

Abstract
Cataracts have a high incidence and prevalence rate worldwide, and they are the leading cause of blindness. Lens epithelial cell (LEC) apoptosis is often analysed in cataract research since it is the pathological basis of cataracts, except for congenital cataract. Chloride channels are present in ocular tissues, such as in trabecular cells, LECs and other cells. They serve an important role in apoptosis and participate in endoplasmic reticulum (ER) stress and oxidative stress. However, their role in the apoptosis of LECs has not been discussed. The present study examined the effects of the chloride channel blocker 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) in human LECs (HLECs) to elucidate the role of NPPB in HLECs and investigate the role and mechanism of chloride channels in cataract formation. HLECs were exposed to NPPB. Cell survival rate was evaluated using Cell Counting Kit-8 assays. Oxidative stress was detected as reactive oxygen species (ROS) in cells by using a ROS assay kit. Apoptosis was examined by assessing mitochondrial membrane potential and using a JC-1 assay kit, and western blot analysis was performed to measure the expression levels of mitochondrial-dependent apoptosis pathway-associated proteins. ER stress was evaluated by determining the intracellular calcium ion fluorescence intensity, and western blot analysis was performed to measure ER stress-associated protein expression. The results revealed that NPPB treatment decreased the viability of HLECs and increased apoptosis. Additionally, NPPB increased intracellular ROS levels, as well as the number of JC-1 monomers and the protein expression levels of B-cell lymphoma-2 (Bcl-2)-associated X and cleaved caspase-3, and decreased Bcl-2 protein expression. NPPB increased intracellular calcium ions, the protein expression levels of activating transcription factor 6, JNK, C/EBP homologous protein and caspase-12, and the phosphorylation of protein kinase R-like endoplasmic reticulum kinase. N-acetylcysteine and 4-phenylbutyric acid inhibited NPPB-induced oxidative stress, ER stress and apoptosis. Therefore, NPPB treatment decreased cell viability and promoted apoptosis of HLECs via the promotion of oxidative and ER stress.
Keywords: 5-nitro-2-(3-phenylpropylamino) benzoic acid, cataract, human lens epithelial cells, apoptosis, endoplasmic reticulum stress, oxidative stress, chloride channel
Introduction
The blindness rate due to cataracts is decreasing every year; however, cataracts remain the leading cause of blindness worldwide, with a high prevalence rate from 1990 to 2010, especially in developing countries (1). Surgery is the only effective cure for cataracts (2). Although the cataract surgery rate is increasing, it needs more cost to achieve improved visual quality through the multifocal lens, and more ophthalmologists are required to perform cataract surgery (3-5). Studies are ongoing, but there are currently no drugs to treat or reverse the formation of cataracts. Therefore, it is urgent and necessary to study the pathogenesis of cataracts and seek effective preventive measures.
Human lens epithelial cells (HLECs) consist of a single layer of epithelial cells located on the anterior surface of the lens (6). HLECs are widely used to investigate the pathogenesis of cataracts (7,8) and were therefore used in the present study. Ion channels are involved in LEC processes, including proliferation, differentiation and apoptosis (9-11). Chloride channels have been widely studied in the apoptosis of various types of cells, such as cardiomyocytes and nasopharyngeal carcinoma cells (12,13). Chloride channels are associated with ER stress and oxidative stress. For example, chloride channels promote ER stress and induce apoptosis in cardiomyocytes and preadipocytes (14,15). Additionally, chloride channels promote oxidative stress and induce apoptosis in nasopharyngeal carcinoma cells and cardiomyocytes (12,16). However, to the best of our knowledge, there are no studies involving HLECs. Therefore, the role of chloride channels in LECs requires further study. Endoplasmic reticulum (ER) stress induces epithelial-to-mesenchymal transition, oxidative stress, apoptosis and autophagy in HLECs, and is an important mechanism of cataract formation (17-19). Additionally, oxidative stress is a known causative factor in cataract formation (20). On one hand, ER stressors, such as thapsigargin and tunicamycin, produce significant levels of ROS and promote apoptosis in colon carcinoma CT26, breast cancer MDA-MB468 cells and neuronal HT22 cells (21,22). On the other hand, ROS induces apoptosis by activating ER stress in acute myeloid leukemia cells (23). ER stress and oxidative stress interact with each other. However, whether chloride channels affect HLEC apoptosis and the corresponding mechanism have not been investigated. 5-Nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) is a non-specific chloride channel inhibitor, which was used in the present study to observe the effects and possible mechanisms of chloride channels in HLECs apoptosis.
