

Summary
Cancer genomes often harbor hundreds of somatic DNA rearrangement junctions, many of which cannot be easily classified into simple (e.g. deletion) or complex (e.g. chromothripsis) structural variant classes. Applying a novel genome graph computational paradigm to analyze the topology of junction copy number (JCN) across 2,778 tumor whole genome sequences, we uncovered three novel complex rearrangement phenomena: pyrgo, rigma, and tyfonas. Pyrgo are “towers” of low-JCN duplications associated with early replicating regions and superenhancers, enriched in breast and ovarian cancers. Rigma comprise “chasms” of low-JCN deletions at late-replicating fragile sites, enriched in gastrointestinal carcinomas. Tyfonas are “typhoons” of high-JCN junctions and fold-back inversions enriched in expressed protein-coding fusions and breakend hypermutation, frequently found in acral, but not cutaneous, melanomas. Clustering of tumors according to genome graph-derived features identified subgroups associated with DNA repair defects and poor prognosis.
Keywords: Cancer genomics, Structural variation, DNA rearrangements, Mutational Processes, Genome graphs, Copy number alterations
Introduction
Cancer genomes are shaped by mutational processes that create both simple and complex structural variants (SVs). Whereas simple SVs (e.g. deletions, duplications, translocations, and inversions) arise through the breakage and fusion of 1 or 2 genomic locations, complex SVs can cause multiple (> 2) DNA junctions in distinct topologies within the reference genome that yield one or more copies of complex rearranged alleles (Maciejowski and Imielinski, 2017). Though several mechanisms have been proposed to explain complex SV patterns (chromothripsis (Stephens et al., 2011), chromoplexy (Baca et al., 2013), templated insertion chains (TIC) (Spies et al., 2017; Li et al., 2020), breakage fusion bridge cycles (BFBCs) (Zakov et al., 2013)), the field has not yet converged to a single algorithmic framework to identify the full spectrum of these patterns in a tumor whole genome sequence. In addition, it is unclear whether some of the clustered rearrangement patterns commonly observed in cancer represent variations on known events (e.g. “amplified” chromothripsis) or as yet uncharacterized event classes (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020; Li et al., 2020). As a result, the mutational processes driving complex SV evolution are still largely unknown.
The detection of variant junctions (each a pair of oriented genomic locations, or breakends, made adjacent in one of four configurations: deletion-like (DEL-like), duplicationlike (DUP-like), inversion-like (INV-like), or translocationlike (TRA-like, Fig. S1A) in whole genome sequencing (WGS) is routine. However, the classification of junctions into events (e.g. deletion, duplication, inversion, translocation) becomes difficult when two or more junctions are near each other. Furthermore, rearrangements and copy number alterations (CNA) are usually analyzed and interpreted separately, despite being facets of a single genome structure. As a result, quantitative details of the copy number state are only loosely integrated with the junction topology when loci are classified into events.
In WGS, CNAs are detected as change-points (Chiang et al., 2009) in sequencing read depth along the genome (e.g. BIC-seq (Xi et al., 2011)) while rearrangements are nominated through the analysis of junction-spanning read pairs (e.g. SvABA (Wala et al., 2018), GRIDSS (Cameron et al., 2017)). CNAs and junctions are, however, intrinsically coupled, since every copy of every non-telomeric segment must have a left (towards smaller coordinates) and a right (towards larger coordinates) neighbor, whether that neighbor is already adjacent on the reference (via a reference or REF junction) or introduced through rearrangement (via a variant or ALT junction) (Medvedev et al., 2010; Greenman et al., 2012). Furthermore, though copy number (CN) is a concept primarily applied to describe the dosage of genomic intervals, a junction may also be present in one or more copies per cell, and thus be associated with a junction copy number (JCN).
Elevated JCN might occur through the focal (e.g. extrachromosomal) amplification or whole chromosome (or genome) duplication of an already rearranged allele (Fig. 1A). The inference of JCN in WGS involves the fitting of a genome graph to read depth and junction data through the application of a junction balance constraint, which requires the CN of every genomic interval (i.e. vertex) to be in balance with the JCN of its neighboring junctions (i.e. edges) (Fig. S1B–C) (Medvedev et al., 2010; Greenman et al., 2012; Oesper et al., 2012; Li et al., 2016; Dzamba et al., 2017; McPherson et al., 2017). We hypothesized that the topology and CN of both vertices and edges on junction-balanced genome graphs might reveal novel classes of complex SV events and mutational processes.
Figure 1. Junction-balanced genome graphs enable complex structural variant characterization.
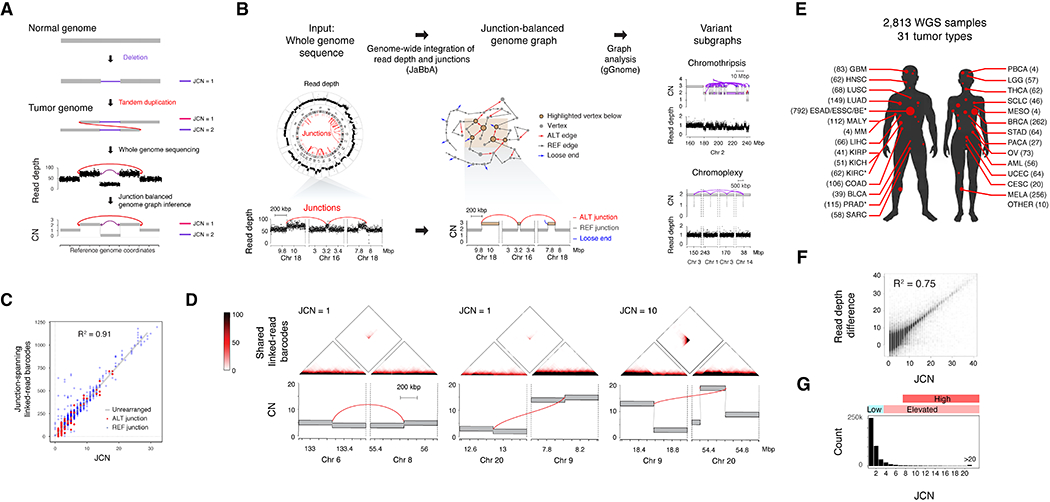
(A) Schematic of elevated junction copy number (JCN) from the duplication of an allele harboring a DEL-like junction, resulting in a characteristic read depth and junction pattern from which a junction-balanced genome graph can be reconstructed. (B) Junction balance analysis (JaBbA) integrates binned WGS read depth data and junctions to estimate JCN and generates coherent models of rearranged genome structure. The topology of JCN can be used to define complex SV events as variant subgraphs. (C) The coverage of junction-spanning 10X linked-read barcodes in HCC1954 (y axis) correlates highly with the JaBbA JCN estimate, obtained through the analysis of HCC1954 short-read WGS (x axis). (D) Top, heatmap of the number of 10X linked-read barcodes shared between each pair of 1 kbp genomic bins. Bottom, JaBbA genome graphs within 400 kbp of the featured junctions. (E) Tumor type sample counts across 2,778 genome graphs. See Table S1 for abbreviations. * marks datasets with multiple samples. (F) Correlation of the purity and ploidy corrected read depth difference vs. JaBbA-fitted JCN. (G) Histogram of JCNs and their associated categories in the cohort. See also Fig. S1 and Tables S1–S3
Results
JaBbA accurately infers junction-balanced genome graphs.
The inference of junction-balanced genome graphs is a computationally challenging combinatorial optimization problem that others have approached using probabilistic graphical models (PGM) and mixed-integer programming (MIP) (Medvedev et al., 2010; Greenman et al., 2012; Oesper et al., 2012; Li et al., 2016; Dzamba et al., 2017; McPherson et al., 2017). A serious impediment to applying these approaches to the study of complex SVs is their inability to produce CN profiles of comparable quality to non-graph based CNA callers (e.g. CONSERTING (Chen et al., 2015), BIC-Seq (Xi et al., 2011) while also inferring JCN across a wide range of tumor purities. The inference of junction-balanced genome graphs is a computationally challenging combinatorial optimization problem that others have approached using probabilistic graphical models (PGM) and mixed-integer programming (MIP) (Medvedev et al., 2010; Greenman et al., 2012; Oesper et al., 2012; Li et al., 2016; Dzamba et al., 2017; McPherson et al., 2017). A serious impediment to applying these approaches to the study of complex SVs is their inability to produce CN profiles of comparable quality to non-graph based CNA callers (e.g. CONSERTING (Chen et al., 2015), BIC-Seq (Xi et al., 2011)) while also inferring JCN across a wide range of tumor purities.
We developed an algorithm, JaBbA, to infer junction-balanced genome graphs with high fidelity. Analogous to previous approaches, we define a (directed) genome graph of vertices representing strands of genomic segments and edges representing a pair of 3’ and 5’ DNA ends that are adjacent in the reference genome (REF edge) or connected through rearrangement (ALT edge). A junction balanced genome-graph assigns every vertex and edge in the graph an integer CN, while enforcing the constraint that the dosage of every interval (vertex) is equal to the sum of the JCN of the incoming (or similarly, outgoing) junctions (Fig. S1B). The MIP optimization solved by JaBbA minimizes the residual between observed read depth and inferred interval dosage through joint assignment of CN to intervals and junctions (Fig. 1B, Fig. S1C, STAR Methods).
JaBbA differs primarily from previous genome graph methods in its robust modeling of WGS read depth noise and explicit accounting of false negative junctions, also called loose ends (see STAR Methods). In our benchmarking experiments, JaBbA inferred JCN with consistently higher fidelity than published genome graph-based methods (ReMixT (McPherson et al., 2017), Weaver (Li et al., 2016), PREGO (Oesper et al., 2012)) across a wide range of tumor purities (Fig. S1D–E). In addition, JaBbA qualitatively outperformed genome graph-based methods and even classic (i.e. non-graph based) CNA callers (BIC-seq (Xi et al., 2011) and CONSERTING (Chen et al., 2015)) in estimating interval CN and change point locations across a wide range of tumor purities (Fig. S1F–I). These results show that JaBbA is the first genome graph SV caller to accurately infer the topology of JCN while approaching (or exceeding) the fidelity of classic CNA callers.
