

Microglial activity is needed to halt nascent vascular calcification.
Abstract
Microglia participate in central nervous system (CNS) development and homeostasis and are often implicated in modulating disease processes. However, less is known about the role of microglia in the biology of the neurovascular unit (NVU). In particular, data are scant on whether microglia are involved in CNS vascular pathology. In this study, we use a mouse model of primary familial brain calcification, Pdgfbret/ret, to investigate the role of microglia in calcification of the NVU. We report that microglia enclosing vessel calcifications, coined calcification-associated microglia, display a distinct activation phenotype. Pharmacological ablation of microglia with the CSF1R inhibitor PLX5622 leads to aggravated vessel calcification. Mechanistically, we show that microglia require functional TREM2 for controlling vascular calcification. Our results demonstrate that microglial activity in the setting of pathological vascular calcification is beneficial. In addition, we identify a previously unrecognized function of microglia in halting the expansion of vascular calcification.
INTRODUCTION
Microglia participate in central nervous system (CNS) development and homeostasis by regulating neural cell numbers, migration of interneurons, as well as promoting connectivity, synapse formation, and pruning (1). Microglia are considered the first line of immune defense in the brain by monitoring the brain parenchyma under homeostatic conditions and resolving cerebral insults (2). However, less is known about the function of microglia at the neurovascular unit (NVU), which is composed of vascular cells (endothelial and mural cells, as well as perivascular fibroblasts and macrophages), associated neurons, and glia (3). Microglia have been implicated in shaping brain vasculature during development (4). In adults, microglial processes directly contact the endothelial basement membrane at the NVU (5), and microglia has been shown to maintain vascular integrity during various insults such as mechanical injury or systemic inflammation (6, 7). Disruption of the NVU is common to many neurodegenerative diseases (3); however, studies on microglial function have mainly focused on brain parenchyma and not the NVU.
Optimal functioning of the NVU, which mediates functional hyperemia, is crucial for cerebral perfusion (3). Blood vessels play an integral role in brain development and provide a niche for brain stem cells. In addition, cerebral vasculature senses the environment and communicates changes to neural tissue, participates in glymphatic clearance, and controls immune quiescence in the CNS (8, 9). Accordingly, dysfunction of the NVU accompanies or may even represent a primary cause of many neurodegenerative diseases (3). In the case of primary familial brain calcification (PFBC), bilateral basal ganglia calcification of blood vessels is a key diagnostic criterion. The pathogenic mechanism points to a compromised NVU (10). PFBC is a clinically and genetically heterogeneous neuropsychiatric disorder (composed of psychiatric signs—anxiety and psychosis, cognitive impairment, and movement disorders—ataxia, dystonia, and parkinsonism) caused by mutations in at least six genes, JAM2, MYORG, PDGFB, PDGFRB, SLC20A2, and XPR1 (10, 11). Notably, recent studies have estimated the minimal prevalence of PFBC ranges from 4 to 6 per 10,000, depending on the causative gene mutation, thus suggesting that PFBC is not a rare disorder and is likely underdiagnosed (12). In addition, basal ganglia calcification is a common radiological finding, estimated in up to 20% of patients undergoing computed tomography (CT) imaging (13). Although the effect of cerebral calcification on the NVU and brain parenchyma is unknown, peripheral vascular calcification can lead to cardiovascular morbidity and mortality (14). Recent studies have suggested the involvement of glia in PFBC pathophysiology (15–17); however, functional studies demonstrating the role of glia in cerebral vessel calcification pathology are absent.
In this study, we investigated the role of microglia in vascular calcification using a mouse model of PFBC. Previously, we described that mouse platelet-derived growth factor B (Pdgfb) hypomorphs (Pdgfbret/ret) develop brain vessel–associated calcifications similar to human PFBC (18). Vascular calcifications in human PFBC and mouse models of PFBC were conspicuously encircled by activated microglia (15, 17, 19). In addition, an osteogenic environment with the surrounding cells expressing osteoclast markers was recently reported (17), although the cellular origin of these cells remains unknown.
Here, we characterize a distinct activation phenotype of microglia encircling vascular calcifications in Pdgfbret/ret mice, coined “calcification-associated microglia” (CAM). Pharmacological ablation of microglia in Pdgfbret/ret mice leads to aggravated calcification of the NVU. In addition, we show in wild-type mice and a mouse model of PFBC that ablation of microglia leads to the development of axonal spheroids containing bone proteins (osteocalcin and osteopontin) in the white matter. Mechanistically, we show that microglia require functional triggering receptor expressed on myeloid cells 2 (TREM2) for controlling vessel-associated calcifications, as genetic deletion of Trem2 in Pdgfbret/ret mice exacerbates vascular calcification. In conclusion, our study shows that microglia play an important role in modifying vascular calcification in the brain and identifies microglia as a potential therapeutic target in PFBC.
RESULTS
Brain vessel–associated calcifications trigger an inflammatory environment
Vascular calcification in PFBC elicits a conspicuous glial reaction (17–19). To gain insights into inflammatory changes accompanying vessel calcifications, we performed transcriptome analysis using RNA sequencing (RNA-seq) on tissue enriched with vessel-associated calcifications isolated from brains of Pdgfbret/ret mice, a mouse model of PFBC. Brain calcifications are autofluorescent (18), which we exploited to manually isolate calcifications from the brains of Pdgfbret/ret animals under a fluorescent stereomicroscope. Noncalcified tissue from the same anatomical region (thalamus/midbrain) was also collected from control animals (Pdgfbret/wt). This brain region is referred to as a “calcification-prone region” (Fig. 1A). We also isolated tissue from the cortex (referred to as a “non–calcification-prone region”) of both Pdgfbret/ret and control animals (Fig. 1A). Principal components analysis (PCA) of transcriptomic data showed that the first PC accounts for variability due to anatomical differences (thalamus/midbrain versus cortex), while the second PC accounts for differences between genotypes (fig. S1A). When comparing calcification- and non–calcification-prone brain regions of Pdgfbret/ret and control animals, we detected 92 and 94 deregulated genes, respectively [false discovery rate (FDR) < 0.05 and fold change > 2; Fig. 1B, fig. S1B, and tables S3 and S4]. Contrasting differentially expressed genes in non–calcification-prone and calcification-prone regions between Pdgfbret/ret and control animals showed that 74 genes were deregulated only in the calcification-prone region (thalamus/midbrain) and that 60 genes were deregulated only in a non–calcification-prone region (cortex) (Fig. 1, C and D, and table S5). The remaining 26 deregulated genes detected in both regions (Fig. 1, C and D, and table S5) concur with previously reported vascular transcriptional alterations due to reduced pericyte numbers in Pdgfbret/ret mice (20). Enrichment analysis of hallmark pathways showed that several inflammatory pathways, such as interferon γ and α pathways, reactive oxygen species pathway, and inflammatory response pathway, are enriched in Pdgfbret/ret mice compared to controls (Fig. 1E). Several pathways were enriched only in calcification-prone regions, such as interleukin-2–signal transducer and activator of transcription 5 signaling, unfolded protein response, and tumor necrosis factor–α signaling via nuclear factor κB (fig. S1C). Various significantly deregulated hallmark pathways (Fig. 1E and fig. S1C) have been associated with activated microglia (e.g., interferon related and complement related) or microglia quiescence (e.g., transforming growth factor–β related) (21, 22). Notably, Cst7, which encodes a cysteine protease inhibitor and is expressed in activated microglia (23), was the top up-regulated gene (3.7-fold) in brain regions with calcifications (Fig. 1B and table S3). The expression of Cst7 was not detected in either genotype in a non–calcification-prone brain region (cortex) or in the calcification-prone region of control animals (fig. S1D and tables S3 and S4). To further explore the gene signature associated with vascular calcification, we performed coexpression network analysis on highly variable genes in the RNA-seq dataset (fig. S1E) and identified six modules of positively correlated genes (fig. S1F and table S6). We found that only one module (M4) was associated with the calcified brain region in the Pdgfbret/ret mice (Fig. 1F). To summarize the node profiles of the M4 module, we examined its intramodular connectivity to identify the most highly connected intramodular hub genes (see Materials and Methods for details). Intuitively, hub genes can be viewed as module representatives that are centrally located in a network representation of correlated genes. Among the hub genes within the M4 module, we identified Cst7 and Itgax (Fig. 1G), genes induced in microglia in neurodegenerative diseases, termed DAM (disease-associated microglia), and in aging (23). Furthermore, in addition to Cst7 and Itgax, several other genes associated to reactive microglia or the DAM signature (e.g., Cd68, Clec7a, and Lpl) were significantly up-regulated in the calcified brain regions of Pdgfbret/ret mice (Figs. 1B and 2A). However, when comparing up-regulated genes in calcification-prone regions in Pdgfbret/ret mice using stringent criteria (FDR < 0.05 and fold change > 2) with the DAM signature, we found an overlap of only six genes (Lpl, Cd68, Tyrobp, Itgax, Cst7, and Clec7a) (fig. S1G). The DAM signature was recently reported to overlap with the “proliferative region–associated microglia” (PAM) signature that defines a subset of microglia found in the white matter during development (24). By comparing the PAM signature with our dataset using the same criteria as for the DAM genes, an overlap of seven genes (Hmox1, Lpl, Cd68, Slc16a3, Lag3, Tyrobp, and Clec7a) was found (fig. S1H). We validated the expression of DAM/PAM signature genes TIMP2 (Fig. 2B), CD68 (Fig. 2C), CLEC7A (Fig. 2, D and E), ITGAX (Fig. 2F), and CST7 (Fig. 2G) in microglia surrounding vessel-associated calcifications with immunohistochemistry. CD68 was expressed by microglia and perivascular macrophages (PVM) throughout the brain (fig. S2A), but its expression was up-regulated in CAM (Fig. 2C). Conspicuous CLEC7A positivity was seen in CAM (Fig. 2D), which showed down-regulation of P2YR12, a homeostatic microglia marker, and up-regulation of Iba1 (Fig. 2E). CLEC7A expression was also seen in a subpopulation of white matter microglial cells (fig. S2B), in agreement with reported expression of CLEC7A in white matter microglia (25). In addition, single microglial cells with normal morphology in the cortex of Pdgfbret/ret mice showed CLEC7A expression (fig. S2C). ITGAX and CST7 were expressed only in CAM and not in microglia in other brain regions or in other cell types. Similarly, TIMP2 was expressed only by microglia surrounding calcifications and in neurons (fig. S2D), as previously reported (26). Thus, CAM exhibited an activation profile overlapping with microglia during aging, neurodegenerative proteinopathies, and development.
