

Transcriptional maps reveal diverse responses of immune and glial cells in spinal cord injury.
Abstract
The innate immune response influences neural repair after spinal cord injury (SCI). Here, we combined myeloid-specific transcriptomics and single-cell RNA sequencing to uncover not only a common core but also temporally distinct gene programs in injury-activated microglia and macrophages (IAM). Intriguingly, we detected a wide range of microglial cell states even in healthy spinal cord. Upon injury, IAM progressively acquired overall reparative, yet diversified transcriptional profiles, each comprising four transcriptional subtypes with specialized tasks. Notably, IAM have both distinct and common gene signatures as compared to neurodegeneration-associated microglia, both engaging phagocytosis, autophagy, and TyroBP pathways. We also identified an immediate response microglia subtype serving as a source population for microglial transformation and a proliferative subtype controlled by the epigenetic regulator histone deacetylase 3 (HDAC3). Together, our data unveil diversification of myeloid and glial subtypes in SCI and an extensive influence of HDAC3, which may be exploited to enhance functional recovery.
INTRODUCTION
Microglia, the resident myeloid cells in the central nervous system (CNS), help maintain CNS homeostasis (1). Under physiological conditions, microglial activities are restrained by checkpoint mechanisms. Upon injury, microglia are activated within minutes and, together with macrophages derived from blood-borne monocytes, constitute the innate immunity, serving as the first line of defense (2). However, poor outcome of CNS injury has led to the view that prolonged inflammatory phenotypes of injury-activated microglia/macrophages (IAM) exacerbate neuronal damage (3).
The innate immunity also influences disease progression of neurodegenerative disorders such as Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS) (4). Transcriptomic changes in disease-associated microglia (DAM) underscore the functional importance of lipid metabolism and phagocytosis, exemplified by the induction of apolipoprotein E (Apoe) and lipoprotein lipase (Lpl) (5). A two-step gene regulation program has been proposed for DAM activation, first a down-regulation of microglial checkpoint genes independent of Trem2 (an AD risk gene) and followed by a Trem2-dependent process (5). An important question is whether IAM and DAM share hallmark genes and regulatory mechanisms.
Immune cell activation requires large-scale gene reprogramming, which involves chromatin modifications to facilitate coordinated access of transcription factors (TFs) to specific genomic loci. Histone acetylation plays an essential role in macrophage activation (6), and our recent work has identified a robust up-regulation of histone deacetylase 3 (HDAC3) in IAM after spinal cord injury (SCI) (7). Pharmacological inhibition of HDAC3 with a small-molecule inhibitor with CNS availability resulted in global inflammatory suppression, along with improved neuroprotection and functional recovery after SCI (7). However, it remains to be determined whether HDAC3 inhibition affects all IAM or only a subset of myeloid population, whether other glial cells are also affected, and what gene pathways are altered.
As microglia respond to injury signals within minutes (8), a fast cell type–specific isolation method is imperative to preserve the in vivo gene signatures. However, traditional methods using fluorescence-activated cell sorting (FACS) (9, 10) or immunopanning (11) require lengthy preparations that can introduce transcriptional artifacts, e.g., induction of immediate-early genes (IEGs) and proinflammatory genes (e.g., Cd86) (12). Here, we performed RNA sequencing (RNA-seq) using the INTACT (isolation of nuclei tagged in specific cell types) method based on immunopurification of green fluorescent protein (GFP)–tagged nuclei from snap-frozen tissues (13). This was complemented with single-cell RNA-seq (scRNA-seq) on spinal cord tissues without prior FACS, thus providing unbiased information on transcriptional signatures in different cell types (14). Our analyses unveil a highly diversified transcriptional response in myeloid and other glial populations after SCI and an extensive influence of HDAC3.
RESULTS
Temporally distinct transcriptional profiles in IAM after SCI
To purify nuclei of myeloid cells, we generated Cx3cr1CreER INTACT mice (13, 15) (Fig. 1A and fig. S1A). We administered tamoxifen at 3 days and 1 day before thoracic T8 dorsal column transection (7, 16) to initiate GFP tagging of nuclear envelope of myeloid cells. Immunohistochemistry (IHC) confirmed near complete overlap of GFP+ cells with cells expressing IBA1 (a myeloid marker) and ~70% overlap with cells expressing Tmem119 (a microglia-specific marker) at the lesion site at 7 days post injury (dpi) (fig. S1B).
Fig. 1. Transcriptional dynamics in IAM after SCI.
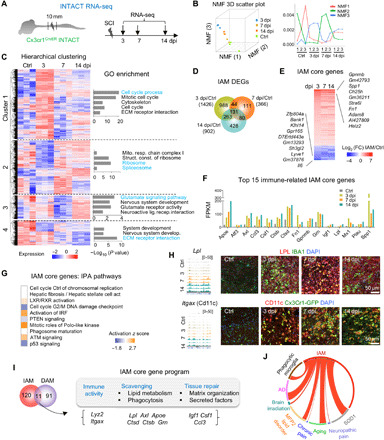
(A) Experimental paradigm. (B) Left: NMF three-dimensional (3D) scatter plot. Right: NMF line plot revealing three metagenes representing distinct transcriptional programs. Y axis, expression of NMF metagenes. (C) Hierarchical clustering identified four gene clusters in IAM. Heatmap of gene expression levels shown as row normalized z-score values. Enriched GO terms shown on right. (D) Venn diagram displaying unique and overlapping DEGs in IAM. (E) Heatmap showing transcriptional changes of 131 core IAM genes common to all time points after SCI relative to control microglia. FC, fold change. Top 10 up- and down-regulated genes in IAM are listed. (F) Absolute expression levels of 15 selected significantly regulated immune system–related genes in the core IAM gene program [fragments per kilobase of exons per million mapped reads (FPKM) averaged from triplicates for each time point]. (G) Pathway enrichment of the IAM core genes by IPA, ranked by adjusted P value. IPA, ingenuity pathway analysis. (H) RNA-seq read coverage tracks (left) and IHC images (right) showing expression of LPL and CD11c in myeloid cells (green) after SCI. (I) Venn diagram showing overlap of IAM and DAM signature genes: 11 genes grouped by distinct functions during progressive IAM activation. (J) Circos plot of connectivity map of IAM gene signatures with gene sets of activated microglia in CNS disease models. Lines represent pairwise dataset overlaps.
Spinal cord segments encompassing injury epicenter and flanking areas were dissected at 3, 7, and 14 dpi from SCI mice and control mice with laminectomy only. RNA-seq was performed on triplicate complementary DNA (cDNA) libraries prepared from immunopurified GFP-labeled myeloid cell nuclei for each experimental condition (table S1). For specificity control, we performed quantitative reverse transcription polymerase chain reaction (qRT-PCR) on the INTACT libraries, which verified high expression levels of the myeloid marker Gpr84, but negligible expression of other cell type markers (fig. S1C), a finding confirmed by subsequent RNA-seq (fig. S1D). Robust induction of Tnf (a classic myeloid activation marker) was detected after SCI, while expression of Gpr84 and Itgam (CD11b) remained stable (fig. S1E).
RNA-seq data revealed much higher transcriptional levels of microglial markers (Hexb, Tgfbr1, and P2ry12) than those of macrophage markers (Cfp, Crip1, Cxcl13, Pf4, Prg4, Saa3, and Sytl1) or monocyte markers (Ccl22, Ccr1, Cxcl2, Ly6c1, and Plcb1) (17–19) at all three time points (fig. S1F). Hence, our INTACT samples comprised mainly microglial nuclei, with a smaller contribution from infiltrating macrophages or nonparenchymal perivascular and meningeal macrophages (12, 20, 21). This is consistent with the fact that our tamoxifen injection scheme favors labeling of microglia over peripherally derived macrophages because the peak time of macrophage infiltration occurs at ~5 to 7 dpi (22–24).
Dimension reduction of the INTACT RNA-seq data by Uniform Manifold Approximation and Projection (UMAP) showed a clear separation between different experimental groups, indicating reproducibility of our approach (fig. S2A). We also performed unsupervised k-means clustering analysis using 20 UMAP components of the INTACT data and found that the resulting four clusters matched the four experimental groups, indicating temporally distinct gene expression patterns (fig. S2A). Non-negative matrix factorization (NMF) (25) on the basis of three metagenes further confirmed divergent transcriptional states corresponding to progressive stages of IAM activation (Fig. 1B). Notably, both UMAP and NMF analyses showed that the 3-dpi samples were further separated from the rest, signifying a more divergent transcriptional state at this early stage of IAM activation.
To detect global transcription patterns during IAM activation, we performed hierarchical gene clustering according to expression patterns, which identified four main gene clusters with distinct characteristics (Fig. 1C and table S1). Cluster 1 genes, predominantly up-regulated at 3 dpi, were enriched for Gene Ontology (GO) terms related to cell proliferation, migration, and interaction with extracellular matrix (ECM). Cluster 2 genes, mostly down-regulated at 3 dpi, regulated cell metabolism (mitochondrial respiratory chain complex) and RNA processes (ribosome and spliceosome). Cluster 3 genes, down-regulated at 7 dpi but up-regulated at 14 dpi, concerned neuronal interaction (glutamate signaling and nervous system development). Cluster 4 genes, mostly down-regulated across all three time points, were involved in nervous system development and ECM receptor interaction.
To further identify coregulated gene modules in IAM activation, we applied weighted gene coexpression network analysis (WGCNA) (26), which identified 103 modules, 13 of which displayed a significant enrichment for one or multiple gene clusters mentioned above (fig. S2, B and C). For instance, the “yellow” gene module contained 955 coexpressed genes controlling cell cycle, motility, and metabolism, and concordantly, cluster 1 and 2 genes were enriched. The “blue” module contained 2286 coexpressed genes linked to kinase activity and biopolymer metabolic process, and cluster 1 and 3 genes were enriched. Together, these data converged on the conclusion that IAM progressively activate temporally distinct gene programs to govern myeloid cell proliferation, migration, metabolism, neuronal interaction, and ECM organization.
IAM share a common core gene program in response to SCI
We next identified the differentially expressed genes (DEGs) in IAM relative to homeostatic microglia, which revealed 1426 DEGs at 3 dpi, 366 at 7 dpi, and 902 at 14 dpi (Fig. 1D and table S1). Shared across all time points were 131 core IAM genes (104 up-regulated and 27 down-regulated) that were highly enriched for phagosome and lipid metabolism pathway genes, e.g., Apoe and Lpl (both DAM markers) (5); Axl, Ctsb, Ctsd (cathepsin B and D, two lysosomal cysteine proteases), Grn (enhances endocytic activity), and Abca1 and Abcg1 [two cholesterol transporter genes that facilitate cholesterol efflux to ApoE in astrocytes and microglia (27)] (Fig. 1, E and F, and table S1). Consistently, pathway enrichment analyses revealed activation of LXR/RXR [liver X receptor/retinoic acid receptor, which regulates lipid and cholesterol metabolism (28)], phagosome maturation, phospholipid efflux, cholesterol metabolism, and statin pathway in IAM (Fig. 1G and fig. S2, D and E).
A second main enriched GO category for IAM core genes concerned immunity, e.g., activation of interferon regulatory factor (IRF) pathway, Toll-like receptor signaling, cytokine receptor interaction, and tumor necrosis factor (TNF) signaling (Fig. 1G and fig. S2, D and E); while a third category involved ECM organization, e.g., Spp1 and Fn1 (Fibronectin), which facilitates wound healing (29), and secreted factors such as chemokines (Ccl3 and Csf1) and trophic factors (Igf1) (Fig. 1F). IAM also up-regulated genes to promote myeloid cell recruitment [urokinase plasminogen activator (uPA/Plau) and receptor uPAR/Plaur (30)], protect neurons (Gpnmb), as well as genes with lesser-known functions in microglia, e.g., voltage-gated Ca2+ channel activity genes (Cacnab2, Hcn1, Slc24a2, and Trpc5), all down-regulated in IAM (Fig. 1F and fig. S2E). IHC confirmed a robust up-regulation of LPL and integrin αX (Itgax; CD11c) in ~75% of IBA1+ or Cx3cr1-GFP+ myeloid cells at the injury site at all three time points (Fig. 1H).
