

Fully functional, tumor-specific, SLAMF7-expressing cytotoxic CD4 T cells directly mediate immunity against human cancer.
Abstract
CD4 T cells have been implicated in cancer immunity for their helper functions. Moreover, their direct cytotoxic potential has been shown in some patients with cancer. Here, by mining single-cell RNA-seq datasets, we identified CD4 T cell clusters displaying cytotoxic phenotypes in different human cancers, resembling CD8 T cell profiles. Using the peptide-MHCII-multimer technology, we confirmed ex vivo the presence of cytolytic tumor-specific CD4 T cells. We performed an integrated phenotypic and functional characterization of these cells, down to the single-cell level, through a high-throughput nanobiochip consisting of massive arrays of picowells and machine learning. We demonstrated a direct, contact-, and granzyme-dependent cytotoxic activity against tumors, with delayed kinetics compared to classical cytotoxic lymphocytes. Last, we found that this cytotoxic activity was in part dependent on SLAMF7. Agonistic engagement of SLAMF7 enhanced cytotoxicity of tumor-specific CD4 T cells, suggesting that targeting these cells might prove synergistic with other cancer immunotherapies.
INTRODUCTION
Antigen-driven activation of naïve CD4 T cell precursors leads to their expansion and differentiation into a wide spectrum of functionally defined subsets (1). The diverse functional polarization of CD4 T cells [e.g., T helper cell 1 (Th1), Th2, Th17, and regulatory T cell (Treg)] was proven to affect tumor immunity distinctly, playing a notable role in the patients’ clinical outcome (2–9). While several studies and clinical trials have highlighted the helper activities of CD4 T cells (2, 3, 5–9), the evaluation of direct cytolytic functions of these cells against cancer has been demonstrated in some patients.
CD4 T cells with cytotoxic functions (Th-CTX) have been found in certain viral infections (10–14), malaria (15), and autoimmune disorders (16–18). Their presence was associated with either the escalation of the disease (19) or the induction of protective immunity. However, the highly variable combinations of surface biomarkers used to define Th-CTX [including CRTAM (20), CX3CR1 (11), CD103 (21), NKG7 (22), CD29 (19), CD38 (23), and NKG2D (24)], without the assessment of their direct cytolytic activity, render comparisons on the functionality of this unique cell subset difficult across different studies. Furthermore, the differentiation pathway of Th-CTX remains poorly understood. Various transcription factors including T-bet (T-box transcription factor TBX21), Eomes (11, 25–27), Runx3, ThPOK, (28), Hobit (29), and Blimp-1 (30) have been suggested as the upstream inducers for cytolytic-related functions of Th-CTX.
In cancer, most of the data on Th-CTX stem from preclinical models of solid tumors. The in vivo differentiation of naïve tumor-specific CD4 T cells into Th1 cytolytic T cells was sufficient to induce the regression of established tumors in a major histocompatibility complex class II (MHCII)–restricted manner (31). In addition, adoptive transfer of CD4 T cells in combination with CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4) blockade (32) or agonistic anti-CD137 antibody immunotherapy (33) into lymphopenic mice resulted in potent rejection of large vascularized melanomas, independent of other immune cells and in an MHCII-restricted manner.
In patients, Th-CTX were initially reported in patients with B-CLL (B-cell chronic lymphocytic leukemia) (34). More recently, cytotoxic CD4 T cells were identified in four patients with melanoma undergoing anti–CTLA-4 treatment. Antigen-specific CD4 T cells recognized tumor cell lines and exerted direct lysis of autologous NY-ESO-1+ (New York esophageal squamous cell carcinoma 1) melanoma cells in an MHCII-dependent fashion (25). Similarly, in vitro OX40 engagement by three human-derived antigen-specific CD4 T cell lines exhibited an enhanced antitumor function against melanoma cell lines and a higher EOMES and T-bet expression, arguing for a tumoricidal capacity as observed in the mouse counterpart (26). Recently, the cytolytic function of CD4 T cells was associated with an interleukin-2 (IL-2)/Blimp-1/granzyme B axis, whereby Tregs acted as suppressors by sequestering IL-2 from the tumor bed. In this setting, T-bet expression was required for the polyfunctionality but not for the cytotoxicity of tumor-reactive CD4 T cells (30). Furthermore, it was shown that basal in situ MHCII expression, but not MHCI, was sufficient to segregate responders from nonresponders in advanced melanoma patients treated with an anti–PD-1 (Programmed cell death protein 1) antibody (35), arguing for a potential direct contribution of cytotoxic CD4 T cells to tumor elimination in patients. Another study in bladder cancer recently reported on highly heterogeneous CD4 T cell transcriptomic states within tumors, with cytotoxic signatures being predictive of response to PD-1 blockade (36).
Here, we aimed at assessing the presence, rate, and cytotoxic function of tumor-specific Th-CTX directly in patients with cancer. We capitalized on published single-cell transcriptomic analyses of patient samples, integrated with the direct phenotypic and functional characterization of clonal, tumor-specific CD4 T cell populations, using peptide-MHCII (pMHCII) multimers and a novel high-throughput single-cell cytotoxicity assay in picowell arrays. The direct tumor cell killing by cytolytic tumor-specific CD4 T cells in the arrays was monitored in a high-throughput manner by combining multichannel time-lapse microscopy with deep neural networks. Our work provides a deep characterization of human Th-CTX in cancer and supports their role in tumor immunity.
RESULTS
The transcriptomic signature of tumor-infiltrating cytolytic CD4 T cells from patients with melanoma
To define a putative signature of cytolytic CD4 T cells, we mined a publicly available single-cell RNA sequencing (scRNA-seq) dataset from 32 tumor biopsies of patients with advanced melanoma (n = 12,850 single cells in total) (37). We used these data to generate a robust transcriptional map of Th-CTX and to determine potential candidate markers linked with their cytotoxic function in cancer.
Within the total tumor-infiltrating lymphocytes (TILs) (n = 8162), the CD3+CD4+CD8− population was defined as “CD4 T cells” (n = 2333, 28.6%), while the CD3+CD8+CD4− expressing population, used as control, was annotated as “CD8 T cells” (n = 5829, 71.4%) (Fig. 1A). Using an unsupervised clustering of the CD4 T cell population, further diversity was found within this group with the identification of four major cell states (Fig. 1B). Clusters were annotated on the basis of the expression of known genes. One cluster showed high expression of FOXP3 (forkhead box P3), TIGIT (T-cell immunoreceptor with Ig and ITIM domains), and IL-2RA and was defined as CD4_Regulatory cluster (n = 1109). Another cluster was enriched in genes linked with memory and survival such as IL-7R, T cell factor 7, and CCR7 and was referred to as CD4_Memory cluster (n = 599). A third cluster demonstrated increased expression of genes implicated in proliferation such as MKI67, ZWINT, and TYMS, suggesting that it represents a CD4_Cycling cluster (n = 180). The fourth cluster was enriched in genes linked to effector/cytolytic functions in CD4 T cells such as PRF1, GZMA, GZMB, GZMH, GZMK, GZMM, GNLY, FASLG, NKG7, SLAMF7, PLEK, and KLRG1 and was annotated as CD4_Cytolytic cluster (n = 445) (Fig. 1B and fig. S1A). Notably, each of the clusters contained cells from different patients, indicating that clusters and expression states are conserved across patients and do not represent patient-specific subpopulations or batch effects, although they do vary in their relative proportions (Fig. 1C).
Fig. 1. The transcriptomic signature of tumor-infiltrating cytolytic CD4 T cell from patients with melanoma.
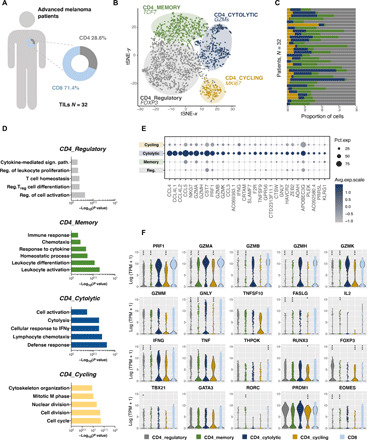
(A) Frequency of CD4 and CD8 TIL among all CD3+ T cells collected from 32 patients with melanoma. (B) t-distributed stochastic neighbor embedding (tSNE) two-dimensional (2D) plot of scRNA-seq data representing all CD3+CD4+CD8− T cells. Colors correspond to the four clusters discriminating the cytolytic (blue, n = 445), memory (green, n = 599), cycling (yellow, n = 180), and regulatory (gray, n = 1109) subsets. (C) The relative CD4 cluster abundance within the CD3+CD4+CD8− T cells in each patient. (D) GO-based analysis illustrating enrichment of pathways observed in each of the four distinct CD4 clusters. (E) Bubble plot of the top 30 up-regulated genes in the CD4_Cytolytic cluster. (F) Violin plots showing the distribution of gene expression levels for selected cytolysis related and master transcription factor genes across the four CD4 T cell clusters (regulatory in gray, memory in green, cytolytic in blue, and cycling in yellow) as compared to tumor-infiltrating CD8 T cells (in light blue). Statistical power was assessed using Wilcoxon test for violin plot graphs.
Further, gene ontology (GO) enrichment analysis of the gene signature of each cluster supported the distinct role of these CD4 T cell populations (Fig. 1D). The CD4_Regulatory signature was enriched in pathways related to general leukocyte homeostasis as well as Treg differentiation. The CD4_Memory cluster was enriched in pathways promoting the activation and proliferation of leukocytes and associated with response to cytokines and migration of cells. The CD4_Cycling signature demonstrated a major cytoskeleton and nuclear reorganization bias, linked with cell cycle and division. The lytic granule genes identified in the scRNA-seq analysis were exclusively up-regulated in the CD4_Cytolytic signature, at similar expression levels as in the CD8 cluster (Fig. 1, E and F), with further enrichment of an extensive defense response axis, mostly built on cytolysis related GO pathways. The observed CD4_Cytolytic cluster specifically showed higher expression of genes previously linked to effector/cytolytic functions in CD4 T cells, such as CRTAM (20), NKG7 (22), KLRG1 (38), and CST7 (Cystatin F) (Fig. 1E). Only a modest FasL (Fas Ligand) increase, but no increase of TRAIL death receptor/ligand pathway, was apparent in the CD4_Cytolytic cluster (Fig. 1, E and F).
The cytolytic CD4 T cell cluster included also an increased expression of CCL5, CCL4, and CCL4L1/2 (Fig. 1E), which was additionally evidenced by the enrichment of the lymphocyte chemotaxis and cellular response to interferon-γ (IFNγ) pathways in the GO-based analysis (Fig. 1D). None of the known master transcription factors was specifically overexpressed in the CD4_Cytolytic cluster (Fig. 1, E and F). RUNX3 (RUNX family transcription factor 3) and BLIMP-1 (B lymphocyte-induced maturation protein-1) (30) did not correlate with a cytolytic potential. A modest enrichment of EOMES, an upstream inducer of cytolytic-related molecules, in combination with T-bet (11, 25–27), was observed within the CD4_Cytolytic cluster, despite general low transcript expression.
Overall, these results suggest the in vivo presence of Th-CTX CD4 T cells infiltrating tumor tissues of patients and provide detailed gene signatures characterizing them.
Direct ex vivo detection of tumor-specific cytolytic CD4 T cells in patients with melanoma
To gain insight into the cytolytic features of antigen-specific CD4 T cells directly ex vivo and to validate the gene signature obtained by the scRNA-seq analysis (Fig. 1), we used peptide-MHCII multimers (39). We quantified and isolated NY-ESO-187–99/DR7-specific CD4 T cells ex vivo from both peripheral blood lymphocytes (PBLs; nine samples) and TILs/tumor-infiltrated lymph nodes (TILNs) (eight samples), respectively. We observed homogeneous CD4 T cell frequencies in PBLs in comparison to more variable infiltration within the TIL/TILN samples (Fig. 2A). Similarly, the presence of NY-ESO-187–99/DR7 CD4 T cells was detected in eight of nine blood samples in contrast to the tumor, where five of eight samples showed multimer+ cells (Fig. 2B). Because of the limited number of cells that could be recovered after flow cytometry–based sorting, we focused on the assessment of selected genes that were either enriched within the scRNA-seq CD4_Cytolytic cluster (Fig. 1D) or previously associated in the literature with cytolytic capacity in CD4 T cells (28, 30). Granzyme B was higher in NY-ESO-187–99/ DR7 CD4 T cells present in the blood compared to the tumor bed, while perforin, granzyme A, and BLIMP-1 showed an opposite distribution pattern. RUNX3 expression was relatively scarce but stable in the two groups, similarly to granulysin. If compared to multimer-negative circulating CD4 T cells and to CD8 T cell subsets (naïve and non-naïve CD8 T cells) isolated from the same patients, NY-ESO-187–99/DR7 CD4 T cells show cytolytic-related transcript similar to antigen-primed CD8 T cells (fig. S1B). Overall, the direct ex vivo detection of tumor-specific CD4 T cells displaying cytolytic-related transcripts not only in the circulation but also within the tumor argues for their potential functional role in vivo (Fig. 2C).
