

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal chronic lung disease characterized by progressive scarring of the lung tissue, leading to respiratory failure. There is no cure for IPF, and current anti-fibrotic treatments modestly arrest its further progression. IPF prevalence and incidence increase with age, which is a recognized risk factor. Intense clinical and basic research over the last fifteen years has shown that hallmarks of accelerated aging are present in the lungs of patients with IPF. Different cell types in IPF lungs exhibit premature hallmarks of aging, including telomere attrition and cellular senescence. In this Review, we discuss recent insights into the mechanisms behind these age-related alterations and their contribution to the development of lung fibrosis. We focus on the genetic and molecular basis of telomere attrition in alveolar type II epithelial cells, which promote cellular senescence and lung fibrosis. Mechanistically, senescent cells secrete pro-fibrotic factors that activate scar-forming myofibroblasts. Ultimately, senescent alveolar epithelial cells lose their regenerative capacity, impeding fibrosis resolution. In addition, mitochondrial dysfunction is strongly associated with the appearance of senescent epithelial cells and senescent myofibroblasts in IPF, which persist in the fibrotic tissue by adapting their metabolic pathways and becoming resistant to apoptosis. We discuss emerging novel therapeutic strategies to treat IPF by targeting cellular senescence with the so-called senotherapeutics.
Keywords: Aging, Cellular senescence, Telomere attrition, Mitochondrial dysfunction, Myofibroblasts, Lung fibrosis, IPF
1. Lung aging
Aging is a progressive decline in biological and physiological functions characterized by a reduced capacity of organs to respond and adapt to environmental challenges. Aging is genetically determined but highly modulated by the environment [1] since external interventions, such as dietary restriction, can improve health during aging and extend lifespan in various animal models [2] and presumably in humans [3]. Age can be seen as a risk factor in a plethora of common chronic diseases [4], including cardiovascular and respiratory diseases, hypertension, cancer, diabetes mellitus, osteoporosis and osteoarthritis [5]. Strikingly, in the developed countries, over half the population 65 year old or older present more than one of those chronic diseases [6] and the prevalence of multiple conditions is rapidly increasing as the aged population grows. Lung pathologies are no exception and the prevalence of several chronic lung diseases (chronic obstructive pulmonary disease (COPD), most forms of lung cancer and idiopathic pulmonary fibrosis, (IPF)) has been found to increase considerably with age [7]. Chronic respiratory diseases are now the third leading cause of death in the US population over 65 years old (just behind heart disease and cancer). To understand how aging contributes to the development and progression of chronic lung diseases such as IPF, it is important to elucidate the molecular mechanisms of lung aging. While our understanding of aging biology has made tremendous progress in the last two decades, the molecular mechanisms linking aging with pulmonary diseases remain poorly understood.
Several changes in the morphology and the physiology of the lung appear with age, even in the absence of any disease. As we age, narrowing of the intervertebral spaces modifies the structure and shape of the thoracic cavity reducing the optimal lung volume. Aging is associated with reduced muscle strength that modifies the inspiratory and expiratory movements [8]. Also related with muscle atrophy, cough frequency is decreased in the elderly population. Particulate clearance from the lung through the mucociliary escalator is adversely affected and associated with ciliary dysfunction [9]. Aging lungs show increased lung compliance and reduced arterial partial pressure of oxygen (PO2) due to terminal airway closure and reduced cardiac output (caused by the decline in stroke volume and muscle O2 extraction) [10]. Age is also associated with a reduction in the lung elastic recoil, which may be associated with structural and functional shifts in the extracellular matrix (ECM) of the lung parenchyma and loss of alveolar surface area [11]. Lung diffusing capacity in older adults declines as result of a reduction in the alveolar-capillary surface area available for gas exchange, caused by both a decrease in number of pulmonary alveoli (increased pulmonary alveolar size with age) as well as a decrease in the density of pulmonary capillaries [12]. Numerous other age-related physiological changes could also influence lung injury responses, including altered baroreceptor activity and decreased adrenergic control of cardiovascular function [13]. Concomitantly, the immune system changes with age systemically (“inflammaging”, including a less active immune response from both the innate and adaptive immune systems) increasing the susceptibility to infections in the elderly [14].
2. IPF is an age-related chronic lung disease
Idiopathic Pulmonary Fibrosis is a progressive interstitial fibrotic lung disease, which accounts for up to 80 % of all interstitial lung diseases (ILD) [15]. IPF is a chronic, irreversible and fatal lung disease characterized by scarring and thickening of the interstitial lung tissue leading to dyspnea and, ultimately, respiratory failure [16]. IPF presents a mean survival of 3–5 years after diagnosis [17]. Two new FDA-approved therapies, pirfenidone and nintedanib, are modestly effective at reducing the decline of lung function over one-year follow-up [18–20]. New meta-analysis and extended 3-year survival studies show that pirfenidone has a survival benefit in an IPF cohort [21]. However, these novel anti-fibrotic therapies are still in their infancy and are not widely prescribed to patients with a milder or stable progression of the disease due to significant side effects [22,23].
The incidence of IPF changes moderately by continent. Based on large database studies over a decade, Europe presents an incidence of 5–9 cases per 100,000 person-year, 1–4 per 100,000 per year in Asia while North America shows an incidence of 9–30 cases per 100,000 people [24]. Incidence and mortality of IPF are rising consistently during the last decades. A UK study estimates that IPF incidence had increased annually by 11 % between 1991 and 2003 [25]). Aside from genetic mutations (mucin 5B, telomerase genes, surfactant proteins), environmental risk factors for IPF are smoking and occupational dust inhalations (farming, construction, etc.) [26]. IPF is more prevalent in males. Gastroesophageal reflux disease and chronic viral infections (hepatitis C and Epstein-Barr virus) have been suggested as other potential risk factors for IPF [27,28]. Notably, IPF increases its prevalence and incidence with age and typically occurs in the sixth and seventh decades of life. The risk of interstitial lung disease is 7 times higher in those aged over 70 than in their forties. [29]. Raghu et al. in 2006 estimated that the prevalence of IPF increased from 4 to 227 per 100,000 persons in the population 75 years or older compared to people younger than 35 years old [30]. The same team reviewed their epidemiology data a decade later and found that the estimated prevalence had now increased as high as 400 cases per 100,000 people in patients over 65 years old [31]. Several studies have confirmed the increasing trend of IPF prevalence with age [15]. As a result, IPF is now recognized as an age-related lung disease.
