

An integrative structural approach reveals crucial roles of specific subunits in the plant TPLATE/TSET complex.
Abstract
Eukaryotic cells rely on endocytosis to regulate their plasma membrane proteome and lipidome. Most eukaryotic groups, except fungi and animals, have retained the evolutionary ancient TSET complex as an endocytic regulator. Unlike other coatomer complexes, structural insight into TSET is lacking. Here, we reveal the molecular architecture of plant TSET [TPLATE complex (TPC)] using an integrative structural approach. We identify crucial roles for specific TSET subunits in complex assembly and membrane interaction. Our data therefore generate fresh insight into the differences between the hexameric TSET in Dictyostelium and the octameric TPC in plants. Structural elucidation of this ancient adaptor complex represents the missing piece in the coatomer puzzle and vastly advances our functional as well as evolutionary insight into the process of endocytosis.
INTRODUCTION
Eukaryotic life strongly depends on a dynamic exchange of proteins and lipids between its different organelles, a feature already present in the last eukaryotic common ancestor (1). This exchange is mitigated by a wide array of protein complexes that regulate membrane shaping and coating at distinct locations. On the basis of the protocoatomer hypothesis, a number of complexes involved in vesicle trafficking, including the TSET complex (TSET), the adaptin protein complex 1 (AP1) to AP5, and the coat protein complex I (COPI), share a common origin (2). Through time, diversification, specialization, and loss-of-function events occurred within various branches of the tree of life, but many structural leitmotifs (i.e., building blocks), as well as fundamental mechanisms, remain shared (1, 3). To understand vesicle trafficking and its various adaptations, mechanistic insight into multiple coating complexes across different species is vital. Structural and functional understanding of COPI and most AP complexes is available as they have been well studied in animal and yeast cells (4, 5). However, our knowledge concerning the ancient TSET complex remains very limited. TSET is broadly present among different eukaryotic supergroups but was hidden from previous studies because of its absence in the metazoa and fungi (6). It was therefore described as a “jotnarlog” by analogy to the ancient hidden world Jotunheim in the Norse mythology (7).
TSET and its counterpart in plants, the TPLATE complex (TPC) are formed by TSPOON (LOLITA), TSAUCER (TASH3), TCUP (TML), TPLATE, TTRAY1 (TWD40-1), TTRAY2 (TWD40-2), and in the case of TPC, supplemented by two AtEH/Pan1 proteins. TSET/TPC are stoichiometrically uniform (1:1) complexes as determined by quantitative mass spectrometry (MS) or blue native polyacrylamide gel electrophoresis (6, 8). In analogy to other coatomer complexes from the AP:clathrin:COPI group, similar building blocks are present in TPC/TSET (2, 3, 9–11). The smallest and medium subunits, LOLITA and TML, respectively, both contain a longin domain, which is, in the case of TML, extended with a μ-homology domain (μHD). This μHD is a unique constituent of TPC as it is absent in the TSET complex of Dictyostelium (6). Two large subunits TASH3 and TPLATE are formed by an α-solenoid domain (fig. S1, A and B). The solenoid domains are C-terminally extended by an appendage domain that contains unique features in TPC. The canonical appendage domain (platform/sandwich) is in the case of TPLATE conserved but extended by an additional anchor domain, while in TASH3, its appendage domain is exchanged for an SH3 domain. The core is associated with two TWD40 proteins that consist of two β-propellers followed by an α-solenoid domain, a key signature motif in the eukaryotic evolution and the emergence of protocoatomer complexes (2). The additional AtEH/Pan1 subunits in Arabidopsis TPC are the most structurally characterized members. They unite accessory protein interactions and membrane targeting via their EPS15 Homology (EH) domains while allowing dimerization through their coiled-coil regions (12, 13).
Originally, TPC was described as a major adaptor module for clathrin-mediated endocytosis, but recent insight has also implicated the AtEH/Pan1 subunits as initiators of actin-mediated autophagy (14). As a central player in endocytosis and autophagy, TPC associates with a plethora of endocytic accessory proteins, cargo proteins, and autophagy-related proteins (8, 12, 13, 15). TPLATE, along with the AtEH/Pan1 subunits, has been shown to play a major role in these intermolecular protein-protein interactions. Interactions among the TPC subunits remain, however, scarce. Next to its function as a protein interaction hub, TPC localizes to membranes to fulfill its role. The molecular nature of the TPC membrane interaction remains, however, largely unknown. Currently, only the EH domains of the AtEH/Pan1 proteins have been shown to directly interact with anionic phospholipids, determinants of the electrostatic signature of the plant plasma membrane (12, 16).
To unravel TPC’s complex function, molecular organization, and its possible direct interaction with the plasma membrane, we used an integrative structural approach using chemical cross-linking, comparative protein structure models, and experimentally determined structures. These data were translated into three-dimensional representations or spatial restraints and were combined using the integrative modeling platform (IMP) (17). On the basis of these restraints, an ensemble of structures was calculated satisfying the input data. We validated the generated TPC structure by a variety of protein-protein interaction assays. Novel structural insight allowed positioning of all TPC subunits with high precision inside the complex. We show, in planta, that TPLATE and its appendage domain are essential for complex formation because of its central location within the complex. Moreover, the interaction between the AtEH/Pan1 proteins and the TML μHD provides evolutionary insight into the difference between the hexameric TSET complex and octameric TPC. Besides providing insights into the molecular architecture of TPC, we reveal a direct interaction with negatively charged phospholipids and orient the complex relative to the plasma membrane, providing the first mechanistic insight into its role in endocytosis.
RESULTS AND DISCUSSION
The TPC architecture reveals a central position of the TPLATE subunit
To gain understanding of the intersubunit arrangement and to generate novel insight into the functions of plant TPC, an integrative structural approach was implemented using the integrative modeling platform (18). The integrative modeling platform consists of a five-step process that starts with data gathering and combining experimental data, theoretical models, and physical principles. The second step is model representation. Input data are translated into restraints and/or representations. In the third step, different models are generated and scored. This is followed by the fourth step where good-scoring models have to be clustered and evaluated on the basis of the input data. The final step is the validation of the obtained structural model by data not used in the previous modeling steps (17).
To inform the relative orientation and arrangement of the TPC subunits, we used cross-linking MS (XL-MS). TPC was purified, via tandem affinity purification, from Arabidopsis cell cultures expressing the TML or AtEH1/Pan1 subunit fused with the immunoglobin G/streptavidin (GS) tag that consists of two immunoglobulin G–binding domains of protein G and a streptavidin-binding peptide. Both purifications yielded pure complexes and allowed the identification of individual TPC subunits via MS and silver staining SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1A and data file S2). After purification, on-bead cross-linking was performed with various bissulfonsuccinimidyl suberate (BS3) concentrations followed by on-bead digest and MS analysis (Fig. 1B). BS3 was used as a cross-linker chemically linking protein amine moieties within a Cα spacing of approximately 25 Å. In total, 12 XL-MS datasets of two different baits at 1.2 and 5 mM BS3 were generated. A similar cross-linking profile was observed for all experimental conditions (Fig. 1C). Last, the datasets were merged, resulting in a final dataset of 30 inter- and 89 intrasubunit cross-links (Fig. 1C, data file S4).
Fig. 1. On-bead BS3 cross-linking reveals a highly interlinked complex.
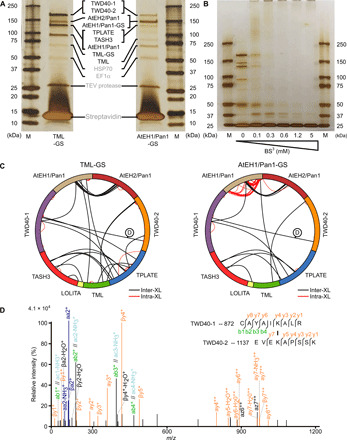
(A) Tandem affinity purification of TPC using TML- and AtEH1/Pan1-tagged subunits. The purified complex was analyzed by silver stain on a 4 to 20% SDS-PAGE. All TPC subunits, except LOLITA, were identified on the basis of MS (data file S8) and are indicated between both gels. M, marker; HSP70, heat shock protein 70. EF1, elongation factor 1; TEV, Tobacco Etch Virus. (B) Tandem affinity–purified TML-GS before and after cross-linking with various concentrations of BS3, analyzed by silver staining on a 4 to 20% SDS-PAGE gel. The vast size of TPC, with an expected molecular weight of 914 kDa, is manifested by the loss of individual subunits and the accumulation of proteins unable to penetrate the stacking gel. (C) Cross-linking analysis following tandem purification of TML- and AtEH1/Pan1-tagged subunits expressed in PSB-D cell cultures visualized by Xvis. Each analysis originates from a total of six experiments and combines 1.2 and 5 mM BS3 datasets. (D) An example of the fragmentation spectrum of the intersubunit cross-link between TWD40-1 and TWD40-2, as indicated in (C).
Given the evolutionary relationship between coatomer complexes, comparative structure models could be generated for most structured parts of TPC (47% of the TPC sequences; fig. S1 and table S1). In addition, a structure was recently solved for both EH domains of AtEH1/Pan1 by nuclear magnetic resonance (NMR)/x-ray crystallography (3% of the TPC sequences) (12). On the basis of the published interaction between LOLITA-TASH3, we performed protein-protein docking of LOLITA and the TASH3 trunk domains (amino acid residues 104 to 894) using the ClusPro2.0 docking algorithm (8, 19). The best scoring model revealed an almost identical orientation of the LOLITA longin domain and the TASH3 trunk domain compared to other coatomer complexes (fig. S1C). The same approach enabled positioning of the TML longin domain with respect to the trunk domain of TPLATE (amino acid residues 1 to 467; fig. S1C).
Input information to calculate a TPC structure included predicted stoichiometry, chemical cross-links, protein-protein docking data, comparative structure models, and the two experimentally solved EH domains. Comparative models and experimentally solved structures (covering, in total, 50% of the TPC sequences) were represented as rigid bodies, while linker or indeterminate regions were described as flexible beads of different sizes ranging from 1 to 50 amino acid residues per bead (fig. S1C and table S1). After randomization of the position of all subunits, the Metropolis Monte Carlo algorithm was used to search for structures satisfying the input restraints. An ensemble was obtained containing 4234 models satisfying excluded volume restraints, sequence connectivity, and at least 98% of chemical cross-links (good-scoring models). The ensemble was then analyzed and validated in a four-step protocol (20). Using this protocol, we tested the thoroughness of sampling, performed structural clustering of the models, and estimated the sampling precision (fig. S2, A to E). All four convergence tests were passed, and the analysis showed that the precision of the generated TPC structure is 39 Å as defined by the root mean square fluctuation of the dominant cluster containing 95% of all good-scoring models (fig. S2D). This value represents the average fluctuation of the individual protein residues or beads in three-dimensional space across the ensemble of models present in the dominant (highest-scoring) cluster.