Materials and methods
Materials
HLECs were purchased from the American Type Culture Collection. The following additional reagents were used in the present study: NPPB (Abmole Bioscience Inc.), DMSO, 4-phenylbutyric acid (4-PBA; Sigma-Aldrich; Merck KGaA), low-glucose DMEM (HyClone; Cytiva), FBS (Gibco; Thermo Fisher Scientific, Inc.), Cell Counting Kit-8 (CCK-8) assays (cat. no. C0040), RIPA lysis buffer (cat. no. P0013C), SDS-PAGE Sample Loading Buffer 5X (cat. no. P0015), Ca2+ indicator dye Fluo-4 AM kit (cat. no. S1060), reactive oxygen species (ROS) Assay kit (cat. no. S0033M), N-acetylcysteine (NAC; cat. no. ST1546-10g), enhanced bicinchoninic acid (BCA) protein assay kits (cat. no. P0009), JC-1 probes (cat. no. C2006) and antibodies against caspase-3 (1:1,000; cat. no. AC030) (all from Beyotime Institute of Biotechnology), primary antibodies against protein kinase R-like endoplasmic reticulum kinase (PERK; 1:2,000; cat. no. 33247), phosphorylated (p)-PERK (1:1,000; cat. no. 12814), B-cell lymphoma-2 (Bcl-2; 1:500; cat. no. 48496), Bcl-2-associated X (BAX; 1:1,000; cat. no. 48690), C/EBP homologous protein (CHOP; 1:1,000; cat. no. 49418), activating transcription factor 6 (ATF6; 1:1,000; cat. no. 32008), JNK (1:3,000; cat. no. 48615), caspase-12 (1:1,000; cat. no. 48277), β-actin (1:10,000; cat. no. 21338), HRP-conjugated goat anti-mouse IgG secondary antibody (1:10,000; cat. no. L3032) and HRP-conjugated goat anti-rabbit IgG secondary antibody (1:10,000; cat. no. L3042) (all from Signalway Antibody LLC), immobilon western chemiluminescent HRP substrate (cat. no. WBKLS0500; EMD Millipore).
Cell culture
HLECs are located in the inner surface of the lens capsule and are characterized by a flat and irregular polygonal-shaped morphology (6). HLECs were maintained and cultured in DMEM with 10% FBS in a humidified incubator with 5% CO2 at 37°C.
Cell viability assay
NPPB was dissolved in DMSO to form a 0.1 M stock solution. The solution was stored at <−20°C in the dark until use. NPPB was diluted to concentrations of 10-200 µM in serum-free DMEM on the day of the experiment. HLECs were transferred to 96-well plates (100 µl/well) at a density of 0.5×104 cells/well. Cells were incubated for 24 h and treated with 10, 50, 100 and 200 µM of NPPB for 24 h at 37°C. CCK-8 solution (10 µl) was added to each well for 30 min to 1 h at 37°C. The absorbance was expressed as the optical density and was measured at 450 nm using an Infinite M200 Pro microplate reader (Tecan Group, Ltd.).
NAC and 4-PBA with NPPB treatment protocol
NAC was dissolved in double-distilled H2O to form a 1 M stock solution. 4-PBA was dissolved in PBS to form a 500 mM stock solution. Cells were incubated for 24 h and were then treated with 50 µM NPPB with NAC (500 µM) or 4-PBA (20 µM) for 24 h at 37°C.
Intracellular reactive oxygen species (ROS) measurement
The intracellular ROS level was determined using a ROS assay kit and dichloro-dihydro-fluorescein diacetate (DCFH-DA). According to the manufacturer's protocol, the cells were cultured in 96-well plates for 24 h at 37°C and were then washed twice with serum-free medium. Medium containing 10 µM DCFH-DA was added. The cells were incubated in a cell incubator for 20 min at 37°C. Light was avoided during procedures and incubation. After incubation, the cells were washed three times with serum-free medium, then observed and photographed using a fluorescence microscope (magnification, ×400; Olympus Corporation). The fluorescence intensity was quantitatively measured using ImageJ software (v1.51; National Institutes of Health).
Mitochondrial membrane potential (ΔΨm) analysis
The JC-1 probe was used to measure apoptosis via mitochondrial depolarization in HLECs. Briefly, cells (80% confluent) were cultured in 96-well plates after treatment with 50 µM NPPB at 37°C for 24 h. The cells were washed three times with PBS, and 50 µl medium and 50 µl JC-1 working solution were added. The cells were incubated at 37°C for 20 min and washed three times with precooled JC-1 solution. The ΔΨm was determined by measuring the fluorescence intensity of red and green fluorescence using a fluorescence microscope (magnification, ×400). Green fluorescence indicated JC-1 monomers. JC-1 monomers appeared in the cytosol after mitochondrial membrane depolarization, which indicated early stage of apoptosis. Red fluorescence indicated JC-1 aggregation and was located on the mitochondria. Mitochondrial depolarization indicated apoptosis, which was reflected by an increase in the green/red fluorescence intensity ratio. The fluorescence intensity was measured using ImageJ software.
Ca2+ measurement
Cytosolic Ca2+ levels were measured using a calcium ion fluorescent probe (Fluo-4 AM) assay kit. Cells were cultured in 96-well plates after treatment with 50 µM NPPB for 24 h at 37°C. The cells were washed three times with PBS, and Fluo-4 AM (final concentration of 1 µM) was added for 1 h at 37°C and washed three times with PBS. To ensure that the Fluo-4 AM was completely converted into Fluo-4 in the cells, another 30-min incubation at 37°C was necessary after washing. The cells were observed and photographed under a fluorescence microscope (magnification, ×400) at 488 nm.
Western blot analysis
The cells were washed three times with PBS and scraped with a rubber spatula. Cells were collected and centrifuged at 157 × g for 5 min at 4°C. The cells were lysed in an appropriate amount of RIPA lysis buffer containing 1% phosphatase and protease inhibitors. The protein concentration was determined by BCA protein assay kit. A total of 20 µg protein was mixed with SDS-PAGE sample loading buffer. The samples were boiled for 5 min and kept at -20°C until use. The proteins were separated via 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes. The membrane was blocked with a blocking solution composed of 5% non-fat dry milk and TBS with 0.1% Tween-20 (TBS-T) for 1 h at 37°C, then washed in TBS-T three times for 5 min each. The membranes were incubated with primary antibodies against BAX, Bcl-2, caspase-3, ATF6, p-PERK, PERK, JNK, CHOP, caspase-12 and β-actin overnight at 4°C. The membranes were washed three times with TBS-T for 10 min each, and incubated with the corresponding biotinylated secondary antibodies for 1 h at 37°C. The membranes were washed three times with TBS-T. After coating with immobilon western chemiluminescent HRP substrate, the membranes were observed and photographed using a microscope equipped with a CCD camera (Tanon Science & Technology Co., Ltd.). The western blotting results were presented as the ratio of the fluorescence intensity of the target protein to β-actin or PERK (for p-PERK) calculated using ImageJ software.