Fig. 1. An inflammatory microenvironment surrounds brain calcifications.
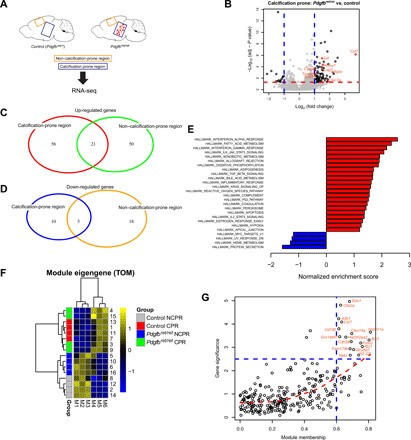
(A) Samples collected for transcriptome analysis. RNA-seq was performed from tissue isolated from two anatomical regions of Pdgfbret/ret and control animals (n = 4). Tissue enriched with brain calcifications was isolated from the thalamus/midbrain region labeled as calcification-prone region. Tissue isolated from the cortex is labeled as non–calcification-prone region. (B) Volcano plot showing deregulated genes in calcification-prone regions in Pdgfbret/ret animals compared to control animals. (C and D) Venn diagrams showing up-regulated (C) and down-regulated (D) genes in calcification-prone and non–calcification-prone regions. (E) Significantly up-regulated (in red) and down-regulated (in blue) pathways identified by gene set enrichment analysis (GSEA) (P < 0.05) in calcification-prone regions in Pdgfbret/ret animals compared to controls. (F) For each module identified by the network analysis, the module eigengene was calculated, which summarizes the expression profile of the module. The M4 module is associated with calcification-prone brain regions in Pdgfbret/ret mice. Heatmap shows column-wise standardized (z score) module eigengene values. CPR, calcification-prone region; NCPR, non–calcification-prone region; TOM, topological overlap matrix. (G) Graphical representation of representative hub genes with a high gene significance and module membership in the M4 module.
Fig. 2. Microglia around vessel calcifications express proteins associated with a DAM/PAM signature.
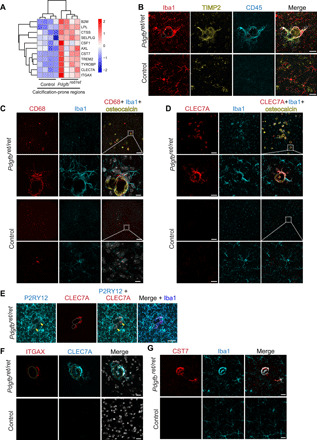
(A) Heatmap showing the expression of selected DAM signature genes significantly up-regulated in calcification-prone regions (thalamus/midbrain) in Pdgfbret/ret animals and controls (n = 4). Data are presented as z score. (B to D) Immunohistochemical validation of proteins associated with the DAM signature. (B) Iba1-positive (in red) microglia adjacent to a calcification (encircled with white dotted lines) are strongly positive for CD45 (in cyan) and positive for TIMP2 (in yellow). (C) Iba1-positive (in cyan) microglia surrounding osteocalcin-positive calcifications (in yellow) are positive for CD68 (in red). (D) Iba1-positive (in cyan) microglia surrounding osteocalcin-positive calcifications (in yellow) are positive for CLEC7A (in red). (E) P2RY12-positive (in cyan) resident microglia around brain calcifications (encircled with white dotted lines) express CLEC7A (in red). Double-positive cells are marked with yellow arrowheads. P2RY12 staining colocalizes with Iba1 (in blue) staining. (F) A subset of CAMs (CLEC7A; in cyan) express ITGAX (in red). Calcifications are surrounded with yellow dashed lines. (G) Microglia (Iba1; in cyan) surrounding brain calcifications express CST7 (in red). Nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI) (in white). n = 3. Scale bars, 100 μm (C and D), 50 μm (E), 20 μm (B, F, and G), and 10 μm (C inset and D inset).
Brain vessel–associated calcifications contain protein aggregates
Because the CAM protein expression profile resembles the DAM signature reported in β-amyloid plaque–associated microglia in Alzheimer’s disease (AD) (Fig. 2) (23) and vascular calcifications contain protein deposits such as amyloid β precursor protein (APP) and amyloid precursor–like protein 2 (15), we next asked whether brain calcifications might also contain amyloid fibrils as β-amyloid plaques. To investigate this possibility, we used thioflavin T and Congo red, standard stains to detect amyloid fibrils in β-amyloid plaques, and a recently developed amyloid-interacting luminescent conjugated oligothiophene (LCO), h-HTAA. Vessel-associated calcifications were thioflavin T and Congo red (fig. S3A) negative but positive for h-HTAA (fig. S3B) that binds protein aggregates before the formation of amyloid fibrils recognized by thioflavin T (27). Thus, vessel-associated calcifications contained aggregated proteins, which lack the β-pleated sheet conformation and structural regularity recognized by thioflavin T or Congo red.
CAM exhibit a distinct phenotype from DAM
Osteopontin (OPN, Spp1), one of the signature genes defining PAM and DAM (23, 24), is deposited in brain calcifications in human PFBC and animal models of PFBC (17). We therefore investigated whether microglia surrounding calcifications express osteopontin. Immunohistochemical visualization of microglia using anti-Iba1 and costaining with osteopontin did not confirm osteopontin expression by microglia (fig. S4, A and B). Instead, a strong osteopontin expression was seen in a subset of glial fibrillary acidic protein (GFAP)–positive, reactive astrocytes surrounding or adjacent to calcifications (fig. S4, A and C). Thus, in contrast to PAM and DAM, CAM do not up-regulate osteopontin expression. Hydroxyapatite, found in vessel-associated calcifications (17), and β-amyloid plaques induce formation of the inflammasome in macrophages and microglia, respectively (fig. S4D) (28, 29). We therefore investigated whether microglia around calcifications express ASC, an adaptor protein for inflammasome-mediated caspase-1 activation. We could not find ASC positivity in microglia surrounding vessel calcifications in Pdgfbret/ret animals (fig. S4E), indicating that vessel-associated calcifications do not lead to inflammasome activation in microglia. Thus, a phenotype of CAM is distinct from DAM, although they share expression of selected signature genes (Fig. 2).
Microglia proliferate around vessel-associated calcifications
To further characterize microglia in Pdgfbret/ret mice, we performed flow cytometry analysis on calcification-prone brain regions using a multicolor flow cytometry panel to distinguish immune cells and activated microglia from normal microglia. We detected eight immune cell clusters both in control and Pdgfbret/ret mice, as visualized by a dimension reduction algorithm uniform manifold approximation and projection (UMAP) (Fig. 3A and fig. S5A) (30). Within the microglial cluster (fig. S5B), we defined three subclusters (homeostatic microglia, phagocytotic microglia, and Ki67+ microglia) of microglia on the basis of median marker expression of seven markers (CD11b, Ki67, F4/80, CD64, CX3CR1, CCR5, and MerkTK) (Fig. 3, B and C). Overall changes in Pdgfbret/ret microglia compared to controls included increased expression of CX3CR1 and reduced CCR5 expression (fig. S5C). We also detected an increase in Ki67+ microglia and the emergence of CX3CR1high, MerTKhigh microglia (phagocytotic microglia) in Pdgfbret/ret animals (Fig. 3, A to C). Using immunohistochemical staining for the proliferation marker Ki67, we confirmed the presence of proliferating microglia (Fig. 3D). Also in vivo labeling of proliferating cells using 5-ethynyl-2′-deoxyuridine (EdU) in control and Pdgfbret/ret mice showed EdU-positive microglial cells around calcifications (Fig. 3E). In addition, we detected proliferating CD45high cells with round morphology surrounding calcifications (fig. S6A), while other cell types of the NVU (endothelial cells, pericytes, and astrocytes) were Ki67 negative (fig. S6, B to D). Brain vasculature in Pdgfbret/ret mice is permissive for leukocyte entry (31); therefore, we investigated whether CD45high cells with a round morphology (fig. S6A) surrounding calcifications could be T cells. Using immunohistochemistry, CD3+ T cells were detected in the brain parenchyma surrounding calcifications (fig. S6E) and in brain regions not prone to vascular calcification, e.g., in corpus callosum (fig. S6F). Some of these T cells were Ki67 positive (fig. S6, E and F, yellow arrowheads). To assess whether T cells trigger the formation of brain calcification, we crossed Pdgfbret/ret mice with Rag1−/− mice, which lack mature T cells. The absence of T cells did not prevent the formation of brain calcifications in Pdgfbret/ret; Rag1−/− mice (fig. S7), indicating that T cells, although present around brain calcifications, do not induce vessel calcification.
