

Earth-abundant manganese catalyst enabled the expedient synthesis of cyclic peptides via activation of inert C─H bonds.
Abstract
Bioorthogonal late-stage diversification of structurally complex peptides bears enormous potential for drug discovery and molecular imaging. Despite major accomplishments, these strategies heavily rely on noble-metal catalysis. Herein, we report on a manganese(I)-catalyzed peptide C─H hydroarylation that enabled the stitching of peptidic and sugar fragments, under exceedingly mild and racemization-free conditions. This convergent approach represents an atom-economical alternative to traditional iterative peptide synthesis. The robustness of the manganese(I) catalysis regime is reflected by the full tolerance of a plethora of sensitive functional groups. Our strategy enabled an expedient access to challenging cyclic peptides by a modular late-stage macrocyclization of structurally complex peptides.
INTRODUCTION
The late-stage diversification of biomolecules is of prime importance in biomolecular chemistry with immediate impact on academia and pharmaceutical industries (1). In this regard, nonnatural amino acids bear substantial potential, as they can modify the conformation of peptides, improve bioactivities, and diminish the hindrance from proteolytic degradation of native peptides (2). This holds particularly true for cyclic peptides, as they generally lack free N- and C-terminal residues that are essential for recognition by most proteolytic enzymes (3, 4). Significant recent momentum was gained through transformative palladium-catalyzed cross-coupling of peptides (5–7). Despite major advances, this approach requires two pre-functionalized substrates, leading to multistep syntheses. As a consequence, late-stage diversification of amino acids and peptides (8, 9) via noble metal-catalyzed C─H activation was developed by Lavilla/Albericio (10, 11), Chen (12–14), Ackermann (15–18), and Yu (19–22), among others (23–27). While this regime enabled the assembly of challenging cyclic peptides, toxic and costly palladium catalysts were required, which, among others, prove detrimental because of their costs and trace metal impurities (28–32). In contrast, 3d transition metal-catalyzed C─H activation has gained considerable recent attention, with major advances by Earth-abundant, nontoxic manganese (33, 34). Thus, manganese-catalyzed C─H activation has proved instrumental for efficient and selective C─H functionalization (35, 36). In sharp contrast, base metal catalysis for peptide diversification and macrocyclization continues to be scarce (37–40). As part of our program on sustainable C─H activation (41, 42), we now report on the first manganese(I)-catalyzed C─H activation for hydroarylations of structurally complex peptides with easily accessible propiolates. Notable features of our findings include (i) racemization-free hydroarylation of peptides by user-friendly manganese catalysis, (ii) facile synthesis of ligated and hybrid peptides, and (iii) an unprecedented strategy for the assembly of cyclic peptides embedded with an electrophilic α,β-unsaturated moiety (Fig. 1).
Fig. 1. Late-stage stitching and macrocyclization toward molecular complexity.

A versatile approach toward site-selective peptide modifications via a ligation and macrocyclization of complex precursors, catalyzed by Earth-abundant manganese.
RESULTS
We commenced our studies by probing various reaction conditions for the envisioned hydroarylation of propiolate 2a with tryptophan derivative 1a (Table 1). Among a representative set of solvents, DME (1,2-dimethoxyethane) and 1,4-dioxane were identified as being optimal (entries 3 and 4). Gratifyingly, lowering the reaction temperature did not diminish the catalyst’s efficacy (entries 7 and 8). Control experiments clearly demonstrated the importance of NaOAc, likely enabling carboxylate-assisted C─H cleavage (42–44), and the essential nature of the MnBr(CO)5 catalyst and the 2-pyridyl directing group (entries 10 to 12). Notably, the peptide backbone was not essential for the C─H activation, because 1-(pyridin-2-yl)-1H-indole was efficiently converted under otherwise identical reaction conditions (entry 13). Changing the propiolate 2a to the more complex serine-derived propiolate 2b called for an elevated reaction temperature to ensure full conversion (entry 16). Notably, the reaction proceeded without erosion of the enantiointegrity of the tryptophan scaffold.
Table 1. Optimization studies for the hydroarylation with tryptophan 1a.
Exploration of solvent, additive, and temperature effects on the hydroarylation of alkynes with tryptophan derivative 1a.

