

Engineered ionizable lipid nanoparticles (LNPs) were developed for the LSEC-specific delivery of RNA therapeutics.
Abstract
Ionizable lipid nanoparticles (LNPs) have been widely used for in vivo delivery of RNA therapeutics into the liver. However, a main challenge remains to develop LNP formulations for selective delivery of RNA into certain types of liver cells, such as hepatocytes and liver sinusoidal endothelial cells (LSECs). Here, we report the engineered LNPs for the targeted delivery of RNA into hepatocytes and LSECs. The effects of particle size and polyethylene glycol–lipid content in the LNPs were evaluated for the hepatocyte-specific delivery of mRNA by ApoE-mediated cellular uptake through low-density lipoprotein receptors. Targeted delivery of RNA to LSECs was further investigated using active ligands. Incorporation of mannose allowed the selective delivery of RNA to LSECs, while minimizing the unwanted cellular uptake by hepatocytes. These results demonstrate that engineered LNPs have great potential for the cell type–specific delivery of RNA into the liver and other tissues.
INTRODUCTION
In recent years, RNA therapeutics have received tremendous attention as a tool to regulate gene expression in patients. These approaches include the regulation of abnormal gene expression by short interfering RNA (siRNA) and mRNA (1–8). There have been two U.S. Food and Drug Administration–approved siRNA therapeutics (patisiran and givosiran) for rare disease treatments. These drugs target the selective degradation of mRNA to reduce the expression of disease-associated proteins in polyneuropathy and acute hepatic porphyria (2, 5, 9). In addition, synthetic mRNA can produce insufficient proteins (e.g., factor IX and vascular endothelial growth factor) in patients to treat various diseases such as hemophilia A and ischemic heart disease (3, 6). Furthermore, in vivo genome editing has been introduced, and multiple studies reported a successful gene editing in mice by the delivery of a single guide RNA with synthetic mRNA encoding CRISPR-Cas9 (10, 11).
To fully realize the potential of RNA therapeutics, an efficient in vivo delivery system is of the utmost importance. Ionizable lipid nanoparticles (LNPs) have been widely used for the systemic delivery of RNA therapeutics. LNPs are mainly composed of ionizable lipid or lipid-like materials with helper lipid, cholesterol, and polyethylene glycol (PEG)–lipid. Previously, various types of ionizable lipid materials were reported for LNP formulations such as C12-200, cKK-E12, and DLin-MC3-DMA (12–14). These LNPs showed an efficient gene silencing in the liver at the dosing level of 0.002 mg of siRNA/kg (14). The mechanism of cellular uptake was well investigated, and adsorption of serum ApoE (apolipoprotein E) on the surface of LNPs was a major effector facilitating the intracellular delivery of LNPs into hepatocytes through low-density lipoprotein (LDL) receptors (15). Although LNPs are particularly advantageous for in vivo delivery, systemic delivery of RNA therapeutics other than liver hepatocytes remains highly challenging.
The liver consists of two major types of cells, including parenchymal cells (e.g., hepatocytes) and nonparenchymal cells [e.g., liver sinusoidal endothelial cells (LSECs), Kupffer cells, and stellate cells] (16–19). Given their physiological differences, a cell type–specific delivery of RNA in the liver requires a precise engineering of LNPs. For example, LSECs have the highest endocytic capacity without a basement membrane, making them the most permeable among liver cells (20). LSECs have many fenestrae, allowing the facile transport of macromolecules between sinusoidal lumen and hepatocytes. Because of the average diameter of liver fenestrae (~100 nm in human and ~140 nm in mouse), only small nanoparticles can penetrate through these holes (21). Chen and coworkers (22) previously reported the effect of particle size on the in vivo potency of LNPs in the liver. They showed that the particle size heavily influenced the gene delivery efficacy of LNPs, as well as the stability of LNPs during blood circulation.
Nanoparticles that cannot penetrate through the fenestrae are taken up by different types of liver cells such as LSECs and Kupffer cells. It was shown that LNPs with larger particle size can be delivered to LSECs and induced Tie2 gene silencing (23). The size and composition of LNPs can help the endothelium-specific delivery; however, the detailed mechanism of cell-specific uptake is not well understood. Therefore, targeting specific cells in the liver requires an alternative strategy other than adjusting the physical size and composition of the LNPs. Ligand incorporation can be an ideal approach for selective delivery of RNA therapeutics to targeted liver cells. LSECs play an important role in scavenger system and express selective receptors such as stabilin receptors, mannose receptors, and Fc-γRIIb2 (17). These receptors are involved in the removal of blood-borne antigens by active endocytosis. For example, phosphorothioate-modified antisense oligonucleotides or hyaluronic acid–conjugated nanoparticles were successfully delivered to LSECs by targeting the scavenger receptors (24, 25).
In this study, we report the engineered LNP formulations for the targeted delivery of RNA into different types of cells in the liver. Hepatocytes and LSECs are two major cells involved in various liver-related diseases. Hepatocytes are often associated with rare diseases in metabolism and endocrine dysfunctions, while LSECs are closely related with chronic liver diseases such as liver fibrosis and cirrhosis (19, 26, 27). It is important to selectively target the liver cells in different diseases, and its selectivity becomes more critical when introducing in vivo gene regulation and editing. Here, ionizable lipids were first synthesized and screened for their pKa (where Ka is the acid dissociation constant) and endolytic activities. Selected lipid candidates were further evaluated for in vitro and in vivo gene expression after formulating LNPs with reporter mRNA [firefly luciferase (FLuc)]. Targeted delivery of LNPs with mRNA (Cre) in different types of cells in the liver was examined by controlling the particle size and PEG-lipid content of LNPs. In addition, engineered LNPs with mannose moieties were prepared for active targeting of LSECs. Optimized condition for the targeted delivery of mRNA to LSECs was lastly confirmed. Additional in vivo gene silencing study revealed that these engineered LNPs provided compatible formulation parameters with siRNA. Moreover, the engineered LNPs with mannose moieties showed more selectivity over conventional LNPs and provided superior potential for active targeting of LSECs in the liver.
RESULTS
Formulation of LNPs
To identify efficient ionizable lipid materials, we synthesized six different ionizable lipids by an epoxide ring-opening reaction with amines (Fig. 1, A and B). Previously, we reported poly(amido-amines), cystamine-bisacrylamide–106 (here reported as 246), for plasmid DNA transfection (28). This allowed enhanced transfection efficiency than a commercial agent in vitro. On the basis of previous study, we chose six different piperazine- or piperidine-containing amine candidates for this study. These amines were reacted under 90°C, together with epoxide that has 10 carbon tails. Before testing pKa and transfection efficiency, resulting ionizable lipids were formulated into LNPs. As in previously reported studies, newly synthesized ionizable lipids (241C10 to 246C10) were formulated into LNPs with dioleoylphosphatidylethanolamine (DOPE), cholesterol, and C16-PEG2000 ceramide (PEG-lipid) as well as mRNA by using a microfluidic-based system (29, 30). As a representative, 246C10 LNPs were imaged with cryo–transmission electron microscopy, and these LNPs had an average diameter of 60 to 100 nm (Fig. 1C).
Fig. 1. Preparation of LNPs derived from developed ionizable lipids.