Statistical analysis
All experiments were repeated 3 times independently. The data are expressed as the mean ± standard deviation. The data were analysed by unpaired Student's t-test and one-way ANOVA followed by Tukey's post-hoc test and Bonferroni's correction. SPSS statistic software 25.0 (IBM Corp.) was used to analyse the data. P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of NPPB on the viability of HLECs
Cells were treated with different concentrations of NPPB (0, 10, 50, 100 and 200 µM) for 24 h. Cell viability was determined via CCK-8 assays. Fig. 1 shows that NPPB significantly inhibited the viability of HLECs in a dose-dependent manner, with an IC50 value of 53.36±0.1% (P<0.01 vs. 0 µM). These results suggested that NPPB decreased cell survival rate and inhibited the viability of HLECs. A concentration of 50 µM NPPB was used in subsequent experiments.
Figure 1.
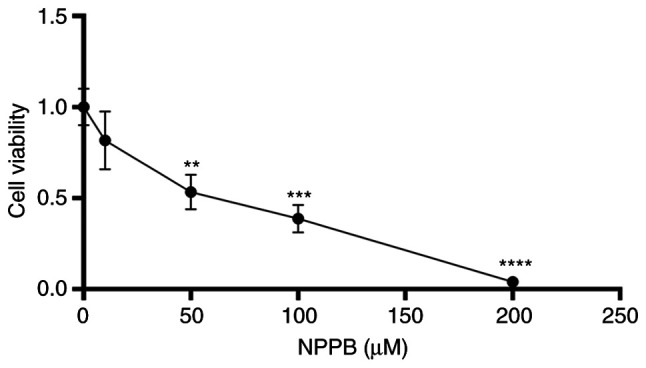
Effect of NPPB on HLEC viability. HLECs were incubated with the indicated concentrations of NPPB for 24 h, and cell viability was determined using Cell Counting Kit-8 assays. The results are presented as the mean ± standard deviation (n=3). **P<0.01; ***P<0.001; ****P<0.0001 vs. vehicle (0 µM). HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid.
Effect of NPPB on mitochondrial apoptosis of HLECs
Red fluorescence indicated that JC-1 accumulated in mitochondria and the cells were alive, while green fluorescence indicated apoptosis. Exposure of HLECs to NPPB (50 µM) for 24 h resulted in the dissipation of the ΔΨm. Green fluorescence increased after JC-1 staining, and the ratio of green to red fluorescence increased, indicating the toxicity of NPPB in mitochondria (Fig. 2). JC-1 aggregated in the mitochondria in the control group, with a ratio of 0.15±0.027, while the NPPB-treated group exhibited a significantly higher ratio (0.59±0.080) since the monomeric form of JC-1 was present in the cytosol. (P<0.001; Fig. 2). Additionally, the expression levels of apoptosis-associated proteins were detected by western blot analysis. NPPB-induced apoptosis was accompanied by a decrease in Bcl-2 expression (0.51±0.07 vs. 0.80±0.16; P<0.05) and a significant increase in BAX (0.92±0.19 vs. 0.57±0.08; P<0.05) and cleaved caspase-3 expression (1.77±0.47 vs. 0.49±0.21; P<0.05) compared with the vehicle group (Fig. 3).
Figure 2.
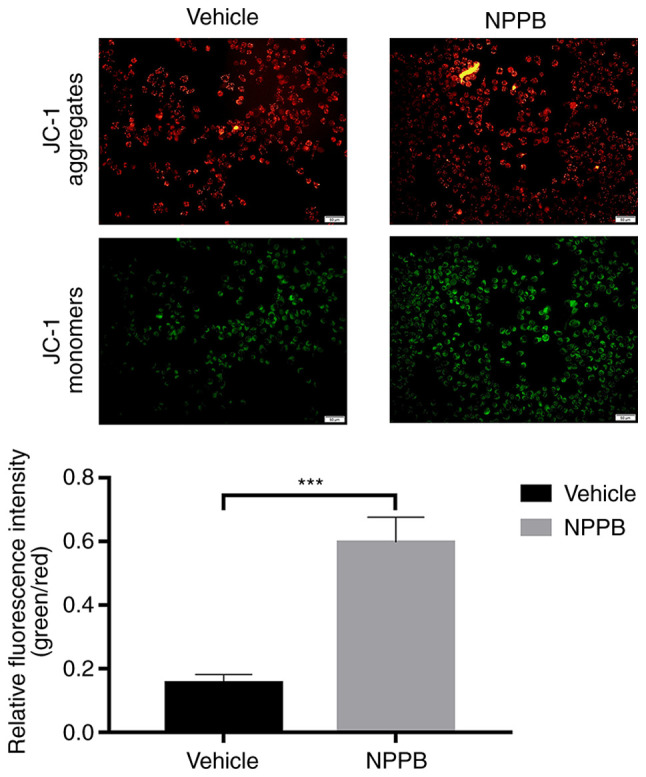
Effect of NPPB on mitochondrial apoptosis of HLECs. The ΔΨm of HLECs treated with 50 µM NPPB was evaluated by JC-1 assays, and the number of cells with low ΔΨm was quantified. The relative green/red fluorescence intensity was determined. Scale bar, 50 µm. The results are presented as the mean ± standard deviation (n=3). ***P<0.001. HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid; ΔΨm, mitochondrial membrane potential.