As lipid metabolism and phagocytosis functions were shared by both IAM and DAM (5), we further compared the two signatures and found 11 overlapping genes involved in immunity, scavenging, and tissue repair (Fig. 1I). Of the stage 1 DAM genes, Apoe, Ctsb, Ctsd, and Lyz2 were up-regulated in IAM, while P2ry12 was down-regulated; of the stage 2 DAM genes, the overlap was larger, with Axl, Csf1, Itgax, and Lpl induced at all time points, while Cst7 and Cd9 (two phagocytic pathway genes) were up-regulated at 7 and 14 dpi (fig. S2F). A notable difference is that the DAM marker genes Trem2 and its signaling adaptor Tyrobp (31) were not significantly up-regulated in IAM. IHC confirmed down-regulation of P2ry12 in Cx3cr1-GFP+ cells after SCI, but Tmem119 expression remained stable (fig. S2G).
In neurodegenerative disorders such as AD and ALS and in multiple sclerosis, a cluster of 28 inflammatory molecules has been shown to be associated with microglial neurodegenerative phenotype (4), and we found that many of them overlapped with IAM core genes, including Apoe, Axl, Clec7a, Csf1, Gpnmb, Itgax, and Spp1. A connectivity map analysis demonstrated a notable overlap of gene signatures of IAM and activated microglia in a variety of neurological conditions (Fig. 1J).
IAM activate overall reparative gene programs
To further understand the sequence of events during the multiphasic immune response, we analyzed time point–specific DEGs in IAM, which revealed that the main molecular events at 3 dpi involved cell cycle and interferon signaling, while migration and ion channel activity were the dominant events at 7 dpi and ECM reorganization at 14 dpi [Fig. 2, A and B, and fig. S3, A to D; also see our recent study (32)]. Comparative pathway analysis showed that the DEGs at 3 dpi were uniquely enriched for cancer metastasis signaling, mitotic kinase, and interferon signaling, whereas at 14 dpi, activation of LXR/RXR and deactivation of synaptic long-term depression, opioid, Gβγ, and calcium signaling were the prominent events (Fig. 2A). Notably, at 7 dpi, IAM down-regulated a variety of ion channel activity genes, including Gaba receptors (Gabrb1, Gabrb3, and Gabra5), voltage-gated sodium channels (Scn3a and Scn9a), and transient receptor potential channel (Trpc5), whereas fewer ion channel genes were up-regulated, e.g., Cacna1a (a calcium channel), P2rx4a (a purinergic receptor), and Trpc4 (Fig. 2B and fig. S3, B to D).
Fig. 2. Transcriptionally distinct gene signatures of IAM exhibit anti-inflammatory profiles.
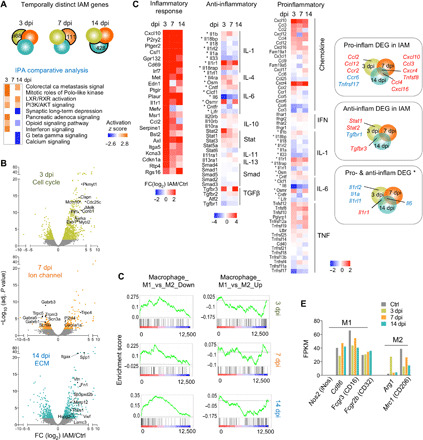
(A) Venn diagrams show temporally distinct DEGs in IAM. Comparative IPA showing pathway enrichment of temporally distinct DEGs. PI3K, phosphatidylinositol 3-kinase. (B) Volcano plots showing DEGs in IAM at successive time points relative to control microglia. Selected top-ranked DEGs (by fold change) of the indicated GO term are highlighted. (C) Heatmaps depicting transcriptional changes of inflammatory genes (grouped by pathways) in IAM at different time points. Asterisks denote genes with both pro- and anti-inflammatory functions. Right: Venn diagrams show significant DEGs of different inflammatory categories. Red denotes up-regulated genes, and blue denotes down-regulated genes. (D) GSEA reveals positive enrichment of Macrophage_M1_vs_M2_Down gene set in IAM, with normalized enrichment score (NES) = 2.49 and false discovery rate q value (FDR) = 0.000 at 3 dpi; NES = 2.09 and FDR = 0.000 at 7 dpi; and NES = 3.06 and FDR = 0.000 at 14 dpi. For Macrophage_M1_vs_M2_Up gene set, inverse enrichment was observed in IAM, with NES = −1.77 and FDR = 0.018 at 3 dpi; NES = −1.07 and FDR = 0.36 at 7 dpi; and NES = −1.68 and FDR = 0.029 at 14 dpi. (E) Bar graphs showing expression levels of macrophage polarization markers, in FPKM averaged from triplicate samples for each time point in IAM.
It is thought that a prolonged inflammatory phenotype could escalate secondary injury (3). Despite up-regulation of many inflammatory response genes, we did not observe a predominant proinflammatory gene profile in IAM (Fig. 2C); among the 57 proinflammatory genes surveyed, only 9 showed significant up-regulation—Ccl2, Ccl12, and Ccr2 at 3 dpi; Ccl4 and Cxcl16 at 7 and 14 dpi; and Cxcl10, Ccl3, Cxcr4, and Tnfsf8 at all three time points—whereas two were down-regulated at 3 dpi (Ccr6 and Tnfrsf17). Regarding anti-inflammatory genes, Tgfbr3 was notably induced at both 3 and 14 dpi, while Tgfbr1 was down-regulated at 3 dpi (Fig. 2C). Interleukin-6 (IL-6), with both neuroprotective and neurotoxic effects, was down-regulated in IAM at all three stages. Last, IL-1 pathway genes were predominantly down-regulated at 3 dpi (e.g., Il1rl2, Il1a, and Il1rl1), except for Il1r1, which was induced at 3 and 14 dpi (Fig. 2C).
Gene set enrichment analyses (GSEA) for macrophage M1 (proinflammatory) and M2 (anti-inflammatory) polarizations (33) showed a positive enrichment for the M1_vs_M2_Down gene set in IAM across all three time points and an inverse enrichment for the M1_vs_M2_Up gene set at 3 and 14 dpi (Fig. 2D). We further examined the expression of classic M1 and M2 genes described in SCI models (3, 22) and found that M1 marker genes iNos, Cd86, Fcgr3 (Cd16), and Fcgr2b (Cd32) were not significantly up-regulated in IAM; whereas M2 marker gene arginase-1 (Arg1) was up-regulated in IAM at 3 dpi, but mannose receptor (Mrc1 and Cd206) was down-regulated at 3 and 14 dpi (Fig. 2E). Together, IAM displayed an overall reparative gene profile.
We further examined the expression of TNF, IL-1a, and C1q, which were described as microglial proinflammatory factors that induce a neurotoxic astrocyte phenotype (A1 polarization) after lipopolysaccharide (LPS) stimulation (11). We found a down-regulation of Il1a and only a trend of up-regulation for C1qa (fig. S3E). Two other classic immune-associated genes, Il1b and Tgfb1, showed no significant changes in IAM (fig. S3E and see table S1 for detailed expression changes).
Reactivation of developmental gene program in IAM
We next asked whether the gene profiles of IAM bore resemblance to the developmental gene programs, as embryonic microglia are also engaged in scavenging of apoptotic cells and clearing of excess synapses. Of the top 100 DEGs during microglia development (17), 30 were significantly regulated in IAM (22 up-regulated and 8 down-regulated) (fig. S4, A and B), indicating partial resemblance of IAM and young microglia. A notable exception was Tmem119, a highly specific microglial marker and developmentally regulated gene (17), which remained unchanged in IAM. We next surveyed microglial homeostatic genes (10) and found that 9 of 38 were significantly changed in IAM, all down-regulated, including Tgfbr1 at 3 dpi and P2ry12, P2ry13, and Siglech at both 3 and 14 dpi (fig. S4C). Last, we analyzed a list of highly expressed genes in microglia in adult healthy spinal cord, grouped into DEGs after SCI (e.g., Tgfbr1, Siglech, P2ry12, and Mef2a) and non-DEGs (e.g., Hexb, Csf1r, and C1qb), many of which overlapped with known microglia-specific markers, but also included previously undescribed markers (fig. S4D).
Usage of alternative splicing in IAM
Intriguingly, we observed that IAM used differential exon usage in a large number of genes compared to steady-state microglia, indicating active alternative splicing (fig. S4, E and F). The 3-dpi IAM harbored the largest number of differential exons (1361), followed by 14 dpi (459) and 7 dpi (125). The directionality of the differential exon usage for most of these exons was identical at different time points, indicating both short- and long-term usage of alternative splicing in IAM, although most exons displayed maximal fold changes at 3 dpi (fig. S4G). Only 15 differential exons were shared by all three time points, and they belonged to genes implicated in CNS disorders (Taf15, Itpr2, and Mdn1), as well as inflammatory regulators (Laptm5, Nlrc3, Myo1d, and Lrba), trophic factors (Igf2), metabolic regulators (Kdm3a and Nadk2), and motility-related genes (Myo9b, Myh9, Inpp4b, and Runx2) (fig. S4, H and I).
Upstream regulators of IAM gene programs are linked to HDAC3
We next searched for upstream regulators of the core IAM gene program, which identified classic inflammatory regulators CSF2 (colony stimulating factor 2), LPS, IL-6, and interferon-γ (IFN-γ), as well as transforming growth factor–β1 (TGFβ1), a critical microglial developmental regulator (10) and an inducer of quiescent and resting-state microglia (fig. S5A) (34). p53 and cyclin-dependent kinase inhibitor CDKN1A were identified as negative regulators, consistent with a high proliferative state for IAM. Comparative IPA (ingenuity pathway analysis) revealed LPS, IFN-γ, and TNF as the top upstream regulators of the IAM DEGs across all activation stages, albeit with a higher activation score at 3 dpi, signifying a heightened inflammatory state at this early stage (fig. S5A).
The IAM gene programs were also associated with specific upstream TFs such as nuclear factor κB (NFκB) and members of the signal transducer and activator of transcription (STAT) and IRF families (Fig. 3A). IRF7, hypoxia-inducible factor 1α, and STAT1 were themselves differentially regulated in IAM relative to control microglial, together with a total of 77 TFs and 19 chromatin remodelers (fig. S5, B to D). We had previously shown an up-regulation of HDAC3 in IAM after SCI and that administration of RGFP966, a specific HDAC3 inhibitor (HDAC3i), at 2, 24, and 48 hours after injury resulted in global immunosuppression along with enhanced functional recovery after SCI (7). We conducted an interactome analysis, which revealed close interaction of HDAC3 with most of the upstream transcriptional regulators of the IAM gene programs (Fig. 3B and fig. S5E). To determine the optimal timing of RGFP966 intervention in SCI, we compared the efficacy of acute versus subacute HDAC3i regimens, which showed a better improvement of BMS (Basso mouse scale) scores by the acute regimen (Fig. 3C), aligned with our previous results (7). We therefore continued our study using the acute HDAC3i treatment.
Fig. 3. HDAC3 inhibition during acute phase promotes functional recovery after SCI.
