

Abstract
Significance: Hypertension has major health consequences, which is associated with endothelial dysfunction. Endothelial nitric oxide synthase (eNOS)-produced nitric oxide (NO) signaling in the vasculature plays an important role in maintaining vascular homeostasis. Considering the importance of NO system, this review aims to provide a brief overview of the biochemistry of members of NO signaling, including GTPCH1 [guanosine 5′-triphosphate (GTP) cyclohydrolase 1], tetrahydrobiopterin (BH4), and eNOS.
Recent Advances: Being NO signaling activators and regulators of eNOS signaling, BH4 treatment is getting widespread attention either as potential therapeutic agents or as preventive agents. Recent clinical trials also support that BH4 treatment could be considered a promising therapeutic in hypertension.
Critical Issues: Under conditions of BH4 depletion, eNOS-generated superoxides trigger pathological events. Abnormalities in NO availability and BH4 deficiency lead to disturbed redox regulation causing pathological events. This disturbed signaling influences the development of systemic hypertension as well as pulmonary hypertension.
Future Directions: Considering the importance of BH4 and NO to improve the translational significance, it is essential to continue research on this field to manipulate BH4 to increase the efficacy for treating hypertension. Thus, this review also examines the current state of knowledge on the effects of eNOS activators on preclinical models and humans to utilize this information for potential therapy.
Keywords: hypertension, eNOS uncoupling, GTPCH1, endothelial nitric oxide synthase
Background
High blood pressure (BP) epidemic is a major public health concern and most patients will require pharmacologic interventions to control their BP. The role of nitric oxide (NO) has its importance in keeping up vascular health as well as for the control of BP. The very important members of the NO system include guanosine 5′-triphosphate (GTP)-cyclohydrolase 1 (GTPCH1) and nitric oxide synthases (NOS), mainly endothelial NOS (eNOS) and some essential cofactors: tetrahydrobiopterin (BH4), calmodulin, heme, flavins, and NADPH. Thus, BP risk management by improving endothelial bioavailability of NO had emerged as a major research goal by many research groups. However, the importance of NO had been highlighted by many key observations in animal models and humans in the past several decades; a lack of understanding still exists as a gap that is supposed to be filled up. This is evidenced by a few clinical trials that were done before to improve the NO bioavailability to control BP. Although these trails were ideally expected to have a positive effect on controlling BP, it ended up with less clinical significance. Therefore, compiling conclusive evidence on experimental, preclinical, and clinical reports on NO signaling and its importance in BP homeostasis is important.
The review is organized as follows. It starts with describing the relationship between the three important members of the NO system, including eNOS, BH4, GTPCH1 and the damping factors of NO bioavailability, mainly, eNOS uncoupling and oxidative stress. Furthermore, the review briefly describes the potential NO signaling modulators and their clinical perspectives.
Hypertension Epidemiology
Hypertension, otherwise known as a condition of elevated BP—is a serious medical condition that significantly increases the risks of myocardial infarction, stroke, cardiac failure, and renal failure if not controlled (45). An estimated 1.13 billion people worldwide have hypertension (120), and it is one of the leading risk factors, reason for all-cause mortality that kills 9 million people each year. As two-thirds of the people with hypertension are living in low- and middle-income countries (120), hypertension could be considered a socioeconomic burden for these countries. The trend of the highest BP level during these past four decades has shifted from high-income countries to low-income countries. Contrastingly, BP has been persistently high in Central and Eastern Europe (23, 76), supporting increased death rates from stroke (98). Given its high burden and the aging of the population, hypertension remains an issue of global concern (12). However, many impressive progressions were made over several decades to improve hypertension detection and treatment, still the strategy to develop primary prevention is a subject of growing interest (67).
Nitric Oxide
Free radical NO is an unorthodox messenger molecule that is gaseous and lipophilic. It is one of the most important signaling molecules in maintaining vascular homeostasis. NO is synthesized from the oxidation and catalytic conversion of l-arginine to citrulline in the presence of molecular oxygen and different cofactors such as BH4, NADPH, flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD). Its concentration within the biological system is regulated by the activity of a family of NOS enzymes (90, 92). Besides, NOS isoform NO can also be produced by xanthine oxidoreductase or cytochrome P450 reductase. Besides, NO can be released nonenzymatically from iron-nitrosyl hemoglobin, S-nitrosylated blood proteins (21). Through its cyclic guanosine monophosphate (cGMP)-dependent mechanisms, NO exerts a wide variety of biological functions but not limited to maintaining homeostasis, including modulation of vascular tone, vasodilation, vascular permeability, antiplatelet, antithrombotic, and anti-inflammatory properties within the vasculature (15). Furthermore, NO produced by eNOS in the endothelium diffuses to vascular smooth muscle cells and activates the soluble guanylyl cyclase-cGMP-dependent protein kinase pathway, eventually leading to vasodilation (81).
NO is generated by NOS in nearly all types of mammalian cells. Chemically, NO is very unstable and is highly reactive with reactive oxygen species (ROS) such as superoxide anions (O2·−). Thus, the amount of NO is not only dependent upon its production by NOS but also by the rate of its inactivation by ROS, including O2·−. NO also undergoes rapid radical/radical or oxidation reactions to produce a plethora of biologically active derivatives. The other free radicals produced by various reactions with free NO are collectively called as reactive nitrogen species (RNS), which also play crucial roles in the maintenance of physiological homeostasis. RNS include NO radicals and related species, including nitric oxide (NO·), nitrogen dioxide (NO2·), nitrosyl (NO+), nitroxide (NO−), nitrous acid (HNO2), dinitrogen trioxide (N2O3), nitronium ion (NO2+), peroxynitrite (ONOO−), nitroso persulfide (SSNO−), and alkyl peroxynitrites (RONOO) (92). These RNS can further produce nitrosothiols, nitrosamine, nitro fatty acids, and so on (66).
eNOS regulation, activation, and eNOS uncoupling
Physiologically important NO is produced by eNOS, which is an important regulator of vascular function. The human eNOS gene is located on chromosome 7q35–36, consisting of 26 exons spanning ∼21 kb with multiple polymorphisms that might confer risk for hypertension. An association between eNOS polymorphism and reduced eNOS expression and activity, as well as increased oxidative stress in the vascular endothelium, has been reported by many research groups (60).
Many kinases were reported to regulate eNOS phosphorylation and NO production, under various stimuli. These kinases include Akt/PKB, cAMP-dependent protein kinase (PKA), and the AMP-activated protein kinase (AMPK). Importantly, AMPK, Akt, ERK1/2, and Ca2+/calmodulin-dependent protein kinase II (CaMK-II) activated Ser1177 phosphorylation, enhancing the specific activity of eNOS, so that NO production is boosted in the absence of a maintained increase in intracellular Ca2+. The other known eNOS phosphorylation that results in activation of eNOS is Ser615 and 633, and the inhibitory phosphorylation is Thr-495. Under the stimulation of vascular endothelial growth factor (VEGF) in endothelial cells, Ser1177 phosphorylation is activated, while Thr-495 phosphorylation is transiently reversed (Fig. 1) (72).
FIG. 1.

Mechanisms by which eNOS activity is regulated in endothelial cells. eNOS is regulated by PKC (phosphorylates at S116); PKC-α (phosphorylates at T495); PKA (phosphorylates at S635); and AMPK, PI3K/AKT (phosphorylates at S1177). eNOS is an obligate homodimer and this dimeric association is mediated by a cysteine-complexed Zn2+ (zinc-tetrathiolate) at the dimer interface. AMPK, 5′ adenosine monophosphate-activated protein kinase; BH2, dihydrobiopterin; BH4, tetrahydrobiopterin; DHFR, dihydrofolate reductase; eNOS, endothelial nitric oxide synthase; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; NO, nitric oxide. Color images are available online.
The promoter region in the eNOS gene encodes transcription of eNOS protein, which plays a critical role in the regulation of eNOS expression. The human NOS3 promoter contains two regulatory regions, positive regulatory domains I and II. A positive regulatory domain I links to a high-affinity Sp1 transcription factor recognition site and binds to three nucleoproteins identified as Sp1 and two variants of Sp3. The positive regulatory domain II forms nucleoprotein complexes with the positively regulating transcription factors Ets-1, Elf-1, YY1, and Sp1, and the inhibitory factor MYC-associated zinc finger protein (58, 87).
The catalytic mechanisms of NOS involve flavin-mediated electron transport from the C-terminal-bound NADPH to the N-terminal heme center where oxygen is reduced and incorporated into the guanidine group of l-arginine giving rise to NO and l-citrulline. Thus, NOS activity requires FMN, FAD, BH4, Ca2+-calmodulin, and heme (37). Another critical determinant of eNOS activity is the availability of BH4 (3). eNOS must be fully saturated with BH4 to completely couple NADPH oxidation to NO production (Fig. 1). Under conditions of limited BH4 availability, eNOS functions in an “uncoupled” state in which NAD(P)H-derived electrons are added to molecular oxygen rather than l-arginine, thereby resulting in the formation of O2·−, instead of NO (89, 91). Therefore, its chemical environment (i.e., presence of superoxide) determines whether NO exerts protective or harmful effects by producing ROS or RNS. In this setting, uncoupled eNOS exacerbates oxidative stress that is initiated by other ROS-generating enzymes (e.g., NADPH oxidase, NOX). This phenomenon is referred to as NOS “uncoupling” and was first demonstrated in purified neuronal NOS (nNOS) (51, 88), and also extended to eNOS (121, 125). eNOS uncoupling requires Ca2+/calmodulin and heme and will be blocked by the specific NOS inhibitor N-nitro-l-arginine methyl ester (L-NAME) (121). ONOO− directly uncouples eNOS by oxidizing and releasing zinc from the zinc/thiolate cluster of eNOS and presumably forming disulfide bonds between the monomers (129).