Fig. 2. Direct ex vivo detection of tumor-specific cytolytic CD4 T cells in patients with melanoma.
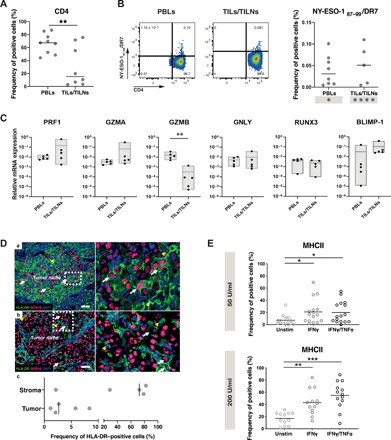
(A) Ex vivo frequency of CD4 T cells among PBLs and TILs/TILNs in patients with melanoma. (B) Representative examples (left) and summarizing graphs (right) of ex vivo pMHCII multimer staining of CD4 T cells specific for NY-ESO-187–99/DR7 obtained in PBL and TIL/TILN of HLA-DR7+ patient samples. (C) Quantitative reverse transcription PCR (RT-PCR) analysis of the indicated transcripts in ex vivo sorted NY-ESO-187–99/DR7 CD4 T cells isolated either from spell peripheral blood (PB) or TILs/TILNs. (D) Multispectral imaging assessment of MHCII expression directly in situ on 18 metastatic melanoma tissues. Representative examples of two cases (a) with expression of HLA-DR quantified to 5.8% of tumor cells within the SOX-10+ tumor niche (white arrows) and (b) lower expression of HLA-DR in 2.07% of the SOX-10+ tumor cells (white arrows). Stromal cells expressed HLA-DR, pointed by yellow arrowheads. The insets illustrate high-power images. The MSI were acquired using a 20× objective of the Vectra 3.0 imaging system. Scale bar, 50 μm. (c) Graph of frequency of HLA-DR–positive cells expressed in percentages, in the tumor SOX-10+ classified tissue category versus the stroma. DAPI, 4′,6-diamidino-2-phenylindole. (E) In vitro MHC II expression by 16 tumor cell lines after IFNγ and TNFα treatment. Statistical power was assessed using unpaired t test (A) and Kruskal-Wallis test (E).
The cytolytic activity of CD4 T cells against tumors requires MHCII expression in cancer cells. Although MHCII is not often constitutively expressed in solid tumors, it is induced in diverse cancer types (40) with around 30% of melanoma cases exhibiting around 10% of MHCII+ cells in the tumor bed (35).
To evaluate the presence of MHCII in melanoma directly in situ, immunostaining for HLA-DR was performed on metastatic melanoma sections of 18 patients. MHCII expression in the tumor cells was detected in 5 of 18 cases, and the percentage of positive tumor cells ranged from 1.2 to 8.4% (Fig. 2D). HLA-DR positivity was also detected on cells within the tumor stroma (Fig. 2D). Accordingly, in vitro experiments using IFNγ or/and tumor necrosis factor–α (TNFα) treatment of melanoma cell lines, to mimic a proinflammatory microenvironment, showed a significant enhancement of MHCII expression after treatment in most of the tested lines (Fig. 2E), arguing for potential Th-CTX–tumor cell interactions in immune-infiltrated tumor regions.
Delayed but direct cytolytic capacity of NY-ESO-187–99/DR7 CD4 T cells against melanoma cell lines
To date, the absence of a functional assessment of their cytolytic capacity has limited the study and the comparison of Th-CTX cells across different models (11, 20, 22, 23, 29). To tackle this issue, we isolated tumor-specific CD4 T cell clones from ex vivo multimer+ sorted cells from patient samples. To discriminate helper (Th) versus cytolytic (Th-CTX) profiles, we used the standard 4 hours 51Chromium release cytotoxic assay on each clone (fig. S2A), in the presence of tumor cells either pretreated with IFNγ or stably CIITA (Class II Major Histocompatibility Complex Transactivator) transduced, to increase the MHCII expression. The CIITA-transduced cell lines showed a more homogeneous and high expression of MHCII, as compared to wild-type (WT) or IFNγ-treated tumor cells (Fig. 3A). As expected, no specific lysis of NY-ESO-1− cells occurred in the absence of the specific peptide, independently of the MHCII expression levels on target cells (Fig. 3B). A high level of lysis, up to 46% at the highest 30:1 E:T (effector-to-target) ratio, of CIITA-transduced tumor cells loaded with the cognate peptide was observed (Fig. 3B). Significantly, lower levels of specific lysis were measured when IFNγ-treated and WT tumor cells were used as targets (Fig. 3B). The use of an HLA-matched, endogenously expressing NY-ESO-1 tumor cell line coincubated with the CD4 T cell clones showed similar but delayed specific lysis occurring after 24 hours in a proportion of the tested clones, demonstrating that peptide-specific CD4 T cell clones recognize a naturally processed epitope on tumor cells (Fig. 3C). This effector capacity was not limited to one epitope, as similar assays conducted on additional CD4 T cell clones specific for the Mage-A3111–125/DP4 and Mage-A3243–258/DP4 epitopes showed a similar pattern of lysis (fig. S2B).
Fig. 3. Delayed but direct cytolytic capacity of NY-ESO-187–99/DR7 CD4 T cells against melanoma lines.
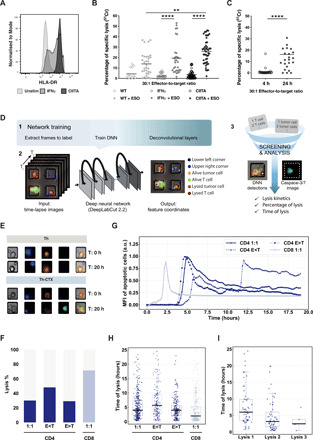
(A) Representative histogram of MHCII expression of a WT (light gray), IFN𝛾-treated (dark gray), and CIITA-transduced (black) tumor cell lines. FMO is shown as dashed line. (B) Cumulative analysis of 51Chromium release assays conducted with tumor cell lines transduced with CIITA, IFN𝛾 treated, or unmanipulated, pulsed or not with a specific peptide, and coincubated with PB (peripheral blood)–derived CD4 T cell clones. (C) Summary of the specific lysis obtained using a tumor cell line endogenously expressing NY-ESO-1 cocultured with PB-derived NY-ESO-187–99/DR7–specific CD4 T cell clones. Statistics were performed using nonparametric unpaired Kruskal-Wallis test (B) and matched-pairs signed-rank test (C). P values of <0.05 were considered statistically significant. (D) Single-cell tumor recognition assay using time-lapse imaging microscopy in picowell grids: Schematic illustration of the data analysis with deep learning approaches including the network training (1), video analysis for features extraction (2), and data screening and MFI analysis. DNN, deep neural network. (E to H) Fluorescently labeled CIITA–transduced tumor cell line pulsed with a specific peptide was cocultured with PB-derived NY-ESO-187–99/DR7 CD4 T cells in a PDMS picowell array. (E) Representative time series data at 0 and 20 hours of an enlarged view of two picowells showing CD4 T cells (blue), tumor cells (red), and the caspase-3/7 pathway reagent identifying cells undergoing apoptosis (green). (F) Cumulative analysis of average percentage of specific lysis, (G) real-time dynamic MFI of the tumor apoptosis, (H) the time of the lysis defined at a single cell level, and (I) time of the consecutive lytic events in wells with target cells outnumbering T cells. PB-derived NY-ESO-1157–165 /A2 CD8 T cells were used as positive control. a.u., arbitrary units.
Next, to estimate the cytotoxicity kinetics of individual cytolytic CD4 T cells, a novel high-throughput single-cell tumor recognition assay was developed. This consists of a time-lapse imaging platform for profiling dynamic cell-cell interactions at the single-cell level. It combines large arrays of microfabricated picowells [made of polydimethylsiloxane (PDMS)] with automated time-lapse fluorescence microscopy. The microfabricated chip consists of 12 columns and 18 rows of blocks, and each of the block contains 99 square picowells, thus providing, at full capacity, 21,384 wells in total (see fig. S2D for the chip configuration). The picowells (35 μm by 35 μm by 50 μm) that have a confinement volume less than 65 pl allow close interaction between a few effector and target cells in spatially confined volumes. At the same time, their microarray configuration provides a potent tool to monitor cytotoxicity of thousands of T cells simultaneously at the single-cell level (41, 42). To perform the assay, suspensions of prelabeled CIITA-transduced tumor cells (PKH26, red) and tumor-specific T cells (CellTrace Violet, blue) were sequentially and manually loaded into the picowells, followed by immersion in media containing the specific peptide as well as a live-dead green apoptotic staining cocktail. The phenotype and state of the cells in each well were determined with four channels [bright field, AF488, tetramethyl rhodamine isothiocyanate (TRITC), and BV421] imaged at a 10-min interval for 24 hours. In particular, viable T cells are labeled with blue (BV421), viable and dead target cells in red (TRITC), and red and green (TRITC + AF488), respectively (Fig. 3D). The picowell array–based time-lapse microscopy generates a large amount of imaging data. To perform automated quantitative analysis of independent T cell tumor cell interactions, acquired images were processed by using an adapted version of DeepLabCut v2.2, an open-source deep learning toolset (43). The algorithm enables discriminating the diverse E:T ratios (e.g., discarding the empty/overcrowded wells or wells with only one cell type) and to analyzing the specificity of target cell lysis induced by tumor antigen–specific CD4 T cells. The deep convolutional network can efficiently extract the coordinates of the key features, including well boundaries, viable, and apoptotic tumor or T cells, in the entire video based on manually annotated data from a few frames (see Materials and Methods and fig. S2E for a representative movie). Tumor-specific CD8 T cells were used as positive cytolytic controls (Fig. 3, F to H, and fig. S2C). Before testing cytotoxicity of individual antigen-specific T cells and to gain insight on the overall cytotoxic capacity of CD4 T cells, we used MHCII–expressing targets, loaded with the superantigen Staphylococcus enterotoxin B (SEB). We observed similar capacities of CD4 and CD8 T cells in target cell killing in this setting (fig. S2F).
Next, we moved to tumor-specific T cell evaluation. Results from five independent experiments demonstrated that antigen-specific CD4 and CD8 T cells were able to lyse tumor cells directly at a 1:1 ratio, reaching 30.2 and 71.6% of lysed tumor cells by CD4 or CD8 T cells, respectively (Fig. 3F). Tumor-specific T cells exhibited functional heterogeneity at identical E:T ratios among different picowells. Despite the detected contacts, the interactions between target and CD4 T cells either resulted in specific lysis or failed to induce apoptosis (Fig. 3E). Quantification of the average percentage of tumor apoptosis showed a tendency toward a higher lytic capacity in the presence of an excess of target over CD4 T cells. The observed pattern of lysis “E<T” > “E:T” > “E>T” was in line with previous work performed on CD8 T and natural killer (NK) cells where negative ratios were favorable to increase apoptosis of the targets (44, 45). Further temporal analysis of the onset of tumor cell apoptosis (Fig. 3, G and H) revealed that cytolytic activity of CD4 T cells started after a few minutes of initiation of coculture and persisted until 15 to 20 hours with an average time of 5.3 hours for the 1:1 ratio. Tumor antigen–specific CD8 T cells demonstrated a faster killing window with an average time of 2.4 hours. Further, the fact that “E<T” ratios showed a delayed time of killing but a higher rate of specific lysis implies a burst of T cells’ lytic capacity. Independent kinetics assessment of 121 picowells presenting a single CD4 T cell outnumbered by targets cells revealed sequential fluorescence peaks of the caspase-3/7 reagent, suggesting the serial engagement in multiple killing activities by some Th-CTX (Fig. 3I).