Fig. 3. Microglia proliferate around vessel-associated calcifications.
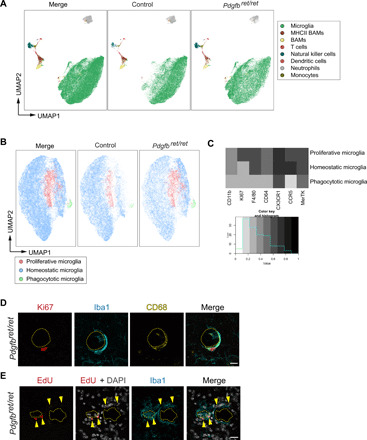
(A to C) Tissue from calcification-prone brain regions of Pdgfbret/ret mice and controls were analyzed using flow cytometry with a panel of antibodies to identify leukocytes and subtypes of brain myeloid cells. (A) Eight subpopulations of immune cells were identified with UMAP analysis. Microglia are depicted in green. BAMs, border-associated macrophages. MHCII, major histocompatibility complex II. (B) Microglia [in green in (A)] were isolated and analyzed separately using UMAP. An increase in proliferating (Ki67+, in pink) microglia and the appearance of phagocytotic (in green) microglia in Pdgfbret/ret mice were observed in contrast to control animals (Pdgfbret/wt). (C) Heatmap showing the expression of markers in different microglial subpopulations used to define subpopulations in (B) using UMAP. n = 3. (D) CD68-positive (in yellow) microglial cell (Iba1, in cyan) surrounding brain calcification (encircled with yellow dashed lines) is Ki67 positive (in red). (E) EdU (in red)–labeled Iba1-positive (in cyan, yellow arrowheads) microglia around brain calcifications (encircled with yellow dashed lines). Nuclei were visualized using DAPI (in white, E). n = 3. Scale bars, 20 μm (D) and 10 μm (E).
Microglia give rise to cathepsin K–expressing cells around vessel calcifications
We have reported previously that a subset of cells surrounding calcifications express osteoclast-associated markers receptor activator of nuclear factor κB (RANK) and cathepsin K (17). In addition, cathepsin K, which is a principal collagen I–degrading protease in bone, is deposited in brain calcifications (17). Here, we show that RANK-expressing cells around calcifications are positive for the microglial marker Iba1 (Fig. 4A). To investigate whether resident microglia give rise to osteoclast-like cells around vessel-associated brain calcifications in Pdgfbret/ret mice, we crossed two inducible Cre lines (Sall1-CreERT2 and Cx3cr1-CreERT2) and a reporter line expressing tdTomato under the Rosa26 promoter (Ai14) with Pdgfbret/ret animals to genetically label microglia. The Cx3cr1-CreERT2 line targets CX3CR1+ macrophages including microglia and “border-associated” macrophages in the CNS (32), whereas the Sall1-CreERT2 line targets resident microglia but not infiltrating immune cells or brain PVM (33). Mice were treated with tamoxifen at 1 month of age to induce tdTomato expression and euthanized at 4 months of age (Fig. 4B). Immunostaining with cathepsin K and Iba1 showed a colocalization with tdTomato both when using the Cx3cr1-CreERT2 (Fig. 4C) or Sall1-CreERT2 (Fig. 4D) line, indicating that resident microglia respond to calcifications by expressing and depositing cathepsin K into vascular calcifications.
Fig. 4. Microglia give rise to cathepsin K–expressing cells around calcifications.
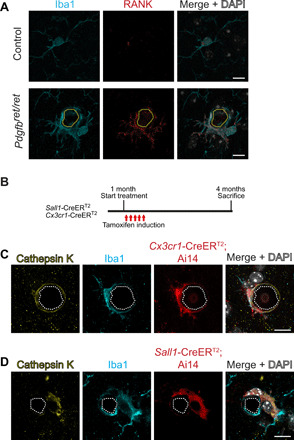
(A) Iba1-positive (in cyan) microglia around brain calcifications (encircled with yellow dotted lines) express the osteoclast marker RANK (in red). (B) Experimental setup of lineage tracing experiments. At 1 month of age, control and Pdgfbret/ret mice positive for Sall1-CreERT2; Ai14 or Cx3cr1-CreERT2; Ai14 were administered tamoxifen for five consecutive days and euthanized at 4 months of age. TdTomato expression was induced using inducible Cre lines where the expression of Cre is driven under Sall1 or Cx3cr1 promoter. (C and D) Cathepsin K staining (in yellow) colocalizes with tdTomato (in red) and Iba1 staining (in cyan) adjacent to calcification (encircled with white dotted lines). Nuclei were visualized using DAPI (in white; A, C, and D). Scale bars, 10 μm.
Pharmacological ablation of microglia intensifies vessel calcification
After establishing that cathepsin K–expressing cells surrounding vascular calcifications are derived from resident microglia (Fig. 4), we asked whether microglia actively participate in calcification-associated pathology. To this end, we depleted microglia in Pdgfbret/ret and control animals for 2 months by using the colony-stimulating factor 1 receptor (CSF1R) inhibitor PLX5622 (Fig. 5A). In contrast to mice fed with control chow, mice fed with chow containing PLX5622 showed a reduction in the number of microglia (fig. S8, A and B). We used two markers, osteocalcin and APP, to detect and quantify vascular calcification (Fig. 5B). We previously reported that cerebral vascular calcifications are positive for bone matrix proteins, including osteocalcin (17). Moreover, a recent study identified that several nonbone proteins, including APP, are enriched in vessel-associated calcifications (15). A noticeable increase in vascular calcification (approximately fourfold) in Pdgfbret/ret mice was seen 2 months after microglial depletion (Fig. 5, B and C). In addition, calcifications appeared heterogeneous in size and shape in PLX5622 chow–fed compared to control chow–fed Pdgfbret/ret mice (Fig. 5B). In microglia-depleted Pdgfbret/ret mice, the staining pattern of APP and osteocalcin was altered. APP and osteocalcin immunostainings were stronger along the periphery and weaker within calcifications. Some calcifications only stained for APP, whereas others immunolabeled only with osteocalcin (Fig. 5B, arrowheads). However, the ratio of APP- and osteocalcin-positive calcifications between PLX5622-treated and control chow–treated Pdgfbret/ret mice was not statistically significant (Fig. 5D). Depletion of microglia did not alter vascular density (Fig. 5E) or modify already impaired vascular permeability to plasma proteins (20, 34) in Pdgfbret/ret mice (fig. S8, D to F). CAMs express cathepsin K (Fig. 4, C and D). Accordingly, long-term microglial ablation should eliminate cathepsin K expression and deposition into calcifications. Microglial depletion reduced cathepsin K deposition in calcifications (Fig. 5, F and G). Consistent with our observation that activated microglia encircling calcifications do not up-regulate osteopontin expression (fig. S4), osteopontin is still deposited in calcifications in Pdgfbret/ret mice after microglial depletion (fig. S8C). Together, these results provide additional evidence that microglia are the principal cell type, giving rise to osteoclast-like cells surrounding calcifications. In addition, our data show that microglia depletion aggravates the vessel-associated calcification phenotype, indicating that microglial activity could modify the pathophysiology of PFBC.
Fig. 5. Pharmacological ablation of microglia aggravates vascular calcification.
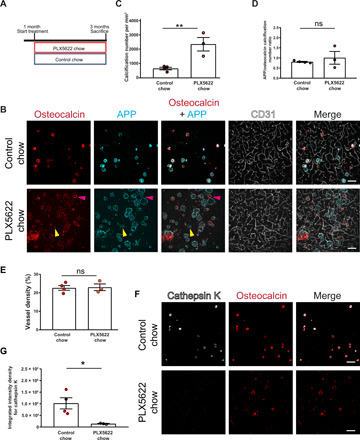
(A) Experimental setup of the pharmacological ablation of microglia. One-month-old mice (Pdgfbret/wt, Pdgfbret/ret, or C57BL6) were fed PLX5622 or control chow for 2 months. Mice were euthanized at 3 months of age. (B) Vessel-associated calcifications, visualized by osteocalcin (in red) and APP staining (in cyan), are increased in Pdgfbret/ret compared to control chow–fed Pdgfbret/ret animals. Blood vessels are visualized using CD31 staining (in white). Note that some calcifications in PLX5622-treated Pdgfbret/ret mice are only positive for APP (yellow arrowheads), whereas others are positive only for osteocalcin (magenta arrowheads). (C) Quantification of calcification number in Pdgfbret/ret mice administered PLX5622 or control chow (unpaired two-tailed t test; **P = 0.0087). (D) Ratio between APP- and osteocalcin-positive calcifications after PLX5622 and control chow treatment. (E) Quantification of vessel density in Pdgfbret/ret mice with administered PLX5622 or control chow (unpaired two-tailed t test; P = 0.8782). (F) Cathepsin K (in white) deposition in calcifications (in red) in Pdgfbret/ret mice is reduced after PLX5622 treatment. (G) Quantification of cathepsin K intensity from immunohistochemical stains in (F) (unpaired two-tailed t test; *P = 0.0276). Control chow, n = 4; and PLX5622 chow, n = 3. Scale bars, 100 μm (B and F). ns, not significant. All data are presented as means ± SEM.