We also validated JaBbA’s estimates of JCN from short-read WGS with 10X Chromium linked-read WGS, which provides a more direct readout of JCN with its high number of long junction-spanning fragments. In the breast cancer cell line HCC1954, JaBbA-derived JCN estimates closely correlated with the coverage of junction-spanning linked-read barcodes (R2 = 0.91, Fig. 1C). This included examples of low JCN (e.g. JCN = 1) junctions connecting both low and high CN intervals, as well as high JCN (JCN = 10) junctions (Fig. 1D). These results show that JCN is a property that can be robustly inferred from short-read WGS and is independent from (interval) CN.
Pan-cancer analysis of junction-balanced genome graphs.
To investigate the topology of JCN across cancers, we assembled a dataset comprising 2,813 short-read WGS tumor or cell line samples spanning 31 primary tumor types (Table S1–S3), including WGS for 539 previously unpublished cases (Table S2). In total, our analysis included 1,648 WGS samples not included in the Pan-Cancer Analysis of Whole Genomes (PCAWG) effort (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020). Application of harmonized pipelines followed by JaBbA (Fig. 1B) yielded 2,778 high quality genome graphs (Fig. 1E, see STAR Methods for sample characteristics and exclusion criteria).
Analyzing junction-balanced genome graph topology, we identified subgraphs associated with previously identified complex rearrangement patterns such as chromothripsis, chromoplexy, and TICs (Fig. 1B, right panel) implementing criteria described in previous publications within our framework (see STAR Methods). Consistent with our 10X Chromium WGS benchmarks (see above) (Fig. 1C), we observed wide variation in inferred JCN across our datasets which correlated with observed read depth changes at breakends (Fig. 1F). While the vast majority of junctions demonstrated low-JCN (JCN ≤ 3), we observed a long tail of junctions with elevated– (JCN > 3) and high-JCN (JCN > 7) (Fig. 1G).
Low-JCN junctions cluster into towers and chasms.
To distinguish between complex SV patterns associated with low-JCN vs. high-JCN junctions, we identified junction clusters based on their overlapping footprints on the reference and labeled each cluster as high- / low- JCN on the basis of its highest copy junction (high-JCN thresholded at > 7). Considering clusters harboring three or more junctions, we found low-JCN clusters were significantly more likely to be dominated (> 90% representation of that type) by DEL-like (P < 2.2 × 10−16, z-test, logistic regression) or DUP-like junctions (P < 2.2 × 10−16) (Fig. S2A).
To rigorously nominate clusters of low copy DUP-like and DEL-like junctions within each tumor sample, we identified genomic tiles (1 Mbp width, 500 kbp stride) enriched in low-JCN of a given type (e.g. DEL-like) relative to a calibrated gamma-Poisson background model (see STAR Methods). In each analysis, non-outlier tiles harbored apparent simple deletions or duplications (Fig. 2A–B). In contrast, outlier bins in the DUP-like model resembled “towers” of low-JCN DUP-like junctions (Fig. 2A, top right) that we named pyrgo (πύργος, Greek meaning tower). Outlier loci in the DEL-like models comprised subgraphs of interval CN “chasms” flanked by low-JCN DEL-like junctions whose interval CN often reached 0 (Fig. 2B, top left). We named these patterns rigma (ρήγμα, Greek meaning rift).
Figure 2. Rigma and pyrgo are novel patterns of clustered low copy rearrangements.
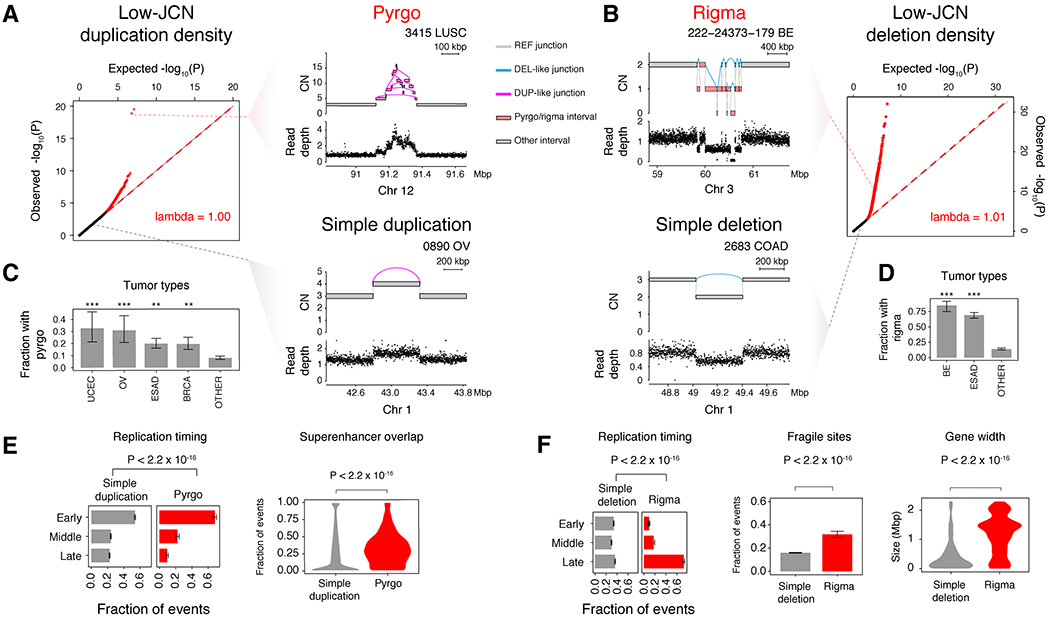
(A) Quantile-quantile (Q-Q) plot of observed vs. expected −log10(P) quantiles obtained from a gamma-Poisson model of low-JCN DUP-like junction density across genomic bins (see STAR Methods). Red dots indicate sample-specific model outliers (FDR<0.5). Top right, an example of a pyrgo-associated outlier window with a high density of low-JCN DUP-like rearrangements within a sample. Bottom right, a non-outlier window containing a simple duplication event. (B) Right, Q-Q plot similar to (A), but low-JCN DEL-like junctions. Top left, an example of a rigma-associated outlier window containing a high density of low-JCN DEL-like junctions within a sample. Bottom left, a non-outlier window containing a simple deletion event. (C-D) Fraction of samples within tumor types that harbor pyrgo and rigma, respectively. Significantly enriched tumor types (compared to all others) are marked by asterisks (Fisher’s exact test). Significance levels: *** (P < 1 × 10−3), ** (P < 0.01), * (P < 0.05). (E-F) Association of events with replication timing (ordinal logistic regression), superenhancers (Hnisz et al., 2013) (logistic regression), known fragile sites (Kumar et al., 2019) (logistic regression), and gene width (Wilcoxon rank-sum test, Bonferroni corrected P-values < 0.05). Error bars on bar plots represent 95% confidence intervals on the Bernoulli trial parameter. See also Fig. S2
We found a significantly increased burden of pyrgo in endometrial, ovarian, breast, and esophageal adenocarcinoma (ESAD) (Bonferroni P < 0.05, Fisher’s Exact test, Fig. 2C), while an excess of rigma was found in Barrett’s esophagus (BE) cases and ESAD (Bonferroni P < 0.05, Fig. 2D). Compared to simple duplications, pyrgo were significantly enriched in early replicating regions (P < 2.2 × 10−16, OR = 1.36, z-test, ordered logistic regression) and superenhancers as defined by (Hnisz et al., 2013) (P < 2.2 × 10−16 OR = 8.09, z-test, logistic regression) (Fig. 2E). In contrast, rigma were significantly enriched in late replicating regions (P < 2.2 × 10−16, OR = 1.5, ordered logistic regression), fragile sites (P < 2.2 × 10−16, OR = 2.45, z-test, logistic regression),6 and long genes (>P < 2.2 × 10−16, RR = 1.31, z-test, log-linear regression) relative to simple deletions (Fig. 2F). These genomic distribution patterns indicate that rigma and pyrgo are driven by distinct mutational processes and/or forces of somatic selection from those driving simple deletions and duplications.
Recurrent hotspots of rigma and pyrgo.
To nominate loci that were recurrently targeted by pyrgo and rigma across independent patients, we employed fishHook (Imielinski et al., 2017) while correcting for genomic covariates (replication timing, gene width, and superenhancer status). Applying fishHook to find recurrent pyrgo hotspots across 8,642 non-overlapping previously annotated superenhancer regions (Hnisz et al., 2013) (see STAR Methods), we found 16 loci were significantly mutated above background (FDR < 0.25) in the setting of a calibrated model fit (λ = 1.03, Fig. S2B).
This included a superenhancer associated with the oncogene MYC, targeted by pyrgo in 14 cases spanning 6 cancer types, including 7 esophageal adenocarcinomas (ESAD) (Fig. S2C). Additional recurrent targets of pyrgo included manually annotated Sanger Cancer Gene Census (CGC) genes (Sondka et al., 2018) associated with published GISTIC amplification peaks (Fig. S2B (Zack et al., 2013)), with the MYC gene body being the most enriched. Interestingly, pyrgo enrichment around superenhancers was found even outside of pyrgo hotspots (Fig. S2D, P = 2.2 × 10−16, OR = 1.5, z-test, Bayesian logistic regression). We also note that the minority (33%) of pyrgo intersected CGC genes.