The release of cytolytic molecules is mainly responsible for the cytolytic potential of tumor-specific CD4 T cells
To understand the molecular pathways involved in the cytotoxicity of CD4 T cells, cytokines and cytolytic related molecules produced by NY-ESO-187–99/DR7 CD4 T cell clones were analyzed after a 6-hour coculture with titrated concentrations of peptides. All tested clones presented a relatively low level of perforin, granzyme K, and granzyme M without significant differences between Th and Th-CTX. In contrast, both populations showed high production of granzyme A with around 78.8 or 87.5% in Th or Th-CTX, respectively. Granzyme B and granulysin loads were significantly increased within Th-CTX CD4 T cell clones in comparison to the helper ones (Fig. 4A). Notably, granzyme B expression was also significantly enhanced at the transcriptomic level, suggesting a potential key role of this granzyme in the lytic capacity of the CD4 T cells (Fig. 4B). These results were in line with the observed significant enrichment of lytic granules within the CD4_Cytolytic cluster in the ex vivo scRNA-seq analysis (Fig. 1D). Accordingly, tumor recognition assays performed in the presence of concanamycin (CMA), an inhibitor of the vacuole proton type adenosine triphosphatase pump, leading to the degradation of the perforin present within the vacuoles containing granzymes, exhibited a marked decrease of lysis (Fig. 4C and fig. S2B). Cytotoxicity was contact dependent, because the use of a transwell completely abolished target cell killing (fig. S3A, left graph). In line with these observations, CD107a was up-regulated during the cytotoxic process in Th-CTX (fig.S3A, right graphs) and the use of the membrane-soluble Compound 20, a specific granzyme B inhibitor, significantly reduced target cell lysis (fig. S3B). As a complete abrogation of lysis was not achieved neither upon CMA nor Compound 20 exposure, a possible role of alternative apoptotic pathways was considered, including Fas and TRAIL. However, no significant differences in the Fas or TRAIL expression were detected between Th and Th-CTX clones (fig. S3C).
Fig. 4. The release of cytolytic molecules is mainly responsible for the cytolytic potential of tumor-specific CD4 T cells.
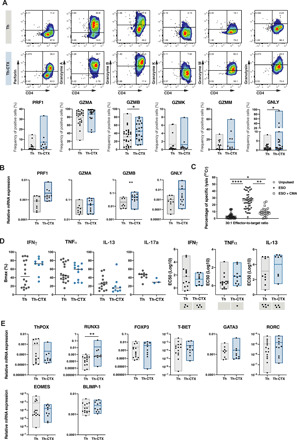
PB-derived NY-ESO-187–99/DR7–specific CD4 T cell clones were discriminated as Th or Th-CTX based on results obtained with a standard 4-hour 51Chromium release assay. (A) Representative example (top) and cumulative analysis (bottom) of the production of perforin (PRF1), granzymes (GZMA, GZMB, GZMK, and GZMM), and granulysin (GNLY) by NY-ESO-187–99/DR7–specific CD4 T cell clones. (B) Quantitative RT-PCR (qRT-PCR) analysis of the indicated transcripts in PB-derived NY-ESO-187–99/DR7 CD4 T cell clones. (C) Standard 4-hour 51Chromium release assay was conducted with a CIITA-transduced tumor cell line pulsed or not with a specific peptide. Release of perforin was blocked by a 2-hour pretreatment of antigen-specific CD4 T cells with CMA (100 nM). Results are expressed as percentage of specific lysis of the target cells at 30:1 E:T ratio. Statistics were performed using nonparametric unpaired Kruskal-Wallis test. (D) Average dose-response curves and Bmax (maximal response), EC50 values of IFNγ, TNFα, IL-13, and IL-17a induced by titrated concentrations of NY-ESO-187–99 peptide by PB-derived NY-ESO-187–99/DR7–specific CD4 T cell clones. (E) Quantitative RT-PCR analysis of the indicated transcripts in PB-derived NY-ESO-187–99/DR7 CD4 T cell clones. All statistics for RT-PCR data were performed using two-way analysis of variance (ANOVA) with Sidak’s post hoc test.
To define whether the cytotoxic and helper potentials were mutually exclusive, we profiled all clones for their capacity to secrete Th-prototypic cytokines for Th1 (IFNγ and TNFα), Th2 (IL-13), and Th17 (IL-17a) profiles (Fig. 4D). Bmax (maximum cytokine production) values showed a generally higher type 1 cytokine (IFNγ and TNFα) production compared to IL-13 and IL-17a, with this tendency being enhanced in the Th-CTX (Fig. 4D). Median effective concentration (EC50) of IFNγ, TNFα, and IL-13 production showed similar values in all clones tested, irrespective of their cytotoxic capacities. EC50 of IL-17a could not be assessed as several clones were negative for this cytokine. The cytotoxic function was not linked to the expression of a specific master transcription factor, because no distinction was observed among the clones tested, with the exception of RUNX3 up-regulation in Th-CTX cells in comparison to the helper clones (Fig. 4E). These results suggest a polyfunctional phenotype of cytolytic tumor-specific CD4 T cell clones with the concomitant production of cytokines, in addition to their acquisition of cytolytic functions. Hence, the ultimate cytotoxic potential might depend on additional intrinsic differences within the CD4 T cells themselves, such as variable T cell receptor (TCR) repertoires and affinities, or distinct expression of activating and inhibitory receptors.
TCR repertoires of Th and Th-CTX CD4 T cells are polyclonal
We sequenced the TCR α and β chains of 32 NY-ESO-187–99/DR7 CD4 T cell clones isolated from three peripheral blood mononuclear cells (PBMCs) of patients and defined as either Th or Th-CTX based on the cytotoxic assays presented in Fig. 3. NY-ESO-187–99/DR7 CD4 T cell clones showed diverse clonally distributed TCRs, with the presence of 16 distinct clonotypes. However, variability in the number of clonotypes was high across patients, with highest polyclonality observed for patient LAU1293 (Fig. 5A). The relative abundance of each clonotype as a whole, within Th or Th-CTX clones, was also variable in the three patients (Fig. 5B). For each patient, shared but also distinct TCR α and β chains were identified among Th and Th-CTX (Fig. 5, A and C). Notably, the assessment of the MFI (mean fluorescence intensity) membrane expression of TCRαβ showed similar values in all clones tested, irrespective of their cytotoxic capacities (fig. S3D), arguing for a minor impact of the TCR in functional divergence.
Fig. 5. TCR repertoires of Th and Th-CTX CD4 T cells are polyclonal.
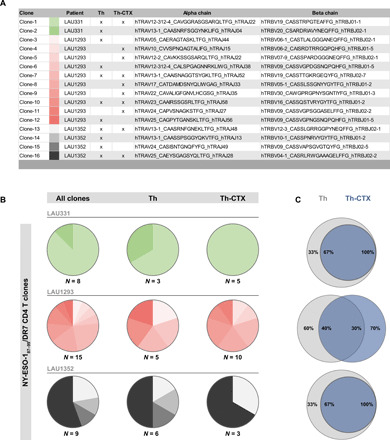
PB-derived NY-ESO-187–99/DR7–specific CD4 T cell clones were discriminated as Th or Th-CTX based on results obtained with a standard 4-hour 51Chromium release assay. (A) Table listing the diverse clonotypes found in PB-derived NY-ESO-187–99/DR7 CD4 T cell clones from three patients with melanoma (LAU331, n = 8 clones; LAU1293, n = 15 clones; LAU1352, n = 9 clones). (B) Pies illustrating the relative abundance of each clonotypes as a whole (left), within Th (middle), or Th-CTX (right) clones. (C) Venn diagrams showing the distribution of unique and shared clonotypes frequencies between Th (gray) or Th-CTX (blue) clones.
SLAMF7 as candidate to increase cytolytic potential of tumor-specific CD4 T cells
To screen for potential surface markers of Th-CTX, we exploited the CD4_Cytolytic cluster that we determined in the scRNA-seq dataset. We identified the SLAMF7 (Signaling lymphocytic activation molecule F7) as being predominantly expressed in the CD4_Cytolytic cluster and, to a lesser extent, PD-1 as being mainly expressed in cycling and cytolytic clusters (Fig. 6A). Homophilic SLAMF7 interactions in NK cells have been reported to lead to the activation of their natural cytotoxicity (46). Moreover, the engagement of the SLAMF7 axis was associated with the partial rescue of effector functions in NK and CD8 T cells in patients with lupus erythematosus and multiple myeloma, correlating with beneficial clinical effects (46, 47). In addition, the expression of PD-L1 (Programmed death-ligand 1) and PD-L2 in melanoma cell lines in a proinflammatory environment (fig. S4) led us to investigate the SLAMF7 and PD-1 distribution in correlation to cytolysis-related molecules.
Fig. 6. SLAMF7 as potential candidate to increase cytolytic potential of tumor-specific CD4 T cells.
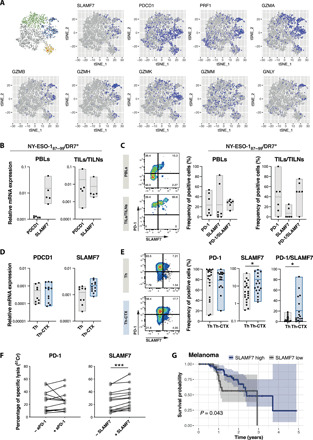
(A) tSNE 2D plot of scRNA-seq data representing the four CD4+ T cell clusters discriminating the cytolytic (blue), memory (green), cycling (yellow), and regulatory (gray) subsets and their expression of selected genes (SLAMF7, PDCD1, PRF1, GZMA, GZMB, GZMH, GZMK, GZMM, and granulysin). (B) Quantitative RT-PCR analysis of the indicated transcripts in ex vivo–sorted NY-ESO-187–99/DR7 CD4 T cells from either PB or TILs/TILNs. (C) Representative examples and cumulative analysis of direct ex vivo NY-ESO-187–99/DR7 CD4 T cell multimer staining combined with the evaluation of SLAMF7 and PD-1 expression of either PB or TILs/TILNs from patients with melanoma. (D) qRT-PCR analysis of the indicated transcripts in PB-derived NY-ESO-187–99/DR7 CD4 T cell clones discriminated as Th or Th-CTX. (E) Representative examples and cumulative analysis of SLAMF7 and PD-1 expression in PB-derived NY-ESO-187–99/DR7 CD4 T cell Th and-Th-CTX clones. (F) Standard 4-hour 51Chromium release assay, conducted with target tumor cell lines transduced with CIITA pulsed with the specific peptide in the presence or absence of Nivolumab (10 μg/ml) (left) or agonistic anti-SLAMF7 antibody (5 μg/ml) (right) and coincubated with antigen-specific CD4 T cell clones. Results are expressed as percentage of specific lysis of the target cells at 30:1 E:T ratios. (G) Kaplan-Meier plot depicting the survival probability and 95% confidence interval over time (years) for melanoma patients with high (blue) and low (black) SLAMF7 expression (data originated from TCGA). The difference between the two survival curves was assessed using a log-rank test. Statistics were performed using two-way ANOVA with Sidak’s post hoc test (B to D), nonparametric Mann-Whitney test (E), and Wilcoxon test (F).
In addition to SLAMF7 and, to a lower extent, of PD-1 (PDCD1), Th-CTX also expressed GZMB, GZMH, and GNLY (Fig. 6A). Next, we verified the expression of these molecules at a protein level in patient samples ex vivo. As shown in Fig. 6 (B and C), both SLAMF7 and PD-1 expression, even if heterogeneously, were detected directly ex vivo in NY-ESO-187–99/DR7–specific CD4 T cells from both PBLs and TILs/TILNs. PD-1 expression tended to be lower within circulating CD4 T cells in comparison to the tumor bed, at both transcriptomic and proteomic levels. In contrast, SLAMF7 transcripts were comparable in both populations. A tendency toward lower SLAMF7 single-positive cells but higher double-positive SLAMF7+PD-1+ CD4 T cells in TILs/TILNs was observed, potentially reflecting the higher activation state of these cells in the tumor microenvironment.
Similar results were obtained by analyzing tumor-specific CD4 T cell clones, where expression of SLAMF7 and PD-1 was quite variable among patients with a range of expression of 6 to 61.5% and 23.1 to 75%, respectively. While PD-1 expression was high in both Th and Th-CTX clones, SLAMF7 and double-positive PD-1+SLAMF7+ were significantly enriched in cytolytic CD4 T cell clones (Fig. 6, D and E). SLAMF7 expression could be induced in vitro by exposure to recombinant IL-12 or to the histone deacetylase (HDAC) inhibitor Entinostat, previously reported to induce cytotoxicity in CD8 T cells (48) and to up-regulate cytotoxic-related genes in CD4 T cells (49), respectively (fig. S3E). Notably, EAT-2 (EWS/FLI1 activated transcript), the requisite adaptor protein for SLAMF7 to mediate activation signaling, at least in NK cells, was higher in Th-CTX than Th clones (fig. S3F).
