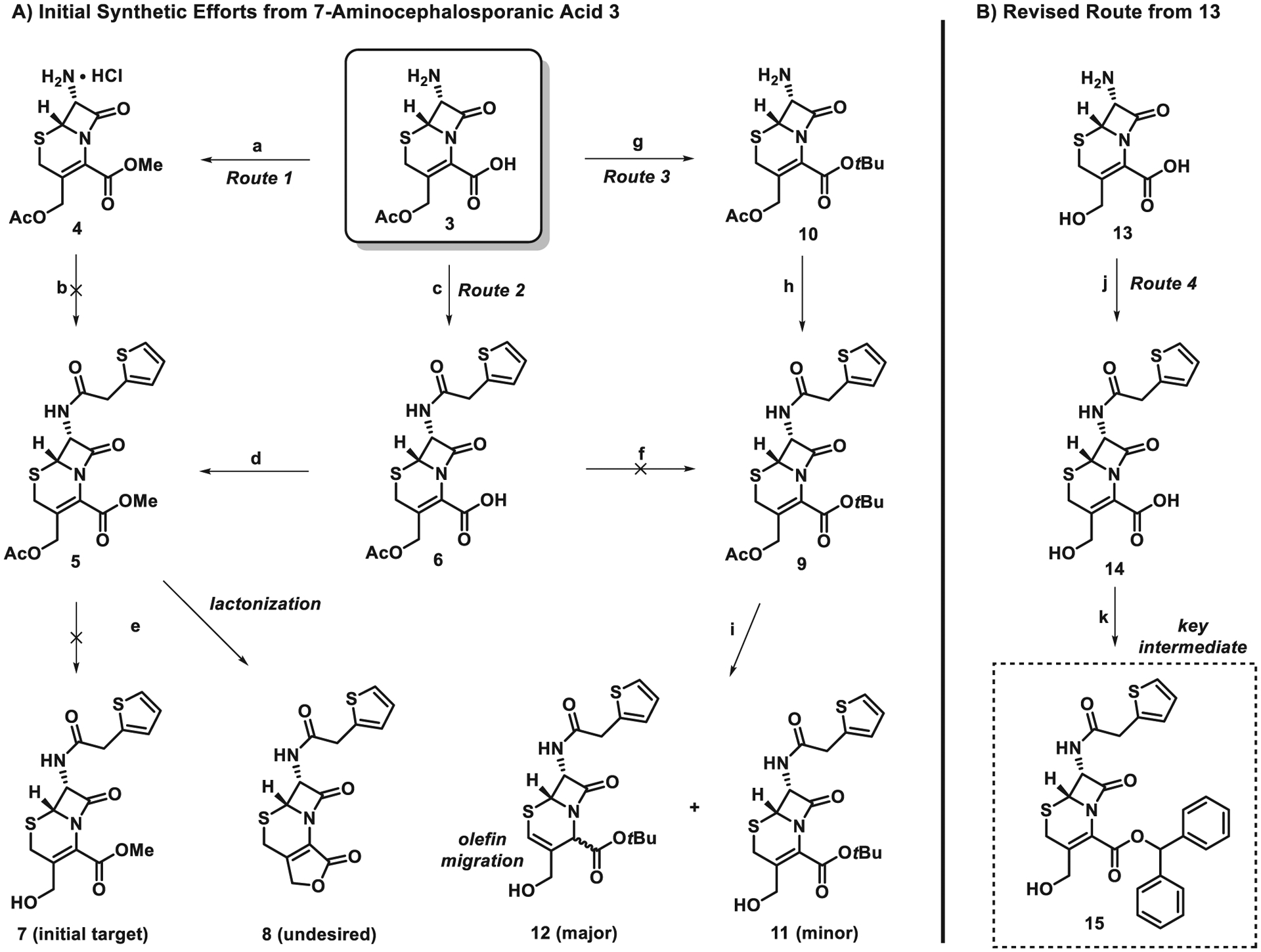
Synthetic efforts to modify the cephalosporin moiety. Reagents and conditions: (a) SOCl2 MeOH, reflux, 5 h, 90%; (b) thiophene-2-acetic acid, EDCI, HOBt, NMM, CH3CN, rt; (c) thiophene-2-acetyl chloride, N,O-bis(trimethylsilyl)acetamide, CH3CN, 85%; (d) TMSCHN2, PhMe/MeOH (v/v = 4:1), 98%; (e) K2CO3, MeOH/H2O (v/v = 5:1), 0 °C, 62%; (f) tert-Butyl acetate, BF3•Et2O, rt; (g) tert-Butyl acetate, BF3•Et2O, rt, 77%; (h) thiophene-2-acetyl chloride, pyridine, CH2Cl2, rt, 67%; (i) K2CO3, MeOH/H2O (v/v = 5:1), 0 °C; (j) thiophene-2-acetyl chloride, N,O-bis(trimethylsilyl)acetamide, CH3CN, 75%; (k) Diphenyldiazomethane, CH2Cl2, 0 °C, 5 h, 72%.