| Entry | R | Solvent | Temperature (°C) | Yield (%) |
| 1 | R1 | toluene | 100 | 37 |
| 2 | R1 | DCE | 100 | 83 |
| 3 | R1 | DME | 100 | 97 |
| 4 | R1 | 1,4-dioxane | 100 | 98 |
| 5 | R1 | DMF | 100 | – |
| 6 | R1 | H2O | 100 | 43 |
| 7 | R1 | 1,4-dioxane | 80 | 97 |
| 8 | R1 | 1,4-dioxane | 60 | 97 |
| 9 | R1 | 1,4-dioxane | 40 | 58 |
| 10 | R1 | 1,4-dioxane | 60 | 85* |
| 11 | R1 | 1,4-dioxane | 60 | –† |
| 12 | R1 | 1,4-dioxane | 60 | –‡ |
| 13 | R1 | 1,4-dioxane | 60 | 97§ |
| 14 | R2 | 1,4-dioxane | 60 | 86 |
| 15 | R2 | 1,4-dioxane | 80 | 90 |
| 16 | R2 | 1,4-dioxane | 100 | 95 |
Reaction conditions: 1a (0.15 mmol), 2 (0.23 mmol), MnBr(CO)5 (10 mol %), NaOAc (30 mol %), solvent (0.6 ml), 16 hours. Yields of isolated product.
*Without NaOAc.
†Without MnBr(CO)5.
‡Boc-Trp-OMe (0.15 mmol) in lieu of 1a.
§1-(pyridin-2-yl)-1H-indole (0.15 mmol) in lieu of 1a.
With the optimal reaction conditions in hand, we envisioned a straightforward approach for the synthesis of peptides via manganese(I)-catalyzed C─H activation for the stitching of peptidic fragments. This approach would offer a convergent ligation toward large peptides in sharp contrast to tedious traditional iterative peptide syntheses. Thus, a plethora of di- and tripeptides were efficiently synthesized in an atom-economical manner under exceedingly mild reaction conditions that did not jeopardize the tolerance of sensitive functional groups on the side chain of amino acids (Fig. 2). In addition, under the optimized reaction conditions, internal alkynes 2e and 2f proved viable substrates leading to tryptophan derivatives bearing trisubstituted olefins 3e and 3f. Furthermore, nitrogen-containing amino acids, such as tryptophan 3d and protected arginine 3n, were also well tolerated, albeit in the latter case with diminished E/Z ratio. In addition, aliphatic alcohols and phenols, found in serine, threonine, and tyrosine, were fully tolerated, selectively furnishing the desired tripeptides 3j, 3k, and 3l. Strongly coordinating and oxidation-prone thioether, present in methionine, did not affect the robustness of the manganese(I) catalysis, as tripeptide 3m was obtained in 88% yield. Likewise, diketopiperazine derivatives, found in a plethora of natural products, 1o and 1p were efficiently converted to the desired tripeptides 3o and 3p in good yields.
Fig. 2. Small peptide assembly by manganese catalysis.

(A) Synthesis of dipeptides via a C─H peptidic coupling. (B) Disubstituted alkynes 2e and 2f as coupling partner. (C) Synthesis of tripeptides via a C─H peptidic coupling featuring various functional groups. (D) Ligation of brevianamide F and fellutanine A analogs.
Encouraged by the outstanding versatility of the manganese(I) catalysis manifold, we investigated whether more complex peptides could be viable substrates. Thus, tetra- and pentapeptides were readily accessed in excellent yields, showcasing the translational impact such method could have to biological sciences (Fig. 3). Thus, under mild and racemization-free reaction conditions, complex peptides containing tryptophan, tyrosine, protected serine, threonine, and cysteine could be efficiently assembled. The ability to stitch fragments that bear two distinct functions is of utmost importance since new entities arise, featuring multifunctional molecules. Thus, hybrid peptides were selectively prepared bearing natural products including terpenes and sugars, as well as fluorescent tag coumarin in good to excellent yields.
Fig. 3. Synthesis of complex hydrid molecules via C─H hydroarylation.

(A) Synthesis of complex via a C─H coupling strategy. (B) C─H stitching of biomolecules toward hydrid architectures.