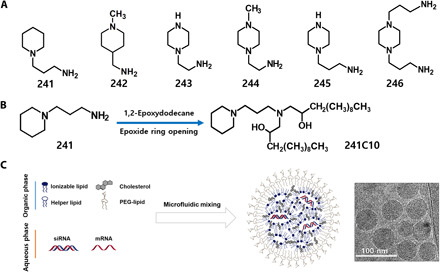
(A) Six different piperidine- and piperazine-containing amines used in this study. (B) Ionizable lipids were synthesized by epoxide ring opening with amines and epoxide containing 10 carbon tails. (C) LNPs were prepared via a microfluidic system with ionizable lipids, helper lipid, cholesterol, and PEG-lipid. Prepared LNPs were examined with cryo–transmission electron microscopy for their size and structural analysis.
Physicochemical properties of ionizable LNP candidates
Next, we evaluated the pKa of LNPs. LNPs were prepared with mRNA formulations. pKa of each LNPs was determined by 2-(p-toluidino)-6-napthalene sulfonic acid (TNS) assay (31–33). Formulated LNPs were titrated from pH range of 2.5 to 11.0 in 0.5 pH increments. The measured fluorescence was normalized by (fluorescence − minimum fluorescence) divided by the (maximum fluorescence − minimum fluorescence). The normalized fluorescence was evaluated by a curve fit analysis, and a fluorescence titration S curve was generated (fig. S3, A and B). From the resulting curves, pKa of LNPs was calculated (pH at 50% of maximum fluorescence) (fig. S3C). Anionic TNS interacts with positively charged ionizable lipids and becomes lipophilic. As the pH value approaches the pKa value of the each LNPs, TNS becomes less lipophilic and more water molecules can quench TNS fluorescence. Therefore, “S”-shaped curve with pKa of 6.5 to 7.0 represents that these LNPs have a good chance to interact well with endosomal membrane and escape endosomes upon acidification. Among ionizable lipid candidates, 244C10, 245C10, and 246C10 resulted in a sharp sigmoidal-shaped curve with corresponding pKa values of 5.5, 5.6, and 6.75, respectively.
Characterization and evaluation of ionizable LNP candidates in vitro
To evaluate the efficacy of ionizable lipid candidates to deliver RNA in cells, we formulated six different LNPs with mRNA encoding firefly luciferase (mFLuc). Before cellular transfection using HeLa cells, formulated LNPs (241C10 to 246C10) were incubated with apolipoprotein E3 (apoE3) at the concentration of 0.1 μg/ml for 10 min at room temperature. apoE3 is a major isoform of ApoE in human (34). As shown in fig. S4 (A to D), ApoE adsorption resulted in the increase in size of LNPs, confirmed by dynamic light scattering (DLS) measurement. It is well known that lipoproteins are associated with LNPs in blood and enhance the cellular uptake of LNPs via LDL receptors (LDLRs). HeLa cells are known to express LDLR on their surface (15). Six hours after LNP treatments, cells were lysed with Bright-Glo reagent and the relative intensity of luciferase expression was evaluated. As shown in Fig. 2A, 244C10, 245C10, and 246C10 showed substanial luciferase expression and 246C10 LNPs showed the highest luminescent expression. In serum-containing media, the role of ApoE was not significant (Fig. 2B), whereas in serum-free media, ApoE incubation resulted in the increase in luciferase expression (Fig. 2C). Although preincubation of 244C10 and 245C10 LNPs with ApoE slightly enhanced the FLuc expression (100-fold), preincubation of 246C10 LNPs with ApoE showed the marked increase in FLuc expression by 400-fold. To further investigate the cellular uptake mechanism of LNPs, intracellular uptake of LNPs was inhibited with anti-LDLR antibody (Fig. 2D). The significant inhibition was observed only with 246C10 LNP formulation. These findings suggested that cellular uptake of 246C10 LNPs was mainly mediated by interaction between ApoE and LDLR.
Fig. 2. In vitro evaluation of ionizable lipid candidates.
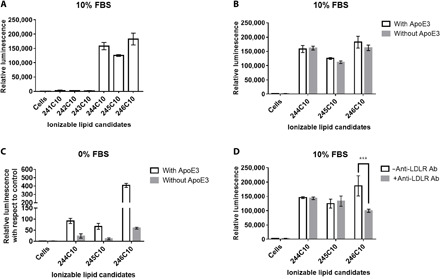
Ionizable lipid candidates were formulated into LNPs with mFLuc, and their efficacy was evaluated in vitro. After incubation with ApoE, LNPs were treated to HeLa cells (96 wells) at mRNA dose of 100 ng (n = 4). (A) Expression of mFLuc after transfection of HeLa cells in the presence of serum and (B) in the presence or absence of ApoE. (C) ApoE rescues the transfection efficiency of 246C10 in the absence of serum. (D) Pretreatment of anti-LDLR antibody (Ab) to HeLa cells decreased luciferase expression in 246C10 LNP–treated HeLa cells. P > 0.05, ***P < 0.001 as compared with antibody-pretreated cells. FBS, fetal bovine serum.
In vivo evaluation of ionizable LNP candidates for biodistribution and gene regulation
In addition to the in vitro evaluation of ionizable lipid candidates, we evaluated ionizable lipid candidates in vivo. LNPs with high transfection ability in previous data were pooled and formulated with mFLuc to confirm their biodistribution and FLuc expression. Sizes of mFLuc-loaded 244C10, 245C10, 246C10 LNPs were validated before injection. The mean particle size of LNPs remained relatively similar (60 to 70 nm). Formulated LNPs were intravenously injected to C57BL/6 mice. Luminescence was evaluated 3 hours after injection via live-animal and ex vivo organ imaging. As shown in Fig. 3, 244C10, 245C10, and 246C10 LNPs mediated the liver-specific delivery and expression of mFLuc. Like the in vitro cellular study, 246C10 LNPs showed the highest luciferase expression in the liver. In addition, ex vivo imaging of five different organs (liver, kidney, lung, spleen, and heart) supported the liver tissue–specific expression of mFLuc using 246C10 LNPs.
Fig. 3. In vivo evaluation of ionizable lipid candidates (mFLuc).
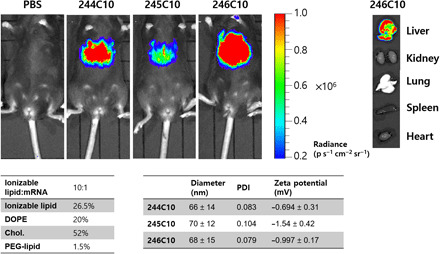
Ionizable lipid candidates were formulated with mFLuc. mFLuc-loaded LNPs were injected to C57BL/6 mice at mRNA dose of 0.1 mg/kg. Three hours after injection, bioluminescence was analyzed. 244C10- to 246C10-formulated LNPs resulted in potent luciferase expression. Ex vivo organ image showed that LNPs were mostly uptaken into liver. p, photons; PDI, polydispersity index.
To figure out the impact of ApoE on the cellular uptake of 246C10 LNPs in vivo, we also evaluated luciferase expression in ApoE knockout mice (fig. S5, A to D). By injecting ApoE-associated 246C10 LNPs in the ApoE knockout mice, luciferase expression was substantially increased in the liver. The result was consistent with in vitro cellular uptake results and demonstrated that ApoE association could facilitate the delivery of 246C10 LNPs to the liver.