Applying fishHook to analyze rigma recurrence across 18,794 unique protein-coding genes, we found 17 genes significantly mutated above background (FDR < 0.25, λ = 1.03) even after correcting for replication timing, gene width, and fragile site status (Fig. S2E). Among the top loci surviving false discovery correction, we found previous hotspots of genomic fragile sites (FHIT, WWOX, and MACROD2) (Zack et al., 2013; Bignell et al., 2010; Beroukhim et al., 2010; Iliopoulos et al., 2006; Fungtammasan et al., 2012; Cheng et al., 2017) and bona fide tumor suppressors (e.g. CDKN2A, Fig. S2E–F). Nevertheless, only 12% of rigma outside of known fragile sites were associated with a known cancer gene. FHIT rigma loci in esophageal adenocarcinoma and Barrett’s esophagus demonstrated a visually recurrent topology, often targeting the central exon in the gene (Fig. S2G).
Rigma gradually accumulate deletions in trans.
We investigated whether rigma were distinct from chromothripsis, another clustered rearrangement pattern associated with DNA loss (Stephens et al., 2011). Compared to rigma, chromothripsis events were one to two orders of magnitude larger in size (chromothripsis median size: 45.07 Mbp; rigma median size: 0.53 Mbp) (Fig. 3A, upper panel, P < 2.2 × 10−16, Wilcoxon rank-sum test). Analyzing the allelic structure that is latent in the JaBbA graphs, we searched each chromothripsis– or rigma-associated subgraph for a single path or allele that held the most ALT junctions in cis (see STAR Methods). We found that chromothripsis subgraphs usually yielded alleles that placed a higher proportion of the event-associated junctions in cis relative to rigma (Fig. 3A, lower panel, P < 2.2 × 10−16, Wilcoxon rank-sum test). These results indicate that rigma-associated DEL-like junctions are more likely to occur in trans.
Figure 3. Rigma junctions evolve gradually and in trans.
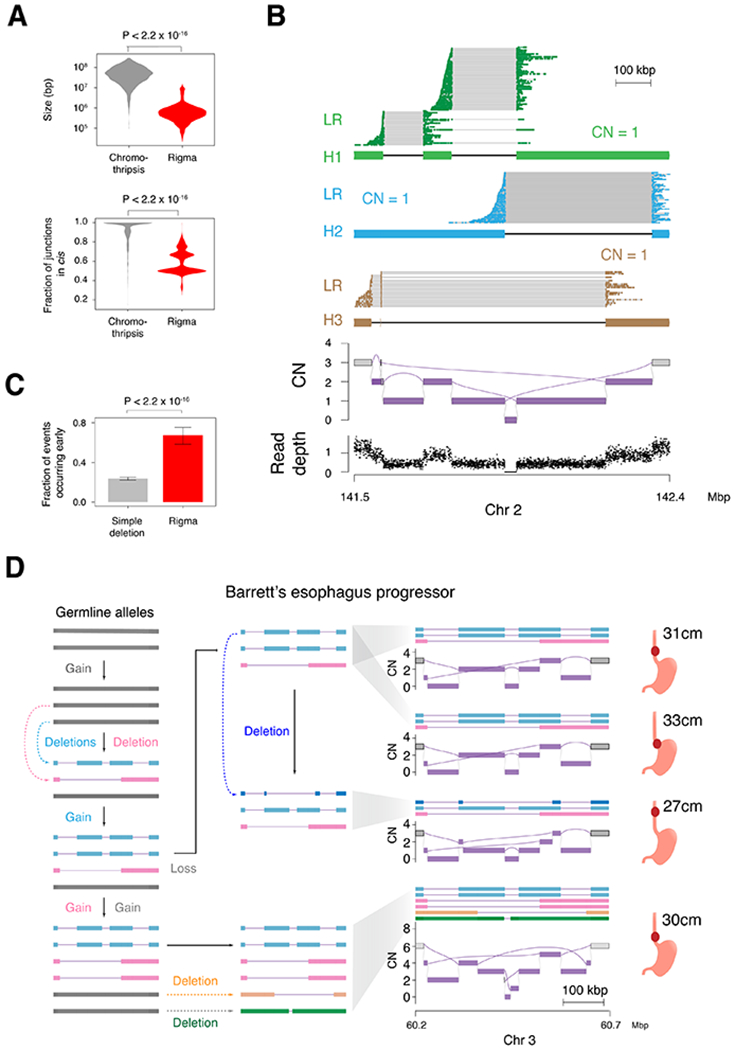
(A) Top, comparison of the total genomic territory covered by chromothripsis and rigma footprints. Bottom, the fraction of rearrangements that occur in cis (i.e. on the same predicted haplotype) when the longest possible contigs are inferred from the JaBbA graph. (B) Linked-read (LR) sequencing and local allelic deconvolution of a rigma found in a lung adenocarcinoma cell line (NCI-H838, see STAR Methods) demonstrates DEL-like junctions occur on separate haplotypes (i.e. in trans). (C) A comparison of the fraction of events that occur early (i.e. in multiple samples from the same patient) for simple deletions and rigma in the BE cohort, which has a median of 4 samples per patient across 80 patients. (D) Reconstruction of haplotypes across multiple samples from a single case in the BE WGS dataset. P-values obtained by Wilcoxon rank-sum test in (A) and Fisher’s exact test in (C). RD, read depth. Error bars on bar plots represent 95% confidence intervals on the Bernoulli trial parameter. See also Fig. S3
We then asked whether rigma junctions were likely to reside in the same clone, or as single DEL-like junctions in separate competing clones. We generated simulations in which one aneuploid clone harboring a 5-junction rigma (“rigma clone”) in FHIT was mixed with random proportions of 5 near-identical clones, each harboring a single DEL-like junction (taken from another FHIT rigma). Applying JaBbA across 250 simulations with varying rigma clone fractions, we only detected a rigma when the rigma clone reached 50% or greater cancer cell fraction (Fig. S3A–B). Interestingly, we found similar results for analogous clone-mixing experiments for pyrgo (Fig. S3C–D), indicating that pyrgo junctions also likely occur in the same cell. Taken together, these results indicate that JaBbA-derived SV patterns belong to the same dominant or ancestral cancer clone, and also that rigma junctions were likely to reside in the same clone, but on different alleles.
To validate the latter, we generated 10X Chromium WGS for a rigma-harboring Cancer Cell Line Encyclopedia (CCLE) cell line in our cohort (NCI-H838). Integration of long range contiguity data in the linked-read barcodes enabled unambiguous deconvolution of alleles in the JaBbA genome graph (see STAR Methods). Our allelic reconstruction supported the existence of three linear alleles in trans orientation (Fig. 3B, see STAR Methods), each with a CN of 1. Of the four DEL-like junctions associated with this rigma, all but one pair were in trans.
Given the enrichment of rigma in BE and ESAD, we probed whether these events occurred early or late in esophageal cancer evolution. Analyzing 340 multi-regionally and longitudinally sampled biopsies taken from 80 BE cases, we found that rigma were significantly more likely to be found in two or more biopsies relative to simple deletions (P < 2.2 × 10−16, OR = 6.69, Fisher’s exact test) (Fig. 3C). Reconstructing the allelic evolution in an early rigma case, we found evidence for successive accumulation of DEL-like junctions, with DEL-like junctions appearing on alleles that had already harbored a previous deletion (Fig. 3D). These results suggest that rigma represent a gradual SV mutational process that targets late replicating fragile sites and occurs early in BE / ESAD evolution.
Subgraphs of high-JCN junctions reveal genomic typhoons.
We then sought to investigate the rearrangement patterns associated with high-JCN junctions (JCN > 7) in our genome graphs. A priori, a junction at such an extreme of JCN may evolve through a double minute, BFBC, or an as yet undescribed mechanism for duplicating already rearranged DNA. To characterize classes of amplification events associated with these high-JCN junctions, we first identified 12,588 subgraphs harboring an interval CN of at least twice ploidy among the 2,487 unique genome graphs (by patient) (Fig. 4A), then identified among these amplicons (amplified clusters within a genome) those that harbor at least one junction with JCN > 7. We annotated the resulting 1,703 high-JCN amplicons according to several features: 1) the maximum JCN normalized by the maximal interval CN, 2) the summed JCN associated with fold back inversion junctions (INV-like junctions that terminate and begin at nearly the same location in the genome) relative to the maximal interval CN, and 3) the number of junctions with elevated JCN (JCN>3) (see STAR Methods).
Figure 4. Analysis of amplified subgraphs identifies tyfonas.
