

Abstract
Organ fibrosis is a lethal outcome of autoimmune rheumatic diseases such as systemic sclerosis. Myofibroblasts are scar-forming cells that are ultimately responsible for the excessive synthesis, deposition and remodelling of extracellular matrix proteins in fibrosis. Advances have been made in our understanding of the mechanisms that keep myofibroblasts in an activated state and control myofibroblast functions. However, the mechanisms that help myofibroblasts to persist in fibrotic tissues remain poorly understood. Myofibroblasts evade apoptosis by activating molecular mechanisms in response to pro-survival biomechanical and growth factor signals from the fibrotic microenvironment, which can ultimately lead to the acquisition of a senescent phenotype. Growing evidence suggests that myofibroblasts and senescent myofibroblasts, rather than being resistant to apoptosis, are actually primed for apoptosis owing to concomitant activation of cell death signalling pathways; these cells are poised to apoptose when survival pathways are inhibited. This knowledge of apoptotic priming has paved the way for new therapies that trigger apoptosis in myofibroblasts by blocking pro-survival mechanisms, target senescent myofibroblast for apoptosis or promote the reprogramming of myofibroblasts into scar-resolving cells. These novel strategies are not only poised to prevent progressive tissue scarring, but also have the potential to reverse established fibrosis and to regenerate chronically injured tissues.
Organ fibrosis is a lethal outcome of autoimmune rheumatic diseases such as systemic sclerosis (SSc, also known as scleroderma) and rheumatoid arthritis (RA)1–4. In SSc and RA, autoimmune responses result in chronic inflammation and tissue injury, ultimately leading to the development of fibrosis, mostly in the lungs (in the form of interstitial lung disease (ILD)) and in the skin1,2,4,5. ILD is associated with high morbidity and mortality and is the leading cause of death in patients with rheumatic disease-associated fibrotic disorders worldwide3,6. ILD is characterized by progressive scarring of the lungs, which severely distorts the normal tissue architecture, resulting in respiratory failure and, ultimately, in death7. Nintedanib, an anti-fibrotic drug approved for the treatment of lung fibrosis in patients with idiopathic pulmonary fibrosis (IPF), has now also been approved for the treatment of lung fibrosis in SSc-associated ILD following the positive results of the SENSCIS trial8. The efficacy of pirfenidone, another FDA-approved treatment for IPF, in patients with SSc is currently being investigated in clinical trials9–12. Although both drugs reduce the rate of progression of IPF, they cannot halt disease progression8,13. Despite advances in therapy, destroyed lung architecture and function cannot be regained with currently available treatments and organ transplantation often remains the only option for patients14. Thus, an improved understanding of the pathobiological mechanisms promoting the progression of organ fibrosis in autoimmune rheumatic diseases is crucial to provide cellular and molecular targets for novel therapies that not only halt fibrosis, but also restore lost tissue function.
Excessive scarring of skin and lung tissues in rheumatic disease-associated fibrotic disorders is promoted by fibrogenic cells. These so-called myofibroblasts are the ultimate culprits responsible for excessive synthesis, deposition and remodelling of extracellular matrix (ECM) proteins — most notably fibrillar collagens15,16. Whereas substantial progress has been made over the past 40 years in understanding the molecular mechanisms that promote myofibroblast formation from different precursor cells17, considerably less is known about the mechanisms that promote their survival and persistence in fibrotic disorders. Although myofibroblast differentiation was initially regarded as a terminal state for fibroblastic cells, it is now clear that the myofibroblast phenotype can be plastic and reversible. Thus, the term ‘activation’ is used in this Review to describe myofibroblast phenotypic transition.
In this Review, we discuss advances in our knowledge of the mechanisms that keep myofibroblasts in an activated state, control myofibroblast functions and help myofibroblasts evade apoptosis. We focus on pathways and processes that are relevant for the development, progression and persistence of rheumatic disease-associated fibrotic disorders. However, the fundamental processes leading to persistent fibrosis are remarkably universal, so examples from different organ systems are also provided. Furthermore, we discuss novel therapeutic strategies aimed at specifically inducing myofibroblast apoptosis or, alternatively, promoting their deactivation with the aim of re-educating fibrogenic myofibroblasts into scar-resolving cells that reverse the damage inflicted by fibrosis.
Fibroblastic cell types
The generic term fibroblast is often used to describe collagen-producing, heterogeneous cell populations that reside in soft connective tissue, including mesenchymal stromal (or stem) cells18, pericytes19 and bona fide fibroblasts with different phenotypes20–23. Because specific molecular markers are lacking and the phenotypic features of these cell types are malleable, one could use the term fibroblast to describe the cells of a tissue that remain after all other cells have been classified according to their unique criteria. The following sections focus on fibroblasts and myofibroblasts, which differ in several ways (TABLE 1). Myofibroblasts are important cells that orchestrate the integration of the complex dynamic biochemical and biophysical cues present within tissue undergoing repair, with the aim of restoring tissue integrity and, ideally, homeostasis. Myofibroblasts are not typically found in healthy connective tissues.
Table 1 |.
Characteristics of fibroblasts and myofibroblasts
| Characteristic | Fibroblasts | Myofibroblasts | Refs |
|---|---|---|---|
| Distribution | Ubiquitous in connective tissues | Damaged tissues, fibrosis and cancer | 299 |
| Morphology | Large, flat and elongated (spindleshaped) cells that contain cortical actin microfilaments | Smooth muscle-like cells that contain contractile actin-myosin bundles and have specialized adhesion structures | 15 |
| Origin | Mesoderm | Tissue-resident fibroblasts, epithelial cells, endothelial cells, pericytes and mesenchymal stem cells | 17 |
| Functions | Maintain ECM homeostasis by secreting ECM proteins and matrix-degrading enzymes; proliferate and migrate into the wound bed to deposit granulation tissue during tissue repair | Produce large amounts of type I collagen during tissue repair; contract wound margins to facilitate re-epithelialization and restoration of tissue integrity; their persistence contributes to the pathogenesis of fibrosis and cancer | 1,30,300 |
| Molecular markers | Vimentin+α-SMA− cells that secrete ECM proteins and produce MMPs and TIMPs | Fibronectin ED-A+α-SMA+ cells that synthesize type I collagen and produce matrix crosslinking enzymes | 98 |
| Proliferative capacity | High (during wound healing) | Low | 301 |
| Metabolic activity | Low | High (aerobic glycolysis) | 168 |
| Biomechanical activity | Low | High (hypercontractile phenotype) | 302 |
| Apoptosis sensitivity | Low | High (primed for death) | 86 |
ECM, extracellular matrix; fibronectin ED-A, fibronectin extra domain A; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinase; α-SMA, α-smooth muscle actin.
Fibroblasts in tissue homeostasis.
Historically, fibroblasts residing in healthy tissues were considered to be quiescent or dormant, a belief that was partly based on the misperception that the ECM is a permanent scaffold that exists to support cells and only undergoes turnover following tissue injury or during disease. However, normal connective tissue ECM is a highly complex and dynamic structure that is continuously being synthesized, degraded and remodelled by resident fibroblasts24. These fibroblast activities enable the adaption of normal organ structures and functions to changing physical demands. For example, in athletes who are training, physiological connective tissue remodelling enables the heart to resist increasing mechanical loads and the lungs to increase in capacity25,26.
To fulfil their role as homeostats, fibroblasts have developed numerous, complex, positive and negative feedback mechanisms that enable them to sense and respond to changes in the biomechanical state, integrity and biochemical composition of the ECM. For example, weakening of the ECM by matrix metalloproteinase (MMP)-mediated degradation triggers fibroblasts to synthesize and deposit ECM and to secrete ECM crosslinking enzymes such as lysyl oxidases, leading to a mechanically reinforced ECM27,28. Moreover, fibroblasts orient ECM fibres during normal tissue turnover, which contributes to the mechanical stability of organs and tissues29. These activities are carefully orchestrated and seem to function seamlessly under physiological conditions; however, in acute traumatic conditions, during chronic inflammation or when repeated tissue microdamage occurs, fibroblasts cannot maintain homeostasis and the activity of professional repair cells — myofibroblasts — is required.
Myofibroblasts in tissue repair.
In conjunction with preceding tissue inflammation, changes in ECM biochemistry and/or mechanics that exhaust the homeostatic capacity of fibroblasts cause them to become activated and to phenotypically transition into myofibroblasts17 (FIG. 1a). The definition of a myofibroblast is equally as broad as that of a fibroblast and is still under debate30,31; however, all myofibroblasts share their common spatial association with fibrillar collagen, the formation of contractile actin–myosin bundles and the exertion of contractile forces within the ECM15,32. In addition, myofibroblasts are often characterized by the expression of α-smooth muscle actin (α-SMA) in stress fibres, which confers on them their highly contractile phenotype33. No known unique cell surface markers exist that distinguish myofibroblasts from other mesenchymal cells, define subsets of myofibroblasts with distinct physiological repair functions or discriminate ‘good’ wound-healing myofibroblasts from ‘bad’ pro-fibrotic myofibroblasts in fibrosis31. Beneath their surface, however, single-cell analysis has revealed that heterogeneous (myo)fibroblast populations have different gene expression and transcription profiles that undergo dynamic changes during tissue repair and fibrosis in various organs34–40. Despite the justified excitement around single-cell sequencing and the representation of the resulting complex data in relatively easy-to-grasp t-distributed stochastic neighbour embedding (t-SNE) cluster plots, one should bear in mind that myofibroblast heterogeneity and plasticity are essential for our body to react to unpredictable injury scenarios. Thus, attempts to classify fibroblast and myofibroblast populations might result in the same conclusion as that reached by researchers in the macrophage field41 — that classifying cells by their functions is preferable to classification by molecular markers.
Fig. 1 |. Origin, functions and fate of myofibroblasts during tissue repair and fibrosis.
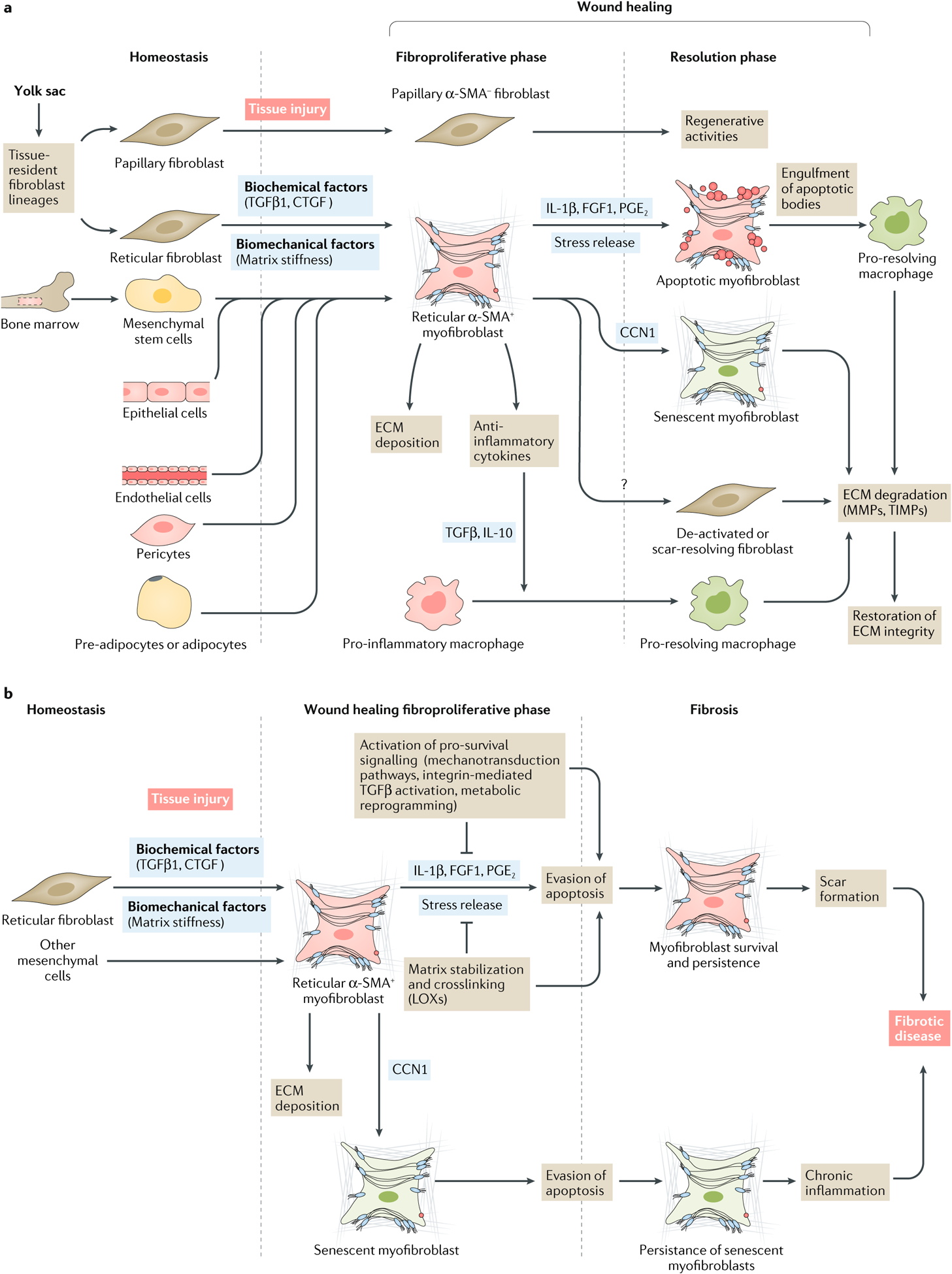
a | Activated myofibroblasts are important orchestrators of wound healing. During tissue injury, fibroblasts can differentiate into myofibroblasts. In the skin, two dermal fibroblast populations (reticular and papillary), which are derived from a common embryonic origin, have distinct functions during tissue repair. Whereas papillary fibroblasts do not express α-smooth muscle actin (α-SMA) and are involved in the regeneration of skin structures such as hair follicles, reticular fibroblasts differentiate into collagen-producing α-SMA+ myofibroblasts by the action of biomechanical (extracellular matrix (ECM) stiffness) and biochemical factors such as transforming growth factor-β1 (TGFβ1) and connective tissue growth factor (CTGF). In specific conditions and experimental models, myofibroblasts can also arise from epithelial and endothelial cells via epithelial-to-mesenchymal transition or endothelial-to-mesenchymal transition, respectively, and from mesenchymal stem cells, pericytes and pre-adipocytes or adipocytes. Myofibroblasts are characterized by increased synthesis of ECM proteins and by the expression of α-SMA, which confers a contractile phenotype that promotes wound closure. Activated myofibroblasts also produce anti-inflammatory cytokines such as TGFβ1 and IL-10, thereby shifting macrophages towards an ECM-degrading phenotype. During the resolution of wound healing, myofibroblasts can follow several different cell fates. Myofibroblasts can die via apoptosis, a process induced by ECM softening (stress release) or soluble pro-apoptotic factors such as IL-1β, fibroblast growth factor 1 (FGF1) and prostaglandin E2 (PGE2). Alternatively, they can deactivate, become scar-resolving fibroblasts or become senescent via the action of CCN family member 1 (CCN1); these cell types all participate in ECM degradation in partnership with pro-resolving macrophages. b | Myofibroblasts and senescent myofibroblasts can also escape apoptosis and continue to remodel tissue beyond repair, which results in pathological scarring and the development of fibrotic disease. LOXs, lysyl oxidases; MMPs, matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinases.