Figure 3.

Effect of NPPB on apoptosis in HLECs. HLECs were incubated with 50 µM NPPB for 24 h, and the expression levels of the apoptosis markers BAX, Bcl-2 and cleaved caspase-3 were determined by western blotting. β-actin was used as an internal control. The results are presented as the mean ± standard deviation (n=3). *P<0.05. HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid; Bcl-2, B-cell lymphoma-2; BAX, Bcl-2-associated X.
Effect of NPPB on ROS in HLECs
NPPB induced ROS generation in HLECs. NPPB (50 µM) significantly increased intracellular ROS levels (3.33±0.71 vs. 0.88±0.17; P<0.01) compared with the vehicle group (Fig. 4).
Figure 4.

Effect of NPPB on intracellular ROS production in HLECs. HLECs were treated with 50 µM NPPB for 24 h. ROS were stained with a dichloro-dihydro-fluorescein diacetate fluorescent probe and examined using ImageJ software. Scale bar, 50 µm. The results are presented as the mean ± standard deviation (n=3). **P<0.01. HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid; ROS, reactive oxygen species.
Effect of NPPB on ER stress and caspase-dependent apoptosis in HLECs
Mitochondria and the ER are major reservoirs of intracellular Ca2+. The imbalance of calcium ion is closely associated with mitochondrial apoptosis and ER stress (24). Therefore, whether NPPB induced apoptosis and ER stress by perturbing intracellular Ca2+ homeostasis was further explored. NPPB stimulation significantly increased Ca2+ levels in the cytosol in HLECs (7.0±0.53 vs. 0.46±0.09; P<0.0001) compared with the vehicle group (Fig. 5).
Figure 5.

Effect of NPPB on intracellular Ca2+ dysregulation in HLECs. HLECs were treated with the indicated concentrations of NPPB for 24 h, and cytosolic Ca2+ was measured with Fluo-4 AM, a Ca2+ indicator dye, and examined using ImageJ software. Scale bar, 50 µm. The results are presented as the mean ± standard deviation (n=3). ****P<0.0001. HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid.
The expression levels of ER stress-associated proteins were detected by western blot analysis, including p-PERK (1.37±0.03 vs. 1.01±0.16; P<0.05), ATF6 (1.09±0.14 vs. 0.86±0.03; P<0.05), JNK (1.13±0.28 vs. 0.63±0.03; P<0.05), CHOP (1.04±0.04 vs. 0.80±0.04; P<0.01) and caspase-12 (1.07±0.02 vs. 0.87±0.08; P<0.05) were upregulated following NPPB (50 µM) treatment compared with the vehicle group (Fig. 6). Quantification of the western blotting results indicated that NPPB significantly induced ER stress and the unfolded protein response (UPR) in HLECs.
Figure 6.

Effect of NPPB on ER stress-associated markers. ER stress markers and ER stress-associated caspase-dependent apoptosis markers ATF6, JNK, CHOP, caspase-12, PERK and p-PERK were measured via western blotting. β-actin was used as an internal control. The results are presented as the mean ± standard deviation (n=3). *P<0.05; **P<0.01. HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid; ER, endoplasmic reticulum; ATF6, activating transcription factor 6; CHOP, C/EBP homologous protein; p-PERK, phosphorylated protein kinase R-like endoplasmic reticulum kinase.
Effects of NAC and 4-PBA in NPPB-treated HLECs
The ROS scavenger NAC (500 µM) and the ER stress inhibitor 4-PBA (20 µM) were used to co-treat cells with 50 µM NPPB at 37°C for 24 h. NAC and 4-PBA significantly decreased the levels of the green-to-red fluorescence ratio in JC-1 staining compared with NPPB alone (NAC + NPPB, 4.53±0.70 and 4-PBA + NPPB, 3.47±0.25 vs. NPPB, 8.81±2.10; both P<0.01; Fig. 7A). Additionally, compared with NPPB alone, NAC and 4-PBA with NPPB decreased the expression levels of BAX (NAC + NPPB, 1.34±0.07 and 4-PBA + NPPB, 1.47±0.12 vs. NPPB, 1.75±0.11; P<0.01 and P<0.05, respectively) and cleaved caspase-3 (NAC+NPPB, 0.69±0.20 and 4-PBA + NPPB, 0.69±0.24 vs. NPPB, 1.77±0.47; both P<0.01) and increased the expression levels of Bcl-2 (NAC + NPPB, 1.16±0.11 and 4-PBA + NPPB, 1.14±0.13 vs. NPPB, 0.82±0.12; both P<0.05) compared with the NPPB group (Fig. 7B). Furthermore, NAC and 4-PBA significantly decreased NPPB-induced ROS (NAC + NPPB, 1.21±0.19 and 4-PBA + NPPB, 1.10±0.21 vs. NPPB, 12.32±1.62; both P<0.0001; Fig. 7C), Ca2+ levels (NAC + NPPB, 4.53±0.70 and 4-PBA + NPPB, 3.47±0.25 vs. NPPB, 8.81±2.10; both P<0.05; Fig. 7D) in the cytosol and the level of ER stress, including the levels of p-PERK (NAC + NPPB, 1.19±0.20 and 4-PBA + NPPB, 1.19±0.12 vs. NPPB, 2.00±0.41; both P<0.05), ATF6 (NAC + NPPB, 0.89±0.03 and 4-PBA+NPPB, 0.85±0.11 vs. NPPB, 1.70±0.07; both P<0.0001,), JNK (NAC + NPPB, 2.74±0.15 and 4-PBA + NPPB, 2.80±0.16 vs. NPPB, 3.50±0.17; both P<0.05), CHOP (NAC+NPPB, 1.85±0.13 and 4-PBA + NPPB, 1.72±0.04 vs. NPPB, 2.25±0.19; P<0.05 and P<0.01, respectively) and caspase-12 (NAC + NPPB, 1.91±0.06 and 4-PBA + NPPB, 1.75±0.07 vs. NPPB, 2.29±0.08; P<0.05 and P<0.01, respectively) (Fig. 7E). Therefore, NPPB activated ROS production and induced the UPR pathway, which likely promoted apoptosis.