To establish the role of SLAMF7 and PD-1 in modulating the cytolytic function of CD4 T cells, standard 51Chromium release assays were performed, in the absence or presence of either an anti–PD-1 antibody (Nivolumab) or an agonistic SLAMF7 antibody. Expression of SLAMF7 on target cells was verified, given the homophylic nature of this receptor. Melanoma cell lines showed variable but consistent SLAMF7 expression with a mean of 27% positive cells (fig. S4B). While the percentage of specific lysis was not enhanced by the blockade of the PD-1/PD-L1 axis, engagement of SLAMF7 was able to trigger a modest but consistent significant increase of the lytic capacity of tumor-specific CD4 T cell clones (Fig. 6F), arguing for its therapeutic potential.
Data mining of The Cancer Genome Atlas (TCGA) RNA-seq dataset using SLAMF7 as a query revealed statistically significant association with favorable prognosis in melanoma (Fig. 6G), as well as in the other tumor types considered in the section below (fig. S5A). Although SLAMF7 may be expressed by various tumor-infiltrating immune cell types other than CD4 T cells, primarily NK cells, the association with favorable clinical outcome warrants further studies on the prognostic value of SLAMF7+ CD4 TILs. This correlative analysis was not possible in our patients’ cohort given the small sample size, the heterogeneity in terms of disease stage and treatment. However, notably, by further mining the TCGA RNA-seq dataset using neural cell adhesion molecule 1 (NCAM1), CD16, or NCR1 (Natural Cytotoxicity Triggering Receptor 1) as a query, no positive correlation was observed for NK cells and survival. Instead, a negative correlation was observed between NCAM1 and SLAMF7, suggesting that the main source of SLAMF in the tumor might not be NK cells (fig. S5, B and C).
Distribution of cytolytic CD4 T cells across cancer types and their relatedness to other T helper clusters
To verify to which extent our observations on cytotoxic CD4 T cells are conserved across tumor types, we investigated three additional published scRNA-seq datasets from breast cancer (BC) (50), head and neck cancer (51), and hepatocellular carcinoma (52), applying the same analysis pipeline as in Fig. 1. Th-CTX were observed in all tumor types analyzed, at population frequencies of 19.1, 2.8, 21.3, and 39.6%, respectively (Fig. 7A). Moreover, transcriptomic comparison of the differentially expressed genes between the CD4_Cytolytic cluster and the other CD4 T cell clusters in these datasets demonstrated similar profiles, with the up-regulation of GZMA, CCL4/L2, and CCL5 in all cases. Other genes including GZMB, GZMK, IFNG, CST7, NKG7, EOMES, and CRTAM were up-regulated in Th-CTX in three of four cancer types. SLAMF7 was enriched in melanoma, BC, and hepatocellular carcinoma but not in head and neck cancer (Fig. 7B).
Fig. 7. Distribution of cytolytic CD4 T cells across cancer types and their relatedness to other T helper clusters.
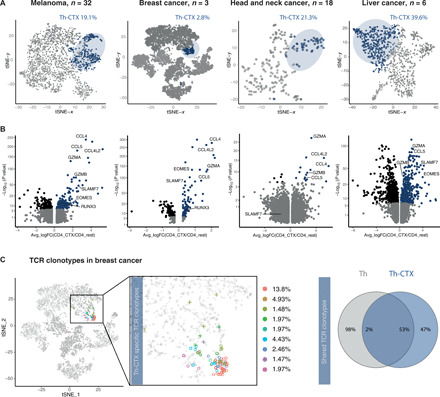
(A) tSNE 2D plot of scRNA-seq data representing the CD4+ T cell clusters discriminating the cytolytic (dark blue) and noncytolytic (gray) subsets across patients with melanoma, BC, head and neck cancer, and hepatocellular carcinoma. (B) Volcano plot analysis of gene expression changes in melanoma, BC, head and neck cancer, and hepatocellular carcinoma patients from Th-CTX compared to other Th clusters. Statistical power was assessed using Wilcoxon test [gray, P ≥ 0.01; blue, log fold change (logFC) > 0.25 and P < 0.01; black, logFC ≤ 0.25 and P < 0.01]. (C) 2D tSNE of scRNA-seq data representing the most abundant TCR clonotypes identified in the CD4_Cytolytic cluster in patients with BC, their relative abundance (left), and Venn diagram showing the distribution of unique and overlapping TCR sequences and their relative abundance across Th-CTX and non–Th-CTX (right).
We next analyzed single-cell TCR sequences available for the patients with BC to evaluate the clonotype distribution in the different clusters and specifically within the CD4_Cytolytic cluster. Results showed the presence of 21 distinct clonotypes within the CD4_Cytolytic cluster, suggesting the emergence of cytolytic CD4 T cells from a polyclonal population (Fig. 7C). Relative abundance of the CD4_Cytolytic cluster–specific TCR clonotypes revealed a heterogeneous distribution, with one preferably expanded clonotype (13.8%, clonotype red in Fig. 7C). This clonotype was among the most abundant detected in the entire CD4 T cell population. Moreover, 2% of clonotypes within the CD4_Cytolytic cluster was shared with the entire CD4 T cell population, similarly to our observations on tumor-specific CD4 T cell clones (Fig. 5). Together, these results support the existence of cytotoxic CD4 T cells among TILs in various human tumor types.
DISCUSSION
The main finding in this work is the frequent existence of fully active, tumor-specific cytotoxic CD4 T cells in patients with cancer. Further, we identified the therapeutically targetable SLAMF7 molecule as a direct regulator of tumor-specific CD4 T cell cytotoxicity, highlighting a potential clinical relevance of triggering tumor-infiltrating CD4 T cell killers in immunotherapy. By building on preexisting knowledge on Th-CTX in cancer, we provide novel evidence on contact- and granule-dependent, delayed CD4 T cell cytotoxicity, visualized in real time and at the single-cell level.
Our unbiased scRNA-seq analysis in different patient datasets showed that Th-CTX share similarities with classical cytotoxic lymphocytes (10, 20). The direct ex vivo analysis of sorted tumor-specific CD4 T cells confirmed the presence of cytolytic-related transcripts in samples from both peripheral blood and TILs/TILNs. Notably, granzyme B expression was significantly down-regulated in the tumor bed compared to the peripheral blood. This might be due to an accumulation of Tregs in the TME (tumor microenvironment), which would sequestrate the IL-2 necessary to induce a cytolytic CD4 T cell profile, as recently reported (30). We also found preferential expression of PLEK, a gene previously reported in precursors of Th-CTX (22), opening the question of the differentiation trajectories of these cells in vivo. The high levels of KLRG1 and the low levels of CD27 and CD28 (53), together with the significantly higher levels of CRTAM, argue for a late differentiation stage and potential long-term persistence of Th-CTX in vivo. In this regard, the association of CADM1 expressing dendritic cells and CRTAM on CD8 T cells was reported as critical for the accumulation of antigen-specific CTLs and their subsequent proliferation in the draining LNs, suggesting a potential role of this molecule also in the priming and maintenance of Th-CTX cells. Moreover, the recently reported expansion of a cytotoxic CD4 T cell cluster in supercentenarians hints for a role of this population in immune surveillance in elderly healthy donors.
In parallel to the cytolytic-related molecules, IFNγ, CCL4, and CCL5 were also enriched in Th-CTX cells. Recently, intratumoral NK cells producing CCL5 have been associated with the recruitment of cross-priming cDC1s (54). Thus, it is tempting to speculate that tumor-infiltrating Th-CTX might also promote cDC1 migration to sustain CD8 T cell functions.
The main drivers of the cytolytic potential in human CD4 T cells remain unknown. One determinant might be the affinity of the TCR, as reported for CD8 T cells, where the affinity and binding strength of a TCR to its cognate pMHC correlates with T cell effector functions (55). Previous TCR repertoire analyses of CD4 T cells featuring cytolytic functions (22, 36) demonstrated a marked clonal expansion. Similarly, one TCR clonotype was significantly enriched and restricted to the Th-CTX cluster in the BC patient dataset, and it was also one of the most abundant clonotypes across all CD4 T cells. The detection of a total of 21 different clonotypes within the cytolytic CD4 T cell cluster and the absence of unique TCRs in our helper and cytotoxic CD4 T cell clones rather suggest the emergence of cytolytic CD4 T cells from an oligo/polyclonal population.
Alternatively, the concerted up- and down-regulation of master TFs (transcription factors) might be the key for the establishment of a stable Th-CTX phenotype. Several studies showed the induction of Th-CTX from Th2 (56), Th17, Treg (33, 57), or, more often, Th1-polarized T cell populations (28, 53, 58). We observed the up-regulation of EOMES and the down-regulation of ThPOK in the cytolytic CD4 cluster and a significant enrichment of RUNX3 expression among Th-CTX. These results are in line with the previously reported cross-regulation of RUNX3 and ThPOK during the acquisition of cytotoxic functions by human Th1 lymphocytes in the context of CMV infection (28) and in the reprogramming of CD4 T cells into intestinal intraepithelial cytolytic effectors (16, 30). Runx3 is a key factor in the development of the CD8 T cell lineage, by orchestrating the downstream activation of T-bet and Eomes (59, 60), which have also been suggested as defining cytolytic CD4 T cells (11, 25–27). However, no difference in T-bet expression was observed in our comparison between helper versus cytotoxic CD4 T cells, suggesting for a dispensable role of this factor in Th-CTX functions. In agreement with our observations, T-bet deficient CD4 TILs in anti–CTLA-4–treated melanoma-bearing mice exhibited similar tumor rejection potency as their WT counterparts. These results illustrate the requirement of T-bet in IFNγ production but not in the cytolytic potential of this population, which was associated with a Blimp-1/granzyme B axis (30). Intriguingly, the transcription factor homolog of Blimp-1, called Hobit, was also associated with a cytotoxic potential in CD4 T cells during human cytomegalovirus infection (29). Yet, it was not specifically enriched in our dataset. Recently, it was reported that mCMV (murine cytomegalovirus)–infected mice deficient for the HDACs 1 and 2 in CD4 T cells developed a cytotoxic CD4 T cell program controlled by IFNγ–Janus kinase 1/2–signal transducers and activators of transcription 1 signaling. Both murine and human CD4 T cells treated with short-chain fatty acids, known as metabolites acting as HDAC inhibitors, or Entinostat, up-regulated cytotoxic-related genes (49). Overall, these observations suggest a potential network of transcription factors and epigenetic tuning (49) in the induction of Th-CTX.
Furthermore, the coexpression of an array of activating/inhibitory receptors might be necessary for a full cytotoxic potential profile. In both the scRNA-seq datasets and in isolated patient CD4 T cells, we identified SLAMF7 and, to a lower extent, PD-1, in Th-CTX cells. A humanized anti-SLAMF7 antibody (Elotuzumab) and SLAMF7-CAR T were already successfully used in the treatment of patients with multiple myeloma. Reported mechanisms potentially involved in the clinical efficacy imply the activation of NK cells, antibody-dependent cellular cytotoxicity, and inhibition of SLAMF7 homophilic interactions in the bone marrow niche (46, 47). Instead, the relevance of SLAMF7 targeting in Th-CTX remains to be addressed in cancer. Our results showing an unleashing of the cytotoxic potential of CD4 T cells after agonistic engagement of SLAMF7 open new therapeutic perspectives that warrant rapid testing in combination settings in patients with cancer. SLAMF7+ cytolytic CD4 T cells and their lytic function were previously described as a causal pathogenic link in the fibroinflammatory condition immunoglobulin G4–related disease, through the lysis of MHCII+ endothelial cells (61). In that regard, although we and others reported the expression of MHCII on tumor cells, the observed high frequency of MHCII-expressing stromal cells directly in situ in tumors further illustrates the need to evaluate the interactions of Th-CTX with these accessory cells, as previously shown in patients with multiple sclerosis (62) or in the context of helper CD4 T cell–mediated regulation of angiogenesis in tumors (63, 64).