Next, we investigated the biodistribution of mRNA-loaded LNPs. Given the fact that injected LNPs (<100 nm) were mostly distributed to the liver, the question was which types of liver cells mostly uptake these LNPs. Previously, Chen et al. (22) reported on the size-dependent gene silencing of LNPs in hepatocytes, following intravenous injection. Upon entering systemic circulation, LNPs (<100 nm) were mostly accumulated in the liver. In particular, LNPs with sizes of 45 to 80 nm showed significant silencing in hepatocytes. To figure out which types of liver cells were mostly interacting with the formulated LNPs, we prepared Cre mRNA (mCre)–loaded LNPs with their size under 100 nm. mCre was formulated into LNPs, and mCre dose of 0.5 mg/kg was injected into mice carrying a loxP-flanked stop cassette. Once a stop cassette is deleted, mice can express robust tdTomato fluorescence. Therefore, delivery of functional mCre allows the facile identification of cells transfected with formulated LNPs. As shown in Fig. 4A, robust tdTomato fluorescence was detected exclusively in the liver. To further confirm the cell type–specific gene edition in the liver, we extracted a mouse liver 2 days after injection and evaluated tdTomato fluorescence by the histology observation of liver tissue. Compared to phosphate-buffered saline (PBS)–injected mice, strong tdTomato fluorescence was detected in hepatocytes (Fig. 4B). To further examine transfection efficiency in different types of liver cells, we labeled hepatocytes, LSECs, and Kupffer cells with specific antibodies. Then, tdTomato fluorescence–positive cells were evaluated by histological analysis using the MetaMorph image software. As shown in Fig. 4C, 80% of hepatocytes, 40% of endothelial cells, and 10% of Kupffer cells exhibited the positive results of tdTomato fluorescence. In addition to the histological evaluation, tdTomato fluorescence–positive cells were also confirmed by flow cytometry analysis (fig. S9B). Transfection efficiency in hepatocytes was the highest among different types of liver cells.
Fig. 4. In vivo evaluation of ionizable lipid candidates (mCre).
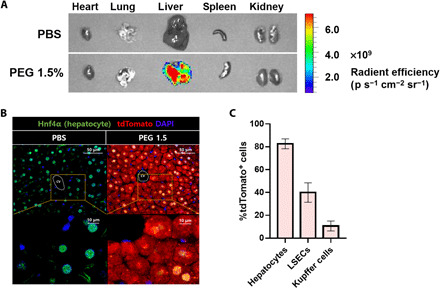
Ionizable lipid candidates were formulated with mCre. mCre-loaded LNPs were injected to LSL-tdTomato at mRNA dose of 0.5 mg/kg. Two-days after injection, tdTomato fluorescence was analyzed. (A) Ex vivo organ image showed that LNPs were mostly uptaken into the liver. (B) Liver histology image showed significant tdTomato fluorescence in the hepatocyte. cv, central vein. (C) Transfection efficiency in hepatocytes, LSECs, and Kupffer cells was confirmed by histological analysis (n = 5). DAPI, 4,6-diamidino-2-phenylindole.
Targeted delivery of mRNA-loaded LNPs to different types of liver cells by controlling their size and PEG-lipid content
After establishing LNP delivery to hepatocytes, we examined the targeted delivery of mRNA-loaded LNPs to different types of liver cells by controlling their size and PEG-lipid content. Among these cells, we focused on LSECs, where many of liver-related diseases originated. First, we hypothesized that as LSECs reside in the liver blood wall, LNPs that cannot penetrate the fenestrae will be uptaken into the LSECs. We prepared various LNPs loaded with mCre, and their size was ranging from 50 to 100 nm by varying PEG-lipid (%) (Fig. 5, A and B). For a change in the mole ratio of PEG-lipid, the change must be replaced with the mole ratio of other components. Mole ratio of PEG-lipid was varied by expending that of cholesterol. We have chosen cholesterol because, first, cholesterol has the least effect on endosomal release compared to other lipids (35). Second, cholesterol accounts for a large mole ratio among the lipid compositions, so this small change only accounts for less than 10% of the original, which will have very little impact. LNPs with mCre (0.5 mg/kg) were intravenously injected into mice. Two-days after injection, the liver was extracted and tdTomato-positive cells were confirmed by histology evaluation and flow cytometry analysis.
Fig. 5. In vivo delivery of mRNA-loaded LNPs to different types of liver cells by controlling their size and PEG-lipid content.

(A) Formulation ratio between lipid components. PEG-lipid was changed from 1.0 to 3.0% by expending that of cholesterol. (B) Resulting size and encapsulation efficiency of mRNA-loaded LNPs with different PEG-lipid density. (C) LSL-tdTomato mice were injected intravenously with mCre (0.5 mg/kg). After 2 days of injection, tdTomato fluorescence–positive liver cells were confirmed by histological analysis (n = 5).
Figure 5C and fig. S9 (A to C) show that LNPs with 68 nm (1.5% PEG-lipid) exhibited potent tdTomato fluorescence in hepatocytes. Smaller LNPs with less than 100 nm in size could easily penetrate the fenestrae upon entering the liver blood vessel and showed maximum expression of tdTomato fluorescence in hepatocytes at PEG-lipid content of 1.5% with 60 nm in size. However, reduced tdTomato fluorescence throughout the liver cells was observed with LNP formulations with 3% PEG-lipid. The observed phenomenon of reduced tdTomato fluorescence in higher PEG-lipid content may be attributed to the reduced ApoE-mediated cellular uptake of LNPs (36). PEG-lipid content strongly affects the ApoE adsorption on the LNPs, and it was confirmed by ApoE enzyme-linked immunosorbent assay (ELISA) (fig. S6). LNPs with 1.0, 1.5, 3.0, and 5.0% PEG-lipid were prepared. Prepared LNPs were incubated with ApoE and purified by ultracentrifugation. We then evaluated the amount of adsorbed ApoE by ELISA. Assuming that protein adsorption is governed by the surface area of LNP, ApoE adsorption per LNP particle was decreased when PEG-lipid was higher than 3%. This indicated that higher PEG content hindered the ApoE adsorption, thus resulting in the decrease of luciferase expression in mFLuc-loaded LNP–injected mice (fig. S7).
Potent tdTomato fluorescence in LSECs was observed for LNPs with 100 nm in size (1.0% PEG-lipid) that is similar to the diameter of fenestrae. However, the uptake of LNPs by LSECs was decreased correspondingly with the reduction in LNP size (60 nm). It is likely that more LNPs are directed to hepatocytes through fenestrae rather than interacting with LSECs for cellular entry. Again, with higher PEG-lipid content of 3% or more, a poor ApoE association would be the cause of reduced tdTomato fluorescence. These results confirmed the importance of size and PEG-lipid content of LNPs governing their delivery efficacy to hepatocytes and LSECs. Unfortunately, LNPs with 1.0% PEG-lipid content not only showed increased LNP delivery into LSECs but also resulted in potent cellular uptake by hepatocytes. It would be difficult to achieve LSEC-specific delivery of RNA by controlling size and PEG-lipid content of LNPs. Therefore, a different strategy was needed.