The robustness and mild nature of the manganese(I) catalysis regime is clearly reflected on the gram-scale synthesis of tryptophan derivatives bearing the valuable electrophilic α,β-unsaturated ester (Fig. 4A). Thereafter, we envisioned a strategy for accessing SH-free cysteine-containing peptides bearing the reactive olefin that could engage in complementary postsynthetic manipulation (Fig. 4B). To this end, cystine-containing peptide 6 was used in a twofold C─H hydroarylation, where the disulfide acts as atom-economical protecting group of the thiol moiety. Subsequent, disulfide reduction swiftly yielded the free cysteine-containing peptide. Furthermore, the dimeric peptide 6 is destined for sequential C─H activation process, governed by the judicious choice of the reaction conditions and stoichiometry. Thus, highly functionalized tetrapeptide 8 was obtained in 67% overall yield after a sequential C─H hydroarylation and C─H allylation.
Fig. 4. Synthetic applications of the C─H hydroarylation.

(A) Gram-scale synthesis. (B) Late-stage manipulation on the hydroarylated peptides.
Inspired by the remarkable versatility of our manganese(I)-catalyzed C─H activation, we probed whether the manganese(I) catalysis can be used for the synthesis of challenging cyclic peptides. To avoid detrimental di- and oligomerization, we used high dilution conditions. Gratifyingly, under these conditions, a plethora of cyclic peptides were thereby obtained with excellent chemoselectivity furnishing 15- to 22-membered macrocycles (Fig. 5A). To exploit the full potential of our manganese(I)-catalyzed macrocyclization, we performed the challenging macrocyclization using either a linker at C terminus (Trp→C terminus) or the native N terminus (Trp→N terminus) to attach the key propiolate motif. Moreover, we recognized that the native functional groups of the side chains of the amino acids can be used for the installation of the propiolate moiety. Hence, cyclic peptides 10m and 10n were selectively obtained in good to excellent yields (Trp→Ser). Moreover, the traceless removal of the N-pyridyl group was accomplished by a selective methylation/hydrogenation protocol giving rise to NH-free tryptophan-containing cyclic peptide 11 (Fig. 5B).
Fig. 5. Manganese-catalyzed macrocyclization and removal of the directing group.
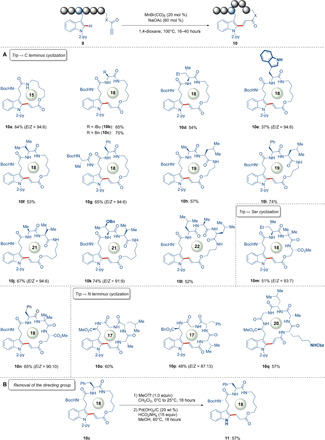
(A) Access to cyclic peptides of various ring sizes using the native C terminus, N terminus, and the serine side chain. (B) Selective methylation/hydrogenation protocol for the traceless removal of the directing group. wt %, weight %.
DISCUSSION
Last, the thus-obtained cyclic peptides showed considerable anticancer activities against HCT116 cells. Small changes in the peptidic backbone had profound effect on the biocativity, as showcased by the enhanced activity of 10h over 10j. Further demonstrating the translational nature of the late-stage C─H activation regime (Fig. 6).
Fig. 6. Anticancer activity of cyclic peptides 10j and 10h against HCT116 cells.

**P < 0.01 and ***P <0.001. ns, not significant. A.U., arbitrary units.
In summary, we have developed an unprecedented manganese(I)-catalyzed C─H hydroarylation with structurally complex peptides. The chemo- and position-selective peptide diversification was characterized by an excellent functional group tolerance with a plethora of sensitive groups under mild and epimerization-free reaction conditions. We also assembled hydrid multifunctional molecules by stitching peptides with natural products and sugars. This robust method paved the way for a highly efficient access to cyclic peptides via a C─H macrocyclization regime.