Figure 7.

NAC and 4-PBA inhibit the effects of NPPB in HLECs. NPPB-treated cells were co-treated with NAC (500 µM) and 4-PBA (20 µM). (A) Relative green/red fluorescence intensity was determined using ImageJ software. (B) Expression levels of apoptosis markers BAX, Bcl-2 and cleaved caspase-3 were determined by western blotting. β-actin was used as an internal control. (C) Reactive oxygen species were stained with a dichloro-dihydro-fluorescein diacetate fluorescent probe and examined using ImageJ software. (D) Cytosolic Ca2+ was measured with Fluo-4 AM, a Ca2+ indicator dye, and examined using ImageJ software. (E) ER stress markers and ER stress-associated caspase-dependent apoptosis markers ATF6, JNK, CHOP, PERK and p-PERK were measured by western blotting. β-actin was used as an internal control. Scale bar, 50 µm. The results are presented as the mean ± standard deviation (n=3). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. HLEC, human lens epithelial cell; NPPB, 5-nitro-2-(3-phenylpropylamino) benzoic acid; Bcl-2, B-cell lymphoma-2; BAX, Bcl-2-associated X; ER, endoplasmic reticulum; ATF6, activating transcription factor 6; CHOP, C/EBP homologous protein; p-PERK, phosphorylated protein kinase R-like endoplasmic reticulum kinase; NAC, N-acetylcysteine; 4-PBA, 4-phenylbutyric acid.
Discussion
Investigation of LEC apoptosis is the main method to study the pathogenesis of cataracts (25). Chloride channels serve important roles in numerous cellular aspects, especially apoptosis (26-29). The non-specific inhibitor of chloride channels NPPB is commonly used to inhibit the chloride current and to study the function of voltage-gated chloride channels (30). Voltage-gated chloride channels are involved in the occurrence of some diseases, such as cardiovascular and nervous system diseases and nasopharyngeal carcinoma (29,31,32). However, their role in cataracts has not been widely discussed. Therefore, NPPB was used in the present study to investigate whether chloride channels were involved in the pathogenesis of cataracts. If NPPB was associated with the apoptosis of LECs, further studies may examine which voltage-gated chloride channels may be involved in the formation of cataracts. NPPB is widely used to study apoptosis mechanisms (30,33,34). Souktani et al (35) demonstrated that NPPB promoted cardiomyocyte apoptosis. However, Malekova et al (30) indicated that NPPB served a protective role in H2O2-induced cardiomyocyte apoptosis. To the best of our knowledge, the mechanism of chloride channel mediation of LEC apoptosis has not been previously studied. Therefore, NPPB was used in the present study to investigate the role of chloride channels in LEC apoptosis. The current results revealed that NPPB inhibited cell viability in a concentration- dependent manner. The results of CCK-8 assays indicated that the IC50 of NPPB in LECs at 24 h was ~50 µM. Therefore, a concentration of 50 µM was used in subsequent experiments. NPPB increased the number of JC-1 monomers and the expression levels of BAX and cleaved caspase-3, and decreased Bcl-2 expression. Therefore, it was concluded that NPPB induced HLEC apoptosis via a mitochondrial-dependent pathway. Similar results were obtained in basilar artery smooth muscle cells and human bronchial epithelium (36,37). The effects of chloride channels on apoptosis are distinct in different cell types. Chloride channels have been shown to be cardioprotective in ischemic preconditioning of isolated hearts; however, the inhibitor of chloride channels NPPB prevented the appearance of H2O2-induced apoptosis of pheochromocytoma cells (38,39). Chloride channel activation in intestinal epithelial cells leads to apoptosis via activation of caspase 3 (40). The reason for the different effects may be associated with the various experimental conditions and models used.
Cataract pathogenesis is closely associated with oxidative stress (41). To the best of our knowledge, the present study revealed for the first time that NPPB induced oxidative stress in LECs and increased the production of intracellular ROS, and that NAC inhibited NPPB-induced production of ROS. ROS promote apoptosis in a variety of ways, such as via ER stress, PI3K/AKT signalling, the Foxo3a/TRIM69/p53 regulatory network and the calmodulin-like-protein 3-dependent JNK1/2 and ERK1/2 signalling pathways (17,42-44). In the present study, 4-PBA also inhibited the production of NPPB-induced ROS, suggesting that oxidative stress and ER stress interacted and influenced each other.