The direct assessment of the cytotoxic potential of Th-CTX demonstrated their primary dependence on cytotoxic granules, similar to classical cytotoxic lymphocytes (10), however with a different kinetic. Classical cytotoxic lymphocytes are known to form a lytic synapse with target cells within few minutes after the initial cell contact, resulting in the rapid apoptosis of the targets (65). These timing discrepancies might be due to the weaker affinity of CD4 TCRs than those of CD8 T cells. Alternatively, this might be attributed to the absence of a TCR-pMHC–stabilizing function of the CD4 coreceptor (66), as opposed to the CD8 molecule. Further, CD8 CTLs have demonstrated a capacity to kill outnumbering target cells, indicating a highly dynamic expression of the TCRs at the surface of the cell (65). In line with CD8 T cell data, our observations in the nanoscale array argue for the capacity of some CD4 T cells to lyse multiple targets, highlighting the potential presence of “superkiller cells” (65) also within the CD4 T cell population. Future evolution of these types of platforms will allow the retrieval of the cell(s) of interest. Invaluable phenotypic, transcriptomic, and TCR analyses directly on patients’ blood or tumor tissue T cells will provide ultimate knowledge on the function of these cells.
Overall, we show, through an integrated large-scale transcriptomic analysis and direct cell functional profiling down to the antigen-specific clonal and single-cell resolution level, that tumor-specific Th-CTX CD4 T cells represent an alternative/synergistic population to classical CD8 T cells, which carry cytotoxic capacities in addition to their helper/regulatory function. MHCI loss or down-regulation is a major tumor escape mechanism (67), which might be counteracted by the therapy-induced activation of MHCI-independent Th-CTX. Moreover, adoptive cell transfer therapy (ACT) of autologous, primary, or genetically modified CD4 T cells was shown to be both safe and effective in patients with metastatic solid tumors (2, 3, 9), highlighting the attractive potential of ACT using tumor-specific Th-CTX cells for personalized next-generation immunotherapy.
MATERIALS AND METHODS
scRNA-seq data source and processing
All the scRNA-seq data of the patients with melanoma were retrieved from the publication of Sade-Feldman et al. (37) [processed and normalized data were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) series GSE120575]. Cells from clusters G5 to G11 (n = 12,850), which correspond to T/NKT/NK cells according to the original study, were selected for the downstream analyses. Of these cells, CD4 T cells were identified as CD3E+CD4+ CD8A−CD8B− and CD8 T cells as CD3E+CD4−CD8A+/CD8B+.
Following Seurat 2.3.4 pipeline, the normalized data were first scaled using Seurat method ScaleData. Variable genes (n = 533) were identified using Seurat method FindVariableGenes with the parameters x.low.cutoff = 0.01, x.high.cutoff = 14, y.cutoff = 1.5. The dimensionality reduction was performed using Seurat method RunPCA. The first 10 principal components were used to compute tSNE embedding using Seurat method RunTSNE with the parameter seed.use = 12345. Four clusters of CD4 T cells were identified by shared nearest neighbor clustering using Seurat method FindClusters, with the parameter resolution = 0.3. Gene markers of these clusters were identified using Seurat method FindAllMarkers with parameters min.pct = 0.2, logfc.threshold = 1, only.pos = TRUE.
scRNA-seq data from patients with BC were retrieved from Azizi et al. (50). scRNA-seq data from patients with head and neck cancer were retrieved from Puram et al. (51). scRNA-seq data from patients with hepatocellular carcinoma were retrieved from (52). For the BC dataset, an additional quality control of cells was applied, because the downloaded dataset contained unprocessed matrix of UMI (unique molecular identifier) counts. In particular, cells having (i) less than 600 or more than 1500 genes, (ii) less than 5000 or more than 30,000 UMIs, (iii) more than 15% of UMIs from mitochondrial protein-coding genes, and (iv) more than 50% of UMIs from ribosomal protein–coding genes were excluded. Data were normalized using a standard approach with the Seurat method NormalizeData.
In the head and neck cancer dataset, only T cells from primary tumors were kept for the downstream analyses. For both BC and head and neck cancer datasets, the identification of CD4/CD8 T cells was identical to the approach described above (i.e., identification of CD3+CD4+CD8− cells followed by clustering and cluster annotation based on known markers), while for the hepatocellular carcinoma, there was no additional filtering on CD4 and CD8 cells at the transcriptional level, because cells were sorted with fluorescence-activated cell sorter (FACS), only four outliers were removed. In total, we identified 7348 CD4 T cells in the BC dataset, 207 CD4 T cells in head and neck cancer dataset, and 1159 CD4 T cells in hepatocellular carcinoma. Within those CD4 T cells, CD4_CTX cluster contained 203, 44, and 459 cells in BC dataset, head and neck cancer dataset, and hepatocellular carcinoma dataset, respectively.
TCR sequencing of T cells from BC dataset was downloaded from NCBI GEO series GSE114724 (GSM3148580 and GSM3148584). TCR was successfully sequenced for 99% of CD4 T cells. In our notation, clonotype is uniquely identified by its productive α and β chain sequences. Cells of the same clonotype, therefore, have the same α and β chain sequences.
Gene ontology
Lists of the top 30 genes of each CD4 cluster were tested for overrepresentation in GO gene sets linked to biological processes using the Panther online tool (68, 69). Overrepresentation analyses for each gene set were performed using default settings using a Fisher’s exact test. A false discovery rate–corrected P value threshold of 0.05 was used (see complete data in tables S3 to S6).
Lymphocyte isolation from peripheral blood and tumor tissues
PBL and TILs or TILNs from patients with stage III/IV melanoma were obtained from the Department of Oncology, University Hospital [CHUV (Centre hospitalier universitaire vaudois), Lausanne, Switzerland; NCT00112229, NCT00002669, and NCT00002763; table S1], under the approval of the Lausanne University Hospital’s Institutional Review Board. Blood was diluted with phosphate-buffered saline (PBS), and mononuclear cells were purified by density gradient centrifugation using Lymphoprep (STEMCELL). Isolated lymphocytes (from PBL, preimmunization TIL, or preimmunization TILN) were cryo-preserved in 90% fetal calf serum (FCS) (Gibco) and 10% dimethyl sulfoxide (DMSO) (Sigma-Aldrich) and stored in liquid nitrogen until further use.
Generation of enriched antigen-specific CD4 T cells and antigen-specific CD4 T cell clones
Antigen-specific CD4 T cells were evaluated either directly ex vivo or after one round of in vitro stimulation using fluorescent pMHCII multimers provided by the Peptide and Tetramer Core Facility, UNIL (University of Lausanne)–CHUV, Epalinges, Switzerland. CD4 T cells were incubated with optimal concentrations of fluorescent pMHCII multimers in 8% RPMI culture medium for 45 min at room temperature following the addition of fluorescently labeled anti-CD3 and anti-CD4 antibodies and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific). Multimer-positive CD4 T cell populations were sorted using a FACSAria II or III sorter (BD Biosciences). Where indicated, multimer-negative CD4 T cells, naïve CD8 T cells (CD45RA+/CCR7+), and antigen-experienced CD8 T cells (CD45RA−/CCR7+, CD45RA−/CCR7−, and CD45RA+/CCR7−) were also sorted from the same patient. Unstained cultures from the same patients or nonspecific HLA-matched pMHCII stainings were used as negative controls. Clones were obtained from T cells sorted from patients’ PBMCs by limiting dilution (0.5 cell per well) in Terasaki plates and cultured in RPMI medium with 8% HS (human serum) and recombinant human IL-2 (rh–IL-2) (100 U/ml), phytohemagglutinin (1 μg/ml), and 10,000 irradiated allogenic feeder cells per well. Where indicated, CD4 T cell clones were exposed for 24 hours to rh–IL-12 (20 ng/ml; Peprotech) or 2 μM Entinostat (MS-275, Selleckchem), before evaluation of SLAMF7 expression by flow cytometry.
Synthetic peptides
Peptides were synthesized at the Peptide and Tetramer Core Facility, UNIL-CHUV, Epalinges, Switzerland using INTAVIS synthesizers, using F-moc (fluorenylmethoxycarbonyl) for transient NH2-terminal protection. All peptides were >90% pure as indicated by analytic mass spectrometry and high-performance liquid chromatography. Lyophilized peptides were diluted in pure DMSO at 10 mg/ml, or aliquots of 1 mg/ml in 10% DMSO were prepared in PBS and stored at −80°C.
Tumor cell lines
Melanoma cell lines were established from metastatic surgery specimens from patients with melanoma from the Department of Oncology UNIL-CHUV and Ludwig Institute for Cancer Research, University of Lausanne (70) or provided by the Division of Immunology, Transplantation and Infectious Diseases, San Raffaele Scientific Institute, Milan. Tumor cell lines were cultured in 10% FCS RPMI.
Assessment of MHCII and SLAMF7 expression on melanoma tumor cell lines
Where indicated, tumor cell lines were treated with IFNγ (Peprotech), TNFα (Peprotech), or both IFNγ and TNFα at 50 or 200 U/ml for 48 hours before the flow cytometry analysis. Tumor cell lines were systematically labeled with fluorescein isothiocyanate (FITC) anti–HLA-DR, clone L243 (Abcam), BV421 anti–PD-1 (BioLegend), phycoerythrin (PE)–Cy7 anti-SLAMF7 (BioLegend), PE-Cy7 anti–PD-L1 (BD Biosciences), and PE anti–PD-L2 (BD). Data were acquired on LSR II (BD Biosciences) and analyzed using FlowJo software (v.10.4.2).
Multiplex fluorescent IHC staining of metastatic melanoma tissue sections
Immunostainings were performed on 4-μm formalin-fixed paraffin-embedded tissue sections from 18 melanoma cases, provided by the Institute of Pathology at the Lausanne University Hospital, CHUV. These cases were unrelated to the samples used for CD4 T cell functional evaluation, precluding any correlative analyses between immunohistochemistry (IHC) results and CD4 T cell functional assessment in PBMCs/TILNs. Chromogenic IHC was performed using HLA-DR, MHCII (clone CR3/43, dilution 1:100; reference no. M0775) from Dako on the Ventana BenchMark XT system from Roche. For the multiplex fluorescent IHC on the Ventana Discovery Ultra Autostainer from Roche, apart from HLA-DR, SOX-10 (SRY-Box Transcription Factor 10), a melanoma marker (clone BC34, dilution 1:100; reference no. ACI 3099 A, C) from Biocare Medical and PD-L1 primary antibody (clone E1L3N, dilution 1:200: reference no. 13684S) from Cell Signaling were used, followed by incubation with secondary horseradish peroxidase–labeled antibodies. Tissue sections were counterstained with Spectral DAPI (4′,6-diamidino-2-phenylindole) from Akoya for 4 min. For the fluorescent detection with Opal dyes, an Opal 7-color Automation IHC kit (from Akoya, reference no. NEL821001KT) was used. An adjacent section was stained with hematoxylin and eosin (Ventana HE 600 system) for morphological reappraisal and to assist IHC interpretation. The multiplex fluorescent panel was developed by ensuring that all antibodies stained robustly in a monoplex protocol, identifying only the target antigen of interest. A spectral library was created, preparing single-plex spectral control slides and an autofluorescence slide from the melanoma samples. Annotations were placed selectively in SOX-10+ areas, covering approximately two-thirds of the tissue sections, using the Phenochart 1.0.12 software from Akoya, a whole-slide contextual viewer used for navigation around slides for high-resolution multispectral acquisition. Multispectral images (MSI) were acquired using the Vectra 3.0 automated quantitative pathology imaging system from PerkinElmer. The whole-tissue slides were prescanned at a ×10 magnification, and approximately 20 to 40 regions of interest images (number of fields in the PhenoptR analysis excel sheet) obtained with the 20× objective were selected for the acquisition of high power (20×) MSI. In the Vectra, the scale bars of a 20× image were set as follows: 1 pixel is calculated as (0.5 μm) × (0.5 μm) = 0.25 μm2. To get the area in square millimeters, the area in pixels is divided by 4,000,000 as 1 mm2 = 1,000,000 μm2, and therefore, 0.25/1,000,000 is equal to 1/4,000,000. For the data processing, algorithms for each case were designed, and the analysis was performed using the inForm 2.4.1 software from Akoya. Regarding the frequency of HLA-DR+ cells, in the segmented tumor regions, it is calculated from SOX-10+ cells. In the segmented stromal region, it is calculated from the total number of cells given by the PhenoptR reports platform. The data were further treated with the Phenoptr Reports, an R-based addin for RStudio from Akoya.