Microglia ablation leads to bone protein containing axonal spheroids in white matter
After 2 months of chronic microglial depletion in control (C57BL6, Pdgfbret/wt) and Pdgfbret/ret mice (Fig. 5A), we observed conspicuous osteocalcin staining, indicative of calcification in the internal capsule, thalamus, and striatum adjacent to white matter fiber tracts (Fig. 6A, dotted yellow areas). However, linear osteocalcin-positive structures were not vessel associated (Fig. 6B and movie S1). Negativity for Alizarin red indicated that the observed white matter deposits were not calcified in three-month-old mice (fig. S8G, top pink panel). However, this result could be due to the young age of the mice. We previously showed that vascular calcifications in Pdgfbret/ret mice do not bind Alizarin red (i.e., are calcified) until 4 months of age (18). Accordingly, spheroidal deposits on vessels in the thalamus were Alizarin red negative (fig. S8G, mid blue panel, arrowheads), whereas occasional spheroids in the midbrain showed Alizarin red positivity (fig. S8G, bottom orange panel). We further characterized deposits in the white matter that appeared after microglial depletion using classical histochemical stains. Deposits were periodic acid–Schiff (PAS) positive, indicating the presence of glycoproteins (Fig. 6C, black arrowheads). The Bielschowsky silver stain revealed the presence of brown to black spheroids in the white matter, indicative of degenerating neurites (Fig. 6D, white arrowheads). In addition, costaining of osteocalcin with APP, a marker for neuronal injury, and with myelin basic protein (MBP) showed colocalization within axonal spheroids (Fig. 6, E and F). Furthermore, APP-positive spheroids in the white matter were also positive for another bone protein, osteopontin (Fig. 6G). Thus, prolonged microglial depletion using the CSF1R inhibitor PLX5622 in adult mice leads to white matter injury, characterized by the formation of axonal spheroids, which immunolabel with APP and MBP as well as osteopontin, indicating the accumulation of bone proteins.
Fig. 6. Microglia ablation leads to bone protein–containing axonal spheroids in white matter.
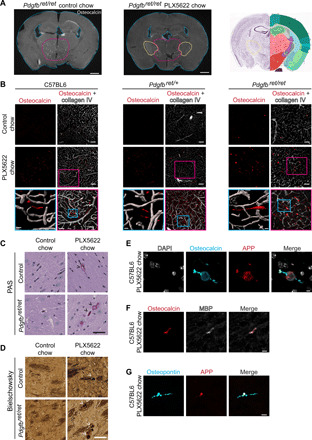
(A) Coronal sections of Pdgfbret/ret mouse brains fed with control chow or chow containing PLX5622. Mice treated with PLX5622 exhibited conspicuous osteocalcin staining (in white) in the internal capsule (yellow dotted lines) in addition to calcifications in the thalamus (encircled with pink dotted lines). Coronal mouse brain section (Allen Adult Mouse Brain Atlas) depicts the location of analyzed brain sections (photo credit: Allen Institute). (B) Linear inclusions, positive for osteocalcin (in red), in white matter are not vessel associated in C57BL6 (left), Pdgfbret/wt (middle), and Pdgfbret/ret (right) mice. Blood vessels are visualized using the anti–collagen IV antibody (in white). (C) White matter deposits appearing in Pdgfbret/ret and control mice (Pdgfbret/wt) after PLX5622 treatment are positive for PAS (black arrowheads). (D) Dystrophic neurites exhibiting axonal spheroids (indicated by white arrowheads) in white matter after PLX5622 treatment in Pdgfbret/ret and control (Pdgfbret/wt) mice. (E to G) White matter deposits appearing after microglial depletion are positive for osteocalcin (in cyan) and APP (in red) (E), osteocalcin (in red) and MBP (in white) (F), and OPN (in cyan) and APP (in red) (G). Nuclei were visualized using DAPI (E, in white). C57BL6 control chow and PLX5622 chow, n = 5. Pdgfbret/ret and Pdgfbret/wt control chow, n = 4; and PLX5622 chow, n = 3. Scale bars, 1000 μm (A), 100 μm (B), 50 μm (B pink inset, C, and D), 10 μm (B blue inset, F, and G), and 5 μm (E).
Microglia control vascular calcification in a TREM2-dependent manner
In addition to microglial depletion using a pharmacological CSF1R inhibitor, we used a genetic approach to impair microglial function in Pdgfbret/ret mice by crossing Pdgfbret/ret mice with Trem2−/− mice. Deletion of TREM2 leads to reduced microglial functionality (35). Furthermore, TREM2 expression is necessary for microglia to achieve the full DAM signature in a mouse model of AD (23). However, Trem2 is expressed in two brain-resident myeloid populations, microglia and PVM (36). We performed immunohistochemical detection of CD206+ PVM and observed that PVM are located around arteries and veins on the vascular tree and are not present at the capillary bed (fig. S9A), which is the calcified vascular bed in PFBC. Thus, we conclude that PVM are unlikely to play a role in a calcification process at the capillaries. We noticed an altered pattern of calcification in Pdgfbret/ret; Trem2−/− mice with vessels frequently encrusted with multiple osteocalcin-positive nodules (Fig. 7A, bottom), yielding the “pearls-on-a-string” phenotype reminiscent of human PFBC (19). This pattern was particularly evident in rostral thalamic regions that show less calcification than caudal thalamus and midbrain. Calcifications in Pdgfbret/ret;Trem2+/+ animals appeared as single nodules (Fig. 7A, bottom) in accordance with the reported phenotype of calcifications in Pdgfbret/ret mice (15, 17, 18). In addition to the altered pattern, vascular calcification was markedly aggravated in Pdgfbret/ret; Trem2−/− and Pdgfbret/ret; Trem2+/− mice (Fig. 7B). Similar to the microglia depletion experiments, we observed altered APP and osteocalcin immunostaining of calcifications in Pdgfbret/ret mice in the absence of one or two Trem2 alleles (fig. S9B). In Pdgfbret/ret; Trem2+/+ mice, calcifications stain uniformly with both antibodies used to visualize calcifications (osteocalcin and APP), whereas in Pdgfbret/ret; Trem2+/− and Pdgfbret/ret; Trem2−/− animals, calcifications showed weak APP or osteocalcin immunopositivity, respectively (fig. S9, B and C). Quantification of vascular calcifications (using positivity for both APP and OCN) in the midbrain of Pdgfbret/ret; Trem2−/− and Pdgfbret/ret; Trem2+/− mice showed an increased calcification (four- to sixfold) compared to Pdgfbret/ret;Trem2+/+ mice (Fig. 7C). The lack of Trem2 in control mice did not lead to brain calcification (fig. S9D), alter vascular density (fig. S9, B and E), or affect the already altered permeability to plasma proteins in Pdgfbret/ret mice (fig. S9F). Thus, functional TREM2 in microglia is necessary to limit the formation of vessel calcifications in Pdgfbret/ret mice. In addition, we observed that microglia surrounding calcifications express CLEC7A, a DAM-associated protein dependent on Trem2, in Pdgfbret/ret;Trem2−/− animals (fig. S9G). Cathepsin K deposition into calcifications is TREM2 dependent; Pdgfbret/ret; Trem2+/− and Pdgfbret/ret; Trem2−/− mice displayed a strongly reduced cathepsin K deposition into calcifications (Fig. 7, D and E). In summary, these data show that cathepsin K expression by CAM is TREM2 dependent and further corroborate that microglia control the growth of vascular calcifications in the brain.
Fig. 7. TREM2-dependent microglial function is crucial in controlling vascular calcification.
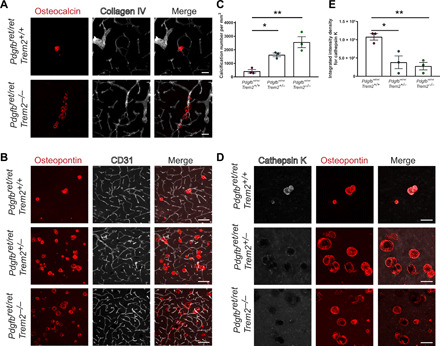
(A) Altered pattern of vessel calcification in Pdgfbret/ret; Trem2−/− animals compared to Pdgfbret/ret; Trem2+/+ animals. Calcifications are visualized using osteocalcin staining (in red) and blood vessels using collagen IV staining (in white). (B) Pdgfbret/ret; Trem2−/− and Pdgfbret/ret; Trem2+/− animals show an increase in calcification load when compared to Pdgfbret/ret; Trem2+/+ animals. Vascular calcification is visualized with osteopontin (in red), and blood vessels are visualized using CD31 staining (in white). (C) Quantification of calcification using immunohistochemical stains [one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison; *P = 0.0270 and **P = 0.0018). (D) Cathepsin K (in white) deposition in calcifications (osteopontin, in red) is reduced in Pdgfbret/ret; Trem2−/− and Pdgfbret/ret; Trem2+/− animals when compared to Pdgfbret/ret; Trem2+/+ animals. (E) Quantification of cathepsin K intensity with immunohistochemical stains (one-way ANOVA with Dunnett’s multiple comparison; *P = 0.0146 and **P = 0.0076). n = 3. Scale bars, 100 μm (B) and 50 μm (A and D). All data are presented as means ± SEM.
