

Abstract
The pyranopterin dithiolene (PDT) ligand is an integral component of the molybdenum cofactor (Moco) found in all molybdoenzymes with the sole exception of nitrogenase. However, the roles of the PDT in catalysis are still unknown. The PDT is believed to be bound to the proteins by an extensive hydrogen bonding network, and it has been suggested that these interactions may function to fine-tune Moco for electron and atom transfer reactivity in catalysis. Here, we use resonance Raman (rR) spectroscopy to probe Moco-protein interactions using heavy atom congeners of lumazine; molecules that bind tightly to both wt-XDH and its Q102G and Q197A variants following enzymatic hydroxylation to the corresponding violapterin product molecules. The resulting enzyme-product complexes possess intense NIR absorption, allowing high quality rR spectra to be collected on wt-XDH and the Q102G and Q197A variants. Small negative frequency shifts relative to wt-XDH are observed for low-frequency Moco vibrational. These results are interpreted in the context of weak hydrogen bonding and/or electrostatic interactions between Q102 and the −NH2 terminus of the PDT, and between Q197 and the terminal oxo of the Mo≡O group. The Q102A, Q102G, and Q197A, Q197E variants do not appreciably affect the kinetic parameters kred, and kred/KD, indicating a primary role for these glutamine residues is to stabilize and coordinate Moco in the active site of XO family enzymes, but not directly affect catalytic throughput. Raman frequency shifts between wt XDH and its Q102A variant suggest the changes in the electron density at the Mo ion that accompany Mo oxidation during electron transfer regeneration of the catalytically competent active site are manifest in distortions at the distant PDT amino terminus. This implies a primary role for the PDT as a conduit for facilitating enzymatic electron transfer reactivity in xanthine oxidase family enzymes.
Keywords: molybdenum cofactor, pyranopterin, molybdopterin, xanthine dehydrogenase, xanthine oxidase, charge transfer, resonance Raman, electronic structure, hydrogen bonding
Graphical Abstract
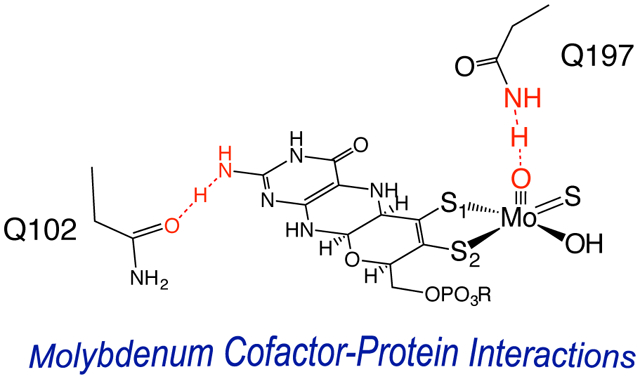
A combination of spectroscopy, kinetics, and computations has been used to evaluate Moco-protein interactions in R. capsulatus xanthine dehydrogenase. Weak hydrogen-bonding interactions between Moco and the active site glutamine residues Q197 and Q102 contribute to anchor the cofactor to the protein. Our work further supports a hypothesis that the PDT component of Moco functions as a conduit for electron transfer in the oxidative half reaction of the enzyme.
INTRODUCTION
R. capsulatus xanthine dehydrogenase (RcXDH) is a pyranopterin molybdenum enzyme1–3 that possesses broad substrate specificity1, 3–4 and a high degree of sequence homology with the mammalian molybdenum hydroxylase, xanthine oxidoreductase (XOR). XDH and XOR belong to the xanthine oxidase (XO) family of pyranopterin molybdenum enzymes, possess nearly identical coordination geometries about the Mo center,5–6 and catalyze the formal oxidative hydroxylation of a variety of purine and aldehyde substrates.1, 3–4, 7–8 XDH, like all molybdenum enzymes except nitrogenase, possesses an MoOn(PDT) (PDT = pyranopterin dithiolene ligand) molybdenum cofactor (Moco) that is comprised of an oxo-molybdenum species coordinated by the ene-1,2-dithiolate side chain of the pyranopterin. The global importance of Moco and the PDT resides in that fact that the Moco biosynthesis pathway traces back to the last universal common ancestor (LUCA) of cells, with obvious implications for the origin of life on Earth.9 The PDT is also referred to as molybdopterin (MPT) in the literature.10 Recent bioinformatics and computational data have been used to show that the pyranopterin in XO family enzymes possesses a larger out-of-plane distortion when compared to pyranopterins found in enzymes that belong to the sulfite oxidase (SO) family of pyranopterin Mo enzymes.11 This study has been used to suggest that the pyranopterin in XDH is in the fully reduced tetrahydro oxidation state (Figure 1).11 Interestingly, the roles of the PDT component of Moco in enzymatic catalysis are still unknown, but it has been suggested to (1) function as an anchor for the molybdenum ion, (2) serve as a conduit for electron transfer between the Mo ion and other redox partners, (3) contribute two redox equivalents for substrate transformations,12 and (4) modulate the Mo reduction potential.1, 10, 13–16
Figure 1.
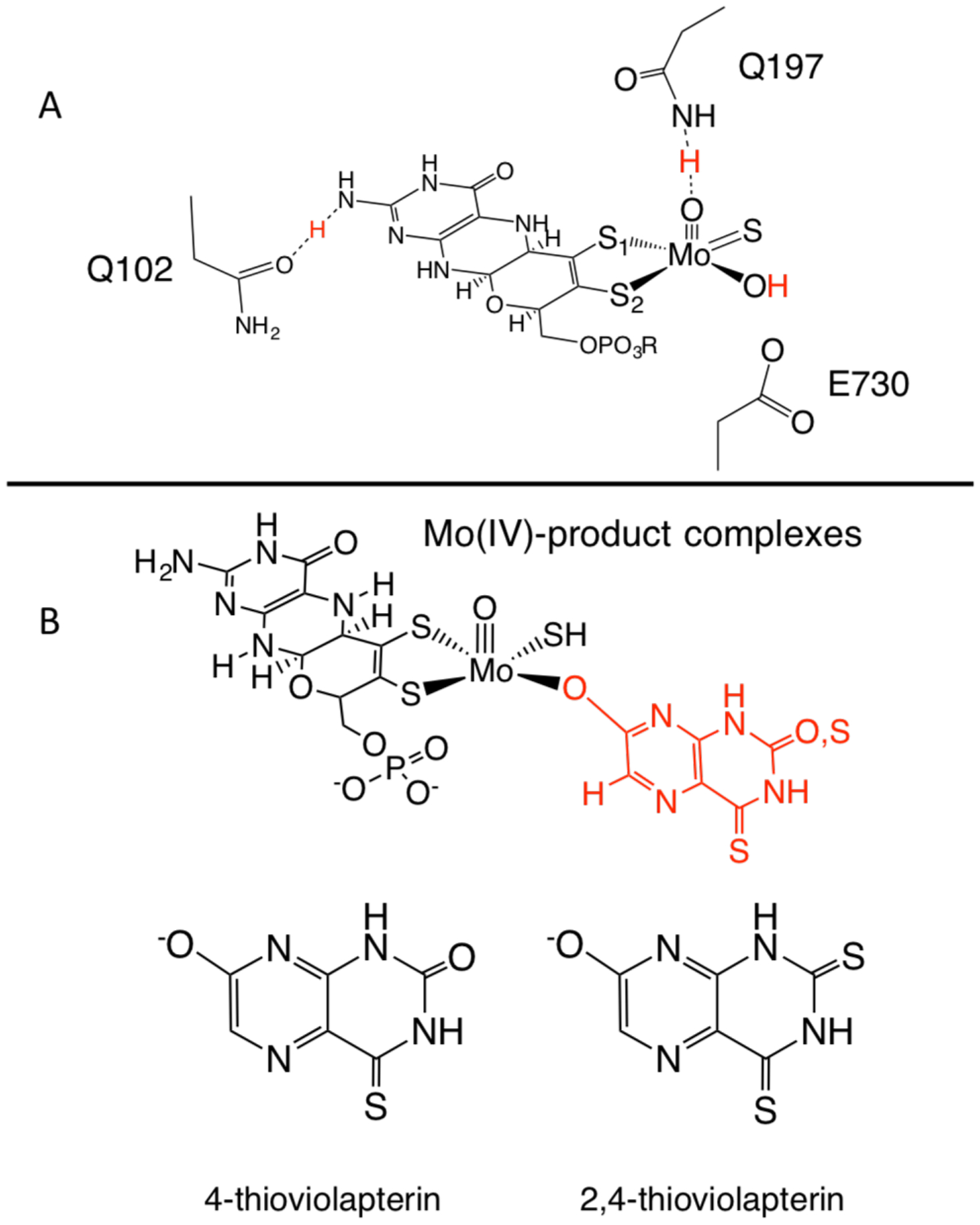
A: Active site of the XDH from R.capsulatus showing Q102 and Q197, which have been suggested to form hydrogen bonds to the amino terminus of the pyranopterin and the terminal oxo ligand bound to Mo. Both Q102 and Q197 are shown as the amide tautomer as opposed to the imide. E730 is catalytically essential and plays a role as an active site base in Mo-OH deprotonation upon nucleophilic attack of the hydroxide ligand on substrate molecules. B: Mo(IV) product complexes with 4-thioviolapterin and 2,4-dithioviolapterin.