3. Cellular and molecular hallmarks of lung aging in IPF
The pathobiology of IPF is deeply linked to aging [32]. All the “hallmarks of aging”, including telomere attrition, genomic instability and epigenetic changes, loss of proteostasis, dysregulated nutrient sensing, mitochondrial dysfunction, stem cell exhaustion, altered cellular communication and cellular senescence, are present in IPF [33]. Age-related perturbations are found in epithelial cells, immune cells and fibroblasts from IPF lungs compared with age-matched cells from normal lungs [34]. Lung alveolar type II epithelial cells (AEC2, ∼ 5 % of the alveolar surface) are surfactant secretory cells that serve as stem cells following lung injury, thus requiring a high-energy turnover. AEC2 cells are very susceptible to age-related changes, especially telomere attrition, mitochondrial dysfunction and mitophagy dysregulation [35], which can directly affect their ability to repair the damaged lungs. Ultimately, pathological aging of AEC2 cells leads to loss of homeostasis [36], stem cell like exhaustion [34] and altered cell-to-cell communication with fibroblasts and immune cells [37]. Lung fibroblasts in IPF has been associated with the acquisition of a senescent phenotype, mitochondrial dysfunction and maladaptive epigenetic responses and attrition of telomeres [38,39]. Supporting this notion that telomere dysfunction is a critical player in the fragility of aged AEC2 in IPF [37], recent studies have shown that genetic ablation of the telomeric repeat-binding factor 1 (TRF1) leads to an age-driven development of lung fibrosis only when it is defective in AEC2 but not in lung fibroblasts [40,41]. Together, age-related perturbations appear to be distinguishable and mechanistically distinct in different cell types in the lungs. How these age-related changes contribute to the development and progression of lung fibrosis in a cell- (spatial control) and/or time- (temporal control) specific manner remains to be investigated in IPF. Then, we can question if IPF is an “accelerated aging” disease or the result of “exaggerated” aging [42]. Several studies have recently linked IPF pathogenesis with lung aging via telomere attrition [41] and mitochondrial dysfunction [35,43], ultimately leading to cellular senescence in AEC2 [41,44–47] and myofibroblasts [39,43,48–50]. In the following sections, we discuss the role of cellular senescence as a major driver of lung fibrosis as well as the molecular basis by which telomere attrition and mitochondrial dysfunction promote cellular senescence in the lungs (Fig. 1).
Fig. 1.
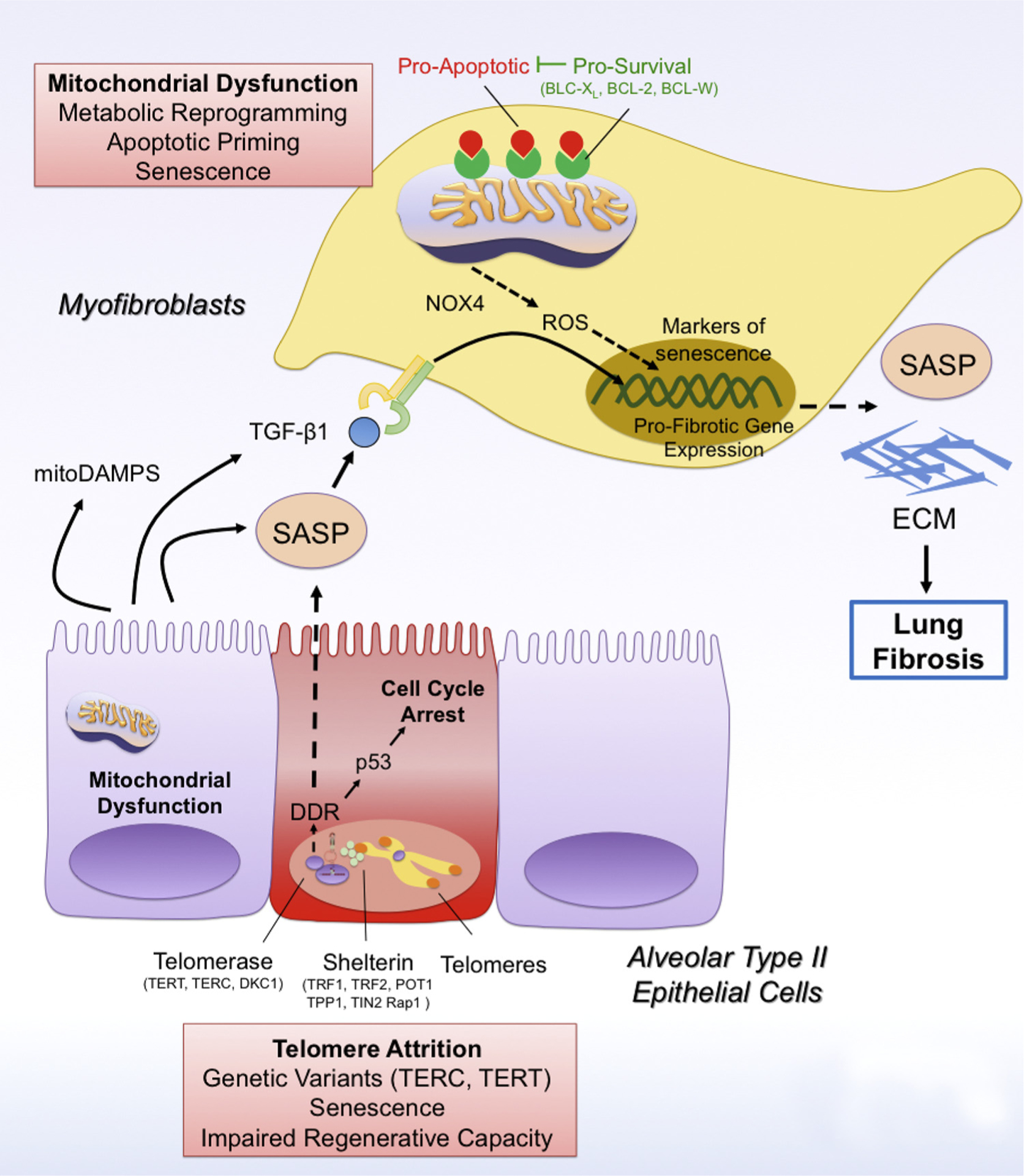
Telomere attrition and mitochondrial dysfunction drive cellular senescence in lung fibrosis.