Generation of CIITA-transduced tumor cell lines
The gene sequence of the human CIITA isoform 3 was provided by W. Reith, Department of Pathology and Immunology, Faculty of Medicine, University of Geneva, Switzerland. The isoform 3 of the CIITA gene was inserted into a promoter modified retroviral vector pQCXIP (Clontech Corp) using the Age I–HF (high-fidelity) and Eco RI–HF restriction enzymes. The resulting hCIITA-pQCXIP plasmid DNA and the packaging plasmid DNA were introduced at ratio 1:1 into XL10-Gold ultracompetent Escherichia coli cells by calcium phosphate precipitation. Forty-eight hours after transfection, the green fluorescent protein reporter expression was examined by using a fluorescence microscope (Promega). Culture medium containing the virus was harvested and filtered to remove cellular debris. A portion of the infectious virions, mixed with polybrene (8 μg/ml) to increase virus absorption within target lines, was added into a culture plate containing around 50 to 70% confluent GEFI and Me275 cell lines. Selection for drug resistance was initiated at 48 hours after infection at 10 μg/ml of blasticidin (Invitrogen). After selection, cell surface MHCII expression was assessed by flow cytometry using an anti–HLA-DR antibody (L243, BioLegend).
Chromium release assay and CD107a degranulation assay
The cytolytic potential of the blood-derived tumor-specific CD4 T cells was assessed against HLA-matched melanoma cell lines (GEFI, GEFI-CIITA, GEFII, Me275, and Me275-CIITA), pretreated or not with IFNγ (50 U/ml; Peprotech) for 48 hours where indicated. Cells were labeled with 51Chromium (Amersham Biosciences) for 1 hour at 37°C. Labeled target cells were then incubated with effectors at the indicated effector to target (30:1,10:1, 3:1, and 1:1) ratio for 4 hours at 37°C, in the presence or absence of a specific peptide. When indicated, addition of an inhibitor of perforin-mediated cytotoxicity, CMA at 100 nM (2 hours before incubation; Sigma-Aldrich), membrane-soluble Compound 20 at 20 μM (30 min before incubation of target cells), a monoclonal anti–PD-1 antibody at 10 μg/ml (Opdivo; Bristol-Myers Squibb, CMA), an anti-SLAMF7 antibody at 5 μg/ml (BioLegend), or a transwell was added during the coculture. Supernatants were collected, and the released 51Cr was counted using a gamma counter (PerkinElmer, Boston, MA). Internal controls were included in each assay to measure the spontaneous release (target cells alone) and the maximal release (target cells with 1 M HCl). The percentage of specific lysis was calculated as follows: (experimental-spontaneous release)/(maximal-spontaneous) × 100. The resulting percentage of specific lysis enables the discrimination of the tumor-specific CD4 T cell clones as classical helpers (Th) or presenting dual helper and cytolytic functions (Th-CTX).
For the CD107a degranulation assay, Th-CTX were cocultured at 1:1 ratio for 4 hours with target cells, pulsed or not with the specific peptide, in the presence or absence of a FITC anti-CD107a antibody (BioLegend). After the coculture, cells were stained with PerCP-Cy5.5 anti-CD4 antibody (BioLegend). CD107a positivity was defined on CD4+ T cells, based on the FMO (fluorescence minus one) control.
High-throughput and time-resolved single-cell tumor recognition assay in picowell arrays
The picowell structures (35 μm by 35 μm by 50 μm) for the confinement of the cells within subnonaoliter volumes were fabricated by a replica molding process with PDMS as previously reported (42). A master mold was first prepared by standard photolithography on a 10.16 cm silicon substrate. A direct laser writer (MLA 150, Heidelberg Instruments) patterned the spin-coated positive photoresist (AZ9221, Microchemicals GmbH), followed by a deep reactive Ion etching (AMS200, Alcatel Adixen). The picowell array was then oxidized by O2 plasma and bonded to the bottom of four-well μ-Slide (ibidi, Germany), forming an assembly for the following cell seeding. Immediately before use in cell-based assays, the picowell array was reoxidized with O2 plasma and subsequently rinsed with cell media. Prestained tumor-specific CD4 or CD8 T cell clones (CellTrace Violet Cell Proliferation Kit, Invitrogen) and HLA-matched tumor cells (PKH26 and MINI26, Sigma-Aldrich) were seeded sequentially into the picowell array. The seeding density was 1 × 105/ml and 1 × 105/ml, respectively. The assembly was incubated shortly after each step of cell seeding (10 min in the cell incubator) to allow sufficient cell deposition in the picowells. The medium was then replaced by 700 μl of 10% FCS RPMI containing specific peptide (5 μg/ml) and 5 μM Incucyte Caspase-3/7 reagent (Sartorius). The prepared assembly was placed inside a microscope stage incubator that can provide essential cell culture conditions. The phenotype of the cells was distinguished with four channels (bright field, AF488, TRITC, and BV421) at 10-min intervals for a duration of 24 hours. Images were acquired using a Nikon Eclipse Ti-E inverted microscope and analyzed using the Nikon NIS-Elements (Advanced Research version 3.10, Nikon) software. The acquired multichannel images were then converted to a video for the following feature extraction, which is done with an adapted version of DeepLabCut v2.2 [available at https://github.com/DeepLabCut/DeepLabCut; original (43)].
Deep learning algorithm for automated and efficient image processing
To efficiently analyze the cell interactions within each well on the well plate, we used deep learning algorithms based on artificial neural network and trained the DeepLabCut to detect the well corners (lower left/upper right) as well as alive tumor cells, lysed tumor cells, alive T cells, and lysed T cells. Taking advantage of the fully convolutional architecture of DeepLabCut, we only annotated 3 × 3 and 1 × 1 well crops from diverse videos. We found that the network generalized well when processing the complete well plates. To train DeepLabCut, six types of features (well lower left corner, well upper right corner, alive tumor cell, lysed tumor cell, alive T cell, and lysed T cell) were labeled in 116 frames (of 3 × 3 or 1 × 1 well patches) selected from 15 videos in total (43). For interference, on novel videos, the trained network predicts the coordinates of all the detection in every frame. These coordinates were then used to implement further screening process in Python to identify the wells with killing. The MFI of lysis was analyzed by measuring the intensity within selected well areas in AF488 channel, and the time of lysis was defined as the maximum of the first derivative of the MFI curves, which represents the sharpest increase of caspase-3/7 activation when cell apoptosis was taking place.
Multiparametric flow cytometry analysis of cytolytic CD4 T cells
Tumor-specific CD4 T clones were stained systematically in PBS, 0.2% bovine serum albumin (BSA), 5 mM EDTA, and 0.2% NaN3 with either (i) FITC anti-CD3 (BioLegend), BV605 anti-CD4 (BioLegend), ECD anti-CD8 (BD), PE-Cy7 anti-SLAMF7 (BioLegend), PerCP-Cy5.5 anti-CD57 (BioLegend), APC (allophycocyanin) anti-CD28 (BioLegend), BV711 anti–PD-1 (BioLegend), PE anti-TCRαβ (BioLegend), and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) or (ii) FITC anti-CD3 (BioLegend), BV605 anti-CD4 (BioLegend), APC anti-TRAIL (BioLegend), PE anti-FasL (BioLegend), and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) at 4°C for 30 min. Data were acquired on LSR II (BD Biosciences) and analyzed using FlowJo software (v.10.4.2).
Direct ex vivo enumeration of NY-ESO-187–99/DR7 CD4 T cells
Enriched CD4 T cells were purified by positive selection using MACS isolation microbeads (Miltenyi) and stained directly ex vivo using a combination of fluorescent pMHCII multimers, as described above and FITC anti-CD3 (BioLegend), BV605 anti-CD4 (BioLegend), PE-Cy7 anti-SLAMF7 (BioLegend), BV711 anti–PD-1 (BioLegend), and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific). Irrelevant multimers were used as negative controls. Data were acquired on LSR II (BD Biosciences) and analyzed using FlowJo software (v.10.4.2).
Intracellular granule and cytokine staining
Tumor-specific CD4 T clones were stimulated with serial dilutions of the specific peptides or left untreated for 6 hours at 37°C in the presence of brefeldin A (10 μg/ml; Sigma-Aldrich). Cells were then stained in PBS, 0.2% BSA, 5 mM EDTA, and 0.2% NaN3 with BV421 anti-CD3 (BioLegend) and BV605 anti-CD4 (BioLegend) at 4°C for 30 min, fixed in PBS 1% formaldehyde, 2% glucose, and 5 mM NaN3 for 20 min at room temperature, and, lastly, stained in PBS, 0.2% BSA, 5 mM EDTA, 0.2% NaN3, and 0.1% saponin (Sigma-Aldrich) with either (i) PE-Cy7 anti-IFNγ (BD Biosciences), PerCP-Cy5.5 anti-TNFα (BioLegend), APC anti–IL-13 (BioLegend), and A700 anti–IL-17a (BioLegend) or (ii) PE anti-perforin (BioLegend), A700 anti-granzyme A (BioLegend), ECD anti-granzyme B (BD Biosciences), PerCP710 anti-granzyme K (eBioscience), APCeF660 anti-granzyme M (eBioscience), and A488 anti-granulysin (eBioscience) for 30 min at 4°C before acquisition on an LSR II SORP (Beckman Coulter) flow cytometer. Percentages of granule/cytokine-positive T cells were analyzed using FlowJo software (v.10.4.2). EC50 values were derived by dose-response curve analysis [log(agonist) versus response] using Prism software. Noncytokine clones, indicated in the added gray boxes, for which an EC50 value could not be determined accurately, were not included in the statistical analyses.
SEB cytotoxicity assay
PBMCs were stained in PBS, 0.2% BSA, 5 mM EDTA, 0.2% NaN3 with FITC anti-CD4 (BioLegend), and ECD anti-CD8a (Beckman Coulter) at 4°C for 30 min and sorted on a FACSAria II sorter (BD Biosciences). After sorting, cells were seeded for 3 days in the presence of aCD3/CD28 beads (Gibco) in RPMI 8% HS and rh-IL-2 (100 U/ml), before using them in a single cell cytotoxicity assay on the high-throughput nanobiosensor (see above). Individual target cells were seeded in the nanowells, and then they were incubated with SEB (1 μg/ml; Sigma-Aldrich) for 30 min. Cells were then washed, and individual T cells were seeded and monitored over 24 hours.
RNA purification and RT-qPCR
Total RNA was isolated from either directly ex vivo sorted highly pure NY-ESO-187–99/DR7 CD4 T cells, multimer-negative CD4 T cells, naïve CD8 T cells (CD45RA+/CCR7+), and antigen-experienced CD8 T cells (CD45RA−/CCR7+, CD45RA−/CCR7−, and CD45RA+/CCR7−), stained in PBS, 0.2% BSA, 5 mM EDTA, 0.2% NaN3 with BV421 anti-CD45RA (BioLegend), and Alexa Fluor 700 anti-CCR7 (BioLegend), or tumor-specific CD4 T cell clones using the TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). The final preparation of RNA was considered DNA and protein free if the ratio of spectrophotometer (NanoDrop, Thermo Fisher Scientific, Carlsbad, CA, USA) readings at 260/280 nm was ≥1.7. Isolated mRNA was reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Watford, UK) according to the manufacturer’s protocol. The quantitative polymerase chain reaction (qPCR) was carried out in the Applied Biosystems 7900HT Fast Real-Time PCR Sequence Detection System (Applied Biosystems) with specific primers (hPRF1, 5′-GACTGCCTGACTGTCGAGG-3′ and 5′-TCCCGGTAGGTTTGGTGGAA-3′; hGZMA, 5′-CCCTATCCATGCTATGACCCA- 3′ and 5′-AGTATCGGACCAAGATGCACTAT-3′; hGZMB, 5′-TACCATTGAGTTGTGCGTGGG-3′ and 5′-GCCATTGTTTCGTCCATAGGAGA-3′; hGNLY, 5′-CCTGTCTGACGATAGTCCAAAAA-3′ and 5′-GACCTCCCCGTCCTACACA-3′; hTHPOK, 5′-TGATGGCCTACCTAAGCTCCC-3′ and 5′-CATGTGGCGAGGCAGTTTG-3′; hRUNX3, 5′-AGCACCACAAGCCACTTCAG-3′ and 5′-GGGAAGGAGCGGTCAAACTG-3′; hFOXP3, 5′-GTGGCCCGGATGTGAGAAG-3′ and 5′-GGAGCCCTTGTCGGATGATG-3′; hTBET, 5′-GGTTGCGGAGACATGCTGA-3′ and 5′-GTAGGCGTAGGCTCCAAGG-3′; hGATA3, 5′-GCCCCTCATTAAGCCCAAG-3′ and 5′-TTGTGGTGGTCTGACAGTTCG-3′; hRORC, 5′-CTGGGCATGTCCCGAGATG-3′ and 5′-GAGGGGTCTTGACCACTGG-3′; hEOMES, 5′-CTGCCCACTACAATGTGTTCG-3′ and 5′-GCGCCTTTGTTATTGGTGAGTTT-3′; hBLIMP1, 5′-TGGAAGATGAAAGGGCATTG-3′ and 5′-CATGGCTTGGCTCACAATTT-3′; hSLAMF7, 5′-ACAACCCCTCTTGTCACCATA-3′ and 5′-CCCACATAGTAGATCCCTGAGTC-3′; hPDCD1, 5′-CCAGGATGGTTCTTAGACTCCC-3′ and 5′-TTTAGCACGAAGCTCTCCGAT-3′; hEAT-2, 5′-CCGTGTTTCTCTCTGAAGATTCG-3′ and 5′-AGACAGCGAGTCGATACCAG-3′) using KAPA SYBR FAST qPCR Kits (KAPA Biosystems Inc., MA). All reverse transcription qPCR (RT-qPCR) reactions were performed in duplicate and amplified simultaneously with a nontemplate control blank for each primer pair to control for contamination or for primer dimerization, as well as a housekeeping gene (β2-microglobulin) to normalize and determine the Ct values, using the 2−ΔCt formula.