The first coordination sphere geometry about the oxidized Mo(VI) ion in XDH and XOR is roughly square pyramidal (Figure 1A), with the Mo ion being ligated by the dithiolene side chain of the PDT, an axial oxo ligand, and equatorial sulfido and hydroxide ligands. The coordination sphere is unusual for a metalloprotein in that the Mo ion is not ligated to the protein by any amino acid residues. As a result, hydrogen bonding interactions, including those involving the PDT, likely contribute to stabilizing and coordinating Moco in the active site of XO family enzymes.17 X-ray crystallographic studies suggest that Moco is tightly bound to the protein via fifteen different hydrogen bonding interactions that are highly conserved.5–6, 18–19 These hydrogen bonding interactions between Moco and various protein residues may play a role in fine-tuning PDT and cofactor contributions to enzymatic catalysis.1
Previously, we used 4-thiolumazine and 2,4-dithiolumazine (Figure 1B) as reducing substrates to produce strong near-infrared (NIR) absorbing Mo(IV)-product complexes with both mammalian xanthine oxidase and RcXDH.16, 20 These studies were important since they showed that high quality resonance Raman (rR) spectra could be obtained in order to directly probe Moco coordination in XO and XDH. Since the extremely red-shifted NIR band does not appreciably overlap with the intrinsic absorption features of the reduced enzyme, this results in rR spectra that are devoid of contributions from the highly absorbing 2Fe2S clusters and FAD, which are the additional redox chromophores found in XO family enzymes. In this manuscript, we use a combination of enzyme kinetics, electronic absorption and resonance Raman (rR) spectroscopies, and vibrational frequency calculations to probe the effects of potential hydrogen bonding or electrostatic interactions between Q197 and the Mo≡O group of Moco, and between Q102 and the −NH2 terminus of the pyranopterin in RcXDH.
R. capsulatus XDH is comprised of two subunits, XDHA and XDHB. X-ray crystallographic studies indicate that Q102 is potentially hydrogen bonded (3.09 Å) to the −NH2 terminus of the PDT (Figure 1A),5–6, 18–19 and the putative hydrogen bond between the pyranopterin of Moco and Q102 is the only one that derives from the XDHA subunit. Q102 lies in proximity to one (Fe/S I) of the two 2Fe2S clusters in XDH and is specifically oriented for direct contact with the pterin ring of the PDT. Thus, a hydrogen bonding interaction between Q102 and the PDT may mediate electronic communication between the pyranopterin and Fe/S I as part of an electron transfer chain21 that sequentially removes reducing equivalents from the enzyme. Q102 may also function as an anchor to connect the PDT to the protein. Q197 is poised for a hydrogen bonding interaction (2.78 Å) to the terminal oxo of the Moco Mo≡O moiety and may contribute the stabilizing the coordination of Moco in the protein. Additionally, Q197 may also modulate the Mo redox potential by controlling the effective nuclear charge of the Mo ion during catalysis and the pKa of the equatorial sulfido ligand. Both Q102 and Q197 are highly conserved in all XOR and even the related aldehyde oxidoreductase2–3 (AOX) enzymes, and a primary impetus for this work is to obtain a better understand of the function of the pyranopterin and Moco in XO/XDH and related enzymes through spectral and kinetic probing of wt enzyme and specific Q102 and Q197 variants.
EXPERIMENTAL
Enzyme Preparation and Characterization.
Wild-type R. capsulatus XDH was expressed in E. coli6 and RcXDH variants were produced as reported previously.19 The enzyme activity of the reductive half reaction of XDH was performed using a stopped-flow instrument (Applied Photophysics SX20, United Kingdom). Samples were prepared anaerobically in air-tight syringes using 50 mM Tris, 1 mM EDTA buffer (pH 8,0). Measurements where performed at a constant temperature of 20°C. 10 μM XDH was mixed with different xanthine concentrations (12.5; 25; 50; 125; 250; 500 μM) in the stopped-flow system. The reduction of the FAD was followed with a diode array detector at 465 nm. Data were fitted to a double exponential decay in a time range of 1 s to obtain the observed rate constant. Metal content analysis was performed using an Optima 2100DV inductively coupled plasma-optical emission (ICP-OES) spectrometer (Perkin-Elmer, Fremont, CA). Protein samples were incubated overnight in a 1:1 mixture with 65% nitric acid (Suprapur, Merck, Darmstadt, Germany) at 100 °C. Protein samples were diluted 10-fold with ultrapure water (Millipore) prior to ICP-OES analysis. The multielement standard solution XVI (Merck) was used as a reference. pH assays were conducted at 25°C in 1 ml cuvettes in 100 mM CAPS, 100 mM KCl (pH 9–10); 50 mM Tris, 100 mM NaCl (pH 7–9) and 100 mM MES, 100 mM KCl (pH 5.5–6.5) using 500 μM NAD and varying concentrations of xanthine (25, 50, 75, 100, 200, 500 μM). The reaction was followed for 1 min at 340 nm with a Shimadzu UV-2401PC UV-VIS recording spectrophotometer. Circular dichroism spectroscopy was used to construct thermal denaturation curves of wt XDH and Q102 and Q197 variants at 218 nm and at temperatures ranging from 25°C to 80°C. Signals were recorded using a 0.1 cm cuvette at 1°C/min and a data integration time of 8 sec using a Jasco (J-815) CD-Spectropolarimeter. Protein concentration was adjusted to 1 μM (ε465 = 31600 M−1cm−1).
Preparation of the XDH Enzyme-Product Complexes.