Myofibroblast heterogeneity is associated with functional diversity, which can either reflect distinct myofibroblast lineages and precursors or different activation states of the same cell type. Notably, myofibroblasts can arise from other cell types, such as epithelial cells, endothelial cells, mesenchymal stem cells, pericytes, pre-adipocytes and adipocytes17 (FIG. 1a). However, it is still unclear whether the origin of myofibroblasts translates into specific functions during tissue repair or fibrosis. Evidence of different myofibroblast lineages exists in a variety of organs, including the lungs42–44, heart25, kidney45, liver46, bone marrow47 and skin48–50. In mouse dorsal skin, two dermal fibroblast populations (reticular and papillary) are derived from a common mesenchymal progenitor cell that is present at day 12 during embryonic development51 (FIG. 1a). Remarkably, these two subsets of fibroblasts not only differ in their anatomical position within the same tissue but also exert distinct functions during tissue repair. Genetic tracing has revealed that reticular fibroblasts of the lower dermis of mouse skin are the first to migrate into the wounded sites, where they activate to become collagen-producing α-SMA+ myofibroblasts. By contrast, papillary fibroblasts seem to be essential for new hair follicle formation later on in the healing process and never express α-SMA51. Other lineage markers, such as engrailed 1 and CD26, have been used to reveal how at least two succeeding distinct fibroblast populations contribute to skin development21, normal wound healing and scarring in mouse models52. Whether the same or similar fibroblast lineages contribute differently to the healing of human skin injuries and/or to the development of skin fibrosis in SSc remains elusive. However, one common conclusion of such lineage-tracing studies, despite the use of different genetic markers, is that targeting specific myofibroblast precursor cells has the potential to suppress the development of fibrosis without affecting regenerative fibroblast populations. Notably, distinct fibroblast populations have also been identified by single-cell mRNA analysis in synovium and are thought to have different roles in RA53. Thy-1+ fibroblasts in the synovial sublining promoted and sustained inflammation in a mouse model of arthritis without affecting bone and cartilage, whereas a Thy-1− fibroblast population residing in the synovial lining degraded bone and cartilage ECM without affecting inflammation53.
An alternative, but not mutually exclusive, hypothesis is that myofibroblasts undergo dynamic transitions or exist in a variety of maturation states that differently affect the outcome of normal and fibrotic tissue repair15,54–57. The term ‘proto-myofibroblast’ has been proposed to emphasize that full ‘differentiation’ of fibroblasts into highly contractile α-SMA+ myofibroblasts is preceded by the transition to a collagen-synthesizing phenotype that has α-SMA− stress fibres and relatively lower contractile activity15. Such phenotypic shifts must be considered when interpreting phenotypic and/or transcriptional myofibroblast profiles obtained at single time-points from cell cultures, healing tissues or fibrotic lesions, which do not take into account the history or future of these cells.
Phenotypic shifts have also been reported for highly activated, α-SMA+ myofibroblasts at later stages of tissue repair and fibrosis — a phenomenon called reversal. For example, subsets of α-SMA+ myofibroblasts undergo phenotypic transition into senescent myofibroblasts that express p16INK4a (a cell cycle regulatory protein) during the resolution phase of cutaneous healing58,59. This transition is required to shift the activity of myofibroblasts from ECM production to ECM degradation, and thereby create scar-resolving cells (FIG. 1a). Using single-cell tracing in a mouse model of carbon tetrachloride-induced induced liver fibrosis, myofibroblasts reverted back to a fibroblast-like inactive state in vivo, and fibrosis was resolved once the experimental organ insult was discontinued60,61. Irrespective of the underlying mechanism, the termination of myofibroblast ECM secreting and contractile activities has been proposed as an important step in the resolution of organ fibrosis62,63.
Although the molecular mechanisms that promote the activation of myofibroblasts have been relatively well studied, considerably less is known about the pathways that deactivate myofibroblasts upon successful completion of tissue repair. Once tissue integrity is re-established, myofibroblasts can adopt one of several different functions17 (FIG. 1a). Myofibroblasts can deactivate and return to the low activity state characteristic of fibroblasts in homeostatic tissues. Alternatively, they can assume roles that were not characteristic of their precursor cells, such as becoming scar-resolving or senescent cells. Myofibroblasts can also initiate self-clearance via apoptosis (also known as programmed cell death), or can continue to remodel tissue beyond what is required for repair, resulting in pathological scarring (FIG. 1b). How these different fates are determined and how the underlying cellular mechanisms are temporally and spatially regulated remain unclear. In the following sections, we discuss factors and pathways that promote myofibroblast persistence beyond the resolution phase of tissue repair, particularly the evasion of apoptosis.
Apoptotic pathways
Apoptosis is a physiological process in which cells are genetically programmed to self-destruct64–67. In healthy adult tissue, 50–70 billion cells are cleared every day via apoptosis to balance the rate of cell division in tissues68. Although it seems extremely inefficient to continuously destroy so many cells, apoptosis is an important homeostatic mechanism that controls organ size and function. It is also the main cellular mechanism used to eliminate damaged or infected cells, or to control cells that start dividing uncontrollably69. The cellular and molecular features of apoptotic cells include cell shrinkage, membrane blebbing, pyknosis and cell disassembly into apoptotic bodies in a process called budding70. Apoptotic bodies are subsequently phagocytosed by surrounding cells, including macrophages71. Importantly, the engulfment of cells via efferocytosis is associated with decreased production of pro-inflammatory cytokines such as TNF, and increased production of anti-inflammatory cytokines such as transforming growth factor-β1 (TGFβ1) and IL-10 (REF.72).
Apoptosis is a complex process that is tightly controlled by two interconnected molecular pathways: the intrinsic and extrinsic pathways65 (FIG. 2). The intrinsic pathway of apoptosis is triggered by intracellular death stimuli such as DNA damage, oxidative stress or oncogene activation65, all of which initiate apoptosis by inducing mitochondrial outer membrane permeabilization (MOMP). Accordingly, the intrinsic pathway is also called mitochondrial apoptosis. MOMP occurs as the membrane potential and pH gradient across the inner mitochondrial membrane is impaired, leading to mitochondrial swelling, rupture of the outer mitochondrial membrane and release of pro-apoptotic proteins such as cytochrome c from the intermembrane space into the cytoplasm. Cytoplasmic cytochrome c binds to and activates apoptotic protease activating factor 1 (APAF1) to form the apoptosome, which mediates activation of caspase-9, ultimately leading to cell death via the activation of caspase-3 and caspase-7. Caspase-9-mediated cleavage and activation of the effectors caspase-3 and caspase-7 can be inhibited by the pro-survival inhibitor of apoptosis (IAP) proteins (such as X-linked IAP (XIAP)), thereby preventing apoptosis67. In this intrinsic pathway, the apoptotic threshold of a cell (the likelihood that it will enter apoptosis) is set by dynamic interactions at the mitochondrial outer membrane between distinct members of the BCL-2 family of proteins. These BCL-2 proteins are divided by structure and function into effector, activator, pro-survival and sensitizer proteins64,65.
Fig. 2 |. Intrinsic and extrinsic apoptosis pathways.
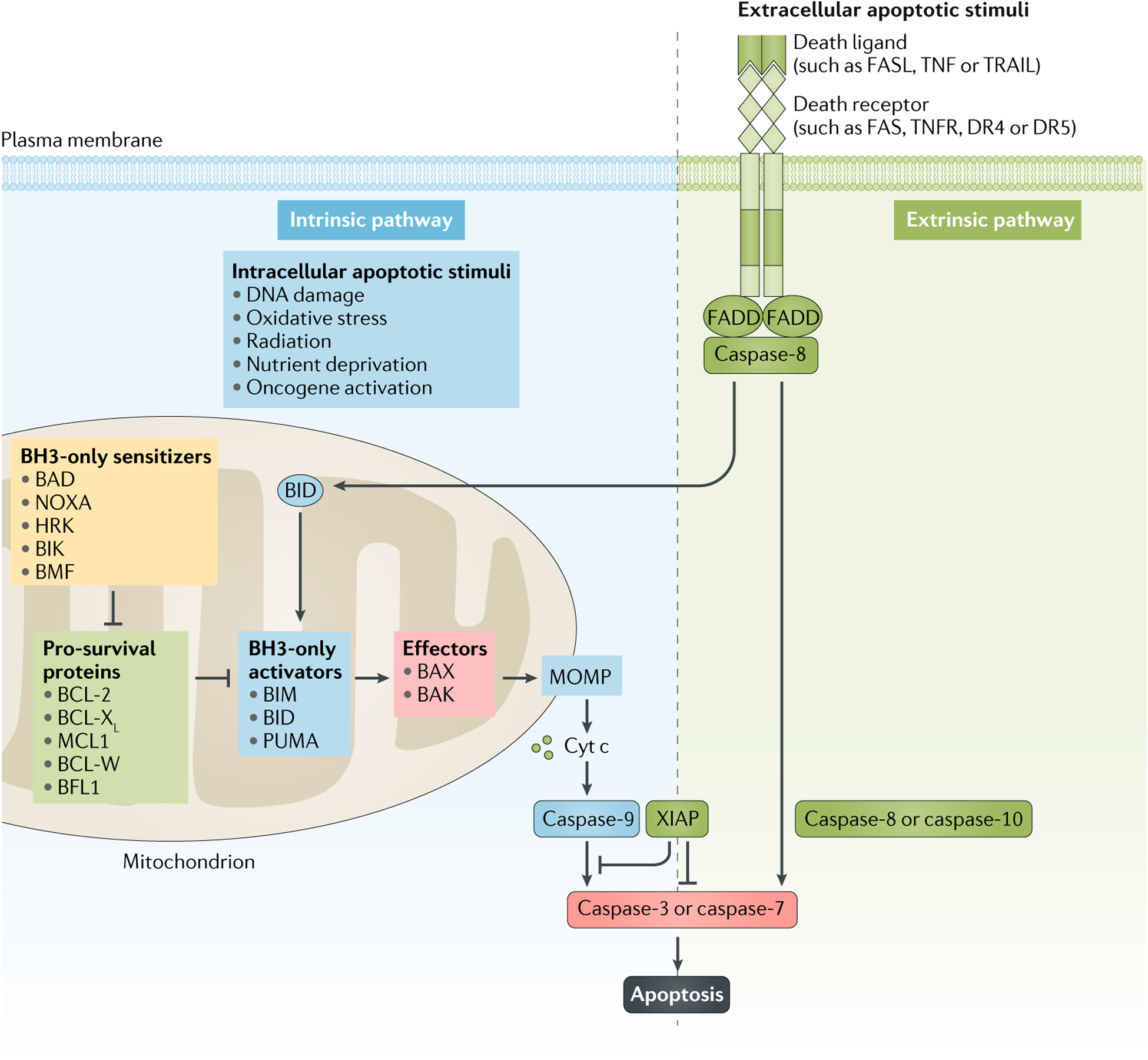
Apoptosis is controlled by two distinct yet connected pathways. The intrinsic pathway is triggered by intracellular death stimuli, such as DNA damage, radiation, nutrient deprivation, oxidative stress or oncogene activation, which induce apoptosis by promoting mitochondrial outer membrane permeabilization (MOMP) and cytochrome c (cyt c)-dependent activation of caspase-9, which then activates caspase-3 and caspase-7. This pathway of apoptosis is tightly controlled by the BCL-2 family of proteins, which are classified on the basis of their structural homology and function into sensitizers, pro-survival proteins, activators and effectors. MOMP is controlled by homo-oligomerization of the effector proteins BCL-associated X protein (BAX) and BCL-2 homologous antagonist/killer (BAK), which are activated by the activator proteins BCL-2-like protein 11 (BCL2L11; also known as BIM), p53-upregulated modulator of apoptosis (PUMA) and BH3-interacting domain death agonist (BID). Pro-survival proteins such as BCL-2, BCL-XL, BCL-W, induced myeloid leukaemia cell differentiation protein MCL1 (MCL1) and BCL-2-related protein A1 (BCL2A1; also known as BFL1) can bind and sequester both activators and effectors, thereby preventing their interaction and the induction of MOMP. Sensitizer proteins (such as BCL-2-associated death promoter (BAD), BCL-2 interaction killer (BIK), BCL-2 modifying factor (BMF), activator of apoptosis harakiri (HRK) and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1; also known as NOXA) are a distinct group of proteins that promote apoptosis by binding and blocking pro-survival proteins, thereby releasing formerly bound activator and effector proteins. The extrinsic pathway is initiated by extracellular ligands such as FAS ligand (FASL), TNF and TNF ligand superfamily member 10 (TNFSF10; also known as TRAIL), which bind to the cell-surface receptors FAS, TNF receptors (TNFRs) and death receptor 4 (DR4) and DR5, respectively. The signal is transmitted via FAS-associated death domain (FADD), which triggers the activation of caspase-8 and caspase-10, ultimately initiating apoptosis through cleavage and activation of pro-caspase-3 and pro-caspase-7. BH3, BCL-2 homology domain 3; XIAP, X-linked inhibitor of apoptosis protein.
Each BCL-2 family member contains one or more of the four conserved BCL-2 homology domains: BH1, BH2, BH3 and BH4 (REF.65). BCL-associated X protein (BAX) and BCL-2 homologous antagonist/killer (BAK) are multi-domain effector proteins that initiate apoptosis via MOMP by forming pores in the mitochondrial membrane. The activities of these effectors are tightly regulated by BH3-only activator proteins, such as BCL-2-like protein 11 (BCL2L11; also known as BIM), p53-upregulated modulator of apoptosis (PUMA) and BH3-interacting domain death agonist (BID). Binding of activators to effectors initiates MOMP, whereas MOMP can be prevented by multi-domain pro-survival proteins such as BCL-2, BCL-XL, BCL-W, induced myeloid leukaemia cell differentiation protein MCL1 (MCL1) and BCL-2-related protein A1 (BCL2A1; also known as BFL1), which can directly bind and inhibit both effectors and activators. MOMP can also be induced in cells even in the presence of pro-survival proteins if another set of BH3-only sensitizer proteins are highly expressed. Sensitizers, such as BCL-2-associated death promoter (BAD), phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1; also known as NOXA), activator of apoptosis harakiri (HRK), PUMA (which is also an activator), BCL-2-interacting killer (BIK) and BCL-2 modifying factor (BMF), indirectly promote apoptosis by blocking pro-survival proteins and thereby freeing activators and effectors to initiate MOMP73.
The extrinsic, or death-receptor, pathway of apoptosis is activated by the binding of extracellular death ligands such as FAS ligand (FASL), TNF ligand superfamily member 10 (TNFSF10; also known as TRAIL) and TNF (FIG. 2). Typical cell-surface death receptors are FAS, TNF receptors, death receptor 4 (DR4) and DR5 (REF.66). In this pathway, the pro-apoptotic signal is transmitted via a multiprotein death-inducing signalling complex, in which the adaptor proteins FAS-associated death domain (FADD), TNF receptor type 1-associated DEATH domain (TRADD) and regulated IRE1-dependent decay (RIDD) assemble to activate caspase-8 and caspase-10. Both caspases then initiate apoptosis via cleavage and activation of pro-caspase-3 (FIG. 2).
Interestingly, the extrinsic and intrinsic apoptotic pathways can also be cross-activated via BID74, an activator protein that can be activated via the extrinsic pathway through caspase-8-mediated proteolytic cleavage. Once cleaved, truncated BID triggers the intrinsic pathway of apoptosis by directly binding to and activating the effector proteins BAX and BAK in the mitochondrial membrane. In the following section we elaborate on how the intrinsic and extrinsic apoptosis pathways are regulated in myofibroblasts during tissue repair and fibrosis.
Myofibroblast apoptosis
Clearance of myofibroblasts in tissue repair.
In seminal studies in the 1990s, Gabbiani and colleagues proposed that the majority of myofibroblasts undergo apoptosis upon completion of tissue repair. They supported this concept by demonstrating the accumulation of fragmented DNA in α-SMA+ cells, and of α-SMA+ apoptotic bodies in the phagolysosomes of phagocytic cells, days after the complete closure of skin wounds in rats75,76. Subsequent studies suggested that activated myofibroblasts become ‘addicted’ to growth factor receptor-mediated pathways that enable their survival during tissue repair, but promote apoptosis upon ‘withdrawal’. For example, pro-survival growth factors, such as TGFβ1 or platelet-derived growth factor (PDGF), are released by platelets and macrophages at an early stage of the inflammatory phase of wound healing; the depletion of these growth factors during wound healing can potentially induce myofibroblast apoptosis77,78.
Such growth factor ‘dependence’ is one plausible mechanism limiting myofibroblast survival during wound healing. Alternatively, pro-apoptotic cytokines released during late stages of tissue repair might selectively induce apoptosis of myofibroblasts by directly activating cell death signalling pathways or by inhibiting pro-survival pathways, which regulate the activation of the intrinsic and/or extrinsic pathways of apoptosis in myofibroblasts. For example, fibroblast growth factor 1 (FGF1) induces caspase-3-mediated apoptosis in activated myofibroblasts from skin granulation tissue by inhibiting the phosphorylation of pro-survival protein kinase B (PKB, also known as AKT) and the related focal adhesion kinase (FAK) signalling pathway; by contrast, FGF1 does not induce apoptosis in fibroblasts from healthy skin79,80. Similarly, IL-1β and hepatic growth factor (HGF) induce caspase-dependent apoptosis in mouse lung myofibroblasts by inhibiting FAK77,81. In the liver, targeted apoptosis of myofibroblasts occurs during liver regeneration, in which hepatic stellate cells upregulate the pro-apoptotic FAS receptor and are rapidly eliminated through the extrinsic pathway of apoptosis82,83. Together, the results of these studies77,81–83 provide evidence that the activation state of myofibroblasts increases their susceptibility to apoptosis during the resolution phase of the wound healing programme.