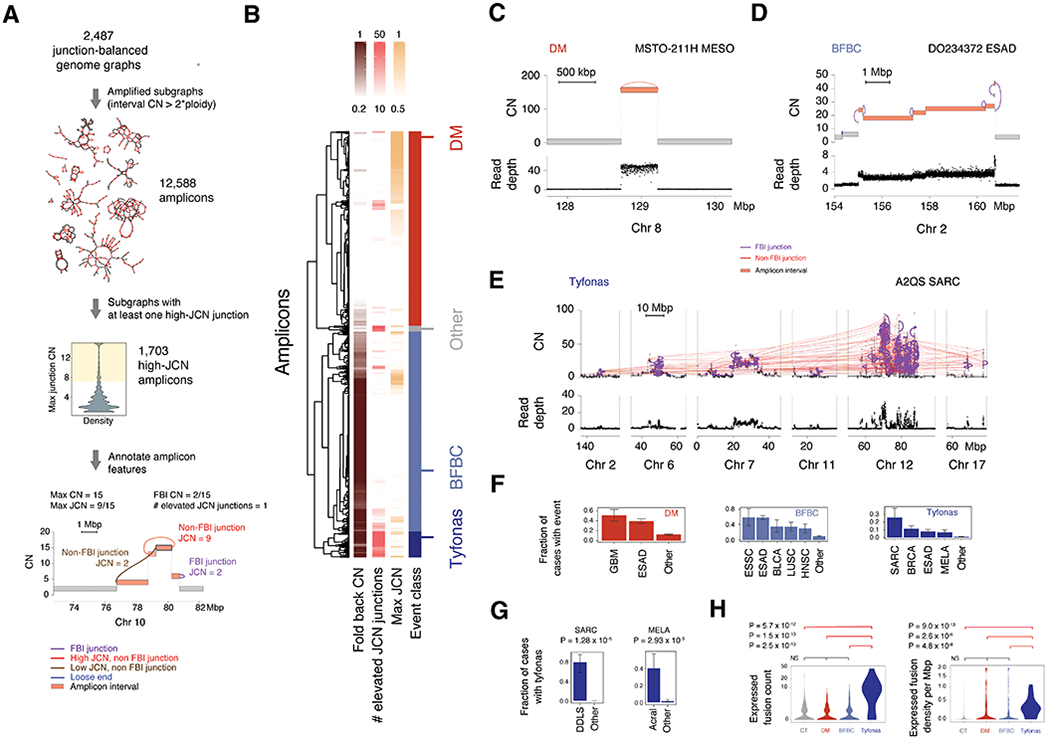
(A) Framework to identify features of complex amplicons through the analysis of amplified subgraphs. Each subgraph is annotated for three features: number of elevated (JCN>3) junctions, maximum JCN relative to the highest vertex CN in the subgraph, and total JCN of all fold back junctions relative to maximum JCN. (B) Hierarchical clustering of subgraph features reveals three stable clusters, representing distinct amplification SV classes: (C) double minutes, (D) BFBCs, and a new event pattern, (E) tyfonas. Top track in C-E is the JaBbA-estimated CN. Bottom, normalized read depth data. (F) Tumor types significantly enriched in each amplicon class. Significance determined by Fisher’s exact test comparing each tumor type against all others. All enrichments with FDR < 0.25 are shown. “Other” category denotes non-significant tumor types in the respective analysis. (G) Tyfonas enrichment in melanoma and sarcoma subtypes. (H) Enrichment of expressed protein-coding fusion transcripts by count (left) and density per Mbp of event territory (right) in tyfonas relative to other amplicon types and chromothripsis (Wilcoxon rank-sum test). Error bars on bar plots represent 95% confidence intervals on the Bernoulli trial parameter. See also Fig. S4
Clustering and classification of amplicons (Fig. 4B) on the basis of these three features yielded three stable clusters (Fig. S4A–B) (see STAR Methods). Upon visual inspection, the first group, harboring low fold-back JCN but high maximal JCN, contained amplicons comprising a single high-JCN junction forming a high CN circular path in the graph (Fig. 4C) as well as more complex cyclic patterns spanning multiple discontiguous loci, consistent with a double minute. The second group, demonstrating high fold-back JCN (> 0.5), a low burden of elevated-JCN junctions (< 26), and a “stair step” pattern of copy gains, was consistent with a BFBC (Fig. 4D) (Zakov et al., 2013). The third group contained both high fold-back JCN (≥0.50) and a significant burden of elevated JCN junctions (≥26). Upon visual inspection, these amplicons comprised dense webs of elevated JCN junctions across subgraphs compris > 100 Mbp of genomic material and often reaching CNs higher than 50 (Fig. S4C). We dubbed these extremely large amplicons, which did not fit in previously defined categories, tyfonas (τύφωνας, Greek meaning typhoon) (Fig. 4E).
Tyfonas are distinct from BFBCs and double minutes.
Comparing amplicon features, we found that tyfonas had significantly larger genomic mass (summed interval width weighted by CN), maximum interval CN, junction burden, and JCN entropy (based on the histogram of JCN in the amplicon) than either double minutes or BFBCs (P < 2.2. × 10−16 , Wilcoxon rank-sum test) (Fig. S4C). All three amplicon classes frequently intersected CGC cancer genes residing within GISTIC pan-cancer amplification peaks (Fig. S4D). EGFR was the most frequent target of double minutes. BFBCs were most frequently implicated in ERBB2, CCND1, and CCNE1 amplification. Tyfonas were associated with MDM2 and CDK4. While double minutes were enriched in glioblastoma and small cell lung cancer, BFBCs were enriched in esophageal squamous cell cancer, lung squamous cell cancer, and head and neck squamous cell cancer (FDR < 0.25, Fisher’s exact test, Fig. 4F). In contrast, tyfonas events were enriched in sarcoma, breast cancer, and melanoma (FDR < 0.25).
Analyzing tumor subtypes, we found tyfonas were common in dedifferentiated liposarcomas (80%) and acral melanomas (40%) but rarely seen (< 2%) in fibrosarcomas and cutaneous melanomas (Fig. 4G). The specificity of tyfonas for these tumor types was much higher than for liposarcoma-like or amplified chromothripsis, which are recently proposed PCAWG categories that also showed substantial overlap with BFBC and double minutes (Fig. S4E–K) (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020). 80% of dedifferentiated liposarcomas harbored a tyfonas on chromosome 12q, suggesting that tyfonas represent the genomic footprint of supernumerary ring chromosomes in this tumor type (Reimann and Fletcher, 2008). Given the enrichment of tyfonas events in acral melanoma, an immunotherapy-responsive melanoma with a low SNV burden (Shoushtari et al., 2016), we hypothesized that tyfonas events may provide an alternate source of neoantigens through the generation of expressed protein-coding fusions. Analyzing the subset of our samples with RNA-seq data, we found that tyfonas were significantly enriched with expressed fusion transcripts relative to double minutes, BFBCs, and even chromothripsis (CT) (Fig. 4H, left panel), even after accounting for the larger genomic footprint of tyfonas (Fig. 4H, right panel)
Tyfonas breakends are enriched in non-APOBEC SNV.
To further distinguish tyfonas from other complex SV types, we analyzed patterns of somatic SNVs around breakends. As expected, a breakend-centered coordinate system (Fig. 5A, see STAR Methods) revealed a robust peak of hypermutation within the first 1 kbp of breakends on the positive side of the axis. Comparing the intensity of these patterns between amplicon types revealed an enrichment of breakend hypermutation around tyfonas relative to BFBC, double minutes, and baseline (i.e. those not associated with a high-JCN amplicon) junctions, even after correcting for CN as a covariate (P < 2.2 × 10−16, RR = 1.64, z test, gamma-Poisson regression; Fig. S5).
Figure 5. Tyfonas are enriched in breakend hypermutation outside of SBS 2/13.
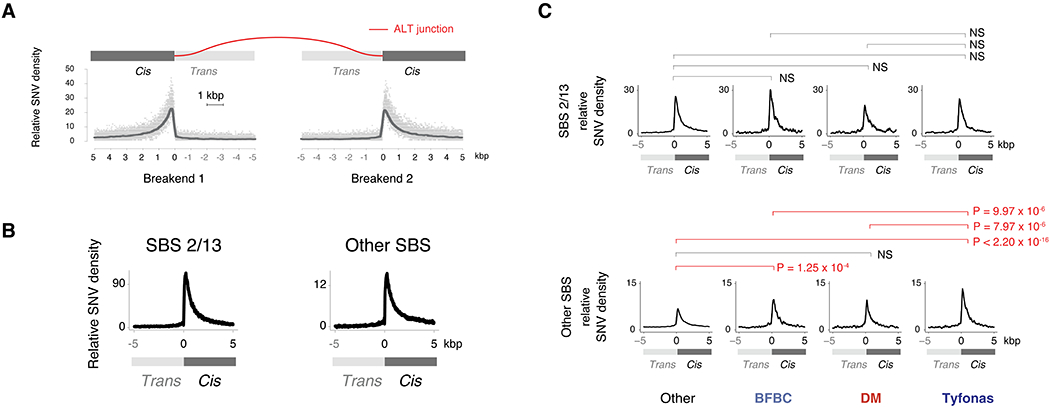
(A) Breakend-centered coordinate system to analyze mutational patterns near junctions. Top, the cis (+ coordinates) sides of the breakend (i.e. attached to the junction) have been fused across the rearrangement junction (red-colored line), while the trans (− coordinates) sides are disconnected from this derivative allele. Bottom, relative SNV density is the CN corrected and 101 bp smoothed density of SNVs at every base pair on this axis normalized to the average SNV density between −5 and 0 kbp. (B) Relative SNV density of SBS 2/13 and all other SBS associated contexts near breakends demonstrates a peak in the first 1 kbp on the cis side of the breakend. (C) Normalized breakend density for SBS 2/13 (top) and other SBS (bottom) SNVs for each of amplified event types. Enrichment P-values obtained from Wald test by gamma-Poisson regression (see STAR Methods). Significance determined by Bonferroni correction at a threshold of < 0.05. See also Fig. S5
Breakend hypermutation has previously been attributed to the activity of APOBEC cytidine deaminases (Roberts et al., 2013) and associated with COSMIC signatures SBS 2 / 13 (Alexandrov et al., 2020). However, we observed breakend hypermutation both within and outside of SBS 2/13 (Fig. 5B). Surprisingly, we found no significant differences in SBS 2/13-driven breakend hypermutation between tyfonas, BFBCs, double minutes, and baseline junctions (Fig. 5C, top row). In contrast, we found that tyfonas were enriched in non-SBS 2/13 breakend hypermutation relative to baseline (P < 2.2 × 10−16, RR = 1.60), BFBCs (P = 9.94 × 10−6, RR = 1.33), and double minutes (P = 7.97 × 10−6, RR = 1.40, Fig. 5C, bottom row). We obtained similar results with alternate APOBEC definitions (TpC contexts and GC-strand coordinated “kataegis” clusters). These results indicate that tyfonas breakends are preferentially exposed to a mutational process that does not satisfy current definitions of APOBEC mutagenesis.
Chromosomal integration of a small cell lung cancer tyfonas.