Microglia depletion or functional modulation alters astrocyte reactivity but not the neurotoxic-like profile around calcifications
We next explored whether microglia also modify the strong astrocyte reactivity around calcifications (17, 18). We had shown previously that astrocytes encircling brain calcifications exhibit a neurotoxic response [e.g., C3 and lipocalin-2 (LCN2) expression] and an unusual reactive phenotype (e.g., podoplanin expression) (17). We investigated whether astrocyte reactivity and the expression of neurotoxic markers are altered after modifying the number and function of microglia in Pdgfbret/ret mice. Microglia ablation using PLX5622 or compromised function (Trem2 genotype) in Pdgfbret/ret mice resulted in an altered staining pattern of GFAP and podoplanin, proteins expressed by reactive astrocytes surrounding calcifications (fig. S10, A and B). In Pdgfbret/ret mice treated with PLX5622, GFAP reactivity showed a diffuse pattern, most likely because of an increased density of calcifications, compared to mice that received control chow (fig. S10, A, E, and G). Furthermore, similar alterations in GFAP staining were observed in Pdgfbret/ret mice with impaired microglial function: Pdgfbret/ret; Trem2+/− and Pdgfbret/ret; Trem2−/− (fig. S10B). Podoplanin staining in reactive astrocytes surrounding calcifications was markedly reduced (fig. S10, A and B). We quantified podoplanin and GFAP staining intensity and calculated the staining intensity ratio, which showed a trend toward a reduction in Pdgfbret/ret mice treated with PLX5622 (fig. S10C) and a significant reduction in Pdgfbret/ret; Trem2+/− and Pdgfbret/ret; Trem2−/− mice (fig. S10D) compared to Pdgfbret/ret mice. Notably, we did not detect a reduction in the expression of neurotoxic signature markers LCN2 and C3 by reactive astrocytes surrounding calcifications after microglia depletion in Pdgfbret/ret mice (fig. S10, E and F). We quantified LCN2 and C3 and GFAP staining intensity and calculated the staining intensity ratio, which showed no significant difference between PLX5622-treated and nontreated Pdgfbret/ret mice (fig. S10, G and H). Together, these results indicate that microglia modulate astrocyte reactivity but are not required to evoke a neurotoxic-like astrocyte phenotype in response to vessel calcification.
DISCUSSION
Our results demonstrate that in addition to already known microglial functions, microglia activity in the context of vascular calcification is beneficial by identifying incipient harmful calcification. We have used a pharmacological approach in a mouse model for PFBC to modify microglial numbers and a genetic approach to modify microglial function. Both approaches enhance the calcification of the NVU (Figs. 5 and 7). Vascular calcification, resulting in increased pulse wave velocity and decreased end-organ perfusion and damage, is very common with aging as a major risk factor (37). Unexpectedly, studies investigating vascular calcifications in the brain are rare (38). Therefore, the consequences of calcification on vascular function at the NVU, including neurovascular coupling, have not been studied. As noted, brain calcification is a common incidental CT finding (13). It is plausible that under homeostatic conditions, i.e., without underlying vascular pathology, microglia limit vascular calcification during aging, which should be addressed by future studies. Vascular calcification is commonly observed in neurodegenerative diseases such as AD (39), which warrants further investigation to determine whether microglial dysfunction contributes to vascular pathology in AD. Thus, in addition to parenchymal surveillance and the removal of various injurious stimuli, microglia could remove calcium phosphate precipitates and halt calcification of the NVU (fig. S11).
Microglial activation is insult dependent (22); however, some microglial activation gene signatures are shared (e.g., Spp1 and Clec7a) by several neurodegenerative proteinopathies, developmental stages, and aging-related changes (23, 24). We observed that CAMs up-regulate CLEC7A (Fig. 2, D and E) but not osteopontin (OPN; gene name, Spp1) (fig. S4, A to C) expression, indicating that calcifications elicit a microglial response similar to but distinct from DAM/PAM and aging microglia. CLEC7A is still expressed by CAM in the absence of Trem2 (fig. S9F), which is necessary for the induction of the DAM signature in microglia (23). Furthermore, osteopontin is deposited in vessel calcifications after microglial depletion (fig. S8C). We detected a strong osteopontin expression in a subset of GFAP-positive astrocytes surrounding calcifications (fig. S4C), similar to osteopontin expression in reactive astrocytes after a stab wound injury (40). Osteopontin is highly expressed in injured tissues, including brain diseases (e.g., Parkinson disease and multiple sclerosis). One of the biological functions of osteopontin is to act as a hydroxyapatite binding mineral chaperone to inhibit the formation of hydroxyapatite crystals (41). The induction of osteopontin by different cells (e.g., microglia, astrocytes, and neurons) in response to various brain insults could prevent injury and ectopic calcification in the parenchyma or at the NVU. Ectopic soft-tissue calcifications in the periphery and in the brain, which are induced by excitotoxic insults, have a similar lamellar structure with darker-appearing lamellae rich in osteopontin (42). Notably, Spp1−/− mice develop severe secondary neurodegeneration accompanied by increased brain calcification in response to excitotoxic insults (43).
We show that cathepsin K deposition in vascular calcifications is decreased after altering microglia numbers or function in Pdgfbret/ret mice (Figs. 5F and 7E). Accordingly, cathepsin K–expressing cells surrounding vessel calcifications derive from resident microglia (Fig. 4, B and C). In the bone, cathepsin K is expressed and secreted by osteoclasts and represents the primary enzyme for degrading type I collagen (44). Our work and that of others have documented collagen I in brain calcifications (17, 45). Thus, cathepsin K activity could be necessary for degrading extracellular matrix (ECM) required for calcium phosphate precipitation. Microglia-specific modification of cathepsin K expression should clarify the role of cathepsin K in vascular calcification. Microglial activity that impedes vessel calcification could trigger a coordinated activation of several pathways in protein and hydroxyapatite degradation. However, it is currently unclear whether microglia remove hydroxyapatite present in vascular calcifications (17). Macrophages have been shown to remove various crystals, including hydroxyapatite, which leads to activation of the NLRP3 inflammasome pathway (29). Our studies show that although vessel calcifications elicit strong microglial reactivity in vivo, they do not activate the inflammasome (fig. S4E). Further studies are needed to understand how microglia sense and remove vascular calcification and whether this activates intracellular pathways as those evoked by self-derived damage-associated molecular patterns via pattern recognition receptors. In addition to altered cathepsin K deposition, we observed an altered deposition of APP and osteocalcin into calcifications in Pdgfbret/ret crossed with Trem2−/− or Trem2+/− or after microglia depletion (Figs. 5B and 7B). This finding indicates that microglial activity not only impedes the growth of calcifications but also modifies matrix composition of calcifications.
In this study, we observed that functional TREM2 is required to halt vessel calcification in a mouse model of PFBC (Fig. 7). TREM2 deficiency also leads to an altered vessel calcification pattern in Pdgfbret/ret mice (Fig. 7A), similar to that described in human autopsy cases. Vascular calcification in PFBC is sometimes described as pearls on a string because of numerous, tiny spherical calcifications that encrust almost the entire abluminal side of capillaries (19, 46). Calcifications appear as single nodules (15–18, 47) in mouse models of PFBC. Variants in a microglia-specific gene, TREM2, have been associated with an increased risk for AD (2). Studies in mouse models of AD have linked functional TREM2 to the development of a specific microglia activation state, DAM (23, 48), and to maintaining microglial metabolic fitness as well as a sustained microglial response to Aβ plaque–induced pathology (49). It has been proposed that although microglia are efficient in phagocytosing protein aggregates in the early stages of AD, over time, they lose their efficiency (2). Therefore, it is plausible that by crossing Pdgfbret/ret with Trem2+/− or Trem2−/−, vascular calcification is accelerated because of microglial exhaustion (2). It is noteworthy that microglia encircling calcifications contain numerous phagocytotic vesicles (15), and also, our study identified a population of phagocytotic microglia in the calcified brain regions in Pdgfbret/ret mice (Fig. 3, B to E). Further studies are needed to understand whether TREM2-driven microglial function is initiated because of altered proteostasis and calcification at the NVU or neuronal death occurring because of altered vessel function. These processes, however, are not necessarily mutually exclusive.
Several lines of evidence indicate that cell-autonomous defects in microglia lead to brain disease and cerebral calcification (50). Microglia have also emerged as a disease modifier in a wide range of neurodegenerative diseases (e.g., AD and frontotemporal dementia) (2). Of six genes implicated in PFBC (JAM2, PDGFB, PDGFRB, SLC20A2, XPR1, and MYORG), microglia express JAM2, PDGFB, SLC20A2, and XPR1. Although microglia express PDGFB, they do not express the receptor PDGFRB. Thus, it is unlikely that PDGFB or PDGFRB haploinsufficiency in PFBC causes cell-autonomous microglial dysfunction. Phosphate importer SLC20A2 and exporter XPR1 are expressed ubiquitously in the brain, and their specific role in microglia has not been investigated. However, it has been reported that in zebrafish, xpr1b, an ortholog of XPR1, is crucial for the differentiation of tissue-resident macrophages and microglia (51). In mice, the full knockout of Xpr1 is embryonic lethal (52), but whether the absence of Xpr1 affects microglia development has not been assessed. Thus, further studies are needed to dissect the role of PFBC genes in microglial function.
In this study, on the basis of positivity for osteocalcin, we found by serendipity that microglial ablation by chronic CSF1R inhibition using PLX5622 for 2 months resulted in localized axonal damage to fiber tracts of the internal capsule and adjacent thalamic and striatal areas (Fig. 6). Positive staining for osteocalcin and osteopontin coincided with the presence of dystrophic neurites exhibiting spheroid formation (Fig. 6, E to G), similar to the pathology described in patients with leukoencephalopathy caused by CSF1R mutations (50). These patients exhibit brain calcification in white matter regions and a reduction in microglia in affected regions. It is plausible that axonal spheroids become calcified during the course of the disease. Further studies are needed to understand the relationship between the appearance of axonal spheroids positive for bone proteins and white matter calcification. We observed that inclusions within dystrophic neurites stain positive for MBP (Fig. 6G), a protein secreted by oligodendrocytes, indicating a disrupted homeostasis in oligodendrocytes. Previous studies on white matter microglia have shown that microglia promote myelinogenesis during early development, providing evidence for a role in optimizing oligodendrocyte function (53). Thus, further studies are needed to ascertain whether axonal damage is directly caused by a reduction in microglia or the absence of microglia has an effect on another cell type (e.g., oligodendrocytes), which may enhance spheroid formation in neurites.