TCR sequencing
mRNA was isolated from CD4 T cell clones derived from patients’ PBMCs using the Dynabeads mRNA DIRECT Purification Kit (Thermo Fisher Scientific/Ambion). First-strand cDNA was synthesized using oligo(dT) (Promega) and the Superscript III (Thermo Fisher Scientific). TCR sequencing was performed as previously described in (71).
Survival analysis using TCGA data
The correlation between the SLAMF7 expression level and survival of patients with cancer was performed by using TCGA RNA-seq data available on the Human Protein Atlas website (www.proteinatlas.org). We obtained [FPKM (Fragments Per Kilobase of sequence per Million mapped)] values of SLAMF7 in samples from melanoma (N = 102 patients), BC (N = 1075 patients), head and neck squamous cell carcinoma (N = 499), and liver hepatocellular carcinoma (N = 365 patients), as well as vital status and survival time information for each patient. The classification of patients into high or low expression level of SLAMF7 was performed by using a different cutoff for each cancer type (i.e., we used the default cutoff value proposed in the Human Protein Atlas): The cutoff was 0.77 FPKM for melanoma, 1.5 FPKM for BC, 6.63 FPKM for head and neck cancer, and 1.33 FPKM for liver cancer. The difference between the high- and the low-SLAMF7 survival curves was assessed using a log-rank test separately for each cancer type.
Statistical analysis
Statistical analyses were performed as indicated in the legend for each experiment using GraphPad Prism (version 8.2.1). For all analyses, a P < 0.05 was statistically significant and labeled by *, <0.05 by **, <0.001 by ***, and <0.0001 by ****. Not statistically significant differences were left unlabeled.
Acknowledgments
We are grateful to the patients for dedicated collaboration and healthy donors for blood and tissue donations. We thank H. Maby-El Hajjami for excellent assistance with clinical data. We sincerely thank L. Queiroz, P. Reichenbach, and A. Cornu for technical assistance. We thank D. Comte for insightful discussions on SLAMF7. We thank the expertise and assistance of the flow cytometry facility at the University of Lausanne. We acknowledge the Ecole Polytechnique Fédérale de Lausanne and the Center of MicroNano Technology facilities for the fabrication of the picowell arrays. Funding: We thank the following funding that supported this work: the ISREC Foundation (to A.C., Y.-C.L., and H.A.), the MEDIC Foundation (to C.J.), the Fondazione San Salvatore (to C.J.), the Swiss National Science Foundation (PRIMA PR00P3_179727 to C.J., SNF 310030-182735 to P.R., and 31003A_173156 to D.G.), the Olga Mayenfish Foundation (to C.J.), the Chan Zuckerberg Initiative (to A.M.), the Taiwan EPFL PhD scholarship (to Y.-C.L.), and the Ludwig Institute for Cancer Research. This project has received funding from the Société Académique Vaudoise-Fonds Novartis Consumer Health (to C.J. and H.A.). Author contributions: A.C., Y.-C.L., X.L., M.S., M.C., G.A.R., P.G., J.S., R.G., G.E., and K.I. designed the research, performed the experiments, analyzed and interpreted the results, and edited the manuscript. M.B. and T.W. performed scRNA-seq data analysis and edited the manuscript. L.d.L. supervised and interpreted the IHC analysis and edited the manuscript. A.H. supervised the TCR sequencing analysis and edited the manuscript. D.G. designed and supervised the scRNA-seq data analysis and edited the manuscript. A.M. and H.A. designed and supervised the single-cell cytotoxicity assay and edited the manuscript. M.P.P., W.R., G.C., and D.E.S. provided samples, generated reagents, and edited the manuscript. P.R. and C.J. designed the research and supervised the entire study, analyzed and interpreted results, and wrote the manuscript. All authors reviewed and approved the final version of the manuscript. Competing interests: G.C. has received grants and research support or is coinvestigator in clinical trials by BMS, Celgene, Boehringer Ingelheim, Roche, Iovance, and Kite. G.C. has received honoraria for consultations or presentations by Roche, Genentech, BMS, AstraZeneca, Sanofi-Aventis, Nextcure, and GeneosTx. G.C. has patents in the domain of antibodies and vaccines targeting the tumor vasculature as well as technologies related to T cell expansion and engineering for T cell therapy. G.C. holds patents around TEM1 antibodies and receives royalties from the University of Pennsylvania regarding technology licensed to Novartis. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabe3348/DC1
REFERENCES AND NOTES
- 1.Sallusto F., Lanzavecchia A., Heterogeneity of CD4+ memory T cells: Functional modules for tailored immunity. Eur. J. Immunol. 39, 2076–2082 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Hunder N. N., Wallen H., Cao J., Hendricks D. W., Reilly J. Z., Rodmyre R., Jungbluth A., Gnjatic S., Thompson J. A., Yee C., Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med. 358, 2698–2703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran E., Turcotte S., Gros A., Robbins P. F., Lu Y. C., Dudley M. E., Wunderlich J. R., Somerville R. P., Hogan K., Hinrichs C. S., Parkhurst M. R., Yang J. C., Rosenberg S. A., Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fridman W. H., Pagès F., Sautès-Fridman C., Galon J., The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Tran E., Ahmadzadeh M., Lu Y. C., Gros A., Turcotte S., Robbins P. F., Gartner J. J., Zheng Z., Li Y. F., Ray S., Wunderlich J. R., Somerville R. P., Rosenberg S. A., Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kreiter S., Vormehr M., van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J. C., Tadmor A. D., Schoenberger S. P., Huber C., Türeci Ö., Sahin U., Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linnemann C., van Buuren M. M., Bies L., Verdegaal E. M. E., Schotte R., Calis J. J. A., Behjati S., Velds A., Hilkmann H., El Atmioui D., Visser M., Stratton M. R., Haanen J. B. A. G., Spits H., van der Burg S. H., Schumacher T. N. M., High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21, 81–85 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Ott P. A., Hu Z., Keskin D. B., Shukla S. A., Sun J., Bozym D. J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., Chen C., Olive O., Carter T. A., Li S., Lieb D. J., Eisenhaure T., Gjini E., Stevens J., Lane W. J., Javeri I., Nellaiappan K., Salazar A. M., Daley H., Seaman M., Buchbinder E. I., Yoon C. H., Harden M., Lennon N., Gabriel S., Rodig S. J., Barouch D. H., Aster J. C., Getz G., Wucherpfennig K., Neuberg D., Ritz J., Lander E. S., Fritsch E. F., Hacohen N., Wu C. J., An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Y. C., Parker L. L., Lu T., Zheng Z., Toomey M. A., White D. E., Yao X., Li Y. F., Robbins P. F., Feldman S. A., van der Bruggen P., Klebanoff C. A., Goff S. L., Sherry R. M., Kammula U. S., Yang J. C., Rosenberg S. A., Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J. Clin. Oncol. 35, 3322–3329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown D. M., Lampe A. T., Workman A. M., The differentiation and protective function of cytolytic CD4 T cells in influenza infection. Front. Immunol. 7, 93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiskopf D., Bangs D. J., Sidney J., Kolla R. V., de Silva A. D., de Silva A. M., Crotty S., Peters B., Sette A., Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc. Natl. Acad. Sci. U.S.A. 112, E4256–E4263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma S., Weiskopf D., Gupta A., Donald B. M., Peters B., Sette A., Benedict C. A., Cytomegalovirus-specific CD4 T cells are cytolytic and mediate vaccine protection. J. Virol. 90, 650–658 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez-Martinez A., Perdomo-Celis F., Acevedo-Saenz L., Rugeles M. T., Velilla P. A., Cytotoxic CD4+ T-cells during HIV infection: Targets or weapons? J. Clin. Virol. 119, 17–23 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Appay V., The physiological role of cytotoxic CD4+ T-cells: The holy grail? Clin. Exp. Immunol. 138, 10–13 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuji M., Romero P., Nussenzweig R. S., Zavala F., CD4+ cytolytic T cell clone confers protection against murine malaria. J. Exp. Med. 172, 1353–1357 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reis B. S., Rogoz A., Costa-Pinto F. A., Taniuchi I., Mucida D., Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nat. Immunol. 14, 271–280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peeters L. M., Vanheusden M., Somers V., van Wijmeersch B., Stinissen P., Broux B., Hellings N., Cytotoxic CD4+ T cells drive multiple sclerosis progression. Front. Immunol. 8, 1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duftner C., Goldberger C., Falkenbach A., Würzner R., Falkensammer B., Pfeiffer K. P., Maerker-Hermann E., Schirmer M., Prevalence, clinical relevance and characterization of circulating cytotoxic CD4+CD28– T cells in ankylosing spondylitis. Arthritis Res. Ther. 5, R292–R300 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y., Feng Y., Liu H., Ye H., Guo C., Feng J., Dai S., Zheng X., CD4+CD29+T cells are blamed for the persistent inflammatory response in ulcerative colitis. Int. J. Clin. Exp. Pathol. 8, 2627–2637 (2015). [PMC free article] [PubMed] [Google Scholar]
- 20.Takeuchi A., Badr M. E. S. G., Miyauchi K., Ishihara C., Onishi R., Guo Z., Sasaki Y., Ike H., Takumi A., Tsuji N. M., Murakami Y., Katakai T., Kubo M., Saito T., CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 213, 123–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glennie N. D., Yeramilli V. A., Beiting D. P., Volk S. W., Weaver C. T., Scott P., Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J. Exp. Med. 212, 1405–1414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patil V. S., Madrigal A., Schmiedel B. J., Clarke J., O’Rourke P., de Silva A. D., Harris E., Peters B., Seumois G., Weiskopf D., Sette A., Vijayanand P., Precursors of human CD4+ cytotoxic T lymphocytes identified by single-cell transcriptome analysis. Sci. Immunol. 3, eaan8664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burel J. G., Apte S. H., Groves P. L., Klein K., McCarthy J. S., Doolan D. L., Reduced Plasmodium parasite burden associates with CD38+ CD4+ T cells displaying cytolytic potential and impaired IFN-γ production. PLOS Pathog. 12, e1005839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang D., Tian Z., Zhang M., Yang W., Tang J., Wu Y., Ni B., NKG2D+CD4+ T cells kill regulatory T cells in a NKG2D-NKG2D ligand–dependent manner in systemic lupus erythematosus. Sci. Rep. 7, 1288 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitano S., Tsuji T., Liu C., Hirschhorn-Cymerman D., Kyi C., Mu Z., Allison J. P., Gnjatic S., Yuan J. D., Wolchok J. D., Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol. Res. 1, 235–244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirschhorn-Cymerman D., Budhu S., Kitano S., Liu C., Zhao F., Zhong H., Lesokhin A. M., Avogadri-Connors F., Yuan J., Li Y., Houghton A. N., Merghoub T., Wolchok J. D., Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J. Exp. Med. 209, 2113–2126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qui H. Z., Hagymasi A. T., Bandyopadhyay S., St. Rose M. C., Ramanarasimhaiah R., Ménoret A., Mittler R. S., Gordon S. M., Reiner S. L., Vella A. T., Adler A. J., CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J. Immunol. 187, 3555–3564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serroukh Y., Gu-Trantien C., Hooshiar Kashani B., Defrance M., Vu Manh T. P., Azouz A., Detavernier A., Hoyois A., Das J., Bizet M., Pollet E., Tabbuso T., Calonne E., van Gisbergen K., Dalod M., Fuks F., Goriely S., Marchant A., The transcription factors Runx3 and ThPOK cross-regulate acquisition of cytotoxic function by human Th1 lymphocytes. eLife 7, e30496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oja A. E., Braga F. A. V., Remmerswaal E. B. M., Kragten N. A. M., Hertoghs K. M. L., Zuo J., Moss P. A., van Lier R. A. W., van Gisbergen K. P. J. M., Hombrink P., The transcription factor hobit identifies human cytotoxic CD4+ T cells. Front. Immunol. 8, 325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sledzinska A., de Mucha M. V., Bergerhoff K., Hotblack A., Demane D. F., Ghorani E., Akarca A. U., Marzolini M. A. V., Solomon I., Vargas F. A., Pule M., Ono M., Seddon B., Kassiotis G., Ariyan C. E., Korn T., Marafioti T., Lord G. M., Stauss H., Jenner R. G., Peggs K. S., Quezada S. A., Regulatory T cells restrain Interleukin-2- and Blimp-1-dependent acquisition of cytotoxic function by CD4+ T cells. Immunity 52, 151–166.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y., Akpinarli A., Maris C., Hipkiss E. L., Lane M., Kwon E. K. M., Muranski P., Restifo N. P., Antony P. A., Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 207, 651–667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quezada S. A., Simpson T. R., Peggs K. S., Merghoub T., Vider J., Fan X., Blasberg R., Yagita H., Muranski P., Antony P. A., Restifo N. P., Allison J. P., Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 207, 637–650 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akhmetzyanova I., Zelinskyy G., Littwitz-Salomon E., Malyshkina A., Dietze K. K., Streeck H., Brandau S., Dittmer U., CD137 agonist therapy can reprogram regulatory T cells into cytotoxic CD4+ T cells with antitumor activity. J. Immunol. 196, 484–492 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Porakishvili N., Kardava L., Jewell A. P., Yong K., Glennie M. J., Akbar A., Lydyard P. M., Cytotoxic CD4+ T cells in patients with B cell chronic lymphocytic leukemia kill via a perforin-mediated pathway. Haematologica 89, 435–443 (2004). [PubMed] [Google Scholar]