MATERIALS AND METHODS
General procedure A: Late-stage C─H hydroarylation on peptides
A suspension of peptide 1 or 6 (0.15 mmol, 1.0 equiv), propiolate 2 (0.23 mmol, 1.5 equiv), MnBr(CO)5 [10 mole percent (mol %)], and NaOAc (30 mol %) in 1,4-dioxane (0.25 M) was stirred at 100°C for 16 hours under N2. After cooling to ambient temperature, CH2Cl2 (10 ml) was added, and the mixture was concentrated in vacuo. Purification by column chromatography on silica gel afforded the desired products 3 to 8.
General procedure B: Late-stage C─H macrocyclization of peptides
A suspension of peptide 9 (0.15 mmol, 1.0 equiv), MnBr(CO)5 (20 mol %), and NaOAc (60 mol %) in 1,4-dioxane (30 ml) was stirred at 100°C for 16 hours under N2. After cooling to ambient temperature, CH2Cl2 (10 ml) was added, and the mixture was concentrated in vacuo. Purification by column chromatography on silica gel afforded the desired product 10.
CellTiter Blue assay (viability assay)
The assay was performed according to the manufacturer’s protocol (Promega, Madison, USA). Briefly, 1 × 104 HCT-116 cells were seeded into 96-well, black-walled tissue culture plates (Corning, NY, USA). After 24-hour incubation at 37°C, 5% CO2, the medium was replaced by 100 μl of medium containing the experimental compounds 10h and 10j. One hour before the selected time points (24 hours/48 hours/72 hours), 10 μl of resazurin was added to each well. Reduced resazurin (resorufin) was measured using the Victor Multilabel Plate Reader (PerkinElmer Inc., Waltham, USA) at 560-nm excitation/590-nm emission wavelength. Every condition was tested in triplicates, and the assay was performed three times. The combined data of three independent experiments are shown as ±SEM.
Acknowledgments
Funding: Generous support by the DFG (Gottfried-Wilhelm-Leibniz award to L.A. and SPP1807) and the Onassis Foundation (fellowship to N.K.) is gratefully acknowledged. Author contributions: Conceptualization: N.K. Methodology: N.K. Investigation: N.K., F.K., G.S., S.F., and T.D.O. Writing—original draft: N.K. Writing—review and editing: N.K., F.K., G.S., S.F., T.D.O., L.-C.C., and L.A. Funding acquisition: N.K. and L.A. Resources: L.-C.C. and L.A. Supervision: L.A. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabe6202/DC1
REFERENCES AND NOTES
- 1.Blakemore D. C., Castro L., Churcher I., Rees D. C., Thomas A. W., Wilson D. M., Wood A., Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Räder A. F. B., Weinmüller M., Reichart F., Schumacher-Klinger A., Merzbach S., Gilon C., Hoffman A., Kessler H., Orally active peptides: Is there a magic bullet? Angew. Chem. Int. Ed. 57, 14414–14438 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Smolyar I. V., Yudin A. K., Nenajdenko V. G., Heteroaryl rings in peptide macrocycles. Chem. Rev. 119, 10032–10240 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Itoh H., Inoue M., Comprehensive structure–activity relationship studies of macrocyclic natural products enabled by their total syntheses. Chem. Rev. 119, 10002–10031 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Dhanjee H. H., Saebi A., Buslov I., Loftis A. R., Buchwald S. L., Pentelute B. L., Protein–protein cross-coupling via palladium–protein oxidative addition complexes from cysteine residues. J. Am. Chem. Soc. 142, 9124–9129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leroux M., Vorherr T., Lewis I., Schaefer M., Koch G., Karaghiosoff K., Knochel P., Late-stage functionalization of peptides and cyclopeptides using organozinc reagents. Angew. Chem. Int. Ed. 58, 8231–8234 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Vinogradova E. V., Zhang C., Spokoyny A. M., Pentelute B. L., Buchwald S. L., Organometallic palladium reagents for cysteine bioconjugation. Nature 526, 687–691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang W., Lorion M. M., Shah J., Kapdi A. R., Ackermann L., Late-stage peptide diversification by position-selective C−H activation. Angew. Chem. Int. Ed. 57, 14700–14717 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Noisier A. F. M., Brimble M. A., C–H functionalization in the synthesis of amino acids and peptides. Chem. Rev. 114, 8775–8806 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Preciado S., Mendive-Tapia L., Albericio F., Lavilla R., Synthesis of C-2 arylated tryptophan amino acids and related compounds through palladium-catalyzed C–H activation. J. Org. Chem. 78, 8129–8135 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Ruiz-Rodríguez J., Albericio F., Lavilla R., Postsynthetic modification of peptides: Chemoselective C-arylation of tryptophan residues. Chem. A Eur. J. 16, 1124–1127 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Wang B., Nack W. A., He G., Zhang S.