In conclusion, we describe an unrecognized role of microglia in brain vascular calcification. In addition, we show that functional microglia are important to prevent calcification of the NVU in neurodegenerative diseases with a compromised NVU. Proposed mechanisms by which microglia control vascular calcification include the removal of apoptotic cells and spontaneous calcium phosphate precipitates as well as the prevention of nucleation of hydroxyapatite by controlling proteostasis of the ECM and/or the secretion of anticalcifying proteins or molecules.
MATERIALS AND METHODS
Mice
Mice used in this study were 1 to 5 months old. Both sexes were used for experiments. The following mouse strains were used: C57BL6/J (Charles River Laboratories), B6.Pdgfbtm3Cbet (Pdgfbret/ret) (18, 54), Cx3cr1-CreERT2 (55), Sall1-CreERT2 (33), B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Ai14) (Jackson Laboratory, stock no. 007914), APP/PS1 (56), Rag1−/− (57), and Trem2−/− (58). This study was carried out in accordance with study protocols approved by the Cantonal Veterinary Office Zurich (permit numbers ZH196/2014 and ZH151/2017).
In vivo treatments
Tamoxifen treatment for the genetic labeling of Cx3Cr1- and Sall1-expressing cells
For tamoxifen-inducible labeling of Cx3cr1- and Sall1-expressing cells in respective genotypes (Cx3cr1-CreERT2; Ai14Tg/wt; Pdgfbret/ret or Pdgfbret/wt and Sall1-CreERT2; Ai14Tg/wt; Pdgfbret/ret or Pdgfbret/wt), 4-week-old animals were treated for five consecutive days via oral gavage with 2 mg of tamoxifen (Sigma-Aldrich, catalog no. T5648) dissolved in corn oil. Mice were euthanized at 4 months of age.
Pharmacological ablation of microglia
Oral CSF1R inhibitor, PLX5622 (Plexxikon Inc.), was formulated in AIN-76A standard chow by Research Diets (New Brunswick, NJ) at 1200 parts per million. Control mice received AIN-76A chow without PLX5622. PLX5622 chow and control chow were provided by Plexxikon Inc. under a material transfer agreement. One-month-old mice were fed with chow containing PLX5622 or control chow for 2 months and euthanized at the age of 3 months.
EdU treatment
Three-month-old mice were injected with EdU (50 mg/kg; Sigma-Aldrich, catalog no. 900584) intraperitoneally for three consecutive days and euthanized on the following day.
Antibodies
Primary antibodies used for immunofluorescence staining are listed in table S1. All secondary antibodies (suitable for multiple labeling) labeled with various fluorophores (Alexa Fluor 488, Cy3, and DyLight 649) made in donkey (anti-rabbit, anti-rat, and anti-goat) or in goat (anti-chicken Cy3) were purchased from Jackson ImmunoResearch. Antibodies used for flow cytometry analysis are listed in table S2.
Histochemistry and immunohistochemistry
Immunohistochemistry was performed according to methods described previously (17). For EdU detection, the Click-iT EdU Alexa Fluor 555 imaging kit (Thermo Fisher Scientific) was used, and slices were treated according to the manufacturer’s instructions. Immunohistochemical stainings were imaged with a confocal microscope [Leica SP5 or SP8; 20× numerical aperture (NA), 0.7; 40× NA, 1.25; and 63× NA, 1.4] or stereomicroscope (Zeiss Axio Zoom.V16; 1× NA, 0.25). For stains that exhibited salt-and-pepper noise, a median filter of 5 × 5 × 5 was applied to eliminate noise. Images were analyzed using the image processing software Imaris 9.2.0. (Bitplane) and Adobe Illustrator CS6.
For histochemistry, mouse brains were collected and embedded in paraffin. Two-micrometer-thick tissue sections were stained with PAS, hematoxylin and eosin, or the Bielschowsky silver stain using standard protocols. For Alizarin red staining, sections were deparaffinized and rehydrated, as well as incubated for 1 hour in 1% Alizarin red solution (pH 9.0) followed by 1 hour in 1% Alizarin red solution (pH 6.4) at room temperature. Stained paraffin sections were scanned with NanoZoomer HT (Hamamatsu Photonics), equipped with a 20× objective (UPlanSApo; NA, 0.75; Olympus). Images were analyzed using Digital Image Hub software (SlidePath) and Adobe Illustrator CS6.
Detection of aggregated proteins
Paraffin-embedded 6-μm-thick brain sections were used from Pdgfbret/ret and controls of 1 year of age or older. As a positive control, paraffin-embedded sections from APP/PS1 mice (56) were used. Sections were deparaffinized and hydrated, followed by a 30-min incubation with LCO h-HTAA (27) at room temperature. For thioflavin T (Sigma-Aldrich, T3516) staining, deparaffinized sections were processed according to the manufacturer’s instructions. Congo red staining was performed according to standard protocol. Stained sections were imaged with a confocal microscope (Leica SP5; 20×; NA, 0.7) or Axioplan (Zeiss; 20×; NA, 0.5). Images were analyzed with image processing software Imaris (Bitplane).
Quantification of immunofluorescent stainings
Images for quantification of brain calcifications, cathepsin K intensity, vessel density, and astrocyte reactivity were acquired using a 20× objective (NA, 0.7; Leica SP5) 42 z-stacks with a 1.48-μm step and 512 × 512 pixel resolution. For quantification of total brain calcification, z-stacks of APP staining and osteocalcin were summed in Fiji (ImageJ). Imaris software was used to quantify calcification using the function “surfaces.” Quantification of calcifications was performed in mid-midbrain, which shows the smallest interindividual variation in calcification load (17). Cathepsin K intensity density was calculated using Fiji (ImageJ) software. Vessel density was quantified using CD31 staining. The analysis was performed with a vessel density plugin (version1.1; with minor modifications) in Fiji (ImageJ) software. Astrocyte reactivity was quantified using GFAP, podoplanin, LCN2, and C3 staining in Fiji (ImageJ). The LCN2 and C3 signal intensity was first masked to GFAP staining to eliminate nonastrocytic expression or deposition. Signal intensity was then normalized to the GFAP signal. Two technical replicates were quantified for each animal.
Images used for quantification of extravasated fibrinogen were acquired using a 20× objective (NA, 0.75, Leica SP8), seven z-stacks with a 1.04-μm step, and 1024 × 1024 pixel resolution. CD31 staining marking blood vessels was thresholded and despeckled, afterward subtracted from the thresholded fibrinogen staining. Image subtraction was performed to eliminate any signal inside the vessel lumen. Quantification of extravasated fibrinogen was performed using Fiji (ImageJ) software.
Microglia were quantified on images acquired using a 20× objective (NA, 0.7; Leica SP5), 41 z-stacks with a 0.64-μm step, and 1024 × 1024 pixel resolution. Quantification of microglia was performed using the morpholibJ package in Fiji.
Flow cytometry analysis
Mice were deeply anesthetized using a mixture of ketamine and xylazine and perfused transcardially using ice-cold phosphate-buffered saline (PBS). Subsequently, mouse brains were dissected into non–calcification-prone brain regions, i.e., cortex, hippocampus, and cerebellum, and calcification-prone regions, i.e., thalamus, midbrain, and pons. Brain cell suspensions were prepared by cutting the tissue into small pieces, followed by collagenase type IV treatment. Dissociated tissue was passed through an 18-gauge syringe to obtain a homogeneous cell suspension and further enriched with a Percoll gradient. Samples were then passed through a 70-μm filter, followed by red blood cell lysis and antibody staining. Flow cytometric analysis was carried using FACSymphony (BD Biosciences) and analyzed with FlowJo and R software.
High-dimensional analysis
Raw data were preanalyzed with FlowJo, subsequently transformed in MATLAB using cyt3, and percentile-normalized in R. Dimensionality reduction was achieved by UMAP. FlowSOM was used for automated and expert-guided cell clustering. Median marker expression was projected onto UMAP to generate a heatmap of median expression values.
Isolation of vessel calcifications and RNA-seq
Mice were deeply anesthetized and transcardially perfused with ice-cold PBS. Brains were removed, placed in RNAlater Stabilization Solution (Thermo Fisher Scientific, catalog no. AM7020), and 1-mm coronal sections were cut using a brain matrix (RBMA-200C, World Precision Instruments). Calcifications were detected on the basis of their autofluorescence using a fluorescent stereomicroscope (Zeiss Axio Zoom.V16) and were surgically removed together with surrounding tissue. Cortical sections were also removed as examples of non–calcification-prone regions. RNA was isolated with a micro RNA kit (Qiagen) according to the manufacturer’s instructions. The concentration of RNA and sample purity were assessed using a 2100 Bioanalyzer (Agilent) and RNA 6000 Pico Kit (Agilent). RNA samples were poly(A)-enriched, and libraries were prepared using the Illumina TruSeq Stranded RNA kit. RNA was sequenced on an Illumina platform HiSeq 4000 at the Functional Genomic Center Zurich (UZH, ETH). The Illumina single-read approach (1 × 125 base pair) was used to generate raw sequencing reads with a depth of 20 million to 30 million reads per sample.