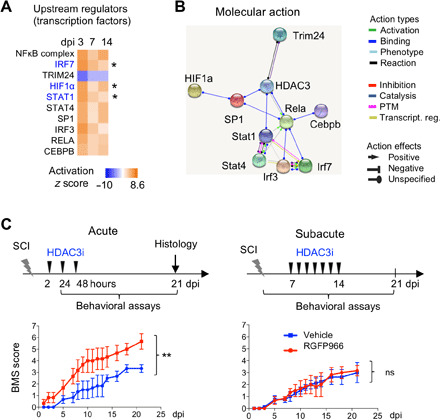
(A) Comparative IPA showing TFs predicted to be upstream regulators of the IAM gene programs at different activation stages. Genes denoted in blue (also marked by asterisks) are themselves differentially expressed between IAM and control microglia. (B) Interactome of HDAC3 with the top predicted upstream transcriptional regulators of IAM activation. (C) SCI functional recovery study with HDAC3 inhibitor (HDAC3i) RGFP966, comparing acute versus subacute treatment regimens as shown on top. Bottom: Locomotor recovery shown by the BMS scores (n = 3 mice for each condition). Two-way analysis of variance (ANOVA) with Bonferroni correction. **P = 0.002. ns, not significant.
scRNA-seq reveals expansion and contraction of different cell type clusters after SCI
To better understand glial cell heterogeneity and the effects of HDAC3i on their responses to SCI, we performed scRNA-seq (10X Genomics) on injured spinal cord tissues at 5 dpi (Fig. 4A). We chose a T8 contusion, an SCI model used in our previous HDAC3 study (7). Adult spinal cord tissues including both the injury epicenter and the adjacent areas were used to capture homeostatic microglia and astrocyte populations for comparison. We also included scRNA-seq data from uninjured adult spinal cord as control (CTRL) reference (35). After applying quality cutoffs, we analyzed a total of 9937 cells (4576 in CTRL, 2276 in SCI, and 3085 in SCI_HDAC3i) (fig. S6A and table S2).
Fig. 4. scRNA-seq reveals distinct expansion and contraction of glial cell types after SCI.
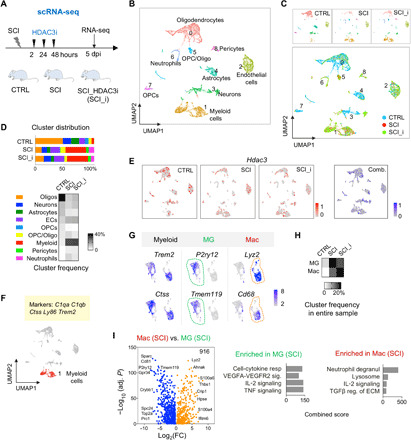
(A) Experimental paradigm of scRNA-seq. (B) UMAP plot showing nine cell type clusters identified in adult spinal cord. n = 9937 cells integrated from three experimental conditions. (C) UMAP plots comparing cluster representation in three experimental conditions, labeled with different colors. (D) Top: Graphs showing the relative distribution of each cluster in different conditions. Bottom: Heatmap showing cluster frequency. ECs, endothelial cells. (E) UMAP plots showing heterogeneous expression of Hdac3 across different clusters in each condition and in combined dataset. (F) UMAP plot of myeloid cells (cluster 1) highlighted in red. Highly expressed marker genes are listed above. Cluster 1 comprises 191 myeloid cells in CTRL, 849 in SCI, and 1169 in SCI_HDAC3i. (G) UMAP plots showing expression of pan-myeloid, microglial (MG), and macrophage (Mac) marker genes in myeloid cells. (H) Frequency heatmap of microglia and macrophage populations, relative to all cells in the entire sample. (I) Volcano plot showing 916 DEGs between infiltrating macrophages and activated microglia in SCI. n = 512 microglia and 337 macrophages in SCI sample. Right: Enriched GO terms by Enrichr.
Unsupervised clustering revealed nine distinct cell clusters, visualized by UMAP (36) (Fig. 4B). Oligodendrocytes (marked by Cldn11 and Stmn4) constituted the largest group, followed by myeloid cells (C1qa and Trem2), endothelial cells (Cldn5 and Ly6c1), neurons (Meg3 and Snhg11), astrocytes (Aqp4 and Atp1a2), neurotrophils (Slpi and G0S2), oligodendrocyte precursor cells (OPCs; C1ql1 and Lhfpl3), and pericytes (Ndufa412 and Higd1b) (Fig. 4B and fig. S6, B and C). We also identified a transitional cell cluster expressing both oligodendrocyte and OPC markers (cluster 5; Bcas1 and Pllp). Different cell types had comparable transcript counts [unique molecular identifiers (UMIs)], although myeloid cells had slightly higher and astrocytes slightly lower UMI counts (fig. S6D).
Cluster frequency comparison showed that the immune (myeloid cells and neutrophils) and the vascular (endothelial cells and pericytes) populations were greatly expanded in SCI, while other cell types were proportionally reduced (Fig. 4, C and D), reflecting ongoing neuroinflammation, angiogenesis, and neural tissue damage at 5 dpi. The OPC/Oligo transitional cluster was also expanded, reflecting OPC activation in response to SCI-induced demyelination. HDAC3i treatment after SCI did not cause a major shift in the cell cluster distribution compared to vehicle treatment, except for a smaller neutrophil cluster and a modest expansion of neuronal and oligodendrocyte clusters (Fig. 4D). Notably, cell type frequencies can only be approximated because of potential bias from cell fragility and capture efficiency for different cell types. Hdac3 transcripts were detected in all clusters with high heterogeneity on single-cell levels but showing no overt changes after SCI (Fig. 4E); hence, posttranscriptional regulation likely accounts for the robust up-regulation of HDAC3 protein at the injury site, as shown by IHC (fig. S6E).
Transcriptional identities of activated microglia and macrophages are maintained in SCI
We first analyzed in detail the myeloid population, which can be clearly partitioned into microglia and macrophage clusters, indicating their inherent transcriptional differences even in injury settings (Fig. 4, F and G, and fig. S6F). While both populations shared many common marker genes (C1qa, C1qb, Ctss, Ly86, and Trem2), certain genes were predominantly expressed in macrophages (e.g., Lyz2, Cd68, Thbs1, S100a4, and S100a6), whereas others remained exclusively expressed in microglia (e.g., P2ry12 and Tmem119) (Fig. 4G).
The myeloid cell population was greatly expanded after SCI, constituting ~37% of all cells in our sample at 5 dpi, with 60% being microglia and 40% macrophages (Fig. 4H). HDAC3i resulted in a modest reduction of microglial expansion (from 22 to 17% of all cells) but a more robust macrophage expansion (from 15 to 21%) (Fig. 4H).
Despite similarities between activated microglia and infiltrating macrophages in regard to morphology and expression of common marker genes, we identified 916 DEGs between the two populations after SCI, with microglia expressing higher levels of genes associated with vascular endothelial growth factor (VEGF) signaling (Adamts1, Itgb5, Jun, and Mef2c), cytokine response (Itgam, Tnf, and Ccr5), IL-2 signaling (Serpine1, Ets1, and Icam1), and TNF signaling (Vcam1, Fos, and Il1b), whereas macrophages expressed higher levels of genes concerning lysosome formation (Cxcl1, CD93, and Arg1), IL-2 signaling (Cdkn1b, Cxcr4, and Plac8), and TGFβ regulation of ECM (Lgals3 and Apoe) (Fig. 4I and fig. S6F). Notably, IL-2 signaling was engaged in both populations, but different individual genes were involved.
HDAC3 inhibition exerts different effects on microglia and macrophages in SCI
We next separately analyzed transcriptomes of microglia and macrophages, comparing SCI versus CTRL (to identify injury response genes) and SCI versus SCI_HDAC3i (to assess HDAC3 dependence in the injury setting) (table S2). We found 390 injury responsive genes in microglia (111 up-regulated and 279 down-regulated), 55 of which showed HDAC3 dependence and were linked to proliferation (Kif23 and Aurkb), chemokine/cytokine activity (Ccl22 and Pf4), and the Ptf1a-related regulatory pathway (Prox1) (Fig. 5, A and B). An additional 168 genes (outside the microglial injury response program) also showed HDAC3 dependence, mostly down-regulated in SCI versus SCI_HDAC3i, consistent with closed chromatin configurations by HDAC3 activity, and they were involved not only in chemotaxis and chemokine pathways but also in iron transporter, phospholipase activity, and calmodulin-dependent protein kinase pathways, indicating HDAC3’s role in regulating these essential cellular processes in microglia (Fig. 5B). Because only few macrophages were present in CTRL spinal cord, the vast majority of macrophages in SCI thus originated from the periphery, and we identified 98 injury response genes and 167 HDAC3-dependent genes (mostly down-regulated by HDAC3) in macrophages, with only 10 overlapping genes (Fig. 5C).
Fig. 5. Microglia and macrophages display distinct transcriptional responses and HDAC3 influences in SCI.
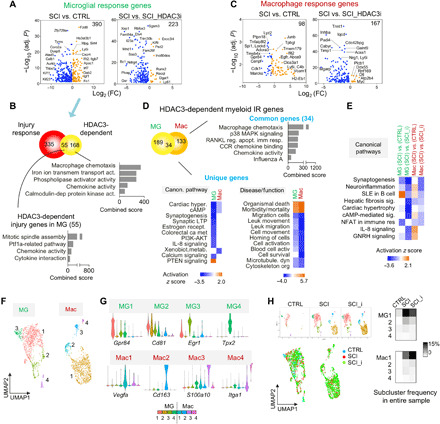
(A) Volcano plots showing microglial DEGs of the indicated comparisons. n = 187 microglia in CTRL, 512 in SCI, and 510 in SCI_HDAC3i. (B) Venn diagram showing overlap of microglial injury response DEGs and HDAC3-dependent DEGs. Bottom: Pathway enrichment analyses of 55 overlapping genes and 168 HDAC3-dependent genes. (C) Volcano plots showing macrophage DEGs of the indicated comparisons. n = 4 macrophages in CTRL, 337 in SCI, and 659 in SCI_HDAC3i. (D) Top: Venn diagram and Enrichr pathway enrichment analysis of HDAC3-dependent genes shared by activated microglia or macrophages in SCI. Bottom: Comparative IPA showing enriched canonical pathways or diseases/functions for HDAC3-regulated genes unique to microglia or macrophages. (E) Comparative IPA of different sets of DEGs revealing distinct pathway enrichment for activated microglia and macrophages in SCI, both heavily dependent on HDAC3. (F) Unbiased subclustering analysis identified four microglia (MG1 to MG4) and four macrophage subclusters (Mac1 to Mac4), shown as UMAP plots. n = 1209 microglia and 1000 macrophage cells from all conditions combined. (G) Violin plots showing expression of marker genes for myeloid subclusters. (H) UMAP plots showing relative representation of myeloid subclusters in different conditions. Right: Frequency heatmaps showing myeloid subclusters relative to all cells in the entire sample in each condition.
Microglia and macrophages shared a small proportion (~20%) of HDAC3-dependent genes that concerned chemotaxis, chemokine activity, receptor activator of nuclear factor kappa-Β ligand (RANKL), and p38 mitogen-activated protein kinase (MAPK) signaling (Fig. 5D), concordant with an immunomodulatory function of HDAC3 (7). The unique HDAC3-dependent genes in microglia concerned synaptogenesis, synaptic long-term potentiation, phosphatidylinositol 3-kinase/AKT signaling, and granulocyte-macrophage colony-stimulating factor signaling, whereas in macrophages, HDAC3-dependent genes were specifically involved in xenobiotic metabolism, leukocyte migration, and cell survival (Fig. 5D). Together, gene signature comparison underscored synaptogenesis-related functions for microglia and neuroinflammation and IL-8 signaling for macrophages, both heavily influenced by HDAC3 activity (Fig. 5E).
Heterogeneity and diversified injury responses of myeloid cells in SCI
Unbiased clustering analyses further separated myeloid cells into eight distinct subclusters (MG1 to MG4 and Mac1 to Mac4), each defined by marker genes with relative high specificities (Fig. 5, F and G, and fig. S7, A and B). SCI resulted in a marked expansion of MG1 and MG2, a small expansion of MG3, but a contraction of MG4 at 5 dpi relative to all cells in the samples, and these changes were slightly mitigated by HDAC3i (Fig. 5H). Among macrophages, Mac1 occupied the largest share, and it was further expanded by HDAC3i after SCI (Fig. 5H).