-Y., Chen G., Palladium-catalyzed trifluoroacetate-promoted mono-arylation of the β-methyl group of alanine at room temperature: Synthesis of β-arylated α-amino acids through sequential C–H functionalization. Chem. Sci. 5, 3952–3957 (2014). [Google Scholar]
- 13.Zhang S.-Y., Li Q., He G., Nack W. A., Chen G., Stereoselective synthesis of β-alkylated α-amino acids via palladium-catalyzed alkylation of unactivated methylene C(sp3)–H bonds with primary alkyl halides. J. Am. Chem. Soc. 135, 12135–12141 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Feng Y., Chen G., Total synthesis of celogentin C by stereoselective C–H activation. Angew. Chem. Int. Ed. 49, 958–961 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Wu J., Kaplaneris N., Ni S., Kaltenhäuser F., Ackermann L., Late-stage C(sp2)–H and C(sp3)–H glycosylation of C-aryl/alkyl glycopeptides: Mechanistic insights and fluorescence labeling. Chem. Sci. 11, 6521–6526 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schischko A., Kaplaneris N., Rogge T., Sirvinskaite G., Son J., Ackermann L., Late-stage peptide C–H alkylation for bioorthogonal C–H activation featuring solid phase peptide synthesis. Nat. Commun. 10, 3553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bauer M., Wang W., Lorion M. M., Dong C., Ackermann L., Internal peptide late-stage diversification: Peptide-isosteric triazoles for primary and secondary C(sp3)−H activation. Angew. Chem. Int. Ed. 57, 203–207 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Schischko A., Ren H., Kaplaneris N., Ackermann L., Bioorthogonal diversification of peptides through selective ruthenium(II)-catalyzed C–H activation. Angew. Chem. Int. Ed. 56, 1576–1580 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Liu T., Qiao J. X., Poss M. A., Yu J.-Q., Palladium(II)-catalyzed site-selective C(sp3)−H alkynylation of oligopeptides: A Linchpin approach for oligopeptide–drug conjugation. Angew. Chem. Int. Ed. 56, 10924–10927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G., Shigenari T., Jain P., Zhang Z., Jin Z., He J., Li S., Mapelli C., Miller M. M., Poss M. A., Scola P. M., Yeung K.-S., Yu J.-Q., Ligand-enabled β-C–H arylation of α-amino acids using a simple and practical auxiliary. J. Am. Chem. Soc. 137, 3338–3351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He J., Li S., Deng Y., Fu H., Laforteza B. N., Spangler J. E., Homs A., Yu J.-Q., Ligand-controlled C(sp3)–H arylation and olefination in synthesis of unnatural chiral α–amino acids. Science 343, 1216–1220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong W., Zhang G., Liu T., Giri R., Yu J.-Q., Site-selective C(sp3)–H functionalization of Di-, Tri-, and tetrapeptides at the N-terminus. J. Am. Chem. Soc. 136, 16940–16946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terrey M. J., Holmes A., Perry C. C., Cross W. B., C–H olefination of tryptophan residues in peptides: Control of residue selectivity and peptide–amino acid cross-linking. Org. Lett. 21, 7902–7907 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Zhan B.-B., Li Y., Xu J.-W., Nie X.-L., Fan J., Jin L., Shi B.-F., Site-selective δ-C(sp3)−H alkylation of amino acids and peptides with maleimides via a six-membered palladacycle. Angew. Chem. Int. Ed. 57, 5858–5862 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Xu J.-W., Zhang Z.-Z., Rao W.-H., Shi B.