Bioinformatics analysis
Quantification of RNA-seq data was performed using kallisto. Briefly, target transcript sequences were obtained from ENSEMBL (GRCm38.p6), and the abundance of transcripts was quantified using kallisto 0.44.0 with sequence-based bias correction. All other parameters were set to default when running kallisto. Kallisto’s transcript-level estimates were further summarized at the gene level using tximport 1.8.0 from Bioconductor.
For downstream analysis, genes of low abundance were filtered out, and unwanted variation was estimated using the RUVr functionality from the RUVseq 1.16.0 package within Bioconductor. The number of factors of unwanted variation estimated from the data was set to 3, and the genes_by_samples matrix of residuals was obtained from a first-pass quasi-likelihood negative binomial generalized log-linear regression of the counts on biological covariates using the edgeR package from Bioconductor.
Differential expression analysis was performed using DESeq2 1.22.0 from Bioconductor, with estimated factors of unwanted variation included as additional covariates in the design formula. Significant genes were identified using FDR < 0.05 and fold change > 2.
Coexpression network analysis was performed on the combination of 16 samples (4 groups × 4 replicates) using weighted correlation network analysis (WGCNA) R package. Briefly, normalized count data obtained from RUVseq were first adjusted for mean-variance trend using the regularized log transformation of DESeq2 1.22.0 from Bioconductor. To exclude uninformative genes from coexpression analysis in an unbiased manner, 867 highly variable genes were selected on the basis of the overall distribution of coefficients of variation. Subsequently, a signed weighted network of highly variable genes was constructed using biweight midcorrelation with beta = 23 as soft thresholding power. Hierarchical clustering of the topological overlap matrix dissimilarity further revealed six modules of positively correlated genes. Module eigengene was defined as the first PC of the expression matrix of the corresponding module. Module membership was defined per gene as the Pearson correlation of gene expression and module eigengene. In the context of module membership, gene significance was defined as the geometric mean of the absolute value of pairwise Welch’s t statistics comparing Pdgfbret/ret CPR (calcification-prone region) to Pdgfbret/ret NCPR (non–calcification-prone region), control NCPR, and control CPR. To identify hub genes associated with brain calcification, we computed module membership and gene significance, as defined above, for each gene belonging to the module associated with brain calcification (i.e., M4).
Gene set enrichment analysis (GSEA) was performed using the fgsea 1.8.0 package from Bioconductor with signal-to-noise ratio as defined in (59) as gene-level statistic. Before GSEA, mouse genes were converted to human orthologs using biomaRt 2.38.0 from Bioconductor. If a human ortholog was associated with more than one mouse gene, then the mouse gene with the maximum mean expression was selected using the collapseRows functionality within the WGCNA R package. Signaling pathways analyzed by GSEA were obtained from the Hallmark gene sets of the MSigDB. Gene signature of DAM and PAM were obtained from literature (23, 24). Heatmaps were generated using the pheatmap R package, with clustering distance and method set to Euclidean and ward.D2, respectively.
Statistical analysis
Quantified values are represented as means ± SEM. The following statistical tests were performed with Prism8 software (GraphPad). Normality was assessed using a Shapiro-Wilk test. The following tests were used to calculate statistical significance: Student’s t test (unpaired and two tailed), one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison, or Mann-Whitney two-tailed test. P values < 0.05 were considered significant.
Acknowledgments
Imaging was performed with equipment maintained by the Center for Microscopy and Image Analysis, University of Zurich, and RNA-seq was performed at the Functional Genomics Center Zurich, University of Zurich and ETH. Flow cytometry analysis was carried out at the Cytometry Facility at the University of Zurich. We thank Plexxikon for providing PLX5622, F. Franzoso for technical help, and S. Hornemann for discussions. Funding: This work was supported by the Swiss National Science Foundation (grants 31003A_159514 and 310030_188952 to A.K. as well as PP00P3_170626 and BSGI0_155832 to M.G.), the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 819229) to M.G., and the Synapsis Foundation and the Choupette Foundation (grant 2019-PI02), Fonds zur Förderung des akademischen Nachwuchses (Zurich University), the Leducq Foundation (grant 14CVD02), the Swiss Heart Foundation, The Swiss Cancer League (grant KLS-3848-02-2016) to A.K., and Forschungskredit und Stiftung für Forschung an der Medizinischen Fakultät der Universität Zürich (grant FK-16-034) to Y.Z. Author contributions: Y.Z. and A.K. conceived the study, designed the experiments, and wrote the manuscript. Y.Z., A.K., S.S., and S.G.U. performed the experiments and analyzed and interpreted the data. S.N. and M.D. analyzed and interpreted the bioinformatics data. J.S. and U.M. performed the experiments. K.P.R.N. and M.C. contributed with reagents. E.J.R. and M.G. analyzed and interpreted the data. A.K. supervised the study. Competing interests: M.C. received research support from Alector, Amgen, Ono, and Pfizer for activities not related to the findings described in this publication. M.C. is a scientific advisory board member of Vigil, Cell Signaling Technology, and Bluefin and has a patent to TREM2 pending. M.C. is an inventor an unpublished patent application to this work filed by Washington University no. 62/981,827 filed on February 26, 2020. All other authors declare that they have no competing interests. Data and materials availability: RNA-seq data, both raw data and gene-by-sample matrix of estimated counts, were deposited in Gene Expression Omnibus (GEO) under accession number GSE135449. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabc4898/DC1
REFERENCES AND NOTES
- 1.Thion M. S., Ginhoux F., Garel S., Microglia and early brain development: An intimate journey. Science 362, 185–189 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Hickman S., Izzy S., Sen P., Morsett L., El Khoury J., Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iadecola C., The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 96, 17–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fantin A., Vieira J. M., Gestri G., Denti L., Schwarz Q., Prykhozhij S., Peri F., Wilson S. W., Ruhrberg C., Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116, 829–840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathiisen T. M., Lehre K. P., Danbolt N. C., Ottersen O. P., The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 58, 1094–1103 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Haruwaka K., Ikegami A., Tachibana Y., Ohno N., Konishi H., Hashimoto A., Matsumoto M., Kato D., Ono R., Kiyama H., Moorhouse A. J., Nabekura J., Wake H., Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 10, 5816 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nimmerjahn A., Kirchhoff F., Helmchen F., Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Ransohoff R., Engelhardt B., The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Rafii S., Butler J. M., Ding B. S., Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westenberger A., Balck A., Klein C., Primary familial brain calcifications: Genetic and clinical update. Curr. Opin. Neurol. 32, 571–578 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Cen Z., Chen Y., Chen S., Wang H., Yang D., Zhang H., Wu H., Wang L., Tang S., Ye J., Shen J., Wang H., Fu F., Chen X., Xie F., Liu P., Xu X., Cao J., Cai P., Pan Q., Li J., Yang W., Shan P. F., Li Y., Liu J. Y., Zhang B., Luo W., Biallelic loss-of-function mutations in JAM2 cause primary familial brain calcification. Brain 143, 491–502 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Nicolas G., Charbonnier C., Campion D., Veltman J. A., Estimation of minimal disease prevalence from population genomic data: Application to primary familial brain calcification. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177, 68–74 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Deng H., Zheng W., Jankovic J., Genetics and molecular biology of brain calcification. Ageing Res. Rev. 22, 20–38 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Chen N. X., Moe S. M., Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 14, 228–237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nahar K., Lebouvier T., Andaloussi Mäe M., Konzer A., Bergquist J., Zarb Y., Johansson B., Betsholtz C., Vanlandewijck M., Astrocyte-microglial association and matrix composition are common events in the natural history of primary familial brain calcification. Brain Pathol. 30, 446–464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao X. P., Cheng X., Wang C., Zhao M., Guo X. X., Su H. Z., Lai L. L., Zou X. H., Chen X. J., Zhao Y., Dong E. L., Lu Y. Q., Wu S., Li X., Fan G., Yu H., Xu J., Wang N., Xiong Z. Q., Chen W. J., Biallelic mutations in MYORG cause autosomal recessive primary familial brain calcification. Neuron 98, 1116–1123.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Zarb Y., Weber-Stadlbauer U., Kirschenbaum D., Kindler D. R., Richetto J., Keller D., Rademakers R., Dickson D. W., Pasch A., Byzova T., Nahar K., Voigt F. F., Helmchen F., Boss A., Aguzzi A., Klohs J., Keller A., Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain 142, 885–902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keller A., Westenberger A., Sobrido M. J., García-Murias M., Domingo A., Sears R. L., Lemos R. R., Ordoñez-Ugalde A., Nicolas G., da Cunha J. E., Rushing E. J., Hugelshofer M., Wurnig M. C., Kaech A., Reimann R., Lohmann K., Dobričić V., Carracedo A., Petrovic I., Miyasaki J. M., Abakumova I., Mäe M. A., Raschperger E., Zatz M., Zschiedrich K., Klepper J., Spiteri E., Prieto J. M., Navas I., Preuss M., Dering C., Jankovic M., Paucar M., Svenningsson P., Saliminejad K., Khorshid H. R., Novakovic I., Aguzzi A., Boss A., Le Ber I., Defer G., Hannequin D., Kostic V. S., Campion D., Geschwind D. H., Coppola G., Betsholtz C., Klein C., Oliveira J. R., Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet. 45, 1077–1082 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Miklossy J., Mackenzie I. R., Dorovini-Zis K., Calne D. B., Wszolek Z. K., Klegeris A., McGeer P. L., Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol. 109, 643–653 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Armulik A., Genove G., Mae M., Nisancioglu M. H., Wallgard E., Niaudet C., He L., Norlin J., Lindblom P., Strittmatter K., Johansson B. R., Betsholtz C., Pericytes regulate the blood-brain barrier. Nature 468, 557–561 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Qin Y., Garrison B. S., Ma W., Wang R., Jiang A., Li J., Mistry M., Bronson R. T., Santoro D., Franco C., Robinton D. A., Stevens B., Rossi D. J., Lu C., Springer T. A., A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 174, 156–171.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman B. A., Srinivasan K., Ayalon G., Meilandt W. J., Lin H., Huntley M. A., Cao Y., Lee S. H., Haddick P. C. G., Ngu H., Modrusan Z., Larson J. L., Kaminker J. S., van der Brug M. P., Hansen D. V., Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T. K., David E., Baruch K., Lara-Astaiso D., Toth B., Itzkovitz S., Colonna M., Schwartz M., Amit I., A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Li Q., Cheng Z., Zhou L., Darmanis S., Neff N. F., Okamoto J., Gulati G., Bennett M. L., Sun L. O., Clarke L. E., Marschallinger J., Yu G., Quake S. R., Wyss-Coray T., Barres B. A., Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101, 207–223.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hart A. D., Wyttenbach A., Perry V. H., Teeling J. L., Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain Behav. Immun. 26, 754–765 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fager N., Jaworski D. M., Differential spatial distribution and temporal regulation of tissue inhibitor of metalloproteinase mRNA expression during rat central nervous system development. Mech. Dev. 98, 105–109 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Klingstedt T., Aslund A., Simon R. A., Johansson L. B., Mason J. J., Nyström S., Hammarström P., Nilsson K. P., Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org. Biomol. Chem. 9, 8356–8370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heneka M. T., Kummer M. P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T. C., Gelpi E., Halle A., Korte M., Latz E., Golenbock D. T., NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin C., Frayssinet P., Pelker R., Cwirka D., Hu B., Vignery A., Eisenbarth S. C., Flavell R. A., NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc. Natl. Acad. Sci. U.S.A. 108, 14867–14872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.L. McInnes, J. Healy; J. Melville, UMAP: Uniform Manifold Approximation and Projection for dimension reduction. arXiv:1802.03426 [stat.ML] (9 February 2018).