Active targeting of LSECs using ligand-incorporated LNPs
Upon entering the blood stream, nanoparticles rapidly absorb serum proteins on their surface. The effects of protein corona heavily control the biodistribution of nanoparticles. For some LNPs, ApoE adsorption is essential for the cellular uptake of LNPs. Akinc et al. (15) have described the role of ApoE in the in vivo uptake of LNPs by hepatocytes. They compared the uptake of LNPs by hepatocytes in wild-type and ApoE knockout mice. The activity of LNPs was remarkably reduced in ApoE knockout (ApoE−/−) mice compared to wild-type mice. Since the major cell population in the liver is hepatocytes and they overexpress LDLR, an alternative targeting ligand is necessary for LSEC-specific delivery of RNA other than the ApoE. Previously, specific targeting ligands were evaluated for enhancing the selective delivery of LNPs to target cells (15). Kumar et al. (36) showed decreased activity of LNPs with higher PEG-lipid content in hepatocytes due to reduced association of ApoE with LNPs. They showed that LNPs with 5% PEG-lipid have a lower binding affinity with ApoE compared to LNPs with 1.5% PEG-lipid. By introducing unique targeting ligands to the highly PEGylated LNPs, ApoE-mediated cellular uptake can be blocked and selective delivery of LNPs can be achieved.
Among various liver cells, LSECs are substantial targets for RNA therapeutics because LSECs are closely related to fetal liver diseases resulting from LSECs-derived gene products (17, 19). To achieve the selective delivery of LNPs to LSECs, we introduced LSEC-specific ligands to highly PEGylated LNPs. Mannose receptor is one of the receptors that specifically expressed on human and mice LSECs (17). Mannose-conjugated nanoparticles have been investigated in various targeted delivery studies, and these nanoparticles have been proven to be safe (37, 38). To introduce mannose moieties on the surface of LNPs, we used mannose-PEG lipid (Fig. 6A). Since mannose is conjugated to PEG, when mannose-PEG lipid is formulated into LNPs together with other lipid components, mannose can be exposed to the outer layer of LNPs. First, we formulated LNPs with mannose-PEG lipid with different content and tested whether introduction of mannose-PEG lipid can alter the cellular uptake of LNPs as compared to that of original LNPs in the absence of ApoE.
Fig. 6. In vitro evaluation of receptor-mediated cellular uptake of mannose-LNPs.
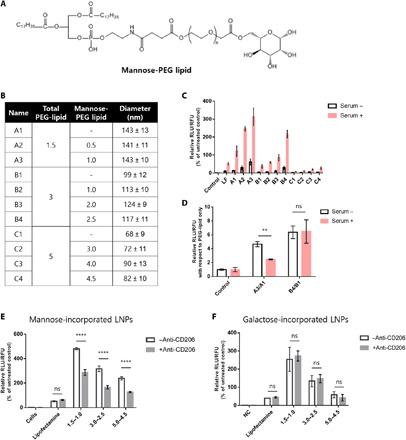
(A) Structure of mannose-PEG lipid. (B) Formulation details. (C) HepG2 cells were incubated with LNPs at 25 ng of mFLuc (n = 4). Mannose-lipid–incorporated LNPs allowed enhanced cellular internalization compared to LNPs with PEG-lipid only. (D) Relative FLuc expression of mannose-incorporated LNPs with respect to that of LNPs with PEG-lipid only. RLU, relative luminescence units; RFU, relative fluorescence units. Pretreatment of anti-CD206 resulted in decreased mRNA expression in LNPs with mannose-PEG lipid–treated HepG2 cells (E) but not in LNPs with PEG-galactose–treated HepG2 cells (F). ns, not significant: P > 0.05; **P < 0.01 and ****P < 0.0001 as compared with LNPs formulated with PEG-lipid only.
Mannose-PEG lipid was incorporated from 0.5 to 4.5% to LNPs with total 1.5, 3.0, and 5.0% PEG-lipid (Fig. 6B). Prepared LNPs with mFLuc were treated to the HepG2 cells with serum-containing media or serum-free media. Luciferase level was evaluated after 24 hours. Since HepG2 cells are known to express mannose receptor (CD206), mannose-mediated cellular uptake can be quantitatively evaluated (39, 40). Figure 6C shows that the uptake of mannose-incorporated LNPs into HepG2 cells via CD206 was enhanced with increasing the content of mannose-PEG lipid in LNPs. Incorporation of mannose increased LNP uptake into HepG2 in the presence or absence of serum. To further evaluate the role of mannose incorporation in the uptake of LNPs by HepG2 cells, the mRNA expression level of mannose-incorporated LNPs was divided by that of LNPs with PEG-lipid only (Fig. 6D). Unexpectedly, incorporation of mannose to LNPs with 3.0% PEG-lipid showed a similar increase in mRNA expression level in the presence or absence of serum. On the contrary, for 1.5% PEG-lipid, incorporation of mannose did not result in an increase in mRNA expression level in the presence of serum compared to that in the absence of serum. In the presence of serum, uptake of mannose-incorporated LNPs with 1.5% of PEG-lipid could be simultaneously internalized by ApoE- and mannose-mediated pathway. Therefore, mannose/CD206-mediated LNP uptake was less significant. However, higher PEG-lipid content (3%) could allow reduced absorption of ApoE and mediate LNP uptake mainly by mannose and CD206 interaction. To further figure out whether LNPs with mannose were internalized via CD206-mediated pathway, we performed a cellular uptake inhibition experiment (Fig. 6, E and F). Anti-CD206 antibody was pretreated to the cells, and LNPs with mannose-PEG lipid or galactose-PEG lipid were incubated with HepG2 cells. Galactose is the one of mannose enantiomers and adopted as a control. The significant decrease in luciferase expression was confirmed when LNPs were incorporated with mannose-PEG. In contrast, mRNA expression of LNPs with galactose moieties was not inhibited by anti-LDLR antibody. This result indicated that mannose moieties could play an important role in CD206-mediated intracellular uptake of LNPs.
Next, to evaluate the selective targeting of mannose-incorporated LNPs to LSECs, we performed in vivo evaluation with two different mRNAs (mFLuc and mCre). LNPs were formulated with 3.0% PEG-lipid (with 2.5% mannose-PEG lipid). Since extrahepatic macrophages also express CD206, we first confirmed the liver-specific delivery of mFLuc-loaded LNPs with mannose. In addition, we compared a ligand-dependent mRNA delivery efficiency of LNPs using mannose-PEG lipid with that of galactose-PEG lipid. As shown in fig. S8, LNPs with mannose-PEG lipid and galactose-PEG lipid resulted in slightly bigger size than LNPs with PEG-lipid only. This phenomenon was previously reported among saccharide-conjugated nanoparticles (41, 42). The prepared mFLuc-loaded LNPs were injected intravenously into C57BL/6 mice at a dose of 0.1 mg/kg. Whole-body image showed substantial bioluminescence in the mice liver with mannose-incorporated LNPs and supported the liver tissue–specific expression of mFLuc.