Since we were unable to prevent methyl ester 5 from undergoing lactone formation to 8 in the previous route, we designed a third route to utilize the more sterically encumbered tert-butyl ester to avoid the undesired lactonization reaction (Scheme 1A). 7-ACA (3) was treated with tert-butyl acetate and boron trifluoride to afford the corresponding tert-butyl ester 10 in 77% yield.30 Following the installation of the desired tert-butyl ester group, 10 was then acylated with thiophene-2-acetyl chloride to form amide 9 in 67% yield following a literature protocol.31 The deacetylation of 9 has been reported with the use of an enzyme32; however, we did not pursue this avenue. Instead, we attempted the deacylation of 9 with potassium carbonate in methanol/water; however, this reaction led to an inseparable mixture of 11 and 12. Although we were able to observe very small amounts of the desired compound 11 by NMR, the olefin migration product 12 was the major product. Despite this route overcoming the lactonization issue from the previous route, the olefin migration prevented this synthetic pathway from moving forward.
At this stage, we decided to pursue the synthesis of a suitable cephalosporin moiety from 13 in a fourth route (Scheme 1B). The amine of 13 was selectively acylated with thiophene-2-acetyl chloride in the presence of N,O-bis(trimethylsilyl)acetamide to afford amide 14 in 75% yield. Diphenyldiazomethane was then utilized to selectively functionalize the carboxylic acid of 14 to yield ester 15 in 72%.33 We found compound 15 to be quite stable and we were encouraged to find that lactonization (to undesired compound 8) did not occur on this substrate.
With alcohol 15 in hand, we focused our attention on coupling 15 with HP 1 to yield carbonate-linked conjugate 16 (Scheme 2) upon treatment with triphosgene. Initially, we planned to condense 15 with triphosgene to yield an initial chloroformate intermediate, followed by treatment of HP 1 to yield the target carbonate 16. Unfortunately, this experimental approach proved unsuccessful as was initial treatment of 1 with triphosgene followed by the addition of alcohol 15. Interestingly, when a stoichiometric quantity of triphosgene dissolved in dichloromethane was added dropwise to a mixture of alcohol 15 and HP 1 in the presence of pyridine and triethylamine, the target carbonate 16 was observed in 72% yield on 270 mg scale.