To understand the functional diversity, we performed pathway enrichment analyses (Fig. 6, A and B), which revealed that MG1 took up largely immune-related functions, expressing higher levels of cytokines (Il1a, Il1b, and Cxcl10) and immune response genes [Tnf, Ccl3, and Nfkbia (NFκB inhibitor α)], and engaging NFκB and TNFα signaling. MG2 specialized in phagocytosis and lipid metabolism, expressing autophagy and lysosome genes (Ctsd, Ctsl, Cd81, Cd68, and Tyrobp), and engaging TyroBP (TYRO protein tyrosine kinase-binding protein) network and TGFβ signaling. MG3 was defined by brain-derived neurotrophic factor signaling and induction of IEGs—Fos, Jun, and Egr1 (a transcriptional regulator of cell proliferation and survival), thus revealing a more intrinsically responsive nature. MG4 was marked by proliferation genes (Cdk1, Top2a, Nusap1, and Humg2) and p53 signaling, signifying a proliferative state.
Fig. 6. Functional specialization and trajectory of microglia transformation in SCI.
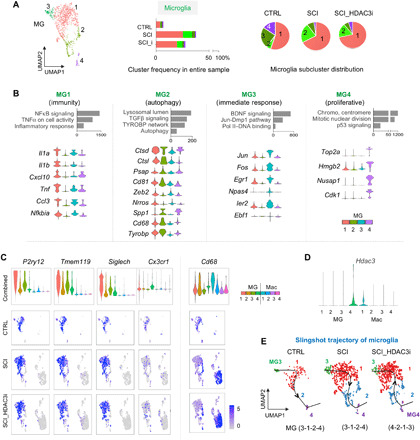
(A) Left: UMAP plot of microglia subclusters (MG1 to MG4). Middle: Graphs showing relative abundance of MG1 to MG4 as compared to all cells in the entire sample in each condition. Right: Pie charts showing relative proportions of MG1 to MG4 among microglia in each condition. n = 187 microglia in CTRL, 512 in SCI, and 510 in SCI_HDAC3i. (B) Enriched GO terms associated with the top 30 up-regulated marker genes for each microglia subcluster. Bottom: Violin plots showing expression of selected marker genes related to the pathway ontologies indicated above. (C) Violin and UMAP plots show heterogeneous expression patterns of microglial markers genes and phagocytic marker Cd68. (D) Expression of Hdac3 in myeloid subclusters, shown by violin plot. Note a preferential expression of Hdac3 in MG4. (E) Single-cell trajectory analysis of microglia transformation under each condition using Slingshot software platform. HDAC3i influences lineage trajectories of microglia, with the starting point shifted from MG3 to MG4.
Unexpectedly, MG1 to MG4 were all represented in microglia in the CTRL sample, with the immune-focused MG1 occupying the largest share (55%), followed by MG3 (25%), MG4 (15%), and MG2 (5%) (Fig. 6A). This indicated a diverse range of cell states for microglia even in healthy spinal cord. Expression levels of microglial homeostatic genes P2ry12, Tmem119, and Siglech were highest in MG1 and lowest in MG2 (Fig. 6C), supporting MG1 as representative of a homeostatic population and MG2 of a reactive one. Concordantly, phagocytic marker Cd68 was more prominently expressed in MG2. Notably, SCI led to an expansion of both MG1 and MG2 at 5 dpi, both in abundance and relative proportion among microglia, with MG2 gaining the most share (from 5 to 22%), followed by MG1 (from 55 to 67%); in contrast, the share of MG3 shrank after SCI (from 25 to 8%, despite an overall gain in abundance), so was the share of MG4 (from 16 to 2%) (Fig. 6A). HDAC3 inhibition slightly shifted microglial representation back toward the steady state by reducing the proportion of MG2 (22 to 17%) while expanding the MG4 share (2 to 5%) (Fig. 6A). Notably, Hdac3 expression was not uniform among microglia but more prominent in the proliferative MG4 (Fig. 6D), echoing HDAC3 dependence of proliferation-related genes in microglia (see Fig. 5B). Expression of another microglia marker, Cx3cr1, was also more prominent in MG4 (Fig. 6C).
We further examined the expression patterns of 14 microglial injury response genes identified by scRNA-seq that intersected with the IAM core signature genes identified by INTACT RNA-seq (fig. S7C), and indeed, most were represented in the reactive microglia subclusters, e.g., Igf1 in MG2, Ifit2, Nes, and Slfn5 in MG3 and Adam8, Cxcr4, Fn1, and Gas2 in MG4, while Lgals3, Lyz2, and Spp1 in both MG2 and MG4 and Apoc1, Bank1, and Cd300lf were uniformly expressed. IHC confirmed an up-regulation of the matrix proteins secreted phosphoprotein 1/osteopontin (SPP1) and fibronectin (FN) at the injury site (fig. S7D). Together, microglia displayed diversified activation states, and single-cell trajectory analysis mapped microglia along a continuous trajectory, progressing from the immediate responsive MG3 toward the homeostatic MG1, followed by the reactive MG2, and ending in the proliferative MG4 (i.e., MG3-1-2-4, Fig. 6E). HDAC3i shifted microglial transformation toward a new trajectory (i.e., MG4-2-1-3), consistent with a role of HDAC3 in controlling both proliferation and chemokine/RANKL activation.
Macrophage transcriptional responses bear hallmarks of IAM and DAM gene signatures
Among the largely peripherally derived macrophages, Mac1 constituted ~75%, with a higher expression for Apoe, Vegfa, Pla1g7, Ecm1, Spp1, Lgals1, and Thbs1, and an enrichment for pathways of iron and collagen binding, angiogenesis, lipid metabolism, and phagocytosis (Fig. 7, A and B, and fig. S8, A and B). Mac2 was defined by chemokine and NOD (nucleotide-binding oligomerization domain-containing protein 1) signaling, expressing higher levels of Cxcl2, Ccl7, C4b, Ccl12, Ccl2, and Cd163, and major histocompatibility class II–related genes (Cd74 and H2-A2) that mark M1 macrophage activation. Mac3 was marked by prostaglandin synthesis and RAGE (receptor for advanced glycation endproducts) activation (a pattern recognition receptor that activates inflammatory function of innate immunity), expressing Plac8, Vim, Gda, Ifitm3, Lgals3, Ly6c3, S100a10, and Ms4a6b. Mac4 gene profile was characterized by the expression of genes related to ECM interaction and Hippo-Merlin signaling, e.g., Ifgal, Itga4, Ace, Cd44, Pglyrp1, and Eno3.
Fig. 7. Diversified transcriptional responses and functional specialization of macrophages in SCI.
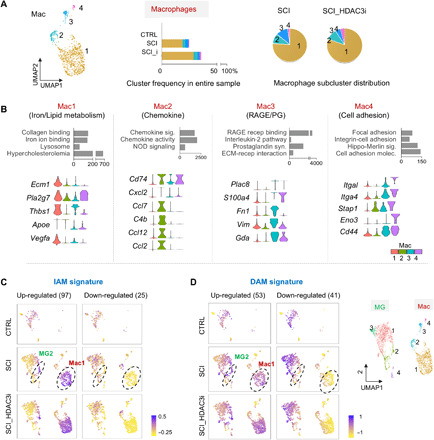
(A) Left: UMAP plot of macrophage subclusters (Mac1 to Mac4). Middle: Graphs showing relative abundance of Mac1 to Mac4 as compared to all cells in the entire sample in each condition. Right: Pie charts showing relative proportions of Mac1 to Mac4 among macrophages in each condition. n = 4 macrophages in CTRL, 337 in SCI, and 659 in SCI_HDAC3i. (B) Enriched GO terms associated with the top 30 up-regulated marker genes of each macrophage subcluster. Bottom: Violin plots show expression of selected marker genes related to the pathway ontologies indicated above. (C and D) Enrichment of IAM core genes (C) or DAM signature genes (D), separated as up- and down-regulated groups relative to control microglia, plotted as signature scores on the UMAPs of myeloid cells under different conditions. Notably, 9 of the 131 IAM core genes identified by INTACT RNA-seq were not detectable in scRNA-seq. Results showed that the IAM and DAM signature genes were predominantly enriched in subclusters MG2 and Mac1. Right: Reference UMAP plot of myeloid subclusters.
To relate cell type–specific injury responses as revealed by scRNA-seq to the core IAM gene signatures captured by INTACT RNA-seq from myeloid populations at different time points, we evaluated their convergence (fig. S8C). As mentioned above, among the 131 IAM core genes, 14 intersected with the microglial injury response gene set (390 DEGs between MG-SCI and MG-CTRL), while 32 intersected with the genes differentially expressed in influxed macrophages relative to homeostatic microglia (922 DEGs between Mac-SCI and MG-CTRL); 9 genes were common to both. Notably, these nine common genes were predominantly enriched in macrophages, in particular Mac1, and only a small subset of microglia (i.e., MG2) (fig. S8, D and E). Intriguingly, this enrichment pattern was also observed for the entire IAM core genes (Fig. 7C and fig. S8F), thus highlighting functional convergence of Mac1 and MG2 in engaging phagocytosis and debris clearance.
We next revisited the DAM gene signatures [representing largely microglial responses due to low content of infiltrating macrophage in AD (37)], and they interestingly also showed an enrichment pattern predominantly in Mac1 (Fig. 7D). For instance, among the common 11 signature genes shared by both IAM and DAM, 5 were predominantly expressed in macrophages (Apoe, Ctsd, Ctsb, Igf1, and Lyz2), 3 in microglia (Ccl2 and Csf1 in MG1 and Lpl in MG4), and only 2 (Itgax and Axl) were uniformly expressed, albeit at lower levels (fig. S9A). Trem2 and Tyrobp, two DAM marker genes, were widely expressed in both microglia and macrophages (fig. S9B), but their expression levels remained stable after SCI, echoing our INTACT RNA-seq data (see fig. S2F).
To further appreciate myeloid diversity, we reexamined M1 and M2 genes and found that for M1 genes, Cd86 and Fcgr3 (Cd16) were largely associated with microglia, whereas Fcgr2b (Cd32) was associated with macrophages; for M2 genes, Arg1 was exclusively expressed in MG4 and Mrc (CD206) in Mac2 (fig. S10A). All of these markers showed no significant transcriptional changes after SCI, echoing bulk INTACT RNA-seq results. With respect to the microglial proinflammatory factors that can induce A1 polarization of astrocytes, Tnf and Il1a were associated with microglia, while C1qa was associated with both microglia and a subset of macrophages (fig. S10B). The classic myeloid marker genes Ptprc (Cd45) and Itgam (Cd11b) showed ubiquitous but low expression in myeloid cells, while Cd83 was predominantly expressed in microglia (fig. S10C). Last, the 55 HDAC3-dependent microglial injury response genes, particularly those down-regulated genes, were predominantly enriched in MG4 (fig. S10D).
Transcriptional responses of neuronal and ependymal populations in SCI in dependence of HDAC3
We next examined cluster 3, initially assigned as neuronal cluster (Fig. 8A), but zoom-in analysis revealed a distinct ependymal cell population separate from the three neuronal groups, based on the expression of ependymal markers Sox2, Foxj1, and Riiad1 (Fig. 8, B and C, and fig. S11, A to C). There were 1838 DEGs between ependymal cells and neurons, regulating ion transporter activity, IL-11 pathway, and RAGE pathway, with 618 genes up-regulated in ependymal cells, including Tmem212 [an ependymal marker (35)], Ccd190, Dnah6, and Smim5, and 1220 genes down-regulated, including Gda, Lyz2, and Napsa (fig. S11, D and E).
Fig. 8. Injury response genes and HDAC3 dependence in ependymal cells after SCI.