- 35.Rodig S. J., Gusenleitner D., Jackson D. G., Gjini E., Giobbie-Hurder A., Jin C., Chang H., Lovitch S. B., Horak C., Weber J. S., Weirather J. L., Wolchok J. D., Postow M. A., Pavlick A. C., Chesney J., Hodi F. S., MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 10, eaar3342 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Oh D. Y., Kwek S. S., Raju S. S., Li T., McCarthy E., Chow E., Aran D., Ilano A., Pai C. C. S., Rancan C., Allaire K., Burra A., Sun Y., Spitzer M. H., Mangul S., Porten S., Meng M. V., Friedlander T. W., Ye C. J., Fong L., Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 181, 1612–1625.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sade-Feldman M., Yizhak K., Bjorgaard S. L., Ray J. P., de Boer C. G., Jenkins R. W., Lieb D. J., Chen J. H., Frederick D. T., Barzily-Rokni M., Freeman S. S., Reuben A., Hoover P. J., Villani A.-C., Ivanova E., Portell A., Lizotte P. H., Aref A. R., Eliane J.-P., Hammond M. R., Vitzthum H., Blackmon S. M., Li B., Gopalakrishnan V., Reddy S. M., Cooper Z. A., Paweletz C. P., Barbie D. A., Stemmer-Rachamimov A., Flaherty K. T., Wargo J. A., Boland G. M., Sullivan R. J., Getz G., Hacohen N., Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munier C. M. L., van Bockel D., Bailey M., Ip S., Xu Y., Alcantara S., Liu S. M., Denyer G., Kaplan W.; PHIIDO Study group, Suzuki K., Croft N., Purcell A., Tscharke D., Cooper D. A., Kent S. J., Zaunders J. J., Kelleher A. D., The primary immune response to Vaccinia virus vaccination includes cells with a distinct cytotoxic effector CD4 T-cell phenotype. Vaccine 34, 5251–5261 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Baumgaertner P., Costa Nunes C., Cachot A., Maby-el Hajjami H., Cagnon L., Braun M., Derré L., Rivals J. P., Rimoldi D., Gnjatic S., Abed Maillard S., Marcos Mondéjar P., Protti M. P., Romano E., Michielin O., Romero P., Speiser D. E., Jandus C., Vaccination of stage III/IV melanoma patients with long NY-ESO-1 peptide and CpG-B elicits robust CD8+ and CD4+ T-cell responses with multiple specificities including a novel DR7-restricted epitope. Onco. Targets. Ther. 5, e1216290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thibodeau J., Bourgeois-Daigneault M.-C., Lapointe R., Targeting the MHC class II antigen presentation pathway in cancer immunotherapy. Onco. Targets. Ther. 1, 908–916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merouane A., Rey-Villamizar N., Lu Y., Liadi I., Romain G., Lu J., Singh H., Cooper L. J. N., Varadarajan N., Roysam B., Automated profiling of individual cell-cell interactions from high-throughput time-lapse imaging microscopy in nanowell grids (TIMING). Bioinformatics 31, 3189–3197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liadi I., Roszik J., Romain G., Cooper L. J. N., Varadarajan N., Quantitative high-throughput single-cell cytotoxicity assay for T cells. J. Vis. Exp. , e50058 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathis A., Mamidanna P., Cury K. M., Abe T., Murthy V. N., Mathis M. W., Bethge M., DeepLabCut: Markerless pose estimation of user-defined body parts with deep learning. Nat. Neurosci. 21, 1281–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Khazen R., Müller S., Lafouresse F., Valitutti S., Cussat-Blanc S., Sequential adjustment of cytotoxic T lymphocyte densities improves efficacy in controlling tumor growth. Sci. Rep. 9, 12308 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasconcelos Z., Müller S., Guipouy D., Yu W., Christophe C., Gadat S., Valitutti S., Dupré L., Individual human cytotoxic T lymphocytes exhibit intraclonal heterogeneity during sustained killing. Cell Rep. 11, 1474–1485 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Malaer J. D., Mathew P. A., CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am. J. Cancer Res. 7, 1637–1641 (2017). [PMC free article] [PubMed] [Google Scholar]
- 47.Guo H., Cruz-Munoz M.-E., Wu N., Robbins M., Veillette A., Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol. Cell. Biol. 35, 41–51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barbey C., Baumgaertner P., Devevre E., Rubio-Godoy V., Derre L., Bricard G., Guillaume P., Luescher I. F., Liénard D., Cerottini J. C., Romero P., Rufer N., Speiser D. E., IL-12 controls cytotoxicity of a novel subset of self-antigen-specific human CD28+ cytolytic T cells. J. Immunol. 178, 3566–3574 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Preglej T., Hamminger P., Luu M., Bulat T., Andersen L., Göschl L., Stolz V., Rica R., Sandner L., Waltenberger D., Tschismarov R., Faux T., Boenke T., Laiho A., Elo L. L., Sakaguchi S., Steiner G., Decker T., Bohle B., Visekruna A., Bock C., Strobl B., Seiser C., Boucheron N., Ellmeier W., Histone deacetylases 1 and 2 restrain CD4+ cytotoxic T lymphocyte differentiation. JCI Insight 5, e133393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azizi E., Carr A. J., Plitas G., Cornish A. E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M., Choi K., Fromme R. M., Dao P., Mc Kenney P. T., Wasti R. C., Kadaveru K., Mazutis L., Rudensky A. Y., Pe’er D., Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308 e36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puram S. V., Tirosh I., Parikh A. S., Patel A. P., Yizhak K., Gillespie S., Rodman C., Luo C. L., Mroz E. A., Emerick K. S., Deschler D. G., Varvares M. A., Mylvaganam R., Rozenblatt-Rosen O., Rocco J. W., Faquin W. C., Lin D. T., Regev A., Bernstein B. E., Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng C., Zheng L., Yoo J.-K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J. Y., Zhang Q., Liu Z., Dong M., Hu X., Ouyang W., Peng J., Zhang Z., Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Appay V., Zaunders J. J., Papagno L., Sutton J., Jaramillo A., Waters A., Easterbrook P., Grey P., Smith D., McMichael A. J., Cooper D. A., Rowland-Jones S. L., Kelleher A. D., Characterization of CD4+ CTLs ex vivo. J. Immunol. 168, 5954–5958 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Bottcher J. P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N. C., Sahai E., Zelenay S., Sousa C. R. E., NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allard M., Couturaud B., Carretero-Iglesia L., Duong M. N., Schmidt J., Monnot G. C., Romero P., Speiser D. E., Hebeisen M., Rufer N., TCR-ligand dissociation rate is a robust and stable biomarker of CD8+ T cell potency. JCI Insight 2, e92570 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lancki D. W., Hsieh C. S., Fitch F. W., Mechanisms of lysis by cytotoxic T lymphocyte clones. Lytic activity and gene expression in cloned antigen-specific CD4+ and CD8+ T lymphocytes. J. Immunol. 146, 3242–3249 (1991). [PubMed] [Google Scholar]
- 57.Vignali D. A. A., Collison L. W., Workman C. J., How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Leeuwen E. M., Remmerswaal E. B. M., Vossen M. T. M., Rowshani A. T., Wertheim-van Dillen P. M. E., van Lier R. A. W., ten Berge I. J. M., Emergence of a CD4+CD28– granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J. Immunol. 173, 1834–1841 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Cruz-Guilloty F., Pipkin M. E., Djuretic I. M., Levanon D., Lotem J., Lichtenheld M. G., Groner Y., Rao A., Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 206, 51–59 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Intlekofer A. M., Takemoto N., Wherry E. J., Longworth S. A., Northrup J. T., Palanivel V. R., Mullen A. C., Gasink C. R., Kaech S. M., Miller J. D., Gapin L., Ryan K., Russ A. P., Lindsten T., Orange J. S., Goldrath A. W., Ahmed R., Reiner S. L., Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, 1236–1244 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Della-Torre E., Bozzalla-Cassione E., Sciorati C., Ruggiero E., Lanzillotta M., Bonfiglio S., Mattoo H., Perugino C. A., Bozzolo E., Rovati L., Arcidiacono P. G., Balzano G., Lazarevic D., Bonini C., Falconi M., Stone J. H., Dagna L., Pillai S., Manfredi A. A., A CD8α− subset of CD4+SLAMF7+ cytotoxic T cells is expanded in patients with IgG4-related disease and decreases following glucocorticoid treatment. Arthritis Rheumatol. 70, 1133–1143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Axelrod M. L., Cook R. S., Johnson D. B., Balko J. M., Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 25, 2392–2402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maehara T., Kaneko N., Perugino C. A., Mattoo H., Kers J., Allard-Chamard H., Mahajan V. S., Liu H., Murphy S. J. H., Ghebremichael M., Fox D., Payne A. S., Lafyatis R., Stone J. H., Khanna D., Pillai S., Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Invest. 130, 2451–2464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tian L., Goldstein A., Wang H., Ching Lo H., Sun Kim I., Welte T., Sheng K., Dobrolecki L. E., Zhang X., Putluri N., Phung T. L., Mani S. A., Stossi F., Sreekumar A., Mancini M. A., Decker W. K., Zong C., Lewis M. T., Zhang X. H. F., Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544, 250–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiedemann A., Depoil D., Faroudi M., Valitutti S., Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. U.S.A. 103, 10985–10990 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Artyomov M. N., Lis M., Devadas S., Davis M. M., Chakraborty A. K., CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc. Natl. Acad. Sci. U.S.A. 107, 16916–16921 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garrido F., Aptsiauri N., Doorduijn E. M., Garcia Lora A. M., van Hall T., The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.The Gene The Gene Ontology Consortium , The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neubert N. J., Tillé L., Barras D., Soneson C., Baumgaertner P., Rimoldi D., Gfeller D., Delorenzi M., Fuertes Marraco S. A., Speiser D. E., Broad and conserved immune regulation by genetically heterogeneous melanoma cells. Cancer Res. 77, 1623–1636 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Bobisse S., Genolet R., Roberti A., Tanyi J. L., Racle J., Stevenson B. J., Iseli C., Michel A., le Bitoux M. A., Guillaume P., Schmidt J., Bianchi V., Dangaj D., Fenwick C., Derré L., Xenarios I., Michielin O., Romero P., Monos D. S., Zoete V., Gfeller D., Kandalaft L. E., Coukos G., Harari A., Sensitive and frequent identification of high avidity neo-epitope specific CD8+ T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 9, 1092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabe3348/DC1