We then examined the ability of LSEC-specific gene editing using mCre. mCre-loaded LNPs with mannose were injected to mice at a dose of mRNA at 0.5 mg/kg. As shown in Fig. 7A, ex vivo organ images also confirmed robust liver-specific tdTomato fluorescence. Next, cell type–specific fluorescence was quantified by imaging tissue sections. Notable tdTomato fluorescence was detected along the liver vessel (LSECs), and Kupffer cells rarely expressed tdTomato fluorescence (Fig. 7B). We further quantified this phenomenon by counting tdTomato fluorescence–positive cells with MetaMorph image software. As shown in Fig. 7C, 15% of hepatocytes, 70% of LSECs, and 15% of Kupffer cells were tdTomato fluorescence positive. tdTomato fluorescence–positive cells were also confirmed by flow cytometry analysis (fig. S9D). Notably, compared with LNPs with 3.0% PEG-lipid, mannose-incorporated LNPs showed the substantial decrease of tdTomato fluorescence in hepatocytes. While some discussion exists about the expression of CD206 on the surface of mice Kupffer cells, in our data, incorporation of mannose seemed to have little effect on the uptake of the LNPs by the Kupffer cells. These results demonstrated a highly effective targeting strategy to LSECs using mannose-incorporated LNPs.
Fig. 7. Targeted in vivo genome editing in LSECs by mannose-LNPs (mCre).

Ionizable lipid candidates were formulated with mCre. mCre-loaded LNPs were injected to LSL-tdTomato mice at mRNA dose of 0.5 mg/kg. After 2 days of injection, tdTomato fluorescence was analyzed. (A) Ex vivo organ image showed that LNPs with mannose were mostly uptaken into the liver. (B) Liver histology image showed significant tdTomato fluorescence along the liver vessel. α-SMA, α–smooth muscle actin. (C) Transfection efficiency of mannose-LNPs was evaluated among hepatocytes, LSECs, and Kupffer cells by histological analysis (n = 5).
Next, we confirmed the selective targeting of siRNA-loaded LNPs with mannose to LSECs. LNPs were formulated with 1.5% PEG-lipid (with 1.0% mannose-PEG lipid) and 3.0% PEG-lipid (with 2.5% mannose-PEG lipid) for targeting LSECs and compared the FVIII gene silencing efficiency to that of LNPs with PEG-lipid only. As shown in Fig. 8 (A and C), similar to that of mRNA-loaded LNPs, LNPs with mannose-PEG lipid resulted in slightly bigger size than LNPs with PEG-lipid only. The prepared LNPs with siFVIII were injected into C57BL/6 mice at the dose of 0.5 mg/kg. After 2 days of injection, blood was collected and the level of FVIII protein in serum was validated. As shown in Fig. 8B, addition of mannose-PEG lipid into LNPs with 3% total PEG-lipid content showed a more enhanced FVIII inhibition in LSECs. On the contrary, for 1.5% PEG-lipid, incorporation of mannose to LNPs was less significant. This could be due to high ApoE adsorption at lower PEG content that facilitated the ApoE/LDLR-mediated uptake of LNPs. In addition, we further compared the LNP uptake using mannose-PEG lipid with that of galactose-PEG lipid. As expected, galactose-incorporated LNPs showed similar diameter of mannose-incorporated LNPs. However, in vivo study revealed that significant FVIII inhibition was only observed in mannose-incorporated LNPs. To determine whether addition of mannose-PEG lipid could decrease the LNP uptake into hepatocytes, LNPs were formulated with siFVII and injected into C57BL/6 mice at the dose of 0.2 mg/kg. As shown in Fig. 8D, addition of mannose-PEG lipid reduced the uptake of LNPs with 1.5 and 3.0% PEG-lipid, whereas addition of galactose-PEG lipid was less significant.
Fig. 8. Selective in vivo gene silencing in LSECs using mannose-LNPs.
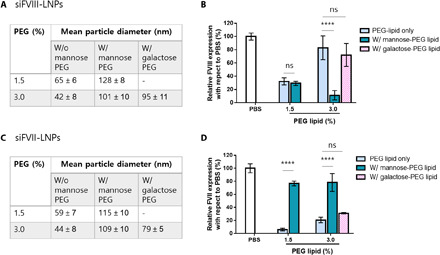
C57BL/6 mice were injected with siFVII (0.2 mg/kg) or siFVIII (0.5 mg/kg; n = 3 to 5). (A and C) Formulation details. (B) Incorporation of mannose to LNPs with 3.0% of PEG-lipid increased the delivery of siRNA to LSECs but incorporation of galactose did not. (D) Incorporation of mannose to LNPs with 1.5 and 3.0% PEG-lipid reduced the delivery of siRNA to hepatocytes, whereas incorporation of galactose to LNPs with 3.0% PEG-lipid did not. (****P < 0.0001 as compared between sugar moiety–incorporated LNPs and LNPs with PEG-lipid only; ns: P > 0.05).
DISCUSSION
A platform technology that delivers RNA therapeutics to targeted cells has great potential for treating various liver diseases associated with different types of cells in the liver. In particular, LSECs participate in initiation and progression of chronic liver diseases by change in their pathological characteristics in response to chemical toxins, cytokines, and inflammatory stimuli (17, 19). Previous studies have shown that targeting endothelial sphingosine-1-phosphate receptor-1 and Bax in LSECs could also reduce apoptosis of LSECs and eventually reduced acute hepatic failure (43, 44). The conjugation of hyaluronan, KLGR peptide, and mannose resulted in increased uptake of nanoparticles into LSECs compared to nonconjugated groups. Moreover, previously reported studies have confirmed that LNPs can be delivered to different tissue by changing LNP formulations. Some of these formulations showed increased mRNA delivery to LSECs without targeting ligands (45). Nevertheless, its selectivity remained not well understood (37).
To translate the targeted delivery to LSECs, ApoE-dependent cellular uptake must be replaced by alternative ligand selection and incorporation. Upon entering blood circulation system, LNPs can be readily delivered to hepatocytes and LSECs by ApoE-dependent cellular uptake. However, delivery efficacy of LNPs in LSECs is significantly less than that of hepatocytes (23). Here, we demonstrated a targeted delivery of RNA to LSECs by incorporating active ligands to LNPs. The mannose ligand can interact with specific receptors (CD206) expressed on LSECs. We have shown that incorporation of mannose to LNPs with high PEG-lipid content (3%) allowed selective delivery of mRNA/siRNA only to LSECs through mannose-CD206 interaction. It is likely that, when PEG-lipid content is high enough, the ApoE-mediated cellular uptake is mostly reduced, while mannose-CD206 interaction becomes predominant for the cellular entry of LNPs. Moreover, mannose-incorporated LNPs showed bigger physical size than the diameter of the fenestrae. This additionally reduced the uptake of LNPs to hepatocytes and allowed more selective uptake into the LSECs. Collectively, our results successfully demonstrated the targeted delivery of RNA into different types of cells in the liver by controlling physical size of LNPs, PEG-lipid content, and incorporation of active targeting ligands. Overall, this approach may offer new strategies to develop cell type–specific delivery of RNA in the liver and other tissues.
MATERIALS AND METHODS
Materials
241 to 244 and 246 amines were purchased from Sigma-Aldrich, and 245 amine was purchased from Tokyo Chemical Industry (TCI). 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and C16-PEG ceramide (PEG-lipid) were purchased from Avanti Polar Lipids. Cholesterol was purchased from Sigma-Aldrich. DSPE-PEG-mannose and DSPE-PEG-galactose were purchased from Biochempeg. Firefly luciferase mRNA (mFLuc) and mCre were purchased from TriLink BioTechnologies. d-luciferin and Bright-Glo reagent were purchased from Promega.