L-NAME inhibits ONOO−-mediated eNOS uncoupling. In addition, myeloperoxidase-derived hypochlorous acid (HOCl) also uncouples eNOS via ONOO−-mediated oxidation of the zinc/thiolate center and zinc release (Fig. 2) (125). Thus, this transformation of eNOS makes it from a protective enzyme to a contributor to oxidative stress. Other biochemical mechanisms proposed to be involved in eNOS uncoupling are the increase of endogenous asymmetric dimethylarginine, l-arginine deficiency and oxidative stress (79). Besides, eNOS is regulated by a complex pattern of post-translational modifications. These modifications include S-nitrosation, glutathionylation, and persulfidation, which were reported to impact eNOS activity. In general, exogenous NO causes inhibition of eNOS activity in vascular endothelial cells. This is because exposure of NO free radicals or NO donor causes S-nitrosation of eNOS and diminishes its activity (93). The exogenous NO also can decrease NOS activity along with decreasing of eNOS dimer levels. Whereas the presence of the thioredoxin and thioredoxin reductase system could prevent eNOS monomerization and loss of activity, indicating the inhibitory action of NO by disturbing the zinc tetrathiolate cluster at the dimeric interface through S-nitrosylation of the cysteine residues (93). Another study showed that in resting endothelial cells, eNOS is tonically S-nitrosylated and that the enzyme undergoes rapid transient denitrosylation after the addition of the eNOS agonist, VEGF (34). The receptor-mediated decrease in eNOS S-nitrosylation is inversely related to enzyme phosphorylation at Ser1179 (34). Another protein modification that can diminish the production of NO and enhance the superoxide generation is glutathionylation (20). Two highly conserved cysteine residues were identified as sites of S-glutathionylation in the reductase domain of eNOS and found to be critical for redox regulation of eNOS function. Protein thiols can undergo S-glutathionylation, which is considered to be a reversible protein modification that involves cellular signaling and adaptation. In eNOS, this S-glutathionylation of eNOS reversibly decreases NOS activity with an increase in O2·− and provides a pivotal switch providing redox regulation of cellular signaling, endothelial function, and vascular tone (20). DNA methylation in the eNOS promoter region is also involved in the cell-specific expression of the human eNOS gene (18), which allows the eNOS to be specifically expressed on endothelial cells. This methylation pattern exhibits a dramatic difference between endothelial and nonendothelial cell types, including vascular smooth muscle cells. Methylation exhibits a marked decrease in the synergistic action of Sp1, Sp3, and Ets1 on eNOS promoter activity and the addition of methyl-CpG-binding protein 2 further reduced the transcriptional activity of methylated eNOS constructs (18).
FIG. 2.
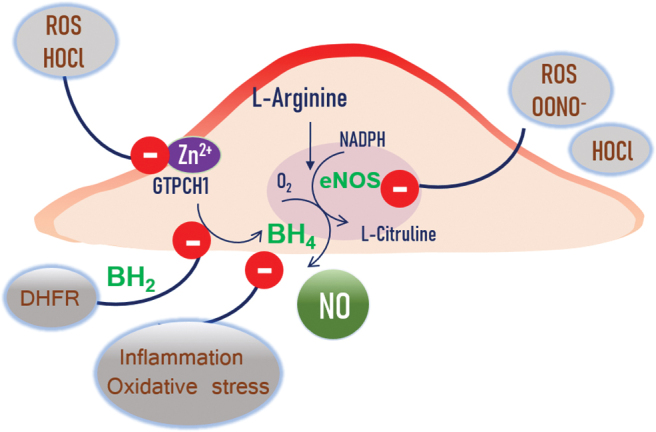
Inhibitory factors of NO. production. DHFR recycles the BH4 to BH2 and further inhibits the eNOS activity. ROS and HOCl target the GTPCH1 enzyme and release the Zn ion, and the BH4 production is inhibited. Inflammation, oxidative stress, peroxynitrite, and HOCl all can target eNOS and reduce the NO. production. GTPCH1, guanosine 5′-triphosphate (GTP)-cyclohydrolase 1; HOCl, hypochlorous acid; O2·−, superoxide; ONOO−, peroxynitrite; ROS, reactive oxygen species. Color images are available online.
eNOS uncoupling has been implicated in a variety of experimental and clinical vascular disease states, including diabetes (114), hypertension (68), abdominal aortic aneurysms (43), and overt atherosclerosis (112). The role of eNOS uncoupling in vascular diseases is best exemplified by the fact that overexpression of eNOS in ApoE knockout (ApoE-KO/eNOS-Tg) mice does not inhibit, but accelerates atherosclerosis with increased O2·− production in aortic rings under hypercholesterolemia compared with ApoE-KO mice (112), suggesting that eNOS is uncoupled and contributes to atherogenesis. eNOS uncoupling also presents in hepatic ischemia/reperfusion injury in type 2 diabetic mice (32).
Role of NO in cardiovascular diseases
NO bioavailability indicates the production and utilization of endothelial NO in the vasculature. Importantly, decreased NO bioavailability exerts a critical role in hypertension development (53), a major risk factor for cardiovascular diseases. The endothelium is a central regulator of vascular tone and BP by virtue of its ability to produce NO (31, 110). Reciprocally, NO also clearly affects endothelial function. Endothelial dysfunction is an important risk factor for both hypertension and cardiovascular diseases. A hallmark of endothelial dysfunction is the loss of the protective actions of NO-NO bioavailability, due to a reduction in its synthesis by eNOS and an increase in scavenging by ROS (3, 33). Decreased NO bioavailability has been also widely described in vascular diseases such as atherosclerosis and disease factors, including cigarette smoking and diabetes.
There are three isoforms of NOS, eNOS (NOS III), nNOS (NOS I), and inducible NOS (iNOS, NOS II). The importance of eNOS-derived NO for BP regulation is supported by evidence of systemic hypertension in the eNOS knockout mice (57, 102) and hypotension in eNOS transgenic (eNOS-Tg) animals (86). It was demonstrated that nNOS-derived NO has an important role in the physiological regulation of BP in healthy humans (101). Moreover, in eNOS knockout mice, nNOS compensates eNOS-dependent vasodilation in coronary arteries (55), indicating the importance of NO in the vascular homeostasis. Similar to eNOS in central vascular beds, nNOS mediates vasodilation in peripheral and coronary vascular beds through NO production (99). An early feature of hypertension is abnormal endothelial function or endothelial dysfunction (13, 52). eNOS knockout mice have less NO production under physiological conditions compared with wild-type and iNOS knockout mice. Excessive NO production by iNOS induction results in hypotension and shock during sepsis (47). A single administration of the oral NO supplementation decreases BP and improves endothelial function in hypertensive patients (54). Thrombospondin-4 (Thbs4), which plays an important role in endothelial dysfunction, is involved in NO signaling. Palao et al. showed that Ang II induced impaired vasodilation in mesenteric arteries causing hypertension in WT mice, whereas in Thbs4 knockout mice, vasodilation was preserved indicating the role of Thbs4 in the NO-mediated signaling pathway and thereby in hypertension (87a). In another study, Lindsey et al. showed that G-1, an agonist of G protein-coupled estrogen receptor 1 (GPER), also known as G protein-coupled receptor 30, which induces vasorelaxation by inducing NO from endothelial cells through increasing cAMP signaling in smooth muscle cell (71). Besides, they also showed that GPER protects from AT II-induced hypertension thereby indicating the role of NO in GPER-mediated cardiovascular protection (85).
Mitochondria-associated production of NO
Several vascular diseases such as hypertension, ischemia/reperfusion injury, atherosclerosis, heart failure, cardiac hypertrophy, Alzheimer's disease, Parkinson disease (108), and diabetes (77, 78) are associated with mitochondrial dysfunction due to uncontrolled ROS (103). Thus, detecting an early stage of mitochondrial dysfunction might be a crucial step in treating cardiovascular diseases. Mitochondrial depolarization in endothelial cells activates eNOS leading to NO production by increased phosphorylation of eNOS through the PI3K/Akt pathway (60). Also, drugs such as BMS-191095 and diazoxide protect neurovascular components during ischemic/reperfusion injury through mitochondrial depolarization and NO production (61).
Aging is one of the main risk factors for cardiovascular diseases (48), and impaired mitochondrial respiration is observed in several cardiovascular diseases (96). Even though most of the studies have shown that NO produced by NOS isoforms inhibits mitochondrial respiration by binding with different complexes in mitochondrial respiration, the expression and function of NOS isoforms in mitochondria are controversial until recently (95). This is partly due to the lack of novel high-throughput techniques to measure respiration in isolated mitochondria to exclude the effect of extramitochondrial NOS effects. However, advancements in measuring mitochondrial respiration in isolated mitochondria from various tissues such as the brain, heart, and kidney using extracellular flux assays (e.g., Seahorse XFe96 analyzer) have enabled us to draw more conclusions about NOS isoforms in mitochondria (105). A recent study by Sakamuri et al. showed that a selective nNOS inhibitor ARL-17477 decreased mitochondrial respiration by decreasing S-nitrosylation of mitochondrial protein in freshly isolated mitochondria from both the brain and heart, whereas a selective eNOS inhibitor NIO [N5-(1-iminoethyl)-l-ornithine] has no effect on mitochondrial respiration indicating the role of NO in mitochondrial respiration, thereby in all cardiovascular diseases (95).