Scheme 2.
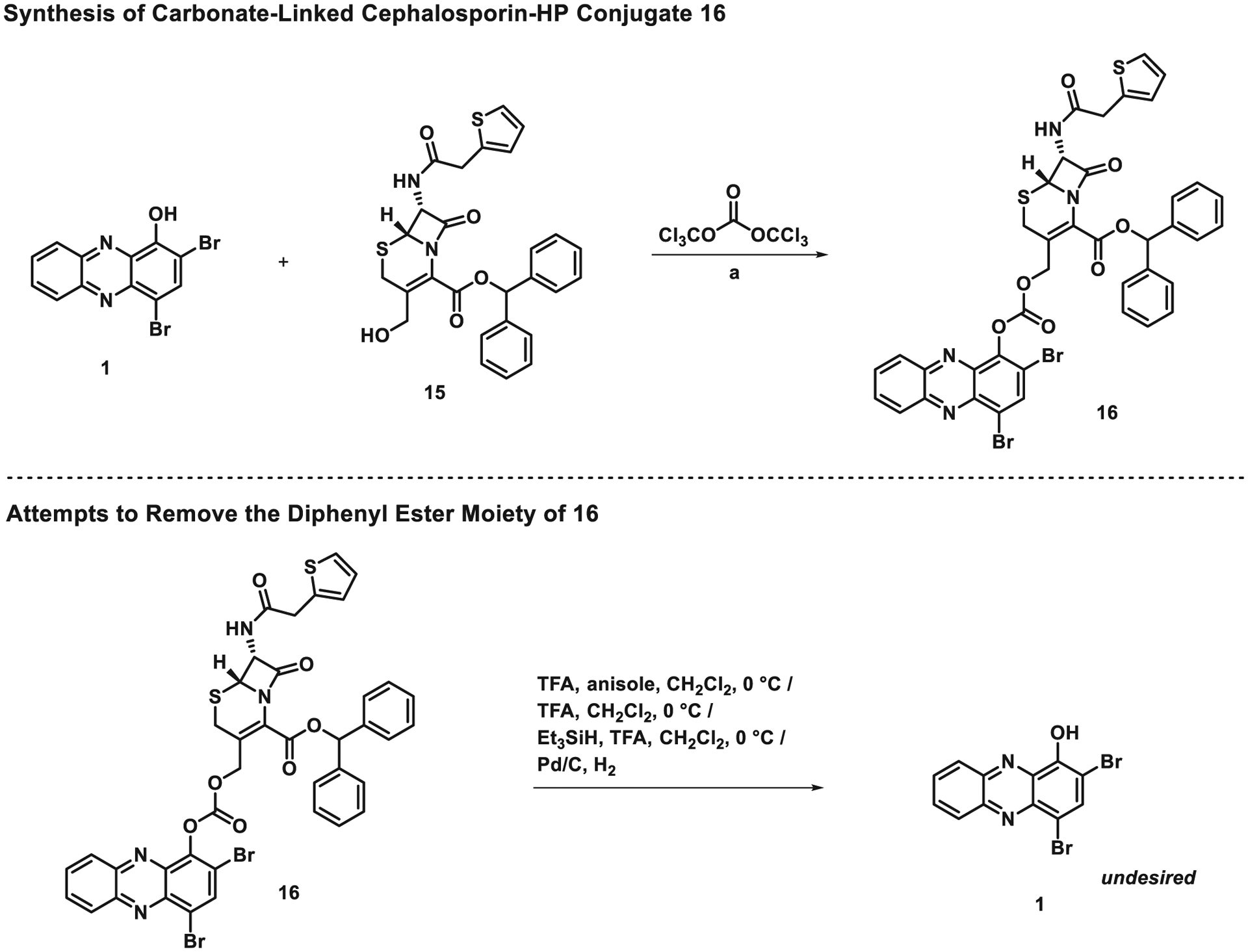
Synthesis of carbonate-linked cephalosporin-HP conjugate 16 and efforts towards removal of the benzhydryl ester moiety. Reagents and conditions: (a) triphosgene, pyridine, triethylamine, CH2Cl2, 0 °C, 2 h, 72%.
With carbonate-linked cephalosporin-HP conjugate 16 in hand, we attempted to remove the benzhydryl ester moiety to target structure 2 using relatively standard trifluoroacetic acid (TFA) based protocols (Scheme 2); however, despite many efforts, we were unable to selectively cleave of the benzhydryl ester moiety as planned. Additional attempts were made to convert 16 to target structure 2 under hydro-genolysis conditions, but those efforts were also met with failure. The only product identified from these experiments was HP 1, which forms following the cleavage of the carbonate linker. In addition, we made several attempts to synthesize target structure 2 directly by reacting 1 with 14 in the presence of triphosgene; however, these attempts were also met with failure and we are currently working around these synthesis problems in an effort to access HP 2 and related analogues.
We were concerned about the overall stability of the carbonate linker in 16 since HP 1, the only observable product from the attempts to remove the benzhydryl ester, would result from carbonate fragmentation. To probe the stability of the carbonate-linker in 16, we evaluated this molecule in an MIC assay against MRSA to determine if this compound possesses any antibacterial activities. We hypothesized that 16 would not demonstrate antibacterial activity as the benzhydryl ester moiety on the cephalosporin scaffold is not a good substrate for a cephalosporinase to cleave the beta-lactam warhead that is required for subsequent carbonate fragmentation and release of active HP 1. When evaluated for antibacterial properties against MRSA BAA-1707, 16 proved inactive at the highest test concentration (MIC > 100 μM). As predicted, this “inactive” antibacterial result can be attributed to the carbonate linker of 16 remaining intact throughout the MIC assay and not liberating the active HP 1 moiety, which has an MIC ~ 1 μM against this MRSA isolate (see Supporting information for an image of this MIC assay).16 Despite the fragmentation of 16’s carbonate during chemical synthesis efforts, this linker proved to be highly robust in antibacterial assays.