TPC is ellipsoidal in shape with dimensions of approximately 150 by 160 by 250 Å (Fig. 2, A and B). The core is organized similarly to the core of the evolutionary-related COPI and AP complexes (9–11). The trunk domains of TPLATE and TASH3 interact C-terminally and embrace the longin domains of the small and medium subunits (Fig. 2, B and C). Comparable to the outer-coat complex of COPI, two TWD40 proteins overarch the core by forming a heterodimer (via their α-solenoid domains), and their N-terminal β-propellers face the same side of the complex (fig. S2F). The AtEH/Pan1 proteins are both attached on one side of TPC and in proximity to the appendage domain of TPLATE, the N-terminal part of TWD40-1, and TML μHD. In line with published data, a dimerization between both AtEH/Pan1 proteins was observed, likely driven by the interaction between their coiled-coil domains as indicated by a high number of cross-links between these two regions (Figs. 1C and 2, B and C) (14). The localization density map for AtEH2/Pan1 and its position in the centroid structure point to a high structural flexibility of this subunit (Fig. 2, A and B).
Fig. 2. The TPC structure reveals TPLATE as highly interconnected and centrally located.
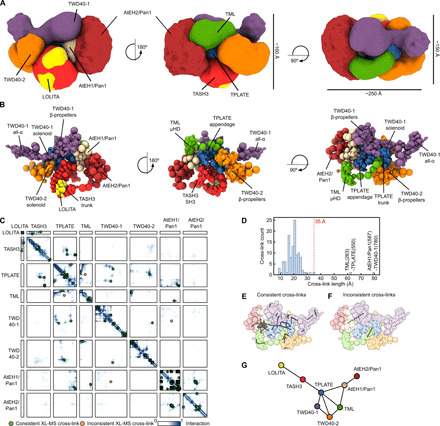
(A) Architecture of TPC as obtained by the integrative modeling platform. The localization of each subunit is defined by a density map, visualized here at a threshold equal to 1/10 of the maximum. The localization density map represents the probability of any volume element being occupied by a given subunit. The approximate dimensions of TPC are 150 × 160 × 250 Å. (B) Architecture of TPC shown as a multiscale centroid structure, i.e., the structure with the minimal sum of root mean square deviations from all the good-scoring models in cluster 1. (C) The residue contact frequency map, calculated over 20 randomly selected models from cluster 1, is depicted by colors ranging from white (0, low frequency) to dark blue (1, high frequency). A contact between a pair of amino acid residues is defined by the distance between bead surfaces below 35 Å. Cross-links are plotted as green dots (consistent cross-links) or as orange dots (inconsistent cross-links). Each box represents the contact frequency between the corresponding pair of TPC subunits. (D) Distance distribution of obtained chemical cross-links in the centroid structure. The dotted red line represents the threshold for the consistent cross-links. Only 2 of 129 observed cross-links are violated (located right of the 35-Å border) in the TPC structure. (E) Consistent cross-links mapped on the centroid structure (gray lines). (F) Inconsistent cross-links mapped on the centroid structure (gray lines). (G) Chain-chain network diagram of the TPC structure. Nodes represent individual TPC subunits, and edges (lines) are drawn between nodes, which are chemically cross-linked. This reveals that TPLATE is a central hub interconnecting other TPC subunits.
Only two cross-links were inconsistent with the generated TPC structure (Fig. 2, C and D). These two cross-links are between AtEH1/Pan1 (K587) and TWD40-1 (K789) and between TML (K283) and TPLATE (K550) (Fig. 2, D to F). A detailed analysis revealed that these cross-links connect disordered parts of the particular TPC subunits, and the inconsistency is therefore likely caused by the coarse-grained representation of these parts, limiting their flexibility.
The proposed molecular architecture of TPC together with the obtained chemical cross-links revealed the central position of TPLATE inside the complex connecting the core subunits with more auxiliary ones (Fig. 2G). The TPLATE subunit can thus be seen as a hub with all its domains (trunk, appendage, and anchor) forming an extensive network of interactions with other TPC subunits (Fig. 2, B and C).
Yeast and in planta interaction data validate the obtained structure
Yeast two-hybrid (Y2H), Y3H, and in planta interaction methods were used to validate the obtained structure. To corroborate the protein-protein docking approach, we performed detailed Y2H mapping of the TASH3-LOLITA interaction. We confirmed that the TASH3 trunk domain is necessary and sufficient for LOLITA binding (Fig. 3A). The interaction is consistent with the obtained structure of TPC. To further expand the yeast interaction landscape beyond TASH3-LOLITA, we combined a Y2H matrix of all subunits with a third nontagged subunit, extending it to a Y3H. In contrast to previously published data, a novel weak interaction between TASH3 and TPLATE could be identified in the Y2H matrix, which was strengthened upon the additional expression of a nontagged LOLITA (8). This interaction was also present between LOLITA and TPLATE, in the presence of TASH3 (Fig. 3B). In accordance with the model (Fig. 2G), we can conclude that LOLITA solely interacts with the trunk domain of TASH3 and that TASH3 interacts with TPLATE independently of LOLITA. Next to TASH3-TPLATE-LOLITA, a number of additional interactions were observed in Y3H. These interactions originate because the third subunit bridges two other subunits, interacting with a very low affinity or without a natural interaction surface at all. TWD40-1 interacts with TASH3 but only in the presence of TPLATE, and the autoactivation of AtEH2/Pan1 is reduced in the presence of an untagged TML subunit, indicating that they can interact. Both additional interactions are in line with and further validate the TPC structure (Fig. 2).
Fig. 3. Validation of TPC structure by heterologous and in planta interaction assays.
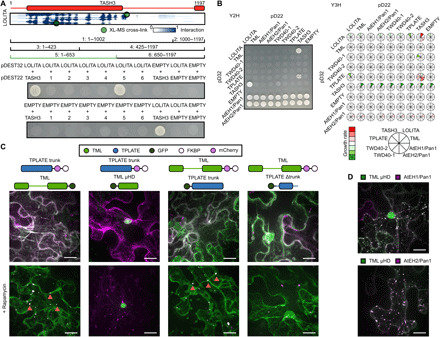
(A) Elucidation of the TASH3 interaction domain with the LOLITA subunit. Several TASH3 truncations (annotated as 1 to 6) were tested for their ability to interact with full-length LOLITA. None of the TASH3 constructs showed autoactivation when mated with an empty vector. Fragment 5, consisting of the trunk of TASH3, interacts strongly with LOLITA. The picture is representative of eight independent colonies. (B) Left: Representative Y2H matrix of all TPC subunits as well as with empty vectors used as autoactivation control. TASH3 interacts with both LOLITA and TPLATE. Both AtEH/Pan1 proteins strongly autoactivate. Right: Schematic visualization of the expansion of the Y2H with additional TPC subunit constructs without DNA binding or activation domain. Stronger interactions are indicated in green, and weakened interactions as compared to Y2H are indicated in red. Full images can be found in data file S6. (C) Representative Z-stack projected images of epidermal N. benthamiana cells transiently expressing various GFP-fused TML and TPLATE constructs, as well as mCherry-FKBP–fused TML and TPLATE constructs together with the MITO-TagBFP2-FRB* anchor. The used constructs are indicated above the image. Rapamycin induces relocalization of mCherry-FKBP–fused bait constructs to the mitochondrial anchor. The TPLATE trunk domain is sufficient for the TML-TPLATE interaction, and TML μHD is not involved in this interaction, as it displays no colocalization. Arrows indicate colocalization of both interacting constructs at the mitochondrial anchor. (D) TML μHD is recruited to AtEHs/Pan1-induced autophagosomes. The used constructs are indicated above the image. Scale bars, 10 μm.
To address the TML-TPLATE interaction, we used the recently developed knocksideway in plants (KSP) assay (21). KSP uses the ability of rapamycin to change the localization of a bait protein and its interacting partner via heterodimerization of the FK506-binding protein (FKBP) and the FKBP rapamycin-binding domain of mammalian target of rapamycin (mTOR) (FRB). Using KSP, it was previously shown that full-length TPLATE can relocalize together with full-length TML. Furthermore, this tool allowed visualization of the ternary interaction between LOLITA, TASH3, and TPLATE (21). To this end, we transiently coexpressed various full-length and domain constructs of TML and TPLATE fused either to FKBP-mCherry or green fluorescent protein (GFP), together with mitochondria-targeted FRB. We observed that the TPLATE trunk domain is sufficient for the TML-TPLATE interaction and found that the interaction is not driven by the μHD of TML (Fig. 3C).
A distinctive feature of TPC compared to the TSET complex in Dictyostelium is the presence of two AtEH/Pan1 proteins. In Arabidopsis, AtEH/Pan1 proteins play a dual role, where on one hand, they drive actin-mediated autophagy, and on the other hand, they bind auxiliary endocytic adaptors as well as the plasma membrane (12, 14). Distinctive features of the AtEH/Pan1 subunits such as autophagy-interacting motifs and a common domain organization are shared with their evolutionary counterparts in animals (Eps15/Eps15R) and yeast (Ede1p and Pan1p). We previously showed the ability of AtEH/Pan1 proteins to recruit other TPC subunits to autophagosomes, which are formed upon overexpression of AtEH/Pan1 proteins in Nicotiana benthamiana (14). Here, we took advantage of this method to visualize interactions and quantitatively analyzed autophagosomal recruitment of different TPC subunits, independently for each AtEH/Pan1 subunit (fig. S3). Our quantitative analysis revealed clear differences between the recruitment of distinct TPC subunits. We found that LOLITA and TWD40-2 are the least recruited subunits by both AtEH/Pan1 proteins, in accordance with minimal contacts observed between these TPC subunits (Fig. 2A). On the contrary, TWD40-1 showed the strongest recruitment upon overexpression of AtEH1/Pan1 consistently with proximity of these two subunits in the TPC structure. Last, in the case of AtEH2/Pan1, TML and TWD40-1 were preferentially recruited, again in accordance with their position in the TPC structure. We hypothesize that the observed differences reflect on pairwise interactions between subunits of the hexameric TSET complex and the AtEH/Pan1 proteins. On one hand, the subunits that exhibit no or a limited number of direct interactions are recruited to the autophagosomes, probably only after being built into the endogenous complex. On the other hand, the subunits that can directly interact with AtEH/Pan1 proteins are recruited to autophagosomes via their respective interacting domains, independently of complex assembly.