Evasion of apoptosis by myofibroblasts in fibrosis.
The shift from physiological tissue repair to pathological organ fibrosis in the context of autoimmune disease involves molecular signalling pathways that prevent the termination of myofibroblast activities. By contrast to their counterparts in wound healing, which disappear at the end of physiological repair, the persistence of myofibroblasts in fibrotic disease has led to the suggestion that these cells are apoptosis-resistant84,85. Studies from the past 2 years have shown that, rather than resisting apoptosis, myofibroblasts are actually poised to self-destruct in fibrotic conditions. The molecular basis for the pro-apoptotic tendencies of myofibroblasts seems to centre around an increase in mitochondrial apoptotic priming in these cells86,87 (FIG. 3). Mitochondrial priming refers to the closeness of mitochondria to the apoptosis threshold, and is controlled by the relative expression of effector, activator, pro-survival and sensitizer proteins from the BCL-2 family88–90. Importantly, apoptotic priming is not determined by the expression of individual pro-apoptotic or anti-apoptotic proteins, and cell survival is not a binary ‘yes’ or ‘no’ decision. Instead, cell survival is determined by an intricate dynamic molecular rheostat, in which the relative balance between multiple pro-apoptotic and anti-apoptotic BCL-2 family proteins instructs cells as to whether to survive or undergo apoptosis (FIG. 3). For example, pro-apoptotic BH3-only proteins can be induced transcriptionally and/or post-translationally by cytotoxic stress signals, thereby increasing mitochondrial priming73,89. When mitochondrial priming is high enough to cross the apoptotic threshold, MOMP and subsequent apoptosis will occur.
Fig. 3 |. Mitochondrial priming of myofibroblasts.
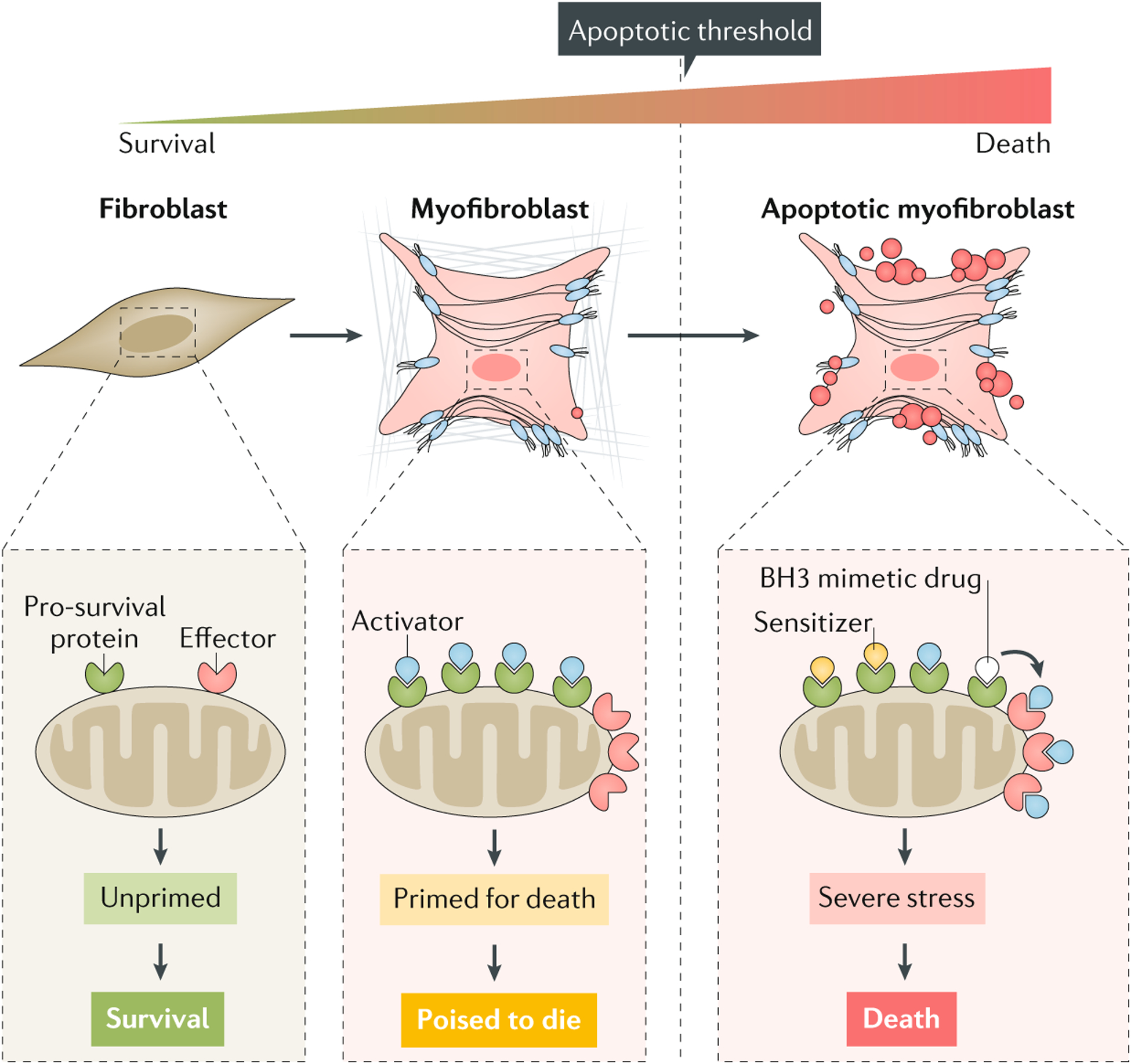
Mitochondrial priming refers to the proximity of mitochondria to the apoptosis threshold and is determined by the relative expression of pro-apoptotic (effectors, activators and sensitizers) and pro-survival members of the BCL-2 family of proteins. ‘Unprimed’ fibroblasts express low amounts of activators and/or effectors, leading to an apoptosis-resistant phenotype. Conversely, high expression of sensitizers, activators and/or effectors results in high mitochondrial priming, ultimately triggering cytochrome-c-mediated mitochondrial outer membrane permeabilization and apoptosis. However, myofibroblasts can survive with high mitochondrial priming if a pro-survival mechanism is activated. In this cellular state, known as ‘primed for death’, cells are poised to die and become dependent on one or more pro-survival proteins to sequester pro-apoptotic proteins and ensure survival. BCL-2 homology domain 3 (BH3) mimetic drugs can trigger the intrinsic pathway of apoptosis in primed-for-death myofibroblasts by binding to pro-survival proteins, resulting in the release of activator proteins, which can then bind to and activate effector proteins.
However, cells can still survive with a high degree of mitochondrial priming if a pro-survival mechanism is activated. Typically, cells with high mitochondrial priming upregulate anti-apoptotic proteins that sequester pro-apoptotic BH3-only proteins, thus preventing MOMP91–93. In myofibroblasts, upregulation of anti-apoptotic BCL-2 proteins such as BCL-XL in response to the extracellular environment enable survival despite these cells being primed for death86. Nevertheless, cells primed for death become dependent on anti-apoptotic proteins for survival, and the inhibition of important pro-survival proteins therapeutically (with, for example, the so-called BH3 mimetic drugs) can rapidly induce apoptosis in such cells91 (FIG. 3). In the following section, we discuss molecular mechanisms that promote the survival of myofibroblasts that are primed for death in fibrosis.
Myofibroblast survival mechanisms
The list of factors known to activate myofibroblasts in vitro, during normal tissue repair and during organ fibrosis is ever growing and has been extensively reviewed elsewhere4,17,20,31,94–101. In this Review, we focus on biomechanical and biochemical factors that not only promote myofibroblast activation, but also have pro-survival functions.
Biomechanical myofibroblast survival factors.
A crucial factor that enables myofibroblasts primed for death to survive is the specific biomechanical conditions that they generate around themselves in the form of fibrotic ECM. As a result of excessive collagen deposition and incremental myofibroblast contractions, fibrotic ECM becomes stiffer than the ECM of healthy organs. In SSc, the formation of stiff fibrotic scars changes the mechanical properties of the skin, kidney, heart and lungs, which directly affects organ function102. For example, the mechanical obstacle of stiff scar tissue reduces the pumping function of the heart and disturbs electrical transmission. Likewise, stiffening of lung tissue not only causes failure to promote proper gas exchange in the alveolar space, but also reduces distensibility and thus overall lung capacity102. In addition to impeding the function of organs that depend on their biomechanical properties, tissue stiffening has a profound influence on the progression of fibrosis by promoting the biomechanical activation of pro-fibrotic TGFβ1 and of myofibroblasts26,103. This biomechanical positive feedback loop establishes a vicious cycle that eventually perpetuates fibrosis26,102,104–106 (FIG. 4). The concept that increased ECM stiffness contributes to the progression of fibrosis, in addition to being its consequence, has intensified mechanobiology research in the context of fibrotic disorders, which has revealed important regulatory elements102,103,107–110 (discussed below).
Fig. 4 |. Molecular control of myofibroblast activation and survival.
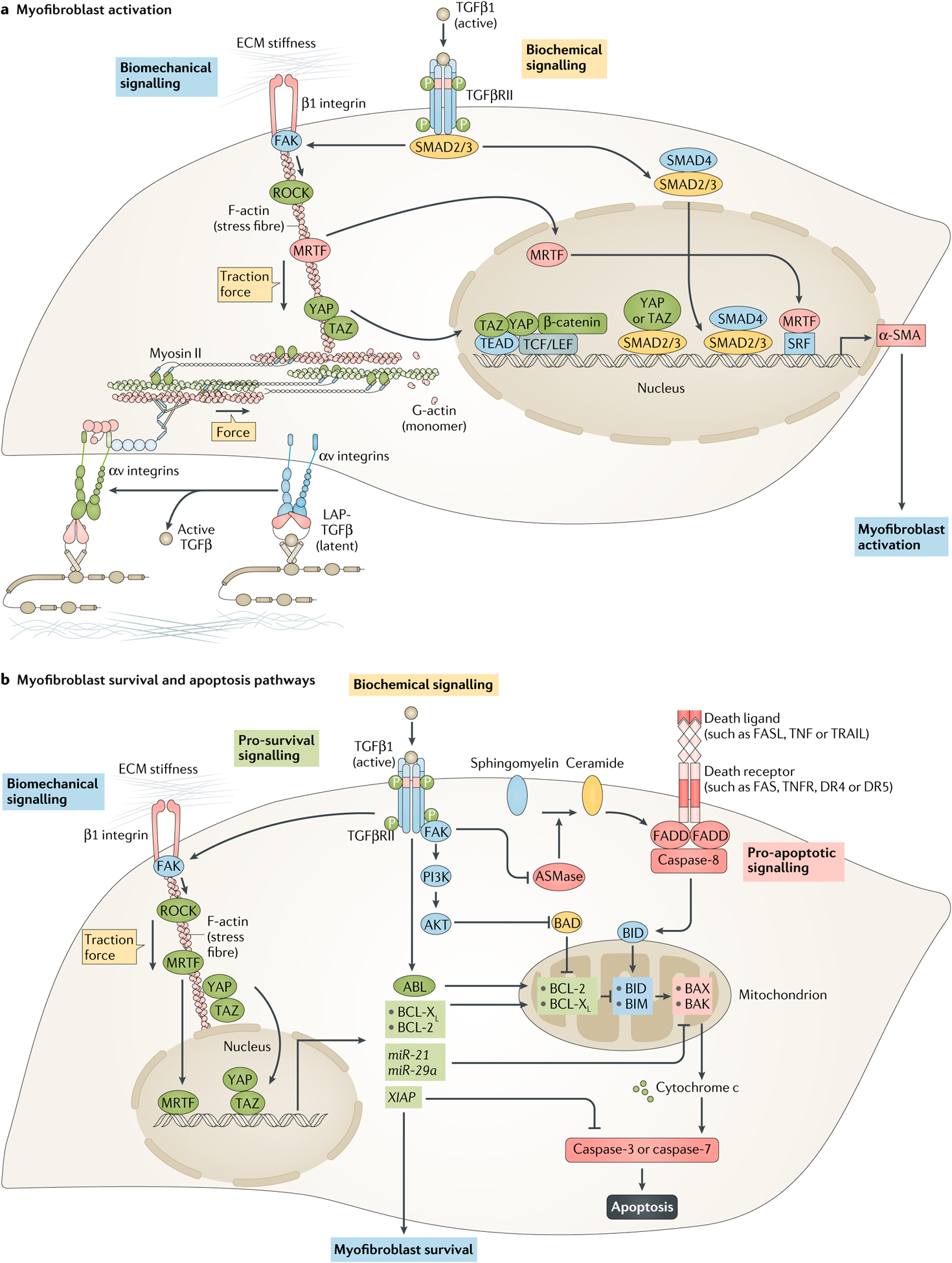
a | Myofibroblast activation is jointly promoted by biomechanical and biochemical cues. Biomechanical signalling induced by extracellular matrix (ECM) stiffness activates mechanotransduction pathways that directly control α-smooth muscle actin (α-SMA) transcription. These pathways involve β1 integrin, focal adhesion kinase (FAK) and RHO-associated protein kinase (ROCK), together with increasing traction forces in fibroblasts. Increased actomyosin activity causes the nuclear translocation of myocardin-related transcription factor (MRTF), as well as the transcriptional co-activators yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ), which regulate α-SMA expression by binding and activating other transcription factors, such as serum response factor (SRF), TEA domain family member (TEAD), T cell factor/lymphoid enhancer-binding factor (TCF/LEF) and β-catenin. ECM stiffness and mechanical forces also regulate force-dependent activation of latent transforming growth factor-β1 (TGFβ1) by increasing resistance to traction forces generated by fibroblasts. In this mechanism, extracellular latent TGFβ1 (TGFβ1 with its latency-associated peptide (L AP)) is released from latent TGFβ1 binding protein stores when αv integrins respond to mechanical pulling forces. Once activated, TGFβ1 binds to TGFβ receptors and promotes canonical mothers against decapentaplegic homolog 3 (SMAD3) activation. Activated SMAD3 binds to SMAD4 and translocates to the nucleus, where it binds to SMAD-binding elements in the promoters of fibrogenic genes, such as ACTA2 (encoding α-SMA). Together, myofibroblast activation is controlled by both the TGFβ–SMAD pathway, as well as biomechanical pathways such as integrin–FAK–ROCK–MRTF–YAP–TAZ signalling. b | Mitochondria in activated myofibroblasts contain large amounts of pro-apoptotic factors, which force these cells to activate pro-survival mechanisms to ensure survival. The intrinsic pathway of apoptosis is directly inhibited by TGFβ1 via activation of ABL signalling, which increases the amount of the pro-survival proteins BCL-2 and BCL-XL. TGFβ1 also blocks the intrinsic pathway by inhibiting the pro-apoptotic protein BCL-2-associated death promoter (BAD) via the FAK–PI3K–AKT signalling pathway, and TGFβ1-mediated FAK signalling blocks the extrinsic pathway of apoptosis by inhibiting sphingomyelinase (ASMase), an enzyme that controls FAS-mediated apoptosis by generating ceramide from sphingomyelin. Biomechanical signalling similarly inhibits the intrinsic pathway via the biomechanically regulated TGFβ–FAK–YAP1–TAZ–BCL-XL and TGFβ–ROCK–MRTF–BCL-2 pathways. Moreover, ECM stiffness induces expression of the microRNAs miR-21 and miR-29a, which promote the survival of myofibroblasts by increasing the expression of pro-survival BCL-2 proteins. Both ECM stiffness and TGFβ1 can also block apoptosis by increasing amounts of X-linked inhibitor of apoptosis protein (XIAP), a direct caspase inhibitor. AKT, pro-survival protein kinase B; BAK, BCL-2 homologous antagonist/killer; BAX, BCL-associated X protein; BID, BH3-interacting domain death agonist; BIM, BCL-2-interacting mediator of cell death (also known as BCL2L11); PI3K, phosphoinositide 3-kinase; TGFβRII, transforming growth factor-β receptor II.