In conclusion, we have synthesized a carbonate-linked HP-cephalosporin conjugate that demonstrates significant improvements in stability compared to an ether-linked conjugate analogue from our previous studies. Despite our progress, additional efforts are required to generate the carboxylic acid version of 16, which we hypothesize to be an ideal substrate for cephalosporinase-mediated processing and HP release and translational efforts. We believe that optimized HP-cephalosporin conjugate prodrugs could dramatically impact the treatment of antibiotic-resistant infections in humans and we look forward to continued advances in this area.
Supplementary Material
Acknowledgments
We acknowledge the National Institute of General Medical Sciences of the National Institutes of Health for providing financial support for this work (R35GM128621 to R.W.H.). High-resolution mass spectra for synthesized analogues were obtained from the University of Florida’s Mass Spectrometry Research and Education Center and supported by NIH S10 OD021758-01A1.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2020.127515.
References
- 1.Lewis K The science of antibiotic discovery. Cell. 2020;181:29–45. [DOI] [PubMed] [Google Scholar]
- 2.Abouelhassan Y, Garrison AT, Yang H, Chávez-Riveros A, Burch GM, Huigens RW III. Recent progress in natural-product-inspired programs aimed at addressing antibiotic resistance and tolerance. J Med Chem. 2019;62:7618–7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler MS, Paterson DL. Antibiotics in the clinical pipeline in October 2019. J Antibiot. 2020;73:329–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fitzpatrick MA. Real-world antibiotic needs for resistant Gram-negative infections. Lancet Infect Dis. 2020;20:1108–1109. [DOI] [PubMed] [Google Scholar]
- 5.Munita JM, Arias CA. Mechanisms of antibiotic resistance. Microbiol Spectr. 2016;4 VMBF-0016–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher JF, Mobashery S. Endless resistance. Endless antibiotics? MedChemComm. 2016;7:37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature. 2016;529:336–343. [DOI] [PubMed] [Google Scholar]
- 8.Cooper MA, Shlaes D. Fix the antibiotics pipeline. Nature. 2011;472:32. [DOI] [PubMed] [Google Scholar]
- 9.Liu K, Huigens RW III. Instructive advances in chemical microbiology inspired by nature’s diverse inventory of molecules. ACS Infect Dis. 2020;6:541–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh CT, Wencewicz T. Antibiotics: Challenges, Mechanisms. Washington, DC: ASM Press; 2016. [Google Scholar]
- 11.Borrero NV, Bai F, Perez C, et al. Phenazine antibiotic inspired discovery of potent bromophenazine antibacterial agents against Staphylococcus aureus and Staphylococcus epidermidis. Org Biomol Chem. 2014;12:881–886. [DOI] [PubMed] [Google Scholar]
- 12.Garrison AT, Bai F, Abouelhassan Y, Paciaroni NG, Jin S, Huigens RW III. Bromophenazine derivatives with potent inhibition, dispersion and eradication activities against Staphylococcus aureus biofilms. RSC Adv. 2015;5:1120–1124. [Google Scholar]
- 13.Garrison AT, Abouelhassan Y, Kallifidas D, et al. Halogenated phenazines that potently eradicate biofilms, MRSA persister cells in non-biofilm cultures and Mycobacterium tuberculosis. Angew Chem Int Ed. 2015;54:14819–14823. [DOI] [PubMed] [Google Scholar]
- 14.Garrison AT, Abouelhassan Y, Norwood VM, et al. Structure-activity relationships of a diverse class of halogenated phenazines that targets persistent, antibiotic-tolerant bacterial biofilms and Mycobacterium tuberculosis. J Med Chem. 2016;59:3808–3825. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Abouelhassan Y, Burch GM, et al. A highly potent class of halogenated phenazine antibacterial and biofilm-eradicating agents accessed through a modular Wohl-Aue synthesis. Sci Rep. 2017;7:2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrison AT, Abouelhassan Y, Kallifidas D, et al. An efficient Buchwald-Hartwig/reductive cyclization for the scaffold diversification of halogenated phenazines: Potent antibacterial targeting, biofilm eradication and prodrug exploration. J Med Chem. 2018;61:3962–3983. [DOI] [PubMed] [Google Scholar]