The TML μHD bridges membrane and TPC subunits
One of the most notable differences between TSET in Dictyostelium and plant TPC is the presence of a C-terminal μHD in TML, which was evolutionarily lost in the TCUP subunit of Amoebozoa. As previously hypothesized, concomitantly with this loss in Dictyostelium, the TCUP subunit lost some of its functions and the connection to other subunits (6). A tight interaction between TML and the AtEH/Pan1 proteins is present as high-temporal resolution spinning disc data show an identical recruitment to the plasma membrane of AtEH proteins and the core subunits of TPC (22). This complements the data obtained in this study via Y3H and in planta protein-protein interaction assays that TML associates with the AtEH/Pan1 proteins (Fig. 2B and fig. S3B). Previously published data hinted at a specific role of TML μHD in this interaction, as a C-terminal truncation of 18 amino acids resulted in the loss of AtEH/Pan1 proteins but not other TPC subunits (Fig. 4A) (8). To further corroborate this link, we transiently overexpressed TML μHD with either AtEH1/Pan1 or AtEH2/Pan1 in N. benthamiana. In line with previously shown data, the AtEH/Pan1 subunits are present on autophagosomes. The μHD did not alter the AtEH/Pan1 localization but was recruited by both AtEH/Pan1 proteins to the autophagosomes (Fig. 3D and fig. S3). Quantification of the recruitment, by comparing the cytoplasmic signal versus colocalization with AtEH/Pan1, revealed a significant recruitment in comparison to the more distant LOLITA subunit. In conclusion, the longin domain of TML interacts with the trunk domain of TPLATE and is coupled via a long flexible linker to its μHD that is able to associate with both AtEH/Pan1 proteins. In plants, μHD therefore acts as a bridge between the TPC hexamer and the AtEH/Pan1 subunits.
Fig. 4. TML μHD bridges other TPC subunits with the negatively charged lipid bilayer.
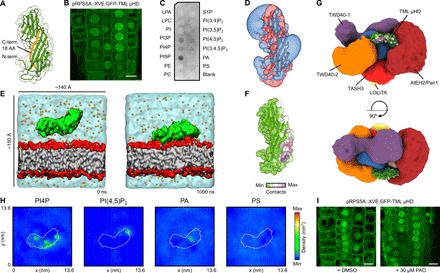
(A) Comparative model of the μHD of TML. The C-terminal 18–amino acid (AA) truncation as published before is shown in orange (3). (B) Inducibly expressed (48 hours) GFP-TML μHD in Arabidopsis epidermal root cells shows clear plasma membrane recruitment next to cytoplasmic and nuclear localization. Scale bar, 10 μm. (C) PIP strip binding of TML μHD obtained by heterologous expression in E. coli shows clear binding to negatively charged phospholipids. (D) Electrostatic potential around the structure of TML μHD. The electrostatic potentials are represented by means of positive (transparent blue) and negative (transparent red) isosurfaces at +2 and −2 kT/e, respectively. (E) CG-MD simulations of TML μHD with a complex negatively charged membrane. The initial conditions are shown on the left side (0 ns) and the membrane-bound protein on the right side (1000 ns). TML μHD is colored in green, acyl chains are gray, headgroup atoms are red, sodium atoms are orange, and water molecules are transparent cyan. (F) Mean number of μHD-lipid contacts of the dominant membrane orientation mapped onto the protein structure. The contacts were defined as the number of phosphate groups within 0.8 nm of protein atoms. (G) TML μHD superimposed onto the structure of TPC. The localization density of TML is not shown for sake of clarity. TML μHD is colored according to the contacts with the complex membrane. (H) Two-dimensional density (over last 500 ns) of different lipid molecules in the leaflet adjacent to TML μHD shows preferential clustering of PI4P. (I) The 30-min addition of PAO (a PI4 kinase inhibitor) resulted in complete loss of TML μHD membrane localization. Scale bar, 10 μm. DMSO, dimethyl sulfoxide; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PIP, phosphoinositide phosphate; PI(4,5) P2, phosphoinositide 4,5-bisphosphate; phosphoinositide 3,4,5-trisphosphate, PI(3,4,5)P3; PA, phosphatidic acid; PS, phosphatidylserine; S1P, sphingosine 1-phosphate.
μHDs are a common feature among vesicle trafficking complexes and are not only known to be involved in both accessory proteins and cargo interactions but have also been shown to directly interact with lipid membranes (23–25). An unequal expression of TML and TPLATE in a double-complemented, double-mutant Arabidopsis line resulted in the dynamic recruitment of TML to the plasma membrane without the presence of TPLATE but not the other way around (22). Therefore, we hypothesized that TML μHD might provide simultaneous membrane recruitment and association with the AtEH/Pan1 subunits. To further elucidate its role in TPC, we N-terminally fused the μHD of TML to GFP and inducibly expressed it in Arabidopsis and imaged it via confocal microscopy. Next to a nuclear and cytoplasmic localization, the TML μHD was clearly recruited to the plasma membrane (Fig. 4B). To rule out that the recruitment occurs through other auxiliary interactions, we analyzed the protein-lipid interaction in vitro. We heterologously expressed and purified the domain as an N-terminal glutathione S-transferase fusion in Escherichia coli. Using a protein-lipid overlay assay, we confirmed that μHD is able to bind negatively charged lipids (Fig. 4C).
Comparative modeling of the TML μHD structure revealed several positively charged patches indicating one or multiple possible binding modes toward a negatively charged lipid bilayer (Fig. 4D). To further address the TML μHD–lipid interaction, we performed extensive coarse-grained molecular dynamics (CG-MD) simulations, as this approach was shown to be a highly efficient tool to predict the membrane-bound state of peripheral membrane proteins (26). The simulated system contained one molecule of TML μHD, water molecules, ions, and a complex lipid bilayer with the composition of charged lipids corresponding to the plant plasma membrane (27). We carried out 20 independent calculations with different starting velocities resulting in a total of 20 μs of simulation time. In all replicas, we observed that TML μHD was quickly recruited to the lipid bilayer and remained stably bound for the remaining simulation time (Fig. 4E and fig. S4A). Analysis of contacts between protein residues and phosphate atoms of the lipid bilayer revealed three possible orientations of TML μHD toward the membrane with one orientation being slightly dominant (Fig. 4F and fig. S4B). We then positioned TML μHD into the integrative TPC structure on the basis of the TML localization density, the position of μHD in the centroid structure of TPC, and the observed cross-link with AtEH1/Pan1. We found that the dominant membrane-interacting mode is compatible with simultaneous membrane binding and association of TML with other TPC subunits (Fig. 4G and fig. S4C). We, therefore, hypothesize that TML μHD acts as a bridge between TPC subunits and the plant plasma membrane. This hypothesis is in agreement with the fact that in the absence of μHD, TPC is unable to be recruited to the plasma membrane (PM) and the interaction with the AtEH/Pan1 proteins is lost (8).
To further characterize the TML μHD interaction with the complex lipid bilayer, we monitored a two-dimensional distribution of different lipid molecules in the lipid leaflet adjacent to the protein during our CG-MD simulations. We observed that TML μHD causes strong clustering of phosphoinositide 4-phosphate (PI4P) molecules and, to a lesser degree, phosphoinositide 4,5-bisphosphate [PI(4,5)P2] molecules. We did not observe substantial clustering of other phospholipid molecules (Fig. 4H). Given the fact that PI4P was described to control the plasma membrane identity in plant cells, we speculated that PI4P could be a main driving force for the recruitment of TML to the plasma membrane (28). To test the involvement of PI4P, the inducible fluorescently tagged μHD construct was subjected to phenyl arsine oxide (PAO) treatment, which specifically affects PI4P levels in the plant plasma membrane (28). Short-term treatment (30 min) at low concentrations (30 μM) abrogated the membrane localization of the μHD (Fig. 5I). This strongly supports our conclusions based on the CG-MD results and the role of PI4P in the TML-membrane interaction.
Fig. 5. The TPLATE appendage domain is crucial for complex assembly.
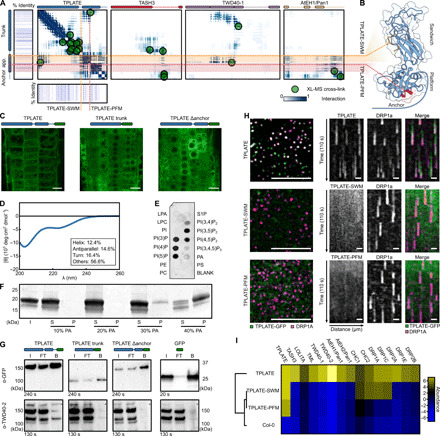
(A) Residue frequency contact map between TPLATE and selected TPC subunits. The subdomains of the TPLATE appendage, sandwich (orange) and platform (red), form an interaction hub. Cross-links are indicated as green dots. Evolutionary conservation of TPLATE is shown around the TPLATE contact map. (B) Structural features of the TPLATE appendage and anchor. Mutated residues in the sandwich (TPLATE-SWM) and platform (TPLATE-PFM) domains are indicated in orange and red, respectively. The anchor domain is shown in light blue. (C) In vivo localization of overexpressed TPLATE truncations as analyzed by confocal microscopy. The used construct is indicated above the image, and the green rectangle depicts GFP. Scale bar, 10 μm. (D) CD of TPLATE anchor. Secondary structure shown in the inset. (E) Lipid strip of TPLATE anchor shows a binding preference for charged phospholipids. (F) TPLATE anchor liposome binding comparing various concentrations of PA (10 to 40%). I, input; S, supernatant; P, pellet. (G) Coimmunoprecipitation assay comparing TPLATE with the truncated versions. Only in the case of the full-length protein an interaction with TWD40-2, as indicated by a star, could be observed. The used constructs are indicated above the image, and the green rectangle depicts GFP. I, input; FT, flow-through; B, bound. (H) Left: TPLATE (green) and DRP1a (purple) are corecruited at the PM, whereas TPLATE-SWM and TPLATE-PFM mutants are not present in the same the focal plane as DRP1a. Scale bar, 10 μm. (I) Hierarchical clustered heatmap after coimmunoprecipitation with anti-GFP, as detected by MS. Mutations in the appendage domain result in the loss of TPC subunit interactions.