Preparation of ionizable lipids
Ionizable lipids were synthesized by epoxide ring-opening reaction. Reaction was conducted under solvent-free environment. To a solution of epoxide with 10 carbon tails (C10) (1,2-epoxydodecane; Sigma-Aldrich), piperazine- and piperidine-containing amines were added at a ratio of 1.2*N:1 epoxide/amine, where N is the number of secondary amine*1 + number of primary amine*2. For example, to synthesize 246C10, to a 5-ml vial, 1 ml of epoxide (4.65 mmol, 4.8 equivalents) was added and 200 μl of amine (0.97 mmol, 1 equivalent) was added dropwise. The reaction solution was stirred at 300 rpm for 3 days at 90°C. Ionizable lipids were isolated as light yellow oil in 70% yield using flash column chromatography system (CombiFlash). Representative nuclear magnetic resonance data and tandem mass spectrometry spectra for 246C10 are shown in figs. S1 and S2.
Preparation of ionizable LNPs
Ionizable LNPs were prepared as previously described (29, 30). Briefly, lipid components and RNAs were first dissolved in organic phase and aqueous phase to achieve a 1:3 volume ratio. For siRNA delivery, ionizable lipids, cholesterol, DSPC, and PEG-lipid were dissolved in ethanol. siRNA was diluted in 50 mM sodium acetate. The final ionizable lipids:siRNA weight ratio was 7.5:1. For mRNA delivery, ionizable lipids, cholesterol, DOPE, and PEG-lipid were dissolved in ethanol, and mRNA was diluted in 10 mM citrate. The final ionizable lipids:mRNA weight ratio was 10:1. Prepared solutions were mixed via a microfluidic mixing device at a flow rate of 12 ml/min to obtain LNPs. Then, prepared LNPs were dialyzed against PBS using 3500 MWCO (Molecular weight cut-off) dialysis cassettes (Life Technologies) overnight. LNPs were characterized for their size and RNA therapeutics encapsulation efficiency.
For confirming characterization of LNPs, LNPs were dialyzed to RNA dose of 0.001 mg/ml with PBS. DLS was used to confirm mean particle diameter, polydispersity index, and zeta potential. RNA therapeutics encapsulation efficiency was confirmed by RiboGreen Assay (Life Technologies) (29, 30).
TNS assay
pKa of formulated LNPs was evaluated by TNS assay as described previously (31–33). Briefly, pH buffers from 2.5 to 11.0 in 0.5 increments were prepared by adjusting a solution of 20 mM sodium phosphate, 25 mM citrate, 20 mM ammonium acetate, and 150 mM NaCl with 0.1 N NaOH and 0.1 N HCl. Each pH buffer was added to a black 96-well plate in 100 μl. In addition, 300 μM stock solution of TNS was added to the above pH buffer in the final concentration of 6 μM. Last, formulated LNPs were added to the mixture solution in the final concentration of 25 μM. Fluorescence intensity was obtained by Tecan at an excitation of 325 nm and an emission of 435 nm. The measured fluorescence was normalized by (fluorescence − minimum fluorescence) divided by the (maximum fluorescence − minimum fluorescence). The normalized fluorescence was evaluated by a curve fit analysis, resulting in a fluorescence titration S curve. pKa of each LNPs was calculated as the pH value where half of the maximum fluorescence is reached.
In vitro evaluation of ionizable lipid candidates
HeLa cells were prepared in a white 96-well plate 16 hours before transfection at 80% confluency. Ionizable lipid candidates were formulated into LNPs with mFLuc, together with DOPE, cholesterol, and PEG-lipid by pipetting. Molar ratio between lipid components was 26.5:20:52:1.5. For dialysis, three times volume of PBS was added to the LNPs solution. For ApoE association, mRNA-loaded LNPs were incubated with ApoE3 (0.1 μg/ml) for 10 min at room temperature and treated to prepared HeLa cells at mRNA dose of 0.25 μg/ml. Six hours later, cells were lysed with Bright-Glo reagents and relative luminescence was read on a Tecan plate reader. For inhibition of LDLR receptor, HeLa cells were treated with anti-LDLR antibody (5 μg/ml) at 37°C for 1 hour and the medium was replaced with a serum-containing medium. mFLuc-loaded LNPs were treated to prepared HeLa cells and incubated for 3 hours. After incubation, the medium was replaced and incubated for 24 hours in serum-containing media. Twenty-four hours later, cells were lysed with Bright-Glo reagents and luminescence intensity was read on a Tecan plate reader. Cell viability was measured using Pierce Coomassie Plus (Thermo Fisher Scientific). Luminescence level was normalized to cell viability.
In vitro evaluation of CD206-dependent uptake
HepG2 cells were prepared in a 96-well plate 16 hours before transfection at 80% confluency. 246C10 were formulated into LNPs with mFLuc, together with DOPE, cholesterol, PEG-lipid, and mannose-PEG lipid by pipetting. mFLuc-loaded LNPs were treated to prepared HepG2 cells in the presence or absence of serum. After 3 hours, medium was replaced with serum-containing medium. Twenty-four hours later, cells were lysed with Bright-Glo reagents and relative luminescence was read on a Tecan plate reader. Cell viability was measured by Pierce Coomassie Plus (Thermo Fisher Scientific). Luminescence level was normalized to cell viability. For inhibition of mannose- or galactose-incorporated LNP uptake, HepG2 cells were treated with anti-CD206 antibody at 37°C for 3 hours and the medium was replaced with a serum-containing medium. mFLuc-loaded LNPs were treated to prepared HepG2 cells. Three hours later, the medium was replaced and incubated for 24 hours in serum-containing media.
In vivo evaluation of ionizable lipid candidates
Procedures for animal studies were approved by the Institutional Animal Care and Use Committee at the Ewha Womans University. Female C57BL/6 mice (7 to 10 weeks old) were purchased from ORIENT BIO. LNPs for in vivo imaging were formulated with mRNA encoding luciferase (mFLuc). Ionizable lipids:RNA weight ratio was 10:1, and molar ratio between lipids was 26.5:20:52:1.5 (ionizable lipid, DOPE, cholesterol, and PEG-lipid). Formulated LNPs were administered intravenously to mice at a dose of mFLuc at 0.1 mg/kg. Three hours later, d-luciferin was injected intraperitoneally and incubated for 15 min. Luciferase expression of whole-body and ex vivo organ imaging was confirmed by an IVIS (PerkinElmer).
To confirm ApoE-dependent uptake of LNPs by the liver, 6-week-old male ApoE−/− mice were obtained from ORIENT BIO. mFLuc-LNPs were incubated with ApoE (0.1 mg/kg) overnight at 4°C and injected to mice. Three hours later, bioluminescence was compared with that of mFLuc-LNP–injected mice (without incubation of ApoE).