- 17.Abouelhassan Y, Zhang Y, Jin S, Huigens RW III. Transcript profiling of MRSA biofilms treated with a halogenated phenazine eradicating agent: A platform for defining cellular targets and pathways critical to biofilm survival. Angew Chem Int Ed. 2018;57:15523–15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huigens RW III, Abouelhassan Y, Yang H. Phenazine antibiotic-inspired discovery of bacterial biofilm-eradicating agents. ChemBioChem. 2019;20:2885–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu R, Miller PA, Vakulenko SB, Stewart NK, Boggess WC, Miller MJ. A synthetic dual drug sideromycin induces Gram-negative bacteria to commit suicide with a Gram-positive antibiotic. J Med Chem. 2018;61:3845–3854. [DOI] [PubMed] [Google Scholar]
- 20.Barraud N, Kardak BG, Yepuri NR, et al. Cephalosporin-3’-diazeniumdiolates: Targeted NO-donor prodrugs for dispersing bacterial biofilms. Angew Chem Int Ed. 2012;51:9057–9060. [DOI] [PubMed] [Google Scholar]
- 21.Desgranges S, Ruddle CC, Burke LP, et al. β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. RSC Adv. 2012;2:2480–2492. [Google Scholar]
- 22.Phelan RM, Ostermeier M, Townsend CA. Design and synthesis of a β-lactamase activated 5-fluorouracil prodrug. Bioorg Med Chem Lett. 2009;19:1261–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blau L, Menegon RF, Ferreira EI, et al. Synthesis and total 1H- and 13C-NMR assignment of cephem derivatives for use in ADEPT approaches. Molecules. 2008;13:841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang X, Cai T, Wang PG. Synthesis of beta-lactamase activated nitric oxide donors. Bioorg Med Chem Lett. 2003;13:1687–1690. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Lambert P, Zhao L, Wang D. Synthesis and antibacterial activity of dual-action agents of a β-lactam antibiotic with cytotoxic agent mitozolomide or temozolomide. Eur J Med Chem. 2002;37:323–332. [DOI] [PubMed] [Google Scholar]
- 26.Smyth TP, O’Donnell ME, O’Connor MJ, St Ledger JO. β-Lactamase-dependent pro-drugs—recent developments. Tetrahedron. 2000;56:5699–5707. [Google Scholar]
- 27.Jungheim LN, Shepherd TA, Kling JK. Synthesis of a cephalosporin-doxorubicin antitumor prodrug: a substrate for an antibody-targeted enzyme. Heterocycles. 1993;35:339–348. [Google Scholar]
- 28.Gaurav K, Kundu S, Srivastava R. Synthesis and in vitro antibacterial activity of some novel cephem antibiotics. Int J Pharm Pharm Sci. 2012;4(Suppl. 3):659–667. [Google Scholar]
- 29.Brulé C, Grugier J, Brans A, et al. 2-Nitrobenzyl esters of penam and cephem derivatives as inhibitors of penicillin-binding proteins. Asian J Org Chem. 2013;2:653–661. [Google Scholar]
- 30.Grigan N, Musel D, Veinberg GA, Lukevics E. A simple preparative method for tert-butyl protection of aminocephalosporanic acid. Synth Commun. 1996;26:1183–1185. [Google Scholar]
- 31.Doherty JB, Ashe BM, Barker PL, et al. Inhibition of human leukocyte elastase. 1. Inhibition by C-7-substituted cetphalosporin tert-butyl esters. J Med Chem. 1990;33:2513–2521. [DOI] [PubMed] [Google Scholar]
- 32.Keltjens R, Vadivel SK, Vroom E, Klunder AJH, Zwanenburg B. A new convenient synthesis of 3-carboxycephems starting from 7-aminocephalosporanic acid (7-ACA). Eur J Org Chem. 2001:2529–2534. [Google Scholar]
- 33.Ghavami A, Labbé G, Brem J, et al. Assay for drug discovery: Synthesis and testing of nitrocefin analogues for use as β-lactamase substrates. Anal Biochem. 2015;486:75–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.