Although the absence of μHD still allows plasma membrane recruitment in Dictyostelium, TPC strongly depends on its presence to recruit the complex to the plasma membrane in plants. On the one hand, other domains might be involved in the direct plasma membrane binding of Dictyostelium TSET. These domains can still retain their membrane interaction in TPC, but here, they might only stabilize the membrane binding. Possible candidates might be the N-terminal β-propellers of the TTRAY/TWD40 subunits. Analogously, the N-terminal β-propellers in COPI are located close to the lipid bilayer and involved in cargo recognition (4, 9). On the other hand, it is also possible that similarly to COPI or AP-1, Dictyostelium TSET is recruited to the membrane indirectly by interacting with a small guanosine triphosphatase from the Arf family (4, 9, 11). SecG, a protein that was copurified with Dictyostelium TSET, is homologous to animal Arf guanyl nucleotide exchange factors and supports such a hypothesis (6).
The C-terminal domains of TPLATE are essential for complex assembly
Next to μHD, a second plant-specific modification of TSET is present at the C terminus of the TPLATE subunit. In the plant TPLATE subunit, the appendage domain is followed by a 115–amino acid extension, which we termed anchor. Our cross-linking analysis revealed that the appendage and anchor domain form a hub for intracomplex interactions (Fig. 5A). Comparative modeling revealed a platform-sandwich subdomain organization of the appendage domain, while no model could be obtained for the anchor domain (Fig. 5B).
To address a potential role of the anchor and appendage domain, we generated GFP-tagged TPLATE truncation constructs lacking both the appendage and anchor domain or only lacking the anchor domain and observed their localization in Arabidopsis root epidermal cells (Fig. 5C and fig. S5D). Both constructs showed strictly cytoplasmic localizations in contrast to the full-length protein that was present in the cytoplasm as well as on PM and the cell plate. This is consistent with previously published data (8, 29). Given the loss of localization of the truncated TPLATE construct without the anchor, we hypothesized that the anchor domain could be directly involved in lipid binding. Because of the absence of a reliable homology model, we heterologously expressed the anchor domain in E. coli. The anchor domain eluted as a high–molecular weight protein during size exclusion chromatography, but size exclusion chromatography multiangle laser light scattering confirmed its expected molecular weight (fig. S5A). This suggested that the anchor domain is loosely folded and may contain disordered regions. The circular dichroism (CD) spectrum further revealed a mostly unstructured (~70%) protein with only a very low percentage of β sheets (~15%) and α helices (~10%) (Fig. 5D). The sequence of the anchor domain contains a highly charged region with a stretch of lysine residues. Charged unstructured regions have been implicated in membrane binding and mediating nanodomain organization (30). As a first proxy for membrane binding, a lipid-protein overlay assay was performed (Fig. 5E). The recombinantly expressed anchor domain displayed a strong preference for charged phosphoinositides. Together with the fact that the anchor domain is highly unstructured, this preference suggested a nonspecific charge-driven interaction. To further elaborate on this possibility, we performed liposome-binding assays. Comparing liposomes composed of neutral phospholipids, phosphatidylcholine (PC), and phosphatidylethanolamine (PE) to liposomes enriched with 5% PI(4,5)P2, 10% PI4P, or 10% phosphatidic acid (PA), we only observed binding to liposomes loaded with 10% charged phospholipid molecules. Increasing the PI(4,5)P2 concentration to 10% also resulted in very clear binding of the anchor domain (fig. S5, B and C). We next performed liposome binding assays with increasing concentrations of PA, as PA represents the simplest charged phospholipid. Higher concentrations of PA (up to 40%) resulted in a stronger protein binding corroborating the nonspecific charge-driven interaction (Fig. 5F). To test the lipid binding capacity of the anchor domain in planta, we expressed the domain as an N-terminal GFP fusion in Arabidopsis. However, only a cytoplasmic localization was observed (fig. S5E). Together with the central position of the TPLATE subunit and the fact that the anchor domain is not easily surface accessible in the TPC structure (Fig. 2A), we speculated that the anchor domain does not primarily serve as the membrane-targeting module but mostly as a protein interaction hub. Consistently, we observed several cross-links between the anchor domain and other TPC subunits (Fig. 5A). To further investigate the role of the anchor domain, we performed a coimmunoprecipitation assay comparing full-length TPLATE with the truncated versions, described earlier, and probed it for its ability to interact with the other TPC subunits (Fig. 5G). Only in case of a full-length protein an interaction with TWD40-2 (as a proxy for complex assembly) could be observed. The TPC structure combined with the coimmunoprecipitation approach thus favors a role for the anchor domain in protein-protein interactions rather than protein-lipid interactions. Although, we cannot exclude that the anchor domain, as being intrinsically disordered, can still interact simultaneously with both lipids and other TPC subunits, especially when extended. However, such flexibility of the anchor domain is limited in the integrative TPC structure because of the coarse-grained representation of unstructured parts.
Next to the anchor domain, the TPC structure revealed a vast number of contacts between the appendage domain of TPLATE and both TWD40-1 and AtEH1/Pan1 (Fig. 5A). The TPLATE appendage likely has a similar bilobal organization as known appendage domains in other coatomer complexes, consisting of sandwich and platform subdomains (Fig. 5B). Appendage domains were initially appointed a crucial role as auxiliary protein interaction platforms. Recent evidence based on electron microscopy in both AP-2 and COPI also hints at a role in coat formation due to proximity between the appendage domain and the N-terminal β-propeller of clathrin or α-COP (31–33). To assess the role of the appendage domain in TPC formation, we made mutations in the evolutionary most conserved stretches of the platform (orange) and sandwich (red) subdomains (Fig. 5, A and B), respectively named TPLATE-PFM (PlatForm Mutant) and TPLATE-SWM (SandWich Mutant). To obtain complemented lines, we transformed the GFP-tagged mutation constructs into heterozygous TPLATE transferred DNA (T-DNA) insertion lines. Expression of all constructs was validated by Western blot (fig. S5F). After extensive screening, no homozygous insertion line could be identified. Segregation analysis of heterozygous insertion lines revealed that both appendage mutants were unable to complement the TPLATE mutation, confirming the requirement of the appendage domain for TPLATE to function (Table 1). In addition, no membrane localization of both TPLATE-SWM and TPLATE-PFM constructs was observed when combined with the styryl dye FM4-64 (fig. S5G). To compare the membrane recruitment, we crossed both lines with the dynamin-related protein 1A (DRP1A) endocytic marker. Spinning-disk confocal microscopy revealed dynamic endocytic spots containing DRP1A in all TPLATE lines. Full-length TPLATE localized in the same focal plane of the DRP1A endocytic foci, indicating membrane recruitment. This was, however, not the case for TPLATE-SWM and TPLATE-PFM mutants (Fig. 5H). Given the central position of the TPLATE subunit in the TPC structure (Fig. 2G), we compared the interactome of TPLATE with TPLATE-SWM/PFM mutations. Coimmunoprecipitation combined with MS analysis revealed the inability of these mutated TPLATE isoforms to interact with any other TPC complex subunits (Fig. 5I and fig. S5H; data file S5). As revealed by XL-MS and the integrative TPC structure, the appendage domain of TPLATE is in close contact with its trunk domain to position β-propellers of TWD40-1 close to the heterotetrameric core (Fig. 2, B and C). β-Propellers are known for their ability to interact with both auxiliary proteins and the plasma membrane. We hypothesize that the correct orientation of the appendage by interacting with the trunk domain and the TWD40 proteins is crucial for proper complex assembly and function of the TPLATE complex at the plasma membrane.
Table 1. Functionality analysis of the TPLATE appendage mutants.
Segregation ratios of the progeny of tplate heterozygous mutants expressing TPLATE-SWM and TPLATE-PFM appendage mutation constructs driven from the functional pLAT52 promotor, as analyzed by polymerase chain reaction analysis. In all T-DNA insertion offspring lines, a 1:1 ratio of T-DNA versus wild type (WT) was observed in both constructs because of male sterility. Three individual transgenic lines were analyzed for each construct. The χ2 test was used to test whether the segregation ratio deviated from 1:1. χ2 0.05 (1) = 3.841.
| Mutant lines | T-DNA | WT | Total | χ2 |
| TPLATE-SWM-1 | 13 | 11 | 24 | 0.167 |
| TPLATE-SWM-2 | 12 | 12 | 24 | 0.000 |
| TPLATE-SWM-3 | 13 | 11 | 24 | 0.167 |
| Total | 38 | 34 | 72 | 0.174 |
| TPLATE-PFM-1 | 11 | 13 | 24 | 0.167 |
| TPLATE-PFM-2 | 8 | 14 | 22 | 1.637 |
| TPLATE-PFM-3 | 12 | 12 | 24 | 0.000 |
| Total | 31 | 39 | 70 | 0.914 |
In conclusion, we implemented a highly multidisciplinary approach to structurally characterize the evolutionary ancient TSET/TPLATE complex. By combining diverse experimental methods together with the integrative modeling platform, we demonstrate that the TPLATE subunit forms a central hub in TSET/TPC creating a vast array of protein-protein interactions and thus being indispensable for TSET/TPC assembly. The appendage domain and the plant-specific anchor domain play herein a vital role. We could also link other specific features of TPC, namely, the AtEH/Pan1 proteins with the μHD of TML. Existence of the interaction between muniscins, which contain μHD evolutionarily related to TML μHD (6,34), and EH domain–containing proteins in yeast and mammalian cells points to the presence of this link already in the last eukaryotic common ancestor. It is therefore plausible to speculate that the ancestral TPLATE complex was an octamer and that the AtEH/Pan1 subunits were lost in the Amoebozoa lineage, concomitant with the loss of μHD. Furthermore, our data clearly point to a direct interaction between the complex and the plasma membrane without the need of any additional protein factors. The generated model for the TPC architecture suggests many structural similarities between TSET/TPC and other coatomer complexes like COPI and AP2-clathrin. It will be of interest to further investigate whether TSET/TPC can form higher-ordered structures when bound to a membrane (i.e., a coat) similarly to other coatomers. As the integrative modeling approach is inherently an iterative process, the obtained model of the TPC structure can be further improved as new experimental data become available.
MATERIALS AND METHODS
Molecular cloning
Primers used to generate TPLATE appendage substitution fragments (SWM/PFM) are listed in table S2 and were generated by mutagenesis polymerase chain reaction (PCR) from the pDONR plasmid containing full-length TPLATE (35) by combining sewing and mutation primers, after which the product was introduced by recombination in pDONR221 via Gateway BP (Invitrogen). All entry clones were confirmed by sequencing. Expression constructs were obtained by combining the generated entry clones. The TPLATE appendage–substituted entry clones were combined with pDONRP4-P1r-Lat52 (29), and pDONRP2-P3R-EGFP (29) in the pB7m34GW backbone (36) via an LR reaction (Invitrogen). TPLATE truncation constructs and full length were cloned in a similar way, but a pH3.3 promoter was used instead. TML μHD was amplified from full-length TML with a stop codon (8), cloned into pDONR221 via BP clonase (Invitrogen) and recombined with an RPS5A::XVE promoter and an N-terminal GFP in a pB7m34GW backbone (36) via an LR reaction (Invitrogen), and verified by restriction digest.