The reducing substrates 4-thiolumazine20, 22–24 and 2,4-dithiolumazine20 were synthesized as described previously. The reduced XDHred-4-thioviolapterin product complex was prepared by addition of the reducing substrate 4-thiolumazine (8.3 mM, 15 μL) to oxidized RcXDH (143 μM, 60 μL) in BICINE buffer (50mM, pH=8.3, 120 μL). This reaction mixture was subsequently incubated for 5 minutes at room temperature under aerobic conditions to ensure complete substrate turnover, and then made anaerobic by bubbling with a stream of nitrogen gas for 15 minutes. The anaerobic reaction mixture was then treated with 15 μL of an anaerobic 0.4 M sodium dithionite solution to produce the reduced Mo(IV)-P species. The Mo(IV)-P species for the enzyme variants were produced in the same manner, with the initial concentrations of XDH Q197A as 185 μM and XDH Q102G as 170 μM. The production of reduced wt XDHred-2,4-thioviolapterin product complex complex with 2,4-thiolumazine was performed in a similar manner, as were the reduced enzyme-product complexes with the Q197A and Q102G variants. Spectrophotometric measurements were used to confirm the formation of XDHred-4-thioviolapterin (wtred-4-TV), Q197Ared-4-thioviolapterin (Q197Ared-4-TV), Q102Gred-4-thioviolapterin (Q102Gred-4-TV), XDHred-2,4-thioviolapterin (wtred-2,4-TV), Q197Ared-2,4-thioviolapterin (Q197Ared-2,4-TV) and Q102Gred-2,4-thioviolapterin (Q102Gred-2,4-TV) through the appearance of the characteristic metal-to-ligand charge transfer (MLCT) band in the near-infrared (NIR) region of the spectrum (720–740 nm).
Electronic Absorption Spectroscopy.
Electronic absorption spectra were collected on a double-beam Hitachi U-4100 UV-vis-NIR spectrophotometer (Hitachi High-Technology Corporation) capable of scanning a wavelength region between 185 and 3200 nm. The samples were measured in a 1 cm path length, black-masked, quartz cuvette (Starna Cells, Inc.) equipped with a Teflon stopper. The instrument was calibrated with reference to the instrument’s 656.10 nm deuterium line.
Resonance Raman Spectroscopy.
Room temperature solution resonance Raman spectra were collected on a commercial DXR Smart Raman Instrument (Thermo Fisher Scientific Inc.). The aqueous buffered samples were sealed in 1.5–1.8 mm diameter capillary tubes and mounted onto a capillary tube holder in the 180° back scattering accessory chamber. A 780 nm diode laser was used as the excitation source and the excitation power was 140 mW. A buffer background and an external standard sample (Na2SO4) were collected before the enzyme data collection. Instrument spectral precision is listed at ±0.25cm−1. Raman shifts were calibrated externally against the standard Na2SO4 peak at 992 cm−1, and internally against the high frequency 1293 cm−1 4-TV and 1296 cm−1 2,4-TV vibrations of the bound product molecules. A cubic spline function was fit to the 1293 cm−1 4-TV and 1296 cm−1 2,4-TV vibrational bands of the product molecules, respectively, to determine peak maxima. The Raman shift calibrated against these high frequency vibrations results in an estimated error of ~ ±0.3 cm−1 across the 4-TV data sets and ~ ±0.2 cm−1 across the 4-TV data sets. Subtraction of the buffer background was performed to yield the final resonance Raman spectrum.
Computational Details.
Spin-restricted gas phase geometry optimizations, vibrational frequency calculations, and TDDFT excited state calculations for the XDHred-product complexes were performed at the density functional theory (DFT) level of theory using the Gaussian 09W suite.25 All calculations used the B3LYP hybrid exchange-correlation functional. A 6–31G* basis set was used for all light atoms, and the LANL2DZ basis set, which includes an effective core potential, was used for Mo. Electron density difference maps (EDDMs) were generated using GaussSum (version 2.1.6).26–27 Computed vibrational modes were plotted using ChemCraft (version 1.7).28 CASSCF calculations, employing an 8-electron, 8-orbital active space, were performed using the ORCA software suite (version 3.0.3).29–30 A B3LYP hybrid functional was used in conjunction with the def2-TZVPP basis set for Mo and S atoms, and def2-SVP for all light atoms. The resulting molecular orbital depictions were rendered in ChemCraft.
RESULTS and DISCUSSION
Electronic Absorption Spectroscopy.
Room temperature electronic absorption spectra of reduced wt, Q197A, and Q102G RcXDH Mo-product (Mo(IV)-P) complexes with 4-TV are shown in Figure 2. The corresponding electronic absorption spectra for the Mo(IV)-P complexes with 2,4-TV are virtually identical and are given in Figure S1. We note that the absorption energy and band shape of the Mo(IV)→product MLCT transition show that the Mo(IV)-P CT complexes that are formed with 4-TV and 2,4-TV are virtually unaffected by the potentially H-bonding variants Q197A and Q102G (Figure 1). This provides evidence that Q197A and Q102G do not adversely affect product binding; an expected result considering that both Q197 and Q102 are remote from the substrate binding pocket.
Figure 2.
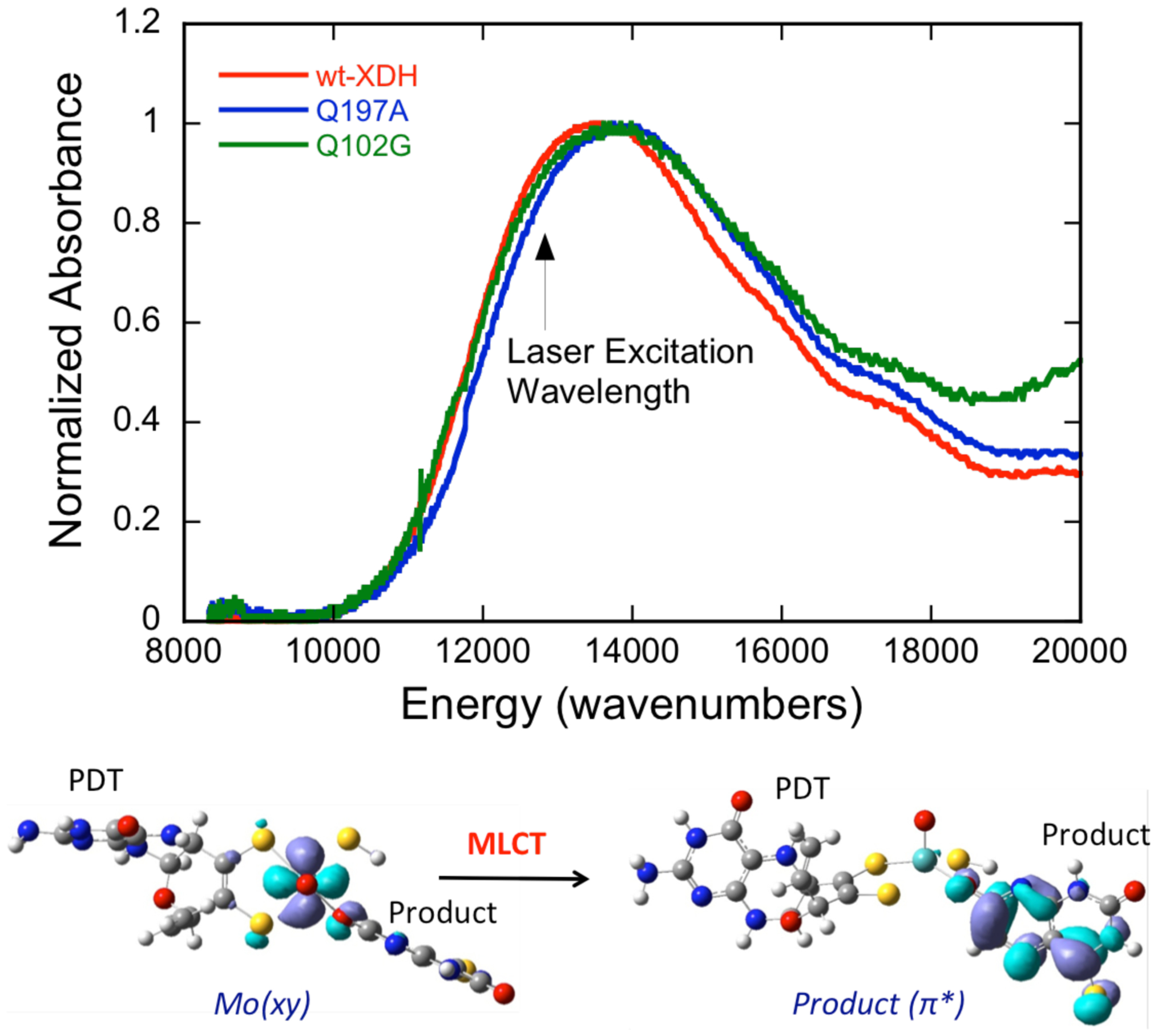
Top: Normalized electronic absorption spectra for wt XDH and the Q197A and Q102G variants using 4-thiolumazine as reducing substrate that clearly show the low energy NIR MLCT transition. Laser excitation wavelength for the resonance Raman experiments is shown using the black arrow. Bottom: CASSCF description of the MLCT transition as Mo(xy) → product (π*).