The ER is a central organelle used in a series of important biological processes to maintain the stability of the intracellular environment. The present study revealed that NPPB increased intracellular Ca2+ and the levels of ER stress-associated proteins, such as p-PERK, PERK, ATF6, JNK and CHOP, which indicated that NPPB significantly induced ER stress in HLECs. Additionally, NPPB increased the expression levels of caspase-12, which indicated that NPPB significantly induced apoptosis via ER stress and the mitochondrial pathway. NAC and 4-PBA decreased intracellular Ca2+ and the expression levels of ER stress-associated proteins, therefore attenuating NPPB-induced ER stress in HLECs. The homeostasis of Ca2+ in HLECs is very important since its balance maintains clarity of the lens (45). When ER stress occurs, the ER releases Ca2+, increasing the cytosolic Ca2+ levels (46). Ca2+ dysregulation further promotes apoptotic cell death (47,48). Therefore, chloride channels serve important roles in maintaining lens transparency, inhibiting ER stress and protecting lens cells.
In conclusion, the present study demonstrated that NPPB induced HLEC apoptosis. NPPB inhibited cell viability and induced ROS, ER stress and apoptosis. The current findings provide strong evidence that chloride channels may serve an important role in the pathogenesis of cataracts.
Acknowledgments
Not applicable.
Funding Statement
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
All authors designed the present study. LN, JZ, KL and YW performed the experiments. LN, XL, JZ and YZ confirmed the authenticity of the data. LN, YW, XL, KL and YL analysed the data and prepared the figures. LN, XL, JZ and YS drafted the initial manuscript. XL, JZ and YZ reviewed and revised the manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Referencves
- 1.Khairallah M, Kahloun R, Bourne R, Limburg H, Flaxman SR, Jonas JB, Keeffe J, Leasher J, Naidoo K, Pesudovs K, et al. Number of people blind or visually impaired by cataract worldwide and in world regions, 1990 to 2010. Invest Ophthalmol Vis Sci. 2015;56:6762–6769. doi: 10.1167/iovs.15-17201. [DOI] [PubMed] [Google Scholar]
- 2.Ibrahim N, Pozo-Martin F, Gilbert C. Direct non-medical costs double the total direct costs to patients undergoing cataract surgery in Zamfara state, Northern Nigeria: A case series. BMC Health Serv Res. 2015;15:163. doi: 10.1186/s12913-015-0831-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundström M, Goh PP, Henry Y, Salowi MA, Barry P, Manning S, Rosen P, Stenevi U. The changing pattern of cataract surgery indications: A 5-year study of 2 cataract surgery databases. Ophthalmology. 2015;122:31–38. doi: 10.1016/j.ophtha.2014.07.047. [DOI] [PubMed] [Google Scholar]
- 4.Lin JC, Yang MC. Cost-effectiveness comparison between monofocal and multifocal intraocular lens implantation for cataract patients in Taiwan. Clin Ther. 2014;36:1422–1430. doi: 10.1016/j.clinthera.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Resnikoff S, Felch W, Gauthier TM, Spivey B. The number of ophthalmologists in practice and training worldwide: A growing gap despite more than 200,000 practitioners. Br J Ophthalmol. 2012;96:783–787. doi: 10.1136/bjophthalmol-2011-301378. [DOI] [PubMed] [Google Scholar]
- 6.Rabsilber TM, Auffarth GU. Lens epithelial cells. In: Schmidt-Erfurth U, Kohnen T, editors. Encyclopedia of Ophthalmology. Springer; Berlin, Heidelberg: 2016. pp. 1–2. [Google Scholar]
- 7.Lu B, Christensen IT, Yu T, Wang C, Yan Q, Wang X. SUMOylation evoked by oxidative stress reduced lens epithelial cell antioxidant functions by increasing the stability and transcription of TP53INP1 in age-related cataracts. Oxid Med Cell Longev. 2019;2019:7898069. doi: 10.1155/2019/7898069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong Y, Sun Y, Rong X, Li D, Lu Y, Ji Y. Exosomes from adipose-derived stem cells attenuate UVB-induced apoptosis, ROS, and the Ca2+ level in HLEC cells. Exp Cell Res. 2020;396:112321. doi: 10.1016/j.yexcr.2020.112321. [DOI] [PubMed] [Google Scholar]
- 9.Nebe B, Kunz F, Peters A, Rychly J, Noack T, Beck R. Induction of apoptosis by the calcium antagonist mibefradil correlates with depolarization of the membrane potential and decreased integrin expression in human lens epithelial cells. Graefes Arch Clin Exp Ophthalmol. 2004;242:597–604. doi: 10.1007/s00417-004-0886-y. [DOI] [PubMed] [Google Scholar]
- 10.Meissner A, Noack T. Proliferation of human lens epithelial cells (HLE-B3) is inhibited by blocking of voltage-gated calcium channels. Pflugers Arch. 2008;457:47–59. doi: 10.1007/s00424-008-0514-5. [DOI] [PubMed] [Google Scholar]