Constructs used in the Y2H of the TASH3 truncations were amplified from full-length TASH3 coding sequence by adding gateway sites; the primers are listed in table S2 and cloned with the help of BP Gateway (Invitrogen) in pDONR221 and verified by sequencing. Entry clones were transformed in a pDEST22 expression vector via an LR gateway reaction (Invitrogen) and checked via restriction digest.
TPLATE Δtrunk was amplified from full-length TPLATE (35), after which it was introduced by recombination in pDONR221 via Gateway BP (Invitrogen). Primers are listed in table S2.
Expression constructs used for KSP were cloned by combining a 35S promoter, entry vectors used in this study, as well as FKBP and FRB entry vectors (21) from full-length pDONR plasmids. Expression constructs were verified by restriction digest.
Tandem affinity purification (TAP) and XL-MS
Before cross-linking, TML and AtEH1/Pan1 subunits were expressed in Arabidopsis cell cultures grown in continous dark (Plant Systems Biology-Dark; PSB-D) with a C-terminal GS tag and subsequently purified based on an established protocol (37). After the purification, the beads were washed with phosphate-buffered saline (PBS) and spun down for 3 min at 500 rpm. Fresh BS3 cross-linker (A39266, Thermo Fisher Scientific) dissolved in PBS was added and incubated on a rotating wheel for 30 min at room temperature. Excess cross-linker was quenched at room temperature for 30 min with 50 mM NH4HCO3. For SDS-PAGE analysis, proteins were boiled from the beads using a mixture of loading dye, PBS, and reducing agent. For further MS analysis, proteins were subsequently reduced with 5 mM dithiothreitol (DTT) and acetylated in the dark with 15 mM iodoacetamide. Next, the beads were washed with 50 mM NH4HCO3 and incubated overnight at 37°C with Trypsin/LysC (V5071, Promega). The supernatant was removed from the beads and desalted with Monospin C18 columns (Agilent Technologies, A57003100) as described in (38).
The peptides were redissolved in 20-μl loading solvent A [0.1% trifluoroacetic acid (TFA) in water/acetonitrile (ACN) (98:2, v/v)] of which 10 μl was injected for liquid chromatography–tandem MS (LC-MS/MS) analysis on an Ultimate 3000 RSLCnano system in-line connected to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Trapping was performed at 10 μl/min for 4 min in loading solvent A on a 20-mm trapping column [made in-house, 100-μm internal diameter (ID), 5-μm beads, C18 Reprosil-HD, Dr. Maisch, Germany]. The peptides were separated on an in-house produced column (75 μm × 400 mm), equipped with a laser-pulled electrospray tip using a P-2000 Laser Based Micropipette Puller (Sutter Instruments), packed in-house with ReproSil-Pur basic 1.9-μm silica particles (Dr. Maisch). The column was kept at a constant temperature of 40°C. Peptides eluted using a nonlinear gradient reaching 30% MS solvent B [0.1% formic acid (FA) in water/acetonitrile (2:8, v/v)] in 105 min, 56% MS solvent B in 145 min, and 97% MS solvent B after 150 min at a constant flow rate of 250 nl/min. This was followed by a 10-min wash at 97% MS solvent B and reequilibration with MS solvent A (0.1% FA in water). The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the 16 most abundant ion peaks per MS spectrum. Full-scan MS spectra [375 to 1500 mass/charge ratio (m/z)] were acquired at a resolution of 60,000 in the Orbitrap analyzer after accumulation to a target value of 3,000,000. The 16 most intense ions above a threshold value of 13,000 were isolated (isolation window of 1.5 m/z) for fragmentation at a normalized collision energy of 28% after filling the trap at a target value of 100,000 for maximum 80 ms. MS/MS spectra (145 to 4085 m/z) were acquired at a resolution of 15,000 in the Orbitrap analyzer. The S-lens radio frequency level was set at 50, and precursor ions with unassigned, single-, and double-charge states were excluded from fragmentation selection.
The raw files were processed with the MaxQuant software (version 1.6.10.43) (39) and searched with the built-in Andromeda search engine against the Araport11plus database. This is a merged database of the Araport11 protein sequences (www.Arabidopsis.org) and sequences of all types of non-Arabidopsis contaminants possibly present in AP-MS experiments. These contaminants include the cRAP protein sequences, a list of proteins commonly found in proteomics experiments, which are present either by accident or by unavoidable contamination of protein samples (The Global Proteome Machine, www.thegpm.org/crap/). In addition, commonly used tag sequences and typical contaminants, such as sequences derived from the resins or the proteases used, were added. Search parameters can be found in data file S1.
The MaxQuant proteingroups file (data file S2) was filtered for two peptide identifications, and “only identified by site,” “reverse,” and “contaminants” were removed. Proteins were ranked by descending intensity-based absolute quantification (iBAQ) values, showing that the eight TPC subunits have the highest iBAQ values, and are thus the most abundant proteins in the samples. Therefore, a custom database consisting of the eight TPC protein sequences was made to use in the pLink2.0 program (40). Used parameters can be found in data file S3. The identified cross-links can be found in data file S4. The fragmentation spectra of the obtained cross-links were manually checked, and intracross-links within 20 amino acids were removed.
Multiple sequence alignment
Homologs of the TPLATE subunit were taken from published data (6). To identify additional TPLATE subunits homologs, the predicted proteins of each genome were searched using BLASTP (41) with Arabidopsis TPLATE as an input sequence. Used databases were GenBank (www.ncbi.nlm.nih.gov/genbank/), Joint Genome Institute (https://genome.jgi.doe.gov/portal/), EnsemblPlants (https://plants.ensembl.org/index.html), and Congenie (http://congenie.org/start). Multiple alignment was constructed with the mafft algorithm in the einsi mode (42) and manually normalized on the protein sequence of Arabidopsis TPLATE using the Jalview program (43).
Integrative structure determination of TPC
The integrative modeling platform (IMP) package version 2.12 was used (18) to generate the structure of TPC. Individual TPC subunits were built on the basis of the experimental structures determined by x-ray crystallography and NMR spectroscopy (12) or comparative models created with MODELLER 9.21 (44) based on the related structures detected by HHPred (45) and RaptorX (46). Domain boundaries, secondary structures, and disordered regions were predicted using the PSIPRED server (47) by DomPRED (48), PSIPRED (49) and DISOPRED (50).
The domains of TPC subunits were represented by beads of varying sizes, 1 to 50 residues per bead, arranged into either a rigid body or a flexible string of beads (loop regions). Regions without an experimental structure or a comparative model were represented by a flexible string of large beads corresponding to 50 residues each.
For protein-protein docking, a part of a trunk domain of TASH3 or TPLATE was used as a receptor, and the entire structure of LOLITA or the longin domain of TML was used as a ligand. Computational rigid-body docking of the respective receptor and ligand was performed using ClusPro2.0 (19) with default parameters without restraining the interaction site. Docked pairs were then described as a single rigid body for each pair. In total, TPC was represented by 12 rigid bodies and 93 flexible bodies (table S1).
One hundred nineteen unique intra- and intermolecular BS3 cross-links obtained by MS were used to construct the scoring function that restrained the distances spanned by the cross-linked residues. The excluded volume restraints were applied to each 10-residue bead. The sequence connectivity restraints were used to enforce proximity between beads representing consecutive sequence segments.
After randomization of position of all the subunits, the Metropolis Monte Carlo algorithm was used to search for structures satisfying input restraints. The sampling produced a total of 1,000,000 models from 50 independent runs, each starting from a different initial conformation of TPC. A total of 4234 good-scoring models satisfying at least 98% of chemical cross-links were selected for further analysis. To analyze sampling convergence, exhaustiveness, and precision, the four-step protocol (20) was used. The residue contact frequency map was calculated according to Algret et al. (51).
Y2H assay
Expression vectors were transformed via heat shock in MaV203. Transformed yeast was grown for 2 days at 30°C. Eight colonies were picked up, grown overnight in liquid SD–Leu/−Trp medium, and diluted to optical density at 600 nm (OD600) 0.2 before 10 μl was plated on SD–Leu/−Trp and SD–Leu/−Trp/-His with 50 mM 3AT, grown for 2 days, after which the plates were imaged.
Y3H assay
All TPC subunits were recombined from available gateway entry clones (8) in pDEST22 and pDEST32 expression vectors and transformed via heat shock in both the PJ69-4a and PJ69-4α yeast strains. They were plated out and a single representative colony was picked out and put in culture. A and α strains were mated and cultured in SD–Leu/−Trp and spotted to analyze the Y2H matrix. The liquid cultures were super transformed, via heat shock, with all TPC subunits (cloned in pAG416GPD) and cultured in SD–Leu/−Trp/–Ura. Cultures were grown for 2 days and were diluted to OD600 0.2, and 10 μl was plated on SD–Leu/−Trp/–Ura and SD–Leu/−Trp/–Ura/-His and grown for 3 days at 30°C, after which the plates were imaged (data file S6).
Autophagosomal recruitment and KSP assay
N. benthamiana plants were grown in a growth room or greenhouse with long-day conditions. Transient expression was performed by leaf infiltration according to (52). Transiently transformed N. benthamiana were imaged 2 days after infiltration using a PerkinElmer UltraVIEW spinning-disk system, attached to a Nikon Ti inverted microscope and operated using the Volocity software package. Images were acquired on an ImagEM charge-coupled device (ccd) camera (Hamamatsu C9100-13) using frame-sequential imaging with a 60× water immersion objective [numerical aperture (NA), 1.20]. Specific excitation and emission windows were used; a 488-nm laser combined with a single band-pass filter (500 to 550 nm) for GFP, 561-nm laser excitation combined with a dual band-pass filter (500 to 530 nm and 570 to 625 nm) for mCherry and 405-nm laser excitation combined with a single band-pass filter (454 to 496 nm) for TagBFP2. Z-stacks were acquired in sequential-frame mode with a 1-μm interval. Images shown are Z-stack projections. For the KSP assay, an FKBP-tagged protein as well as Mito-FRB and a GFP-tagged subunit were infiltrated. Forty-eight hours after infiltration, N. benthamiana leaves were infiltrated with 1 μM rapamycin (Sigma-Aldrich). A stock solution was prepared by diluting a corresponding amount in DMSO (dimethyl sulfoxide). Before the infiltration, final concentrations of the chemicals were diluted in Milli-Q water. Leaves were imaged in a 20- to 45-min time window. Autophagosomal recruitment as well as KSP images were analyzed on the basis of the signal in mitochondria versus the cytoplasm (21).