Telomere attrition due to mutations in telomere genes (TERC, TERT) leads to the activation of the DNA-Damage Response (DDR) pathway, which drives p53-mediated cell cycle arrest and cellular senescence of type II alveolar epithelial cells (AEC2). In addition, PINK1 deficiency in AEC2 cells leads to mitochondrial dysfunction and in these cells. Senescent AEC2 cells are characterized by a senescence-associated secretory phenotype (SASP), which can spread cellular senescence to other AEC2 cells and myofibroblasts. In addition, the SASP has pro-fibrotic factors such as TGFβ1 and mitoDAMPs (such as IL6, mtDNA), which promote the activation of scar-forming myofibroblasts and the development of lung fibrosis. In addition, mitochondrial dysfunction can also drive cellular senescence in myofibroblasts in a NAPDH oxidase 4 (NOX4)/reactive oxygen species (ROS)- and p16-dependent manner by preventing mitophagy and promoting metabolic reprogramming in these cells. Damaged mitochondria in senescent myofibroblasts are characterized by increased levels of pro-apoptotic factors, thus increasing their apoptotic priming. Accordingly, senescent myofibroblasts upregulate expression of pro-survival mitochondrial proteins such as BCL-2, BCL-XL and BCL-W, which block pro-apoptotic signaling and ensure survival of senescent myofibroblasts.
3.1. Cellular senescence in IPF
During homeostasis, cellular senescence prevents the accumulation and proliferation of damaged cells, thus contributing to their clearance as part of the normal self-renewal process [51]. Senescent cell accumulation also occurs during embryonic development, tissue repair, chronic infections, or in response to certain medications and radiation exposure [52]. Senescent cells are characterized by irreversible cell-cycle arrest and loss of proliferative activity. In this state, cells change their morphology (becoming bigger with flattened appearance) and show higher resistance to apoptosis. Cellular senescence is triggered in response to numerous stressors, including telomere attrition (replicative senescence), DNA-damage, exposure to genotoxic agents, oxidative stress, mitochondrial dysfunction, nutrient deprivation and oncogene activation. On the molecular level, p53 and the retinoblastoma (RB) family of proteins promote cell cycle arrest in senescent cells via cyclin-dependent kinase (CDK) inhibitors such as p16, p15, p21 and p27 [53]. Of note, senescent cells are metabolically active and secrete a particular set of proinflammatory cytokines (e.g. IL1-beta and IL-6), chemokines (e.g. IL8), growth factors, and proteases, known as the senescence-associated secretory phenotypes (SASP) [54,55]. SASP can induce bystander cells to undergo senescence, a process called paracrine senescence.
In IPF, cellular senescence has been reported in AEC2 cells [56] and lung fibroblasts [39], a cellular phenotype associated with the secretion of a pro-fibrotic SASPs repertoire of cytokines and growth factors [57,58] that promote maladaptive wound healing responses [59] (Fig. 1). Senescent IPF fibroblasts secrete exaggerated amounts of extracellular matrix components, promoting the development of lung fibrosis [39]. The senescent phenotype of IPF lung fibroblasts associates with increased levels of both p16 and p21, a phenotype not driven by TGFβ1 [60]. While p21 expression is driven by telomere shortening-induced p53 activity, p16 is upregulated by telomere-independent, currently unknown mechanisms. More recently, single cell RNA sequencing (scRNA-seq) studies have shown an enrichment of senescent AEC2 cells in lung tissue from IPF patients, which may activate pro-fibrotic myofibroblasts by secreting TGFβ1 [61–63]. Moreover, one of the consequences of cellular senescence is the exhaustion of AEC2, the alveolar stem cells, leading to reduce regenerative capacity of these cells in the context of fibrogenic injury [64]. In addition, recent reports show that senescent bone marrow-derived mesenchymal stem cells (B-MSCs) from IPF patients can induce senescence in normal-aged fibroblasts, suggesting a possible link between senescent B-MSCs and the late onset of the disease [65]. The pathological role of senescent cells in lung fibrosis has been recently demonstrated in mouse models using pharmacological and genetic means [45]. Genetic ablation of p16-positive cells results in marked reduction on the expression of pro-fibrotic and pro-inflammatory cytokines, leading to improved lung function and fitness even in the absence of fibrotic reversal [49]. Administration of the senolytic drug quercetin, which induces apoptosis of senescent cells, has been shown to mitigate the progression of established pulmonary fibrosis in aged mice [66]. Having established the pathological role of senescent cells in lung fibrosis, here we discuss two mechanisms driving cellular senescence in IPF: telomere attrition and mitochondrial dysfunction.
3.2. Telomere attrition in IPF
Telomeres are repetitive DNA sequences found at the end of chromosomes that prevent chromosome fusion and confer protection to encoding DNA during chromosome replication [67]. Telomeres gradually shorten during each cell cycle due to incomplete replication of the telomere DNA, the so-called end replication problem. The telomeric DNA is incompletely replicated during each cell cycle by the telomerase complex, comprised of telomerase reverse transcriptase (TERT), telomerase RNA (TERC), and dyskerin (DKC1). Thus, telomere length associates with cellular age. Ultimately, telomere shortening leads to the so-called replicative senescence, which was first described in cultured fibroblasts in 1961 by Hayflick and Moorhead [68,69]. In this mechanism, short telomeres are sensed by shelterin, a six-subunit protein complex (comprising Telomeric Repeat Factor 1 (TRF1), Telomeric Repeat Factor 2 (TRF2), repressor/activator protein 1 (RAP1), TRF1-interacting nuclear protein 2 (TIN2), TIN2-interacting protein 1 (TPP1) and protection of telomeres 1 (POT1)) that activates the DNA damage response pathway (DDR) and induction of cellular senescence.
In familial cases of IPF, Armanios et al. first identified genetic variants encoding proteins relevant in telomere biology (TERT, TERC) [70], indicating that telomere shortening or dysfunction can be an important disease mechanism in IPF pathogenesis. More recently, a large re-sequencing study investigating genetic risk factors of IPF [71] has confirmed that rare variants in telomerase constitute a risk to develop both familial [70,72] and sporadic IPF [73–75], which associates with the development of cellular senescence in IPF. Further, IPF patients with shortened telomeres in leukocytes associate with worse survival after lung transplantation [76].
The molecular mechanisms that link telomere shortening and dysfunction to the development of pulmonary fibrosis are still poorly understood. Telomere shortening is significant in AEC2 cells and leukocytes in IPF [70], but not in lung senescent myofibroblasts [39]. These findings led to hypothesize that mutations in telomere biology drive cellular senescence in AEC2 cells [37], which promote the development of fibrosis by secreting pro-fibrotic factors and subsequent activation of scar-forming myofibroblasts (Fig. 1). This hypothesis has been investigated and recently confirmed in mice in which telomere dysfunction through genetic ablation of the shelterin proteins TRF1 or TRF2 in AEC2 cells leads to age-dependent DNA damage, cellular senescence and lung fibrosis, confirming that telomere shortening is a major disease driver of lung fibrosis [40,41]. More recently, the senescent phenotype of AEC2 cells has been also associated with diminished regenerative capacity due to reduced proliferative capacity [64]. Together, telomere shortening in AEC2 appears to be a driver mechanism of lung fibrosis by promoting AEC2 senescence, which is characterized by the secretion of pro-fibrotic mediators and reduced regenerative capacity. Besides genetic variants in telomere-maintenance genes, oxidative stress characterized by increased production of reactive oxygen species (ROS) have been shown to contribute to telomere shorting. Telomeres are sensitive to damage by oxidative stress, although the molecular mechanisms linking ROS with telomere attrition remain poorly understood [77,78]. Recent studies have shown that ROS induce the formation of 8-oxoguanine at telomeres but not other parts of the genome [79]. This telomeric 8-oxoguanine oxidative lesion leads to telomere fragility and shorting by inhibiting DNA repair processes. This mechanism has not yet been investigated in the context of lung fibrosis.