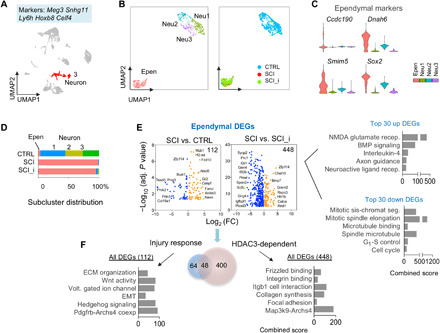
(A) UMAP plot showing all cells in adult spinal cord, with neuronal cluster highlighted in red. Highly expressed marker genes are indicated above. The neuronal cluster contained 942 cells in total: 638 in CTRL, 86 in SCI, and 218 in SCI_HDAC3i. (B) Left: UMAP plot of neuronal subclusters distinguishes an ependymal cell population (Epen) from the eneuronal population (Neu1 to Neu3). Right: UMAP plot showing the four subclusters color-coded for each condition. (C) Expression of marker genes for the ependymal subcluster, shown as violin plots. (D) Bar graphs of subcluster distributions revealed ependymal expansion but neuronal depletion after SCI. (E) Volcano plots showing DEGs of ependymal cells in SCI versus CTRL (injury response) and SCI versus SCI_HDAC3i (HDAC3 dependence). Total number of DEGs is indicated at the top right corner. n = 21 ependymal cells in CTRL, 85 in SCI, and 207 in SCI_HDAC3i. (F) Left: Enriched ontology terms and pathways associated with ependymal injury response DEGs, or HDAC3-regulated genes in reactive ependymal cells. Right: Enriched ontology terms and pathways associated with the top 30 up- or down-regulated HDAC3-dependent genes in reactive ependymal cells (SCI versus SCI_HDAC3i). EMT, epithelial-mesenchymal transition.
SCI resulted in a substantial contraction of neuronal populations but a marked ependymal expansion with a reactive gene signature linked to ECM organization, voltage-gated ion channels, as well as Wnt, Hedgehog, and platelet-derived growth factor B signaling (Fig. 8, D to F). HDAC3i affected transcription of 448 genes in ependymal cells, with the majority (78%) repressed by HDAC3, again obeying the general principle of gene repression by histone deacetylation, and only 48 genes overlapped with ependymal reactive genes (Fig. 8F). HDAC3-regulated genes in ependymal cells controlled Frizzled binding, integrin binding, and collagen biosynthesis; the top 30 up-regulated genes in SCI versus SCI_HDAC3i were related to NMDA glutamate receptor, bone morphogenetic protein signaling, IL-4, and axon guidance, whereas the top 30 down-regulated genes mainly concerned cell cycle (Fig. 8F).
Effects of SCI on fibroblast and astrocyte populations and the impact of HDAC3 inhibition
Astrocytes (cluster 4) expressed classic astrocytic markers, including Aqp4 (aquaporin 4, a water channel expressed on astrocyte vascular end feet), Agt (angiotensinogen), Scl6a11 [GAT3, a GABA (γ-aminobutyric acid) reuptake transporter], and Atp1a2 and Atp1b2 (subunits of Na+/K+ adenosine triphosphatase that restore extracellular K+ homeostasis following neuronal depolarization) (Fig. 9A). The gene expression in the spinal cord astrocytes of our samples obeyed the border between telencephalon astrocytes (expressing Mfge8) and nontelencephalon astrocytes (expressing Agt) (35). The astrocyte cluster could be further partitioned into six subclusters: Astro1 to Astro4 expressing astrocyte-specific genes such as Slc6a9 [GLYT1, a glycine transporter (35)], Slc7a10, and Slc19a12 (both amino acid transporters supporting neuronal activity), a fibroblast subcluster, and an intermediate Astro/Oligo subcluster expressing both markers Aqp4 and Mag (Fig. 9, B and C, and fig. S12, A to D). SCI caused a major contraction of Astro1 to Astro4, an emergence of fibroblasts, but a depletion of the Astro/Oligo subcluster, indicating its vulnerably to injury environment (Fig. 9, B and D).
Fig. 9. Glial transcriptional responses and HDAC3 dependence in SCI.
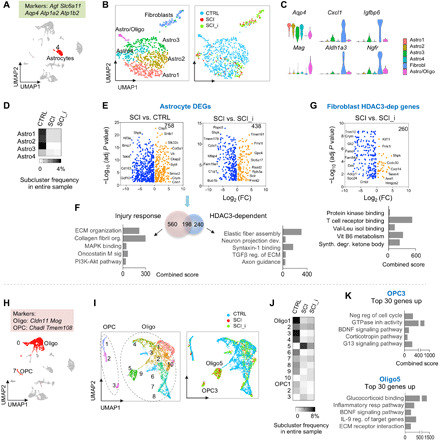
(A) UMAP plot, astrocyte cluster highlighted in red, and highly expressed marker genes indicated on top. n = 905 astrocytes. (B) UMAP plot of astrocyte cluster showing six subclusters. Right: Subclusters color-coded for each condition. (C) Violin plots showing expression of marker genes for astrocyte subclusters. (D) Frequency heatmap showing astrocyte subcluster proportions relative to all cells in entire sample in each condition. (E) Volcano plots showing DEGs in astrocytes (Astro1 to Astro4) of the indicated comparisons. n = 621 astrocytes in CTRL, 76 in SCI, and 121 in SCI_HDAC3i. (F) Venn diagram and pathway enrichment of injury response genes and HDAC3-regulated genes in reactive astrocytes. (G) Volcano plot and pathway enrichment of HDAC3-dependent DEGs in fibroblasts in SCI. n = 28 in SCI and 57 in SCI_HDAC3i. (H) UMAP plot, oligodendrocyte, and OPC clusters highlighted in red. Highly expressed marker genes are indicated above. n = 2815 oligodendrocytes and 318 OPCs. (I) UMAP plot showing subclusters of OPC and oligodendrocytes. Right: Subclusters color-coded for each condition. (J) Frequency heatmap showing subclusters proportion relative to all cells in entire sample in each condition. (K) Ontology and pathway enrichment of the top 30 DEGs in subcluster OPC3 (versus other OPC subclusters) and Oligo5 (versus other Oligo subclusters). GTPase, guanosine triphosphatase; BDNF, brain-derived neurotrophic factor.
The four astrocyte subclusters were evenly distributed at steady state, albeit with a smaller share for Astro4 (Fig. 9D). Astrocytes exhibited distinct expression features in SCI, expressing 638 injury response genes linked to ECM interaction (collagen fibril organization, collagen binding, and miR targets in ECM receptors) and platelet-derived growth factor receptor binding (Fig. 9, E and F, and table S2). HDAC3 exerted a large transcriptional influence on reactive astrocytes, totaling 438 genes concerning ECM organization, Wnt pathway, Ras activation, and axon guidance, and among them, 198 overlapped with the astrocytic injury response genes (Fig. 9F).
The fibroblast subcluster, with a transcriptional profile indicating mesenchymal origin, expressed higher levels of Aldh1a3, Cxcl1, Igfbp6, and Ngfr [nerve growth factor (NGF) receptor] (Fig. 9C). HDAC3 controlled 260 genes in reactive fibroblasts, mostly down-regulated, and they regulated vitamin B6 metabolism, ketone bodies, and valine-leucine binding (Fig. 9G).
We next surveyed chondroitin sulfate proteoglycan (CSPG) gene family members, which revealed that Cspg4 (NG2) was only sparsely expressed in astrocytes, while OPCs and pericytes appeared to be the main cellular sources for NG2 (fig. S12E). Cspg5, Bcan, Ncan, and Vcan were also expressed more abundantly in OPCs than in astrocytes, while Cd44 was predominantly expressed in neutrophils (fig. S12E).
Reaction of oligodendrocytes and OPCs to SCI and HDAC3 inhibition
OPCs could be further divided into 3 subclusters and oligodendrocytes into 10 subclusters (Fig. 9, H and I, and fig. S12F). While most showed a substantial contraction after SCI, two new subclusters—OPC3 and Oligo5—emerged, both enriched for BDNF and corticosteroid signaling, hinting at common environmental cues (Fig. 9K). Compared to other OPCs, OPC3 assumed a gene profile enriched for G13 signaling, guanosine triphosphatase inhibitor activity, and negative regulation of cell cycle, whereas Oligo5 took up the tasks of ECM interaction and inflammatory response (Fig. 9K). HDAC3i led to a modest shift of subcluster distribution, with a smaller expansion of OPC3 and Oligo5 after SCI (Fig. 9J).
DISCUSSION
Upon activation, microglia and macrophages undergo rapid morphological, immunological, and physiological transformations. Our INTACT RNA-seq study unveiled the transcriptional changes that underlie the progressive IAM transformations, while scRNA-seq analysis revealed refined myeloid subtype classifications. We also documented diversification of other glial cells and a wide influence of HDAC3, arising either directly or indirectly from altered injury milieu.
Sequential INTACT RNA-seq on myeloid populations showed that IAM progressively activate temporally distinct gene programs to control proliferation and motility at 3 dpi, axon chemoattraction and ion channel activity at 7 dpi, and ECM reorganization at 14 dpi. Ion channel activities of microglia have been implicated in aiding pathologies of neurodegenerative disorders, and electrophysiological alterations in activated microglia can influence proliferation, migration, ramification, phagocytosis, cytokine secretion, and production of reactive oxygen and nitrogen species (38).
We also revealed a core IAM gene program spanning different time points, which showed commonalities but also differences compared to the DAM signatures. The converging IAM and DAM gene signatures highlighted lipid metabolism, scavenging, and ECM organization as central microglial activities under diverse CNS pathologies. Despite commonalities, there are notable differences: e.g., Trem2 drives microglial phenotypes in AD, but neither Trem2 nor its intracellular adaptor Tyrobp was significantly up-regulated in IAM. Traumatic injury and neurodegeneration differ in pathological triggers, blood-brain barrier integrity, disease foci spread, and time frame. Moreover, infiltrating macrophages are abundant in injured spinal cord but not in AD brains (37). The time points are also rather different: For the DAM study (5), scRNA-seq studies were conducted on immune cells isolated by FACS from whole AD brains at 1, 3, 4, and 8 months, revealing a progressive increase of plaque-associated microglia, a small population compared to the much larger homeostatic microglia population; in contrast, our INTACT RNA-seq study was conducted at 3, 7, and 14 dpi, providing a defined temporal trajectory of the IAM gene program over the course of days.
Our data endorse a persistent reparative function for IAM, supported by the following: (i) Phagocytic genes (Lpl, Cst7, and Cd9) remained induced at 14 dpi, signifying ongoing debris clearing; (ii) ECM genes and trophic factors were highly expressed; and (iii) the anti-inflammatory hallmark gene set was enriched at all stages. Notably, earlier reports on up-regulation of M1 genes and down-regulation of M2 genes were based on microarray analysis on whole spinal cord tissue (3); our INTACT RNA-seq revealed no overt changes of M1/M2 genes in myeloid cells; and our scRNA-seq further unveiled their heterogeneous expression patterns in myeloid subclusters, for instance, MG2 and Mac1 were the main myeloid subclusters that were enriched for IAM and DAM signatures genes. Our data therefore underscore a more nuanced view of neuroinflammation than the conceptual dichotomy of pro- versus anti-inflammatory phenotypes. In addition, a large number of exons displayed differential splicing in IAM, and consistently, genes grouped in cluster 2 were enriched for spliceosome. Future studies are needed in regard to the function and involvement of RNA-binding regulatory proteins, as well as comparisons of alternative exon usage between DAM and IAM.
The scRNA-seq data revealed four transcriptional subtypes of microglia: an immunity-focused MG1, a reactive MG2, an immediate response MG3, and a proliferative MG4. Contrary to the assumption of a uniform population of homeostatic microglia, all four microglial subtypes were already present in uninjured spinal cord, indicating a variety of cell states at baseline. Although homeostatic marker genes were down-regulated in IAM as a whole after SCI (signifying release from homeostatic checkpoints), the activated microglia population was not uniform, with MG2 experiencing the largest expansion after SCI, but the homeostatic MG1 was also expanded. Our trajectory study mapped the microglia transformation along a continuous trajectory, starting from MG3 (likely a source population), progressing toward MG1 and MG2, and ending in MG4. The current study provided a snapshot of microglial heterogeneity at 5 dpi; it is not clear whether individual microglial cells can switch from one subtype to another or whether proliferation largely accounts for subtype expansion. Future longitudinal tracking of different microglial subclusters would help to resolve the fluidity of functional states over time.