CASSCF calculations have been used to probe the nature of the near-infrared (NIR) charge transfer transition observed in these Mo(IV)-P CT complexes. The CASSCF results support an MLCT assignment for the lowest energy transition in the XDHred-product complexes Mo(IV)-4-TV and Mo(IV)-2,4-TV, with a Mo(xy) → product(π*) one-electron promotion being responsible for generating the MLCT excited state (Figure 2, bottom). Transition energies have also been computed for the MLCT state in Mo(IV)-4-TV (Ecalc: 12,800 cm−1; Eexp: 13,600 cm−1) and Mo(IV)-2,4-TV (Ecalc: 12,700 cm−1; Eexp: 13,100 cm−1), providing additional support for the assignment of this band as an Mo(xy) → product(π*) MLCT transition. The orbital nature of the MLCT state relative to the low-spin d2 Mo(IV) ground state configuration is important, since the MLCT state possesses hole character on the Mo ion. As such, the MLCT state closely mimics the Mo(IV) → Mo(V) electron transfer process that occurs in the oxidative, electron transfer half reactions of XO family enzymes to ultimately regenerate the catalytically competent oxidized Mo(VI) state (Figure 1A). Importantly, the only appreciable PDT contributions to the Mo(xy) donor orbital (Figure 2, bottom) are localized on the ene-1,2-dithiolate sulfur atoms. This bonding interaction has been described previously in the small molecule analog complex Tp*MoO(bdt) (Tp* = hydrotris(3,5-dimethylpyrazol-1-yl)borate; bdt = benzene-1,2-dithiolate).31–32 Removal of an electron from the donor orbital is anticipated to result in molecular distortions within the S-Mo-S dithiolene fragment, leading to resonance Raman enhancement of low-frequency Mo-dithiolene core vibrations31–32 that may be kinematically coupled to other low-frequency Moco modes. In this way, resonance Raman spectroscopy of wt XDH and its Q197A, and Q102G variants provides a means of making detailed low-frequency Moco vibrational assignments and sensitively probing specific Moco - protein interactions.
Resonance Raman Spectroscopy.
Low frequency (200 – 400 cm−1) vibrational Raman spectra for reduced Mo(IV)-4-TV and Mo(IV)-2,4-TV forms of wt, Q197A, and Q102G Rc XDH were collected on resonance with the NIR MLCT band and are presented in Figure 3. These data are also summarized in Table 1. The relative intensities and frequencies of vibrational Bands A-D in enzyme-product complexes of wt, Q102G, and Q197A XD are observed to be very similar, and. this indicates that the respective potential energy distributions for these normal modes are essentially identical. Using substrates that can be converted to Mo(IV)-P complexes with NIR absorbing MLCT transition is important, since it dramatically reduces the spectral overlap with the strongly absorbing 2Fe2S centers and FADH, which form part of the electron transfer chain in all XO family enzymes, including XDH. We note that XDHox, XDHred, the 4-thiolumazine and 2,4-dithiolumazine substrates, and the 4-TV and 2,4-TV products do not produce resonantly enhanced Raman vibrations with laser excitation at 780 nm (12,821 cm−1). Thus, the rR data presented in Figure 3 derive solely from the reduced Mo-P complex. Since the low frequency rR spectra of Mo(IV)-4-TV and Mo(IV)-2,4-TV in Figure 3 are very similar, this further indicates that (1) no gross changes have occurred in the pyranopterin or product binding sites as a function of these specific mutations, (2) these modes do not have dominant contributions from the Mo bound product, (3) they represent modes primarily associated with Moco, and (4) the observed vibrational frequency shifts are observed to be small.20
Figure 3.
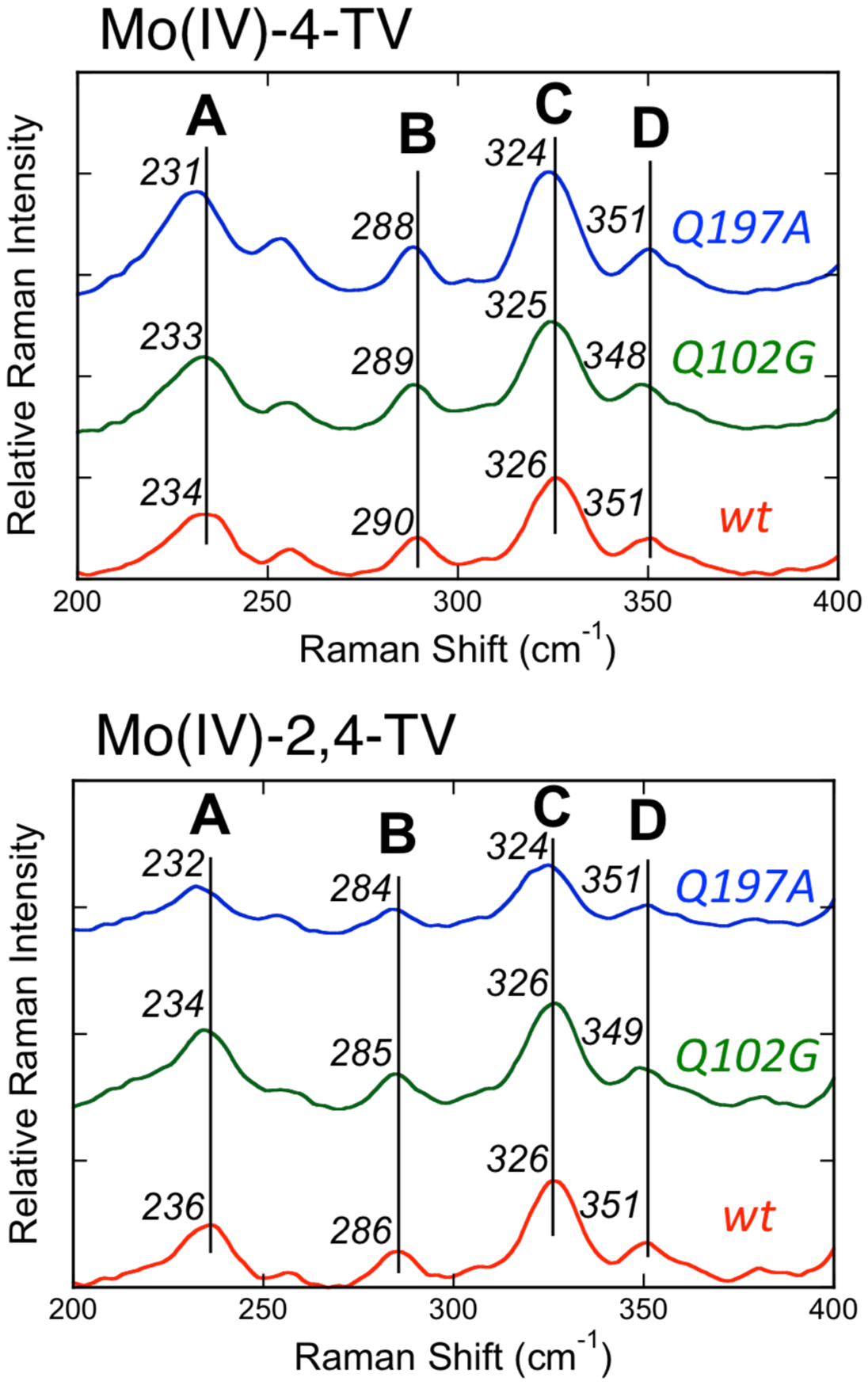
Low frequency resonance Raman spectra for wt, Q102G, and Q197A Mo(IV)-4-TV (top) and Mo(IV)-2,4-TV (bottom). Data was collected using 780nm excitation on resonance with the Mo(IV)-P MLCT band.