3.3. Mitochondrial dysfunction, bioenergetics and apoptosis in IPF
Mitochondrial dysfunction is characterized by reduced bioenergetic capacity, release of ROS, accumulation of NADH, and deregulated mitochondrial biogenesis and apoptosis. Of note, these processes lead to the acquisition of a senescence phenotype and SASP [80,81]. In IPF, dysfunctional mitochondria have been reported in both AEC2 cells, lung fibroblasts and alveolar macrophages [35,36,82]. We and others have shown that dysfunctional mitochondrial in AEC2 cells is driven, at least in part, by reduced expression of PTEN-induced putative kinase 1 (PINK1) (Fig. 1) [35]. PINK1 is a master regulator of mitochondrial homeostasis and plays a key role in mitophagy. PINK1 gets trapped in the outer membrane of damaged mitochondria where it can recruit and activate Parkin, a E3 ubiquitin ligase that promotes the degradation of those damaged mitochondria. Accordingly, PINK1-deficient mice present dysregulated homeostasis (mitochondria fusion and fission dynamics), accumulation of dysfunctional mitochondria, produce higher levels of mitochondrial ROS and susceptible to develop lung fibrosis compared to control mice [35]. Interestingly, PINK1 expression decreases with ER stress and age, thus increasing the predisposition of older lungs to develop fibrosis [83]. Mechanistically, decreased PINK1 expression in AEC2 leads to a senescent phenotype characterized by the secretion of pro-fibrotic TGFβ1 and other SASPs and mitoDAMPs (such as IL6, mtDNA) concomitantly with higher expression of p16 and p21 and higher susceptibility to apoptosis [35,57]. The role of PINK1 in lung fibroblasts is less understood, but recent reports indicate that TGFβ1-induced myofibroblast differentiation associates with reduced PINK1 expression and deregulated mitochondrial biology [84]. Myofibroblasts require energy for the synthesis and contraction of the ECM, which is sustained by metabolic reprogramming characterized by increased mitochondrial glycolysis [85]. Reduced activity of the energy sensor AMP-activated protein kinase (AMPK) drives mitochondrial dysfunction and metabolic reprograming in lung myofibroblasts, and restoration of AMPK activity with metformin restores mitochondrial biogenesis and treats established lung fibrosis in mice [86]. Mechanistically, mitochondrial biogenesis associates with restoration of myofibroblast apoptosis, suggesting that mitochondrial dysfunction underlies the apoptosis-resistant phenotype of myofibroblasts. In this line, we and others have recently shown that dysfunctional mitochondria in myofibroblasts are also characterized by increased levels of pro-apoptotic molecules in the outer mitochondrial membrane, increasing the readiness of myofibroblasts to activate mitochondrial apoptosis [87,88]. The molecular basis for this heightened mitochondrial apoptotic priming in myofibroblasts remains poorly understood, although augmented mitochondrial ROS has been proposed as major pro-apoptotic signaling molecules. Indeed, mitochondrial ROS signaling has been associated with the acquisition of a senescent phenotype in myofibroblasts, and that inhibition of the ROS-producing enzyme NADPH oxidase 4 protein (NOX4) reverses established lung fibrosis by targeting senescent myofibroblast for apoptosis [43]. Senescent myofibroblasts can still evade apoptosis by activating pro-survival mechanisms, which prevent pro-death signaling in dysfunctional mitochondria to execute the intrinsic pathway of apoptosis (Fig. 1) [87,89]. We and others have shown that the anti-apoptotic proteins BCL-2 and BCL-XL are upregulated in senescent myofibroblasts and sequester pro-apoptotic molecules in damaged mitochondria, ensuring myofibroblast survival besides being primed for death. Thus, myofibroblasts and senescent myofibroblasts become addicted to specific anti-apoptotic proteins for survival, and their inhibition with the so-called BH3 mimetic drugs unleashes the mitochondrial pathway of apoptosis in these cells [90]. Therapeutic strategies targeting pro-survival mechanisms in senescent myofibroblasts with senolytics are currently investigated for anti-fibrotic therapy, as discussed below.
4. Targeting cellular senescence in IPF: senolytics and senomorphics
A growing body of evidence associates aging and cellular senescence with the development and progression of IPF. Advances in cellular senescence and lung fibrosis research have led to the identification of novel potential targets and therapeutic approaches to treat lung fibrosis. In this section, we discuss the therapeutic potential of pharmacological strategies aimed at inducing apoptosis of senescent cells with senolytics or reversing the senescent phenotype with the so-called senomorphics (Fig. 2).
Fig. 2.