Myofibroblast activation culminates in the expression of α-SMA and an associated increase in contractile force (FIG. 4a). Intracellularly, α-SMA protein production is controlled by TGFβ1-induced signalling through TGFβ control elements and mothers against decapentaplegic homolog (SMAD)-binding elements in the ACTA2 (encoding α-SMA) promoter111. Extracellularly, the amount of TGFβ1 activation from latent precursor complexes is dependent on both the biomechanical state of the ECM112 and the transmission of cellular forces by integrins binding to latent TGFβ1 (REFS113–115). In addition to indirect biomechanical stimulation through stress-dependent TGFβ1 activation, the ACTA2 promoter contains CArG boxes that bind both to myocardin-related transcription factors (MRTFs) and to serum-response factor (SRF), which positively regulate ACTA2 transcription116. MRTF shuttling from the cytosol to the nucleus is controlled by the polymerization state of the actin cytoskeleton (FIG. 4a). In cells under low stress, MRTFs bind to available monomeric G-actin; under high stress, G-actin polymerizes into F-actin stress fibres and releases MRTFs, which travel to the nucleus and upregulate ACTA2 transcription in conjunction with SRF117,118. In addition, the mechanosensitive transcriptional co-activators yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) positively regulate myofibroblast differentiation from several types of cells33,119–129 (FIG. 4a).
Whereas ECM stiffening is a pro-myofibroblastic stimulus, the release from extracellular and intracellular stress that occurs during wound healing results in the loss of contractile features and reduced myofibroblast activation102,103. In fact, it has been proposed that resolution of stiff scar tissue and the associated tension release serve as important signals for myofibroblasts to terminate their actions and/or to apoptose once normal tissue repair is completed. For example, in vitro, myofibroblast apoptosis can be induced by the relaxing of mechanically restrained fibroblast-populated collagen lattices130,131. In vivo, releasing mouse skin wounds from extracellular strain likewise causes myofibroblast apoptosis and, conversely, inhibiting the release of biomechanical stress in healing tissue by splinting the wound inhibits myofibroblast apoptosis and leads to hypertrophic scarring in rodents132–134.
The molecular pathways that link stress release to myofibroblast apoptosis are still unclear, but potentially involve the inhibition of pro-survival mechanotransduction pathways. Stiffness-activated myofibroblasts undergo rapid apoptosis in vitro upon inhibition of mechanotransduction, and integrin-mediated stress perception and FAK-mediated mechanosignalling are important components of myofibroblast survival on stiff ECM86,135 (FIG. 4b). By contrast, blockade of force-dependent mechanosignalling neither interferes with the homeostatic functions of fibroblasts nor causes apoptosis when fibroblasts are plated on soft ECM, further suggesting that the myofibroblast cellular state seems to prime these cells for apoptosis.
Myofibroblasts activated by ECM stiffness are primed for apoptosis by death signals such as the apoptosis activator protein BIM; upregulation of BIM creates a requirement for tonic expression of anti-apoptotic proteins such as BCL-XL to ensure myofibroblast survival and persistence86. BCL-XL expression in stiffness-activated myofibroblasts seems to be controlled by YAP and TAZ, the translocation of which to the nucleus is induced by integrin-mediated and FAK-mediated mechanotransduction pathways86 (FIG. 4b). Whereas the evasion of apoptosis by dermal myofibroblasts in skin fibrosis is controlled by the mechanically regulated FAK–YAP–TAZ–BCL-XL pathway86, myofibroblast survival in lung fibrosis is mechanically controlled by RHO-associated protein kinase (ROCK)–MRTF–BCL-2 signalling126. In addition, TGFβ1–ROCK–MRTF signalling blocks mitochondrial apoptosis by increasing concentrations of XIAP136–140, which directly inhibits caspases (FIG. 4b). The results of these studies86,126 support the concept that biomechanical signalling induced by ECM stiffness promotes myofibroblast survival by the upregulation of specific BCL-2 family pro-survival factors in myofibroblasts that are primed for death.
In addition to directly affecting mitochondrial priming, biomechanical signalling can also promote myofibroblast resistance to apoptosis via indirect mechanisms. ECM stiffness induces the expression of the microRNAs miR-21 and miR-29a, which promote the survival of stiffness-activated myofibroblasts by increasing the expression of BCL-2 and reducing the expression of BAX134,139 (FIG. 4b). MiR-21 is known to be pro-fibrotic in many organs and fibroblastic cells140–145. Curiously, miR-21 is also involved in maintaining the contractile phenotype of myofibroblasts that have been mechanically primed on pathological scar-stiff polymer substrates, even weeks after moving and exposing the same cells to soft substrate cultures134. This phenomenon has been termed ‘mechanical memory’95, and it will be interesting to see the results of future investigations into how mechanical priming and priming for death are coordinated.
Extracellular myofibroblast survival factors.
Autocrine production of TGFβ1 and the vasoactive peptide endothelin 1 (ET1) mediates resistance to apoptosis in cultured SSc and IPF fibroblasts via increased activation of the FAK–AKT signalling pathway146–150. In promoting myofibroblast survival, the activation of FAK is dependent on β1 integrin77,135,137,149. FAK activation requires auto-phosphorylation at tyrosine 397, which creates a binding site for the Src homology 2 (SH2) domains of the p85 regulatory subunit of phosphoinositide 3-kinase (PI3K) that, in turn, leads to activation of the pro-survival PI3K–AKT signalling pathway150–152. Alternatively, AKT activation by TGFβ1 in lung myofibroblasts can occur in a FAK-independent, p38 mitogen-activated protein kinase (MAPK)-dependent manner153. AKT activation results in phosphorylation and inhibition of the sensitizer protein BAD77,154,155, thereby inhibiting myofibroblast apoptosis at a crossover point between the extrinsic and intrinsic apoptosis pathways (FIG. 4b). Furthermore, TGFβ1-mediated activation of the transcription factor ABL results in increased amounts of the pro-survival protein BCL-2 and decreased amounts of the effector protein BAX156. In addition, PDGF can activate cancer-associated myofibroblasts, while simultaneously increasing their mitochondrial priming and sensitivity to apoptosis157. Similarly, the survival of PDGF-activated myofibroblasts in the liver is dependent on BCL-XL158, indicating an important role for this pro-survival protein in promoting the survival of fibrogenic myofibroblasts, potentially across different organs. Taken together, the intrinsic mitochondrial pathway is directly inhibited by TGFβ1 and PDGF signalling via transcriptional regulation of pro-survival BCL-2 family proteins, leading to decreased mitochondrial priming in myofibroblasts.
In addition to regulating myofibroblast survival through the intrinsic apoptotic pathway, FAK signalling is also important in blocking the extrinsic apoptotic pathway in response to pro-fibrotic cytokines86,159. Both TGFβ1 and ET1 block the extrinsic pathway of apoptosis in SSc and IPF myofibroblasts by inhibiting FAS–FASL signalling77,154,160–162. The pro-apoptotic activities of the FAS–FASL pathway in myofibroblasts are mostly mediated by ceramide generation via the activation of acidic sphingomyelinase163. In SSc myofibroblasts, autocrine TGFβ1 signalling inhibits sphingomyelinase, thereby protecting these cells from FASL-induced apoptosis despite similar expression levels of FAS receptor compared with healthy fibroblasts154,160,164 (FIG. 4b). In IPF myofibroblasts, sensitivity to FASL-induced apoptosis is similarly reduced, albeit through different molecular mechanisms, including increased levels of soluble FAS, which is produced as an alternatively spliced variant of FAS and functions as decoy receptor for FASL162, and upregulation of decoy receptor 3, which binds to FASL and inhibits FASL-induced apoptosis165.
In addition, reduced sensitivity of fibrotic fibroblasts to FASL-induced apoptosis can be mediated by down-regulation of cyclooxygenase-2 (COX2), which normally promotes apoptosis in fibroblasts by producing prostaglandin E2 (PGE2)166, a lipid mediator derived from the metabolism of arachidonic acid. Exogenous administration of PGE2 restores the sensitivity of fibrotic lung myofibroblasts to FASL-induced apoptosis by decreasing AKT signalling166,167. PGE2 treatment also suppresses the caspase inhibitor XIAP, thereby enhancing FASL-mediated apoptosis in lung myofibroblasts and lending support for a potential mechanistic role for XIAP in myofibroblast evasion of apoptosis161,167. Interestingly, ECM stiffening in fibrotic disorders participates in a positive feedback loop with myofibroblasts that leads to suppression of COX2 expression and PGE2 production, which suggests that the fibrotic biomechanical microenvironment itself might be sufficient to promote myofibroblast survival through this pathway105. Thus, diminished PGE2 production and/or responsiveness contribute to myofibroblast apoptosis resistance in fibrotic disorders.
Intracellular myofibroblast survival factors.
Metabolic reprogramming is emerging as an important mechanism of myofibroblast survival. Myofibroblasts are cells with a high metabolic rate that require energy for the synthesis and contraction of the ECM. Notably, although metabolic reprogramming of myofibroblasts rewires their pro-survival pathways to promote their persistence168, the molecular mechanisms promoting myofibroblast survival via metabolic reprogramming remain poorly understood. YAP–TAZ activity is also controlled by metabolic and energy-sensing pathways169, suggesting that the metabolic status and reprogramming of myofibroblasts might control the activation of mechanosensitive pro-survival pathways.
Myofibroblast bioenergetics are sustained by the induction of glycolysis and increased mitochondrial oxygen consumption, which support their aerobic glycolysis phenotype169. In one study170, this high-energy status of myofibroblasts was associated with fragmented and damaged mitochondria in IPF fibroblasts, which potentially contribute to the increased mitochondrial priming of these cells via poorly understood mechanisms. Loss of activity of the energy sensor AMP-activated protein kinase (AMPK) promoted mitochondrial dysfunction and metabolic reprogramming in myofibroblasts. Accordingly, activation of AMPK by treatment with metformin restored the sensitivity of myofibroblasts to apoptosis, indicating that mitochondrial biogenesis is required to induce apoptosis in primed-for-death myofibroblasts. More importantly, metformin reversed established lung fibrosis in experimental models by promoting myofibroblast deactivation and/or apoptosis170.
Myofibroblast senescence.
In addition to avoiding apoptosis, myofibroblasts can persist by becoming senescent (FIG. 1). Growing evidence suggests that progressive fibrosis might result from accumulation of pro-fibrotic senescent myofibroblasts, which are prevalent in age-related fibrotic disorders such as IPF171–173. Cellular senescence was initially defined as cell-cycle arrest and first described in human lung fibroblasts by Hayflick in 1965 (REF.174). The main function of cellular senescence is to prevent the division of damaged cells as a protective mechanism against tumorigenesis. Senescence locks cells in a permanent cell cycle arrest by increasing expression of cell cycle and tumour suppressor inhibitors in response to excessive stressors, such as telomeric dysfunction (replicative senescence), oncogene activation and genotoxic or oxidative stresses175. These processes are promoted by senescence-activating signalling pathways that include the cyclin-dependent kinase inhibitors p53, p16 or p21 (REFS176,177).
Because senescent cells are also destined for elimination, cellular senescence is conceptually similar to apoptosis, albeit operating via different clearance mechanisms. Senescent cells are characterized by the acquisition of a senescence-associated secretory phenotype178. This phenotype is molecularly controlled by transcription factor NF-κB signalling and characterized by the secretion of pro-inflammatory cytokines and chemokines such as IL-6, CC-chemokine ligand 2 (CCL2) and TGFβ1, which collectively trigger a pro-inflammatory response that leads to the elimination of senescent cells by immune cells179. The senescence-associated secretome also comprises pro-inflammatory mediators and growth factors involved in tissue fibrogenesis (such as IL-1α, IL-1β, IL-8, various CCLs, TGFβ1, PDGF and IL-6), proteins involved in ECM turnover (such as fibronectin, collagens and MMPs) and ECM proteins178,180. Both senescence-associated ECM and cytokines can ‘spread’ senescence between pro-inflammatory immune cells and stromal cells in mouse models of repair and fibrosis177,181. Myofibroblast senescence has been implicated in physiological wound healing, tissue regeneration and fibrosis. During liver regeneration, the transition of hepatic stellate cells that have activated to become myofibroblasts into a senescent phenotype facilitates their elimination by natural killer (NK) cells and limits the progression of liver fibrosis182. Similarly, induction of myofibroblast senescence by the matricellular protein CCN1 limits skin and liver scarring by inducing secretion of MMPs that degrade the fibrotic scar183,184.
Senescent myofibroblasts have also been implicated in the development of lung fibrosis172,185–189. In senescent lung myofibroblasts isolated from patients with IPF, p16 and p21 are upregulated in a TGFβ1-independent manner and seem to promote fibrosis by fostering a pro-inflammatory and pro-fibrotic microenvironment190. Indeed, cellular senescence in IPF myofibroblasts might be induced in response to telomere dysfunction as part of the DNA damage response172. In human fibroblasts, telomere shortening triggers p53-mediated p21 upregulation, whereas p16 expression is upregulated in a telomere-independent and DNA damage-independent manner191. In addition, suppression of telomerase expression is sufficient to promote α-SMA expression, suggesting that replicative senescence is associated with the acquisition of the myofibroblast phenotype192–194. By contrast, increased telomerase activity promotes the proliferation and survival of α-SMA− fibroblasts193. Accordingly, mice deficient in telomerase reverse transcriptase have decreased proliferation of α-SMA− fibroblasts and are protected from bleomycin-induced lung fibrosis192,195. Notably, myofibroblast senescence can occur in the absence of shortened telomeres196, suggesting that mechanisms other than replicative senescence might be behind this pathological phenotype during tissue fibrogenesis. For example, the pro-senescence factor CCN1 is induced by TGFβ1 in lung fibroblasts, suggesting that the acquisition of a senescent myofibroblast phenotype can be caused by telomere-independent mechanisms196. Importantly, genetic approaches have been used to ablate p16+ senescent cells in mouse lungs and kidneys, thereby mitigating fibrosis197. These results197 support the idea of using drugs that selectively induce the death of senescent cells (senolytic drugs) for the treatment of organ fibrosis186,198,199. However, care must be taken as senescent (myo)fibroblasts also seem to have beneficial functions in promoting normal tissue repair and in impeding fibrosis180. For example, inducing myofibroblast senescence by miR-34a inhibited lung fibrosis in mouse models of disease173. Timing — as always — seems to be critical for success when therapeutically interfering with senescence.
Resolution of fibrosis
Treatments that halt fibrosis are not necessarily effective in restoring organ function because fibrotic ECM contains biomechanical and biochemical factors that promote myofibroblast activation. To regain function, such mis-instructive and ‘diseased’ ECM needs to be removed and/or replaced with ‘healthy’ ECM in a controlled manner63,200. Although originally considered to be irreversible201, established fibrosis can regress in both mouse models of disease and human fibrotic diseases – particularly in the liver202. Although, human livers at late stages of cirrhosis remain untreatable, clear evidence exists that treatment of hepatitis C infection at early-to-mid stages of cirrhosis results in spontaneous reversion of liver fibrosis in humans203,204; ECM degradation is an important event in this process205,206. Likewise, liver fibrosis reverses spontaneously in mouse models of liver injury induced by alcohol intoxication, carbon tetrachloride or bile duct ligation60,207,208. Fibrosis can also regress in organs with lower regenerative capacity than the liver, such as the lungs63,209. In humans, lung fibrosis resolves substantially in patients with acute respiratory syndrome210,211, mimicking the biology of mouse models of acute lung injury induced by injection of bacterial wall saccharides212. Although lung fibrosis resolves spontaneously in mouse models of fibrogenic injury such as intratracheal administration of bleomycin, asbestos fibres or fluorescein isothiocyanate209, the progressive lung fibrosis seen in patients with IPF or SSc-associated ILD seems to be generally irreversible without treatment213. However, the results of randomized controlled trials in SSc-associated ILD consistently show subsets of patients with improved pulmonary function214,215, suggesting that tissue regeneration or regression of pre-existing fibrosis occurs. Clinical studies have also shown that skin fibrosis can regress in patients with SSc both spontaneously and after treatment216,217. Likewise, mouse skin fibrosis caused by subcutaneous injection of bleomycin resolves shortly after the fibrogenic stimulus is removed218. Overall, the capacity for fibrosis resolution might be organ-specific and differ depending on the nature of the fibrogenic stimulus, as well as host-specific factors including genetic susceptibility, age, sex and immunocompetence96.