- 11.Chimote AA, Adragna NC, Lauf PK. Ion transport in a human lens epithelial cell line exposed to hyposmotic and apoptotic stress. J Cell Physiol. 2010;223:110–122. doi: 10.1002/jcp.22015. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Shen M, Guo X, Wang B, Xia Y, Wang N, Zhang Q, Jia L, Wang X. Volume-sensitive outwardly rectifying chloride channel blockers protect against high glucose-induced apoptosis of cardiomyocytes via autophagy activation. Sci Rep. 2017;7:44265. doi: 10.1038/srep44265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou C, Tang X, Xu J, Wang J, Yang Y, Chen Y, Chen L, Wang L, Zhu L, Yang H. Opening of the CLC-3 chloride channel induced by dihydroartemisinin contributed to early apoptotic events in human poorly differentiated nasopharyngeal carcinoma cells. J Cell Biochem. 2018;119:9560–9572. doi: 10.1002/jcb.27274. [DOI] [PubMed] [Google Scholar]
- 14.Shen M, Wang L, Wang B, Wang T, Yang G, Shen L, Wang T, Guo X, Liu Y, Xia Y, et al. Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: Involvement of CHOP through Wnt. Cell Death Dis. 2014;5:e1528. doi: 10.1038/cddis.2014.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang YY, Huang XQ, Zhao LY, Sun FY, Chen WL, Du JY, Yuan F, Li J, Huang XL, Liu J, et al. ClC-3 deficiency protects preadipocytes against apoptosis induced by palmitate in vitro and in type 2 diabetes mice. Apoptosis. 2014;19:1559–1570. doi: 10.1007/s10495-014-1021-0. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Gao H, Yang X, Liang X, Tan Q, Chen Z, Zhao C, Gu Z, Yu M, Zheng Y, et al. The apoptotic effect of Zoledronic acid on the nasopharyngeal carcinoma cells via ROS mediated chloride channel activation. Clin Exp Pharmacol Physiol. 2018;45:1019–1027. doi: 10.1111/1440-1681.12979. [DOI] [PubMed] [Google Scholar]
- 17.Zhou S, Yang J, Wang M, Zheng D, Liu Y. Endoplasmic reticulum stress regulates epithelial-mesenchymal transition in human lens epithelial cells. Mol Med Rep. 2020;21:173–180. doi: 10.3892/mmr.2019.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma TJ, Lan DH, He SZ, Ye Z, Li P, Zhai W, Chen WQ, Huang Y, Fu Y, Sun A, et al. Nrf2 protects human lens epithelial cells against H2O2-induced oxidative and ER stress: The ATF4 may be involved. Exp Eye Res. 2018;169:28–37. doi: 10.1016/j.exer.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Smith AJ, Ball SS, Bowater RP, Wang N, Michael Wormstone I. Sulforaphane promotes ER stress, autophagy, and cell death: Implications for cataract surgery. J Mol Med (Berl) 2017;95:553–564. doi: 10.1007/s00109-016-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braakhuis AJ, Donaldson CI, Lim JC, Donaldson PJ. Nutritional strategies to prevent lens cataract: Current status and future strategies. Nutrients. 2019;11:1186. doi: 10.3390/nu11051186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880–1891. doi: 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song Q, Gou WL, Zhang R. FAM3A attenuates ER stress-induced mitochondrial dysfunction and apoptosis via CHOP-Wnt pathway. Neurochem Int. 2016;94:82–89. doi: 10.1016/j.neuint.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Wang G, Wu W, Yao S, Han X, He D, He J, Zheng G, Zhao Y, Cai Z, Yu R. Camalexin induces apoptosis via the ROS-ER stress-mitochondrial apoptosis pathway in AML cells. Oxid Med Cell Longev. 2018;2018:7426950. doi: 10.1155/2018/7426950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Wang C, Meng Q, Liu Z, Huo X, Sun P, Sun H, Ma X, Peng J, Liu K. Targeting P-glycoprotein and SORCIN: Dihydromyricetin strengthens anti-proliferative efficiency of adriamycin via MAPK/ERK and Ca2+-mediated apoptosis pathways in MCF-7/ADR and K562/ADR. J Cell Physiol. 2018;233:3066–3079. doi: 10.1002/jcp.26087. [DOI] [PubMed] [Google Scholar]
- 25.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/MCB.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng JW, Wang XG, Ma MM, Lv XF, Liu J, Zhou JG, Guan YY. Integrin β3 mediates cerebrovascular remodelling through Src/ClC-3 volume-regulated Cl(-) channel signalling pathway. Br J Pharmacol. 2014;171:3158–3170. doi: 10.1111/bph.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang W, Huang L, Zhao D, He JZ, Sharma P, Liu J, Gramolini AO, Ward ME, Cho HC, Backx PH. Swelling-activated Cl- currents and intracellular CLC-3 are involved in proliferation of human pulmonary artery smooth muscle cells. J Hypertens. 2014;32:318–330. doi: 10.1097/HJH.0000000000000013. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Li H, Yang L, Deng Z, Luo H, Ye D, Bai Z, Zhu L, Ye W, Wang L, Chen L. The ClC-3 chloride channel associated with microtubules is a target of paclitaxel in its induced-apoptosis. Sci Rep. 2013;3:2615. doi: 10.1038/srep02615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Zhang S, Fan H, Zhong Z, Li X, Jin X, Chang Q. ClC-3 chloride channel in hippocampal neuronal apoptosis. Neural Regen Res. 2013;8:3047–3054. doi: 10.3969/j.issn.1673-5374.2013.32.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malekova L, Tomaskova J, Novakova M, Stefanik P, Kopacek J, Lakatos B, Pastorekova S, Krizanova O, Breier A, Ondrias K. Inhibitory effect of DIDS, NPPB, and phloretin on intracellular chloride channels. Pflugers Arch. 2007;455:349–357. doi: 10.1007/s00424-007-0300-9. [DOI] [PubMed] [Google Scholar]