To more broadly probe the long-range structure of tyfonas amplicons, we selected a tyfonas-harboring small cell lung cancer cell line (NCI-H526) from the Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019b) for further profiling with short read WGS, Bionano optical mapping, Hi-C, and fluorescence in situ hybridization (FISH) (Fig. 6A). The WGS and BioNano-derived JaBbA graph (Fig. 6B, see STAR Methods) resolved all but 4 of the tyfonas-associated loose ends in the original WGS reconstruction, an improvement attributable to the additional sensitivity of optical mapping in repetitive regions (Fig. S6A). The reconstructed NCI-H526 tyfonas co-amplified MYCN (chr 2) to 68 and MYB (chr 6) to 47 intact copies, consistent with the marked over-expression of both genes (>99th percentile) relative to more than 1000 other cancer cell lines in the CCLE RNA-seq dataset (Fig. 6C).
Figure 6. Chromosomal integration of a MYB / MYCN -associated tyfonas in a small cell lung cancer cell line.
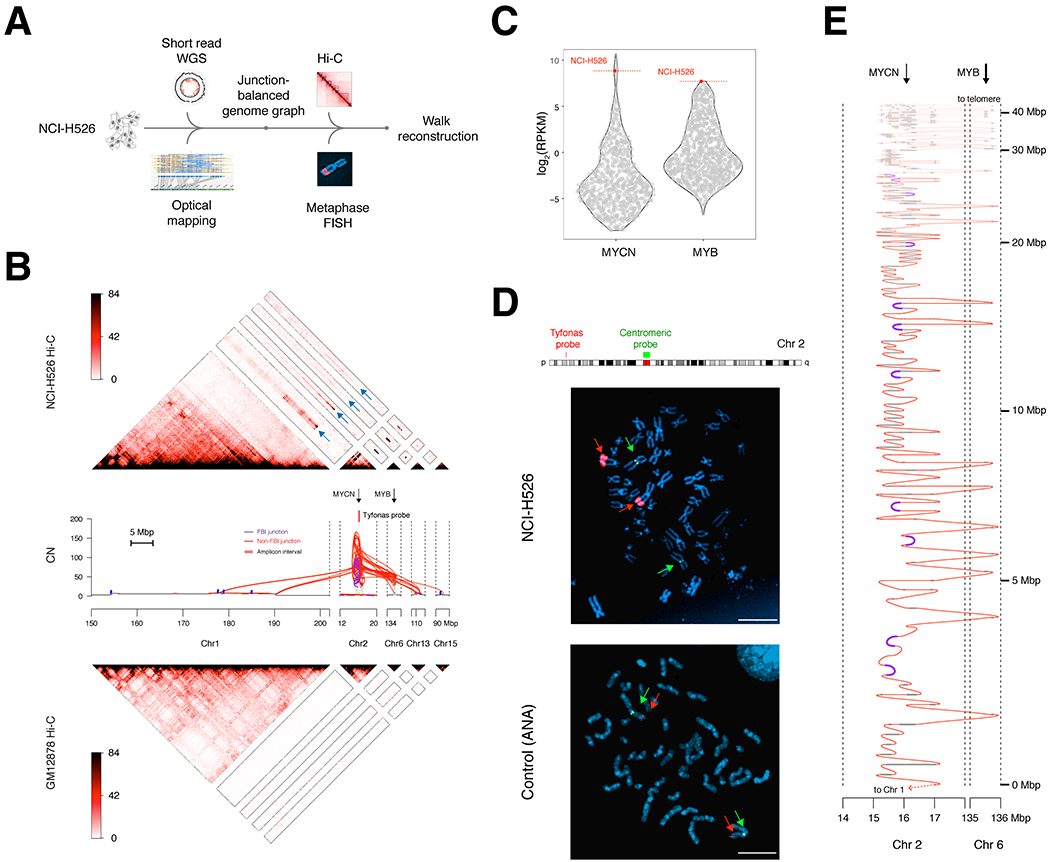
(A) Multi-platform profiling of a tyfonas in the small cell lung cancer cell line NCI-H526. (B) Top, heatmap of Hi-C in cell line NCI-H526 demonstrating MYCN and MYB co-amplification and contiguity with chromosome 1. Blue arrows on the Hi-C heatmap highlight the pixels supporting contiguity of tyfonas fragments with chromosome 1. Middle, JaBbA graph of the NCI-H526 tyfonas, where junction input was derived from short-read WGS and Bionano genomics optical mapping. The locations of MYCN, MYB, and the tyfonas BAC probes used for FISH experiments are shown. Bottom, Hi-C profiling of the normal cell line GM12878, obtained from (Rao et al., 2014). (C) Relative expression of MYCN and MYB across the Cancer Cell Line Encyclopedia (Ghandi et al., 2019b). (D) Metaphase FISH of NCI-H526 using a chromosome 2 probe targeting the center of the tyfonas near MYCN (red) and a second probe targeting the chromosome 2 centromere (green) (scale bars, 5 μm). (E) Candidate reconstruction of a linear allele spanning the tyfonas amplicon. The coordinates of the reconstructed allele (y axis) are shown using a nonlinear axis which begins at a junction adjacent to chromosome 1, and repeatedly threads between the MYCN and MYB genes, which are both amplified by the event. See also Fig. S6
We performed metaphase FISH using two sets of BAC probes targeting one of the most amplified portions of the tyfonas locus on chromosome 2p (Fig. 6D, Fig. S6B). We observed two homogeneously staining NCI-H526 foci of highly amplified probe signal, each with equal intensity, located on two linear chromosomal fragments that were discontiguous with the chromosome 2 centromere. To further derive the long-range architecture of the tyfonas locus, we generated Hi-C of NCI-H526. Hi-C 3D contacts identified a set of 5 Mbp bins on chromosome 1q as the genome-wide most proximal regions to the highly amplified tyfonas “seed region” on chromosome 2 (Fig. S6C). These bins were contiguous along a >40 Mbp piece of chromosome 1q (Fig. S6D), nominating this region as the most likely chromosomal integration site. These patterns were visually apparent in the NCI-H526 Hi-C contact map as foci of inter-chromosomal pixels, absent from the diploid cell line GM12878, connecting highly amplified tyfonas intervals on chromosomes 2, 6, 13, and 15 to abroad region of chromosome 1q (Fig. 6B).
Taken together, these results indicate that the amplified segments of the NCI-H526 tyfonas were arranged in cis in a linear allele that was integrated into chromosome 1q prior to a chromosomal duplication. To resolve this linear structure, we inferred a haplotype with multiplicity two that maximally accounted for all the copies of intervals and junctions in the tyfonas (Fig. 6E, see STAR Methods). This reconstructed allele threaded back and forth between many copies of MYB and MYCN, integrating many copies of fold back inversion junctions in cis.
Graph-derived events define distinct tumor clusters.
Tallying normalized junction counts across 13 event categories and 2,487 patients, we found 14 stable clusters using standard model selection metrics (Fig. 7A, Fig. S7A–B, Table S3). Most clusters were dominated by 1-3 event types (e.g. CT = chromothripsis, BR = BFBC, rigma, DDT = deletion, duplication, TIC) with the exception of two: QUIET (few events) and SPRS (sparse, miscellaneous events).
Figure 7. Genome graph-derived features define biologically distinct patient groups.