Evaluation of LNPs delivery in LSL (loxP-STOP-loxP)–tdTomato mice
Procedures for animal studies were approved by the Institutional Animal Care and Use Committee at the Yonsei University. LSL-tdTomato mice (007914) were purchased from the Jackson laboratory. To confirm liver cell type–dependent gene editing, LNPs were prepared with mCre. Eight-week-old mice were intravenously injected at a dose of 0.5 mg/kg. After 2 days, mice were anesthetized by isoflurane and the liver was perfused with 25 ml of 1× PBS. The liver was collected in a 50-ml conical tube and digested with Liberase (Sigma-Aldrich) at room temperature for 45 min. Digested cells were filtered through a 40-μm mesh. Then, cells were stained with specific antibodies according to the manufacturer’s protocol. The antibodies used for specific liver cell type detection were APC (Allophycocyanin)–labeled anti-CD31 (BioLegend), fluorescein isothiocyanate–labeled anti-CD45 (BioLegend), and PE/cy7-labeled anti-CD68 (BioLegend), and PE-labeled anti-CD47 (BioLegend) was used for tdTomato compensation. Last, hepatocytes and nonparenchymal cells were analyzed by NovoCyte 2060R (ACEA). Representative flow cytometry gate strategy is shown in fig. S10.
For immunofluorescence, isolated livers were fixed with 4% paraformaldehyde. After embedding paraffin, embedded tissue was sectioned (5 μm) using microtome. Then, the sections incubated in pH 6.0 buffer (Dako, S1699) for heat-induced epitope retrieval (HIER). After HIER, the sections were stained with 4,6-diamidino-2-phenylindole, α–smooth muscle actin (Abcam, ab5694), Hnf4α (Hepatocyte nuclear factor 4 alpha) (R&D Systems, PP-H1415-00), and F4/80 (Abcam, ab6640). Fluorescence was detected by fluorescent microscopy (EVOS FL). tdTomato fluorescence–positive cells were digitally counted with a MetaMorph microscopy automation and image analysis software.
In vivo screening of FVII and FVIII activity
To study size-dependent LNP delivery to specific liver cells, we prepared siRNA against FVII-loaded (gift from Alnylam) and FVIII-loaded (ON-TARGETPlus SMARTpool siRNA, purchased from Dharmacon) LNPs. LNPs were complexed together with siRNA and lipid components. Ionizable lipids:DSPC:cholesterol:PEG-lipid molar ratio was 42.5:13:42.5:3, and PEG-lipid mole percent was decreased to 1.5% by expending that of cholesterol. For exogenous ligand-dependent targeting study, mannose-PEG lipid was added in 0.5 to 2.5 mole percent to formulation. siRNA-loaded LNPs were injected intravenously, and 2 days (siFVIII) or 3 days (siFVII) later, blood was collected via cheek bleeding. Blood was mixed with 0.1 M sodium citrate, and the plasma was separated from the cells. FVII or FVIII levels in blood were determined using the Coaset FVII or Coaset FVIII chromogenic assay (Chromogenix, MA) according to the manufacturer’s protocol with a standard curve obtained from PBS-injected mice.
Statistical analysis
All data are shown as means ± SD. The data were analyzed by one-way analysis of variance (ANOVA) and two-way ANOVA. P value of <0.05 was considered significant.
Acknowledgments
Funding: This research was supported by the National Research Foundation of Korea (NRF): 2020R1A2C2004364, 2018R1A5A2025286, 2019M3A9H1103786, 2018M3A9H3020844, and 2016-2017 Solvay Scholarship. Author contributions: M.K. designed the experiments and wrote the paper. M.J. contributed to the FLuc mRNA and siRNA delivery animal study. S.H., Y.C., and K.T.N. contributed to the LSL-tdTomato animal study. H.J. contributed to the synthesis of 246C10. H.J. and Y.S. contributed to the 246C10 formulation optimization study. J.P. and H.A.W. contributed to the perfusion experiment. K.L. supervised the specialized techniques. H.L. conceived and supervised the project and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper maybe requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabf4398/DC1
REFERENCES AND NOTES
- 1.Jin J., Wu X., Yin J., Li M., Shen J., Li J., Zhao Y., Zhao Q., Wu J., Wen Q., Cho C. H., Yi T., Xiao Z., Qu L., Identification of genetic mutations in cancer: Challenge and opportunity in the new era of targeted therapy. Front. Oncol. 9, 263 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kristen A. V., Ajroud-Driss S., Conceição I., Gorevic P., Kyriakides T., Obici L., Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 9, 5–23 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J. B., Karmali P. P., Chivukula P., Verma I. M., Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U.S.A. 114, E1941–E1950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ray K. K., Stoekenbroek R. M., Kallend D., Leiter L. A., Landmesser U., Wright R. S., Wijngaard P., Kastelein J. J. P., Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins: Prespecified secondary end points in ORION 1. Circulation 138, 1304–1316 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Agarwal S., Simon A. R., Goel V., Habtemariam B. A., Clausen V. A., Kim J. B., Robbie G. J., Pharmacokinetics and pharmacodynamics of the small interfering ribonucleic acid, givosiran, in patients with acute hepatic porphyria. Clin. Pharmacol. Ther. 108, 63–72 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Gan L. M., Lagerström-Fermér M., Carlsson L. G., Arfvidsson C., Egnell A.-C., Rudvik A., Kjaer M., Collén A., Thompson J. D., Joyal J., Chialda L., Koernicke T., Fuhr R., Chien K. R., Fritsche-Danielson R., Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 10, 871 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahin U., Karikó K., Türeci Ö., mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Cheng Q., Wei T., Jia Y., Farbiak L., Zhou K., Zhang S., Wei Y., Zhu H., Siegwart D. J., Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 30, e1805308 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Wood H., FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 14, 570 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Finn J. D., Smith A. R., Patel M. C., Shaw L., Youniss M. R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J., Shah R. R., Shah A., Ling D., Growe J., Pink M., Rohde E., Wood K. M., Salomon W. E., Harrington W. F., Dombrowski C., Strapps W. R., Chang Y., Morrissey D. V., A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 22, 2227–2235 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Yin H., Song C.-Q., Suresh S., Wu Q., Walsh S., Rhym L. H., Mintzer E., Bolukbasi M. F., Zhu L. J., Kauffman K., Mou H., Oberholzer A., Ding J., Kwan S.-Y., Bogorad R. L., Zatsepin T., Koteliansky V., Wolfe S. A., Xue W., Langer R., Anderson D. G., Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 35, 1179–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love K. T., Mahon K. P., Levins C. G., Whitehead K. A., Querbes W., Dorkin J. R., Qin J., Cantley W., Qin L. L., Racie T., Frank-Kamenetsky M., Yip K. N., Alvarez R., Sah D. W. Y., de Fougerolles A., Fitzgerald K., Koteliansky V., Akinc A., Langer R., Anderson D. G., Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. U.S.A. 107, 1864–1869 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cullis P. R., Hope M. J., Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 25, 1467–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong Y. Z., Love K. T., Dorkin J. R., Sirirungruang S., Zhang Y., Chen D., Bogorad R. L., Yin H., Chen Y., Vegas A. J., Alabi C. A., Sahay G., Olejnik K. T., Wang W., Schroeder A., Lytton-Jean A. K. R., Siegwart D. J., Akinc A., Barnes C., Barros S. A., Carioto M., Fitzgerald K., Hettinger J., Kumar V., Novobrantseva T. I., Qin J., Querbes W., Koteliansky V., Langer R., Anderson D. G., Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 111, 3955–3960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akinc A., Querbes W., De S., Qin J., Frank-Kamenetsky M., Jayaprakash K. N., Jayaraman M., Rajeev K. G., Cantley W. L., Dorkin J. R., Butler J. S., Qin L., Racie T., Sprague A., Fava E., Zeigerer A., Hope M. J., Zerial M., Sah D. W. Y., Fitzgerald K., Tracy M. A., Manoharan M., Koteliansky V., de Fougerolles A., Maier M. A., Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 18, 1357–1364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R., Oteiza A., Sørensen K. K., Court P. M., Olsen R., Smedsrød B., Svistounov D., Role of liver sinusoidal endothelial cells and stabilins in elimination of oxidized low-density lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G71–G81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sørensen K. K., Simon-Santamaria J., McCuskey R. S., Smedsrød B., Liver sinusoidal endothelial cells. Compr. Physiol. 