- 31.Ganapathi SB, Wei SG, Zaremba A, Lamb FS, Shears SB. Functional regulation of ClC-3 in the migration of vascular smooth muscle cells. Hypertension. 2013;61:174–179. doi: 10.1161/HYPERTENSIONAHA.112.194209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Ma W, Zhu L, Ye D, Li Y, Liu S, Li H, Zuo W, Li B, Ye W, Chen L. ClC-3 is a candidate of the channel proteins mediating acid-activated chloride currents in nasopharyngeal carcinoma cells. Am J Physiol Cell Physiol. 2012;303:C14–C23. doi: 10.1152/ajpcell.00145.2011. [DOI] [PubMed] [Google Scholar]
- 33.Sun L, Dong Y, Zhao J, Yin Y, Tong B, Zheng Y, Xin H. NPPB modulates apoptosis, proliferation, migration and extra- cellular matrix synthesis of conjunctival fibroblasts by inhibiting PI3K/AKT signaling. Int J Mol Med. 2018;41:1331–1338. doi: 10.3892/ijmm.2017.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myssina S, Lang PA, Kempe DS, Kaiser S, Huber SM, Wieder T, Lang F. Cl- channel blockers NPPB and niflumic acid blunt Ca(2+)-induced erythrocyte 'apoptosis'. Cell Physiol Biochem. 2004;14:241–248. doi: 10.1159/000080333. [DOI] [PubMed] [Google Scholar]
- 35.Souktani R, Ghaleh B, Tissier R, d'Anglemont de Tassigny A, Aouam K, Bedossa P, Charlemagne D, Samuel J, Henry P, Berdeaux A. Inhibitors of swelling-activated chloride channels increase infarct size and apoptosis in rabbit myocardium. Fundam Clin Pharmacol. 2003;17:555–561. doi: 10.1046/j.1472-8206.2003.00175.x. [DOI] [PubMed] [Google Scholar]
- 36.Qian Y, Du YH, Tang YB, Lv XF, Liu J, Zhou JG, Guan YY. ClC-3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis. 2011;16:468–477. doi: 10.1007/s10495-011-0584-2. [DOI] [PubMed] [Google Scholar]
- 37.Cheng G, Shao Z, Chaudhari B, Agrawal DK. Involvement of chloride channels in TGF-beta1-induced apoptosis of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1339–L1347. doi: 10.1152/ajplung.00121.2007. [DOI] [PubMed] [Google Scholar]
- 38.Bozeat ND, Xiang SY, Ye LL, Yao TY, Duan ML, Burkin DJ, Lamb FS, Duan DD. Activation of volume regulated chloride channels protects myocardium from ischemia/reperfusion damage in second-window ischemic preconditioning. Cell Physiol Biochem. 2011;28:1265–1278. doi: 10.1159/000335858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuo W, Zhu L, Bai Z, Zhang H, Mao J, Chen L, Wang L. Chloride channels involve in hydrogen peroxide-induced apoptosis of PC12 cells. Biochem Biophys Res Commun. 2009;387:666–670. doi: 10.1016/j.bbrc.2009.07.075. [DOI] [PubMed] [Google Scholar]
- 40.Claud EC, Lu J, Wang XQ, Abe M, Petrof EO, Sun J, Nelson DJ, Marks J, Jilling T. Platelet-activating factor-induced chloride channel activation is associated with intracellular acidosis and apoptosis of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1191–G1200. doi: 10.1152/ajpgi.00318.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Periyasamy P, Shinohara T. Age-related cataracts: Role of unfolded protein response, Ca2+ mobilization, epigenetic DNA modifications, and loss of Nrf2/Keap1 dependent cytoprotection. Prog Retin Eye Res. 2017;60:1–19. doi: 10.1016/j.preteyeres.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Li H, Liu Y. MicroRNA-378a regulates the reactive oxygen species (ROS)/phosphatidylinositol 3-kinases (PI3K)/AKT signaling pathway in human lens epithelial cells and cataract. Med Sci Monit. 2019;25:4314–4321. doi: 10.12659/MSM.916881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rong X, Rao J, Li D, Jing Q, Lu Y, Ji Y. TRIM69 inhibits cataractogenesis by negatively regulating p53. Redox Biol. 2019;22:101157. doi: 10.1016/j.redox.2019.101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia Y, Qin Q, Fang CP, Shen W, Sun TT, Huang YL, Li WJ, Deng AM. UVB induces apoptosis via downregulation of CALML3-dependent JNK1/2 and ERK1/2 pathways in cataract. Int J Mol Med. 2018;41:3041–3050. doi: 10.3892/ijmm.2018.3478. [DOI] [PubMed] [Google Scholar]
- 45.Duncan G, Jacob TJ. Calcium and the physiology of cataract. Ciba Found Symp. 1984;106:132–152. doi: 10.1002/9780470720875.ch8. [DOI] [PubMed] [Google Scholar]
- 46.Hofer AM, Curci S, Machen TE, Schulz I. ATP regulates calcium leak from agonist-sensitive internal calcium stores. FASEB J. 1996;10:302–308. doi: 10.1096/fasebj.10.2.8641563. [DOI] [PubMed] [Google Scholar]
- 47.Bahar E, Kim H, Yoon H. ER Stress-mediated signaling: Action potential and Ca(2+) as key players. Int J Mol Sci. 2016;17:1558. doi: 10.3390/ijms17091558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krebs J, Agellon LB, Michalak M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem Biophys Res Commun. 2015;460:114–121. doi: 10.1016/j.bbrc.2015.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.