The current study also advanced our understanding of the effects of HDAC3 inhibition in SCI: (i) Microglia and macrophages responded differently to the HDAC3i, but the common HDAC3-dependent genes in both populations regulate chemotaxis, chemokine activity, RANKL, and p38 MAPK signaling, thus echoing our earlier finding of an immunosuppressive function of HDAC3i (7). (ii) Hdac3 expression was not uniform among microglia, but predominantly in the proliferative MG4. Single-cell trajectory studies with pseudotime inference revealed that HDAC3i resulted in a shift to MG4 as the starting point of microglia transformation, consistent with HDAC3-dependent genes being enriched in MG4 and linked to proliferation. Hence, we identified a subpopulation of microglia that is highly proliferative and heavily influenced by HADC3 activity. (iii) scRNA-seq revealed that Hdac3 was widely expressed in several cell types in adult spinal cord. Our results showed widespread influence of HDAC3i on transcriptional states of not only myeloid cells but also other glial cells.
Our INTACT RNA-seq studies were conducted in the transection SCI model and the scRNA-seq studies in the contusion model. Although both are traumatic injuries and likely trigger converging transcriptional responses in glial cells, in hindsight, it would have been useful to conduct studies using the same SCI model with matching time points to facilitate direct comparison, particularly for cell compositions. In addition, while our study represents a first step to unravel the molecular underpinning of the complexity of immune response and glial activation after SCI, additional immunostaining and functional validations are needed.
In summary, we found a wide range of activation states in microglia, both at steady state and after SCI. Activated microglia and infiltrating macrophages progressively acquired overall reparative, but highly diversified transcriptional profiles, each comprising four distinct transcriptional subtypes with specialized tasks. The injury responses and functional diversifications of immune and glial cells are influenced by the chromatin regulator HDAC3, which may be exploited to enhance neural repair.
MATERIALS AND METHODS
Mice
All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai. Cx3cr1CreER mice expressing tamoxifen-inducible Cre recombinase (CreER) under the control of the endogenous Cx3cr1 promoter (15) were obtained from the Jackson laboratory (Cx3cr1tm2.1(cre/ERT2)Litt; JAX #021160). The Cx3cr1CreER mice were crossed with Rosa26-loxP-STOP-loxP-INTACT reporter mice [Gt(ROSA)26Sortm5(CAG-Sun1/sfGFP)Nat; JAX #021039]; in the INTACT reporter allele, the C terminus of Sun1, a nuclear membrane protein, is tagged with two tandem copies of superfolder GFP and six copies of the Myc epitope (13). Cx3cr1CreER/+ Rosa26INTACT/+ mice were healthy and fertile, and tamoxifen (100 mg/kg; Sigma-Aldrich, T5648) was injected intraperitoneally to activate INTACT-GFP reporter expression. The Cx3cr1GFP reporter line (39) was obtained from the Jackson laboratory (Cx3cr1tm1Litt; JAX #005582). All mouse strains were backcrossed and maintained on a C57BL/6J genetic background.
Genotyping primers for Cx3cr1CreER mice are as follows: C-F1, AAGACTCACGTGGACCTGCT; M-R1, CGGTTATTCAACTTGCACCA; and WT-R2, AGGATGTTGACTTCCGAGTTG [band sizes: wild type (WT) is 695 base pairs (bp) and Cre allele is 300 bp]; for Rosa26INTACT mice: C-F1, GCACTTGCTCTCCCAAAGTC; WT-R1, CATAGTCTAACTCGCGACACTG; and M-R2, GTTATGTAACGCGGAACTCC (WT is 557 bp and INTACT reporter allele is ~300 bp); for Cx3cr1GFP mice: WT, GTCTTCACGTTCGGTCTGGT; C, CCCAGACACTCGTTGTCCTT; and M, CTCCCCCTGAACCTGAAAC (WT is 410 bp and GFP allele is ~500 bp).
Spinal cord transection injury model
For INTACT SCI studies with a T8 transection model, 6- to 8-week-old Cx3cr1CreER/+ Rosa26INTACT/+ mice were used. Mice were anesthetized using isofluorane (Baxter Healthcare, NDC 10019-360-40) immediately before start of surgeries. Dorsal column transection injuries were performed as previously described (7). Briefly, the lamina of T8 spinal segment was exposed and removed, and the dorsal column was transected bilaterally using Iris microscissors (Fine Science Tools, 15000-00), with maximum depth reaching ~0.8 mm. For sham control, only T8 laminectomy was performed. All animals received subcutaneous injection of saline (1 ml), Baytril (10 mg/kg), and buprenorphine (0.05 mg/kg) every day for the first week following surgery. For INTACT purification, each time point (sham, 3, 7, and14 dpi) was composed of spinal cords from three animals pooled together. In total, 9 animals were used per time point, making it a total of 36 animals to obtain n = 3 for each time point.
Purification of INTACT GFP+ nuclei
Affinity immunopurification of INTACT-GFP-tagged nuclei was performed as previously described (13). Animals were euthanized in a CO2 chamber, and spinal cords (from 5 mm rostral to 5 mm caudal to injury site) were rapidly dissected on ice and pooled from three animals for each sample. Nuclei were separated from cellular debris using an iodixanol gradient, wherein the nuclei separated out at the interphase between the 30 and 40% phases. We obtained between 1.5 million and 2 million nuclei per purification, which were then incubated with 10 μl of rabbit monoclonal anti-GFP antibody (0.2 mg/ml; Life Technologies, G10362). Protein G Dynabeads (Invitrogen, 10004D) were added to this mixture and incubated for 20 min for magnetic separation of GFP-positive nuclei, and the solution was then passed through a 20-μm filter (CellTrics, Sysmex, Partec GmbH, 04-0042-2315). All steps were performed at 4°C.
Total nuclei yields and the percentage of GFP+ nuclei were calculated as previously described (13). Specifically, from the initial ~2 million nuclei obtained per preparation, an aliquot was saved and stained with 4′,6-diamidino-2-phenylindole (DAPI). About 9% nuclei from this fraction were GFP+, as counted by either fluorescence microscopy or using hemocytometer chambers. In the final bead-bound fraction, we obtained 95 to 98% purity of GFP+ cells. These nuclei were stored at −80°C for subsequent nuclear RNA extractions, qRT-PCRs, and RNA-seq.
RNA extraction, library preparation, and sequencing
GFP+ bead-bound nuclei were resuspended in buffer RLT for RNA extraction using the QIAGEN RNeasy Micro Kit, along with the recommended on-column deoxyribonuclease digestion (QIAGEN, 74004). RNA quality and concentrations were measured using an Agilent Bioanalyzer (Agilent RNA 6000 Pico, 5067-1513), and samples having RNA integrity number (RIN) scores higher than 7 were used. Total RNA was converted to cDNA and amplified using the Ovation RNA-Seq System V2 (Nugen, 7102). All samples underwent single primer isothermal amplification (Ovation RNA-Seq System V2, NuGen, 7102-08) before library preparation [NEBNext Ultra DNA Library Prep Kit for Illumina; New England Biolabs (NEB), 7370S]. Three independent samples for each condition were barcoded using the NEBNext Index primers (NEB, E7335S), pooled together as one sample, and run on the Illumina platform HiSeq 2500 (Rapid Mode, 2 × 50 bp) (Macrogen). RNA-seq generated 40 to 80 million paired reads per library (table S1). For qRT-PCR, first-strand cDNA synthesis was performed using the SuperScript III First-Strand Synthesis System, followed by second-strand cDNA synthesis (Invitrogen, 18080-051).
Quantitative real-time RT-PCR
Quantitative PCR was performed using PerfeCTa SYBR Green FastMix Rox (Quanta Biosciences) in an ABI 7900HT quantitative PCR system (Applied Biosystems). Tfrc or Gapdh was used as housekeeping genes. Sequences of primers used are as follows: Gpr84, CTCCTGCTACCATGAGTCTGT (forward) and GTGCAGTAGAGTAGATCAGCCA (reverse); Itgam, ATGGACGCTGATGGCAATACC (forward) and TCCCCATTCACGTCTCCCA (reverse); and Tnf, CCCTCACACTCAGATCATCTTC (forward) and GCTACGACGTGGGCTACAG (reverse).
IHC and microscopy
Sham control animals (laminectomy only) and mice subjected to SCI were euthanized and perfused using ice-cold 4% paraformaldehyde (Thermo Fisher Scientific, AC416785000) in phosphate-buffered saline (PBS). Spinal cords at the injury site were dissected, postfixed overnight, and embedded in O.C.T. compound (Tissue-Tek) (Thermo Fisher Scientific, 4585). Sections were cut at 12-μm thickness and collected onto SuperFrost Plus slides (VWR, 48311-703) and stored at −20°C. The sections were washed with 1× PBS, incubated with blocking buffer with 5% normal donkey serum (Jackson ImmunoResearch, 017-000-121) and 0.3% Triton X-100 (Acros Organics, 21568-2500) in PBS for an hour at room temperature before adding primary antibodies diluted in antibody dilution buffer with 1% bovine serum albumin (Fisher BioReagents, BP9703100) and 0.3% Triton X-100 in PBS, and slides were incubated overnight at 4°C. Primary antibodies and dilutions used are as follows: anti-GFP chicken (Aves Labs, GFP-1020) (1:1000), anti-IBA1 rabbit (Wako Laboratory Chemicals, 019-19741) (1:500), anti-IBA1 goat (Novus Biologicals, NB100-1028) (1:500), anti-CD11b goat (MyBioSource, MBS420973) (1:300), anti-CD45 rat (BD Biosciences, 550539) (1:50), anti-CD68 rat (Bio-Rad, MCA1957GA) (1:300), anti-CD11c Armenian hamster (Thermo Fisher Scientific, N418) (1:300), anti-CD206 goat (R&D Systems, AF2535) (1:400), anti-TMEM119 rabbit (Abcam, ab209064) (1:200), anti-P2ry12 rat (BioLegend, 848001) (1:100), anti-LPL rabbit (Bioss Antibodies, bs-1973r) (1:500), anti-Spp1 rabbit (ProteinTech, 25715-1-AP) (1:500), and anti-Fn1 rabbit (EMD Millipore, ab2033) (1:300). Subsequently, Alexa Fluor 488–, Alexa Fluor 594–, Alexa Fluor 647–conjugated secondary antibodies (1:300, Jackson ImmunoResearch) were added, and DAPI (1:1000; Invitrogen, D1306) was used for counterstaining. Slides were mounted using Fluoromount-G (Southern Biotech, 0100-01), and images were captured with Zeiss microscopes (Axio Imager.A2 with AxioCam MRm).
Differential gene and exon expression analysis
Quality of sequencing reads was assessed using fastQC (40). Reads were mapped against the mouse genome (GRCm38) and ribosomal RNA sequences using ContextMap version 2.7.9 (41), using the Burrows-Wheeler Aligner (42) as short-read aligner and default parameters. Number of read fragments per gene was determined from the mapped RNA-seq reads using featureCounts (strand-specific for stranded libraries, and nonstrand-specific otherwise) (43). Gene expression was quantified in terms of fragments per kilobase of exons per million mapped reads (FPKM).
Differential gene expression analysis was performed using edgeR (44). For each pairwise differential gene expression analysis, only genes with at least an average of 10 reads per sample were evaluated. P values were adjusted for multiple testing using the method by Benjamini and Hochberg (45), and genes with an adjusted P < 0.01 were considered significantly differentially expressed.
Differentially expressed exons were determined with DEXSeq version 1.20.2 (46). Exons were considered differentially expressed if they had a P < 0.01 (adjusted for multiple testing using the method of Benjamini and Hochberg), an absolute log2 fold change ≥ 1, and an exon base mean value ≥ 30 (= exon read count normalized by sequencing depth and averaged across the compared samples).
WGCNA gene coexpression network analysis
A WGCNA (26, 47–51) was applied to the RNA-seq data (control + IAM at three injury stages) to identify gene modules with coordinated expression patterns in microglia using R package WINA (26). With default settings, we identified 103 modules, each of which was assigned a distinct color code. For each module in the coexpression network, we searched the MSigDB gene annotation database for enriched functional pathways.