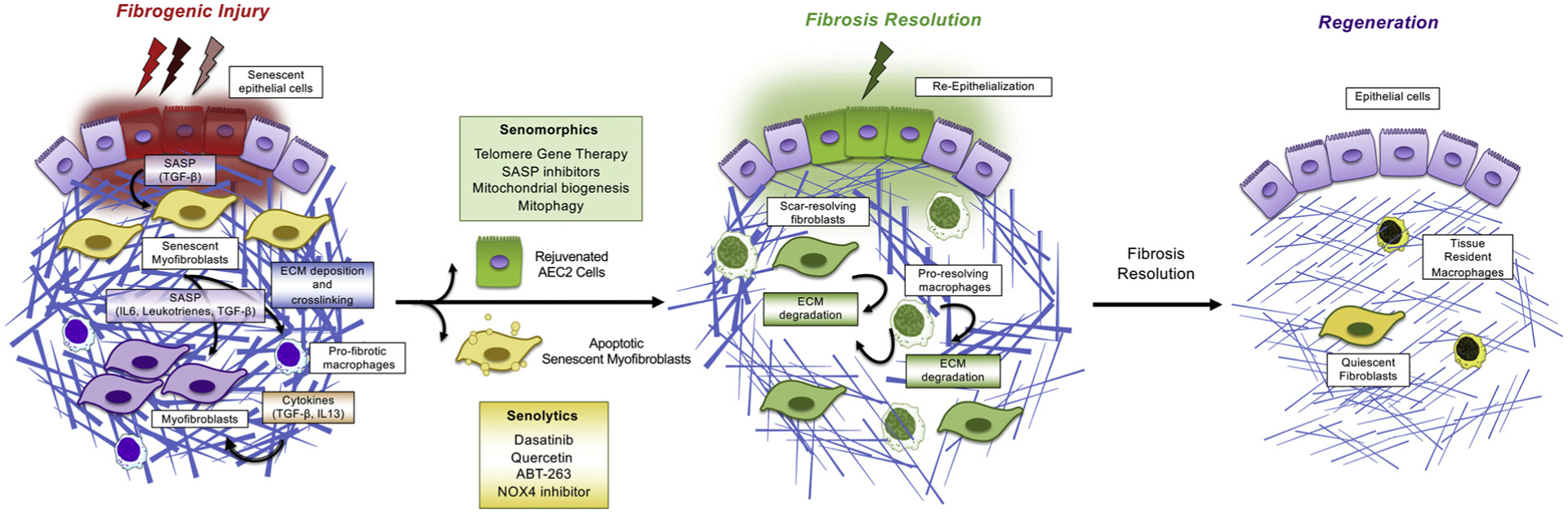
Emerging novel therapeutic strategies to treat IPF by targeting cellular senescence. IPF is a fatal progressive lung disease characterized by cellular senescence and excessive deposition of ECM proteins. Therapeutic targeting of senescent cells is emerging as a novel strategy to treat lung fibrosis. Clearance of senescent cells with the so-called senolytics (Dasatinib, Quercetin, ABT-263, NOX4 inhibitor) have been shown to mitigate lung fibrosis in mouse models and are now entering clinical trials. Alternatively, the so-called senomorphics have been also shown to treat lung fibrosis by interfering with detrimental SASP components (IL-6, TGFβ or leukotrienes). In addition, agents that improve mitochondrial biogenesis or activate mitophagy, such as activators of PINK1 or Sirt3, can potentially treat lung fibrosis by preventing the generation of senescent cells due to mitochondrial dysfunction. Senomorphics also include agents that can revert the senescent phenotype such as telomere gene therapy. Targeting shortened telomeres in senescent alveolar epithelial cells has been similarly shown to treat lung fibrosis in mice, suggesting that telomerase gene therapy may lead to rejuvenation of AEC2 cells and regeneration of the fibrotic lungs. Both senolytics and senomorphics have the potential to break the pro-fibrotic feedback loop between cellular senescence, SASP, myofibroblast activation, matrix deposition, and may initiate mechanisms of fibrosis resolution and tissue regeneration. Mechanisms of fibrosis resolution following anti-aging therapy remain poorly understood but likely involve matrix degradation mechanisms such as activation pro-resolving macrophages, ECM-resolving fibroblasts and alveolar re-epithelialization.
Senolytics
The so-called senolytic drugs induce apoptosis of senescent cells and have been shown to mitigate age-related disorders [91,92], fibrosis [49] and further diseases [93] in mouse models. The mechanism of action of these drugs is not fully understood but certainly target survival mechanisms that keep senescent cells alive. A seminal study published by Zhu et al. in 2015 [94] used a hypothesis-driven, bioinformatics-based approach to determine potential senolytic drugs. It was found that Dasatinib (a tyrosin kinase inhibitor well known for its role in treating myeloproliferative syndromes) and Quercetin (a flavonol component) and in particular its combination (DQ), improved age-related morbidity in mice. DQ therapy reduces senescence in both alveolar epithelial cells [45] and fibroblasts [49], and improves pulmonary outcomes in mice [45,49]. While the mechanisms by which these agents mediate selective apoptosis of senescent cells remain obscure - multiple cellular effects have been described -, their potential clinical usefulness has been pushed forward. Only recently, the first-in-man open-label pilot study with senolytics in IPF and in human in general has been published [95], showing that senolytics are safe and may improve physical function in patients with IPF, as evaluated by 6-min walk test. Additionally, DQ therapy has been recently shown to eliminate senescent cells and reduce experimental lung fibrosis [49,96]. However, senolytic therapy for the treatment of IPF must be tested carefully. Given the low number of AEC2 cells in the IPF lungs [64], elimination of senescent AEC2 cells may leave fibrotic lungs without any stem cells reservoir, preventing lung tissue repair. Thus, specific targeting of senescent myofibroblasts may represent a more selective strategy to treat lung fibrosis. In this regard, interference with the altered redox balance by targeting NOX4 enzyme has been shown to induce senescent myofibroblast apoptosis and reverses established lung fibrosis in mice [43]. The dual NOX1/4 inhibitor GKT137831 is going to be tested in a placebo-controlled, multicenter, randomized trial in IPF (NCT03865927). Myofibroblasts and senescent cells are primed for death due to increased mitochondrial priming, requiring expression of pro-survival BCL-2 proteins to ensure survival. This knowledge of mitochondrial priming has paved the way to investigate the therapeutic efficacy of BH3 mimetic drugs. This class of drugs directly targets pro-survival BCL-2 proteins including BCL-2, BCL-XL or MCL-1. Senescent cells have been recently shown to rely of BCL-W and BCL-XL for survival, suggesting that drugs targeting these pro-survival proteins may serve as senolytics agents [89]. Accordingly, we and others have shown that the BH3 mimetic drugs ABT 263 (blocking BCL-2, BCL-XL and BCL-W), ABT 737 (blocking BCL-XL and BCL-W) and A1331852 (blocking BCL-XL) induce apoptosis in myofibroblasts and senescent cells and treats fibrosis in mouse models [89,97–99] (Fig. 2). BH3 mimetics have not yet been investigated in clinical trials.
Senomorphics
The so-called senomorphics refer to therapeutic agents that can modulate the activities and functions of senescent cells without inducing their apoptosis [100]. It includes agents that can prevent the acquisition of a senescent phenotype by young cells, revert the senescent phenotype or interfere with detrimental SASP components or senescence-activated signaling pathways. Recent studies have shown that senomorphic agents can treat lung fibrosis by modulating the activity of senescent cells. For instance, targeting pro-fibrotic SASP components such as IL-6, TGFβ or leukotrienes has been shown to treat lung fibrosis in mouse models [58,101,102]. Likewise, targeting senescence-related pathways such as JAK-STAT mitigate lung fibrosis in mice [103]. In addition, agents that improve mitochondrial biogenesis or activate mitophagy, such as activators of PINK1 or Sirt3, can potentially treat lung fibrosis by preventing the generation of senescent cells due to mitochondrial dysfunction [56]. More recently, gene therapy has been also exploited as a therapeutic strategy to revert the senescent phenotype of AEC2s in lung fibrosis. As discussed above, genetic variants in telomere genes in AEC2 cells in IPF associates with cellular senescence, age-associated stem cell deficiency and reduced regenerative capacity [104]. Regenerative capacity may be regained by the therapeutic increase of telomere lengths in IPF AEC2s. In this line, recent studies have shown that telomerase adeno-associated virus (AAV) vector-based gene therapy delays aging and reduces age-relating lung disease including pulmonary fibrosis (Fig. 2) [105,106]. These studies provide a proof-of-concept that telomere gene therapy could treat lung fibrosis in IPF patients with short telomeres. This strategy has not yet been tested in humans and further research is needed to ensure that any telomere gene therapy brought into the IPF clinic is safe, selective and effective.