Table 1:
Computed low frequency Moco vibrations in XDHa
| Mo(IV)-(4-TV) | Mo(IV)-(2,4-TV) | |||||
|---|---|---|---|---|---|---|
| DFT Computed Normal Mode Description | Exp | Calc | Exp | Calc | ||
| A: dithiolene envelope folding + Mo≡O rocking + pyranopterin | V28 | 240 | V29 | 244 | ||
| Q197A | 231 | Q197A | 232 | |||
| Q102G | 233 | Q102G | 234 | |||
| wt | 234 | wt | 236 | |||
| B: asymmetric dithiolene ring distortion + Mo-SH stretching + pyranopterin | V31 | 290 | V32 | 292 | ||
| Q197A | 288 | Q197A | 284 | |||
| Q102G | 289 | Q102G | 285 | |||
| wt | 290 | wt | 286 | |||
| C: dithiolene S-Mo-S symmetric core stretch + slight Mo≡O rocking + pyranopterin | V36 | 337 | V36 | 337 | ||
| Q197A | 324 | Q197A | 324 | |||
| Q102G | 325 | Q102G | 326 | |||
| wt | 326 | wt | 326 | |||
| D: dithiolene S-Mo-S asymmetric stretch + Mo-SH stretching | V39 | 357 | V39 | 357 | ||
| Q197A | 351 | Q197A | 351 | |||
| Q102G | 348 | Q102G | 349 | |||
| wt | 351 | wt | 351 | |||
Vibrational frequencies are given in wavenumbers (cm−1).
Low frequency resonance Raman spectra of small molecule analogs for the active sites of pyranopterin Mo enzymes are typically quite simple and display two vibrations. These have been assigned as arising from symmetric S-MoS stretching and bending modes, which have been observed to be mixed due to their similar frequencies.31, 33 The situation is quite different for XDH and XO,20 where six low-frequency vibrations are observed (Figure 3). Here, we consider the experimental rR data for these XDH Mo(IV)-P CT complexes in the context of the small molecule analog resonance Raman data and DFT frequency calculations. This allows us to make quality vibrational frequency assignments (Table 1 and Figure 4) for four of these bands (bands A-D in Figure 4).
Figure 4.
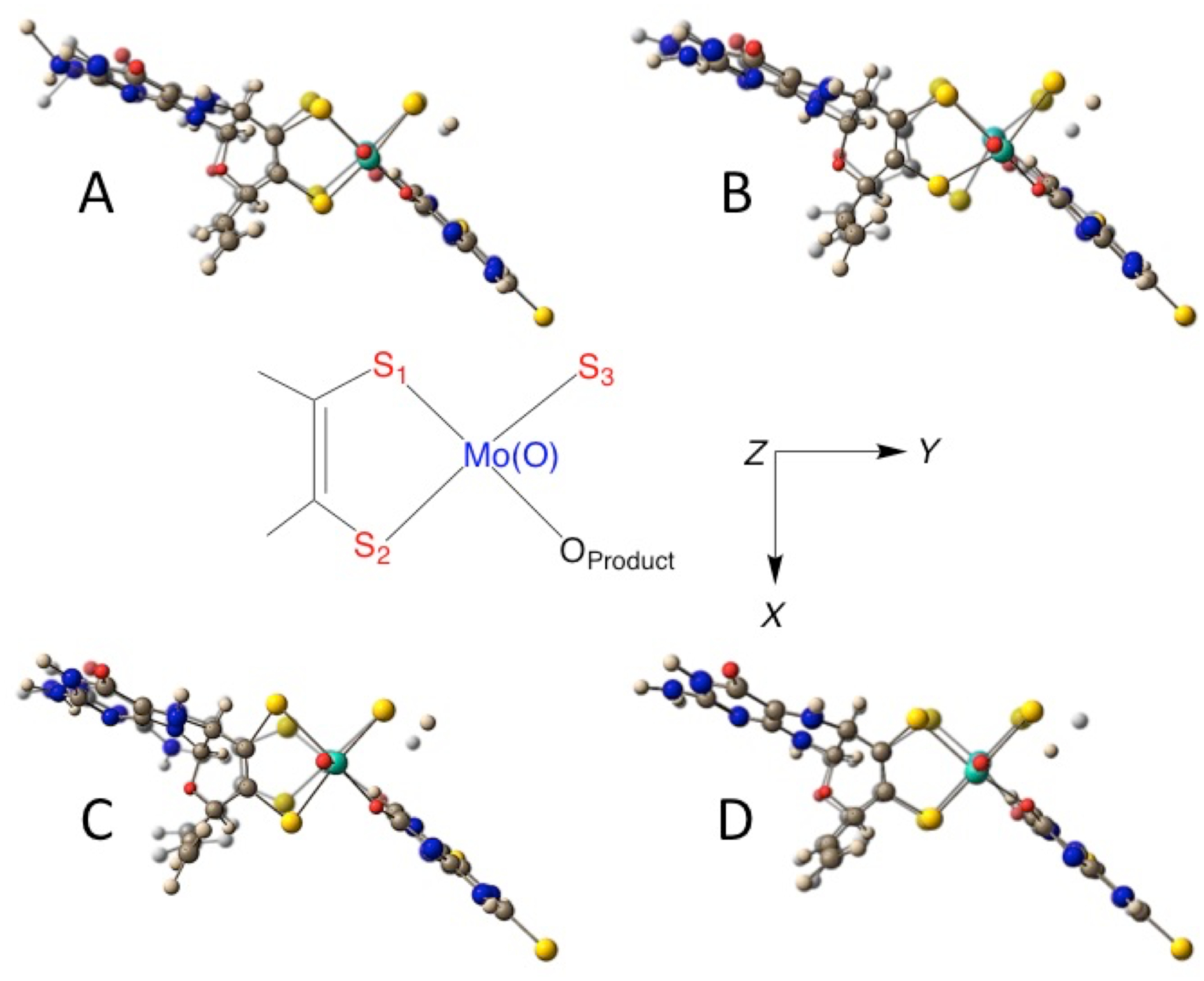
Normal mode depictions for vibrational Bands A-D in the Rc XHD Mo(IV)-2,4-TV enzyme-product intermediate. The dithiolene S donors are labeled S1 and S2, the sulfhydryl S donor is labeled as S3, and the O of the bound product as OProduct. These vibrations are displayed as a superposition of the maximum and minimum normal mode displacements. The molecular orientations are directed looking down the Mo≡O bond, and this is the z-axis.