BH4 Deficiency and eNOS Uncoupling in Hypertension
Cofactor BH4 dose dependently inhibits recombinant eNOS uncoupling in the cell-free system. However, its substrate l-arginine cannot block eNOS uncoupling in the absence of BH4 (121). BH4 cannot be oxidized by ONOO− and cannot prevent eNOS oxidation and uncoupling by ONOO− (129). BH4 is required for eNOS coupling in vivo even in the healthy state (11, 92, 104). Tissue levels of BH4 reduce in disease conditions, including hypertension (30, 64, 128). BH4 availability in the lung controls pulmonary vascular tone, right ventricular hypertrophy, and vascular structural remodeling in a dose-dependent manner under both normoxic and hypoxic conditions. Furthermore, BH4 availability has striking effects on the immediate vasoconstriction response to acute hypoxia. BH4 supplementation improves forearm blood flow in patients and blocks endothelial dysfunction in vessel rings from atherosclerosis (4).
Several studies have demonstrated that BH4 deficiency is responsible for eNOS uncoupling in hypertension (Fig. 3). Landmesser et al. elegantly demonstrated that BH4 reductions due to ROS-mediated BH4 oxidation and eNOS uncoupling are evident in deoxycorticosterone acetate-salt (DOCA-salt) hypertensive mice. Oral BH4 treatment dramatically reduces vascular ROS but increases NO production (eNOS recoupling), and consequently decreases BP in the DOCA-salt mouse model (64).
FIG. 3.

Mechanisms by which coupled eNOS regulates the vascular homeostasis and uncoupled eNOS determines hypertension. Increased bioavailability of BH4 and coupled form of eNOS lead to production of NO, by utilizing the l-arginine as a substrate. The NO. further helps to keep the vessel homeostasis and normal blood pressure. Whereas the uncoupled form of eNOS along with increased BH2 levels helps vessel constriction and leads to high blood pressure. Color images are available online.
eNOS uncoupling triggered by BH4 deficiency also presents in other disease states, since eNOS uncoupling is effectively prevented by coadministration of l-sepiapterin, a BH4 precursor, or folic acid in diabetic animal models (32, 83), Ang II-AAA models (43), and in patients (106). These effects of BH4 are mediated through NO, but not O2·− synthesized by eNOS. Therefore, the presence of adequate BH4 in the endothelium is critical for maintaining “coupled” eNOS in healthy subjects (11) and “recoupling” eNOS in patients with essential hypertension (Fig. 3) (36, 109). These findings support the concept that intracellular BH4 concentrations dictate, at least in part, the balance of NO and O2·− produced by eNOS in healthy and diseased blood vessels.
BH4 deficiency and pulmonary hypertension in the human population
Pulmonary hypertension (PH) is a hemodynamic and pathophysiologic state, which is a complex disease and associated with endothelial dysfunction. The PH condition may further trigger dyspnea, decreased exercise tolerance, and progression to right heart failure (8, 39). Similar to systemic hypertension, aberrant NO system and BH4 deficiency have equal importance in developing PH. Pulmonary endothelium-produced NO has an important role in maintaining vascular homeostasis, and uncoupled eNOS has a pathogenetic role in cardiopulmonary disorders (44).
Chronic hypoxia-induced PH in the newborn pigs has shown evidence of eNOS uncoupling. To test rescuing and rebalancing the NO system in these models, the animals were treated either with l-citrulline or BH4, or both together (29). Strategically, l-arginine is a precursor for l-citrulline and provides an alternate way to increase intracellular l-arginine. Study results showed that when compared with the untreated hypoxic group, pulmonary vascular resistance was lower in hypoxic piglets cotreated with l-citrulline and BH4. Interestingly, these phenomena were not observed in hypoxic piglets treated with BH4 alone. This indicates that the combination therapy of NO precursors and BH4 may offer enhanced therapeutic capacity to ameliorate PH (29). The hph-1 (hyperphenylalaninemia 1) mice exhibit PH as they are deficient in the rate-limiting enzyme for BH4 synthesis. They tend to develop PH phenotype under normoxic conditions without concomitant systemic hypertension (40). Thus, to study if BH4 supplementation can rescue PH, Francis et al. utilized the MCT-induced PH rat model. By supplementing BH4, preventive as well as rescuing experiments were done. The experimental results showed that BH4 intervention restored normal levels of eNOS protein and ameliorated pulmonary vascular muscularization in a dose-dependent manner. Furthermore, BH4 administration also reduced pulmonary artery pressure (40).
In humans, the patients with idiopathic pulmonary fibrosis (IPF), the pulmonary artery expression of eNOS is decreased in parallel to the increased iNOS and ROS as well as nitrotyrosine (97). In another study, BH4 level in serum was found to be low in IPF patients, when compared with the control people. These patients also were found with the absence of GTPCH1 and eNOS expression in their pulmonary arteries (2). Thus, to understand if the intervention of the BH4 could attenuate PH in IPF, the bleomycin-induced pulmonary fibrosis model was utilized. Similar to the human population, these animals also showed an absolute depletion of plasma BH4 levels. The oral supplementation of sepiapterin (BH4 analogue) rebalanced the BH4 level and attenuated bleomycin-induced pulmonary fibrosis, mortality, vascular remodeling, and PH. Sepiapterin also inhibits the endothelial-to-mesenchymal transition induced by fibrotic mediators in pulmonary artery sections (2). The findings of these studies show that rebalancing BH4 is essential to keep the pulmonary homeostasis and greater efficacy of oral BH4 compound in ameliorating the development of PH.
GTPCH1, BH4 Regulation, and Hypertension
BH4 levels in endothelial cells are variable, and a continuous supply is required to maintain its basal levels (Fig. 2). In general, BH4 levels are dictated by GTPCH1, dihydrofolate reductase (DHFR), the enzyme that recycles BH4 in the salvage pathway (17, 26), and oxidative degradation of BH4, namely BH4 oxidation (Fig. 4) (64, 128). GTPCH1 is the first and rate-limiting enzyme in de novo BH4 biosynthesis. GTPCH1 catalyzes GTP to dihydroneopterin triphosphate (126). Dihydroneopterin-3P is again used by 6-pyruvoyl-tetrahydropterin synthase (6-PTS) and produces 6-pyruvoyl-tetrahydropterin and further converted into BH4 by sepiapterin reductase SPR (Fig. 4). Thus, GTPCH1 is critical for the maintenance of BH4 levels since its inhibition leads to a rapid BH4 reduction. GTPCH1 is a homodecameric enzyme consisting of 25-kDa subunits in mammalian cells (117). Zinc ion generates a hydroxyl nucleophile for the attack of imidazole ring carbon atom eight of the substrate, GTP (Fig. 5).
FIG. 4.

GTPCH1 regulation and BH4 synthesis. Cytokines, resveratrol, ARBs, and statins are known to increase the GTPCH1 expression as well as BH4 synthesis. GTP is used as a substrate by GTPCH1 and produces dihydroneopterin-3P. Dihydroneopterin-3P is again used by 6-PTS and produces 6-pyruvoyl-tetrahydropterin and further converted into BH4 by SPR. BH2 is recycled by DHFR into BH4, whereas BH2 is produced by the oxidation of BH4. 6-PTS, 6-pyruvoyl-tetrahydropterin synthase; ARBs, angiotensin receptor blockers; GFRP, GTP cyclohydrolase feedback regulatory protein; SPR, sepiapterin reductase. Color images are available online.
FIG. 5.

GTPCH1 structure and zinc release by oxidants. GTPCH1 is a homodecameric enzyme. Zinc ion generates a hydroxyl nucleophile for the attack of imidazole ring carbon atom eight of the substrate, GTP. Both HOCl and ONOO− react fast with the positively charged zinc atom resulting in its release from zinc-containing proteins. Color images are available online.
Interestingly, the transfer of the GTPCH1 gene in cultured endothelial cells markedly increases BH4 levels without altering the BH4/BH2 ratio in vitro, strongly suggesting that other enzymes in the BH4 synthetic pathway do not become significantly rate limiting even when GTPCH1 is overexpressed (14). GTPCH1 overexpression reverses BH4 deficiency and endothelial dysfunction in carotid arteries of DOCA-salt-induced hypertensive rats and mice in vivo (30, 128). GTPCH1 constitutively expresses in endothelial cells. In cultured human umbilical vein endothelial cells (HUVECs), acute stimulation with cytokines, interferon-γ plus tumor necrosis factor-α, increases GTPCH1 transcription via NF-κB/STAT1 pathways in a distinct but cooperative manner (50, 56). Lipopolysaccharide downregulates GTPCH1 feedback regulatory protein (GFRP) mRNA in HUVECs (119), which inhibits GTPCH1 (122). No information is available regarding the modulation of GTPCH1 activity by phosphorylation in vascular cells. These in vitro findings remain to be confirmed in vivo in vascular disease models. It is also worth noting that the upregulation of GTPCH1 may be a compensatory mechanism during early disease stages that eventually disappear as the disease progresses.
Several studies have shown that, in line with a BH4 reduction and eNOS uncoupling, GTPCH1 activity or protein levels decrease in diabetes and hypertension. Endothelial cells isolated from diabetic BioBreeding rats showed reduced GTPCH1 levels (75), and this effect was reversed by GTPCH1 gene transfer (74, 124). Consistent with the findings, in the diabetes model, aortic GTPCH1 mRNA levels are reduced in a glucocorticoid-induced rat model of hypertension (80). However, whether this decrease is due to the glucocorticoid treatment is unclear (80). In the carotid arteries, GTPCH1 activity is decreased during the late stages of hypertension in DOCA-salt hypertensive rats (i.e., after a 4-week treatment). Arterial gene transfer of human GTPCH1 restores GTPCH1 activity, restores BH4 levels, and normalizes eNOS function in these animals (128). Conversely, hph-1 mice exhibit significantly reduced systemic GTPCH1 expression and BH4 synthesis (73). Initial studies of the vascular phenotype in this model revealed that endothelium-dependent NO-mediated relaxation is only minimally impaired in the aorta, despite a modest but significant increase in BP (24). These findings are consistent with the recent observations in DOCA-salt hypertensive mice that uncoupled eNOS produces H2O2 instead of NO (64), accounting for the minimally impaired relaxation in these conduit arteries (24, 64).