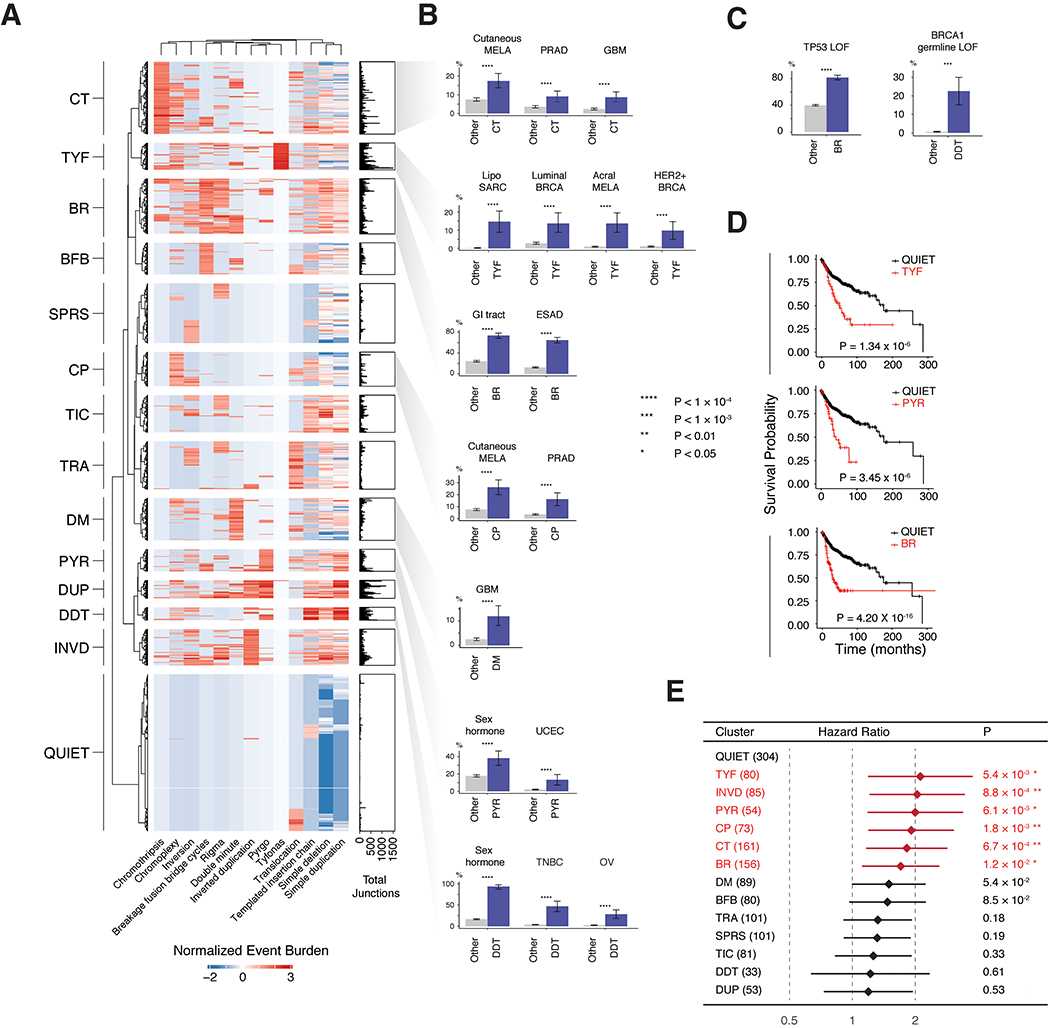
(A) Heatmap of normalized junction burden across 2,487 unique patients yields 14 clusters named after their most prevalent event types (CT, chromothripsis; TYF, tyfonas; BFB, BFBC; BR, BFBC and rigma; SPRS, sparse; CP, chromoplexy; TIC, templated insertion chain; TRA, translocations; DM, double minute; PYR, pyrgo; DUP, various duplications; DDT, deletion, duplication, and TIC; INVD, inverted duplications). (B-C) Significantly enriched tumor types within and outside of selected clusters. Blue (vs. gray) bars indicate the fraction of cases within (vs. outside of) the cluster that have the given feature, e.g. tumor type (B) or mutation (C). Significance in (B) and (C) determined by Bayesian logistic regression (Wald Test), with significant results (FDR < 0.1) for 742 hypotheses in (B) and 4,774 hypotheses in (C) (see STAR Methods). (D) Kaplan-Meier curves across BR, PYR and TYF clusters. P-values obtained via log-rank test. (E) Cox proportional hazard analysis correcting for age, tumor type, sex, metastasis status, tumor mutational burden, SV junction burden, and TP53 status demonstrating confidence bounds on the adjusted hazard ratio for 13 clusters relative to the QUIET cluster. Significantly associated variables (FDR < 0.1, 13 hypotheses) colored in red. Error bars on bar plots represent 95% confidence intervals on the Bernoulli trial parameter. See also Fig. S7 and Tables S3, S4
Consistent with previous reports, the CT cluster was significantly enriched in prostate adenocarcinoma (PRAD, P = 2.05 × 10−5, OR = 1.99, single-sided z-test, Bayesian logistic regression, (Kovtun et al., 2015)) and glioblastoma multiforme (GBM, P = 5.00 × 10−8, OR = 2.61, (Furgason et al., 2015)) (Fig. 7B). Similarly, the CP (chromoplexy) cluster was significantly enriched in PRAD (P = 2.32 × 10−10, OR = 3.18, (Baca et al., 2013)). DDT tumors (defined by high burdens of deletions, duplications, and templated insertion chains) were enriched in triple-negative breast cancer (TNBC) (P < 2.2 × 10−16, OR = 8.80), ovarian cancers (P = 7.03 × 10−16, OR = 6.89), and more broadly sex-hormone driven tumors (P = 3.18 × 10−14, OR = 19.0).
Inspection of the heatmap in Fig. 7A showed that the classes of complex SV introduced in this study (pyrgo, rigma, tyfonas) largely clustered independently from known complex SV types (double minute, BFBC, chromothripsis, chromoplexy). Among these, the BR (BFBC, Rigma dominated) cluster was primarily (60%) composed of ESAD cases (P < 2.2 × 10−16, OR = 6.08) and enriched in gastrointestinal tumors (e.g. esophageal, colorectal, and gastric adenocarcinoma) (P < 2.2 × 10−16, OR = 4.56) (Fig. 7B). The TYF (tyfonas dominated) cluster was enriched in both luminal breast cancer (P = 4.87 × 10−8, OR = 3.25), HER2+ breast cancer (P < 2.64 × 10−9, OR = 4.96), dedifferentiated liposarcoma (P < 2.2 × 10−16, OR = 24.5), and acral melanoma (P < 1.84 × 10−15, OR = 7.40). In contrast, cutaneous melanomas were enriched in the CT cluster. Additional associations are shown in Fig. S7C and Table S4.
We associated somatic or constitutional genotypes in CGC genes with cluster membership, after correcting for tumor subtype as a covariate. More than 20% of cases in the DDT cluster harbored constitutional (P = 1.60 × 10−4, OR = 3.81) loss of function lesions in BRCA1 (Fig. 7C). BR-cluster tumors were also significantly enriched in somatic TP53 mutations (P < 4.71 × 10−7, OR = 2.06). Additional somatic genotype associations (Fig. S7D), included an enrichment of SMC4 (P = 2.33 × 10−3, OR = 3.11) and RAD21 (P = 1.96 × 10−3, OR = 3.27) mutations in the INVD (inverted duplication dominant) cluster, ARID1AM (P = 1.21 × 10−3, OR = 1.86) mutations in the TIC (templated insertion chain dominant) cluster, and KMT2C (P = 2.89 × 10−3, OR = 1.95) mutations in the TRA (translocation-dominated) cluster.
Kaplan-Meier analysis revealed poor survival among novel SV-class dominated clusters (BR, PYR, and TYF) (Fig. 7D, FDR < 0.1, log rank test) as well as several clusters dominated by previously-described SV classes (CP, CT, and INVD) (Fig. S7E). These effects persisted after correcting for clinical and molecular covariates in a Cox regression analysis, with BR (P = 1.17 × 10−2, HR = 1.72, likelihood ratio test, Cox regression), PYR (P = 6.13 × 10−3, HR = 2.01), TYF (P = 5.37 × 10−3, HR = 2.12 , CP (P = 1.76 × 10−3; HR = 1.91), CT (P = 6.69 × 10−4; HR = 1.83), and INVD (P = 8.79 × 10−4; HR = 2.06) clusters each demonstrating reduced survival relative to the QUIET cluster (Fig. 7E, Fig. S7F).
Discussion
Our comprehensive analysis of 2,778 genome graphs establishes JCN topology as a key signal for the classification of complex SVs in human cancer. The regional, genotypic, and tumor type distribution of SV classes nominated in this study (pyrgo, rigma, tyfonas) are distinct from previously identified complex rearrangement patterns (chromothripsis, chromoplexy, BFBC), suggesting that they represent novel mutational processes and/or foci of somatic selection. We anticipate that these findings, combined with JaBbA’s superior performance relative to previous CNA algorithms, will bring the genome graph paradigm to the mainstream of cancer genome analysis.
Our data show that rigma likely arise from early and ongoing accumulation of DEL-like junctions at large and late replicating genes. Though rigma are overrepresented among previously annotated fragile sites defined by cell culture experiments (Schwartz et al., 2006; Fungtammasan et al., 2012; Ohta et al., 1996; Ma et al., 2012), the majority of rigma actually fall outside of these loci. An intriguing possibility is that rigma identified in our study represent previously unannotated in vivo genomic fragile sites. If so, future studies examining somatic rigma patterns as lineage-specific readouts of human genome fragility may shed insight into the biology of aging and developmental disease (e.g. autism, neurodegenerative conditions).
Previous studies have linked fragile sites to replication-transcription collisions in transcriptionally active genes (Helmrich et al., 2011; Wilson et al., 2015; Blin et al., 2019). The significant recurrence of rigma at FHIT and other fragilesite associated genes (e.g. WWOX, MACROD2) even after correcting for covariates suggests that replication timing and gene size do not fully account for their accumulation in hotspots. The preference of rigma for ESAD (and more broadly, GI adenocarcinomas) suggests that somatic rigma patterns in GI cancers may reflect epigenetic states of cell types in the healthy GI epithelium, which may be revealed through further study via single cell approaches or cell sorting and chromatin profiling.
While a subset of pyrgo hotspots (e.g. MYC superenhancers) may be functional and under positive somatic selection, the association of pyrgo with superenhancers is more likely due to a neutral mutational process that predisposes enhancers to (nested) duplications. Recent work suggests that enhancers may form cooperative multivalent chromatin structures (Hnisz et al., 2017), some of which may be prone to characteristic breakage and repair patterns (Canela et al., 2017). Further studies combining WGS and chromatin profiling in the same tumor sample will be required to assess the functional role of pyrgo formation in the evolution of cancer chromatin. More broadly, the explicit consideration of rigma and pyrgo patterns in future statistical background models of SV recurrence will be required to accurately nominate cancer driver loci in large cancer WGS datasets.
Though tyfonas likely represent the footprint of supernumerary rings in dedifferentiated liposarcomas, their prevalence in tumor types (e.g. acral melanomas, breast cancers) not commonly associated with supernumerary rings suggest that the cytogenetic footprint of this event may be more diverse. If our Hi-C, optical mapping, and FISH-based model of tyfonas (Fig. 6E) generalizes, a key question is how these chromosomally integrated loci acquire such a wide breadth of JCN in cis. We speculate that this allelic configuration may require a transient extrachromosomal phase, enabling the progressive self-assembly of many linear fragments ligating and duplicating in parallel across successive cell cycles. These asymmetrically segregating shards may preferentially ligate with their sister shard in end-to-end orientation after each S phase, which may give rise to the tyfonas-associated fold back inversion pattern.
The enrichment of breakend hypermutation across multiple non-APOBEC associated SNV contexts in tyfonas suggests that these junctions may arise in a specific mutagenic cellular environment. Though we cannot rule out the role of APOBEC enzymes in generating these SNV patterns (e.g. within a non-canonical context), they may also arise through error prone Pol-α fill-in or alt-NHEJ repair, both of which are active at unprotected DNA ends and generate mutations across a wide spectrum of trinucleotide contexts (Sfeir and de Lange, 2012; Mirman et al., 2018). We anticipate that future investigations in model systems will resolve some of these exciting mechanistic questions regarding tyfonas genesis.