Band A occurs at 234 cm−1 in Mo(IV)-4-TV, and has previously been assigned as a dithiolene folding + Mo≡O rocking mode. Our work here supports the prior assignment by providing additional evidence that this mode possesses both the Mo≡O rocking and pyranopterin terminal − NH2 twisting character that is observed in the DFT calculated vibrational mode. The Mo≡O rocking contribution to this normal mode is supported by the 2 cm−1 shift in the vibrational frequency as a function of the Q197A substitution. A similar Mo≡O---H hydrogen bond has been postulated to occur between the Mo≡O oxo moiety and an active site Trp from resonance Raman studies on R. capsulatus dimethyl sulfoxide reductase.34 There may also be some product out-of-plane bending character that contributes to this normal mode since there is a small 1–2 cm−1 difference in the Band A vibrational frequency of 4-TV compared to 2,4-TV. A schematic diagram of the normal mode displacements is shown in Figure 4 with the view oriented in a plane orthogonal to the apical Mo≡O bond.
Relatively larger (4 cm−1) spectral shifts are observed between Mo(IV)-4-TV and Mo(IV)-2,4-TV for Band B. Again, we attribute this to a small degree of product character being admixed into this vibrational mode, and this is observed in the DFT computed vibrational mode descriptions. The admixture of product character in this normal mode was also evident in the comparison of wt XOR and wt XDH resonance Raman spectra with 4-TV and 2,4-TV as enzyme bound product molecules.35 In the prior Raman study, the normal mode associated with Band B appeared to possess some product character since this band was also observed to shift by ~ 4 cm−1 as a function of the nature of the product, which is bound to the Mo ion as the enolate tautomer (Figure 1B).35 Here, we find that band B possesses dominant Mo-Sdithiolene and Mo-SH core vibrations (Table 1). The small negative frequency shifts observed relative to wt enzyme in the Q197A and Q102G variants are consistent with additional pyranopterin terminal − NH2 rocking and Mo≡O rocking contributions to this mode that would be affected by changes in hydrogen bonding due to the amino acid substitutions.
The 326 cm−1 mode (Band C) observed for wt Mo(IV)-4-TV XDH was also probed in earlier work that compared wt XOR and wt XDH resonance Raman spectra with both 4-TV and 2,4-TV bound product molecules.20 This mode is assigned as primarily arising from the symmetric Sdithiolene-Mo-Sdithiolene core stretching vibration by analogy to small molecule oxomolybdenum-dithiolene analog compounds31, 33 and this is additionally supported by comparison with our DFT frequency calculations. We note that a small 2 cm−1 shift to lower frequency is observed for this mode in the Q197A variant. Interestingly, inspection of the DFT calculated vibrational mode reveals an additional, albeit small, Mo≡O rocking contribution to this mode. This adds additional support to our hypothesis that the terminal Mo≡O oxo ligand is involved in a weak electrostatic or Mo≡O---H-(NH)-R hydrogen bonding interaction with Q197.
The higher frequency observed for Band D, coupled with its near invariance as function of the nature of the violapterin product bound to the Mo ion, indicates that this is a nearly pure Moco vibration with no bound product character. DFT frequency calculations support this assignment, with asymmetric Sdithiolene-Mo-Sdithiolene + Mo-SS-H stretching contributions dominating in the vibrational mode description. The asymmetric stretching nature of this band is also consistent with its relatively lower resonance Raman enhancement relative to the symmetric stretch of Band C. The frequency of Band D is not sensitive to the Q197A mutation, suggesting a reduction in Mo≡O rocking character contributing to this vibration compared with Band C. This observation is also evident in the computed normal mode description of this vibration. In contrast, the 2–3 cm−1 shift to lower frequency for the Q102G variants with either 4-TV or 2,4-TV as enzyme bound product supports pyranopterin terminal −NH2 character being admixed into this predominantly Mo-S stretching vibration.
The bathochromic shifts observed for vibrational Bands A-D in the Q197A and Q120G variants relative to wt RcXDH point to the elimination of weak Moco-protein hydrogen bonding interactions in these variants that are present in the wt protein. This red shift is most consistent with Moco bending contributions to these normal modes in vicinity of Q197 and Q102, and reflects a reduction in vibrational force constants that accompany the loss of hydrogen bonding. The magnitude of the observed vibrational frequency shifts are in accord with what has been observed for weak hydrogen bonding interactions,36 including weak protein-flavin hydrogen bonding,37 and weak hydrogen bonding interactions between amino acid residues and heme cofactors in metalloproteins.38–40
The observation of multiple low-frequency vibrations in the resonance Raman spectra of these XDH-product intermediates, and their Q197A and Q102G variants, with optical pumping into a Mo(IV)→product charge transfer excited state is important, since only two low-frequency vibrational modes are observed in Tp*MoO(dithiolene) small molecule analog Raman spectra.31–32 However, multiple low-frequency vibrations have also been observed in blue copper proteins and related small molecule analog compounds.41–44 The observation of multiple resonantly enhanced low-frequency vibrations in blue copper proteins has been attributed to a mechanical, or kinematic coupling that mixes low-frequency protein bending modes with a dominant Cu-S vibrational mode that is distorted and resonantly enhanced with optical excitation into a SCys→Cu charge transfer excitation. This vibrational mode mixing was shown to be a function of the potential energy distribution (PED)41, 43 of the Cu–SCys distortions being dispersed over multiple low-frequency normal modes with vibrational frequencies similar to the S-Mo-S stretch. A related allosteric coupling has been suggested to be very important in modulating the magnitude of the electronic coupling matrix element between the T1 Cu and the trinuclear Cu cluster in multicopper oxidases.42 Our Raman data strongly suggest a similar kinematic coupling/vibrational mode mixing is occurring in the reduced RcXDH enzyme-product complexes studied here. Such structural modulation of the electronic coupling by low frequency Moco vibrational modes may serve to increase the electron transfer rate in key pyranopterin molybdenum enzyme catalytic intermediates.44–45 Specifically, bovine XOR and RcXDH20 we now have evidence for long-range kinematic coupling between a symmetric S-Mo-S dithiolene chelate distortion and the pyranopterin, as well as coupling between this S-Mo-S symmetric stretch and the Mo≡O moiety of Moco.
Rapid reaction kinetics of XDH variants.
Site-directed mutagenesis of Q197 and Q102 has been performed in order to gain additional insight into how these glutamine residues affect the XDH reductive half reaction. Specifically, the Q197A and Q197E variants have been produced in order to compare their kinetic parameters with wt enzyme. A series of rapid reaction kinetic experiments has allowed us to obtain values for the rate of substrate reduction, kred, and the equilibrium dissociation constant, KD (Figure S2). The Q197E variant is observed to possess a small, approximately 2-fold reduction in kred when compared with wt enzyme. The different nature of the amino acid side chains in wt, Q197E, and Q197A suggests that these variants may result in the elimination of a hydrogen bonding interaction between the glutamine amide − NH2 and the terminal oxo of the Mo≡O group (Figure 1A). However, we observe that the effects on kred are rather small. Certainly, the simple elimination of a potential hydrogen bond in Q197A shows that hydrogen bonding to Mo≡O does not affect kred. The KD for the Q197E variant with xanthine as substrate is essentially the same as that observed for wt enzyme. Since Q197 is positioned above the Mo≡O group, Q197 does not appear to play a critical role in substrate binding. The KD is also not noticeably affected in the Q197A variant. The pH dependence of Kcat/KM show maximum catalytic efficiencies at pH=8.0 for wt enzyme and pH=7.5 for Q197A (Figure S3). The kinetic parameters obtained for the Q102A and Q102G variants are also quite similar to those of the wt enzyme, indicating that any disruption in a putative electron transfer pathway that involves Q102 mediating electron transfer between Fe/S I and the PDT does not affect overall catalytic efficiency in the reductive half reaction. In summary, our kinetic studies indicate minor changes in kred and kred/KD between wt enzyme and the Q102 and Q197 variants studied here, consistent with Q102 and Q197 possessing weak hydrogen bonding or electrostatic interactions with Moco.