- 31.O. Torok, B. Schreiner, H.-C. Tsai, S. Utz, J. Schaffenrath, S. Nassiri, M. Delorenzi, A. Aguzzi, M. H. Han, M. Greter, B. Becher, A. Keller, Pericytes regulate vascular immune homeostasis in the CNS. Proc. Natl. Acad. Sci. U.S.A, in press (2021). [DOI] [PMC free article] [PubMed]
- 32.Parkhurst C. N., Yang G., Ninan I., Savas J. N., Yates J. R. III, Lafaille J. J., Hempstead B. L., Littman D. R., Gan W.-B., Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buttgereit A., Lelios I., Yu X., Vrohlings M., Krakoski N. R., Gautier E. L., Nishinakamura R., Becher B., Greter M., Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Vanlandewijck M., Lebouvier T., Andaloussi Mae M., Nahar K., Hornemann S., Kenkel D., Cunha S. I., Lennartsson J., Boss A., Heldin C. H., Keller A., Betsholtz C., Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLOS ONE 10, e0143407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulrich J. D., Ulland T. K., Colonna M., Holtzman D. M., Elucidating the role of TREM2 in Alzheimer’s Disease. Neuron 94, 237–248 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Van Hove H., Martens L., Scheyltjens I., De Vlaminck K., Pombo Antunes A. R., De Prijck S., Vandamme N., De Schepper S., Van Isterdael G., Scott C. L., Aerts J., Berx G., Boeckxstaens G. E., Vandenbroucke R. E., Vereecke L., Moechars D., Guilliams M., Van Ginderachter J. A., Saeys Y., Movahedi K., A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22, 1021–1035 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Pescatore L. A., Gamarra L. F., Liberman M., Multifaceted mechanisms of vascular calcification in aging. Arterioscler. Thromb. Vasc. Biol. 39, 1307–1316 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Arnold M. M., Kreel L., Asymptomatic cerebral calcification–a previously unrecognized feature. Postgrad. Med. J. 67, 147–153 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mann D. M., Calcification of the basal ganglia in Down’s syndrome and Alzheimer’s disease. Acta Neuropathol. 76, 595–598 (1988). [DOI] [PubMed] [Google Scholar]
- 40.Sirko S., Irmler M., Gascon S., Bek S., Schneider S., Dimou L., Obermann J., De Souza Paiva D., Poirier F., Beckers J., Hauck S. M., Barde Y. A., Gotz M., Astrocyte reactivity after brain injury: The role of galectins 1 and 3. Glia 63, 2340–2361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunter G. K., Kyle C. L., Goldberg H. A., Modulation of crystal formation by bone phosphoproteins: Structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem. J. 300 ( Pt 3), 723–728 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jahnen-Dechent W., Schafer C., Ketteler M., McKee M. D., Mineral chaperones: A role for fetuin-A and osteopontin in the inhibition and regression of pathologic calcification. J. Mol. Med. (Berl) 86, 379–389 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Maetzler W., Berg D., Funke C., Sandmann F., Stünitz H., Maetzler C., Nitsch C., Progressive secondary neurodegeneration and microcalcification co-occur in osteopontin-deficient mice. Am. J. Pathol. 177, 829–839 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gowen M., Lazner F., Dodds R., Kapadia R., Feild J., Tavaria M., Bertoncello I., Drake F., Zavarselk S., Tellis I., Hertzog P., Debouck C., Kola I., Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J. Bone Miner. Res. 14, 1654–1663 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Fujita D., Terada S., Ishizu H., Yokota O., Nakashima H., Ishihara T., Kuroda S., Immunohistochemical examination on intracranial calcification in neurodegenerative diseases. Acta Neuropathol. 105, 259–264 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi S., Yamadori I., Miki H., Ohmori M., Idiopathic nonarteriosclerotic cerebral calcification (Fahr’s disease): An electron microscopic study. Acta Neuropathol. 73, 62–66 (1987). [DOI] [PubMed] [Google Scholar]
- 47.Wallingford M. C., Chia J. J., Leaf E. M., Borgeia S., Chavkin N. W., Sawangmake C., Marro K., Cox T. C., Speer M. Y., Giachelli C. M., SLC20A2 deficiency in mice leads to elevated phosphate levels in cerbrospinal fluid and glymphatic pathway-associated arteriolar calcification, and recapitulates human idiopathic Basal Ganglia calcification. Brain Pathol. 27, 64–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O’Loughlin E., Xu Y., Fanek Z., Greco D. J., Smith S. T., Tweet G., Humulock Z., Zrzavy T., Conde-Sanroman P., Gacias M., Weng Z., Chen H., Tjon E., Mazaheri F., Hartmann K., Madi A., Ulrich J. D., Glatzel M., Worthmann A., Heeren J., Budnik B., Lemere C., Ikezu T., Heppner F. L., Litvak V., Holtzman D. M., Lassmann H., Weiner H. L., Ochando J., Haass C., Butovsky O., The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ulland T. K., Song W. M., Huang S. C., Ulrich J. D., Sergushichev A., Beatty W. L., Loboda A. A., Zhou Y., Cairns N. J., Kambal A., Loginicheva E., Gilfillan S., Cella M., Virgin H. W., Unanue E. R., Wang Y., Artyomov M. N., Holtzman D. M., Colonna M., TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konno T., Kasanuki K., Ikeuchi T., Dickson D. W., Wszolek Z. K., CSF1R-related leukoencephalopathy. Neurology 91, 1092–1104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meireles A. M., Shiau C. E., Guenther C. A., Sidik H., Kingsley D. M., Talbot W. S., The phosphate exporter xpr1b is required for differentiation of tissue-resident macrophages. Cell Rep. 8, 1659–1667 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu X., Li X., Sun H., Cao Z., Gao R., Niu T., Wang Y., Ma T., Chen R., Wang C., Yang Z., Liu J. Y., Murine placental-fetal phosphate dyshomeostasis caused by an Xpr1 deficiency accelerates placental calcification and restricts fetal growth in late gestation. J. Bone Miner. Res. 35, 116–129 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Hagemeyer N., Hanft K. M., Akriditou M. A., Unger N., Park E. S., Stanley E. R., Staszewski O., Dimou L., Prinz M., Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 134, 441–458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindblom P., Gerhardt H., Liebner S., Abramsson A., Enge M., Hellstrom M., Backstrom G., Fredriksson S., Landegren U., Nystrom H. C., Bergstrom G., Dejana E., Ostman A., Lindahl P., Betsholtz C., Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 17, 1835–1840 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yona S., Kim K. W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., Jung S., Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radde R., Bolmont T., Kaeser S. A., Coomaraswamy J., Lindau D., Stoltze L., Calhoun M. E., Jaggi F., Wolburg H., Gengler S., Haass C., Ghetti B., Czech C., Holscher C., Mathews P. M., Jucker M., Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mombaerts P., Iacomini J., Johnson R. S., Herrup K., Tonegawa S., Papaioannou V. E., RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992). [DOI] [PubMed] [Google Scholar]
- 58.Turnbull I. R., Gilfillan S., Cella M., Aoshi T., Miller M., Piccio L., Hernandez M., Colonna M., Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabc4898/DC1