Because conduit arteries such as the aorta do not regulate total peripheral resistance, the hypertensive phenotype observed in hph-1 mice (24) and the BP-lowering effect observed in BH4-treated DOCA-salt mice (64) may be attributable to changes in the structure and function of resistance arteries (24, 64). However, more recent evidence indicates that BH4 deficiency in DOCA-salt hypertensive mice is secondary to a reduction in GTPCH1 (62). Accordingly, BH4 supplementation or increased BH4 synthesis through adenoviral overexpression of GTPCH1 restores BH4 levels and normalizes eNOS function (64).
Findings from a few in vivo studies of GTPCH1 regulation in vascular disease contrast with those of the acute in vitro studies described above. In particular, a recent study showed that the common GTPCH1 variant, C + 243T, in the 3′-untranslated region predicted diastolic and systolic BP in individuals with extreme BP (primarily in females) as well as renal NO excretion, but not catecholamine secretion (126). The hyperphenylalaninemia (hph-1) mouse model, which is generated by N-ethinyl-N-nitrosourea mutagenesis of the GTPCH1 locus, also displays reduced levels of BH4, NO, catecholamines, and serotonin metabolites (63). In this model, deficient BH4 biosynthesis results in PH, even under normoxic conditions and greatly increases susceptibility to hypoxia-induced PH. Conversely, augmented endothelial BH4 synthesis through targeted transgenic overexpression of GTPCH1 prevents hypoxia-induced PH. Besides, restoring endothelial BH4 levels in hph-1 mice by crossing these animals with GTPCH1 transgenic mice prevents PH.
Several recent publications have confirmed that stachydrine, an active component in Chinese medicine, effectively reversed the Hcy-induced endothelial dysfunction and prevented eNOS uncoupling by increasing the expression of GTPCH1 and DHFR (123). Similarly, metformin, one of the most widely used antidiabetic drugs worldwide, is reported to improve the fluctuating glucose-induced endothelial dysfunction in HUVECs. The protective effect of metformin may be mediated through activation of GTPCH1-mediated eNOS recoupling and inhibition of NADPH oxidase via an AMPK-dependent pathway (5).
Oxidative Stress, Zinc Release, and GTPCH1
GTPCH1 enzyme activity is regulated by several mechanisms that vary among different cell types and include protein expression (5), post-translational modifications, and association with the regulatory GFRP. Zinc is important for maintaining the structure and function of GTPCH1. The human as well as bacterial enzyme was shown to contain an essential zinc ion that bound the sulfhydryl groups of the Cys-110 (-141) and Cys-181 (-212) and His-113 (-143) (Zinc1Cys2His1 complexation) in each active site of the homodecameric enzymes (Fig. 5) (127). The zinc ion is proposed to generate a hydroxyl nucleophile for the attack of imidazole ring carbon atom eight of the substrate, GTP. The zinc ion represents a selective target for oxidants such as ONOO− and HOCl. Both HOCl and ONOO− react fast with the positively charged zinc atom resulting in its release from zinc-containing proteins (Fig. 6). Indeed, the reaction of zinc/thiolate clusters with ONOO− (5.2 × 105 M−1s−1) is at least 1000 times faster than that with cysteine thiols (6 × 102 M−1s−1) and 100 times faster than those of BH4 with ONOO− (6 × 103 M−1s−1) (27).
FIG. 6.

Zinc releases from GTPCH1 by oxidants and polyubiquitination of GTPCH1. Exposure of ONOO− or SIN-1, an NO. donor, both target the GTPCH1-Zn complex and release the Zn ion. High-glucose exposure in endothelial cells triggers the GTPCH1 ubiquitination and inhibits BH4 synthesis. Whereas Mn-SOD overexpression, L-NAME exposure, TEMPO, or uric acid exposure all can inhibit the GTPCH1 ubiquitination triggered by high d-glucose exposure. L-NAME, N-nitro-l-arginine methyl ester; SOD, superoxide dismutase. Color images are available online.
Our published articles have demonstrated that the zinc structure of GTPCH1 represents a selective target for ONOO− (127). We found that (a) exposure of recombinant GTPCH1 to ONOO− or SIN-1, an ONOO− donor, dose dependently inhibited GTPCH1 activity and releases the zinc ion in parallel with increased GTPCH1 ubiquitination; (b) mutation of the zinc-binding residue cysteine 141 significantly reduced GTPCH1 enzyme activity and reduced its half-life, but increased GTPCH1 ubiquitination, indicating an essential role of the zinc ion in maintaining the catalytic activity and stability of GTPCH1; (c) exposure of endothelial cells to d-glucose, but not an osmotic control, inhibited the enzyme but increased ubiquitination of GTPCH1; (d) pharmacological inhibition of ONOO− formation with either Tempo, a potent anti-oxidant, or L-NAME, a nonselective NOS inhibitor, or scavenging of ONOO− with uric acid (a potent ONOO− scavenger) abolished the HG-induced reduction of GTPCH1 protein levels and activity; (e) adenoviral overexpression of Mn-superoxide dismutase blocked HG-enhanced GTPCH1 degradation; (f) GTPCH1 ubiquitination and degradation markedly increased in parallel with decreased GTPCH1 activity; and (g) supplementation with Tempo prevented the hyperglycemia-induced reduction of GTPCH1 activity, oxidative stress, eNOS uncoupling, and atherosclerosis in diabetic mice in vivo (Fig. 6). These data strongly support that oxidation of the zinc-binding structures of GTPCH1 by RNS inactivates the enzyme to trigger its ubiquitination, resulting in eNOS uncoupling and accelerated atherosclerosis in diabetes.
Several recent studies in humans (9, 42, 72, 94, 115) indicate that elevated neutrophils to lymphocyte ratios as a predictor of hypertension and the neutrophil to lymphocyte ratios are correlated with BP variability in hypertensive and normotensive subjects. Elevated white blood cell counts predict an increased incidence of hypertension in the Japanese, especially among females (113). Moreover, neutrophils were the major WBC component contributing to the increased risk for hypertension (113). Consistent with this report, Belen et al. report increased neutrophil to lymphocyte ratio in patients with resistant hypertension (9). Taken together, these studies indicate that neutrophil might play a causative role in the development and progression of hypertension. How neutrophils control BP remains unknown.
Activated neutrophil releases hydrogen peroxide and myeloperoxidase, producing hypochlorite, and secondarily, various chloramines. We have provided evidence supporting that HOCl might be another important oxidant that oxidizes and inactivates GTPCH1 in cardiovascular disease. Our unpublished data demonstrate that exposure of recombinant GTPCH1 to pathologically relevant concentrations of HOCl (10–100 μM) for 30 min dose dependently inhibited GTPCH1 activity and reduced its half-life. HOCl increased the ubiquitination of GTPCH1 in endothelial cells. These results indicate the oxidation of the zinc-binding structures of GTPCH1 by HOCl from myeloperoxidase in neutrophils inactivates the enzyme resulting in BH4 deficiency, with consequent eNOS uncoupling in hypertension.
Potential Pharmacological eNOS/NO Signaling Modulators
NO signaling activators and regulators received widespread attention either as potential therapeutic agents or as preventive agents. Their potential beneficial effects have been largely studied for not just cardiovascular diseases but for many other diseases as well, which include endothelial dysfunction (5), hind limb ischemia (111), cerebral ischemia (6), insulin resistance (82), Parkinson's and Alzheimer's (38), and so on (Table 1). Corticosteroids were shown to exert beneficial effects in the treatment of acute myocardial infarction. Dexamethasone, a corticosteroid, exhibits its cardiovascular protective effects by rapid, nontranscriptional activation of eNOS. Glucocorticoid receptor (GR) is a steroid hormone-activated transcriptional factor. When corticosteroids bind to the GR, it stimulates PI3K/PKA signaling and activates eNOS to trigger its vasorelaxant effect (46). Besides eNOS regulatory elements such as AMPK, PI3K/PKA signaling, Rho/Rho-kinase, the PPAR gamma also is known to regulate the eNOS expression (7). Intriguingly, a huge number of studies showed a key role of statins in inducing the expression of eNOS, which could be implicated in statin-associated cardiovascular disease protection (7). Leptin and low-density lipoprotein receptor deficient double knockout mice develop all the features of the human metabolic syndromes, including insulin resistance and hypertension. When these mice were treated with rosuvastatin for 12 weeks, it normalized BP homeostasis in obese dyslipidemic mice independently of changes in body weight or independent of decreases in plasma cholesterol, whereas rosuvastatin effects were nullified when these animals were treated with inhibition of NOS with L-NAME. This indicates that of these effects, statin effects are mediated by increased activity and expression of eNOS (28).
Table 1.