5. Summary and concluding remarks
Cellular senescence is a hallmark of age-related lung diseases including IPF. A fast-growing body of research links the appearance of senescent cells with the development of lung fibrosis, although the precise mechanisms by which senescent cells drive lung fibrosis remain poorly understood. Hallmarks of aging including telomere shorting, mitochondrial dysfunction and ROS production have been recently shown to drive a senescent phenotype in alveolar type II epithelial cells and myofibroblasts in IPF. In addition, recent studies have identified ECM proteins and pro-fibrotic factors in the SASP of senescent cells, suggesting that these cells are major drivers of IPF pathogenesis. Accordingly, genetic or pharmacological elimination of senescent cells with senolytic drugs have recently shown to ameliorate experimental lung fibrosis. In addition, senomorphics such as telomere-targeting therapies have been shown to rescue cellular senescence in alveolar epithelial cells and treats lung fibrosis in mice. Further, targeting SASP components appears to be another feasible strategy to block detrimental functions of senescent cells in IPF. Together, interest in cellular senescence in IPF has increased exponentially over the last few years. However, the biology of cellular senescence in IPF remains poorly understood. The molecular markers of senescent cells, and why and how these cells acquire their senescent phenotype in the context of IPF remains obscure. Further research is needed to understand the etiology of senescent cells in IPF as well as the mechanisms by which senescent cells and the SAPS promote the development and progression of lung fibrosis. Likewise, the use of senotherapeutics to treat lung fibrosis by eliminating, reversing or treating pathological functions of senescent cells require further research to ensure these emerging therapeutic strategies are both safe and effective in patients with IPF.
Acknowledgements
D.L. gratefully acknowledges funding support from the NIH (grant R01 HL147059-01), the Start-up Package from Massachusetts General Hospital, the Scleroderma Foundation New Investigator Grant, the Scleroderma Research Foundation Investigator-Initiated Research Grant, the American Thoracic Society Foundation/Pulmonary Fibrosis Foundation Research Grant and Sponsored Research Grants from Boehringer Ingelheim, Indalo Therapeutics and Unity Biotechnology. ALM acknowledges the support of the NIH (R01 HL131789-01) and the Aging Institute at the University of Pittsburgh.
Abbreviations:
- AAV
Adeno-associated virus
- AEC2
Alveolar type II epithelial cells
- AMPK
AMP-activated protein kinase
- B-MSCs
Bone marrow-derived mesenchymal stem cells
- COPD
Chronic obstructive pulmonary disease
- DDR
DNA damage response pathway
- DKC1
Dyskerin
- ECM
Extracellular matrix
- IPF
Idiopathic pulmonary fibrosis
- ILD
Interstitial lung diseases
- NOX4
NADPH oxidase 4 protein
- PO2
Partial pressure of oxygen
- POT1
Protection of telomeres 1
- PINK1
PTEN-induced putative kinase 1
- SASP
Senescence-associated secretory phenotypes
- scRNA-seq
Single cell RNA sequencing
- TERT
Telomerase reverse transcriptase
- TRF1
Telomeric Repeat Factor 1
- TRF2
Telomeric Repeat Factor 2
- TERC
Telomerase RNA
- TPP1
TIN2-interacting protein 1
- TIN2
TRF1-interacting nuclear protein 2
- ROS
Reactive oxygen species
- RAP1
Repressor/activator protein 1
- RB
Retinoblastoma
Footnotes
Declaration of Competing Interest
D.L. declares that he has received research funding from Boehringer Ingelheim, Indalo Therapeutics and Unity Biotechnology. Dr. Lagares has a financial interest in Mediar Therapeutics. The company is developing treatments for organ fibrosis. Dr. Lagares’s interests were reviewed and are managed by MGH and Partners HealthCare in accordance with their conflict of interest policies.
References
- [1].Flatt T, A new definition of aging? Front. Genet 3 (2012) 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Anderson RM, Le Couteur DG, de Cabo R, Caloric restriction research: new perspectives on the biology of aging, J. Gerontol. A Biol. Sci. Med. Sci 73 (1) (2017) 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Redman LM, et al. , Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging, Cell Metab. 27 (4) (2018) p. 805–815 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jaul E, Barron J, Age-related diseases and clinical and public health implications for the 85 years old and over population, Front. Public Health 5 (2017) 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Franceschi C, et al. , The continuum of aging and age-related diseases: common mechanisms but different rates, Front. Med. (Lausanne) 5 (2018) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hajat C, Stein E, The global burden of multiple chronic conditions: a narrative review, Prev. Med. Rep 12 (2018) 284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Budinger GRS, et al. , The intersection of aging biology and the pathobiology of lung diseases: a joint NHLBI/NIA workshop, J. Gerontol. A Biol. Sci. Med. Sci 72 (11) (2017) 1492–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lowery EM, et al. , The aging lung, Clin. Interv. Aging 8 (2013) 1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bailey KL, et al. , Oxidative stress associated with aging activates protein kinase Cepsilon, leading to cilia slowing, Am. J. Physiol. Lung Cell Mol. Physiol 315 (5) (2018) L882–L890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Roman MA, Rossiter HB, Casaburi R, Exercise, ageing and the lung, Eur. Respir.J 48 (5) (2016) 1471–1486 [DOI] [PubMed] [Google Scholar]
- [11].Subramaniam K, Kumar H, Tawhai MH, Evidence for age-dependent air-space enlargement contributing to loss of lung tissue elastic recoil pressure and increased shear modulus in older age, J. Appl. Physiol. (1985) 123 (1) (2017) 79–87. [DOI] [PubMed] [Google Scholar]
- [12].Coffman KE, et al. , Age-dependent effects of thoracic and capillary blood volume distribution on pulmonary artery pressure and lung diffusing capacity, Physiol. Rep 6 (17) (2018) p. e13834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mora AL, Rojas M, Aging and lung injury repair: a role for bone marrow derived mesenchymal stem cells, J. Cell. Biochem 105 (3) (2008) 641–647. [DOI] [PubMed] [Google Scholar]
- [14].Canan CH, et al. , Characterization of lung inflammation and its impact on macrophage function in aging, J. Leukoc. Biol 96 (3) (2014) 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martinez FJ, et al. , Idiopathic pulmonary fibrosis, Nat. Rev. Dis. Primers 3 (2017) 17074. [DOI] [PubMed] [Google Scholar]
- [16].Thannickal VJ, et al. , Mechanisms of pulmonary fibrosis, Annu. Rev. Med 55 (2004) 395–417. [DOI] [PubMed] [Google Scholar]
- [17].Collard HR, et al. , Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 168 (5) (2003) 538–542. [DOI] [PubMed] [Google Scholar]