-F., Site-selective alkenylation of δ-C(sp3)–H bonds with alkynes via a six-membered palladacycle. J. Am. Chem. Soc. 138, 10750–10753 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Tran L. D., Daugulis O., Nonnatural amino acid synthesis by using carbon–hydrogen bond functionalization methodology. Angew. Chem. Int. Ed. 51, 5188–5191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy B. V. S., Reddy L. R., Corey E. J., Novel acetoxylation and C−C coupling reactions at unactivated positions in α-amino acid derivatives. Org. Lett. 8, 3391–3394 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Bai Z., Cai C., Sheng W., Ren Y., Wang H., Late-stage peptide macrocyclization by palladium-catalyzed site-selective C−H olefination of tryptophan. Angw. Chem. Int. Ed. 59, 14686–14692 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Li B., Li X., Han B., Chen Z., Zhang X., He G., Chen G., Construction of natural-product-like cyclophane-braced peptide macrocycles via sp3 C–H arylation. J. Am. Chem. Soc. 141, 9401–9407 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Zhang X., Lu G., Sun M., Mahankali M., Ma Y., Zhang M., Hua W., Hu Y., Wang Q., Chen J., He G., Qi X., Shen W., Liu P., Chen G., A general strategy for synthesis of cyclophane-braced peptide macrocycles via palladium-catalysed intramolecular sp3 C−H arylation. Nat. Chem. 10, 540–548 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Tang J., Chen H., He Y., Sheng W., Bai Q., Wang H., Peptide-guided functionalization and macrocyclization of bioactive peptidosulfonamides by Pd(II)-catalyzed late-stage C–H activation. Nat. Commun. 9, 3383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendive-Tapia L., Preciado S., García J., Ramón R., Kielland N., Albericio F., Lavilla R., New peptide architectures through C–H activation stapling between tryptophan–phenylalanine/tyrosine residues. Nat. Commun. 6, 7160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loup J., Dhawa U., Pesciaioli F., Wencel-Delord J., Ackermann L., Enantioselective C−H activation with earth-abundant 3d transition metals. Angew. Chem. Int. Ed. 58, 12803–12818 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., 3d transition metals for C–H activation. Chem. Rev. 119, 2192–2452 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Hu Y., Zhou B., Wang C., Inert C–H bond transformations enabled by organometallic manganese catalysis. Acc. Chem. Res. 51, 816–827 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Liu W., Ackermann L., Manganese-catalyzed C–H activation. ACS Catal. 6, 3743–3752 (2016). [Google Scholar]
- 37.Scamp R. J., deRamon E., Paulson E. K., Miller S. J., Ellman J. A., Cobalt(III)-catalyzed C−H amidation of dehydroalanine for the site-selective structural diversification of thiostrepton. Angew. Chem. Int. Ed. 59, 890–895 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friis S. D., Johansson M. J., Ackermann L., Cobalt-catalysed C–H methylation for late-stage drug diversification. Nat. Chem. 12, 511–519 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Kaplaneris N., Rogge T., Yin R., Wang H., Sirvinskaite G., Ackermann L., Late-stage diversification through manganese-catalyzed C−H activation: Access to acyclic, hybrid, and stapled peptides. Angew. Chem. Int. Ed. 58, 3476–3480 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Ruan Z., Sauermann N., Manoni E., Ackermann L., Manganese-catalyzed C−H alkynylation: Expedient peptide synthesis and modification. Angew. Chem. Int. Ed. 56, 3172–3176 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Ackermann L., Metalla-electrocatalyzed C–H activation by earth-abundant 3d metals and beyond. Acc. Chem. Res. 53, 84–104 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Ackermann L., Carboxylate-assisted ruthenium-catalyzed alkyne annulations by C–H/Het–H bond functionalizations. Acc. Chem. Res. 47, 281–295 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Wang C., Rueping M., Rhenium- and manganese-catalyzed selective alkenylation of indoles. ChemCatChem 10, 2681–2685 (2018). [Google Scholar]
- 44.Liu W., Richter S., Zhang Y., Ackermann L., Manganese(I)-catalyzed substitutive C−H allylation. Angew. Chem. Int. Ed. 55, 7747–7750 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Liang L., Fu S., Lin D., Zhang X.-Q., Deng Y., Jiang H., Zeng W., Ruthenium(II)-catalyzed direct addition of indole/pyrrole C2–H bonds to alkynes. J. Org. Chem. 79, 9472–9480 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabe6202/DC1