Studies Investigating Endothelial Nitric Oxide Synthase Modulators for Targeting Hypertension
| Disease models | eNOS modulator | Effect on NO signaling members | Outcome | References |
|---|---|---|---|---|
| SHR | Rhynchophylline | Rhy activates Src-PI3K/Akt-eNOS signaling pathway | Rhynchophylline ameliorates endothelial dysfunction | (49) |
| 3-MC-mediated murine hypertensive model | 3-Methylcholanthrene | 3-MC reduced the interaction between eNOS and Akt1. Increased interaction between eNOS and caveolin-1 | Simvastatin reduced 3-MC-mediated murine hypertension | (19) |
| eNOS-deficient mice | Spirulina platensis | SP6, a Spirulina peptide exerts endothelium-dependent vasodilation of ex vivo vessels, via PI3K/AKT signaling | In an experimental model of arterial hypertension, SP6 exerted an antihypertensive effect | (16) |
| SHRs | Rosiglitazone | Rosiglitazone increased PPARg expression and activated PI3K/PKB/eNOS signaling | PPARγ agonist could improve endothelial function in the young SHRs | (69) |
| Double KO of LDLR and leptin | Rosuvastatin | Rosuvastatin restores vascular NO signaling | Increased PPARγ expression by rosuvastatin regulates BP, which might be via NO mechanism | (28) |
| ANTU-induced PAH rodent model | Chrysin | Chrysin induces the eNOS level | Chrysin protects against ANTU-induced PAH via increasing eNOS level | (116) |
| L-NAME-induced hypertensive rats | Nebivolol | Nebivolol increased NO, activity and expression of eNOS, p-eNOS, Akt, and p-Akt | Nebivolol treatment reduced systolic blood pressure and ameliorated aortic remodeling | (118) |
| Monocrotaline (MCT)-induced PAH | Liraglutide | Liraglutide has therapeutic effects on MCT-induced PAH, through eNOS/sGC/PKG and Rho kinase signaling | Liraglutide prevented and reversed MCT-induced PAH. | (65) |
| SHRs | Infliximab | Infliximab-enhanced AKT/eNOS phosphorylation reduces I-κB (Iκβ) and in the aorta | Infliximab prevents the increase of both systolic pressure and left ventricle hypertrophy in SHRs | (35) |
| SHRs | Irisin | Irisin increased NO production and eNOS phosphorylation in endothelial cells | Irisin lowers blood pressure through the AMPK-Akt-eNOS-NO signaling pathway | (41) |
| SHRs | Resveratrol | Resveratrol increases intracellular calcium and activates AMPK, and further increases NO production | Resveratrol increases endothelial NO, improves endothelial function, and lowers BP, which depend on calcium-eNOS activation | (70) |
AMPK, AMP-activated protein kinase; BP, blood pressure; eNOS, endothelial nitric oxide synthase; LDLR, low-density lipoprotein receptor; L-NAME, N-nitro-l-arginine methyl ester; NO, nitric oxide; PAH, pulmonary arterial hypertension; sGC-PKG, guanylyl cyclase-cGMP-dependent protein kinase; SHR, spontaneously hypertensive rat.
BH4 and Its Clinical Perspective
BH4 acts as a critical regulator of eNOS function and suggests that BH4 is a rational therapeutic target in vascular disease states, particularly for hypertension. The hph-1 mutant mice have deficient GTPCH1 activity, resulting in lower levels of BH4 content in their tissues. Compared with wild-type mice, the lung peroxide content was increased, whereas the eNOS expression was decreased in hph-1 animals. As they grow and turn to adults these mice tend to develop PH (10). These all indicate the importance of GTPCH1 activity and BH4 level in animal systems to keep the BP homeostasis. BH4 supplementation in preclinical models showed a significant health improvement. In Ang II-induced hypertensive rat models, BH4 supplementation in drinking water significantly prevented the Ang II effects, such as impaired vascular responses to acetylcholine, hypertension, and heart weight index values. On the contrary, BH4 also significantly reduced Ang II-induced iNOS expression and nitrotyrosine and superoxide anion formation in rodents, which indicates that it can reduce the uncoupling effect of NOS (59). However, some clinical trials had been done by several groups, many of the outcomes of these trials were not published or yet to be published. Clinical trial: NCT00435331 (NIH trial identifier number) is one of the very first attempts on PH patients using modified BH4. It is an analogue of BH4, a thermo- and photostable drug named 6R-BH4/sapropterin dihydrochloride, which is used as a drug for phenylketonuric (PKU) patients. The rationale behind this trial was that PKU is also associated with BH4 deficiency and BH4 treatment showed significant outcomes for these patients (Clinical Trial No. NCT00964236) (22). Thus, the purpose of this study is to determine whether the sapropterin treatment has any complementary or additive effects on the existing treatment in patients with pulmonary arterial hypertension (PAH). Another objective of the study was also to evaluate the change in biochemical markers of endothelial dysfunction and eNOS activity (94a). Another set of the study was carried out in patients with systemic hypertension (Clinical Trial No. NCT00325962). Similar to the previous study, this study also intended to collect the information on the efficacy of 6R-BH4 on patients. The baseline eNOS activity and endothelial dysfunction after 8 weeks of treatment in subjects with poorly controlled hypertension were also recorded. However, the 6R-BH4 supplementation to either systemic hypertension or PAH patients did not show significant improvement, and the complete results of the study have not been published yet (1). However, the above clinical trials did not show promising results, a randomized double-blind, placebo-controlled trial showed a positive outcome on hypercholesterolemic patients (25). In this study, hypercholesterolemic patients were given BH4 (400 mg twice daily) for 4 weeks. To determine the effect of BH4 supplement, NO release and O2·− production were measured in human aortic endothelial cells. Using venous occlusion plethysmography, the endothelium-dependent and independent vasodilatation was also assessed. This study concluded that BH4 oral treatment could reverse endothelial dysfunction under hypercholesterolemic settings (25). In another randomized-controlled trial, the patients with type 2 diabetes (T2D) and coronary artery disease were involved to know if improving eNOS protects against ischemia/reperfusion-induced endothelial dysfunction (100). The depletion of physiological levels of l-arginine (eNOS substrate) and BH4 was observed by many preclinical models, and supplying l-arginine (90) or BH4 could abrogate the endothelial dysfunction (92). Here, the patients were provided with l-arginine and BH4. Forearm ischemia was induced in 12 patients with type 2 diabetes and coronary artery disease for 20 min, and 60 min of reperfusion followed by intrabrachial infusion of l-arginine (20 mg/min) and BH4 (500 μg/min). Remarkably, endothelium-dependent vasodilatation (EDV) was significantly reduced at 15 and 30 min of reperfusion following l-arginine and BH4 infusion. Remarkably, after l-arginine and BH4 infusion, EDV was observed to be significantly less reduced at 15 and 30 min of reperfusion compared with the saline infusion (100). Considering the importance of the aforementioned studies, to improve the translational significance, it is essential to continue research in this field to manipulate BH4 to increase the efficacy for treating hypertension.
Conclusion
In this brief review, we have summarized the evidence supporting that oxidation and ubiquitination of GTPCH1 in hypertension, the rate-limiting BH4 synthesis enzyme, GTPCH1, influences oxidative stress caused by eNOS uncoupling, a process that exacerbates oxidative stress in diverse vascular disease states and that the zinc ion in GTPCH1 is sensitive to oxidants such as HOCl and ONOO−. Understanding oxidant-mediated GTPCH1 inactivation, and ubiquitination by oxidants, might help develop new treatment strategies in hypertension that might help to ameliorate the excess vascular diseases associated with the diseases. Besides, the importance of BH4 bioavailability on systemic and PH is well understood, however, implying as therapeutics needs in-depth research. Given that eNOS uncoupling is now considered a common mechanism for endothelial dysfunction and hypertension, the gained information might also increase the understandings of other vascular diseases, including atherosclerosis, hypercholesterolemia, and heart failure.
Abbreviations Used
- 6-PTS
6-pyruvoyl-tetrahydropterin synthase
- AMPK
AMP-activated protein kinase
- BH4
tetrahydrobiopterin
- CaMK-II
calmodulin-dependent protein kinase II
- cGMP
cyclic guanosine monophosphate
- DHFR
dihydrofolate reductase
- DOCA-salt
deoxycorticosterone acetate-salt
- EDV
endothelium-dependent vasodilatation
- eNOS
endothelial nitric oxide synthase
- eNOS-Tg
eNOS transgenic
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- GFRP
GTPCH1 feedback regulatory protein
- GPER
G protein-coupled estrogen receptor 1
- GR
glucocorticoid receptor
- GTPCH1
guanosine 5′-triphosphate (GTP)-cyclohydrolase 1
- HNO2
nitrous acid
- HOCl
hypochlorous acid
- hph-1
hyperphenylalaninemia
- HUVEC
human umbilical vein endothelial cells
- iNOS
inducible NOS
- LDLR
low-density lipoprotein receptor
- L-NAME
N-nitro-l-arginine methyl ester
- N2O3
dinitrogen trioxide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NIO
N5-(1-iminoethyl)-l-ornithine
- nNOS
neuronal NOS
- NO
nitric oxide
- NO+
nitrosyl
- NO2+
nitronium ion
- NO2·
nitrogen dioxide
- NO−
nitroxide
- O2·−
superoxide anion
- ONOO−
peroxynitrite
- PAH
pulmonary arterial hypertension
- PH
pulmonary hypertension
- PKU
phenylketonuric
- RNS
reactive nitrogen species
- RONOO
alkyl peroxynitrites
- ROS
reactive oxygen species
- sGC-PKG
guanylyl cyclase-cGMP-dependent protein kinase
- SHR
spontaneously hypertensive rats
- SOD
superoxide dismutase
- SSNO−
nitroso persulfide
- STAT1
signal transducer and activator of transcription 1
- T2D
type 2 diabetes
- Thbs4
thrombospondin-4
- VEGF
vascular endothelial growth factor
Funding Information
This work was supported by the AHA postdoctoral fellowship award: 19POST34380156 to Tharmarajan Ramprasath and the AHA Career Development award: 19CDA34730035 to Ye Ding.