CG-MD simulation
The structure of TML μHD was mapped into the MARTINI CG representation using the martinize.py script (53–55). The ELNEDYN representation with rc = 0.9 nm and fc = 500 kJ mol−1 nm−2 was used to prevent any undesired large conformational changes during CG-MD simulations (56). The MARTINI CG model for all lipid molecules used in this study was taken from the study by Ingólfsson et al. (57). Lipid bilayer, in total composed of 600 phospholipid molecules, containing POPC:POPE:POPS:POPA:POPI4P:POPI(4,5)P2 (molecular ratio, 37:37:10:10:5:1) was prepared using CharmmGUI Martini Maker (58).
CG-MD simulations were performed in GROMACS v5 (59). The bond lengths were constrained to equilibrium lengths using the LINCS algorithm. Lennard-Jones and electrostatics interactions are cut off at 1.1 nm, with the potentials shifted to zero at the cutoff. A relative dielectric constant of 15 was used. The neighbor list was updated every 20 steps using the Verlet neighbor search algorithm. Simulations were run in the NPT ensemble. The system was subject to pressure scaling to 1 bar using a Parrinello-Rahman barostat, with temperature scaling to 303 K using the velocity-rescaling method with coupling times of 1.0 and 12.0 ps. Simulations were performed using a 20-fs integration time step. Initially, the protein was placed approximately 3.0 nm away from the membrane. Subsequently, the standard MARTINI water and Na+ ions were added to ensure the electroneutrality of the system. The whole system was energy-minimized using the steepest descent method up to the maximum of 500 steps and equilibrated for 10 ns. Production runs were performed for up to 1 μs. The standard GROMACS tools as well as in-house codes were used for the analysis.
Staining and drug treatment for live-cell imaging
FM4-64 (Invitrogen) was stored at 4°C in 2 mM stock aliquots in water and protected from light at all times. Whole Arabidopsis seedlings were incubated with 1/2 MS liquid medium containing 2 μM FM4-64 at room temperature for 15 min before confocal imaging. Colocalization analysis was performed in ImageJ.
PAO treatments were performed for 30 min at room temperature in 1/2 MS liquid medium containing 30 μM PAO (Sigma-Aldrich). PAO was dissolved in DMSO and diluted 1:1000 in liquid MS media before use.
Live-cell imaging of Arabidopsis lines
The subcellular localization of TPLATE and TPLATE motif substitutions was addressed by imaging root meristematic epidermal cells of 4- to 5-day-old seedlings on a Zeiss 710 inverted confocal microscope equipped with the ZEN 2009 software package and using a C-Apochromat 40× water Korr M27 objective (NA, 1.2). Enhanced GFP (EGFP) was visualized with 488-nm laser excitation and 500- to 550-nm spectral detection, and FM4-64 was visualized using 561-nm laser excitation and 650- to 750-nm spectral detection.
TPLATE truncations were imaged on an Olympus FluoView 1000 (FV1000) confocal microscope equipped with a Super Apochromat 60× UPLSAPO water immersion objective (NA, 1.2). EGFP was visualized with 488-nm laser excitation and 500- to 600-nm spectral detection.
Dynamic imaging of TPLATE and TPLATE motif substitutions at the PM was performed in etiolated hypocotyl epidermal cells using a Nikon Ti microscope equipped with an UltraVIEW spinning-disk system and the Volocity software package (PerkinElmer) as described previously (8, 14). Images were acquired with a 100× oil immersion objective (Plan Apo; NA, 1.45). The CherryTemp system (22) was used to maintain the temperature of samples constant at 20°C during imaging.
Seedlings expressing GFP-fused proteins were imaged with 488-nm excitation light and an emission window between 500 and 530 nm in single-camera mode. Seedlings expressing monomeric red fluorescent protein (mRFP)- and tagRFP-labeled proteins were imaged with 561-nm excitation light and an emission window between 570 and 625 nm in single-camera mode or 580 to 630 nm in dual-camera mode. Single-marker line movies were acquired with an exposure time of 500 ms per frame for 2 min. Dual-color lines were acquired sequentially (one-camera mode) with an exposure time of 500 ms per frame.
β-Estradiol induction
β-Estradiol induction of the pRPS5A::XVE:GFP-μHD line was done by transferring 3-day-old seedlings to medium containing β-estradiol (Sigma-Aldrich) or solvent (DMSO) as a control. β-Estradiol concentration used was 1 μM.
Arabidopsis seedling protein extraction
Arabidopsis seedlings were grown for 7 days on 1/2 MS medium under constant light. Seedlings were harvested, flash-frozen, and grinded in liquid nitrogen. Proteins were extracted in a 1:1 ratio, buffer (ml):seedlings (g), in HB+ buffer, as described before (37). Protein extracts were incubated for 30 min at 4°C on a rotating wheel before spinning down twice at 20,000g for 20 min. The supernatant was measured using Qubit (Thermo Fisher Scientific), and equal amounts of proteins were loaded for analysis.
Coimmunoprecipitation assays
Arabidopsis seedling extract, in a 2:1 ratio, buffer (ml):seedlings (g) (see above), was incubated for 2 hours with 20-μl preequilibrated magnetic GFP-beads (Chromotec, gtma-20). After 2 hours, the extract was removed and the beads were washed three times with 1 ml of HB+ buffer. Proteins were eluted using a 20:7:3 mixture of buffer:4× Laemmli sample buffer (Bio-Rad):10× NuPage sample reducing agent (Invitrogen) and incubated for 5 min at 70°C after which they were loaded on SDS-PAGE gels.
SDS-PAGE and Western blot
Antibodies used in this study are listed in table S4. Samples were analyzed by loading on 4 to 20% gradient gels (Bio-Rad), after addition of 4× Laemmli sample buffer (Bio-Rad) and 10× NuPage sample reducing agent (Invitrogen). Gels were transferred to polyvinylidene difluoride or nitrocellulose membranes using the Trans-Blot Turbo system (Bio-Rad). Blots were imaged on a ChemiDoc Imaging System (Bio-Rad). Full gels can be found in data file S7.
In-gel identification of proteins
For MS analysis of proteins bands, the sample was separated on a 4 to 12% gradient NuPAGE gel (Invitrogen) and visualized with colloidal Coomassie Brilliant Blue staining. Gel lanes were cut out. Proteins were processed and digested by trypsin per gel slice (8).
Experimental setup to identify interacting proteins using immunoprecipitation-MS/MS
Immunoprecipitation experiments were performed for three biological replicates as described previously (60), using 3 g of 4-day-old seedlings. Interacting proteins were isolated by applying total protein extracts to α-GFP–coupled magnetic beads (Miltenyi Biotec). Three replicates of TPLATE motif substitution mutants (SWM and PFM) were compared to three replicates of Col-0 and TPLATE-GFP [in tplate(−/−)] as controls.
Identification of proteins using MS/MS
Peptides were redissolved in 15-μl loading solvent A [0.1% TFA in water/ACN (98:2, v/v)] of which 5 μl was injected for LC-MS/MS analysis on an Ultimate 3000 RSLC nano LC (Thermo Fisher Scientific, Bremen, Germany) in-line connected to a Q Exactive mass spectrometer (Thermo Fisher Scientific). The peptides were first loaded on a trapping column made in-house (100 μm ID × 20 mm, 5-μm beads, C18 Reprosil-HD, Dr. Maisch, Ammerbuch-Entringen, Germany), and after flushing from the trapping column, the peptides were separated on a 50-cm μPAC column with C18-endcapped functionality (Pharmafluidics, Belgium) kept at a constant temperature of 50°C. Peptides were eluted by a linear gradient from 99% solvent A′ (0.1% formic acid in water) to 55% solvent B′ [0.1% formic acid in water/acetonitrile, 20/80 (v/v)] in 30 min at a flow rate of 300 nl/min, followed by a 5-min wash reaching 95% solvent B′.
The mass spectrometer was operated in data-dependent, positive-ionization mode, automatically switching between MS and MS/MS acquisition for the five most abundant peaks in a given MS spectrum. The source voltage was 3.5 kV, and the capillary temperature was 275°C. One MS1 scan (m/z 400 to 2000; automatic gain control (AGC) target, 3 × 106 ions; maximum ion injection time, 80 ms), acquired at a resolution of 70,000 (at 200 m/z), was followed by up to five tandem MS scans (resolution, 17,500 at 200 m/z) of the most intense ions fulfilling predefined selection criteria (AGC target, 5 × 104 ions; maximum ion injection time, 80 ms; isolation window, 2 Da; fixed first mass, 140 m/z; spectrum data type, centroid; intensity threshold, 1.3 × 104; exclusion of unassigned, 1, 5 to 8, and >8 positively charged precursors; peptide match, preferred; exclude isotopes, on; dynamic exclusion time, 12 s). The HCD (high-energy collisional dissociation) collision energy was set to 25% normalized collision energy, and the polydimethylcyclosiloxane background ion at 445.120025 Da was used for internal calibration (lock mass).
For the determination of proteins in gel slices, the raw data were searched with MaxQuant (version 1.6.4.0) using standard parameters (data file S1). The obtained identifications can be found in data file S8 (A to J).
To determine the significantly enriched proteins in bait samples versus control samples of the immunoprecipitation-MS/MS experiment, the MaxQuant proteingroups file (data file S5) was uploaded in Perseus software. Reverse, contaminant, and only identified by site identifications were removed, and samples were grouped by the respective triplicates and filtered for minimal two valid values per triplicate. Label-free quantification (LFQ) values were transformed to log2, and missing values were imputated from normal distribution using standard settings in Perseus, width of 0.3 and downshift of 1.8. Next, analysis of variance (ANOVA) [false discovery rate (FDR) = 0.05; S0 = 1] was performed on the logged LFQ values, followed by a post hoc Tukey test (FDR = 0.05; data file S5A). For visualization, a hierarchical clustered heatmap was created in Perseus. For visualization as volcano plots (fig. S5G), t tests were performed using the logged LFQ values for each bait versus control. The significantly different proteins between bait and control were determined using permutation-based FDR. As cutoff, FDR = 0.05; S0 = 1 was applied. Lists of the significantly enriched proteins with each of the baits can be found in data files S5 (B to D).