Fibrosis resolution is characterized by ECM degradation and restoration of the biomechanical and biochemical properties of the ECM. Although it is known that targeted apoptosis of myofibroblasts results in fibrosis resolution in mouse models, the cellular and molecular mechanisms regulating ECM degradation and fibrosis resolution following myofibroblast apoptosis remain largely unknown. Macrophages, fibroblasts, NK cells and neutrophils have been previously implicated in ECM degradation following injury through the secretion of MMPs219. Degradation of fibrotic scar tissue, mostly composed of crosslinked type I collagen fibres, is initiated by collagenases (MMP-1, MMP-2, MMP-8, MMP-13, MMP-14, MMP-15 and MMP-16) and is followed by further cleavage by gelatinases, MMP-2 or MMP-9, as well as by other proteases such as cathepsin K220–222. Following degradation, collagen fragments are taken up by various cell types including fibroblasts and macrophages223. In fibroblasts, collagen fragments are recognized by C-type mannose receptor 2 (also known as ENDO180)224–227, which promotes collagen internalization and lysosomal degradation in a non-phagocytic manner. Alternatively, fibroblasts can also phagocytose large collagen particles via α1β1, α2β1 and α3β1 integrins prior to lysosomal degradation228–232.
Turning activated myofibroblasts into scar-resolving cells.
Whereas several mechanisms are known to deactivate myofibroblasts63, less is known about potential mechanisms that promote the transition of myofibroblasts into scar-resolving cells in vitro and in vivo. In fact, the reprogramming of scar-forming myofibroblasts into scar-resolving cells is an important cellular mechanism for scar resolution during physiological tissue repair (FIG. 1a). Fibroblasts acquire an ECM-degrading phenotype upon YAP–TAZ inhibition, which is associated with an increased expression of ECM-degrading enzymes such as plasmin, MMP-14 and cathepsin K122,231,232. Support for an important role for fibroblasts in degrading fibrillar collagen in vivo comes from studies that found impaired resolution of SSc-like fibrosis in mice with fibroblast-selective deletion of MMP14 (REF.231). In addition, mice deficient in cathepsin K develop increased lung fibrosis compared with wild-type mice, a phenotype that has been explained by decreased collagenolytic activity of fibroblasts232. Conversely, excessive cathepsin K activity of synovial fibroblasts mediates pathological collagen degradation in patients with RA233, indicating that temporal and spatial regulation of fibroblast ECM-degrading enzymes is essential to achieve tissue homeostasis. Concomitantly, expression of the transcription factor PU.1 rapidly causes pro-inflammatory ECM-degrading fibroblasts to become scar-forming pro-fibrotic fibroblasts234. Inhibition of PU.1 reprogrammes pro-fibrotic fibroblasts and promotes fibrosis regression in a mouse model of SSc234, suggesting an attractive novel anti-fibrotic strategy based on stimulating scar-resolving activities in fibroblasts. However, further research is needed to evaluate the plasticity of human pro-fibrotic fibroblasts across the spectrum of inflammatory, homeostatic and fibrotic phenotypes.
Macrophages in myofibroblast survival and fibrosis resolution.
Although fibroblastic cells can phagocytose collagen and other ECM components, they are not considered to be professional phagocytes. Instead, macrophages have been studied as professional phagocytes in the context of scar tissue resolution. In macrophages, the recognition and internalization of degraded collagen products is regulated by the extracellular bridging glycoprotein lactadherin (also known as MFGE8), which mediates collagen uptake and endocytosis via its first discoidin domain235. Accordingly, MFGE8-deficient mice develop severe pulmonary fibrosis after bleomycin-induced lung injury as a result of severe defects in collagen clearance and uptake by macrophages235. Similarly, the collagen receptor mannose receptor C type 1 is involved in collagen uptake by repair-associated macrophages during homeostasis in mice236; however, its role in fibrosis is poorly understood.
Collagen uptake and degradation trigger a rheostat mechanism in which collagen opsonization and endocytosis lead to increased production of collagen-degrading enzymes, supporting a positive feedback loop of ECM degradation237. However, whether these anti-fibrotic ECM degradation mechanisms are reactivated during fibrosis resolution is unclear. In support of this possibility, previous studies have shown that the ECM-degrading activity of macrophages can be induced by apoptotic bodies, thereby promoting the resolution of experimental models of pulmonary or biliary fibrosis in vivo238,239. Furthermore, although pulmonary delivery of TNF to mice does not directly target myofibroblasts for apoptosis, it still triggers lung fibrosis resolution by reprogramming pro-fibrotic CD11c+CD11bvarF4/80+ macrophages to a pro-resolving phenotype240. Thus, modulating macrophage phenotypes might serve as a novel strategy for promoting fibrosis resolution. Other studies suggest that CD11bhiLy6CloF4/80int macrophages are of particular therapeutic interest owing to their capacity to induce myofibroblast apoptosis and to secrete MMPs in the context of liver fibrosis241,242. In addition, NK cells have an important role in the resolution of liver fibrosis by promoting myofibroblast apoptosis; NK cells expressing cell death ligands such as TRAIL and FASL trigger the extrinsic pathway of apoptosis in hepatic stellate cells, ultimately promoting fibrosis resolution243,244. Overall, fibrosis resolution involves the activation of pro-resolving macrophages and NK cells, which promote ECM degradation, collagen uptake and the immunological clearance of myofibroblasts. Targeted therapies aimed at inducing myofibroblast apoptosis or reprogramming the immune system could reactivate these mechanisms in vivo. It will be interesting to see whether complex biomechanical and biochemical macrophage–myofibroblast interactions are involved in promoting myofibroblast and macrophage survival and the resolution of fibrosis31,245–247.
Strategies to reverse fibrosis
Advances in myofibroblast and fibrosis research have led to several exciting new therapeutic approaches and have revealed new potential targets to treat fibrotic disorders, including those associated with SSc1,30,94,248. Targeting myofibroblast apoptosis is emerging as one of these novel therapeutic strategies to reverse established fibrosis86,126,185. In this section, we discuss the molecular mechanisms behind novel therapeutic strategies aimed at inducing myofibroblast apoptosis (TABLE 2). These strategies might not only prevent the progression of organ fibrosis, but also have the potential to reverse established fibrosis.
Table 2 |.
Therapeutic strategies to target myofibroblasts
| Therapy | Target | Effect on myofibroblasts | Mouse models used to show anti-fibrotic effect | Development stage | Refs |
|---|---|---|---|---|---|
| Drugs targeting biomechanical signalling | |||||
| CWHM 12 | αV integrin | Inhibits TGFβ activation | CCL4-induced liver fibrosis; bleomycin-induced lung fibrosis | Preclinical | 115 |
| Abituzumab | αV integrin | Inhibits TGFβ activation | Not tested | Phase II | 303 |
| C8 | αVβ1 integrin | Inhibits TGFβ activation | Bleomycin-induced lung fibrosis; CCL4-induced liver fibrosis | Preclinical | 304 |
| Cilengitide | αVβ3 and αVβ5 integrins | Inhibits mechanotransduction | Bile duct ligation-induced liver fibrosis | Preclinical | 305 |
| 6.3G9 (STX-100) | αVβ6 integrin | Inhibits TGFβ activation | Bleomycin-induced lung fibrosis; irradiation-induced lung fibrosis; bile duct ligation-induced liver fibrosis; DDC-induced liver fibrosis; Alport mouse model Col4A3−/− | Phase II | 306–311 |
| PF-562,271 (VS-6062) | FAK | Inhibits mechanotransduction | Bleomycin-induced lung fibrosis; CCL4-induced liver fibrosis; acute myocardial infarction | Preclinical | 135,312 |
| PF-573,228 | FAK | Inhibits mechanotransduction | Bleomycin-induced lung fibrosis; hypertrophic skin scarring; acute myocardial infarction | Preclinical | 267,269,313 |
| TAE-226 | FAK | Inhibits mechanotransduction | Bleomycin-induced lung fibrosis | Preclinical | 265 |
| VS-4718 | FAK | Inhibits mechanotransduction | Pancreatic ductal adenocarcinoma | Phase II | 266,314 |
| Fasudil (ROCK1 and ROCK2) | ROCK | Inhibits mechanotransduction | Hypoxia-induced lung fibrosis; bleomycin-induced lung fibrosis; HClO-induced skin fibrosis; UUO-induced kidney fibrosis | Preclinical | 315–319 |
| Relaxin | ROCK | Inhibits mechanotransduction | Bleomycin-induced lung fibrosis | Phase III | 271,320 |
| KD025 (ROCK2) | ROCK | Inhibits mechanotransduction | Not tested | Phase II | 321 |
| CCG-1423 | MRTF-Aand MRTF-B | Inhibits mechanotransduction | Peritoneal fibrosis model | Preclinical | 322 |
| CCG-203971 | MRTF-Aand MRTF-B | Inhibits mechanotransduction | Bleomycin-induced lung fibrosis; bleomycin-induced skin fibrosis | Preclinical | 262,323 |
| Verteporfin | YAP and TAZ | Inhibits mechanotransduction | UUO-induced kidney fibrosis | Preclinical | 324 |
| Dimethyl fumarate | YAP and TAZ | Inhibits mechanotransduction | Bleomycin-induced skin fibrosis | Phase I | 264,325 |
| Ac-EEED peptide | α-SMA | Inhibits mechanotransduction | Full-thickness splinted skin wound healing model | Preclinical | 256 |
| Drugs targeting the intrinsic apoptosis pathway | |||||
| ABT-263 (navitoclax) | BCL-XL, BCL-2 and BCL-W/ | Activates intrinsic apoptosis pathway | Bleomycin-induced skin fibrosis; radiation-induced lung fibrosis | Preclinical | 86,275 |
| A-1331852 | bcl-xl | Activates intrinsic apoptosis pathway | Mdr2−/− rodent model of primary sclerosing cholangitis | Preclinical | 158 |
| Drugs targeting the extrinsic apoptosis pathway | |||||
| AT-406 | xiap | Activates extrinsic apoptosis pathway | Bleomycin-induced lung fibrosis | Preclinical | 138 |
| TLY012 | TRAIL | Activates extrinsic apoptosis pathway | Bleomycin-induced skin fibrosis; CCL4-induced liver fibrosis | Preclinical | 285,286 |
| Drugs targeting senescence | |||||
| Dasatinib and quercetin dual therapy | Multiple kinases | Activates apoptotic pathways | Bleomycin-induced lung fibrosis | Phasel | 186,326 |
| ABT-263 and A-1331852 | bcl-xl | Activates intrinsic apoptosis pathway | Radiation-induced lung fibrosis; Mdr2−/− rodent model of primary sclerosing cholangitis | Preclinical | 158,275 |
| GKT137831 | NOX4 | Activates apoptotic pathways | Bleomycin-induced lung fibrosis | Phase II | 185,327 |
α-SMA, α-smooth muscle actin; CCL4, CC-chemokine ligand 4; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; FAK, focal adhesion kinase; HClO, hypochlorous acid; MRTF, myocardin-related transcription factor; NOX4, NADPH oxidase 4; ROCK, RHO-associated protein kinase; TAZ, transcriptional co-activator with PDZ-binding motif; TGFβ, transforming growth factor-β; TRAIL, TNF-related apoptosis-inducing ligand (also known as TNFSF10); UUO, unilateral ureteral obstruction; XIAP, X-linked IAP; YAP, yes-associated protein.
Drugs targeting biomechanical signalling.
ECM remodelling by activated fibroblastic cells contributes to progressive tissue stiffening which, in turn, provides biomechanical feedback control over myofibroblast function and phenotype103,249. Simply put, myofibroblasts adapt their contractile force to the resistance they experience using their mechano-sensing and mechano-transducing cytoskeletal machinery (FIG. 4a). Several molecular elements of this machinery have been considered and tested as therapeutic targets to interrupt the fibrotic cycle by returning myofibroblasts to a relaxed state or inducing their apoptosis120,122–126,129,250–255. For example, direct functional inhibition of α-SMA stress fibres with specific acetylated tetrapeptides (Ac-EEED) reduced myofibroblast contraction and subsequent COL1A1 transcription in vitro256. Therapeutic blockade of myofibroblast integrins using small-molecule inhibitors or biological drugs can potentially have dual effects by reducing αv integrin-mediated TGFβ1 activation and by disturbing myofibroblast mechanosensing114,257–260. Inhibition or deletion of the transcription factor MRTF-A suppresses fibrogenesis in cell culture and the development of fibrosis in the skin, lungs, colon and heart in vivo253,261–263, and therapeutic targeting of TAZ and YAP ameliorates skin fibrosis in mice264. Similarly, inhibition of FAK and ROCK mechanotransduction pathways using small molecules induces myofibroblast apoptosis and has been used to treat fibrosis in various experimental models126,135,147,152,265–271 (TABLE 2).
Drugs targeting the intrinsic apoptosis pathway.
The discovery that apoptotic mitochondrial priming is heightened in pro-fibrotic myofibroblasts has paved the way for these cells to be targeted by pro-apoptotic approaches in fibrotic disorders86,87. Studies over the past few years have demonstrated that inducing apoptosis in myofibroblasts can revert tissue fibrosis in mouse models of disease86,126,160,185, and this approach is now emerging as a novel therapeutic strategy to reverse established fibrosis in human fibrotic diseases. BH3 mimetic drugs can selectively promote the intrinsic apoptotic pathway in myofibroblasts by targeting specific pro-survival BCL-2 family proteins. Importantly, only cells that are primed for death and have increased amounts of pro-apoptotic proteins in their mitochondria are sensitive to these drugs.
BH3 mimetic drugs are small molecules that inhibit interactions between pro-survival proteins and activators or effectors64,65 by directly binding to a shallow, hydrophobic groove in pro-survival BCL-2 family proteins. Differences in the structure of this hydrophobic groove in individual pro-survival BCL-2 family proteins have guided the design and development of selective BH3 mimetic drugs that target specific anti-apoptotic proteins65. Multiple BH3 mimetic drugs have been investigated in preclinical studies and several are currently being tested in clinical trials for various types of cancer. ABT-737 and its orally available analogue ABT-263 (navitoclax) bind to and block BCL-2, BCL-XL and BCL-W with subnanomolar affinity272,273. Treatment with ABT-263 resulted in selective induction of myofibroblast apoptosis in areas of dermal fibrosis and reversion of fibrosis in a mouse model of SSc dermal fibrosis86. ABT-263 also promoted apoptosis in PDGF-activated hepatic stellate cells157,158 and senescent lung fibroblasts274, and in mice, ABT-263 reversed radiation-induced lung fibrosis275. Consistently, the BCL-XL-specific BH3 mimetic drug A-1331852 abrogated biliary liver fibrosis in Mdr2−/− mice by promoting apoptosis of liver myofibroblasts276. As BCL-XL is important in promoting myofibroblast resistance to apoptosis, inhibition of BCL-XL might be a particularly potent therapeutic strategy for SSc and other fibrotic disorders.
However, the potential adverse effects of BH3 mimetic drugs must be considered. For example, human embryonic stem cells have increased mitochondrial priming and low survival ability in vitro compared with fully differentiated somatic cells277, a mechanism that prevents the propagation of genetic mutations by inducing apoptosis in injured stem cells. Although these in vitro findings suggest that BH3 mimetic therapy could potentially disrupt the balance of stem cell homeostasis owing to the high predisposition of these cells for apoptosis, in vivo studies have shown that BH3 mimetic therapy paradoxically rejuvenates prematurely aged mice by inducing apoptosis of senescent haematopoietic stem cells (HSCs)278. Thus, mitochondrial priming seems to be directly regulated by ageing in stem cells; in fact, mitochondrial priming is increased in HSCs from young mice compared with HSCs from aged mice279. However, further research is needed to understand how mitochondrial priming is regulated in tissue-specific stem cells during homeostasis and age-related fibrotic diseases, and what the potential effects of BH3 mimetic therapy might be on stem cell biology in the context of tissue repair and fibrotic disease.
The fact that BH3 mimetic drug ABT-263 causes reversible dose-limiting thrombocytopenia273 has also limited its use in the treatment of haematological malignancies, which require high doses for therapeutic efficacy. The ABT-263 target protein in chronic lymphocytic leukaemia is BCL-2, rather than BCL-XL, a fact that led to the development of BCL-2-specific BH3 mimetic drugs such as ABT-199. ABT-199 does not harm platelets and has been approved by the FDA for the treatment of chronic lymphocytic leukaemia in patients with a specific chromosomal abnormality280,281. Importantly, solid tumours, and particularly cancer-associated fibroblasts, rely on BCL-XL rather than BCL-2 to survive, thus providing enduring interest in using BCL-XL-specific BH3 mimetic drugs (such as ABT-263) to target BCL-XL in desmoplastic tumours282. The risk of thrombocytopenia seems to be manageable in this indication given its reversibility and the high doses needed (above 100 mg/kg) to cause platelet depletion.