Hierarchical clustering of DEGs
Hierarchical clustering was performed on all differentially regulated genes identified between 3, 7, and 14 dpi/Ctrl using the R base function “hclust” with the default settings (complete linkage and Euclidean distance). The dendrogram was cut with a height cutoff value of 4, rendering four main clusters. We then consulted the MSigDB gene annotation database for enriched functional pathways for each cluster. The P values for the functional enrichment test were corrected for multiple tests (Benjamini and Hochberg adjustment).
Enrichr GO analysis
For GO, Kyoto Encyclopedia of Genes and Genomes pathway, and Jensen Tissue database analyses, DEG lists were used as input into the web version of Enrichr software (http://amp.pharm.mssm.edu/Enrichr/) (52). All scales from the Enrichr database are depicted as “combined score” (53), computed by taking log of the P value from the Fisher’s exact test and multiplying it with the z score of the deviation from the expected rank.
GSEA Hallmark analysis
GSEA was performed using the preranked option of the GSEA software (http://software.broadinstitute.org/gsea/index.jsp). Ranking was performed on the basis of log2 fold changes from the differential gene expression analysis. GSEA was performed both for the hallmark gene set and the Coates macrophage datasets (33) available from the GSEA MSigDB database.
QIAGEN Ingenuity Pathway Analysis
Ingenuity software (QIAGEN Bioinformatics, www.ingenuity.com) was used, with P values and fold changes for the genes examined as input data. Comparative analysis for canonical pathways and upstream regulators was also performed. Data are visualized as activation z score.
Circos plot
Previously identified microglia transcriptomes from various mouse models were compared against combined DEGs in IAM relative to control microglia. These were signature gene sets for AD (5xFAD), brain irradiation, MFP2 lipid disorder, chronic pain, aging, neuropathic pain, and ALS [superoxide dismutase 1 (SOD1)] (4). All datasets were cross-compared with the DEGs in IAM. A Circos graph was prepared using the Circos package version 0.69-6.
Spinal cord contusion injury model for scRNA-seq
SCI with 6- to 8-week-old C57BL/6 mice of mixed gender was performed as previously described (32). Briefly, for T8 contusion injury, the lamina of the T8 spinal segment was exposed and removed and an infinite horizon impactor (Precision Systems and Instrumentation, IH-0400) was used to deliver an impact force of 45 kilodynes to the spinal cord. Bladder expression was performed twice a day for the duration of the experiments.
Tissue preparation for scRNA-seq
Animals were perfused, and 10-mm spinal cord tissue (from 5 mm rostral to 5 mm caudal from lesion core) was harvested on ice and rapidly dissociated using a modified papain digestion protocol to achieve single-cell suspension [Miltenyi Neural Tissue Dissociation Kit (P) 130-092-628]. Tissue was harvested in ice-cold Hanks’ balanced salt solution (HBSS) (Thermo Fisher Scientific, 14175095) and chopped into small pieces using a sharp scalpel. Tissue was transferred into 15-ml Falcon tubes containing 1 ml of digestion solution, using a wide-tipped Pasteur pipette. Tubes were incubated for 15 to 20 min at 37°C in a tube rotator under slow continuous rotation. Tissues were slowly triturated using fire-polished glass Pasteur pipettes of decreasing tip diameters (three sizes) 15 times per diameter and incubated at 37°C in the tube rotator for 10 min between each mechanical dissociation step. The final cell suspension was passed through a 70-μm cell strainer, and 10 ml of HBSS (with Ca and Mg; Thermo Fisher Scientific, 24020117) was applied to wash the strainer. The suspension was collected in a 50-ml Falcon tube and centrifuged at 450g at 4°C for 5 min. Supernatant containing dead cells and debris was discarded, and pellet was resuspended in 7 ml of HBSS (without Ca/Mg), mixed with 1.2 ml of fetal bovine serum (FBS) and 3.6 ml of 100% Percoll (GE Healthcare, 45-001-748) [9 parts Percoll + 1 part 10× HBSS (Sigma-Aldrich, H4385) to achieve final Percoll concentration at 30%]. The Percoll cell suspension was overlaid with 1 ml of 10% FBS (500 ml; Sigma-Aldrich, F4135) in RPMI (Thermo Fisher Scientific, 11875101). Tubes were spun at 800g for 15 min at 4°C, and pellet was collected in a new 15-ml tube and resuspended in 0.5 ml of FACS buffer. Red blood cells (RBCs) were lysed by incubating cells with RBC lysis buffer (BioLegend, 420301) for 15 min at room temperature; cells were washed and resuspended in FACS buffer. All steps were performed at 4°C.Cells were counted using an automated cell counter (Thermo Fisher Scientific Countess, AMQAX1000), live/dead measures were made using trypan blue (Gibco, 15250061), and samples with a viability of higher than 70% were used for downstream scRNA-seq analysis.
Single-cell RNA sequencing
scRNA-seq was performed using the Chromium platform (10X Genomics, Pleasanton, CA) with a 3′ gene expression V3 kit and an input of ~10,000 viable cells from a debris-free suspension. Briefly, gel bead-in-emulsions (GEMs) were generated on the sample chip in the Chromium controller. Barcoded cDNA was extracted from the GEMs by post-GEM RT cleanup and amplified for 12 cycles followed by fragmentation of amplified cDNA, end repair, poly(A) tailing, adapter ligation, and 10X-specific sample indexing following the manufacturer’s protocol. Libraries were quantified using QuBit (Thermo Fisher Scientific) and Bioanalyzer (Agilent). Libraries were sequenced in paired-end mode on a NovaSeq instrument (Illumina, San Diego, CA) targeting a depth of 10,000 unique reads per cell. The Cell Ranger Single Cell Software Suite (version 3.0, 10X Genomics) was used to align and quantify sequencing data against the mm10 mouse reference genome. Downstream analyses, such as graph-based clustering and differential expression analysis/visualization, were performed using the Loupe Cell Browser (version 3.1, 10X Genomics) and the Seurat package. Notably, the current scRNA-seq experiments do not provide a read coverage for each cell that could match the coverage depth of bulk RNA-seq as in the INTACT RNA-seq, and the statistical power for low-expressed genes is reduced for scRNA-seq.
Quality control, normalization, and data integration
For quality control, features (genes) expressed in less than five cells and cells expressing less than 500 features were removed. In addition, cells with mitochondrial content >25% were also excluded from further analysis. Cells with high UMI and gene number per cell were filtered out to remove duplets or multiplets. A total of 2523 cells in sample SCI (with vehicle treatment) and 3423 cells in sample SCI_HDAC3i (with acute RGFP966 treatment) were sequenced. We also integrated as a control reference the scRNA-seq data of 7466 cells from uninjured control (CTRL) spinal cord (35). After removal of low-quality cells, outliers, and doublets, we performed our analysis on 4576 cells for CTRL, 2276 cells for SCI, and 3085 cells for SCI_HDAC3i. Next, feature counts were normalized using a LogNormalize method. First, the counts of each cell were divided by the total counts for that cell and multiplied by a scale factor of 10,000. The data were then natural log-transformed. Next, the top 3000 common features across the SCI, CTRL, and SCI-HDAC3i samples were identified as anchors to integrate the three samples. The above analyses were completed using the R package Seurat (54, 55).
Dimensionality reduction, cell clustering, cell marker, and cell type identification
The linear dimensionality reduction of the integrated data was performed using the principal components analysis (PCA), and then, nonlinear dimensionality reduction was performed by the UMAP (36) algorithm using the PCA dimensional reduction as input. Next, cell clusters were identified by a shared nearest neighbor (SNN) modularity optimization-based clustering algorithm using the top 20 principal components. First, k-nearest neighbors (KNNs) of each cell were calculated. The KNN graph was then used to construct the SNN graph by calculating the Jaccard index of the neighborhood overlap between every cell and its 20 nearest neighbors. Last, the modularity function was optimized to determine cell clusters at a resolution of 0.05.
The markers conserved across the three conditions of each cell cluster were identified by comparing the expression levels of that cell cluster and all other clusters. Cell types were then identified by mapping the conserved cell markers with the previous mouse and human cell markers in the PanglaoDB database (56). Cells from major cell types (astrocytes, microglia, oligodendrocytes, and neuron) were reclustered using a higher resolution (0.5) to further analyze the subclusters of each cell type.
The trajectory analysis based on gene expression changes among the cell clusters was performed using the unsupervised Slingshot algorithm (57). Slingshot has two stages. In the first stage, Slingshot infers the key elements of the global lineage structure using a minimum spanning tree method based on clustered cells. In the second stage, Slingshot infers pseudotime variables for cell clusters along each lineage by fitting smooth branching curves to these lineages using simultaneous principal curves. The Slingshot analysis provided 80% confidence on the trajectory results.
Differential expression analysis for scRNA-seq
Differential expression analysis between conditions was performed using the Wilcoxon rank sum test on the genes detected in a minimum fraction of 10% cells in either of the two populations. P value adjustment was performed using the Bonferroni post hoc correction based on the total number of genes tested. DEG lists were generated by filtering all genes by a combined cutoff of fold change > 1.18 and adjusted P < 0.05. GO enrichment analysis was performed on DEGs using the R package GO function (58).
Signature scores
To obtain single-cell gene signature scores for scRNA-seq data, we used lists of selected genes and calculated for each cell in a dataset a signature score with the algorithm developed by Laffy and colleagues (59). The corresponding R package scrabble was obtained from github (“jlaffy/scrabble”).
Quantification and statistical analysis
For each experiment, the numbers of mice used in each cohort and the number of images analyzed from each animal are listed in figure legends. Statistical analyses were performed as indicated or using GraphPad Prism 7 software. Data are presented as means ± SEM. All RNA-seq bar graphs were plotted using mean FPKM values. Sample numbers and adjusted P values and statistical tests used are indicated in the figure legends.