Our study provides a template for integrating short-read WGS with long-range genome profiling to study patterns of somatic junction phase and allelic multiplicity in complex loci. Future studies that utilize long-range whole genome profiles (10X Chromium WGS, Pacific Biosciences, Oxford Nanopore Technologies, Hi-C, Bionano (Sedlazeck et al., 2018)) to phase highly rearranged somatic alleles may provide a more granular classification and mechanistic insight into complex SV mutational processes. Larger short-read meta-analyses spanning >10,000 cancer genome graphs, which may be imminently possible through the combination of our approach and dataset with PCAWG (1,493 additional tumors) (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020), Hartwig Medical Foundation (4000+ cases, (Priestley et al., 2019)), and emerging WGS precision medicine efforts will help link novel features of genome-graph topology with clinical variables, including therapeutic response. This includes future investigations linking the presence of tyfonas to the efficacy of immune checkpoint inhibition in acral melanoma or other tumor types in which these events are found (e.g. small cell lung cancer).
Supplementary Material
Acknowledgments
M.I.thanks Dan Landau for bio-hermeneutic support; Elli Papaemmanuil for consultation in Eλληνικά; Matthew Meyerson, Scott Carter, and Cheng-Zhong Zhang for discussions early in the development of JaBbA; Ron Zeira and Tomasz Imielinski for feedback on the JaBbA mathematical formulation. The authors thank Christopher Black and the NYGC ResComp team for high-performance computing support; Bionano genomics for assistance with optical mapping. M.I., X.Y, J.M.B., A.D., J.R., H.T, E.A., and Z.G. were supported by M.I.’s Burroughs Wellcome Fund Career Award for Medical Scientists, Doris Duke Clinical Foundation Clinical Scientist Development Award, Starr Cancer Consortium Award, National Institutes of Health (NIH) U24-CA15020, and Weill Cornell Medicine Department of Pathology and Laboratory Medicine startup funds. M.I., J.M., T.D.L., X.Y., and J.B. were supported by a Melanoma Research Alliance Team Science Award. K.H. was supported by a NIH/NCI F31 Graduate Research Fellowship (F31-CA232465). M.D. was supported by an NIH F32 training grant given to the Tri-Institutional Computational Biology and Medicine PhD Program. R.B. received funding from the Fund for Innovation in Cancer Informatics. B.J.R., T.P., X.L., P.G., C.S., K.O., M.K., and L.S. are supported by NIH funding (P01-CA9195). The IBM-NYGC Cancer Alliance project was supported in part by a grant from the IBM corporation (IBM Watson Health) to the New York Genome Center, New York Genome Center philanthropic funds and Rockefeller University grant UL1 TR000043 from the National Center for Advancing Translational Sciences (NCATS), and the National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program. T.G.P, P.C.G., K.M.O, C.A.S., B.J.R.,and X.L. were supported by NIH P01 CA91955 and P30 CA015704. M.K.K. and L.PS. were supported by NIH P01 CA91955. Graphical abstract was created with art from BioRender.com under academic license terms.
Footnotes
Declaration of Interests
J.S.R.-F. reports receiving personal/consultancy fees from VolitionRx, Paige.AI, Goldman Sachs, REPARE Therapeutics, GRAIL, Ventana Medical Systems, Roche, Genentech and InviCRO outside of the scope of the submitted work
References
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, PCAWG Mutational Signatures Working Group, Getz G, Rozen SG, Stratton MR. and PCAWG Consortium (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman T and Mishra B (2001). False positives in genomic map assembly and sequence validation. In International Workshop on Algorithms in Bioinformatics 27–40, Springer. [Google Scholar]
- Baca S, Prandi D, Lawrence M, Mosquera J, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald T, Ghandi M, Van Allen E, Kryukov G, Sboner A, Theurillat J-P, Soong T, Nickerson E, Auclair D, Tewari A, Beltran H, Onofrio R, Boysen G, Guiducci C, Barbieri C, Cibulskis K, Sivachenko A, Carter S, Saksena G, Voet D, Ramos A, Winckler W, Cipicchio M, Ardlie K, Kantoff P, Berger M, Gabriel S, Golub T, Meyerson M, Lander E, Elemento O , Getz G, Demichelis F, Rubin M and Garraway L (2013). Punctuated Evolution of Prostate Cancer Genomes. Cell. 153, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr , de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R and Garraway LA. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Henry KTM, Pinchback RM, Ligon AH, Cho Y-J, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q , Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao M-S, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR and Meyerson M (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C , Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, Teh BT, Deloukas P, Yang F, Campbell PJ, Futreal PA and Stratton MR (2010). Signatures of mutation and selection in the cancer genome. Nature 463, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin M, Le Tallec B, Nähse V, Schmidt M, Brossas C, Millot GA, Prioleau M-N and Debatisse M (2019). Transcription-dependent regulation of replication dynamics modulates genome stability. Nat. Struct. Mol. Biol 26, 58–66. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Schröder J, Penington JS, Do H, Molania R, Dobrovic A, Speed TP and Papenfuss AT (2017). GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 27 ,2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon K-R, Pekowska A, Zhang H, Rao SS, Huang S-C, Mckinnon PJ, Aplan PD, Pommier Y, Aiden EL, Casellas R and Nussenzweig A (2017). Genome Organization Drives Chromosome Fragility. Cell 170, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Gupta P, Wang J, Nakitandwe J, Roberts K, Dalton JD, Parker M, Patel S, Holmfeldt L, Payne D, Easton J, Ma J, Rusch M, Wu G, Patel A, Baker SJ, Dyer MA, Shurtleff S, Espy S, Pounds S, Downing JR, Ellison DW, Mullighan CG and Zhang J (2015). CONSERTING: integrating copy-number analysis with structural-variation detection. Nat. Methods 12, 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Demeulemeester J, Wedge DC, Vollan HKM, Pitt JJ, Russnes HG, Pandey BP, Nilsen G, Nord S, Bignell GR, White KP, Børresen-Dale A-L, Campbell PJ, Kristensen VN, Stratton MR, Lingærde OC, Moreau Y and Van Loo P (2017). Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors. Nat. Commun 8, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, Getz G, Jaffe DB, O’Kelly MJT, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M and Lander DS (2009). High-resolution mapping of copy-number alterations with massively parallel sequencing. Nature Methods 6, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z, Ruan J, Gao M, Zhou W, Chen T, Fan X, Ding L, Lee AY, Boutros P, Chen J and Chen K (2017). novoBreak: local assembly for breakpoint detection in cancer genomes. Nat. Methods 14, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghannasiri R, Freeman DE, Jordanski M, Hsieh GL, Damljanovic A, Lehnert E and Salzman J (2019). Improved detection of gene fusions by applying statistical methods reveals oncogenic RNA cancer drivers. Proc. Natl. Acad. Sci. U. S. A 116, 15524–15533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Walradt T, Hu Y, Koren A and Imielinski M (2019). Robust foreground detection in somatic copy number data. bioRxiv. Published online November 20, 2019. 10.1101/847681. [DOI] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES and Aiden EL (2016). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamba M, Ramani AK, Buczkowicz P, Jiang Y, Yu M, Hawkins C and Brudno M (2017). Identification of complex genomic rearrangements in cancers using CouGaR. Genome Res. 27, 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z andEklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol 26, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankell AM, Jammula S, Li X, Contino G, Killcoyne S, Abbas S, Perner J, Bower L, Devonshire G, Ococks E, Grehan N, Mok J, O’Donovan M, MacRae S, Eldridge MD, Tavard S and Fitzgerald RC (2019). The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic. Nature Genetics 51, 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA andMakova KD (2012). A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Research 22, 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furgason JM, Koncar RF, Michelhaugh SK, Sarkar FH, Mittal S, Sloan AE, Barnholtz-Sloan JS and Bahassi BM (2015). Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience 2, 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C , Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA and Sellers WR. (2019a). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, Weck A. d., Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA and Sellers WR (2019b). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman CD, Pleasance ED, Newman S, Yang F, Fu B, Nik-Zainal S, Jones D, Lau KW, Carter N, Edwards PAW, Futreal PA, Stratton MR and Campbell PJ (2012). Estimation of rearrangement phylogeny for cancer genomes. Genome Research 22, 346–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, Biele J, Ding J, Le A, Rosner J, Shumansky K, Marra MA, Gilks CB, Huntsman DG, McAlpine JN, Aparicio S and Shah SP (2014). TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Research 24, 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, Chin SF, Turashvili G, Hirst M, Caldas C, Marra MA, Aparicio S and Shah SP (2012). Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Research 22, 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch A-M, Kakavand H, Alexandrov LB, Burke H, Jakrot V, Kazakoff S, Holmes O, Leonard C, Sabarinathan R, Mularoni L, Wood S, Xu Q, Waddell N, Tembe V, Pupo GM, Paoli-Iseppi RD, Vilain RE, Shang P, Lau LMS, Dagg RA, Schramm S-J, Pritchard A, Dutton-Regester K, Newell F, Fitzgerald A, Shang CA, Grimmond SM, Pickett HA, Yang JY, Stretch JR, Behren A, Kefford RF, Hersey P, Long GV, Cebon J, Shackleton M, Spillane AJ, Saw RPM, López-Bigas N, Pearson JV, Thompson JF, Scolyer RA and Mann GJ (2017). Whole-genome landscapes of major melanoma subtypes. Nature 545, 175. [DOI] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M and Tora L (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Li L, Myers JR and Marth GT (2012). ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Guler G, Han S-Y, Druck T, Ottey M, McCorkell KA and Huebner K (2006). Roles of FHIT and WWOX fragile genes in cancer. Cancer Letters 232, 27–36. [DOI] [PubMed] [Google Scholar]