CONCLUSIONS
The observed pattern of frequency shifts between wt enzyme and the Q197A and Q102G variants is consistent for both Mo(IV)-4-TV and Mo(IV)-2,4-TV enzyme-product complexes, and this has allowed the assignment of low-frequency wt XDH, Q197A, and Q102G vibrational modes. These assignments are consistent with the presence of either weak hydrogen bonding or electrostatic interactions existing between Moco and the glutamines Q102 and Q197 that do not appreciatively affect the kinetic parameters kred or kred/KD (Figure S2, Table S1).
The Mo(IV)→product charge transfer character20 present in the NIR MLCT band for XO and XDH product complexes leads to resonance enhancement of low-frequency vibrations that reflect the nature of Moco distortions that are coupled to a formal one-electron oxidation of the Mo4+-(product) ground state upon forming the Mo5+-(product)1– charge transfer excited state. Remarkably, this effect appears to extend all the way to the −NH2 terminus of the pyranopterin, and supports a hypothesis that the PDT acts as an electron transfer conduit that connects the Mo center to a spinach ferredoxin type 2Fe/2S cluster proximal to the PDT of Moco.
With respect to the postulated roles of the PDT in catalysis, our work suggests that hydrogen bonding interactions involving Q197 and Q102 function to partially anchor Moco to the protein. Our work further supports a hypothesis that the PDT functions as a conduit for electron transfer between the Mo ion and the proximal Fe/S I in XOR, XDH, and other members of the xanthine oxidase family of pyranopterion Mo enzyemes.1, 10,13–15, 20 Future studies will focus on other potential Moco - protein hydrogen bonds in XDH and in other pyranopterin enzymes in a concentrated effort to further define the role of the PDT in pyranopterin Mo enzyme mediated catalysis.
Supplementary Material
Table 2:
Kinetic Parameters of the reductive half reactiona
| Enzyme Variant | kred | KD | Kred/KD |
|---|---|---|---|
| XDH WT | 127.5 ±8.6 | 31.4 ±8.3 | 4.0 |
| XDH Q102G | 112.3 ±4.1 | 54.5 ±6.7 | 2.1 |
| XDH Q102A | 107.9 ±3.3 | 38.8 ±3.8 | 2.8 |
| XDH Q197A | 130.4 ±6.7 | 38.2 ±8.4 | 3.4 |
| XDH Q197E | 48.4 ±1.5 | 28.4 ±4.9 | 1.7 |
Kinetic data were recorded in 50 mM Tris, 1 mM EDTA, pH 8.0.
ACKNOWLEDGMENT
M. L. K. would like to acknowledge the National Institutes of Health (GM-057378) for generous and continued financial support of our work on molybdoenzymes. S. L. thanks the Deutsche Forschungsgemeinschaft for support from grants LE1171/6-2 and LE1171/8.
Footnotes
Supporting Information
Normalized NIR electronic absorption spectra comparing wt Mo(IV)-2,4-TV, Q197A Mo(IV)-2,4-TV, and Q102G Mo(IV)-2,4-TV; enzyme kinetic data; pH dependent comparison of kcat/KM for wild type XDH and variants; electronic absorption spectra for wt XDH and variants; CD spectra for wt XDH and variants; metal content analysis; Cartesian (x,y,z) coordinates for Mo(IV)-4-TV and Mo(IV)-2,4-TV.
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.Kirk ML; Stein B, The Molybdenum Enzymes. In Comprehensive Inorganic Chemistry II (Second Edition), Editors-in-Chief: Jan R; Kenneth P, Eds. Elsevier: Amsterdam, 2013; pp 263–293. [Google Scholar]
- 2.Hille R; Hall J; Basu P, The Mononuclear Molybdenum Enzymes. Chemical Reviews 2014, 114 (7), 3963–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hille R; Nishino T; Bittner F, Molybdenum enzymes in higher organisms. Coordination Chemistry Reviews 2011, 255 (9‚Äì10), 1179–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille R, The Mononuclear Molybdenum Enzymes. Chem. Rev 1996, 96 (7), 2757–2816. [DOI] [PubMed] [Google Scholar]
- 5.Enroth C; Eger B; Okamoto K; Nishino T; Nishino T; Pai E, Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Nat. Acad. Sci. USA 2000, 97 (20), 10723–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Truglio J; Theis K; Leimkuhler S; Rappa R; Rajagopalan K; Kisker C, Crystal structures of the active and alloxanthine-inhibited forms of xanthine dehydrogenase from Rhodobacter capsulatus. Structure 2002, 10 (1), 115–125. [DOI] [PubMed] [Google Scholar]
- 7.Stein B; Kirk M, Electronic structure contributions to reactivity in xanthine oxidase family enzymes. J Biol Inorg Chem 2015, 20 (2), 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stein BW; Kirk ML, Orbital contributions to CO oxidation in Mo-Cu carbon monoxide dehydrogenase. Chemical Communications 2014, 50, 1104–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss MC; Sousa FL; Mrnjavac N; Neukirchen S; Roettger M; Nelson-Sathi S; Martin WF, The physiology and habitat of the last universal common ancestor. Nature Microbiology 2016, 1 (9). [DOI] [PubMed] [Google Scholar]
- 10.Basu P; Burgmayer SJN, Pterin chemistry and its relationship to the molybdenum cofactor. Coordination Chemistry Reviews 2011, 255 (9‚10), 1016–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothery RA; Stein B; Solomonson M; Kirk ML; Weiner JH, Pyranopterin conformation defines the function of molybdenum and tungsten enzymes. Proceedings of the National Academy of Sciences of the United States of America 2012, 109 (37), 14773–14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamson H; Simonov AN; Kierzek M; Rothery RA; Weiner JH; Bond AM; Parkin A, Electrochemical evidence that pyranopterin redox chemistry controls the catalysis of YedY, a mononuclear Mo enzyme. Proceedings of the National Academy of Sciences of the United States of America 2015, 112 (47), 14506–14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matz KG; Mtei RP; Rothstein R; Kirk ML; Burgmayer SJN, Study of Molybdenum(4+) Quinoxalyldithiolenes as Models for the Noninnocent Pyranopterin in the Molybdenum Cofactor. Inorganic Chemistry 2011, 50 (20), 9804–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matz KG; Mtei RP; Leung B; Burgmayer SJN; Kirk ML, Noninnocent Dithiolene Ligands: A New Oxomolybdenum Complex Possessing a Donor Acceptor Dithiolene Ligand. Journal of the American Chemical Society 2010, 132 (23), 7830–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burgmayer SJN; Kim M; Petit R; Rothkopf A; Kim A; BelHamdounia S; Hou Y; Somogyi A; Habel-Rodriguez D; Williams A; Kirk ML, Synthesis, characterization, and spectroscopy of model molybdopterin complexes. Journal of Inorganic Biochemistry 2007, 101 (11–12), 1601–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirk ML, Spectroscopic and Electronic Structure Studies of Mo Model Compounds and Enzymes. In Molybdenum and Tungsten Enzymes: Spectroscopic and Theoretical Investigations, The Royal Society of Chemistry: London, 2016; Vol. 3, pp 13–67. [Google Scholar]
- 17.Schumann S; Saggu M; Moller N; Anker SD; Lendzian F; Hildebrandt P; Leimkuhler S, The mechanism of assembly and cofactor insertion into Rhodobacter capsulatus xanthine dehydrogenase. Journal of Biological Chemistry 2008, 283 (24), 16602–16611. [DOI] [PubMed] [Google Scholar]
- 18.Dietzel U; Kuper J; Doebbler JA; Schulte A; Truglio JJ; Leimkuhler S; Kisker C, Mechanism of Substrate and Inhibitor Binding of Rhodobacter capsulatus Xanthine Dehydrogenase. Journal of Biological Chemistry 2009, 284 (13), 8759–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leimkuhler S; Hodson R; George GN; Rajagopalan KV, Recombinant Rhodobacter capsulatus xanthine dehydrogenase, a useful model system for the characterization of protein variants leading to xanthinuria I in humans. Journal of Biological Chemistry 2003, 278 (23), 20802–20811. [DOI] [PubMed] [Google Scholar]
- 20.Dong C; Yang J; Lehnkuhler S; Kirk ML, Pyranopterin Dithiolene Distortions Relevant to Electron Transfer in Xanthine Oxidase/Dehydrogenase. Inorganic Chemistry 2014, 53 (14), 7077–7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hille R; Anderson RF, Coupled Electron/Proton transfer in complex flavoproteins - Solvent kinetic isotope effect studies of electron transfer in xanthine oxidase and trimethylamine dehydrogenase. J. Biol. Chem 2001, 276 (33), 31193–31201. [DOI] [PubMed] [Google Scholar]
- 22.Gorizdra TE, The synthesis and properties of some derivatives of 2-oxo-4-thioxo- and 2, 4-dithioxo-1h, 3h-pteridines. Chemistry of Heterocyclic Compounds 1969, 5 (5), 677–680. [Google Scholar]
- 23.Schneide Hj; Pfleider W, Pteridines .61. Synthesis and properties of thiolumazines. Chemische Berichte-Recueil 1974, 107 (10), 3377–3394. [Google Scholar]
- 24.Felczak K; Bretner M; Kulikowski T; Shugar D, High-yield regioselective thioation of biologically important pyrimidinones, dihydropyrimidinones, and their ribo, 2′-deoxyribo and 2′–3′ dideoxyribo necleosides Nucleosides Nucleotides 1993, 12 (2), 245–261. [Google Scholar]
- 25.Gaussian 09, R. C. G., Inc, Pittsburgh, PA, 2009. [Google Scholar]
- 26.O’Boyle NMG, Version 2.1.6, Available at http://gausssum.sf.net.