Protein production and purification
TML μHD was cloned into pDEST15 (Gateway). BL21(DE3) cells transformed with the construct were grown at 37°C until OD600 ~0.6 and induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown further at 37°C for 3 hours. Cells were harvested and resuspended in extraction buffer [150 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 1 mM DTT]. The protein was bound on the glutathione sepharose (GE Healthcare) matrix and eluted with extraction buffer, supplemented with 10 mM glutathione.
TPLATE anchor (1062 to 1177) was cloned into the in-house generated pET22b-6xHis-TEV. BL21(DE3) cells transformed with the construct were grown at 37°C until OD600 ~0.4 to 0.6 and induced with 0.4 mM IPTG and grown further at 18°C overnight. Cells were harvested and resuspended in extraction buffer 2 [20 mM Hepes (pH 7.4), 150 mM NaCl, and 1 mM tris-(2-carboxyethyl)fosfine (TCEP)]. The protein was captured on the 5-ml HisTrap HP column (GE Healthcare) and eluted in extraction buffer containing 150 mM imidazole. The protein was further separated from other impurities by strong anion exchange chromatography using a self-packed Source 15Q column. The 6xHis tag was removed by incubating the protein with Tobacco Etch Virus (TEV) protease in a 1:50 (TEV:protease) ratio. After removing TEV by reverse immobilized metal affinity chromatography, the protein was cleaned up using a Superdex 75 Increase 10/300 GL column (GE Healthcare).
Multiangle laser light scattering
Purified His-tagged proteins anchor domain (2.1 mg/ml) was injected onto a Superdex 75 Increase 10/300 GL size exclusion column (GE Healthcare) and equilibrated with 20 mM Hepes (pH 7.4), 150 mM NaCl, and 1 mM TCEP coupled to an online ultraviolet detector (Shimadzu), a mini DAWN TREOS (Wyatt) multiangle laser light scattering detector, and an Optilab T-rEX refractometer (Wyatt) at room temperature. A refractive index increment (dn/dc) value of 0.185 ml/g was used. Band broadening corrections were applied using parameters derived from ribonuclease injected under identical running conditions. Data analysis was carried out using the ASTRA6.1 software.
Circular dichroism
The TPLATE anchor domain was buffer exchanged to PBS using size exclusion chromatography. The protein samples were subsequently spun down for 15 min at 16,200g and degassed for 10 min. Far-ultraviolet CD spectra were recorded using a Jasco J-715 spectropolarimeter (Tokyo, Japan). CD spectra were measured between 200 and 260 nm, using a scan rate of 50 nm/min, a bandwidth of 1.0 nm, and a resolution of 0.5 nm. Six accumulations were taken with samples of TPLATE anchor at 0.2 and 0.4 mg/ml in a 0.1-cm cuvette. The mean residue ellipticity ([θ] in deg·cm2 dmol−1) was calculated from the raw CD data by normalizing for the protein concentration and the number of residues using the following: [θ] = θ × MMn × C × l, with MM, n, C, and l the molecular weight (Da), the number of amino acids, the protein concentration (mg/ml), and the length of the cuvette (cm), respectively. The secondary structure content was estimated using BeStSel (61).
Lipid-binding experiments
For the liposome-binding experiments, either protein-lipid overlay was used according to the manufacturer’s instructions (Echelon Biosciences) or a vesicle cosedimentation assay was used as described in (62).
Statistical analysis
For statistical analysis, the R package in R studio was used. Data were tested for normality and heteroscedasticity, after which the multcomp package was used (63).
Plant material
Transgenic lines expressing truncation constructs of TPLATE are listed in table S3. All the plants are in the Col-0 ecotype. The tplate heterozygous mutant plants, confirmed by genotyping PCR, were transformed by floral dip with various expression constructs of TPLATE substitution motifs fused to GFP under the control of the pLAT52 promoter, similar to the original complementation approach (29). Primary transformants (T1) were selected on 1/2 MS plates supplemented with Basta (10 mg/liter) and selected by genotyping PCR to identify transgenic plants containing the tplate T-DNA insertion. Genotyping PCRs were performed again on T2 transgenic plants expressing TPLATE-SWM or TPLATE-PFM mutations to identify homozygous tplate mutants. Genotyping PCR was performed with genomic DNA extracted from rosette leaves. Genotyping LP (left primer) and RP (right primer) primers for tplate are described before (32), and the primer LBb 1.3 provided by SIGnAL website was used for the T-DNA–specific primer. To obtain dual-marker lines, TPLATE-SWM– or TPLATE-PFM–expressing plants were crossed with 35S::DRP1A-mRFP–expressing plants (64), respectively. Crossed F1 plants were used for imaging.
Visualization of protein structures and data
For the visualization of all protein structures, UCSF Chimera (65) and UCSF ChimeraX (66) were used. Molecular dynamics simulations were visualized with the Visual Molecular Dynamics (VMD) program (67). Cross-linking datasets were visualized by xVis (68). All figures were prepared with the Inkscape program (https://inkscape.org/).
Acknowledgments
We would like to thank the proteomics core facility of VIB for the help and expertise with running all MS experiments. We also express our gratitude to developers of the Linux operating system and open-source programs used in this study, particularly IMP, UCSF Chimera and UCSF ChimeraX, Inkscape, ImageJ, GROMACS, VMD, Gimp, Gnumeric, xVIS, and Rstudio. Funding: This research in the D.V.D. laboratory is supported by the European Research Council T-Rex project number 682436 and by the National Science Foundation Flanders (FWO; G009415N and 3G017919). M.P. is supported by the Czech Science Foundation grant GA19-21758S. J.Wa. was supported by the China Scholarship Council (grant no. 201508440249 to J.W.) and by Ghent University BOF CSC co-funding (grant no. ST01511051). Author contributions: R.P., K.Y., and D.V.D. conceived and planned the experiments. R.P. and K.Y. performed most of the experiments and wrote the paper together with J.Wa. and D.V.D. and with contribution from all coauthors. R.P. and K.Y. performed TAP purification and XL-MS, biochemically characterized TML μHD, and performed live-cell imaging. R.P. performed protein-lipid overlay assays, liposome-binding assays, integrative structural modeling, and molecular dynamics simulations. J.Wa. designed and performed complementation assays and did live-cell imaging. D.E. performed MS analysis. J.Wi. performed the autophagosomal recruitment assay and knocksideway assay. L.D.V. expressed and characterized TPLATE anchor and performed the protein-lipid overlay assay. M.V. generated estradiol-inducible lines and performed Y2H and Y3H. P.G. performed coimmunoprecipitation with the TPLATE truncation constructs. Ev.M. and D.V.D. did live-cell imaging. R.P. and M.K. created comparative models. R.M. helped with TPLATE anchor characterization. J.N., El.M., and B.D.R. performed coimmunoprecipitation and MS analysis of the TPLATE appendage mutants. L.D.V., P.D.B., and R.L. performed and interpreted the CD assay. M.P. helped in designing and performing liposome-binding assays. S.N.S., B.D.R., G.D.J., D.V.D., and R.P. were responsible for experimental design, research supervision, and finalizing the manuscript text. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All data files related to sequence alignments, IMP, and CG-MD are deposited in the Zenodo repository: 10.5281/zenodo.3979550. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD023022 and PXD023051. The IMP structure is deposited in PDB-dev with accession number PDBDEV_00000065.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabe7999/DC1
REFERENCES AND NOTES
- 1.Dacks J. B., Field M. C., Evolution of the eukaryotic membrane-trafficking system: Origin, tempo and mode. J. Cell Sci. 120, 2977–2985 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Field M. C., Sali A., Rout M. P., Evolution: On a bender–BARs, ESCRTs, COPs, and finally getting your coat. J. Cell Biol. 193, 963–972 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rout M. P., Field M. C., The evolution of organellar coat complexes and organization of the eukaryotic cell. Annu. Rev. Biochem. 86, 637–657 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Béthune J., Wieland F. T., Assembly of COPI and COPII vesicular coat proteins on membranes. Annu. Rev. Biophys. 47, 63–83 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Beacham G. M., Partlow E. A., Hollopeter G., Conformational regulation of AP1 and AP2 clathrin adaptor complexes. Traffic 20, 741–751 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirst J., Schlacht A., Norcott J. P., Traynor D., Bloomfield G., Antrobus R., Kay R. R., Dacks J. B., Robinson M. S., Characterization of TSET, an ancient and widespread membrane trafficking complex. eLife 3, e02866 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.More K., Klinger C. M., Barlow L. D., Dacks J. B., Evolution and natural history of membrane trafficking in eukaryotes. Curr. Biol. 30, R553–R564 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Gadeyne A., Sánchez-Rodríguez C., Vanneste S., Di Rubbo S., Zauber H., Vanneste K., Van Leene J., De Winne N., Eeckhout D., Persiau G., Van De Slijke E., Cannoot B., Vercruysse L., Mayers J. R., Adamowski M., Kania U., Ehrlich M., Schweighofer A., Ketelaar T., Maere S., Bednarek S. Y., Friml J., Gevaert K., Witters E., Russinova E., Persson S., De Jaeger G., Van Damme D., The TPLATE adaptor complex drives clathrin-mediated endocytosis in plants. Cell 156, 691–704 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Dodonova S. O., Diestelkoetter-Bachert P., von Appen A., Hagen W. J. H., Beck R., Beck M., Wieland F., Briggs J. A. G., VESICULAR TRANSPORT. A structure of the COPI coat and the role of coat proteins in membrane vesicle assembly. Science 349, 195–198 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Collins B. M., McCoy A. J., Kent H. M., Evans P. R., Owen D. J., Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523–535 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Ren X., Farías G. G., Canagarajah B. J., Bonifacino J. S., Hurley J. H., Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755–767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yperman K., Papageorgiou A. C., Merceron R., De Munck S., Bloch Y., Eeckhout D., Tack P., Evangelidis T., Van Leene J., Vincze L., Vandenabeele P., Potocký M., De Jaeger G., Savvides S. N., Tripsianes K., Pleskot R., Van Damme D., Distinct EH domains of the endocytic TPLATE complex confer lipid and protein binding. bioRxiv 2020.05.29.122911, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sánchez-Rodríguez C., Shi Y., Kesten C., Zhang D., Sancho-Andrés G., Ivakov A., Lampugnani E. R., Sklodowski K., Fujimoto M., Nakano A., Bacic A., Wallace I. S., Ueda T., Van Damme D., Zhou Y., Persson S., The cellulose synthases are cargo of the TPLATE adaptor complex. Mol. Plant 11, 346–349 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Wang P., Pleskot R., Zang J., Winkler J., Wang J., Yperman K., Zhang T., Wang K., Gong J., Guan Y., Richardson C., Duckney P., Vandorpe M., Mylle E., Fiserova J., van Damme D., Hussey P. J., Plant AtEH/Pan1 proteins drive autophagosome formation at ER-PM contact sites with actin and endocytic machinery. Nat. Commun. 