- Imielinski M, Guo G andMeyerson M (2017). Insertions and Deletions Target Lineage-Defining Genes in Human Cancers. Cell 168, 460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P and Saunders CT (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nature Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Korbel J and Campbell P (2013). Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 152, 1226–1236. [DOI] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlic R, Ghosh S, Polak P, Eggan K and McCarroll SA (2014). Genetic variation in human DNA replication timing. Cell 159, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Murphy SJ, Johnson SH, Cheville JC and Vasmatzis G (2015). Chromosomal catastrophe is a frequent event in clinically insignificant prostate cancer. Oncotarget 6, 29087–29096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Nagpal G, Kumar V, Usmani SS, Agrawal P and Raghava GPS (2019). HumCFS: a database of fragile sites in human chromosomes. BMC Genomics 19, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ-K, Park S, Park H, Kim S, Lee J, Lee J, Youk J, Yi K, An Y, Park IK, Kang CH, Chung DH, Kim TM, Jeon YK, Hong D, Park PJ, Ju YS and Kim YT (2019). Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 177, 1842–1857. [DOI] [PubMed] [Google Scholar]
- Li H and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE, Imielinski M, Akdemir KC, Alvarez EG, Baez-Ortega A, Beroukhim R, Boutros PC, Bowtell DDL, Brors B, Burns KH, Campbell PJ, Chen K, Chen K, Cortés-Ciriano I, Dueso-Barroso A, Dunford AJ, Edwards PA, Estivill X, Etemadmoghadam D, Feuerbach L, Fink JL, Frenkel-Morgenstern M, Garsed DW, Gerstein M, Gordenin DA, Haan D, Haber JE, Hess JM, Hutter B, Imielinski M, Jones DTW, Ju YS, Kazanov MD, Klimczak LJ, Koh Y, Korbel JO, Kumar K, Lee EA, Lee JJ-K, Li Y, Lynch AG, Macintyre G, Markowetz B , Martincorena I, Martinez-Fundichely A, Meyerson M, Miyano S, Nakagawa H, Navarro FCP, Ossowski S, Park PJ, Pearson JV, Puiggròs M, Rippe K, Roberts ND, Roberts SA, Rodriguez-Martin B, Schumacher SE, Scully R, Shackleton M, Sidiropoulos N, Sieverling L, Stewart C, Torrents D, Tubio JMC, Villasante I, Waddell N, Wala JA, Weischenfeldt J, Yang L, Yao X, Yoon S-S, Zamora J, Zhang C-Z, Weischenfeldt J, Beroukhim R and Campbell PJ (2020). Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou S, Schwartz DC and Ma J (2016). Allele-Specific Quantification of Structural Variations in Cancer Genomes. Cell Syst 3, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Qiu L, Mrasek K, Zhang J, Liehr T, Quintana LG and Li Z (2012). Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int. J. Mol. Sci 13, 11974–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J. and Imielinski M (2017). Modeling cancer rearrangement landscapes. Current Opinion in Systems Biology 1, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, Skoglund P, Lazaridis I, Sankararaman S, Fu Q, Rohland N, Renaud G, Erlich Y, Willems T, Gallo C, Spence JP, Song YS, Poletti G, Balloux F, van Driem G, de Knijff P, Romero IG, Jha AR, Behar DM, Bravi CM, Capelli C, Hervig T, Moreno-Estrada A, Posukh OL, Balanovska E, Balanovsky O, Karachanak-Yankova S, Sahakyan H, Toncheva D, Yepiskoposyan L, Tyler-Smith C, Xue Y, Abdullah MS, Ruiz-Linares A, Beall CM, Rienzo AD, Jeong C, Starikovskaya EB, Metspalu E, Parik J, Villems R, Henn BM, Hodoglugil U, Mahley R, Sajantila A, Stamatoyannopoulos G, Wee JTS, Khusainova R, Khusnutdinova E, Litvinov S, Ayodo G, Comas D, Hammer MF, Kivisild T, Klitz W, Winkler CA, Labuda D, Bamshad M, Jorde LB, Tishkoff SA, Watkins WS, Metspalu M, Dryomov S, Sukernik R, Singh L, Thangaraj K, Paabo S, Kelso J, Patterson N and Reich D (2016). The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson AW, Roth A, Ha G, Chauve C, Steif A, de Souza CPE, Eirew P, Bouchard-Côté A, Aparicio S, Sahinalp SC and Shah SP (2017). ReMixT: clone-specific genomic structure estimation in cancer. Genome Biol. 18, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Fiume M, Dzamba M, Smith T and Brudno M (2010). Detecting copy number variation with mated short reads. Genome Research 20, 1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D and Lange T. d. (2018). 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polαa-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JV (2010). Genomic mapping: A statistical and algorithmic analysis of the optical mapping system. University of Southern California. [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Loo PV, Ju YS, Smid M, Brinkman AB, Morganella S, Aure MR, Lingjærde OC, Langerød A, Ringnér M, Ahn S-M, Boyault S, Brock JE, Broeks A, Butler A, Desmedt C, Dirix L, Dronov S, Fatima A, Foekens JA, Gerstung M, Hooijer GKJ, Jang SJ, Jones DR, Kim H-Y, King TA, Krishnamurthy S, Lee HJ, Lee J-Y, Li Y, McLaren S, Menzies A, Mustonen V, O’Meara S, Pauporté I, Pivot X, Purdie CA, Raine K , Ramakrishnan K, Rodríguez-González FG, Romieu G, Sieuwerts AM, Simpson PT, Shepherd R, Stebbings L, Stefansson OA, Teague J, Tommasi S, Treilleux I, Eynden G. G. V. d., Vermeulen P, Vincent-Salomon A, Yates L, Caldas C, Veer L. v., Tutt A, Knappskog S, Tan BKT, Jonkers J, Borg A, Ueno NT, Sotiriou C, Viari A, Futreal PA, Campbell PJ, Span PN, Laere SV, Lakhani SR, Eyfjord JE, Thompson AM, Birney E, Stunnenberg HG, Vijver M. J. v. d., Martens JWM, Børresen-Dale A-L, Richardson AL, Kong G, Thomas G and Stratton MR (2016). Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesper L, Ritz A, Aerni SJ, Drebin R and Raphael BJ (2012). Reconstructing cancer genomes from paired-end sequencing data. BMC Bioinformatics 13, S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM and Huebner K (1996). The FHIT Gene, Spanning the Chromosome 3p14.2 Fragile Site and Renal Carcinoma–Associated t(3;8) Breakpoint, Is Abnormal in Digestive Tract Cancers. Cell 84, 587–597. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R and Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Schierz AC, Ward SE, Al-Lazikani B and Pearl FMG (2015). Therapeutic opportunities within the DNA damage response. Nature Reviews Cancer 15, 166–180. [DOI] [PubMed] [Google Scholar]
- Priestley P, Baber J, Lolkema M, Steeghs N, Bruijn E. d., Duyvesteyn K, Haidari S, Hoeck A. v., Onstenk W, Roepman P, Shale C, Voda M, Bloemendal H, Tjan-Heijnen V, Herpen C. v., Labots M, Witteveen P, Smit E, Sleijfer S, Voest E and Cuppen E (2019). Pan-cancer whole genome analyses of metastatic solid tumors. bioRxiv. Published online August 12, 2019. 10.1101/415133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V and Korbel JO (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann JDR and Fletcher CDM (2008). Chapter 37 - Soft-Tissue Sarcomas. In The Molecular Basis of Cancer (Third Edition), (Mendelsohn J, Howley PM, Israel MA, Gray JW and Thompson CB, eds), 471–477. W.B. Saunders. [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G and Gordenin DA (2013). An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nature Genetics 45, ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, Resnick MA and Gordenin DA (2012). Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 46, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS and Swanton C (2016). deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biology 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Zlotorynski E and Kerem B (2006). The molecular basis of common and rare fragile sites. Cancer Letters 232, 13–26. [DOI] [PubMed] [Google Scholar]
- Sedlazeck FJ, Lee H, Darby CA and Schatz MC (2018). Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346. [DOI] [PubMed] [Google Scholar]
- Sfeir A and de Lange T (2012). Removal of Shelterin Reveals the Telomere End-Protection Problem. Science 336, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoushtari AN, Munhoz RR, Kuk D, Ott PA, Johnson DB, Tsai KK, Rapisuwon S, Eroglu Z, Sullivan RJ, Luke JJ, Gangadhar TC, Salama AKS, Clark V, Burias C, Puzanov I, Atkins MB, Algazi AP, Ribas A, Wolchok JD and Postow MA (2016). The efficacy of anti-PD-1 agents in acral and mucosal melanoma. Cancer 122, 3354–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Weng Z, Bishara A, McDaniel J, Catoe D, Zook JM, Salit M, West RB, Batzoglou S and Sidow A (2017). Genome-wide reconstruction of complex structural variants using read clouds. Nature Methods 14, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin M-L, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA and Campbell PJ (2011). Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell. 144, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Schwartz DC, Zhou S and Waterman MS (2006). An algorithm for assembly of ordered restriction maps from single DNA molecules. Proceedings of the National Academy of Sciences 103, 15770–15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala JA, Bandopadhayay P, Greenwald N, O’Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X, Nusbaum C, Campbell P, Getz G, Meyerson M, Zhang C-Z, Imielinski M and Beroukhim R (2018). SvABA: genome-wide detection of structural variants and indels by local assembly. Genome research 28, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Arlt MF, Park SH, Rajendran S, Paulsen M, Ljungman M and Glover TW (2015). Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 25, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, Hadjipanayis AG, Luquette LJ, Kim T-M, Lee E, Zhang J, Johnson MD, Muzny DM, Wheeler DA, Gibbs RA, Kucherlapati R and Park PJ (2011). Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc. Natl. Acad. Sci. U. S. A 108, E1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang C-Z, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M and Beroukhim R (2013). Pan-cancer patterns of somatic copy number alteration. Nature Genetics 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakov S, Kinsella M and Bafna V (2013). An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc. Natl. Acad. Sci. U. S. A 110, 5546–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.