Collectively, targeting apoptotic pathways in activated myofibroblasts with BH3 mimetic drugs reverses established fibrosis in preclinical models of skin, lung and liver fibrosis, suggesting that these drugs could be a selective, safe and potent anti-fibrotic tool to reverse organ fibrosis (TABLE 2). However, further clinical studies are needed to determine the efficacy of BH3 mimetic drugs in human fibrotic diseases.
Drugs targeting the extrinsic apoptosis pathway.
Unlike the growing body of evidence in support of targeting the intrinsic apoptosis pathway in myofibroblasts, less is known about the potential to target the control of myofibroblast apoptosis via the extrinsic pathway. Previous studies have found reduced expression of TNF receptors I and II and reduced sensitivity to FASL-induced and TNF-induced apoptosis in pro-fibrotic fibroblasts160–162,283,284. Although the results of these studies suggest that the extrinsic pathway of apoptosis is generally suppressed in myofibroblasts and cannot be exploited for therapeutic intervention, extrinsic apoptosis can still be triggered via activation of the TRAIL–death receptor pathway. In this mechanism, TGFβ1-mediated myofibroblast activation causes increased expression of DR4 and DR5, which are receptors for TRAIL. Accordingly, selective agonism of DR4 and DR5 with the recombinant TRAIL ligand TLY012 induces apoptosis in myofibroblasts and reverses established liver and skin fibrosis in mouse models of fibrotic disease285,286, highlighting the therapeutic potential of targeting myofibroblasts for apoptosis via activation of the extrinsic apoptosis pathway (TABLE 2).
Drugs targeting senescence.
Senolytic drugs are defined as therapeutic agents capable of inducing apoptosis in senescent cells by targeting pro-survival pathways in these cells175,176,287,288. The first class of senolytic drugs includes dasatinib and quercetin289, which increase lung function and partially reduce lung fibrosis in mouse models of fibrotic disease when administered together (D+Q therapy)186,199,290. In addition, the decreased sensitivity to FASL-induced apoptosis of IPF fibroblasts can be restored by treatment with quercetin291, suggesting that senolytic drugs might be a viable therapeutic option for IPF and other age-related diseases that progress with the accumulation of senescent myofibroblasts. Notably, the mechanism of action behind the senolytic activity of D+Q therapy remains poorly understood. Dasatinib is an ATP-competitive inhibitor that targets several kinases, including BCR-ABL, c-KIT, ephrin receptors, SRC, LYN, FYN and LCK292–294, whereas quercetin is a natural flavonoid that is found in a number of foods and has potent antioxidant effects that modulate the NF-κB and PI3K–AKT pathways295,296. Several studies have also shown that D+Q therapy only removes 30% of senescent cells289, suggesting that it targets an as-yet-unidentified subset of senescent cells.
Advances in understanding the molecular basis for the survival and accumulation of senescent cells have led to the development of a new generation of senolytic agents that promote apoptosis in senescent cells by targeting pro-survival BCL-2 family proteins. Senescent primary human fibroblasts become dependent on BCL-XL and BCL-W expression for survival regardless of what triggered the senescent phenotype (DNA-damage-induced senescence, replicative senescence or oncogene-induced senescence)274. Accordingly, blockade of BCL-XL and BCL-W with the BH3 mimetic ABT-737 induces apoptosis in senescent cells formed following DNA damage in the lungs of mice274. The BCL-XL-specific mimetic A1331852 reduced liver fibrosis in vivo by targeting senescent cholangiocytes for apoptosis158. Furthermore, ABT-263 showed potent senolytic activity in a mouse model of premature ageing278,297. Together, the potent senolytic and anti-fibrotic effects of ABT-263 suggest that the ability of this BH3 mimetic drug to reverse organ fibrosis might be caused by specific ‘fibro-senolytic’ effects on senescent myofibroblasts. Interestingly, the superoxide-generating enzyme NADPH oxidase 4 (NOX4) is induced in senescent lung fibroblasts, and pharmacological inhibition of NOX4 can target senescent fibroblast for apoptosis and reverse established lung fibrosis in aged mice185. Overall, these exciting preclinical data demonstrate the potential efficacy of targeting senescent myofibroblasts for apoptosis with fibro-senolytic agents to reverse established fibrosis. Accordingly, the results of the first-in-human clinical trial evaluating the effects of senolytic drugs on IPF were published in 2019 (TABLE 2). In this pilot study, D+Q therapy for 3 weeks improved physical function (evaluated by the 6-minute walk test and walking speed) in a small cohort of 14 patients with IPF298. Although these data are promising, results should be interpreted with caution and require further investigation.
Conclusions
Organ fibrosis is a lethal outcome of SSc, an autoimmune rheumatic disease characterized by persistent activation of scar-forming myofibroblasts. Intensive research into the mechanisms that promote and maintain myofibroblast activation has revealed novel therapeutic targets and prompted the development of promising first-in-class anti-fibrotic therapies for the treatment of fibrosis — a condition previously thought to be progressive and irreversible. As a result, a growing pipeline of small molecules and biologic drugs are now being tested in multiple phase II clinical trials on the basis of their ability to slow the progression of fibrosis in preclinical models. However, therapies that halt fibrosis are not necessarily effective at restoring organ function, and researchers have been investigating mechanisms of fibrosis resolution and how to reactivate the regenerative capacity of chronically damaged organs in an effort to restore function to fibrotic organs.
In established fibrosis, myofibroblast persistence, rather than activation, seems to be the disease mechanism that prevents tissue regeneration. Myofibroblasts escape death by activating pro-survival mechanisms in response to biomechanical and growth factor signals from the fibrotic microenvironment. Chronic activation of myofibroblasts can ultimately lead to the acquisition of a senescent phenotype. In addition, (senescent) myofibroblasts, rather than being apoptosis-resistant, are primed for apoptosis owing to concomitant activation of cell death signalling pathways. This knowledge of apoptotic priming has paved the way to specifically trigger apoptosis in myofibroblasts by blocking specific pro-survival BCL-2 family proteins. Mechanotherapeutic strategies that reduce the expression of pro-survival proteins by targeting biomechanical signalling and the direct inhibition of BCL-2 family proteins with BH3 mimetic drugs and senolytic agents trigger myofibroblast apoptosis and reverse established fibrosis in mouse models of disease. This growing body of research has set the stage for the development of a second generation of anti-fibrotic therapies that have the potential to reverse established fibrosis and to regenerate chronically injured tissues.
The initial enthusiasm around such drugs has been justified by studies in humans showing that established fibrosis can regress in fibrotic diseases such as liver cirrhosis and dermal SSc. Because ECM degradation is a crucial event in the process of reversing fibrosis, further efforts are needed to understand the cellular and molecular mechanisms that control ECM degradation during fibrosis resolution. Professional phagocytic cells such as macrophages would be expected to have an important role in ECM degradation upon the therapeutic removal of (senescent) myofibroblasts. Alternatively, scar-forming myofibroblasts could potentially be reprogrammed into scar-resolving cells, which would promote ECM degradation by harnessing the natural capacity of fibroblasts to synthesize and degrade ECM. Future research into the mechanisms that promote fibrosis resolution will increase our molecular and cellular understanding of this process and bring potential new therapies to reverse organ fibrosis.
Key points.
Organ fibrosis is a lethal outcome of autoimmune rheumatic diseases such as systemic sclerosis (SSc).
Myofibroblasts are scar-forming cells that are responsible for the excessive synthesis, deposition and remodelling of extracellular matrix proteins in SSc.
Persistent myofibroblast activity leads to progressive tissue fibrosis and distortion of the normal tissue architecture, resulting in organ failure and, ultimately, in death.
The termination of myofibroblast activity is suppressed in fibrotic disease by biomechanical and biochemical cues, which assist myofibroblast escape from apoptosis, thereby halting their elimination.
Evasion of apoptosis results in persistent myofibroblast activation and/or the differentiation of myofibroblasts into a pro-fibrotic or pro-inflammatory senescent phenotype, thereby preventing fibrosis resolution.
Targeting myofibroblast apoptosis and reprogramming these cells to become scar-resolving cells are emerging as novel therapeutic strategies to reverse established fibrosis.
Stress fibres.
Contractile bundles composed of actomyosin filaments.
t-Distributed stochastic neighbour embedding.
A technique for visualizing high-dimensional datasets, often displayed in the form of clusters.
Membrane blebbing.
Protrusions of the cell membrane that occur during apoptosis.
Pyknosis.
Condensation of the nuclear chromatin during apoptosis.
Apoptosome.
A multi-protein complex that mediates the initiation of apoptosis.
Granulation tissue.
New connective tissue that forms during wound healing.
Molecular rheostat.
A system that maintains and controls critical biological processes such as cell death and survival.
CArG boxes.
Repeating [CC(A/T)6GG] DNA sequences present within gene promoters.
Secretome.
The collection of molecules secreted by cells into the extracellular space.
Acknowledgements
The research of B.H. is supported by the Canadian Institutes of Health Research (Foundation Grant 375597). D.L. gratefully acknowledges funding support from the NIH (grant R01 HL147059-01), the Start-up Package from Massachusetts General Hospital, the Scleroderma Foundation New Investigator Grant, the Scleroderma Research Foundation Investigator-Initiated Research Grant, the American Thoracic Society Foundation/Pulmonary Fibrosis Foundation Research Grant and Sponsored Research Grants from Boehringer Ingelheim, Indalo Therapeutics and Unity Biotechnology.
Footnotes
Competing interests
D.L. declares that he has received research funding from Boehringer Ingelheim, Indalo Therapeutics and Unity Biotechnology. D.L. also has a financial interest in Mediar Therpeutics, which is developing treatments for organ fibrosis. D.L.’s interests were reviewed and are managed by MGH and Partners HealthCare in accordance with their conflict of interest policies. B.H. declares no competing interests.
References
- 1.Ho YY, Lagares D, Tager AM & Kapoor M Fibrosis – a lethal component of systemic sclerosis. Nat. Rev. Rheumatol 10, 390–402 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Smolen JS et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 4, 18001 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Bongartz T et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis. Rheum 62, 1583–1591 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allanore Y et al. Systemic sclerosis. Nat. Rev. Dis. Primers 1, 15002 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Trojanowska M & Varga J in Scleroderma (eds Varga J, Wigley F, Allanore Y & Kuwana M) 261–280 (Springer, 2017). [Google Scholar]
- 6.Steen VD & Medsger TA Changes in causes of death in systemic sclerosis, 1972–2002. Ann. Rheum. Dis 66, 940–944 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells AU & Denton CP Interstitial lung disease in connective tissue disease – mechanisms and management. Nat. Rev. Rheumatol 10, 728–739 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Distler O et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N. Engl. J. Med 380, 2518–2528 (2019). [DOI] [PubMed] [Google Scholar]
- 9.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01933334 (2016). [DOI] [PubMed]
- 10.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03068234 (2017). [DOI] [PubMed]
- 11.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03856853 (2019). [DOI] [PubMed]
- 12.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03221257 (2019). [DOI] [PubMed]
- 13.King TE Jr. et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med 370, 2083–2092 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Kumar A, Kapnadak SG, Girgis RE & Raghu G Lung transplantation in idiopathic pulmonary fibrosis. Expert. Rev. Respir. Med 12, 375–385 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C & Brown RA Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol 3, 349–363 (2002). [DOI] [PubMed] [Google Scholar]
- 16.van Caam A, Vonk M, van den Hoogen F, van Lent P & van der Kraan P Unraveling SSc pathophysiology; the myofibroblast. Front. Immunol 9, 2452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinz B et al. The myofibroblast: one function, multiple origins. Am. J. Pathol 170, 1807–1816 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marangoni RG et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. 67, 1062–1073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajkumar VS et al. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res. Ther 7, R1113–R1123 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Philippeos C et al. Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J. Invest. Dermatol 138, 811–825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang D et al. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat. Cell Biol 20, 422–431 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Harper RA & Grove G Human skin fibroblasts derived from papillary and reticular dermis: differences in growth potential in vitro. Science 204, 526–527 (1979). [DOI] [PubMed] [Google Scholar]
- 23.Sorrell JM & Caplan AI Fibroblast heterogeneity: more than skin deep. J. Cell Sci 117, 667–675 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Humphrey JD, Dufresne ER & Schwartz MA Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol 15, 802–812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tallquist MD & Molkentin JD Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol 14, 484–491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinz B & Suki B Does breathing amplify fibrosis? Am. J. Respir. Crit. Care Med 194, 9–11 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Lambert CA, Colige AC, Munaut C, Lapiere CM & Nusgens BV Distinct pathways in the over-expression of matrix metalloproteinases in human fibroblasts by relaxation of mechanical tension. Matrix Biol. 20, 397–408 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Leung DY, Glagov S & Mathews MB Cyclic stretching stimulates synthesis of matrix components by arterial smooth muscle cells in vitro. Science 191, 475–477 (1976). [DOI] [PubMed] [Google Scholar]
- 29.Bonnans C, Chou J & Werb Z Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol 15, 786–801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinz B Myofibroblasts. Exp. Eye Res 142, 56–70 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Pakshir P & Hinz B The big five in fibrosis: macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 68, 91–93 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Gabbiani G, Ryan GB & Majno G Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549–550 (1971). [DOI] [PubMed] [Google Scholar]
- 33.Talele NP, Fradette J, Davies JE, Kapus A & Hinz B Expression of α-smooth muscle actin determines the fate of mesenchymal stromal cells. Stem Cell Rep. 4, 1016–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farbehi N et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 8, e43882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerrero-Juarez CF et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat. Commun 10, 650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie T et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep. 22, 3625–3640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabib T, Morse C, Wang T, Chen W & Lafyatis R SFRP2/DPP4 and FMO1/LSP1 define major fibroblast populations in human skin. J. Invest. Dermatol 138, 802–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyser R et al. Defining the activated fibroblast population in lung fibrosis using single cell sequencing. Am. J. Respir. Cell Mol. Biol 61, 74–85 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Bartoschek M et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun 9, 5150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambrechts D et al. Phenotype moulding of stromal cells in the lung tumour microenvironment. Nat. Med 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park J et al. The Tcf21 lineage constitutes the lung lipofibroblast population. Am. J. Physiol. Lung Cell Mol. Physiol 316, L872–L885 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li R et al. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. eLife 7, e36865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zepp JA et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell 170, 1134–1148 e1110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ & Nguyen TQ Diverse origins of the myofibroblast–implications for kidney fibrosis. Nat. Rev. Nephrol 11, 233–244 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Affo S, Yu LX & Schwabe RF The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol 12, 153–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider RK et al. Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell Stem Cell 20, 785–800 e788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Carlo SE & Peduto L The perivascular origin of pathological fibroblasts. J. Clin. Invest 128, 54–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lynch MD & Watt FM Fibroblast heterogeneity: implications for human disease. J. Clin. Invest 128, 26–35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S, Herault Y, Pavlovic G & Leask A Skin progenitor cells contribute to bleomycin-induced skin fibrosis. Arthritis Rheumatol. 66, 707–713 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Driskell RR et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rinkevich Y et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Croft AP et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu X et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest 128, 2127–2143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rognoni E et al. Fibroblast state switching orchestrates dermal maturation and wound healing. Mol. Syst. Biol 14, e8174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El Agha E et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 20, 261–273 e263 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanchez-Iranzo H et al. Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc. Natl Acad. Sci. USA 115, 4188–4193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jun JI & Lau LF The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol 12, 676–685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Demaria M et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kisseleva T et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl Acad. Sci. USA 109, 9448–9453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuppan D, Surabattula R & Wang XY Determinants of fibrosis progression and regression in NASH. J. Hepatol 68, 238–250 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Jun JI & Lau LF Resolution of organ fibrosis. J. Clin. Invest 128, 97–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glasser SW et al. Mechanisms of lung fibrosis resolution. Am. J. Pathol 186, 1066–1077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Youle RJ & Strasser A The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol 9, 47–59 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Czabotar PE, Lessene G, Strasser A & Adams JM Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol 15, 49–63 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Elmore S Apoptosis: a review of programmed cell death. Toxicol. Pathol 35, 495–516 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagata S Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol 36, 489–517 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Elliott MR & Ravichandran KS Clearance of apoptotic cells: implications in health and disease. J. Cell Biol 189, 1059–1070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jorgensen I, Rayamajhi M & Miao EA Programmed cell death as a defence against infection. Nat. Rev. Immunol 17, 151–164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saraste A & Pulkki K Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res 45, 528–537 (2000). [DOI] [PubMed] [Google Scholar]