Acknowledgments
We thank J. Nathans at the Johns Hopkins University for providing INTACT reagents and protocols, as well as the members of Zou and Friedel laboratories for discussions. Funding: This work was supported by the grants from the National Institutes of Health (NIH; R01/R56 NS073596), the Craig H. Neilsen Foundation (#476516), and the New York State Spinal Cord Injury Research Board (DOH01-C30603GG and DOH01-C30832GG) to H.Z.; NIH grants (R01 AG046170, RF1 AG054014, U01 AG052411, RF1 AG057440, and R01 AG057907) to B.Z.; Deutsche Forschungsgemeinschaft grants (FR2938/7-1 and FR2938/10-1) to C.C.F.; postdoctoral fellowship grants from New York State Spinal Cord Injury Research Board (DOH01-C32634GG) to S.W.; and from the Chinese Scholarship Council to Xiang Zhou. Author contributions: S.W. conducted the study. Xianxiao Zhou analyzed scRNA-seq data. Xiang Zhou contributed to experiments. L.G. and B.Z. performed systems biology analysis. M.-S.F., M.K., and C.C.F. performed RNA-seq mapping and differential gene and exon analyses. A.R. and L.S. provided support for scRNA-seq analyses. S.W., Xianxiao Zhou, R.H.F., and H.Z. designed the project, analyzed the data, and wrote the paper. All authors discussed and helped prepare the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. INTACT RNA-seq and DEXSeq data have been deposited previously (32) at the NCBI Gene Expression Omnibus (GEO), accession number GSE113566. The scRNA-seq data have been deposited under accession number GSE153737.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd8811/DC1
REFERENCES AND NOTES
- 1.Kettenmann H., Hanisch U.-K., Noda M., Verkhratsky A., Physiology of microglia. Physiol. Rev. 91, 461–553 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Beck K. D., Nguyen H. X., Galvan M. D., Salazar D. L., Woodruff T. M., Anderson A. J., Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133, 433–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kigerl K. A., Gensel J. C., Ankeny D. P., Alexander J. K., Donnelly D. J., Popovich P. G., Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O’Loughlin E., Xu Y., Fanek Z., Greco D. J., Smith S. T., Tweet G., Humulock Z., Zrzavy T., Conde-Sanroman P., Gacias M., Weng Z., Chen H., Tjon E., Mazaheri F., Hartmann K., Madi A., Ulrich J. D., Glatzel M., Worthmann A., Heeren J., Budnik B., Lemere C., Ikezu T., Heppner F. L., Litvak V., Holtzman D. M., Lassmann H., Weiner H. L., Ochando J., Haass C., Butovsky O., The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T. K., David E., Baruch K., Lara-Astaiso D., Toth B., Itzkovitz S., Colonna M., Schwartz M., Amit I., A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Grabiec A. M., Krausz S., de Jager W., Burakowski T., Groot D., Sanders M. E., Prakken B. J., Maslinski W., Eldering E., Tak P. P., Reedquist K. A., Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 184, 2718–2728 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Kuboyama T., Wahane S., Huang Y., Zhou X., Wong J. K., Koemeter-Cox A., Martini M., Friedel R. H., Zou H., HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 7, 8641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., Littman D. R., Dustin M. L., Gan W.-B., ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Srinivasan K., Friedman B. A., Larson J. L., Lauffer B. E., Goldstein L. D., Appling L. L., Borneo J., Poon C., Ho T., Cai F., Steiner P., van der Brug M. P., Modrusan Z., Kaminker J. S., Hansen D. V., Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 7, 11295 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butovsky O., Jedrychowski M. P., Moore C. S., Cialic R., Lanser A. J., Gabriely G., Koeglsperger T., Dake B., Wu P. M., Doykan C. E., Fanek Z., Liu L., Chen Z., Rothstein J. D., Ransohoff R. M., Gygi S. P., Antel J. P., Weiner H. L., Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liddelow S. A., Guttenplan K. A., Clarke L. E., Bennett F. C., Bohlen C. J., Schirmer L., Bennett M. L., Münch A. E., Chung W.-S., Peterson T. C., Wilton D. K., Frouin A., Napier B. A., Panicker N., Kumar M., Buckwalter M. S., Rowitch D. H., Dawson V. L., Dawson T. M., Stevens B., Barres B. A., Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haimon Z., Volaski A., Orthgiess J., Boura-Halfon S., Varol D., Shemer A., Yona S., Zuckerman B., David E., Chappell-Maor L., Bechmann I., Gericke M., Ulitsky I., Jung S., Re-evaluating microglia expression profiles using RiboTag and cell isolation strategies. Nat. Immunol. 19, 636–644 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mo A., Mukamel E. A., Davis F. P., Luo C., Henry G. L., Picard S., Urich M. A., Nery J. R., Sejnowski T. J., Lister R., Eddy S. R., Ecker J. R., Nathans J., Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng G. X. Y., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., McDermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., McFarland C., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H., Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkhurst C. N., Yang G., Ninan I., Savas J. N., Yates J. R. III, Lafaille J. J., Hempstead B. L., Littman D. R., Gan W.-B., Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parikh P., Hao Y., Hosseinkhani M., Patil S. B., Huntley G. W., Tessier-Lavigne M., Zou H., Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl. Acad. Sci. U.S.A. 108, E99–E107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett M. L., Bennett F. C., Liddelow S. A., Ajami B., Zamanian J. L., Fernhoff N. B., Mulinyawe S. B., Bohlen C. J., Adil A., Tucker A., Weissman I. L., Chang E. F., Li G., Grant G. A., Hayden Gephart M. G., Barres B. A., New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U.S.A. 113, E1738–E1746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L.-c., Means T. K., El Khoury J., The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautier E. L., Shay T., Miller J., Greter M., Jakubzick C., Ivanov S., Helft J., Chow A., Elpek K. G., Gordonov S., Mazloom A. R., Ma’ayan A., Chua W.-J., Hansen T. H., Turley S. J., Merad M., Randolph G. J.; Immunological Genome Consortium , Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 13, 1118–1128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yona S., Kim K.-W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., Jung S., Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldmann T., Wieghofer P., Jordão M. J., Prutek F., Hagemeyer N., Frenzel K., Amann L., Staszewski O., Kierdorf K., Krueger M., Locatelli G., Hochgerner H., Zeiser R., Epelman S., Geissmann F., Priller J., Rossi F. M. V., Bechmann I., Kerschensteiner M., Linnarsson S., Jung S., Prinz M., Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kroner A., Greenhalgh A. D., Zarruk J. G., Passos Dos Santos R., Gaestel M., David S., TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 83, 1098–1116 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Kigerl K. A., McGaughy V. M., Popovich P. G., Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J. Comp. Neurol. 494, 578–594 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y., Soderblom C., Krishnan V., Ashbaugh J., Bethea J. R., Lee J. K., Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol. Dis. 74, 114–125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee D. D., Seung H. S., Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Wang M., Roussos P., McKenzie A., Zhou X., Kajiwara Y., Brennand K. J., De Luca G. C., Crary J. F., Casaccia P., Buxbaum J. D., Ehrlich M., Gandy S., Goate A., Katsel P., Schadt E., Haroutunian V., Zhang B., Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 8, 104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirsch-Reinshagen V., Zhou S., Burgess B. L., Bernier L., McIsaac S. A., Chan J. Y., Tansley G. H., Cohn J. S., Hayden M. R., Wellington C. L., Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Schultz J. R., Tu H., Luk A., Repa J. J., Medina J. C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D. J., Lustig K. D., Shan B., Role of LXRs in control of lipogenesis. Genes Dev. 14, 2831–2838 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milner R., Crocker S. J., Hung S., Wang X., Frausto R. F., del Zoppo G. J., Fibronectin- and vitronectin-induced microglial activation and matrix metalloproteinase-9 expression is mediated by integrins α5β1 and αvβ5. J. Immunol. 178, 8158–8167 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Blasi F., Carmeliet P., uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 3, 932–943 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Colonna M., Wang Y., TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Zhou X., Wahane S., Friedl M.-S., Kluge M., Friedel C. C., Avrampou K., Zachariou V., Guo L., Zhang B., He X., Friedel R. H., Zou H., Microglia and macrophages promote corralling, wound compaction and recovery after spinal cord injury via Plexin-B2. Nat. Neurosci. 23, 337–350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coates P. J., Rundle J. K., Lorimore S. A., Wright E. G., Indirect macrophage responses to ionizing radiation: Implications for genotype-dependent bystander signaling. Cancer Res. 68, 450–456 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Abutbul S., Shapiro J., Szaingurten-Solodkin I., Levy N., Carmy Y., Baron R., Jung S., Monsonego A., TGF-β signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia 60, 1160–1171 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Zeisel A., Hochgerner H., Lönnerberg P., Johnsson A., Memic F., van der Zwan J., Häring M., Braun E., Borm L. E., La Manno G., Codeluppi S., Furlan A., Lee K., Skene N., Harris K. D., Hjerling-Leffler J., Arenas E., Ernfors P., Marklund U., Linnarsson S., Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I. W. H., Ng L. G., Ginhoux F., Newell E. W., Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Wang Y., Ulland T. K., Ulrich J. D., Song W., Tzaferis J. A., Hole J. T., Yuan P., Mahan T. E., Shi Y., Gilfillan S., Cella M., Grutzendler J., DeMattos R. B., Cirrito J. R., Holtzman D. M., Colonna M., TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eder C., Ion channels in monocytes and microglia/brain macrophages: Promising therapeutic targets for neurological diseases. J. Neuroimmunol. 224, 51–55 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Jung S., Aliberti J., Graemmel P., Sunshine M. J., Kreutzberg G. W., Sher A., Littman D. R., Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.S. Andrews, FastQC: A quality control tool for high throughput sequence data (2010); www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 41.Bonfert T., Kirner E., Csaba G., Zimmer R., Friedel C. C., ContextMap 2: Fast and accurate context-based RNA-seq mapping. BMC Bioinform. 16, 122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 46.Anders S., Reyes A., Huber W., Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emilsson V., Thorleifsson G., Zhang B., Leonardson A. S., Zink F., Zhu J., Carlson S., Helgason A., Walters G. B., Gunnarsdottir S., Mouy M., Steinthorsdottir V., Eiriksdottir G. H., Bjornsdottir G., Reynisdottir I., Gudbjartsson D., Helgadottir A., Jonasdottir A., Jonasdottir A., Styrkarsdottir U., Gretarsdottir S., Magnusson K. P., Stefansson H., Fossdal R., Kristjansson K., Gislason H. G., Stefansson T., Leifsson B. G., Thorsteinsdottir U., Lamb J. R., Gulcher J. R., Reitman M. L., Kong A., Schadt E. E., Stefansson K., Genetics of gene expression and its effect on disease. Nature 452, 423–428 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Labonté B., Engmann O., Purushothaman I., Menard C., Wang J., Tan C., Scarpa J. R., Moy G., Loh Y.-H. E., Cahill M., Lorsch Z. S., Hamilton P. J., Calipari E. S., Hodes G. E., Issler O., Kronman H., Pfau M., Obradovic A. L. J., Dong Y., Neve R. L., Russo S., Kazarskis A., Tamminga C., Mechawar N., Turecki G., Zhang B., Shen L., Nestler E. J., Sex-specific transcriptional signatures in human depression. Nat. Med. 23, 1102–1111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peters L. A., Perrigoue J., Mortha A., Iuga A., Song W.-m., Neiman E. M., Llewellyn S. R., Di Narzo A., Kidd B. A., Telesco S. E., Zhao Y., Stojmirovic A., Sendecki J., Shameer K., Miotto R., Losic B., Shah H., Lee E., Wang M., Faith J. J., Kasarskis A., Brodmerkel C., Curran M., Das A., Friedman J. R., Fukui Y., Humphrey M. B., Iritani B. M., Sibinga N., Tarrant T. K., Argmann C., Hao K., Roussos P., Zhu J., Zhang B., Dobrin R., Mayer L. F., Schadt E. E., A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat. Genet. 49, 1437–1449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang B., Gaiteri C., Bodea L.-G., Wang Z., McElwee J., Podtelezhnikov A. A., Zhang C., Xie T., Tran L., Dobrin R., Fluder E., Clurman B., Melquist S., Narayanan M., Suver C., Shah H., Mahajan M., Gillis T., Mysore J., MacDonald M. E., Lamb J. R., Bennett D. A., Molony C., Stone D. J., Gudnason V., Myers A. J., Schadt E. E., Neumann H., Zhu J., Emilsson V., Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang B., Horvath S., A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., Ma’ayan A., Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., Ma’ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Franzén O., Gan L.-M., Björkegren J. L. M., PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, baz046 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Street K., Risso D., Fletcher R. B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S., Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J., Zhou X., Zhu J., Gu Y., Zhao W., Zou J., Guo Z., GO-function: Deriving biologically relevant functions from statistically significant functions. Brief. Bioinform. 13, 216–227 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Neftel C., Laffy J., Filbin M. G., Hara T., Shore M. E., Rahme G. J., Richman A. R., Silverbush D., Shaw M. L., Hebert C. M., Dewitt J., Gritsch S., Perez E. M., Gonzalez Castro L. N., Lan X., Druck N., Rodman C., Dionne D., Kaplan A., Bertalan M. S., Small J., Pelton K., Becker S., Bonal D., Nguyen Q.-D., Servis R. L., Fung J. M., Mylvaganam R., Mayr L., Gojo J., Haberler C., Geyeregger R., Czech T., Slavc I., Nahed B. V., Curry W. T., Carter B. S., Wakimoto H., Brastianos P. K., Batchelor T. T., Stemmer-Rachamimov A., Martinez-Lage M., Frosch M. P., Stamenkovic I., Riggi N., Rheinbay E., Monje M., Rozenblatt-Rosen O., Cahill D. P., Patel A. P., Hunter T., Verma I. M., Ligon K. L., Louis D. N., Regev A., Bernstein B. E., Tirosh I., Suvà M. L., An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd8811/DC1