References
- 1. Alkaitis MS and Crabtree MJ. Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep 9: 200–210, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Almudever P, Milara J, De Diego A, Serrano-Mollar A, Xaubet A, Perez-Vizcaino F, Cogolludo A, and Cortijo J. Role of tetrahydrobiopterin in pulmonary vascular remodelling associated with pulmonary fibrosis. Thorax 68: 938–948, 2013 [DOI] [PubMed] [Google Scholar]
- 3. Alp NJ and Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 24: 413–420, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Alp NJ, McAteer MA, Khoo J, Choudhury RP, and Channon KM. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler Thromb Vasc Biol 24: 445–450, 2004 [DOI] [PubMed] [Google Scholar]
- 5. An H, Wei R, Ke J, Yang J, Liu Y, Wang X, Wang G, and Hong T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J Diabetes Complications 30: 1017–1024, 2016 [DOI] [PubMed] [Google Scholar]
- 6. Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, and Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest 117: 1961–1967, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Balakumar P, Kathuria S, Taneja G, Kalra S, and Mahadevan N. Is targeting eNOS a key mechanistic insight of cardiovascular defensive potentials of statins? J Mol Cell Cardiol 52: 83–92, 2012 [DOI] [PubMed] [Google Scholar]
- 8. Bazan IS and Fares WH. Pulmonary hypertension: diagnostic and therapeutic challenges. Ther Clin Risk Manag 11: 1221–1233, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belen E, Sungur A, Sungur MA, and Erdogan G. Increased neutrophil to lymphocyte ratio in patients with resistant hypertension. J Clin Hypertens (Greenwich) 17: 532–537, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belik J, McIntyre BA, Enomoto M, Pan J, Grasemann H, and Vasquez-Vivar J. Pulmonary hypertension in the newborn GTP cyclohydrolase I-deficient mouse. Free Radic Biol Med 51: 2227–2233, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, Yokoyama M, Kawashima S, and Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res 97: 864–871, 2005 [DOI] [PubMed] [Google Scholar]
- 12. Bromfield S and Muntner P. High blood pressure: the leading global burden of disease risk factor and the need for worldwide prevention programs. Curr Hypertens Rep 15: 134–136, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cai H and Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000 [DOI] [PubMed] [Google Scholar]
- 14. Cai S, Alp NJ, McDonald D, Smith I, Kay J, Canevari L, Heales S, and Channon KM. GTP cyclohydrolase I gene transfer augments intracellular tetrahydrobiopterin in human endothelial cells: effects on nitric oxide synthase activity, protein levels and dimerisation. Cardiovasc Res 55: 838–849, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Cannon RO, 3rd. Role of nitric oxide in cardiovascular disease: focus on the endothelium. Clin Chem 44: 1809–1819, 1998 [PubMed] [Google Scholar]
- 16. Carrizzo A, Conte GM, Sommella E, Damato A, Ambrosio M, Sala M, Scala MC, Aquino RP, De Lucia M, Madonna M, Sansone F, Ostacolo C, Capunzo M, Migliarino S, Sciarretta S, Frati G, Campiglia P, and Vecchione C. Novel potent decameric peptide of spirulina platensis reduces blood pressure levels through a PI3K/AKT/eNOS-dependent mechanism. Hypertension 73: 449–457, 2019 [DOI] [PubMed] [Google Scholar]
- 17. Chalupsky K and Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 102: 9056–9061, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan Y, Fish JE, D'Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, and Marsden PA. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem 279: 35087–35100, 2004 [DOI] [PubMed] [Google Scholar]
- 19. Chang CC, Hsu YH, Chou HC, Lee YG, and Juan SH. 3-Methylcholanthrene/aryl-hydrocarbon receptor-mediated hypertension through eNOS inactivation. J Cell Physiol 232: 1020–1029, 2017 [DOI] [PubMed] [Google Scholar]
- 20. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, and Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen K, Pittman RN, and Popel AS. Nitric oxide in the vasculature: where does it come from and where does it go? A quantitative perspective. Antioxid Redox Signal 10: 1185–1198, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christ SE, Moffitt AJ, Peck D, and White DA. The effects of tetrahydrobiopterin (BH4) treatment on brain function in individuals with phenylketonuria. Neuroimage Clin 3: 539–547, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collaboration NCDRF. Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet 389: 37–55, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cosentino F, Barker JE, Brand MP, Heales SJ, Werner ER, Tippins JR, West N, Channon KM, Volpe M, and Luscher TF. Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice. Arterioscler Thromb Vasc Biol 21: 496–502, 2001 [DOI] [PubMed] [Google Scholar]
- 25. Cosentino F, Hurlimann D, Delli Gatti C, Chenevard R, Blau N, Alp NJ, Channon KM, Eto M, Lerch P, Enseleit F, Ruschitzka F, Volpe M, Luscher TF, and Noll G. Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart 94: 487–492, 2008 [DOI] [PubMed] [Google Scholar]
- 26. Crabtree MJ, Tatham AL, Hale AB, Alp NJ, and Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem 284: 28128–28136, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Demir M. The relationship between neutrophil lymphocyte ratio and non-dipper hypertension. Clin Exp Hypertens 35: 570–573, 2013 [DOI] [PubMed] [Google Scholar]
- 28. Desjardins F, Sekkali B, Verreth W, Pelat M, De Keyzer D, Mertens A, Smith G, Herregods MC, Holvoet P, and Balligand JL. Rosuvastatin increases vascular endothelial PPARgamma expression and corrects blood pressure variability in obese dyslipidaemic mice. Eur Heart J 29: 128–137, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Dikalova A, Aschner JL, Kaplowitz MR, Cunningham G, Summar M, and Fike CD. Combined L-citrulline and tetrahydrobiopterin therapy improves NO signaling and ameliorates chronic hypoxia-induced pulmonary hypertension in newborn pigs. Am J Physiol Lung Cell Mol Physiol 318: L762–L772, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Du YH, Guan YY, Alp NJ, Channon KM, and Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation 117: 1045–1054, 2008 [DOI] [PubMed] [Google Scholar]
- 31. Dzau VJ and Gibbons GH. Endothelium and growth factors in vascular remodeling of hypertension. Hypertension 18: III115–III121, 1991 [DOI] [PubMed] [Google Scholar]
- 32. Elrod JW, Duranski MR, Langston W, Greer JJ, Tao L, Dugas TR, Kevil CG, Champion HC, and Lefer DJ. eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: a role for eNOS uncoupling. Circ Res 99: 78–85, 2006 [DOI] [PubMed] [Google Scholar]
- 33. Endemann DH and Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol 15: 1983–1992, 2004 [DOI] [PubMed] [Google Scholar]
- 34. Erwin PA, Lin AJ, Golan DE, and Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem 280: 19888–19894, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Filho AG, Kinote A, Pereira DJ, Renno A, dos Santos RC, Ferreira-Melo SE, Velloso LA, Bordin S, Anhe GF, and Moreno Junior H. Infliximab prevents increased systolic blood pressure and upregulates the AKT/eNOS pathway in the aorta of spontaneously hypertensive rats. Eur J Pharmacol 700: 201–209, 2013 [DOI] [PubMed] [Google Scholar]
- 36. Forstermann U. Janus-faced role of endothelial NO synthase in vascular disease: uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol Chem 387: 1521–1533, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Forstermann U and Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J 33: 829–837, 837 a-837d, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Foxton RH, Land JM, and Heales SJ. Tetrahydrobiopterin availability in Parkinson's and Alzheimer's disease; potential pathogenic mechanisms. Neurochem Res 32: 751–756, 2007 [DOI] [PubMed] [Google Scholar]
- 39. Francis BN, Salameh M, Khamisy-Farah R, and Farah R.. Tetrahydrobiopterin (BH4): Targeting endothelial nitric oxide synthase as a potential therapy for pulmonary hypertension. Cardiovasc Ther 36, 2018. DOI: 10.1111/1755-5922.12312 [DOI] [PubMed] [Google Scholar]
- 40. Francis BN, Wilkins MR, and Zhao L. Tetrahydrobiopterin and the regulation of hypoxic pulmonary vasoconstriction. Eur Respir J 36: 323–330, 2010 [DOI] [PubMed] [Google Scholar]
- 41. Fu J, Han Y, Wang J, Liu Y, Zheng S, Zhou L, Jose PA, and Zeng C. Irisin lowers blood pressure by improvement of endothelial dysfunction via AMPK-Akt-eNOS-NO pathway in the spontaneously hypertensive rat. J Am Heart Assoc 5: e003433, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gang L and Yanyan Z. Increased neutrophil to lymphocyte ratio in persons suffering from hypertension with hyperhomocysteinemia. Hypertens Res 39: 606–611, 2016 [DOI] [PubMed] [Google Scholar]
- 43. Gao L, Siu KL, Chalupsky K, Nguyen A, Chen P, Weintraub NL, Galis Z, and Cai H. Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension 59: 158–166, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gielis JF, Lin JY, Wingler K, Van Schil PE, Schmidt HH, and Moens AL. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic Biol Med 50: 765–776, 2011 [DOI] [PubMed] [Google Scholar]