10, 5132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arora D., Abel N. B., Liu C., van Damme P., Yperman K., Eeckhout D., Vu L. D., Wang J., Tornkvist A., Impens F., Korbei B., Van Leene J., Goossens A., De Jaeger G., Ott T., Moschou P. N., Van Damme D., Establishment of proximity-dependent biotinylation approaches in different plant model systems. Plant Cell 32, 3388–3407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platre M. P., Noack L. C., Doumane M., Bayle V., Simon M. L. A., Maneta-Peyret L., Fouillen L., Stanislas T., Armengot L., Pejchar P., Caillaud M. C., Potocký M., Čopič A., Moreau P., Jaillais Y., A combinatorial lipid code shapes the electrostatic landscape of plant endomembranes. Dev. Cell 45, 465–480.e11 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Rout M. P., Sali A., Principles for integrative structural biology studies. Cell 177, 1384–1403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russel D., Lasker K., Webb B., Velázquez-Muriel J., Tjioe E., Schneidman-Duhovny D., Peterson B., Sali A., Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLOS Biol. 10, e1001244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozakov D., Hall D. R., Xia B., Porter K. A., Padhorny D., Yueh C., Beglov D., Vajda S., The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viswanath S., Chemmama I. E., Cimermancic P., Sali A., Assessing exhaustiveness of stochastic sampling for integrative modeling of macromolecular structures. Biophys. J. 113, 2344–2353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winkler J., Mylle E., De Meyer A., Pavie B., Merchie J., Grones P., Van Damme D., Visualizing protein-protein interactions in plants by rapamycin-dependent delocalization. Plant Cell 33, (in press) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J., Mylle E., Johnson A., Besbrugge N., De Jaeger G., Friml J., Pleskot R., Van Damme D., High temporal resolution reveals simultaneous plasma membrane recruitment of TPLATE complex subunits. Plant Physiol. 183, 986–997 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henne W. M., Boucrot E., Meinecke M., Evergren E., Vallis Y., Mittal R., McMahon H. T., FCHo proteins are nucleators of clathrin-mediated endocytosis. Science 328, 1281–1284 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reider A., Barker S. L., Mishra S. K., Im Y. J., Maldonado-Báez L., Hurley J. H., Traub L. M., Wendland B., Syp1 is a conserved endocytic adaptor that contains domains involved in cargo selection and membrane tubulation. EMBO J. 28, 3103–3116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson L. P., Lewis M., Kent H. M., Edeling M. A., Evans P. R., Duden R., Owen D. J., Molecular basis for recognition of dilysine trafficking motifs by COPI. Dev. Cell 23, 1255–1262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto E., Domański J., Naughton F. B., Best R. B., Kalli A. C., Stansfeld P. J., Sansom M. S. P., Multiple lipid binding sites determine the affinity of PH domains for phosphoinositide-containing membranes. Sci. Adv. 6, eaay5736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furt F., König S., Bessoule J.-J., Sargueil F., Zallot R., Stanislas T., Noirot E., Lherminier J., Simon-Plas F., Heilmann I., Mongrand S., Polyphosphoinositides are enriched in plant membrane rafts and form microdomains in the plasma membrane. Plant Physiol. 152, 2173–2187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon M. L. A., Platre M. P., Marquès-Bueno M. M., Armengot L., Stanislas T., Bayle V., Caillaud M.-C., Jaillais Y., A PtdIns(4)P-driven electrostatic field controls cell membrane identity and signalling in plants. Nat. Plants 2, 16089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Damme D., Coutuer S., De Rycke R., Bouget F.-Y., Inzé D., Geelen D., Somatic cytokinesis and pollen maturation in Arabidopsis depend on TPLATE, which has domains similar to coat proteins. Plant Cell 18, 3502–3518 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gronnier J., Crowet J.-M., Habenstein B., Nasir M. N., Bayle V., Hosy E., Platre M. P., Gouguet P., Raffaele S., Martinez D., Grelard A., Loquet A., Simon-Plas F., Gerbeau-Pissot P., Der C., Bayer E. M., Jaillais Y., Deleu M., Germain V., Lins L., Mongrand S., Structural basis for plant plasma membrane protein dynamics and organization into functional nanodomains. eLife 6, e26404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodonova S. O., Aderhold P., Kopp J., Ganeva I., Röhling S., Hagen W. J. H., Sinning I., Wieland F., Briggs J. A. G., 9Å structure of the COPI coat reveals that the Arf1 GTPase occupies two contrasting molecular environments. eLife 6, e26691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kovtun O., Dickson V. K., Kelly B. T., Owen D. J., Briggs J. A. G., Architecture of the AP2/clathrin coat on the membranes of clathrin-coated vesicles. Sci. Adv. 6, eaba8381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paraan M., Mendez J., Sharum S., Kurtin D., He H., Stagg S. M., The structures of natively assembled clathrin-coated vesicles. Sci. Adv. 6, eaba8397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma L., Umasankar P. K., Wrobel A. G., Lymar A., McCoy A. J., Holkar S. S., Jha A., Pradhan-Sundd T., Watkins S. C., Owen D. J., Traub L. M., Transient Fcho1/2·Eps15/R·AP-2 nanoclusters prime the AP-2 clathrin adaptor for cargo binding. Dev. Cell 37, 428–443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Damme D., Bouget F.-Y., Van Poucke K., Inzé D., Geelen D., Molecular dissection of plant cytokinesis and phragmoplast structure: A survey of GFP-tagged proteins. Plant J. 40, 386–398 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Karimi M., Bleys A., Vanderhaeghen R., Hilson P., Building blocks for plant gene assembly. Plant Physiol. 145, 1183–1191 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Leene J., Eeckhout D., Cannoot B., De Winne N., Persiau G., Van De Slijke E., Vercruysse L., Dedecker M., Verkest A., Vandepoele K., Martens L., Witters E., Gevaert K., De Jaeger G., An improved toolbox to unravel the plant cellular machinery by tandem affinity purification of Arabidopsis protein complexes. Nat. Protoc. 10, 169–187 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Van Leene J., Han C., Gadeyne A., Eeckhout D., Matthijs C., Cannoot B., De Winne N., Persiau G., Van De Slijke E., Van de Cotte B., Stes E., Van Bel M., Storme V., Impens F., Gevaert K., Vandepoele K., De Smet I., De Jaeger G., Capturing the phosphorylation and protein interaction landscape of the plant TOR kinase. Nat. Plants 5, 316–327 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Chen Z.-L., Meng J.-M., Cao Y., Yin J.-L., Fang R.-Q., Fan S.-B., Liu C., Zeng W.-F., Ding Y.-H., Tan D., Wu L., Zhou W.-J., Chi H., Sun R.-X., Dong M.-Q., He S.-M., A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 10, 3404 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J., Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katoh K., Rozewicki J., Yamada K. D., MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., Barton G. J., Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Šali A., Blundell T. L., Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Zimmermann L., Stephens A., Nam S.-Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A. N., Alva V., A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Källberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., Xu J., Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buchan D. W. A., Jones D. T., The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 47, W402–W407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bryson K., Cozzetto D., Jones D. T., Computer-assisted protein domain boundary prediction using the DomPred server. Curr. Protein Pept. Sci. 8, 181–188 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Jones D. T., Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Jones D. T., Cozzetto D., DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Algret R., Fernandez-Martinez J., Shi Y., Kim S. J., Pellarin R., Cimermancic P., Cochet E., Sali A., Chait B. T., Rout M. P., Dokudovskaya S., Molecular architecture and function of the SEA Complex, a modulator of the TORC1 pathway. Mol. Cell. Proteomics 13, 2855–2870 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sparkes I. A., Runions J., Kearns A., Hawes C., Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1, 2019–2025 (2006). [DOI] [PubMed] [Google Scholar]
- 53.de Jong D. H., Singh G., Bennett W. F. D., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P., Marrink S. J., Improved parameters for the Martini coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., Marrink S.-J., The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 4, 819–834 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H., The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Periole X., Cavalli M., Marrink S.-J., Ceruso M. A., Combining an elastic network with a coarse-grained molecular force field: Structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 5, 2531–2543 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Ingólfsson H. I., Melo M. N., van Eerden F. J., Arnarez C., Lopez C. A., Wassenaar T. A., Periole X., de Vries A. H., Tieleman D. P., Marrink S. J., Lipid organization of the plasma membrane. J. Am. Chem. Soc. 136, 14554–14559 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Hsu P.-C., Bruininks B. M. H., Jefferies D., de Souza P. C. T., Lee J., Patel D. S., Marrink S. J., Qi Y., Khalid S., Im W., CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem. 38, 2354–2363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 60.De Rybel B., Möller B., Yoshida S., Grabowicz I., Barbier de Reuille P., Boeren S., Smith R. S., Borst J. W., Weijers D., A bHLH complex controls embryonic vascular tissue establishment and indeterminate growth in Arabidopsis. Dev. Cell 24, 426–437 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Micsonai A., Wien F., Bulyáki É., Kun J., Moussong É., Lee Y.-H., Goto Y., Réfrégiers M., Kardos J., BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 46, W315–W322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kooijman E. E., Tieleman D. P., Testerink C., Munnik T., Rijkers D. T. S., Burger K. N. J., de Kruijff B., An electrostatic/hydrogen bond switch as the basis for the specific interaction of phosphatidic acid with proteins. J. Biol. Chem. 282, 11356–11364 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Herberich E., Sikorski J., Hothorn T., A robust procedure for comparing multiple means under heteroscedasticity in unbalanced designs. PLOS ONE 5, e9788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mravec J., Petrášek J., Li N., Boeren S., Karlova R., Kitakura S., Pařezová M., Naramoto S., Nodzyński T., Dhonukshe P., Bednarek S. Y., Zažímalová E., de Vries S., Friml J., Cell plate restricted association of DRP1A and PIN proteins is required for cell polarity establishment in Arabidopsis. Curr. Biol. 21, 1055–1060 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 68.Grimm M., Zimniak T., Kahraman A., Herzog F., xVis: A web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res. 43, W362–W369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bashline L., Li S., Zhu X., Gu Y., The TWD40-2 protein and the AP2 complex cooperate in the clathrin-mediated endocytosis of cellulose synthase to regulate cellulose biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 112, 12870–12875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabe7999/DC1