- [18].Khalil N, et al. , Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis, Eur. Respir. J 53 (3) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mora AL, et al. , Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease, Nat. Rev. Drug Discov 16 (11) (2017) 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fleetwood K, et al. , Systematic review and network meta-analysis of idiopathic pulmonary fibrosis treatments, J. Manag. Care Spec. Pharm 23 (3-b Suppl) (2017) S5–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Margaritopoulos GA, et al. , Pirfenidone improves survival in IPF: results from a real-life study, BMC Pulm. Med 18 (1) (2018) 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Maher TM, et al. , Unmet needs in the treatment of idiopathic pulmonary fibrosis-insights from patient chart review in five European countries, BMC Pulm. Med 17 (1) (2017) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Diamantopoulos A, et al. , The burden of illness of idiopathic pulmonary fibrosis: a comprehensive evidence review, Pharmacoeconomics 36 (7) (2018) 779–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wakwaya Y, Brown KK, Idiopathic pulmonary fibrosis: epidemiology, diagnosis andOutcomes, Am. J. Med. Sci 357 (5) (2019) 359–369. [DOI] [PubMed] [Google Scholar]
- [25].Navaratnam V, et al. , The rising incidence of idiopathic pulmonary fibrosis in the U.K, Thorax 66 (6) (2011) 462–467. [DOI] [PubMed] [Google Scholar]
- [26].Raghu G, et al. , An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management, Am. J. Respir. Crit. Care Med 183 (6) (2011) 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tobin RW, et al. , Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 158 (6) (1998) 1804–1808. [DOI] [PubMed] [Google Scholar]
- [28].Moore BB, Moore TA, Viruses in idiopathic pulmonary fibrosis. Etiology and exacerbation, Ann. Am. Thorac. Soc 12 Suppl 2 (2015) S186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Choi WI, et al. , Risk factors for interstitial lung disease: a 9-year Nationwide population-based study, BMC Pulm. Med 18 (1) (2018) 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Raghu G, et al. , Incidence and prevalence of idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 174 (7) (2006) 810–816. [DOI] [PubMed] [Google Scholar]
- [31].Raghu G, et al. , Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11, Lancet Respir. Med 2 (7) (2014) 566–572. [DOI] [PubMed] [Google Scholar]
- [32].Selman M, et al. , Aging and interstitial lung diseases: unraveling an old forgotten player in the pathogenesis of lung fibrosis, Semin. Respir. Crit. Care Med 31 (5) (2010) 607–617. [DOI] [PubMed] [Google Scholar]
- [33].Gulati S, Thannickal VJ, The aging lung and idiopathic pulmonary fibrosis, Am. J. Med. Sci 357 (5) (2019) 384–389. [DOI] [PubMed] [Google Scholar]
- [34].Mora AL, Bueno M, Rojas M, Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis, J. Clin. Invest 127 (2) (2017) 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bueno M, et al. , PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis, J. Clin. Invest 125 (2) (2015) 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Torres-Gonzalez E, et al. , Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis, Am. J. Respir. Cell Mol. Biol 46 (6) (2012) 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Alder JK, et al. , Telomere dysfunction causes alveolar stem cell failure, Proc. Natl. Acad. Sci. U. S. A 112 (16) (2015) 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bai L, et al. , Glutaminolysis epigenetically regulates antiapoptotic gene expression in idiopathic pulmonary fibrosis fibroblasts, Am. J. Respir. Cell Mol. Biol 60 (1) (2019) 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Alvarez D, et al. , IPF lung fibroblasts have a senescent phenotype, Am. J. Physiol. Lung Cell Mol. Physiol 313 (6) (2017) L1164–L1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Povedano JM, et al. , Mice with pulmonary fibrosis driven by telomere dysfunction, Cell Rep. 12 (2) (2015) 286–299. [DOI] [PubMed] [Google Scholar]
- [41].Naikawadi RP, et al. , Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis, JCI Insight 1 (14) (2016) p. e86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Selman M, Pardo A, Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model, Am. J. Respir. Crit. Care Med 189 (10) (2014) 1161–1172. [DOI] [PubMed] [Google Scholar]
- [43].Hecker L, et al. , Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance, Sci. Transl. Med 6 (231) (2014) p. 231ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Disayabutr S, et al. , miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis, PLoS One 11 (6) (2016) p. e0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lehmann M, et al. , Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo, Eur. Respir. J 50 (2) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Selman M, Lopez-Otin C, Pardo A, Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis, Eur. Respir. J 48 (2) (2016) 538–552. [DOI] [PubMed] [Google Scholar]
- [47].Aoshiba K, Tsuji T, Nagai A, Bleomycin induces cellular senescence in alveolar epithelial cells, Eur. Respir. J 22 (3) (2003) 436–443. [DOI] [PubMed] [Google Scholar]
- [48].Minagawa S, et al. , Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells, Am. J. Physiol. Lung Cell Mol. Physiol 300 (3) (2011) L391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schafer MJ, et al. , Cellular senescence mediates fibrotic pulmonary disease, Nat. Commun 8 (2017) 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yanai H, et al. , Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients, Aging (Albany NY) 7 (9) (2015) 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Munoz-Espin D, Serrano M, Cellular senescence: from physiology to pathology, Nat. Rev. Mol. Cell Biol 15 (7) (2014) 482–496. [DOI] [PubMed] [Google Scholar]
- [52].Regulski MJ, Cellular senescence: what, why, and how, Wounds 29 (6) (2017) 168–174. [PubMed] [Google Scholar]
- [53].Chicas A, et al. , Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence, Cancer Cell 17 (4) (2010) 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Campisi J, Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors, Cell 120 (4) (2005) 513–522. [DOI] [PubMed] [Google Scholar]
- [55].Gorgoulis V, et al. , Cellular senescence: defining a path forward, Cell 179 (4) (2019) 813–827. [DOI] [PubMed] [Google Scholar]
- [56].Mora AL, et al. , Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease, Nat. Rev. Drug Discov 16 (11) (2017) 755–772. [DOI] [PubMed] [Google Scholar]
- [57].Bueno M, et al. , PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses, PLoS One 14 (6) (2019) p. e0218003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wiley CD, et al. , Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis, JCI Insight (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hamsanathan S, et al. , Cellular senescence: the trojan horse in chronic lung diseases, Am. J. Respir. Cell Mol. Biol 61 (1) (2019) 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Barnes PJ, Baker J, Donnelly LE, Cellular senescence as a mechanism and target in chronic lung diseases, Am. J. Respir. Crit. Care Med 200 (5) (2019) 556–564. [DOI] [PubMed] [Google Scholar]
- [61].Changfu Yao, et al. , Senescence of alveolar stem cells drives progressive pulmonary fibrosis, BioRxiv (2019). [Google Scholar]