5, 1751–1774 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Gissen P., Arias I. M., Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 63, 1023–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poisson J., Lemoinne S., Boulanger C., Durand F., Moreau R., Valla D., Rautou P.-E., Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 66, 212–227 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Braet F., Wisse E., Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 1, 1 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wisse E., Jacobs F., Topal B., Frederik P., De Geest B., The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene Ther. 15, 1193–1199 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Chen S., Tam Y. Y. C., Lin P. J. C., Sung M. M. H., Tam Y. K., Cullis P. R., Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Release 235, 236–244 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Khan O. F., Zaia E. W., Yin H., Bogorad R. L., Pelet J. M., Webber M. J., Zhuang I., Dahlman J. E., Langer R., Anderson D. G., Ionizable amphiphilic dendrimer-based nanomaterials with alkyl-chain-substituted amines for tunable siRNA delivery to the liver endothelium in vivo. Angew. Chem. Int. Ed. Engl. 53, 14397–14401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller C. M., Tanowitz M., Donner A. J., Prakash T. P., Swayze E. E., Harris E. N., Seth P. P., Receptor-mediated uptake of phosphorothioate antisense oligonucleotides in different cell types of the liver. Nucleic Acid Ther. 28, 119–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seth P. P., Tanowitz M., Bennett C. F., Selective tissue targeting of synthetic nucleic acid drugs. J. Clin. Invest. 129, 915–925 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burra P., Liver abnormalities and endocrine diseases. Best Pract. Res. Clin. Gastroenterol. 27, 553–563 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Krawczyk M., Müllenbach R., Weber S. N., Zimmer V., Lammert F., Genome-wide association studies and genetic risk assessment of liver diseases. Nat. Rev. Gastroenterol. Hepatol. 7, 669–681 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Jeong H., Lee E.-S., Jung G., Park J., Jeong B., Ryu K. H., Hwang N. S., Lee H., Bioreducible-cationic poly(amido amine)s for enhanced gene delivery and osteogenic differentiation of tonsil-derived mesenchymal stem cells. J. Biomed. Nanotechnol. 12, 1023–1034 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Li B., Luo X., Deng B., Wang J., McComb D. W., Shi Y., Gaensler K. M. L., Tan X., Dunn A. L., Kerlin B. A., Dong Y., An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett. 15, 8099–8107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ball R. L., Hajj K. A., Vizelman J., Bajaj P., Whitehead K. A., Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano Lett. 18, 3814–3822 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Heyes J., Palmer L., Bremner K., MacLachlan I., Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 107, 276–287 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Jayaraman M., Ansell S. M., Mui B. L., Tam Y. K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J. K., Rajeev K. G., Hafez I. M., Akinc A., Maier M. A., Tracy M. A., Cullis P. R., Madden T. D., Manoharan M., Hope M. J., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 51, 8529–8533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alabi C. A., Love K. T., Sahay G., Yin H., Luly K. M., Langer R., Anderson D. G., Multiparametric approach for the evaluation of lipid nanoparticles for siRNA delivery. Proc. Natl. Acad. Sci. U.S.A. 110, 12881–12886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y.-A., Zhou B., Wernig M., Südhof T. C., ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168, 427–441.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kauffman K. J., Dorkin J. R., Yang J. H., Heartlein M. W., De Rosa F., Mir F. F., Fenton O. S., Anderson D. G., Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 15, 7300–7306 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Kumar V., Qin J., Jiang Y., Duncan R. G., Brigham B., Fishman S., Nair J. K., Akinc A., Barros S. A., Kasperkovitz P. V., Shielding of lipid nanoparticles for siRNA delivery: Impact on physicochemical properties, cytokine induction, and efficacy. Mol. Ther. Nucleic. Acids 3, e210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hattori Y., Suzuki S., Kawakami S., Yamashita F., Hashida M., The role of dioleoylphosphatidylethanolamine (DOPE) in targeted gene delivery with mannosylated cationic liposomes via intravenous route. J. Control. Release 108, 484–495 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Ye J., Yang Y., Dong W., Gao Y., Meng Y., Wang H., Li L., Jin J., Ji M., Xia X., Chen X., Jin Y., Liu Y., Drug-free mannosylated liposomes inhibit tumor growth by promoting the polarization of tumor-associated macrophages. Int. J. Nanomedicine 14, 3203–3220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandez N., Alonso S., Valera I., Vigo A. G., Renedo M., Barbolla L., Crespo M. S., Mannose-containing molecular patterns are strong inducers of cyclooxygenase-2 expression and prostaglandin E2 production in human macrophages. J. Immunol. 174, 8154–8162 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Pandya A. D., Jager E., Bagheri Fam S., Hocherl A., Jager A., Sincari V., Nystrom B., Stepanek P., Skotland T., Sandvig K., Hruby M., Maelandsmo G. M., Paclitaxel-loaded biodegradable ROS-sensitive nanoparticles for cancer therapy. Int. J. Nanomedicine 14, 6269–6285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dreaden E. C., Morton S. W., Shopsowitz K. E., Choi J.-H., Deng Z. J., Cho N.-J., Hammond P. T., Bimodal tumor-targeting from microenvironment responsive hyaluronan layer-by-layer (LbL) nanoparticles. ACS Nano 8, 8374–8382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi K. Y., Correa S., Min J., Li J., Roy S., Laccetti K. H., Dreaden E., Kong S., Heo R., Roh Y. H., Lawson E. C., Palmer P. A., Hammond P. T., Binary targeting of siRNA to hematologic cancer cells in vivo using layer-by-layer nanoparticles. Adv. Funct. Mater. 29, 1900018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanoi T., Tamura T., Sano N., Nakayama K., Fukunaga K., Zheng Y.-W., Akhter A., Sakurai Y., Hayashi Y., Harashima H., Ohkohchi N., Protecting liver sinusoidal endothelial cells suppresses apoptosis in acute liver damage. Hepatol. Res. 46, 697–706 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Sano N., Tamura T., Toriyabe N., Nowatari T., Nakayama K., Tanoi T., Murata S., Sakurai Y., Hyodo M., Fukunaga K., Harashima H., Ohkohchi N., New drug delivery system for liver sinusoidal endothelial cells for ischemia-reperfusion injury. World J. Gastroenterol. 21, 12778–12786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paunovska K., Da Silva Sanchez A. J., Sago C. D., Gan Z., Lokugamage M. P., Islam F. Z., Kalathoor S., Krupczak B. R., Dahlman J. E., Nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 31, e1807748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabf4398/DC1