- 27.O’Boyle NM; Tenderholt AL; Langner KM, cclib: A library for package-independent computational chemistry algorithms. Journal of Computational Chemistry 2008, 29 (5), 839–845. [DOI] [PubMed] [Google Scholar]
- 28.ChemCraft; http://www.chemcraftprog.com, a. a.
- 29.Neese F, ORCA, an ab initio, density functional, and semi-empirical program package, University of Bonn, Germany. [Google Scholar]
- 30.Neese F, The ORCA program system. Wiley Interdisciplinary Reviews: Computational Molecular Science 2012, 2 (1), 73–78. [Google Scholar]
- 31.Inscore FE; Knottenbelt SZ; Rubie ND; Joshi HK; Kirk ML; Enemark JH, Understanding the origin of metal-sulfur vibrations in an oxo-molybdenurn dithiolene complex: Relevance to sulfite oxidase. Inorganic Chemistry 2006, 45 (3), 967. [DOI] [PubMed] [Google Scholar]
- 32.Inscore FE; McNaughton R; Westcott BL; Helton ME; Jones R; Dhawan IK; Enemark JH; Kirk ML, Spectroscopic evidence for a unique bonding interaction in oxo-molybdenum dithiolate complexes: Implications for sigma electron transfer pathways in the pyranopterin dithiolate centers of enzymes. Inorganic Chemistry 1999, 38 (7), 1401–1410. [Google Scholar]
- 33.Inscore FE; McNaughton R; Westcott BL; Helton ME; Jones RM; Dhawan IK; Enemark JH; Kirk ML, Spectroscopic Evidence for a Unique Bonding Interaction in Oxo-Molybdenum Dithiolate Complexes: Implications of s Electron Transfer Pathways in the Pyranopterin Dithiolate Centers of Enzymes. Inorg. Chem 1999, 38, 1401–1410. [Google Scholar]
- 34.Bell AF; He X; Ridge JP; Hanson GR; McEwan AG; Tonge PJ, Active site heterogeneity in dimethyl sulfoxide reductase from Rhodobacter capsulatus revealed by Raman spectroscopy. Biochemistry 2001, 40 (2), 440–448. [DOI] [PubMed] [Google Scholar]
- 35.Dong C; Yang J; Leimkuhler S; Kirk ML, Pyranopterin Dithiolene Distortions Relevant to Electron Transfer in Xanthine Oxidase/Dehydrogenase. Inorganic Chemistry 2014, 53 (14), 7077–7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh A; Gangopadhyay D; Nandi R; Sharma P; Singh RK, Raman signatures of strong and weak hydrogen bonds in binary mixtures of phenol with acetonitrile, benzene and orthodichlorobenzene. Journal of Raman Spectroscopy 2016, 47 (6), 712–719. [Google Scholar]
- 37.Schmidt J; Coudron P; Thompson AW; Watters KL; McFarland JT, Hydrogen-Bonding between Flavin and Protein - A Resonance Raman Study Biochemistry 1983, 22 (1), 76–84. [DOI] [PubMed] [Google Scholar]
- 38.Rousseau DL; Song S; Friedman JM; Boffi A; Chiancone E, Heme-heme interactions in a homodimeric cooperative hemoglobin - Evidence from transient Raman-scattering J. Biol. Chem 1993, 268 (8), 5719–5723. [PubMed] [Google Scholar]
- 39.Peterson ES; Friedman JM; Chien EYT; Sligar SG, Functional implications of the proximal hydrogen-bonding network in myoglobin: A resonance Raman and kinetic study of Leu89, Ser92, His97, and F-helix swap mutants. Biochemistry 1998, 37 (35), 12301–12319. [DOI] [PubMed] [Google Scholar]
- 40.Mukai M; Nagano S; Tanaka M; Ishimori K; Morishima I; Ogura T; Watanabe Y; Kitagawa T, Effects of concerted hydrogen bonding of distal histidine on active site structures of horseradish peroxidase. Resonance Raman studies with Asn70 mutants. Journal of the American Chemical Society 1997, 119 (7), 1758–1766. [Google Scholar]
- 41.Qiu D; Kilpatrick LT; Kitajima N; Spiro TG, Modeling Blue Copper Protein Resonance Raman Spectra with Thiolate-Cu(II) Complexes of a Sterically Hindered Tris(pyrazolyl)borate. Journal of the American Chemical Society 1994, 116 (6), 2585–2590. [Google Scholar]
- 42.Augustine AJ; Kragh ME; Sarangi R; Fujii S; Liboiron BD; Stoj CS; Kosman DJ; Hodgson KO; Hedman B; Solomon EI, Spectroscopic studies of perturbed T1 Cu sites in the multicopper oxidases Saccharomyces cerevisiae Fet3p and Rhus vernicifera laccase: Allosteric coupling between the T1 and trinuclear Cu sites. Biochemistry 2008, 47 (7), 2036–2045. [DOI] [PubMed] [Google Scholar]
- 43.Solomon EI; Hadt RG, Recent advances in understanding blue copper proteins. Coord. Chem. Rev 2011, 255 (7–8), 774–789. [Google Scholar]
- 44.McNaughton RL; Helton ME; Cosper MM; Enemark JH; Kirk ML, Nature of the oxomolybdenum-thiolate pi-bond: Implications for Mo-S bonding in sulfite oxidase and xanthine oxidase. Inorg. Chem 2004, 43 (5), 1625–1637. [DOI] [PubMed] [Google Scholar]
- 45.Xie X; Gorelsky SI; Sarangi R; Garner DK; Hwang HJ; Hodgson KO; Hedman B; Lu Y; Solomon EI, Perturbations to the Geometric and Electronic Structure of the CuA Site: Factors that Influence Delocalization and Their Contributions to Electron Transfer. Journal of the American Chemical Society 2008, 130 (15), 5194–5205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.