- 45. Guwatudde D, Nankya-Mutyoba J, Kalyesubula R, Laurence C, Adebamowo C, Ajayi I, Bajunirwe F, Njelekela M, Chiwanga FS, Reid T, Volmink J, Adami HO, Holmes MD, and Dalal S. The burden of hypertension in sub-Saharan Africa: a four-country cross sectional study. BMC Public Health 15: 1211, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, and Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med 8: 473–479, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hallemeesch MM, Janssen BJ, de Jonge WJ, Soeters PB, Lamers WH, and Deutz NE. NO production by cNOS and iNOS reflects blood pressure changes in LPS-challenged mice. Am J Physiol Endocrinol Metab 285: E871–E875, 2003 [DOI] [PubMed] [Google Scholar]
- 48. Han YM, Ramprasath T, and Zou MH. Beta-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp Mol Med 52: 548–555, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hao HF, Liu LM, Pan CS, Wang CS, Gao YS, Fan JY, and Han JY. Rhynchophylline ameliorates endothelial dysfunction via Src-PI3K/Akt-eNOS cascade in the cultured intrarenal arteries of spontaneous hypertensive rats. Front Physiol 8: 928, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hattori Y, Nakanishi N, Kasai K, and Shimoda SI. GTP cyclohydrolase I mRNA induction and tetrahydrobiopterin synthesis in human endothelial cells. Biochim Biophys Acta 1358: 61–66, 1997 [DOI] [PubMed] [Google Scholar]
- 51. Heinzel B, John M, Klatt P, Bohme E, and Mayer B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem J 281(Pt 3): 627–630, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heitzer T, Schlinzig T, Krohn K, Meinertz T, and Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104: 2673–2678, 2001 [DOI] [PubMed] [Google Scholar]
- 53. Hermann M, Flammer A, and Luscher TF. Nitric oxide in hypertension. J Clin Hypertens (Greenwich) 8: 17–29, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Houston M and Hays L. Acute effects of an oral nitric oxide supplement on blood pressure, endothelial function, and vascular compliance in hypertensive patients. J Clin Hypertens (Greenwich) 16: 524–529, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang A, Sun D, Shesely EG, Levee EM, Koller A, and Kaley G. Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am J Physiol Heart Circ Physiol 282: H429–H436, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Huang A, Zhang YY, Chen K, Hatakeyama K, and Keaney JF Jr. Cytokine-stimulated GTP cyclohydrolase I expression in endothelial cells requires coordinated activation of nuclear factor-kappaB and Stat1/Stat3. Circ Res 96: 164–171, 2005 [DOI] [PubMed] [Google Scholar]
- 57. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, and Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377: 239–242, 1995 [DOI] [PubMed] [Google Scholar]
- 58. Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D'Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y, and Marsden PA. Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem 274: 3076–3093, 1999 [DOI] [PubMed] [Google Scholar]
- 59. Kase H, Hashikabe Y, Uchida K, Nakanishi N, and Hattori Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J Hypertens 23: 1375–1382, 2005 [DOI] [PubMed] [Google Scholar]
- 60. Katakam PV, Dutta S, Sure VN, Grovenburg SM, Gordon AO, Peterson NR, Rutkai I, and Busija DW. Depolarization of mitochondria in neurons promotes activation of nitric oxide synthase and generation of nitric oxide. Am J Physiol Heart Circ Physiol 310: H1097–H1106, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Katakam PV, Gordon AO, Sure VN, Rutkai I, and Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol 307: H493–H503, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Katusic ZS and d'Uscio LV. Tetrahydrobiopterin: mediator of endothelial protection. Arterioscler Thromb Vasc Biol 24: 397–398, 2004 [DOI] [PubMed] [Google Scholar]
- 63. Lam AA, Hyland K, and Heales SJ. Tetrahydrobiopterin availability, nitric oxide metabolism and glutathione status in the hph-1 mouse; implications for the pathogenesis and treatment of tetrahydrobiopterin deficiency states. J Inherit Metab Dis 30: 256–262, 2007 [DOI] [PubMed] [Google Scholar]
- 64. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, and Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee MY, Tsai KB, Hsu JH, Shin SJ, Wu JR, and Yeh JL. Liraglutide prevents and reverses monocrotaline-induced pulmonary arterial hypertension by suppressing ET-1 and enhancing eNOS/sGC/PKG pathways. Sci Rep 6: 31788, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leo F, Hutzler B, Ruddiman CA, Isakson BE, and Cortese-Krott MM. Cellular microdomains for nitric oxide signaling in endothelium and red blood cells. Nitric Oxide 96: 44–53, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leung AA, Bushnik T, Hennessy D, McAlister FA, and Manuel DG. Risk factors for hypertension in Canada. Health Rep 30: 3–13, 2019 [PubMed] [Google Scholar]
- 68. Li H, Witte K, August M, Brausch I, Godtel-Armbrust U, Habermeier A, Closs EI, Oelze M, Munzel T, and Forstermann U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol 47: 2536–2544, 2006 [DOI] [PubMed] [Google Scholar]
- 69. Li R, Zhang H, Wang W, Wang X, Huang Y, Huang C, and Gao F. Vascular insulin resistance in prehypertensive rats: role of PI3-kinase/Akt/eNOS signaling. Eur J Pharmacol 628: 140–147, 2010 [DOI] [PubMed] [Google Scholar]
- 70. Li X, Dai Y, Yan S, Shi Y, Li J, Liu J, Cha L, and Mu J. Resveratrol lowers blood pressure in spontaneously hypertensive rats via calcium-dependent endothelial NO production. Clin Exp Hypertens 38: 287–293, 2016 [DOI] [PubMed] [Google Scholar]
- 71. Lindsey SH, Liu L, and Chappell MC. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids 81: 99–102, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu X, Zhang Q, Wu H, Du H, Liu L, Shi H, Wang C, Xia Y, Guo X, Li C, Bao X, Su Q, Sun S, Wang X, Zhou M, Jia Q, Zhao H, Song K, and Niu K. Blood Neutrophil to Lymphocyte Ratio as a Predictor of Hypertension. Am J Hypertens 28: 1339–1346, 2015 [DOI] [PubMed] [Google Scholar]
- 73. McDonald JD and Bode VC. Hyperphenylalaninemia in the hph-1 mouse mutant. Pediatr Res 23: 63–67, 1988 [DOI] [PubMed] [Google Scholar]
- 74. Meininger CJ, Cai S, Parker JL, Channon KM, Kelly KA, Becker EJ, Wood MK, Wade LA, and Wu G. GTP cyclohydrolase I gene transfer reverses tetrahydrobiopterin deficiency and increases nitric oxide synthesis in endothelial cells and isolated vessels from diabetic rats. FASEB J 18: 1900–1902, 2004 [DOI] [PubMed] [Google Scholar]
- 75. Meininger CJ, Marinos RS, Hatakeyama K, Martinez-Zaguilan R, Rojas JD, Kelly KA, and Wu G. Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem J 349: 353–356, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mensah GA. Epidemiology and Global Burden of Hypertension. ESC CardioMed, 3rd ed. Oxford, England: Oxford University Press, 2018 [Google Scholar]
- 77. Merdzo I, Rutkai I, Sure V, Katakam PVG, and Busija DW. Effects of prolonged type 2 diabetes on mitochondrial function in cerebral blood vessels. Am J Physiol Heart Circ Physiol 317: H1086–H1092, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Merdzo I, Rutkai I, Sure VN, McNulty CA, Katakam PV, and Busija DW. Impaired mitochondrial respiration in large cerebral arteries of rats with type 2 diabetes. J Vasc Res 54: 1–12, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Merdzo I, Rutkai I, Tokes T, Sure VN, Katakam PV, and Busija DW. The mitochondrial function of the cerebral vasculature in insulin-resistant Zucker obese rats. Am J Physiol Heart Circ Physiol 310: H830–H838, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mitchell BM, Dorrance AM, and Webb RC. GTP cyclohydrolase 1 downregulation contributes to glucocorticoid hypertension in rats. Hypertension 41: 669–674, 2003 [DOI] [PubMed] [Google Scholar]
- 81. Munzel T, Feil R, Mulsch A, Lohmann SM, Hofmann F, and Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3',5'-cyclic monophosphate-dependent protein kinase [corrected]. Circulation 108: 2172–2183, 2003 [DOI] [PubMed] [Google Scholar]
- 82. Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, and King GL. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes 55: 691–698, 2006 [DOI] [PubMed] [Google Scholar]
- 83. Nie H, Wu JL, Zhang M, Xu J, and Zou MH. Endothelial nitric oxide synthase-dependent tyrosine nitration of prostacyclin synthase in diabetes in vivo. Diabetes 55: 3133–3141, 2006 [DOI] [PubMed] [Google Scholar]
- 84. This reference has been deleted
- 85. Ogola BO, Zimmerman MA, Sure VN, Gentry KM, Duong JL, Clark GL, Miller KS, Katakam PVG, and Lindsey SH. G Protein-coupled estrogen receptor protects from angiotensin ii-induced increases in pulse pressure and oxidative stress. Front Endocrinol (Lausanne) 10: 586, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ohashi Y, Kawashima S, Hirata K, Yamashita T, Ishida T, Inoue N, Sakoda T, Kurihara H, Yazaki Y, and Yokoyama M. Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J Clin Invest 102: 2061–2071, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Oliveira-Paula GH, Lacchini R, and Tanus-Santos JE. Clinical and pharmacogenetic impact of endothelial nitric oxide synthase polymorphisms on cardiovascular diseases. Nitric Oxide 63: 39–51, 2017 [DOI] [PubMed] [Google Scholar]
- 87a. Palao T, Swärd K, Jongejan A, Moerland PD, de Vos J, van Weert A, Arribas SM, Groma G, vanßavel E, Bakker EN. Gene expression and microRNA expression analysis in small arteries of spontaneously hypertensive rats. Evidence for ER stress. PLoS One 10: e0137027, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pou S, Pou WS, Bredt DS, Snyder SH, and Rosen GM. Generation of superoxide by purified brain nitric oxide synthase. J Biol Chem 267: 24173–24176, 1992 [PubMed] [Google Scholar]