- [62].Reyfman PA, et al. , Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis, Am. J. Respir. Crit. Care Med 199 (12) (2019) 1517–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Morse C, et al. , Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis, Eur. Respir. J 54 (2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liang J, et al. , Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice, Nat. Med 22 (11) (2016) 1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cardenes N, et al. , Senescence of bone marrow-derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis, Stem Cell Res. Ther 9 (1) (2018) 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hohmann MS, et al. , Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo, Am. J. Respir. Cell Mol. Biol 60 (1) (2019) 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].O’Sullivan RJ, Karlseder J, Telomeres: protecting chromosomes against genome instability, Nat. Rev. Mol. Cell Biol 11 (3) (2010) 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hayflick L, The limited in vitro lifetime of human diploid cell strains, Exp. Cell Res 37 (1965) 614–636. [DOI] [PubMed] [Google Scholar]
- [69].Hayflick L, Moorhead PS, The serial cultivation of human diploid cell strains, Exp. Cell Res 25 (1961) 585–621. [DOI] [PubMed] [Google Scholar]
- [70].Armanios MY, et al. , Telomerase mutations in families with idiopathic pulmonary fibrosis, N. Engl. J. Med 356 (13) (2007) 1317–1326. [DOI] [PubMed] [Google Scholar]
- [71].Moore C, et al. , Resequencing study confirms that host defense and cell senescence gene variants contribute to the risk of idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 200 (2) (2019) 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stuart BD, et al. , Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening, Nat. Genet 47 (5) (2015) 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tsakiri KD, et al. , Adult-onset pulmonary fibrosis caused by mutations in telomerase, Proc. Natl. Acad. Sci. U. S. A 104 (18) (2007) 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fingerlin TE, et al. , Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis, Nat. Genet 45 (6) (2013) 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dai J, et al. , Telomerase gene mutations and telomere length shortening in patients with idiopathic pulmonary fibrosis in a Chinese population, Respirology 20 (1) (2015) 122–128. [DOI] [PubMed] [Google Scholar]
- [76].Stuart BD, et al. , Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation, Lancet Respir. Med 2 (7) (2014) 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Correia-Melo C, Hewitt G, Passos JF, Telomeres, oxidative stress and inflammatory factors: partners in cellular senescence? Longev. Healthspan 3 (1) (2014) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Coluzzi E, et al. , Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells, PLoS One 9 (10) (2014) p. e110963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fouquerel E, et al. , Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis, Mol. Cell 75 (1) (2019) p. 117–130 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Passos JF, von Zglinicki T, Saretzki G, Mitochondrial dysfunction and cell senescence: cause or consequence? Rejuvenation Res. 9 (1) (2006) 64–68. [DOI] [PubMed] [Google Scholar]
- [81].Wiley CD, et al. , Mitochondrial dysfunction induces senescence with a distinct secretory phenotype, Cell Metab. 23 (2) (2016) 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Larson-Casey JL, et al. , Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis, Immunity 44 (3) (2016) 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bueno M, et al. , ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis, Aging Cell 17 (2) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sosulski ML, et al. , Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFbeta1, Aging Cell 14 (5) (2015) 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bernard K, et al. , Metabolic reprogramming is required for myofibroblast contractility and differentiation, J. Biol. Chem 290 (42) (2015) 25427–25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rangarajan S, et al. , Metformin reverses established lung fibrosis in a bleomycin model, Nat. Med 24 (8) (2018) 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lagares D, et al. , Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis, Sci. Transl. Med 9 (420) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zhou Y, et al. , Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis, J. Clin. Invest 123 (3) (2013) 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yosef R, et al. , Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL, Nat. Commun 7 (2016) 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kuehl T, Lagares D, BH3 mimetics as anti-fibrotic therapy: unleashing the mitochondrial pathway of apoptosis in myofibroblasts, Matrix Biol. 68-69 (2018) 94–105. [DOI] [PubMed] [Google Scholar]
- [91].Baker DJ, et al. , Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders, Nature 479 (7372) (2011) 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Baar MP, et al. , Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging, Cell 169 (1) (2017) p. 132–147 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jeon OH, et al. , Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment, Nat. Med 23 (6) (2017) 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhu Y, et al. , The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs, Aging Cell 14 (4) (2015) 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Justice JN, et al. , Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study, EBioMedicine 40 (2019) 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Hohmann MS, et al. , Quercetin enhances ligand-induced apoptosis in senescent IPF fibroblasts and reduces lung fibrosis in vivo, Am. J. Respir. Cell Mol. Biol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Moncsek A, et al. , Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(−/−)) mice, Hepatology 67 (1) (2018) 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chang J, et al. , Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice, Nat. Med 22 (1) (2016) 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhu Y, et al. , Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors, Aging Cell 15 (3) (2016) 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kim EC, Kim JR, Senotherapeutics: emerging strategy for healthy aging and age-related disease, BMB Rep. 52 (1) (2019) 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Le TT, et al. , Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis, J.Immunol 193 (7) (2014) 3755–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Meng XM, Nikolic-Paterson DJ, Lan HY, TGF-beta: the master regulator of fibrosis, Nat. Rev. Nephrol 12 (6) (2016) 325–338. [DOI] [PubMed] [Google Scholar]
- [103].Milara J, et al. , The JAK2 pathway is activated in idiopathic pulmonary fibrosis, Respir. Res 19 (1) (2018) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Blasco MA, Telomere length, stem cells and aging, Nat. Chem. Biol 3 (10) (2007) 640–649. [DOI] [PubMed] [Google Scholar]
- [105].Tomas-Loba A, et al. , Telomerase reverse transcriptase delays aging in cancer-resistant mice, Cell 135 (4) (2008) 609–622. [DOI] [PubMed] [Google Scholar]
- [106].Povedano JM, et al. , Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]