- 71.Taylor RC, Cullen SP & Martin SJ Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol 9, 231–241 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Morioka S, Maueroder C & Ravichandran KS Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity 50, 1149–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh R, Letai A & Sarosiek K Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol 20, 175–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kantari C & Walczak H Caspase-8 and Bid: caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 1813, 558–563 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Darby I, Skalli O & Gabbiani G Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab. Invest 63, 21–29 (1990). [PubMed] [Google Scholar]
- 76.Desmouliere A, Redard M, Darby I & Gabbiani G Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol 146, 56–66 (1995). [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang HY & Phan SH Inhibition of myofibroblast apoptosis by transforming growth factor β1. Am. J. Respir. Cell Mol. Biol 21, 658–665 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Bostrom H et al. PDGF-A signalling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863–873 (1996). [DOI] [PubMed] [Google Scholar]
- 79.Ishiguro S et al. Basic fibroblast growth factor induces down-regulation of alpha-smooth muscle actin and reduction of myofibroblast areas in open skin wounds. Wound Repair Regen. 17, 617–625 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Ramos C et al. Acidic fibroblast growth factor decreases alpha-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol 291, L871–L879 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Mizuno S, Matsumoto K, Li MY & Nakamura T HGF reduces advancing lung fibrosis in mice: a potential role for MMP-dependent myofibroblast apoptosis. FASEB J. 19, 580–582 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Saile B, Knittel T, Matthes N, Schott P & Ramadori G CD95/CD95L-mediated apoptosis of the hepatic stellate cell. A mechanism terminating uncontrolled hepatic stellate cell proliferation during hepatic tissue repair. Am. J. Pathol 151, 1265–1272 (1997). [PMC free article] [PubMed] [Google Scholar]
- 83.Iredale JP et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest 102, 538–549 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thannickal VJ & Horowitz JC Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc 3, 350–356 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fattman CL Apoptosis in pulmonary fibrosis: too much or not enough? Antioxid. Redox Signal 10, 379–385 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Lagares D et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl Med 9, eaal3765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuehl T & Lagares D BH3 mimetics as anti-fibrotic therapy: unleashing the mitochondrial pathway of apoptosis in myofibroblasts. Matrix Biol. 68–69, 94–105 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Del Gaizo Moore V et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Invest 117, 112–121 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deng J et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Ryan JA, Brunelle JK & Letai A Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl Acad. Sci. USA 107, 12895–12900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Certo M et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Ryan JA, Brunelle JK & Letai A Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl Acad. Sci. USA 107, 12895–12900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chonghaile TN et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334, 1129–1133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Volkmann ER & Varga J Emerging targets of disease-modifying therapy for systemic sclerosis. Nat. Rev. Rheumatol 15, 208–224 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Balestrini JL, Chaudhry S, Sarrazy V, Koehler A & Hinz B The mechanical memory of lung myofibroblasts. Integr. Biol 4, 410–421 (2012). [DOI] [PubMed] [Google Scholar]
- 96.Horowitz JC & Thannickal VJ Mechanisms for the resolution of organ fibrosis. Physiology 34, 43–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Verrecchia F & Mauviel A Transforming growth factor-beta and fibrosis. World J. Gastroenterol 13, 3056–3062 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Serini G et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol 142, 873–881 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leask A & Abraham DJ The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem. Cell Biol 81, 355–363 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Lagares D et al. ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis. Nat. Med 23, 1405–1415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu B, Rockel JS, Lagares D & Kapoor M Ephrins and Eph receptor signalling in tissue repair and fibrosis. Curr. Rheumatol. Rep 21, 23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Santos A & Lagares D Matrix stiffness: the conductor of organ fibrosis. Curr. Rheumatol. Rep 20, 2 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Hinz B, McCulloch CA & Coelho NM Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res 379, 119–128 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Parker MW et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest 124, 1622–1635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu F et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol 190, 693–706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hinz B Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr. Rheumatol. Rep 11, 120–126 (2009). [DOI] [PubMed] [Google Scholar]
- 107.Tschumperlin DJ, Ligresti G, Hilscher MB & Shah VH Mechanosensing and fibrosis. J. Clin. Invest 128, 74–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herrera J, Henke CA & Bitterman PB Extracellular matrix as a driver of progressive fibrosis. J. Clin. Invest 128, 45–53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Burgess JK, Mauad T, Tjin G, Karlsson JC & Westergren-Thorsson G The extracellular matrix – the under-recognized element in lung disease? J. Pathol 240, 397–409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lampi MC & Reinhart-King CA Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci. Transl Med 10, eaao0475 (2018). [DOI] [PubMed] [Google Scholar]
- 111.Hinz B Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol 127, 526–537 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Klingberg F et al. Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J. Cell Biol 207, 283–297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wipff PJ, Rifkin DB, Meister JJ & Hinz B Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol 179, 1311–1323 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reed NI et al. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl Med 7, 288ra279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Henderson NC et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med 19, 1617–1624 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao XH et al. Force activates smooth muscle alpha-actin promoter activity through the Rho signalling pathway. J. Cell Sci 120, 1801–1809 (2007). [DOI] [PubMed] [Google Scholar]
- 117.Speight P, Kofler M, Szaszi K & Kapus A Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat. Commun 7, 11642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Varney SD et al. Hic-5 is required for myofibroblast differentiation by regulating mechanically dependent MRTF-A nuclear accumulation. J. Cell Sci 129, 774–787 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Calvo F et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol 15, 637–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Speight P, Nakano H, Kelley TJ, Hinz B & Kapus A Differential topical susceptibility to TGFbeta in intact and injured regions of the epithelium: key role in myofibroblast transition. Mol. Biol. Cell 24, 3326–3336 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Piersma B et al. YAP1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am. J. Pathol 185, 3326–3337 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Liu F et al. Mechanosignalling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 308, L344–L357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Piersma B et al. YAP1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am. J. Pathol 185, 3326–3337 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Szeto SG et al. YAP/TAZ are mechanoregulators of TGF-beta-Smad signalling and renal fibrogenesis. J. Am. Soc. Nephrol 27, 3117–3128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen H et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun 7, 12564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou Y et al. Inhibition of mechanosensitive signalling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Invest 123, 1096–1108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nardone G et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun 8, 15321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sabra H et al. Beta1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem 292, 19179–19197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Martin K et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun 7, 12502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Grinnell F, Zhu M, Carlson MA & Abrams JM Release of mechanical tension triggers apoptosis of human fibroblasts in a model of regressing granulation tissue. Exp. Cell. Res 248, 608–619 (1999). [DOI] [PubMed] [Google Scholar]
- 131.Niland S et al. Contraction-dependent apoptosis of normal dermal fibroblasts. J. Invest. Dermatol 116, 686–692 (2001). [DOI] [PubMed] [Google Scholar]
- 132.Carlson MA, Longaker MT & Thompson JS Wound splinting regulates granulation tissue survival. J. Surg. Res 110, 304–309 (2003). [DOI] [PubMed] [Google Scholar]
- 133.Hinz B, Mastrangelo D, Iselin CE, Chaponnier C & Gabbiani G Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol 159, 1009–1020 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li CX et al. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat. Mater 16, 379–389 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Lagares D et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis. Rheum 64, 1653–1664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ajayi IO et al. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am. J. Respir. Cell Mol. Biol 49, 86–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Horowitz JC et al. Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int. J. Biochem. Cell Biol 44, 158–169 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ashley SL et al. Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol 54, 482–492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jafarinejad-Farsangi S et al. MicroRNA-29a induces apoptosis via increasing the Bax:Bcl-2 ratio in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity 48, 369–378 (2015). [DOI] [PubMed] [Google Scholar]
- 140.Cavarretta E & Condorelli G miR-21 and cardiac fibrosis: another brick in the wall? Eur. Heart J 36, 2139–2141 (2015). [DOI] [PubMed] [Google Scholar]
- 141.Huang Y, He Y & Li J MicroRNA-21: a central regulator of fibrotic diseases via various targets. Curr. Pharm. Des 21, 2236–2242 (2015). [DOI] [PubMed] [Google Scholar]
- 142.Wang T et al. miR-21 regulates skin wound healing by targeting multiple aspects of the healing process. Am. J. Pathol 181, 1911–1920 (2012). [DOI] [PubMed] [Google Scholar]
- 143.Chen Z, Dai T, Chen X, Tan L & Shi C Activation and regulation of the granulation tissue derived cells with stemness-related properties. Stem Cell Res. Ther 6, 85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Thum T & Lorenzen JM Cardiac fibrosis revisited by microRNA therapeutics. Circulation 126, 800–802 (2012). [DOI] [PubMed] [Google Scholar]
- 145.Chau BN et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl Med 4, 121ra118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jun JB et al. Scleroderma fibroblasts demonstrate enhanced activation of Akt (protein kinase B) in situ. J. Invest. Dermatol 124, 298–303 (2005). [DOI] [PubMed] [Google Scholar]
- 147.Lagares D, Busnadiego O, Garcia-Fernandez RA, Lamas S & Rodriguez-Pascual F Adenoviral gene transfer of endothelin-1 in the lung induces pulmonary fibrosis through the activation of focal adhesion kinase. Am. J. Respir. Cell Mol. Biol 47, 834–842 (2012). [DOI] [PubMed] [Google Scholar]
- 148.Lagares D et al. Endothelin 1 contributes to the effect of transforming growth factor beta1 on wound repair and skin fibrosis. Arthritis. Rheum 62, 878–889 (2010). [DOI] [PubMed] [Google Scholar]
- 149.Kulasekaran P et al. Endothelin-1 and transforming growth factor-beta1 independently induce fibroblast resistance to apoptosis via AKT activation. Am. J. Respir. Cell Mol. Biol 41, 484–493 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kulkarni AA et al. PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLOS ONE 6, e15909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xia H, Nho RS, Kahm J, Kleidon J & Henke CA Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signalling pathway. J. Biol. Chem 279, 33024–33034 (2004). [DOI] [PubMed] [Google Scholar]
- 152.Horowitz JC et al. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 19, 761–771 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Horowitz JC et al. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J. Biol. Chem 279, 1359–1367 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jelaska A & Korn JH Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis. Rheum 43, 2230–2239 (2000). [DOI] [PubMed] [Google Scholar]
- 155.Zhang L, Zhou F & ten Dijke P Signalling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends. Biochem. Sci 38, 612–620 (2013). [DOI] [PubMed] [Google Scholar]
- 156.Karimizadeh E et al. c-Abl silencing reduced the inhibitory effects of TGF-beta1 on apoptosis in systemic sclerosis dermal fibroblasts. Mol. Cell Biochem 405, 169–176 (2015). [DOI] [PubMed] [Google Scholar]
- 157.Rizvi S et al. Platelet-derived growth factor primes cancer-associated fibroblasts for apoptosis. J. Biol. Chem 289, 22835–22849 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Moncsek A et al. Targeting senescent cholangiocytes and activated fibroblasts with B cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 67, 247–259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dodi AE et al. Regulation of fibroblast Fas expression by soluble and mechanical pro-fibrotic stimuli. Respir. Res 19, 91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Santiago B, Galindo M, Rivero M & Pablos JL Decreased susceptibility to fas-induced apoptosis of systemic sclerosis dermal fibroblasts. Arthritis. Rheum 44, 1667–1676 (2001). [DOI] [PubMed] [Google Scholar]
- 161.Tanaka T et al. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur. Respir. J 20, 359–368 (2002). [DOI] [PubMed] [Google Scholar]
- 162.Buhling F et al. Altered expression of membrane-bound and soluble CD95/Fas contributes to the resistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respir. Res 6, 37 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lin T et al. Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J. Biol. Chem 275, 8657–8663 (2000). [DOI] [PubMed] [Google Scholar]
- 164.Samuel GH, Lenna S, Bujor AM, Lafyatis R & Trojanowska M Acid sphingomyelinase deficiency contributes to resistance of scleroderma fibroblasts to Fas-mediated apoptosis. J. Dermatol. Sci 67, 166–172 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Im J, Kim K, Hergert P & Nho RS Idiopathic pulmonary fibrosis fibroblasts become resistant to Fas ligand-dependent apoptosis via the alteration of decoy receptor 3. J. Pathol 240, 25–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang SK et al. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 23, 4317–4326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Maher TM et al. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am. J. Respir. Crit Care Med 182, 73–82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bernard K et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem 290, 25427–25438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Santinon G, Pocaterra A & Dupont S Control of YAP/TAZ activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 26, 289–299 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Rangarajan S et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med 24, 1121–1127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mora AL, Rojas M, Pardo A & Selman M Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov 16, 755–772 (2017). [DOI] [PubMed] [Google Scholar]
- 172.Alvarez D et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell Mol. Physiol 313, L1164–L1173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cui H et al. miR-34a inhibits lung fibrosis by inducing lung fibroblast senescence. Am J Respir. Cell Mol. Biol 56, 168–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hayflick L The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res 37, 614–636 (1965). [DOI] [PubMed] [Google Scholar]
- 175.Munoz-Espin D & Serrano M Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol 15, 482–496 (2014). [DOI] [PubMed] [Google Scholar]
- 176.He S & Sharpless NE Senescence in health and disease. Cell 169, 1000–1011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bird TG et al. TGFbeta inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl Med 10, eaan1230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Coppe JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumour suppression. Annu. Rev. Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chien Y et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mavrogonatou E, Pratsinis H, Papadopoulou A, Karamanos NK & Kletsas D Extracellular matrix alterations in senescent cells and their significance in tissue homeostasis. Matrix Biol. 75–76, 27–42 (2019). [DOI] [PubMed] [Google Scholar]
- 181.Hiebert P et al. Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev. Cell 46, 145–161 e110 (2018). [DOI] [PubMed] [Google Scholar]
- 182.Krizhanovsky V et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kim KH, Chen CC, Monzon RI & Lau LF Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell Biol 33, 2078–2090 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jun J-I & Lau LF The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol 12, 676–685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hecker L et al. Reversal of persistent fibrosis in ageing by targeting Nox4-Nrf2 redox imbalance. Sci. Transl Med 6, 231ra247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Schafer MJ et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun 8, 14532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Schuliga M et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J. Cell Mol. Med 22, 5847–5861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li Y et al. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 55, 35–48 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mora AL, Rojas M, Pardo A & Selman M Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov 16, 755–772 (2017). [DOI] [PubMed] [Google Scholar]
- 190.Barnes PJ, Baker J & Donnelly LE Cellular senescence as a mechanism and target in chronic lung diseases. Am. J. Respir. Crit. Care Med 200, 556–564 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Herbig U, Jobling WA, Chen BP, Chen DJ & Sedivy JM Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513 (2004). [DOI] [PubMed] [Google Scholar]
- 192.Liu T et al. Telomerase regulation of myofibroblast differentiation. Am. J. Respir. Cell Mol. Biol 34, 625–633 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu T, Nozaki Y & Phan SH Regulation of telomerase activity in rat lung fibroblasts. Am. J. Respir. Cell Mol. Biol 26, 534–540 (2002). [DOI] [PubMed] [Google Scholar]
- 194.Razdan N, Vasilopoulos T & Herbig U Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Ageing Cell 17, e12838 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu T et al. Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J. Clin. Invest 117, 3800–3809 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kurundkar AR et al. The matricellular protein CCN1 enhances TGF-beta1/SMAD3-dependent profibrotic signalling in fibroblasts and contributes to fibrogenic responses to lung injury. FASEB J. 30, 2135–2150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Braun H et al. Cellular senescence limits regenerative capacity and allograft survival. J. Am. Soc. Nephrol 23, 1467–1473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mailleux AA & Crestani B Licence to kill senescent cells in idiopathic pulmonary fibrosis? Eur. Respir. J 50, 1701360 (2017). [DOI] [PubMed] [Google Scholar]