- 89. Ramprasath T, Freddy AJ, Velmurugan G, Tomar D, Rekha B, Suvekbala V, and Ramasamy S.. Context-dependent regulation of Nrf2/ARE axis on vascular cell function during hyperglycemic condition. Curr Diabetes Rev 2020. [Epub ahead of print]; DOI: 10.2174/1573399816666200130094512 [DOI] [PubMed] [Google Scholar]
- 90. Ramprasath T, Kumar PH, Puhari SS, Murugan PS, Vasudevan V, and Selvam GS. L-Arginine ameliorates cardiac left ventricular oxidative stress by upregulating eNOS and Nrf2 target genes in alloxan-induced hyperglycemic rats. Biochem Biophys Res Commun 428: 389–394, 2012 [DOI] [PubMed] [Google Scholar]
- 91. Ramprasath T and Selvam GS. Potential impact of genetic variants in Nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Curr Med Chem 20: 4680–4693, 2013 [DOI] [PubMed] [Google Scholar]
- 92. Ramprasath T, Vasudevan V, Sasikumar S, Puhari SS, Saso L, and Selvam GS. Regression of oxidative stress by targeting eNOS and Nrf2/ARE signaling: a guided drug target for cardiovascular diseases. Curr Top Med Chem 15: 857–871, 2015 [DOI] [PubMed] [Google Scholar]
- 93. Ravi K, Brennan LA, Levic S, Ross PA, and Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A 101: 2619–2624, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Regal JF, Lillegard KE, Bauer AJ, Elmquist BJ, Loeks-Johnson AC, and Gilbert JS. Neutrophil depletion attenuates placental ischemia-induced hypertension in the rat. PLoS One 10: e0132063, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94a. Robbins I. 6R-BH4 pulmonary arterial hypertension study. NIH Clinical Trials NCT00435331, 2007–2008. 2013. https://clinicaltrials.gov/ct2/show/NCT00435331?term=NCT00435331&draw=2&rank=1 (accessed February10, 2020)
- 95. Sakamuri S, Sperling JA, Evans WR, Dholakia MH, Albuck AL, Sure VN, Satou R, Mostany R, and Katakam PVG. Nitric oxide synthase inhibitors negatively regulate respiration in isolated rodent cardiac and brain mitochondria. Am J Physiol Heart Circ Physiol 318: H295–H300, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sakamuri S, Sperling JA, Sure VN, Dholakia MH, Peterson NR, Rutkai I, Mahalingam PS, Satou R, and Katakam PVG. Measurement of respiratory function in isolated cardiac mitochondria using Seahorse XFe24 Analyzer: applications for aging research. Geroscience 40: 347–356, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saleh D, Barnes PJ, and Giaid A. Increased production of the potent oxidant peroxynitrite in the lungs of patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 155: 1763–1769, 1997 [DOI] [PubMed] [Google Scholar]
- 98. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, and Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond) 131: 2671–2685, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Seddon MD, Chowienczyk PJ, Brett SE, Casadei B, and Shah AM. Neuronal nitric oxide synthase regulates basal microvascular tone in humans in vivo. Circulation 117: 1991–1996, 2008 [DOI] [PubMed] [Google Scholar]
- 100. Settergren M, Bohm F, Malmstrom RE, Channon KM, and Pernow J. L-arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis 204: 73–78, 2009 [DOI] [PubMed] [Google Scholar]
- 101. Shabeeh H, Khan S, Jiang B, Brett S, Melikian N, Casadei B, Chowienczyk PJ, and Shah AM. Blood pressure in healthy humans is regulated by neuronal NO synthase. Hypertension 69: 970–976, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, and Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 93: 13176–13181, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, Tagkou NM, Tsimpiktsioglou A, Stampouloglou PK, Oikonomou E, Mourouzis K, Philippou A, Vavuranakis M, Stefanadis C, Tousoulis D, and Papavassiliou AG. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann Transl Med 6: 256, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Song P, Ramprasath T, Wang H, and Zou MH. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell Mol Life Sci 74: 2899–2916, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sperling JA, Sakamuri S, Albuck AL, Sure VN, Evans WR, Peterson NR, Rutkai I, Mostany R, Satou R, and Katakam PVG. Measuring respiration in isolated murine brain mitochondria: implications for mechanistic stroke studies. Neuromolecular Med 21: 493–504, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, Luscher T, and Rabelink T. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest 99: 41–46, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. This reference has been deleted
- 108. Sure VN, Sakamuri S, Sperling JA, Evans WR, Merdzo I, Mostany R, Murfee WL, Busija DW, Katakam PVG. A novel high-throughput assay for respiration in isolated brain microvessels reveals impaired mitochondrial function in the aged mice. Geroscience 40: 365–375, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Taddei S, Virdis A, Ghiadoni L, Magagna A, and Salvetti A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 97: 2222–2229, 1998 [DOI] [PubMed] [Google Scholar]
- 110. Taddei S, Virdis A, Ghiadoni L, and Salvetti A. The role of endothelium in human hypertension. Curr Opin Nephrol Hypertens 7: 203–209, 1998 [DOI] [PubMed] [Google Scholar]
- 111. Takahashi N, Shibata R, Ouchi N, Sugimoto M, Murohara T, and Komori K. Metformin stimulates ischemia-induced revascularization through an eNOS dependent pathway in the ischemic hindlimb mice model. J Vasc Surg 61: 489–496, 2015 [DOI] [PubMed] [Google Scholar]
- 112. Takaya T, Hirata K, Yamashita T, Shinohara M, Sasaki N, Inoue N, Yada T, Goto M, Fukatsu A, Hayashi T, Alp NJ, Channon KM, Yokoyama M, and Kawashima S. A specific role for eNOS-derived reactive oxygen species in atherosclerosis progression. Arterioscler Thromb Vasc Biol 27: 1632–1637, 2007 [DOI] [PubMed] [Google Scholar]
- 113. Tatsukawa Y, Hsu WL, Yamada M, Cologne JB, Suzuki G, Yamamoto H, Yamane K, Akahoshi M, Fujiwara S, and Kohno N. White blood cell count, especially neutrophil count, as a predictor of hypertension in a Japanese population. Hypertens Res 31: 1391–1397, 2008 [DOI] [PubMed] [Google Scholar]
- 114. Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, Tsikas D, Ertl G, and Bauersachs J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 56: 666–674, 2007 [DOI] [PubMed] [Google Scholar]
- 115. Wang H, Hu Y, Geng Y, Wu H, Chu Y, Liu R, Wei Y, and Qiu Z. The relationship between neutrophil to lymphocyte ratio and artery stiffness in subtypes of hypertension. J Clin Hypertens (Greenwich) 19: 780–785, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang L, Wang Y, and Lei Z. Chrysin ameliorates ANTU-induced pulmonary edema and pulmonary arterial hypertension via modulation of VEGF and eNOs. J Biochem Mol Toxicol 33: e22332, 2019 [DOI] [PubMed] [Google Scholar]
- 117. Wang S, Xu J, Song P, Wu Y, Zhang J, Chul Choi H, and Zou MH. Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension 52: 484–490, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang Y, Zhang F, Liu Y, Yin S, Pang X, Li Z, and Wei Z. Nebivolol alleviates aortic remodeling through eNOS upregulation and inhibition of oxidative stress in l-NAME-induced hypertensive rats. Clin Exp Hypertens 39: 628–639, 2017 [DOI] [PubMed] [Google Scholar]
- 119. Werner ER, Bahrami S, Heller R, and Werner-Felmayer G. Bacterial lipopolysaccharide down-regulates expression of GTP cyclohydrolase I feedback regulatory protein. J Biol Chem 277: 10129–10133, 2002 [DOI] [PubMed] [Google Scholar]
- 120. WHO. Hypertension Facts. 2019. https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed September13, 2019)
- 121. Xia Y, Tsai AL, Berka V, and Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998 [DOI] [PubMed] [Google Scholar]
- 122. Xie L, Smith JA, and Gross SS. GTP cyclohydrolase I inhibition by the prototypic inhibitor 2, 4-diamino-6-hydroxypyrimidine. Mechanisms and unanticipated role of GTP cyclohydrolase I feedback regulatory protein. J Biol Chem 273: 21091–21098, 1998 [DOI] [PubMed] [Google Scholar]
- 123. Xie X, Zhang Z, Wang X, Luo Z, Lai B, Xiao L, and Wang N. Stachydrine protects eNOS uncoupling and ameliorates endothelial dysfunction induced by homocysteine. Mol Med 24: 10, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Xu J, Wu Y, Song P, Zhang M, Wang S, and Zou MH. Proteasome-dependent degradation of guanosine 5'-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 116: 944–953, 2007 [DOI] [PubMed] [Google Scholar]
- 125. Xu J, Xie Z, Reece R, Pimental D, and Zou MH. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol 26: 2688–2695, 2006 [DOI] [PubMed] [Google Scholar]
- 126. Zhang L, Rao F, Zhang K, Khandrika S, Das M, Vaingankar SM, Bao X, Rana BK, Smith DW, Wessel J, Salem RM, Rodriguez-Flores JL, Mahata SK, Schork NJ, Ziegler MG, and O'Connor DT. Discovery of common human genetic variants of GTP cyclohydrolase 1 (GCH1) governing nitric oxide, autonomic activity, and cardiovascular risk. J Clin Invest 117: 2658–2671, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhao Y, Wu J, Zhu H, Song P, and Zou MH. Peroxynitrite-dependent zinc release and inactivation of guanosine 5'-triphosphate cyclohydrolase 1 instigate its ubiquitination in diabetes. Diabetes 62: 4247–4256, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 128. Zheng JS, Yang XQ, Lookingland KJ, Fink GD, Hesslinger C, Kapatos G, Kovesdi I, and Chen AF. Gene transfer of human guanosine 5'-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension. Circulation 108: 1238–1245, 2003 [DOI] [PubMed] [Google Scholar]
- 129. Zou MH, Shi C, and Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 109: 817–826, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]