- 199.Lehmann M et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J 50, 1602367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sand JM et al. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir. Res 16, 69 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rockey DC, Bell PD & Hill JA Fibrosis–a common pathway to organ injury and failure. N. Engl. J. Med 372, 1138–1149 (2015). [DOI] [PubMed] [Google Scholar]
- 202.Drew L Tipping the balance. Nature 564, S74–S75 (2018). [DOI] [PubMed] [Google Scholar]
- 203.Ellis EL & Mann DA Clinical evidence for the regression of liver fibrosis. J. Hepatol 56, 1171–1180 (2012). [DOI] [PubMed] [Google Scholar]
- 204.Iredale JP et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest 102, 538–549 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.van der Meer AJ & Berenguer M Reversion of disease manifestations after HCV eradication. J. Hepatol 65, S95–S108 (2016). [DOI] [PubMed] [Google Scholar]
- 206.Lee YA & Friedman SL Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antivir. Res 107, 23–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Iredale JP Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Invest 117, 539–548 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Friedman SL Fibrogenic cell reversion underlies fibrosis regression in liver. Proc. Natl Acad. Sci. USA 109, 9230–9231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Moore BB et al. Animal models of fibrotic lung disease. Am. J. Respir. Cell Mol. Biol 49, 167–179 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Marshall R, Bellingan G & Laurent G The acute respiratory distress syndrome: fibrosis in the fast lane. Thorax 53, 815–817 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Burnham EL, Janssen WJ, Riches DW, Moss M & Downey GP The fibroproliferative response in acute respiratory distress syndrome: mechanisms and clinical significance. Eur. Respir. J 43, 276–285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Szarka RJ, Wang N, Gordon L, Nation PN & Smith RH A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J. Immunol. Methods 202, 49–57 (1997). [DOI] [PubMed] [Google Scholar]
- 213.Thannickal VJ, Toews GB, White ES, Lynch JP 3rd & Martinez FJ Mechanisms of pulmonary fibrosis. Annu. Rev. Med 55, 395–417 (2004). [DOI] [PubMed] [Google Scholar]
- 214.Zamora AC et al. Use of mycophenolate mofetil to treat scleroderma-associated interstitial lung disease. Respir. Med 102, 150–155 (2008). [DOI] [PubMed] [Google Scholar]
- 215.Gerbino AJ, Goss CH & Molitor JA Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest 133, 455–460 (2008). [DOI] [PubMed] [Google Scholar]
- 216.Dobrota R et al. Prediction of improvement in skin fibrosis in diffuse cutaneous systemic sclerosis: a EUSTAR analysis. Ann. Rheum. Dis 75, 1743–1748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Merkel PA et al. Patterns and predictors of change in outcome measures in clinical trials in scleroderma: an individual patient meta-analysis of 629 subjects with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 64, 3420–3429 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Beyer C, Schett G, Distler O & Distler JH Animal models of systemic sclerosis: prospects and limitations. Arthritis Rheum. 62, 2831–2844 (2010). [DOI] [PubMed] [Google Scholar]
- 219.Lu P, Takai K, Weaver VM & Werb Z Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol 3, a005058 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Egeblad M & Werb Z New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 (2002). [DOI] [PubMed] [Google Scholar]
- 221.Visse R & Nagase H Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res 92, 827–839 (2003). [DOI] [PubMed] [Google Scholar]
- 222.Fonovic M & Turk B Cysteine cathepsins and extracellular matrix degradation. Biochim. Biophys. Acta 1840, 2560–2570 (2014). [DOI] [PubMed] [Google Scholar]
- 223.McKleroy W, Lee TH & Atabai K Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 304, L709–L721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Madsen DH et al. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/Endo180, cooperate in fibroblast-mediated collagen degradation. J. Biol. Chem 282, 27037–27045 (2007). [DOI] [PubMed] [Google Scholar]
- 225.Wienke D, MacFadyen JR & Isacke CM Identification and characterization of the endocytic transmembrane glycoprotein Endo180 as a novel collagen receptor. Mol. Biol. Cell 14, 3592–3604 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.East L et al. A targeted deletion in the endocytic receptor gene Endo180 results in a defect in collagen uptake. EMBO Rep. 4, 710–716 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Engelholm LH et al. uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J. Cell Biol 160, 1009–1015 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Segal G et al. Involvement of actin filaments and integrins in the binding step in collagen phagocytosis by human fibroblasts. J. Cell Sci 114, 119–129 (2001). [DOI] [PubMed] [Google Scholar]
- 229.Arora PD, Manolson MF, Downey GP, Sodek J & McCulloch CA A novel model system for characterization of phagosomal maturation, acidification, and intracellular collagen degradation in fibroblasts. J. Biol. Chem 275, 35432–35441 (2000). [DOI] [PubMed] [Google Scholar]
- 230.Lee W, Sodek J & McCulloch CA Role of integrins in regulation of collagen phagocytosis by human fibroblasts. J. Cell Physiol 168, 695–704 (1996). [DOI] [PubMed] [Google Scholar]
- 231.Zigrino P et al. Fibroblast-derived MMP-14 regulates collagen homeostasis in adult skin. J. Invest. Dermatol 136, 1575–1583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Buhling F et al. Pivotal role of cathepsin K in lung fibrosis. Am. J. Pathol 164, 2203–2216 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hou WS et al. Cathepsin K is a critical protease in synovial fibroblast-mediated collagen degradation. Am. J. Pathol 159, 2167–2177 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wohlfahrt T et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566, 344–349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Atabai K et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Invest 119, 3713–3722 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Madsen DH et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol 202, 951–966 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Borza CM & Pozzi A Discoidin domain receptors in disease. Matrix Biol. 34, 185–192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Yoon YS et al. PPARγ activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol. 8, 1031–1046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Popov Y et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol 298, G323–G334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Redente EF et al. Tumour necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am. J. Respir. Cell Mol. Biol 50, 825–837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Imamura M, Ogawa T, Sasaguri Y, Chayama K & Ueno H Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology 128, 138–146 (2005). [DOI] [PubMed] [Google Scholar]
- 242.Mitchell C et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am. J. Pathol 174, 1766–1775 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Glassner A et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Invest 92, 967–977 (2012). [DOI] [PubMed] [Google Scholar]
- 244.Radaeva S et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumour necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 130, 435–452 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Shook BA et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362, eaar2971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lodyga M et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-beta. Sci. Signal 12, eaao3469 (2019). [DOI] [PubMed] [Google Scholar]
- 247.Pakshir P et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun 10, 1850 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wynn TA & Ramalingam TR Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med 18, 1028–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Van De Water L, Varney S & Tomasek JJ Mechanoregulation of the myofibroblast in wound contraction, scarring, and fibrosis: opportunities for new therapeutic intervention. Adv. Wound Care 2, 122–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Herum KM, Lunde IG, McCulloch AD & Christensen G The soft- and hard-heartedness of cardiac fibroblasts: mechanotransduction signalling pathways in fibrosis of the heart. J. Clin. Med 6, 53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Follonier Castella L, Gabbiani G, McCulloch CA & Hinz B Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp. Cell Res 316, 2390–2401 (2010). [DOI] [PubMed] [Google Scholar]
- 252.Htwe SS et al. Role of ROCK isoforms in regulation of stiffness induced myofibroblast differentiation in lung fibrosis. Am J Respir Cell Mol Biol 56, 772–783 (2017). [DOI] [PubMed] [Google Scholar]
- 253.Shiwen X et al. A role of myocardin related transcription factor-A (MRTF-A) in scleroderma related fibrosis. PLOS ONE 10, e0126015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Velasquez LS et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl Acad. Sci. USA 110, 16850–16855 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Crider BJ, Risinger GM Jr., Haaksma CJ, Howard EW & Tomasek JJ Myocardin-related transcription factors A and B are key regulators of TGF-β1-induced fibroblast to myofibroblast differentiation. J. Invest. Dermatol 131, 2378–2385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hinz B, Gabbiani G & Chaponnier C The NH2-terminal peptide of alpha-smooth muscle actin inhibits force generation by the myofibroblast in vitro and in vivo. J. Cell Biol 157, 657–663 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Schnittert J, Bansal R, Storm G & Prakash J Integrins in wound healing, fibrosis and tumour stroma: high potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev 129, 37–53 (2018). [DOI] [PubMed] [Google Scholar]
- 258.Schulz JN et al. New developments on skin fibrosis - Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 68–69, 522–532 (2018). [DOI] [PubMed] [Google Scholar]
- 259.Murray IR et al. Alphav integrins on mesenchymal cells regulate skeletal and cardiac muscle fibrosis. Nat. Commun 8, 1118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hinz B It has to be the αv: myofibroblast integrins activate latent TGF-β1. Nat. Med 19, 1567–1568 (2013). [DOI] [PubMed] [Google Scholar]
- 261.Johnson LA et al. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-β-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis 20, 154–165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sisson TH et al. Inhibition of myocardin-related transcription factor/serum response factor signalling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am. J. Pathol 185, 969–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Penke LR, Huang SK, White ES & Peters-Golden M Prostaglandin E2 inhibits α-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J. Biol. Chem 289, 17151–17162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Toyama T et al. Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J. Invest. Dermatol 138, 78–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kinoshita K et al. Antifibrotic effects of focal adhesion kinase inhibitor in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol 49, 536–543 (2013). [DOI] [PubMed] [Google Scholar]
- 266.Jiang H et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med 22, 851–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wong VW et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signalling. Nat. Med 18, 148–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zhao XK et al. Focal adhesion kinase regulates hepatic stellate cell activation and liver fibrosis. Sci. Rep 7, 4032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang J et al. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci. Rep 7, 43146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bond JE et al. Wound contraction is attenuated by fasudil inhibition of Rho-associated kinase. Plast. Reconstr. Surg 128, 438e–450e (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Huang X et al. Relaxin regulates myofibroblast contractility and protects against lung fibrosis. Am. J. Pathol 179, 2751–2765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Van Delft MF et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Tse C et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428 (2008). [DOI] [PubMed] [Google Scholar]
- 274.Yosef R et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun 7, 11190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Pan J et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int. J. Radiat. Oncol. Biol. Phys 99, 353–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Moncsek A et al. Targeting senescent cholangiocytes and activated fibroblasts with Bcl-xL inhibitors ameliorates fibrosis in Mdr2−/− mice. Hepatology 67, 247–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Liu JC et al. High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 13, 483–491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Chang J et al. Clearance of senescent cells by ABT263 rejuvenates aged haematopoietic stem cells in mice. Nat. Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Gutierrez-Martinez P et al. Diminished apoptotic priming and ATM signalling confer a survival advantage onto aged haematopoietic stem cells in response to DNA damage. Nat. Cell Biol 20, 413–421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Souers AJ et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumour activity while sparing platelets. Nat. Med 19, 202–208 (2013). [DOI] [PubMed] [Google Scholar]
- 281.Mullard A 2016 FDA drug approvals. Nat. Rev. Drug Discov 16, 73–76 (2017). [DOI] [PubMed] [Google Scholar]
- 282.Mullard A Pioneering apoptosis-targeted cancer drug poised for FDA approval. Nat. Rev. Drug Discov 15, 147–149 (2016). [DOI] [PubMed] [Google Scholar]
- 283.Vancheri C et al. Different expression of TNF-α receptors and prostaglandin E(2) production in normal and fibrotic lung fibroblasts: potential implications for the evolution of the inflammatory process. Am. J. Respir. Cell Mol. Biol 22, 628–634 (2000). [DOI] [PubMed] [Google Scholar]
- 284.Frankel SK et al. TNF-α sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis. Am. J. Respir. Cell Mol. Biol 34, 293–304 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Park JS et al. Targeting of dermal myofibroblasts through death receptor 5 arrests fibrosis in mouse models of scleroderma. Nat. Commun 10, 1128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Oh Y et al. Systemic PEGylated TRAIL treatment ameliorates liver cirrhosis in rats by eliminating activated hepatic stellate cells. Hepatology 64, 209–223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Ovadya Y & Krizhanovsky V Strategies targeting cellular senescence. J. Clin. Invest 128, 1247–1254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of ageing. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhu Y et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Ageing Cell 14, 644–658 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sellares J & Rojas M Quercetin in idiopathic pulmonary fibrosis: another brick in the senolytic wall. Am. J. Respir. Cell Mol. Biol 60, 3–4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hohmann MS, Habiel DM, Coelho AL, Verri WA Jr. & Hogaboam CM Quercetin enhances ligand-induced apoptosis in senescent IPF fibroblasts and reduces lung fibrosis in vivo. Am. J. Respir. Cell Mol. Biol 60, 28–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Das J et al. 2-Aminothiazole as a novel kinase inhibitor template. Structure–activity relationship studies towards the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1, 3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J. Med. Chem 49, 6819–6832 (2006). [DOI] [PubMed] [Google Scholar]
- 293.Li J et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol 6, 291–299 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Han L, Schuringa JJ, Mulder A & Vellenga E Dasatinib impairs long-term expansion of leukemic progenitors in a subset of acute myeloid leukemia cases. Ann. Haematol 89, 861–871 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Russo GL et al. Quercetin: a pleiotropic kinase inhibitor against cancer. Cancer Treat. Res 159, 185–205 (2014). [DOI] [PubMed] [Google Scholar]
- 296.Williams RJ, Spencer JP & Rice-Evans C Flavonoids: antioxidants or signalling molecules? Free Radic. Biol. Med 36, 838–849 (2004). [DOI] [PubMed] [Google Scholar]
- 297.Zhu Y et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Ageing Cell 15, 428–435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Justice JN et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Darby IA, Laverdet B, Bonte F & Desmouliere A Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol 7, 301–311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Gascard P & Tlsty TD Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 30, 1002–1019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Akamatsu T et al. Direct isolation of myofibroblasts and fibroblasts from bleomycin-injured lungs reveals their functional similarities and differences. Fibrogenesis Tissue Repair 6, 15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hinz B, Celetta G, Tomasek JJ, Gabbiani G & Chaponnier C Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 12, 2730–2741 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02745145 (2019). [DOI] [PubMed]
- 304.Song KH, Cho SJ & Song JY Alphavbeta1 integrin as a novel therapeutic target for tissue fibrosis. Ann. Transl. Med 4, 411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Patsenker E et al. Pharmacological inhibition of integrin alphav beta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology 50, 1501–1511 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Horan GS et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med 177, 56–65 (2008). [DOI] [PubMed] [Google Scholar]
- 307.Wang B et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology 46, 1404–1412 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Peng ZW et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 63, 217–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Puthawala K et al. Inhibition of integrin alpha(v) beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am. J. Respir. Crit. Care Med 177, 82–90 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Hahm K et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am. J. Pathol 170, 110–125 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01371305 (2018). [DOI] [PubMed]
- 312.Fan GP et al. Pharmacological inhibition of focal adhesion kinase attenuates cardiac fibrosis in mice cardiac fibroblast and post-myocardial-infarction models. Cell Physiol. Biochem 37, 515–526 (2015). [DOI] [PubMed] [Google Scholar]
- 313.Zhao XK et al. Focal adhesion kinase regulates fibroblast migration via integrin beta-1 and plays a central role in fibrosis. Sci. Rep 6, 19276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02758587 (2018). [DOI] [PubMed]
- 315.Qi XJ et al. Fasudil, an inhibitor of Rho-associated coiled-coil kinase, attenuates hyperoxia-induced pulmonary fibrosis in neonatal rats. Int. J. Clin. Exp. Pathol 8, 12140–12150 (2015). [PMC free article] [PubMed] [Google Scholar]
- 316.Jiang C et al. Fasudil, a Rho-kinase inhibitor, attenuates bleomycin-induced pulmonary fibrosis in mice. Int. J. Mol. Sci 13, 8293–8307 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Bei Y et al. RhoA/Rho-kinase activation promotes lung fibrosis in an animal model of systemic sclerosis. Exp. Lung Res 42, 44–55 (2016). [DOI] [PubMed] [Google Scholar]
- 318.Baba I, Egi Y, Utsumi H, Kakimoto T & Suzuki K Inhibitory effects of fasudil on renal interstitial fibrosis induced by unilateral ureteral obstruction. Mol. Med. Rep 12, 8010–8020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Nagatoya K et al. Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 61, 1684–1695 (2002). [DOI] [PubMed] [Google Scholar]
- 320.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00704665 (2008). [DOI] [PubMed]
- 321.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02688647 (2019). [DOI] [PubMed]
- 322.Sakai N et al. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 27, 1830–1846 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Haak AJ et al. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J. Pharmacol. Exp. Ther 349, 480–486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Liang M et al. Yap/Taz deletion in Gli+ cell-derived myofibroblasts attenuates fibrosis. J. Am. Soc. Nephrol 28, 3278–3290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02981082 (2019). [DOI] [PubMed]
- 326.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02874989 (2019). [DOI] [PubMed]
- 327.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03865927 (2019). [DOI] [PubMed]